 Dóra Tombácz1
Dóra Tombácz1 Zsolt Csabai1Attila Szűcs1Zsolt Balázs1Norbert Moldován1Donald Sharon2Michael Snyder2
Zsolt Csabai1Attila Szűcs1Zsolt Balázs1Norbert Moldován1Donald Sharon2Michael Snyder2 Zsolt Boldogkői1*
Zsolt Boldogkői1*- 1Department of Medical Biology, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 2Department of Genetics, School of Medicine, Stanford University, Stanford, CA, United States
In this study, we used the amplified isoform sequencing technique from Pacific Biosciences to characterize the poly(A)+ fraction of the lytic transcriptome of the herpes simplex virus type 1 (HSV-1). Our analysis detected 34 formerly unidentified protein-coding genes, 10 non-coding RNAs, as well as 17 polycistronic and complex transcripts. This work also led us to identify many transcript isoforms, including 13 splice and 68 transcript end variants, as well as several transcript overlaps. Additionally, we determined previously unascertained transcriptional start and polyadenylation sites. We analyzed the transcriptional activity from the complementary DNA strand in five convergent HSV gene pairs with quantitative RT-PCR and detected antisense RNAs in each gene. This part of the study revealed an inverse correlation between the expressions of convergent partners. Our work adds new insights for understanding the complexity of the pervasive transcriptional overlaps by suggesting that there is a crosstalk between adjacent and distal genes through interaction between their transcription apparatuses. We also identified transcripts overlapping the HSV replication origins, which may indicate an interplay between the transcription and replication machineries. The relative abundance of HSV-1 transcripts has also been established by using a novel method based on the calculation of sequencing reads for the analysis.
Introduction
Herpes simplex virus type 1 (HSV-1) is a human pathogenic alphaherpesvirus from the Herpesviridae family. Herpes is a lifelong infection, which often has mild or no symptoms. The most common symptoms of viral infection are cold sores. HSV-1 can cause acute encephalitis in immunocompromised patients. According to WHO's first global estimates, worldwide more than 3.7 billion people under the age of fifty are infected with HSV-1 (Looker et al., 2015). The HSV-1 genome is composed of a unique long (UL) and a unique short (US) region, both being bracketed by inverted repeats (IRLs and IRSs, respectively). According to earlier annotations, the HSV-1 DNA contains 89 protein-coding, 10 long non-coding (lnc)RNA genes (Rajčáni et al., 2004; McGeoch et al., 2006; Macdonald et al., 2012; Lim, 2013; Hu et al., 2016) and several micro RNAs (Du et al., 2015).
Herpesvirus genes are expressed in a coordinated temporal cascade and grouped into three kinetic classes, immediate-early (IE), early (E), and late (L) (Harkness et al., 2014). The IE proteins are required for the transcription of both E and L genes. The E genes typically encode proteins that play a role in DNA replication, while the L genes specify the structural components of the virus. Kinetic analysis of the HSV transcriptome faces a significant challenge due to the overlapping nature of the viral genes. The typical architecture of polycistronic units is characterized by varying transcription start sites (TSSs) that are caused by the control of distinct promoters, and shared transcription end sites (TESs). As an example, the following transcripts are produced from a tetracistronic unit: 1-2-3-4, 2-3-4, 3-4, and 4, where “1” represents the most upstream gene, while “4” is the most downstream gene within the given unit. In a recent study on the transcriptome of another alphaherpesvirus, the pseudorabies virus (PRV), we demonstrated that additional gene combinations are also produced mostly in the form of low-abundance RNA molecules (Tombácz et al., 2016). The downstream genes on the polycistronic transcription units are generally thought to be untranslated because the eukaryotic RNAs—with some exceptions (Craigen and Caskey, 1987)—use cap-dependent ribosome-binding sites at their 5′-ends (Merrick, 2004).
It has been demonstrated that a large part of the mammalian genome is transcribed, producing a large variety of non-coding (nc)RNA molecules (Bertone et al., 2004). There is a current debate on whether the genome-wide expression of ncRNAs merely represents transcriptional noise, or whether these molecules might have yet undisclosed functions (Mattick, 2004). The most abundant class of the non-coding transcripts are the lncRNAs (Mattick and Makunin, 2006), which are defined as RNA molecules that exceed 200 nucleotides. A large number of lncRNAs have recently been described in mice and humans (Wilusz et al., 2009). Several genomic regions containing protein-coding genes also encode antisense lncRNAs from the complementary DNA strand. The HSV LAT was described as the first viral lncRNA (Stroop et al., 1984). In a recent study, we have shown that both DNA strands along the entire PRV genome are transcriptionally active (Tombácz et al., 2016). Some evidence suggests that the extensive transcriptional read-throughs produce very long RNA molecules including some containing oppositely oriented ORFs, which have been classified as complex transcripts (Tombácz et al., 2016).
Alternative splicing expands the coding capacity of metazoan genomes by producing multiple messages from a single gene. The splice variants can have similar or antagonistic functions (Boise et al., 1993). Previously, only five HSV-1 genes had been reported to produce spliced transcripts (Rajčáni et al., 2004; Sedlackova et al., 2008).
Massively parallel DNA sequencing technologies have become useful tools for the analysis of the transcriptomes. However, the conventionally applied short-read platforms cannot reliably distinguish between transcript isoforms and overlapping RNA molecules. Currently, the most commonly used long-read technology is the single-molecule real-time (SMRT) sequencing platform developed by Pacific Biosciences (PacBio). This approach is capable of reading cDNAs generated from full-length transcripts in a single sequencing run permitting mapping of the transcript ends with base-pair precision.
Various methods have already been used for the analysis of the herpesvirus transcriptome including microarrays (Stingley et al., 2000), Illumina sequencing (Harkness et al., 2014; Oláh et al., 2015), multi-time-point real-time reverse transcription PCR (qRT-PCR) analysis (Tombácz et al., 2009), and PacBio SMRT sequencing (O'Grady et al., 2016; Tombácz et al., 2016, 2017). Next-generation sequencing platforms have only been used for analyzing the transcriptional activity along the viral genome (Harkness et al., 2014). The PacBio long-read sequencing approach used for transcriptome profiling of PRV has proven to be the most effective platform for the identification of polycistronic RNA molecules, complex transcripts, transcript isoforms, and transcript overlaps. Our multiplatform-based approach has revealed a previously unexplored complexity of the PRV transcriptome (Tombácz et al., 2016).
In this study, we report the application of PacBio long-read sequencing technology for the characterization of the global lytic transcriptome of HSV-1. We used an amplified isoform sequencing (Iso-Seq) protocol that was based on PCR amplification of the cDNAs prior to sequencing. Besides the identification of novel transcripts and transcript isoforms, our intention was to map the TSSs and TESs of the viral RNAs with base pair precision. Long-read sequencing has proved to be efficient in the identification of overlapping and alternatively processed transcripts, as well as other long RNA sequences. Additionally, this method is particularly suitable for the sequencing of small genomes, GC-rich DNAs (HSV-1 has a 68% GC content) and with many repetitive sequences (Tombácz et al., 2014; Scott and Ely, 2015). The PacBio sequencing technique has an additional advantage over the short-read platforms in that if any random errors occurs in the raw reads, they are easily corrected thanks to the exceptionally high consensus accuracy of this platform (Miyamoto et al., 2014). In this report, we reevaluated the currently available datasets concerning the polyadenylated fraction of the HSV-1 transcripts generated in productively infected cells.
Materials and Methods
Cells and Viral Infection
An immortalized kidney epithelial cell line (Vero) isolated from African green monkey was used for the propagation of HSV-1 strain KOS. Vero cells were cultivated in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Gibco Invitrogen) and 100 μl penicillin-streptomycin 10K/10K mixture (Lonza)/ml and 5% CO2 at 37°C. For the preparation of viral stocks, rapidly-growing semi-confluent Vero cells were infected at a multiplicity of infection (MOI) of 1 plaque-forming unit (pfu)/cell, followed by incubation until a complete cytopathic effect was observed. The infected cells were then frozen and thawed three times, followed by centrifugation at 10,000 × g for 15 min. The titer of the virus stock was determined in Vero cells. For the sequencing analysis cells were infected with MOI = 1, incubated for 1 h, followed by removal of the virus suspension and washing with PBS. This was followed by the addition of fresh culture medium to the cells, which were incubated for 1, 2, 4, 6, 8, or 12 h. Samples taken from each experiment were then mixed for the sequencing analysis.
Pacbio RS II Sequencing—The Amplified Iso-Seq Protocol
Synthesis of cDNAs and Preparation of SMRTbell Template
Polyadenylated RNAs were isolated from total RNA by using the Oligotex mRNA Mini Kit (Qiagen) according to the manufacturer's instructions. The cDNA libraries were prepared using anchored oligo-dT primers for the reverse transcription. The cDNA production and the SMRTbell library preparation were performed with the Iso-Seq protocol of PacBio sequencing using the Clontech SMARTer PCR cDNA Synthesis Kit. We carried out either no Size Selection for the analysis of short viral transcripts, or performed SageELF™ and BluePippin™ Size-Selection Systems (Sage Science) for the isolation of long RNA molecules. The SMARTer PCR cDNA Synthesis Kit (Clontech) was used for the generation of the first-strand cDNAs. Subsequently, the single-stranded cDNAs were PCR-amplified using KAPA HiFi Enzyme (Kapa Biosystems) following PacBio's recommendations, which were as follows: initial denaturation was carried at −95°C for 2 min, followed by 16 cycles (the optimal cycle was determined in the optimization step) at −98°C for 20 s (denaturation), −65°C for 15 s (annealing) −74°C for 4 min (extension). The final extension was carried out at −72°C for 5 min. From the non-size selected samples 500 ng of each cDNA sample was applied for the SMRTbell template preparation, using the PacBio DNA Template Prep Kit 2.0. Subsequently, PCR products were pooled then size-selected with the SageELF™ System according to the PacBio's protocol. Size-selected samples were amplified with KAPA enzyme using the conditions as above. The fraction of cDNAs with a size over 5 kb was run on BluePippin™ System to remove the short SMRTbell templates.
