 Marie-Line Joffret1,2,3*†
Marie-Line Joffret1,2,3*† Patsy M. Polston1,2*†
Patsy M. Polston1,2*† Richter Razafindratsimandresy4
Richter Razafindratsimandresy4 Maël Bessaud1,2,3
Maël Bessaud1,2,3 Jean-Michel Heraud4
Jean-Michel Heraud4 Francis Delpeyroux1,2,3
Francis Delpeyroux1,2,3- 1Unité de Biologie des Virus Entériques, Institut Pasteur, Paris, France
- 2Institut National de la Santé et de la Recherche Médicale, Paris, France
- 3WHO Collaborating Center for Research on Enteroviruses and Viral Vaccines, Institut Pasteur, Paris, France
- 4Unité de Virologie, Institut Pasteur de Madagascar, Antananarivo, Madagascar
Human enteroviruses (EV) consist of more than 100 serotypes classified within four species for enteroviruses (EV-A to -D) and three species for rhinoviruses, which have been implicated in a variety of human illnesses. Being able to simultaneously amplify the whole genome and identify enteroviruses in samples is important for studying the viral diversity in different geographical regions and populations. It also provides knowledge about the evolution of these viruses. Therefore, we developed a rapid, sensitive method to detect and genetically classify all human enteroviruses in mixtures. Strains of EV-A (15), EV-B (40), EV-C (20), and EV-D (2) viruses were used in addition to 20 supernatants from RD cells infected with stool extracts or sewage concentrates. Two overlapping fragments were produced using a newly designed degenerated primer targeting the conserved CRE region for enteroviruses A-D and one degenerated primer set designed to specifically target the conserved region for each enterovirus species (EV-A to -D). This method was capable of sequencing the full genome for all viruses except two, for which nearly 90% of the genome was sequenced. This method also demonstrated the ability to discriminate, in both spiked and unspiked mixtures, the different enterovirus types present