PacBio Sequencing
The purified SMRTbell templates were bound to v2 sequencing primers and polymerases by using the DNA/Polymerase Binding Kit P6 (P/N 100-356-300). The conditions for annealing of the primers and binding of polymerase were determined using the PacBio's Binding Calculator in RS Remote. The polymerase-template complexes were bound to magbeads using the PacBio MagBead Binding Kit. The samples were analyzed on an Agilent 2100 bioanalyzer. Sequencing was carried out by using the PacBio RS II sequencer with DNA Sequencing Reagent 4.0 (P/N 100-356-200). The length of each movie was 360 min. We applied the PacBio Iso-Seq protocol (SMRT Analysis version v2.3.0.; Chaisson and Tesler, 2012) for the analysis of the HSV-1 transcriptome.
PCR Analysis
The novel HSV transcripts identified by PacBio sequencing were validated by PCR analysis. SuperScript III reverse transcriptase (Life Technologies) was used to convert the RNA samples to single-stranded cDNAs according to the manufacturer's instructions. The cDNAs were amplified with the Veriti Thermal Cycler (Applied Biosystems), using AccuPrime™ GC-Rich DNA Polymerase (Invitrogen). The PCR reactions ran for 3 min at 95°C (denaturation), followed by 30 cycles at 92°C for 30 s, at 60°C for 30 s (annealing), and at 72°C for 10 s (extension). Finally, a 10 min elongation step ran at 72°C. The primers used in this study are listed in Table S1.
Strand-Specific Real-Time RT-PCR
The relative amounts of transcripts were calculated by real-time reverse transcription (RT)-PCR. Three parallel RT reactions were carried out using 70 ng of total RNA as template, Superscript III enzyme (Life Technologies) and anchored oligo(dT) primers. In order to control for potential DNA contamination, No RT reactions were carried out for each sample by omitting the RT enzyme. The PCR reactions were carried out in a volume of 20 μl with Absolute QPCR SYBR Green Mix (Thermo Scientific) containing 7 μl of cDNA solution diluted 10-fold, 1.5 μl of forward and 1.5 μl of reverse primers (10 μM each). The normalized relative expression ratios (Rx) were calculated by the following formula:
, where E is the amplification efficiency, Ct is the threshold cycle number. The mean expression value of all examined transcripts (total) in a given sample was used as a normalization factor for the transcripts (sample) to obtain Rx values. This method is similar to that used by Mestdagh et al. (2009). However, instead of using the Ct values alone, we used ECt for the calculation of the expression values. The Comparative Quantitation module of the Rotor-Gene Q software v2.3.1 (Qiagen) was used for the determination of Ct values. The efficiency of the reaction and the thresholds were automatically calculated by the software.
Transcript Quantitation Using PacBio Sequencing Reads
The following criteria were set to count the number of reads aligning to each transcript isoform: Reads of Inserts (ROIs) containing poly(A) tails were assigned to a transcript, if the poly(A) site (PAS) of the read was no longer than 10 nts from the PAS of the transcript (in either direction), and if the 5′ end of the read was no longer than 10 nts upstream or 10 nts downstream from the TSS of the transcript isoform. Reads with long trimmed ends (>100 nts), or with many deletions, insertions or mismatches (>5% of the alignment) were discarded. Only the reads that contained a single poly(A) tail were counted. A set of DNA ladders was sequenced to determine the read length preference of the sequencing method. Equimolar molecular-weight size markers (Lambda DNA StyI, Lambda DNA PstI and phiX174 DNA HaeIII, resulting in 50 discrete fragments, between 15 and 19.329 nts) were sequenced along with the viral sequences. The number of reads for each fragment length was determined and their distributions were used to normalize read counts of the HSV-1 transcripts. When assigning reads to transcript isoforms, each read was weighted by the probability of sequencing a read of that given length.
Prediction of cis-Regulatory Sequences of the HSV Genes
The poly(A) signals were predicted by Dragon PolyA Spotter (DPS) (Kalkatawi et al., 2012) using Artificial Neural Network classifier. We determined the common transcription factor binding sites (−100 nts from transcription start) for common binding sites including TATA-box, GC-Box etc., using JASPAR POLII database (Mathelier et al., 2016) and FIMO (Find Individual Motif Occurrences) software (Grant et al., 2011). The threshold was set to p < 0.001. The best hits are linked to the genes and are included in Tables S2, S3.
Results
Analysis of the HSV Transcriptome with PacBio Isoform Sequencing
The annotation of the herpesvirus genome has traditionally been carried out in silico by detection of ORFs and other cis-motifs. Many HSV-1 transcripts have been identified by Northern-blot analysis (Costa et al., 1984; Sedlackova et al., 2008), while the 5′ and 3′ termini of the transcripts have been determined by S1 nuclease mapping (McKnight, 1980; Rixon and Clements, 1982) or by primer extension (Perng et al., 2002; Naito et al., 2005) techniques. For the investigation of the HSV-1 lytic transcriptome, we employed a PacBio polyadenylation sequencing approach based on Iso-Seq template preparation protocol utilizing the switching mechanism at the 5′ end of the RNA template, which produces full-length cDNAs (Zhu et al., 2001). We mapped the ROIs for both the HSV-1 (X14112) and the host genome (NC_023642.1) using the GMAP (Wu and Watanabe, 2005) alignment tool with default parameters. Altogether, 50,166 ROIs were mapped for the viral genome with a mean length of 1,461 nts, representing 7.32 million bases; and 35,089 ROIs were mapped for the host genome with mean length of 1,262 nts, representing 4.43 million nucleotides. Size-selection was applied for the detection of transcripts exceeding 5 kbps. The criteria for accepting sequencing reads as existing transcripts were the presence of at least two independent ROIs which contained the same TSSs and TESs. Likewise, a novel splice site was accepted if the same site occurred in at least two independent ROIs. We ran 17 parallel sequencing reactions for providing independent sequencing reads. Additionally, in many cases, the same TSS, TES, or splice junction were also found in different transcripts detected within the same sequencing reaction, which further enhanced the number of independent ROIs. In most cases, even in low-abundance transcripts, far more than two independent ROIs contained the same transcript ends or splice junctions. Besides the poly(A) tails, a few sequencing reads were also produced from A-rich regions of the transcripts; these non-specific products were discarded from further analysis. A few ROIs possessed both poly(A) and poly(T) tails. This phenomenon was earlier described in human (Sharon et al., 2013) and PRV (Tombácz et al., 2016) transcriptomes that were sequenced using the PacBio technique. Altogether, we could predict TATA boxes for 77 HSV-1 transcripts (Table S2). The promoter regions of the other genes lack typical TATA boxes, but many of them contain GC- and/or CAAT boxes (Table S2).
Novel Putative Protein-Coding Genes
Our investigations revealed 34 novel putative protein-coding genes (Figure 1, Table 1, Table S4), which is a fairly large number in a well-characterized viral genome. Each new gene is embedded into already annotated protein-coding genes. The mRNAs from these 5′ truncated genes are generated by alternative transcription initiation from an intragenic promoter within the larger host gene. Embedded genes are well-known in the herpesviruses, and their number is increasing; for example, the detection of the novel ul36.5 gene by PacBio sequencing has recently been described (Tombácz et al., 2016). The first in-frame AUG triplets are supposed to correspond to the first amino acids of the putative protein molecules. With the exception of seven genes (ul5.5; ul15.5; ul21.5; ul24.5; ul27.2; ul41.5; and ul53.5), the remaining novel genes were expressed at markedly lower levels than the host genes.
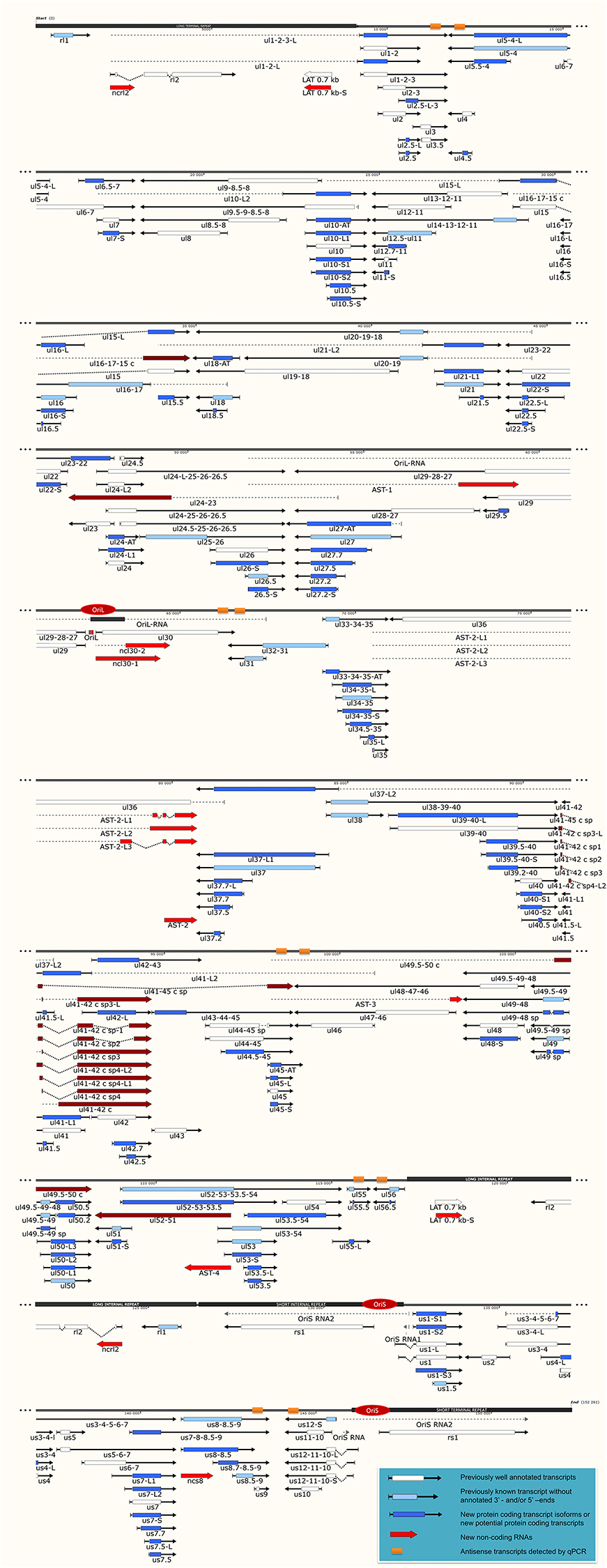
Figure 1. The transcriptome of the herpes simplex virus. The current version of the HSV-1 genome is composed of 115 protein-coding genes and 19 putative non-coding RNAs. The coding transcripts identified earlier are depicted as arrow-rectangles with white boxes (indicating the ORFs), the previously known transcripts (without annotated TSS and TES positions) are illustrated as light blue arrow-rectangles. The novel potential protein coding genes are labeled by dark blue rectangular arrows. The already identified lncRNAs are represented as dark-gray arrow lines, while the novel putative lncRNAs are depicted as red arrow lines. The novel polycistronic transcripts are indicated with dark blue rectangular arrows. The dark-red rectangular arrows show complex transcripts. The long black boxes represent the repeat regions of the HSV genome; red ovals represent the three replication origins. Abbreviations within the name of the transcripts: S, short TSS variant; L, long TSS variant; AT, alternative TES variant.
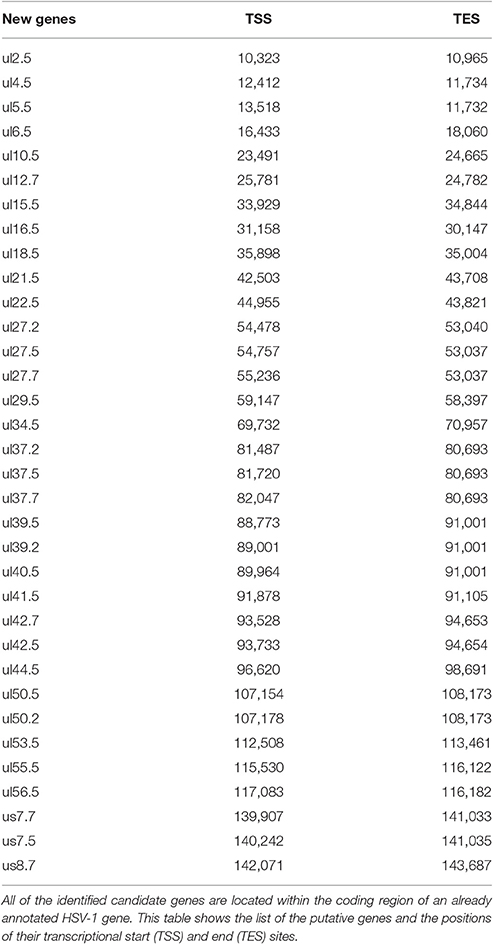
Table 1. Novel putative HSV genes.
Novel Non-coding Transcripts
In this part of our study, we identified 10 novel ncRNAs, including a LAT variant, four antisense (as) and four 3′ truncated transcripts, as well as a putative RNA molecule overlapping the replication origin (Ori-L) of the virus (Figure 1, Table 2). The novel 0.7 kb LAT-S transcript is a TSS isoform of the 0.7-kb LAT (Zhu et al., 1999). We detected polyadenylated asRNAs (termed antisense transcripts, AST) transcribed from the complementary DNA strands (the predicted TATA boxes are presented in Table S2). The existence of ASTs was verified by strand-specific PCR (Figure 2). We also detected lncRNAs, which were produced from protein-coding genes, but their transcriptions were prematurely terminated, and as a result lack stop codons. The potential function of these 3′ truncated RNA molecules remains obscure. The homolog of NCS8 transcript has been described in PRV (Tombácz et al., 2016). Furthermore, we identified a putative Ori-L-overlapping ncRNA with uncertain orientation, TSS and TES. We cannot exclude the possibility that it is not an individual RNA molecule but is rather the upstream region of a putative longer TSS isoform of the UL30 transcript. We did not detect HSV ncRNAs homologous to the PRV CTO-S transcript located in the close vicinity of the Ori-L of this virus (Tombácz et al., 2015). The probable reason for this is that the Ori-L is situated at different genomic loci in the two viruses. We could however detect the Ori-S-overlapping RNA described by Voss and Roizman (1988).
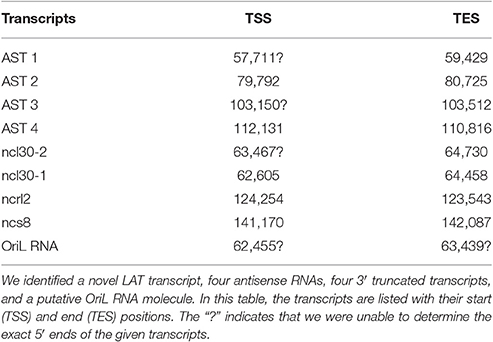
Table 2. Novel non-coding RNAs.
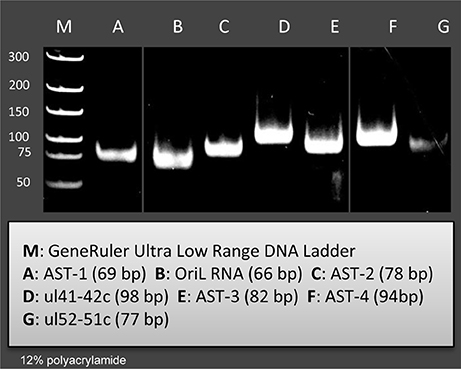
Figure 2. Validation and quantifying of antisense transcripts. Our PacBio analyses identified ten novel ncRNAs. We could confirm the expression of those transcripts with PCR analyses which were not embedded or overlapped with other transcripts. The following novel ncRNAs were detected by PCR (the sizes of PCR amplicons are in brackets): AST-1 (69 nts), (B2) OriL RNA (66 nts), AST-2 (78 nts), ul41-42c (98 nts), AST-3 (82 nts), AST-4 (94 nts), and ul51-52 c (77 nts).
Determination of the 5′ and 3′ Termini of the HSV Transcripts
In this report, we precisely mapped the 5′ and 3′ ends of the HSV transcripts (Table S3) and re-annotated those that had not been well-established earlier. Our amplified Iso-Seq technique is capable of identifying the full-length transcripts without any loss at the terminal sequences. We found that most of the TSSs were located between 28 and 33 nts downstream from the TATA box. Altogether, we were able to identify 46 novel TSSs and 6 TESs in already described transcripts, as well as 16 TSSs in the novel transcripts.
Transcription Start Site Isoforms
PacBio cDNA sequencing uncovered 42 protein-coding and non-coding transcripts with alternative TSSs (Figure 1, Table 3). We could detect putative TATA boxes for six TSS variants; however, TATA-less genes have been reported to be common in eukaryotic organisms (Yang et al., 2007). Similar to PRV, we detected multiple TSSs for the HSV UL10 transcripts, which indicate that the complex regulation of this gene is conserved among herpesviruses. We found that the ul22 and the us7 gene contain two active TATA sequences, and seven genes (ul2; ul6.5; ul12; ul22; ul51; us1; us7) have at least two potential TATA boxes. We identified two transcripts that overlap the Ori-Ss of HSV (Figure 1) at their 5′-UTRs; both are long TSS isoforms of us1 and us12 genes. In contrast to the PRV us1 gene that is located in the IRS, in the HSV only the promoter and a short 5′-UTR of this gene map to the repeat region. The Ori-S-overlapping US1 transcript is homologous to the PRV PTO-US1. Our analysis has revealed extensive minor variations in ~68% of the transcripts. These RNA molecules are controlled by the same promoter and vary in length from 1 to 10 nts at their 5′-ends (Table 3).
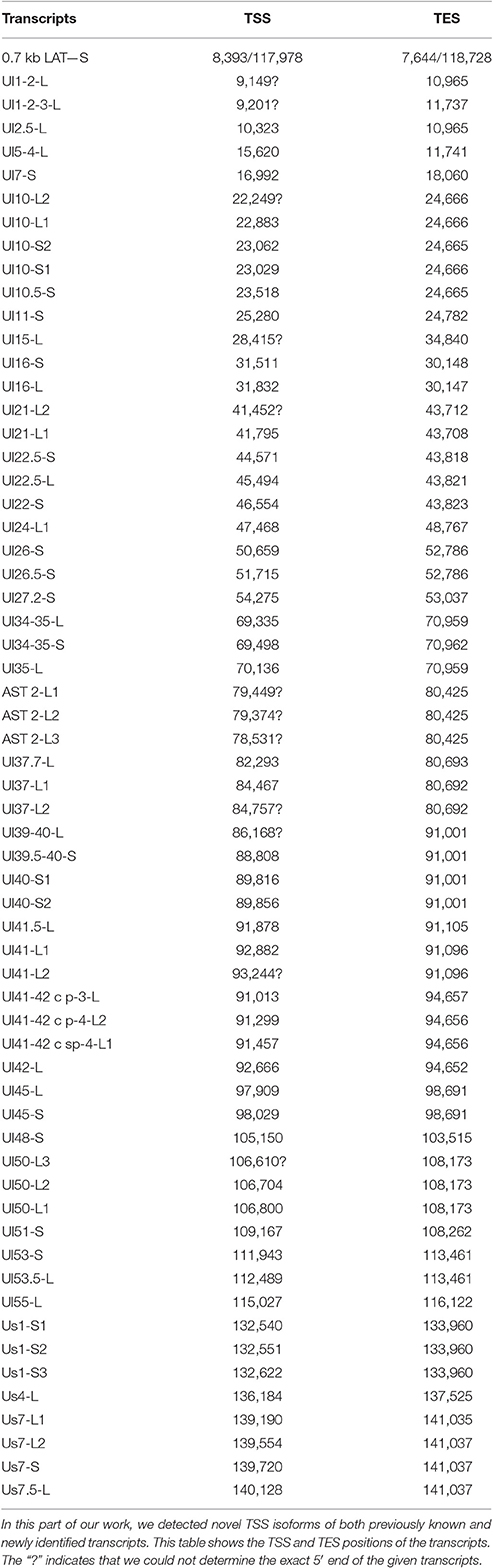
Table 3. Novel TSS isoforms.
Transcription End Site Isoforms
It has earlier been shown that many eukaryotic genes produce TES isoforms using alternative PA signals (Edwalds-Gilbert et al., 1997; Shen et al., 2008; Proudfoot, 2011). We detected six HSV transcripts, which each had two TES isoforms (Figure 1, Table 4). Except for three transcripts, we found that the rest of the abundant RNA molecules containing the same PA signal exhibited considerable polymorphism at their 3′-ends (Table 4).
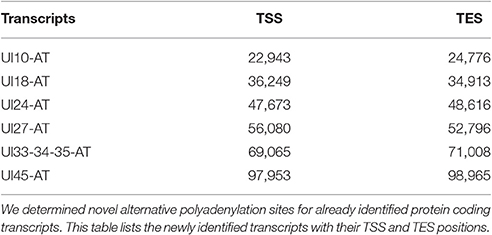
Table 4. Novel TES isoforms.
Splice Isoforms
In this report, we detected 13 novel spliced transcripts (Figures 1, 3, Table 5). The most intricate splicing pattern was found in the ul41-42 and ul41-45 complex transcripts (Figure 3). We identified two splice variants of the AST-2 antisense transcript. We also detected a new splice isoform of the ul49 gene, which was found to be expressed in a much higher level than the non-spliced variant. The UL49 and UL49-48 transcripts are the only ones in which the splicing occurs within an ORF, which is translated (in other cases the splicing takes place in either non-coding transcripts or in the downstream genes in polycistronic transcripts, which are non-translated). The splicing within the ul49 ORF results an in-frame deletion.

Figure 3. Splice isoforms. PCR validation of the splice sites of the UL41-42 complex transcript (A1), AST-2 transcript (B1), and the UL49 RNA (C1). (A2) Shows the coverage of reads across the examined genomic region, the ROIs (gray rectangles) and the schematic representation of the genes (red or black arrows). (B2) Illustrates the expanded view of ROIs, the read coverage along analyzed region and the schematic view of AST-2 and adjacent genes. (C2) Graph shows the read coverage, the ROIs and the schematic representation of the examined region. The numbers on the left side of panels A1, B1, and C1 indicate the sizes of DNA ladders while the numbers on the right side of these panels indicate the sizes of the PCR products amplified from the splice junction regions of the various splice isoforms (see Table 5). (A1) The 1,358-bp band shows the unspliced, while the 694- and the 388-bp bands show the two splice variants of the UL41-42 transcripts. (B1) The 1,566-bp indicates the unspliced, while the 436-bp band show the spliced version of AST-2 transcript. (C1) The 236-bp band shows the unspliced, while the 158-bp band shows the spliced isoform of UL49 mRNA. The colors indicate the poly(A) tails of the transcripts; green: from left to right orientation, red: from right to left orientation. The striped colors indicate the Clontech adaptor (universal primer binding site) used for the PCR amplification. They are labeled as such because they do not map to the PRV genome. The vertical red arrows indicate the precise location of the forward and reverse PCR primers on the HSV genome.
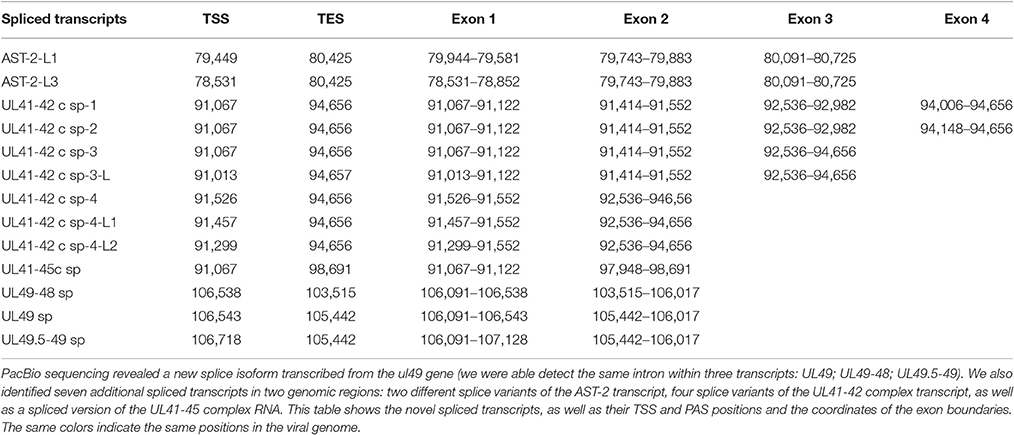
Table 5. Novel splice isoforms.
Novel Polycistronic Transcripts
According to the current concept, the HSV genome is organized in such a way that the downstream genes in a tandem gene cluster are transcribed either as monocistronic transcripts or as downstream genes of polycistronic (bi-, tri-, tetra-, or penta-cistronic) RNA molecules, whereas the upstream genes are expressed exclusively as parts of polycistronic transcripts. Our earlier investigations revealed that several upstream genes of 3′-coterminal gene clusters of the PRV are also transcribed as monocistronic RNA molecules (Tombácz et al., 2016). However, in this study, with the exception of the transcripts expressed from the embedded genes, we only detected novel polycistronic molecules (Table 6) that include transcripts terminated upstream of the co-terminal PA sites. Most of the newly discovered polycistronic RNA molecules are expressed at low levels, which explain why they had previously gone undetected.
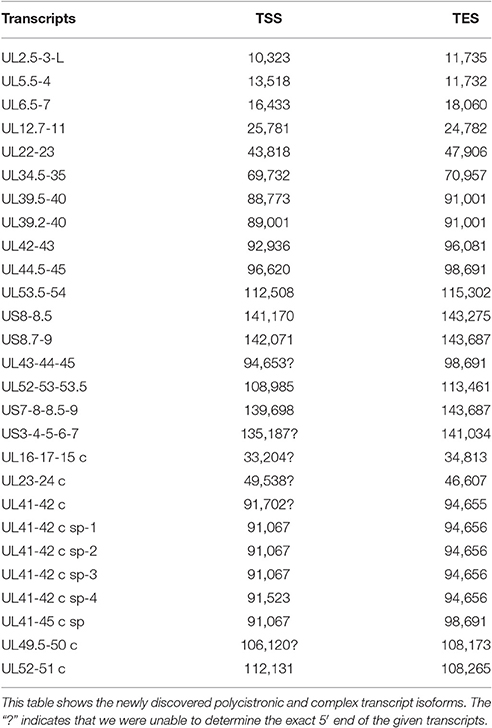
Table 6. Novel polycistronic and complex transcripts.
Complex Transcripts
Complex transcripts are defined as containing at least two genes with opposite orientations. We identified 10 full-length complex transcripts in HSV, seven of which had been generated by alternative splicing of the RNA molecule encoded by the genes within the ul41-45 region (Figure 1, Table 6). Earlier we had detected the homologs of the UL41/42 and the UL52/51 complex transcripts of HSV-1 also in PRV (Tombácz et al., 2016). Our investigations revealed a widespread expression of very long complex transcripts in PRV whose upstream sequences could not be determined even with the long-read PA-Seq technique. We detected more full-length but fewer partial complex transcripts in HSV than in PRV; the reason for the latter may be that in our previous report we also used random primer-based sequencing that allowed for the capture of more distal upstream sequences than the PA-Seq technique alone. The RNA molecules with partial sequences were illustrated as if they were controlled by the promoters of the closest upstream genes and were given ad hoc names accordingly. However, it is possible that they are shorter and initiated by yet unidentified promoters, or even longer and driven by more distal cis-regulatory sequences.
Transcriptional Overlaps between HSV Genes
In this part of the study, we investigated the transcriptional overlaps along the entire HSV genome.
Parallel Overlaps
Tandemly-oriented adjacent HSV genes can either partially or fully overlap one another. We detected 115 novel parallel full transcript overlaps, including the 34 5′ truncated genes, four 3′ truncated genes and 18 adjacent genes, as well as six partial tail-to-head overlaps between tandem genes. This latter kind of overlap is illustrated in Figure 4A, using ul42, ul43, and ul44 genes as examples. As a result of this analysis, we obtained that HSV contains fewer ORF-overlaps between the adjacent tandem genes than does PRV (7 vs. 8). However, there are many more embedded genes were detected in HSV.
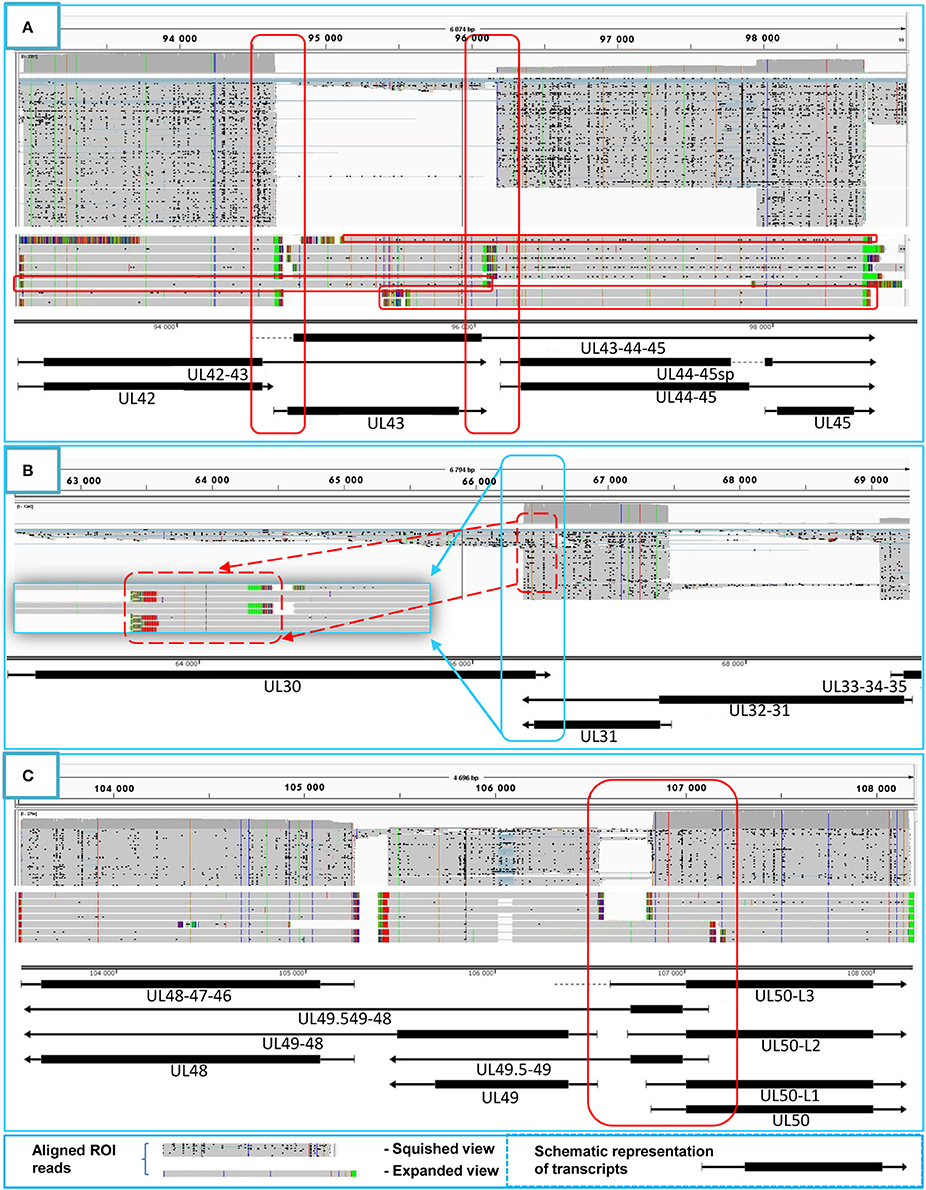
Figure 4. Transcriptional overlaps. Transcriptional overlaps between adjacent genes. (A) Represents the parallel transcriptional overlap exemplified by the ul42, ul43, and ul44 genes, which is a unique genomic region of HSV-1 in the sense that the transcripts from these genes have their own transcription termination sites and only rarely form polycistronic transcripts. (B) Illustrates convergent “hard” transcriptional overlaps using the ul30 and ul31 genes as an example. The TES sequences of these genes are located within the transcribed region of the partner gene. (C) Illustrates the divergent transcriptional overlaps with the example of ul49.5 and ul50 genes. It can be seen that all of the isoforms of the UL50 transcript exhibit a large extent of overlap with the UL49.5 RNA molecules. The panels of this figure show the ROIs (gray color; squished and expanded view) and the coverage of reads across the given genomic regions. The overlapping transcripts are shown at the bottom of the figures. The colors indicate the poly(A) tails of the transcripts: green: from left to right orientation, red: from right to left orientation. The striped colors indicate the Clontech adaptor used for the PCR amplification.
Convergent Overlaps
The tail-to-tail overlaps can be “hard” when all of the adjacent transcripts overlap each other or “soft” when both overlapping and non-overlapping transcripts are produced from the same gene. We detected the UL30/31-32, UL1-2-3/4-5, AST-1/UL37, and the AST-2/UL37 transcripts, which represent the only “hard” overlaps in HSV. The UL30/31-32 overlap is 46 nts longer than the earlier annotation suggests (McGeoch et al., 1988), and it is also present in PRV (Tombácz et al., 2016) (Figure 4B). Furthermore, very short (3–10 nts) intergenic regions were detected between the convergently-oriented gene pairs ul4/ul3.5, ul35-36, ul45-46, AST-3-ul48, AST-1-ul29, which potentially lead to interference between the RNA polymerase molecules when these simultaneously synthesize mRNAs on the neighboring genes, since transcription normally does not stop at the TES.
Divergent Overlaps
Earlier annotations revealed that three out of 13 divergently-oriented HSV gene pairs form head-to-head overlaps with each other (Figure 4C), including 5′-UTR/5′-UTR, the 5′-UTR/ORF, and ORF/ORF overlaps. PacBio analysis revealed alternative TSSs for several genes, which generate overlaps or much longer overlapping regions than published earlier (12 out of 13).
Detection of Antisense RNAs with Sequence-Specific qRT-PCR
In order to determine the existence of additional asRNAs, which are undetected by sequencing due to the potential lack of PA tails or being too long to be sequenced by the PacBio technique, five convergent HSV gene pairs were selected for the analysis of transcriptional activity of the complementary DNA strand by qRT-PCR. The asRNAs are supposed to be produced by transcriptional read-through from the convergent partner genes. We detected varying levels of asRNAs from each examined locus (Figure 5). The standard deviation (SD) values of the three parallel experiments are presented in Table S5. The pairwise comparison of qPCR curves shows that the relative amounts of asRNAs in 8 out of 10 genes are much lower than those of their mRNA partners, except in ul54 gene where the mRNA level only slightly exceeds that of the asRNA, and in the us10 gene where the sense-antisense partners are expressed at a similar rate. The reason for this latter phenomenon could be that the us10 gene is expressed in low level, and therefore even a weak antisense RNA expression can produce a comparable level of transcripts.
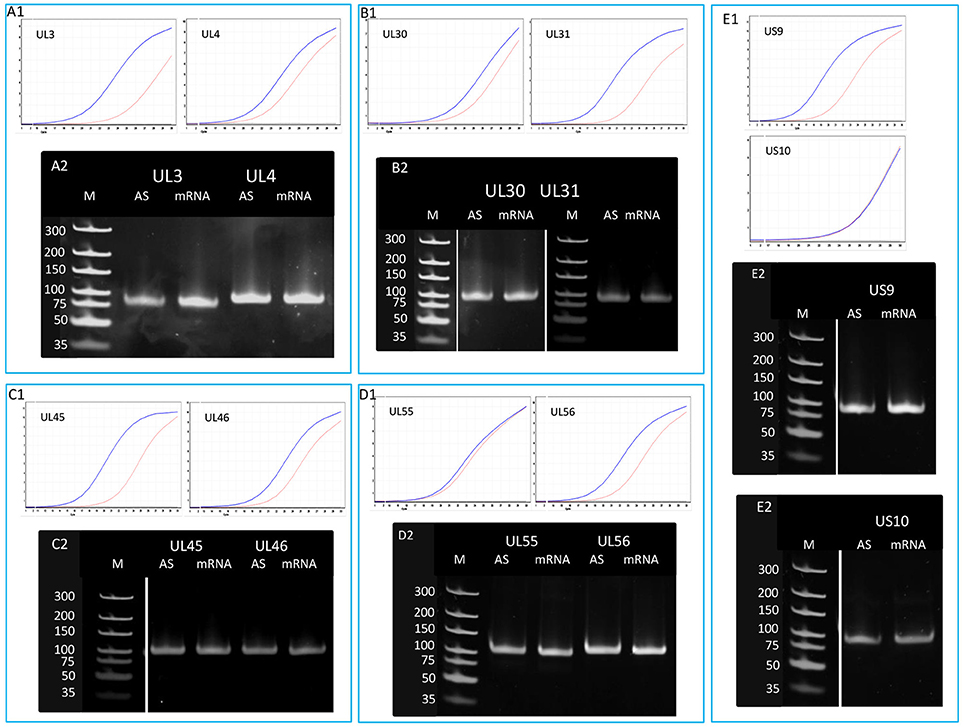
Figure 5. Detection of the antisense RNAs produced from the complementary DNA strands of 10 convergent HSV genes. A quantitative RT-PCR technique was used for the analysis of potential asRNAs derived from the transcriptional read-through between convergent genes. The amplification curves obtained by qRT-PCR are shown to demonstrate the specificity of the amplification. The curves of specific transcripts cross the threshold line within 18.3–22.5 cycles, while the curves of NO-RT controls remain flat. Panel (A1) shows the expression curves of the transcripts produced from plus and minus strands of the ul3 and ul4 convergent gene pair. Panel (A2) shows PCR products of the UL3 and UL4 transcripts and their antisense partners. Panel (B) shows the qRT-PCR amplification curves (B1) and the PCR products (B2) of UL30 and UL31 mRNAs and their asRNA partners. (C1) qRT-PCR curves of UL45 and UL46 mRNAs and the asRNAs produced from the complementary strands of these genes. (C2) PCR amplification products from the ul45-ul46 genomic region. (D1) Amplification curves of the UL55 transcript and its convergent partner UL56, as well as the antisense transcripts produced from the complementary DNA strands. (D2) Gel electrophoresis of the PCR products amplified from cDNAs UL55 and UL56 HSV mRNAs and their antisense partners. Panel (E) illustrates the qRT-PCR expression curves (E1) and the PCR products (E2) of US9-US10 mRNAs and their asRNAs. Gel samples were collected after saturation.
The Expression of Convergent Genes Exhibits an Inverse Pattern
In this part of the study, we observed the phenomenon that high transcriptional activity of a gene is associated with a low level of mRNAs from the convergent genes. We found that the higher the expression of a gene, the lower the expression of the convergent partner was (Figure 6A). Furthermore, we also observed that a gene with a high expression rate has a convergent partner with a low mRNA/asRNA ratio, and conversely, the partners of genes with low expression rates have higher values (Figure 6B).
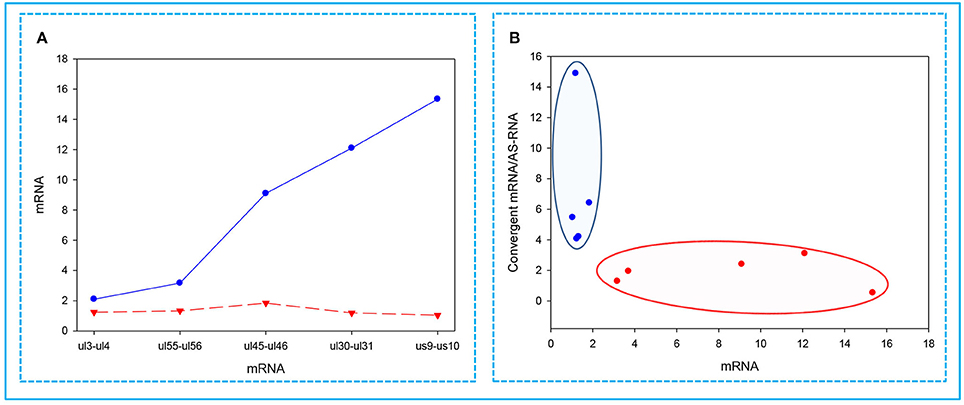
Figure 6. Correlations between the expressions of convergent HSV genes. The relative copy numbers (Rx values) were calculated for the transcripts encoded by five convergent gene pairs: ul3-ul4; ul30-ul31; ul45-ul46; ul55-ul56; us9-us10. (A) The less abundant mRNAs encoded by one of the convergent gene pairs are indicated by a red dashed line, while the more abundant partner mRNAs are depicted by a blue solid line. The figure shows that the higher the relative abundance of a transcript in the total PRV transcriptome, the lower the level of mRNA produced from the convergent gene. (B) Cluster analysis of the Rx values of the examined mRNAs compared to the ratio between the mRNA/antisense transcripts produced by the convergent gene partner. This figure shows that if a high mRNA levels are produced from a gene, then it exerts a decrease of mRNA/asRNA ratio for the convergent gene. The reason for this is that asRNAs are supposed to be produced as a result of transcriptional read-throughs from the oppositely-oriented genes. The blue dots illustrate low-abundance, while the red dots represent the high-abundance mRNAs.
Read Count Normalization by Spike-In DNA
It has been reported in our recent publication (Tombácz et al., 2017) and by others (O'Grady et al., 2016) that library preparation methods for PacBio sequencing enrich and sequence 1–2 kb-long fragments. Consequently, the long (>3 kb) or the short (<700 nts) transcript isoforms are underrepresented in the raw number of reads. To account for this bias, raw read counts were normalized by spike-in DNA (Figure 7). This analysis represents global gene expression along the HSV genome in a sample containing a mixture of RNA molecules collected from six different time points of viral infection. We also calculated the transcriptional activity throughout the HSV genome using the same method (Figure 8).
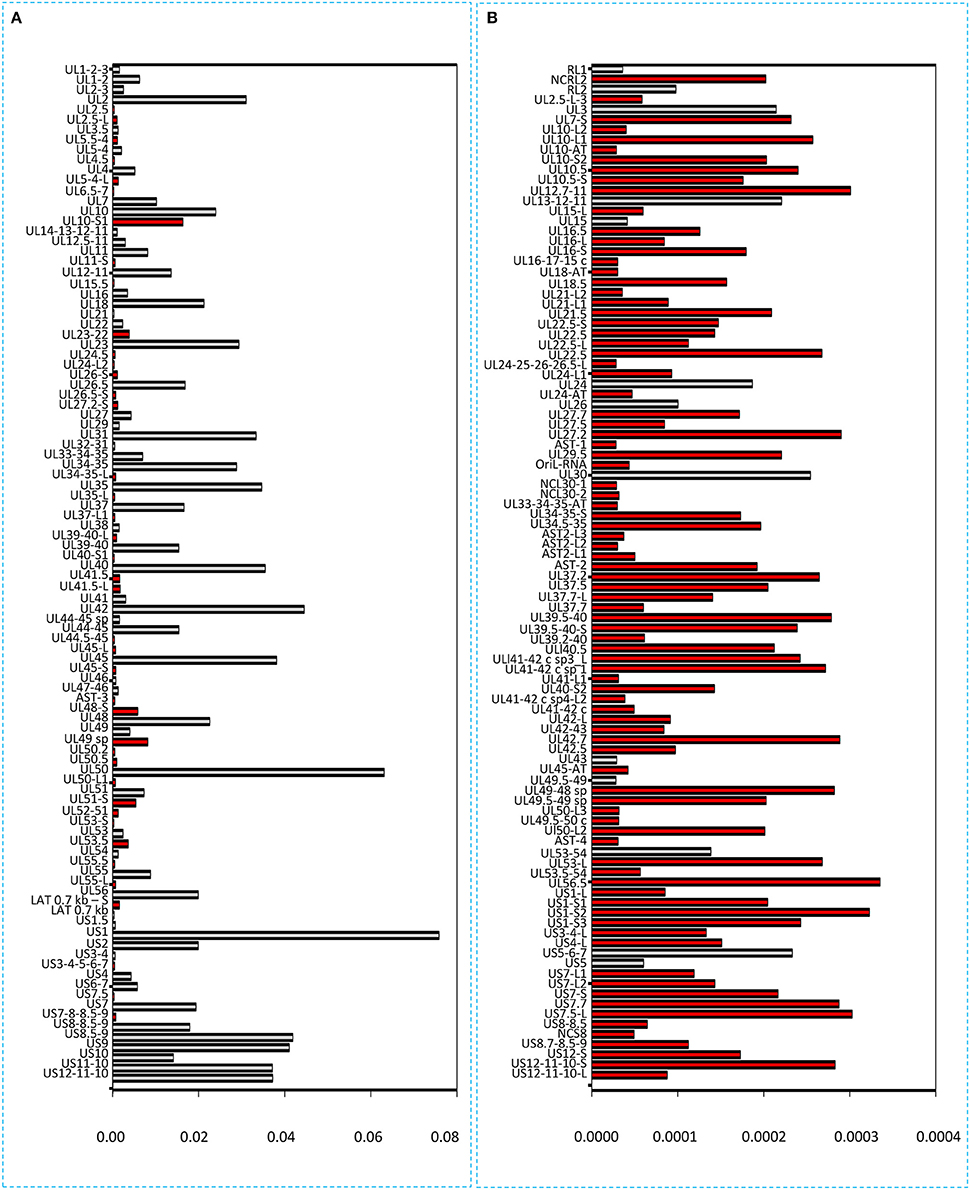
Figure 7. Bar chart representation of the HSV read counts using the marker-normalized values. (A) Horizontal bars represent the normalized read counts of the most abundant 100 HSV-1 transcripts. The novel transcripts are labeled by red. (B) This chart shows the less abundant 100 transcripts (101–200 in rank) of HSV. Red color indicates the newly identified transcripts. The scale (x-axis) of the two parts of the figure is different (200-fold difference). The transcripts are ranked on the basis of their genomic positions.
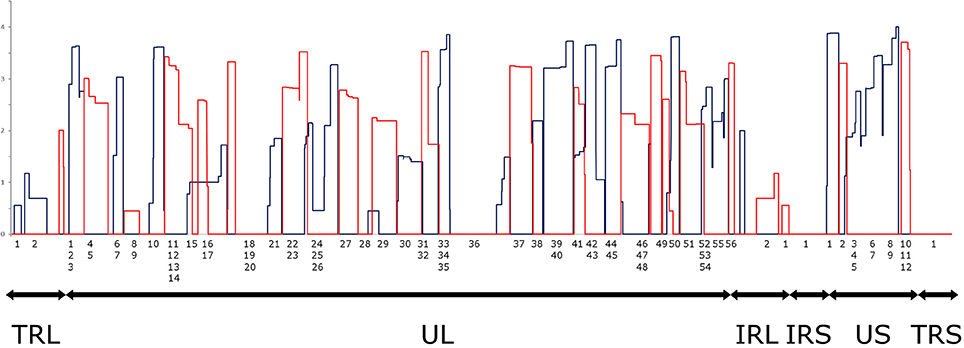
Figure 8. Transcriptional activity throughout the HSV genome. Transcriptional activity was calculated by normalizing the read counts to the read length distributions determined by equimolar molecular-weight size markers. The expression values of overlapping transcripts were summed for each nucleotide. The blue histogram shows the expression values on the plus strand, while the red histogram shows the expression values on the minus strand. All values are shown on a logarithmic scale. The genomic regions are marked under the histogram (TRL, Long Terminal Repeat; UL, Unique Long; IRL, Long Internal Repeat; IRS, Short Internal Repeat; US, Unique Short; TRS, Short Terminal Repeat). Note that an RNA population from several different time points p.i. was sequenced, where late genes are presumably overrepresented.
Discussion
Short-read sequencing has become a common technique for the annotation of transcriptome datasets (Mortazavi et al., 2008; Wang et al., 2009; Djebali et al., 2012). However, these methods are not optimal for resolving transcript structures, especially for the de novo characterization of complex transcript isoforms and long overlapping RNAs. In this study, a PCR-based long-read sequencing method was applied for the analysis of the HSV-1 transcriptome. Our investigations revealed a much higher level of transcriptomic complexity than has been captured by existing annotations. Here we provide a base-pair-precision annotation of already detected but not fully characterized RNA molecules. We have also succeeded in identifying 34 previously undescribed putative protein-coding transcripts, which were all transcribed from the ORF region of already annotated HSV-1 genes. We were able to identify in-frame AUG triplets for all genes in relative close position to the TSSs. Ten putative ncRNAs have also been detected. Four of these were 3′ truncated RNA molecules expressed from the promoter of protein-coding HSV-1 genes, but they were prematurely terminated from an internal PA signal. These transcripts lack stop-codons, and therefore we assume that they are lncRNAs. The homolog of HSV-1 NCS8 transcript has also been described in PRV in a recent report (Tombácz et al., 2016). This study identified many polycistronic and complex transcripts. Two complex transcripts of HSV show homology to previously described PRV transcripts (Tombácz et al., 2016), while the rest of them appear to be unique to HSV. We have also detected pervasive asRNA expression throughout the entire HSV genome with both cDNA sequencing and PCR. The antisense RNAs either have their own promoters, or they are transcribed from the promoter of neighbor or of distal genes as being transcriptional read-through products. The question as to whether the long transcripts are the result of transcriptional noise, if they contribute to the viral proteome, or if they fulfill other roles, remains to be ascertained. Another possibility is that these long transcripts are mere byproducts of a gene regulation mechanism that is based on interference between the transcriptionally active genes (Boldogköi, 2012).
No short ncRNAs homologous to CTO-S or PTO of PRV located near the replication origins were detected in HSV. However, we identified Ori-overlapping HSV transcripts, which were the long TSS variants of genes us1 and us12, as well as a putative short ncRNA overlapping Ori-L. The existence of these transcripts may indicate a crosstalk between the replication and transcription machinery.
Rutkowski and colleagues have recently investigated the translation of HSV-1 transcripts throughout the viral life cycle with a ribosome profiling technique using the RPKM (reads per kilobase per million mapped reads) model (Rutkowski et al., 2015). However, they mapped only the known HSV-1 transcripts, and provided no information from the translational start sites, and therefore we cannot check whether our novel transcripts (e.g., the 5′ truncated or the antisense RNAs) are translated or not.
PacBio long-read sequencing revealed the existence of many transcript isoforms including TSS, TES, and splice variants produced from the same gene. Additionally, as it has been detected in PRV (Tombácz et al., 2016), HSV transcripts also exhibit minor variations in both of their 5′ and 3′ termini.
Our investigations revealed an intricate meshwork of transcriptional read-throughs leading to overlapping RNA molecules. It turned out that HSV genes are transcribed in more combinations than it had been previously thought. The number of asRNAs and the complex transcripts of HSV are likely to be underestimated, because most of them may have been undetected due to their non-polyadenylated nature or because they are too long to be identifiable with even a long-read platform. This hypothesis was tested by examining the transcriptional activity of 10 HSV genes by qRT-PCR, which detected asRNAs in each case, suggesting that the entire HSV genome expresses long overlapping transcripts.
The analysis of antisense transcripts and their mRNA partners has disclosed the interconnected regulation of transcription (Katayama et al., 2005), and in many cases reciprocal patterns of gene expression (Lehner et al., 2002). Additionally, asRNAs have been shown to inhibit the transcription of complementary mRNA in human cells (Izant and Weintraub, 1985). In this work, we show that high mRNA expression from a gene was associated with low mRNA levels produced from the convergent partner. Additionally, high expression rates from genes were also associated with a low mRNA per asRNA ratio for convergent genes. We explain this phenomenon by transcriptional read-throughs, which are believed to result in the collision of the transcriptional machineries. The extensive transcriptional read-throughs and inverse correlations between the expression of convergent genes provide further support to our TIN hypothesis, which claims that genes interact with each other at the level of transcription throughout the entire genome (Boldogköi, 2012).
Finally, in this study, we developed a method for the quantification of RNA molecules based on calculations using ROI reads, which differs from the approach that we published earlier (Tombácz et al., 2017) in two important aspects: first, instead of the non-amplified SMRT method, here we used an amplified Iso-Seq technique, and second, we also sequenced spike-in DNAs for normalizing the size-dependent loading of cDNA molecules into ZMWs of the PacBio SMRT-cells, which allowed for comparison of the relative amounts of transcripts of different lengths.
In summary, our investigations essentially redefine the transcriptome of HSV-1. We demonstrated that HSV-1 exhibits a far higher transcriptional complexity than predicted from in silico ORF-based genome annotations or than those detected by techniques that map the termini of the transcripts. Most of the novel RNA molecules are produced at low levels which hinders their detection by gel-based assays or, because of their overlapping nature, even by PCR. Additionally, our analysis significantly increased the number of already known transcriptional read-throughs between the genes.
Accession Number
Raw sequencing files, processed data files as well as metadata have been submitted to the NCBI GEO repository and can be found with GenBank accession number GSE97785.
Author Contributions
ZBo, DT, and MS conceived and designed the study. DT, ZC, DS, and ZBo performed the experiments. DT, ZBo, AS, NM, ZBa, and DS analyzed the data. ZBo and DT wrote the manuscript. ZBo coordinated the project. All authors took part in the revision, read and approved the final manuscript. Funding acquisition: MS, ZBo, DT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Marianna Ábrahám (University of Szeged) for technical assistance, as well as to Kristin Mars (Pacific Biosciences) for helping to carry out the sequencing work. This study was supported by the European Union and the Hungarian State, co-financed by the European Social Fund in the framework of TÁMOP [4.2.2/B-10/1-2010-0012] and Swiss-Hungarian Cooperation Programme [SH/7/2/8] to ZBo. The work was also supported by the Bolyai János Scholarship of the Hungarian Academy of Sciences to DT and by the Centers of Excellence in Genomic Science—National Institutes of Health [5P50HG00773502] to MS.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01079/full#supplementary-material
Table S1. The list of the primers used in this study.
Table S2. The list of the predicted promoter elements.
Table S3. The list of the predicted poly(A) signals.
Table S4. The of the identified HSV-1 genes.
Table S5. The SD values of the parallel experiments used in the qRT-PCR analysis.
References
Bertone, P., Stolc, V., Royce, T. E., Rozowsky, J. S., Urban, A. E., Zhu, X., et al. (2004). Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242–2246. doi: 10.1126/science.1103388
Boise, L. H., González-García, M., Postema, C. E., Ding, L., Lindsten, T., Turka, L. A., et al. (1993). Bcl-X, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608. doi: 10.1016/0092-8674(93)90508-N
Boldogköi, Z. (2012). Transcriptional interference networks coordinate the expression of functionally related genes clustered in the same genomic loci. Front. Genet. 3:122. doi: 10.3389/fgene.2012.00122
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238
Costa, R. H., Cohen, G., Eisenberg, R., Long, D., and Wagner, E. (1984). Direct demonstration that the abundant 6-kilobase herpes simplex virus type 1 mRNA mapping between 0.23 and 0.27 map units encodes the major capsid protein VP5. J. Virol. 49, 287–292.
Craigen, W. J., and Caskey, C. T. (1987). Translational frameshifting: where will it stop? Cell 50, 1–2. doi: 10.1016/0092-8674(87)90652-0
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Du, T., Han, Z., Zhou, G., Zhou, G., and Roizman, B. (2015). Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc. Natl. Acad. Sci. U.S.A. 112, E49–E55. doi: 10.1073/pnas.1422657112
Edwalds-Gilbert, G., Veraldi, K. L., and Milcarek, C. (1997). Alternative poly(A) site selection in complex transcription units: means to an end? Nucleic Acids Res. 25, 2547–2561. doi: 10.1093/nar/25.13.2547
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Harkness, J. M., Kader, M., and DeLuca, N. A. (2014). Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J. Virol. 88, 6847–6861. doi: 10.1128/JVI.00516-14
Hu, B., Huo, Y., Chen, G., Yang, L., Wu, D., and Zhou, J. (2016). Functional prediction of differentially expressed lncRNAs in HSV-1 infected human foreskin fibroblasts. Virol. J. 13:137. doi: 10.1186/s12985-016-0592-5
Izant, J. G., and Weintraub, H. (1985). Constitutive and conditional suppression of exogenous and endogenous genes by anti-sense RNA. Science 229, 345–352. doi: 10.1126/science.2990048
Kalkatawi, M., Rangkuti, F., Schramm, M., Jankovic, B. R., Kamau, A., Chowdhary, R., et al. (2012). Dragon PolyA Spotter: predictor of poly(A) motifs within human genomic DNA sequences. Bioinformatics 28, 127–129. doi: 10.1093/bioinformatics/btr602
Katayama, S., Tomaru, Y., Kasukawa, T., Waki, K., Nakanishi, M., Nakamura, M., et al. (2005). Antisense transcription in the mammalian transcriptome. Science 309, 1564–1566. doi: 10.1126/science.1112009
Lehner, B., Williams, G., Campbell, R. D., and Sanderson, C. M. (2002). Antisense transcripts in the human genome. Trends Genet. 18, 63–65. doi: 10.1016/S0168-9525(02)02598-2
Lim, F. (2013). HSV-1 as a model for emerging gene delivery vehicles. ISRN Virol. 2013, 1–12. doi: 10.5402/2013/397243
Looker, K. J., Magaret, A. S., May, M. T., Turner, K. M., Vickerman, P., Gottlieb, S. L., et al. (2015). Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE 10:e0140765. doi: 10.1371/journal.pone.0140765
Macdonald, S. J., Mostafa, H. H., Morrison, L. A., and Davido, D. J. (2012). Genome sequence of herpes simplex virus 1 strain KOS. J. Virol. 86, 6371–6372. doi: 10.1128/JVI.00646-12
Mathelier, A., Fornes, O., Arenillas, D. J., Chen, C., Denay, G., Lee, J., et al. (2016). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115. doi: 10.1093/nar/gkv1176
Mattick, J. S. (2004). RNA regulation: a new genetics? Nat. Rev. Genet. 5, 316–323. doi: 10.1038/nrg1321
Mattick, J. S., and Makunin, I. V. (2006). Non-coding RNA. Hum. Mol. Genet. 15, R17–R29. doi: 10.1093/hmg/ddl046
McGeoch, D. J., Dalrymple, M. A., Davison, A. J., Dolan, A., Frame, M. C., McNab, D., et al. (1988). The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69, 1531–1574. doi: 10.1099/0022-1317-69-7-1531
McGeoch, D. J., Rixon, F. J., and Davison, A. J. (2006). Topics in herpesvirus genomics and evolution. Virus Res. 117, 90–104. doi: 10.1016/j.virusres.2006.01.002
McKnight, S. L. (1980). The nucleotide sequence and transcript map of the herpes simplex virus thymidine kinase gene. Nucleic Acids Res. 8, 5949–5964. doi: 10.1093/nar/8.24.5949
Merrick, W. C. (2004). Cap-dependent and cap-independent translation in eukaryotic systems. Gene 332, 1–11. doi: 10.1016/j.gene.2004.02.051
Mestdagh, P., Van Vlierberghe, P., De Weer, A., Muth, D., Westermann, F., Speleman, F., et al. (2009). A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 10:R64. doi: 10.1186/gb-2009-10-6-r64
Miyamoto, M., Motooka, D., Gotoh, K., Imai, T., Yoshitake, K., Goto, N., et al. (2014). Performance comparison of second- and third-generation sequencers using a bacterial genome with two chromosomes. BMC Genomics 15:699. doi: 10.1186/1471-2164-15-699
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Naito, J., Mukerjee, R., Mott, K. R., Kang, W., Osorio, N., Fraser, N. W., et al. (2005). Identification of a protein encoded in the herpes simplex virus type 1 latency associated transcript promoter region. Virus Res. 108, 101–110. doi: 10.1016/j.virusres.2004.08.011
O'Grady, T., Wang, X., Höner zu Bentrup, K., Baddoo, M., Concha, M., and Flemington, E. K. (2016). Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 44:e145. doi: 10.1093/nar/gkw629
Oláh, P., Tombácz, D., Póka, N., Csabai, Z., Prazsák, I., and Boldogkői, Z. (2015). Characterization of pseudorabies virus transcriptome by Illumina sequencing. BMC Microbiol. 15:130. doi: 10.1186/s12866-015-0470-0
Perng, G. C., Maguen, B., Jin, L., Mott, K. R., Kurylo, J., BenMohamed, L., et al. (2002). A novel herpes simplex virus type 1 transcript (AL-RNA) antisense to the 5′ end of the latency-associated transcript produces a protein in infected rabbits. J. Virol. 76, 8003–8010. doi: 10.1128/JVI.76.16.8003-8010.2002
Proudfoot, N. J. (2011). Ending the message: poly(A) signals then and now. Genes Dev. 25, 1770–1782. doi: 10.1101/gad.17268411
Rajčáni, J., Andrea, V., and Ingeborg, R. (2004). Peculiarities of Herpes Simplex Virus (HSV) transcription: an overview. Virus Genes 28, 293–310. doi: 10.1023/B:VIRU.0000025777.62826.92
Rixon, F. J., and Clements, J. B. (1982). Detailed structural analysis of two spliced HSV-1 immediate-early mRNAs. Nucleic Acids Res. 10, 2241–2256. doi: 10.1093/nar/10.7.2241
Rutkowski, A. J., Erhard, F., L'Hernault, A., Bonfert, T., Schilhabel, M., Crump, C., et al. (2015). Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 6:7126. doi: 10.1038/ncomms8126
Scott, D., and Ely, B. (2015). Comparison of genome sequencing technology and assembly methods for the analysis of a GC-rich bacterial genome. Curr. Microbiol. 70, 338–344. doi: 10.1007/s00284-014-0721-6
Sedlackova, L., Perkins, K. D., Lengyel, J., Strain, A. K., van Santen, V. L., and Rice, S. A. (2008). Herpes simplex virus type 1 ICP27 regulates expression of a variant, secreted form of glycoprotein C by an intron retention mechanism. J. Virol. 82, 7443–7455. doi: 10.1128/JVI.00388-08
Sharon, D., Tilgner, H., Grubert, F., and Snyder, M. (2013). A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014. doi: 10.1038/nbt.2705
Shen, Y., Ji, G., Haas, B. J., Wu, X., Zheng, J., Reese, G. J., et al. (2008). Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 36, 3150–3161. doi: 10.1093/nar/gkn158
Stingley, S. W., Ramirez, J. J., Aguilar, S. A., Simmen, K., Sandri-Goldin, R. M., Ghazal, P., et al. (2000). Global analysis of herpes simplex virus type 1 transcription using an oligonucleotide-based DNA microarray. J. Virol. 74, 9916–9927. doi: 10.1128/JVI.74.21.9916-9927.2000
Stroop, W. G., Rock, D. L., and Fraser, N. W. (1984). Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization. Lab. Invest. 51, 27–38.
Tombácz, D., Balázs, Z., Csabai, Z., Moldován, N., Szűcs, A., Sharon, D., et al. (2017). Characterization of the dynamic transcriptome of a herpesvirus with long-read single molecule real-time sequencing. Sci. Rep. 7:43751. doi: 10.1038/srep43751
Tombácz, D., Csabai, Z., Oláh, P., Balázs, Z., Likó, I., Zsigmond, L., et al. (2016). Full-length isoform sequencing reveals novel transcripts and substantial transcriptional overlaps in a herpesvirus. PLoS ONE 11:e0162868. doi: 10.1371/journal.pone.0162868
Tombácz, D., Csabai, Z., Oláh, P., Havelda, Z., Sharon, D., Snyder, M., et al. (2015). Characterization of novel transcripts in pseudorabies virus. Viruses 7, 2727–2744. doi: 10.3390/v7052727
Tombácz, D., Sharon, D., Oláh, P., Csabai, Z., Snyder, M., and Boldogkői, Z. (2014). Strain kaplan of pseudorabies virus genome sequenced by pacbio single-molecule real-time sequencing technology. Genome Announc. 2, 14–15. doi: 10.1128/genomeA.00628-14
Tombácz, D., Tóth, J. S., Petrovszki, P., and Boldogkoi, Z. (2009). Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genomics 10:491. doi: 10.1186/1471-2164-10-491
Voss, J. H., and Roizman, B. (1988). Properties of two 5′-coterminal RNAs transcribed part way and across the S component origin of DNA synthesis of the herpes simplex virus 1 genome. Proc. Natl. Acad. Sci. U.S.A. 85, 8454–8458. doi: 10.1073/pnas.85.22.8454
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wilusz, J. E., Sunwoo, H., and Spector, D. L. (2009). Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 23, 1494–1504. doi: 10.1101/gad.1800909
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Yang, C., Bolotin, E., Jiang, T., Sladek, F. M., and Martinez, E. (2007). Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389, 52–65. doi: 10.1016/j.gene.2006.09.029
Zhu, J., Kang, W., Marquart, M. E., Hill, J. M., Zheng, X., Block, T. M., et al. (1999). Identification of a novel 0.7-kb polyadenylated transcript in the LAT promoter region of HSV-1 that is strain specific and may contribute to virulence. Virology 265, 296–307. doi: 10.1006/viro.1999.0057
Keywords: human herpesvirus 1, herpes simplex virus type 1, transcriptome, transcriptional interference, full-length sequencing, long-read sequencing, Pacific biosciences, transcriptional overlap
Citation: Tombácz D, Csabai Z, Szűcs A, Balázs Z, Moldován N, Sharon D, Snyder M and Boldogkői Z (2017) Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol. 8:1079. doi: 10.3389/fmicb.2017.01079
Received: 20 April 2017; Accepted: 29 May 2017;
Published: 20 June 2017.
Edited by:
Akio Adachi, Tokushima University, JapanReviewed by:
Takayuki Murata, Nagoya University, JapanAnu Susan Charles, Louisiana State University, United States
Copyright © 2017 Tombácz, Csabai, Szűcs, Balázs, Moldován, Sharon, Snyder and Boldogkői. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zsolt Boldogkői, Ym9sZG9na29pLnpzb2x0QG1lZC51LXN6ZWdlZC5odQ==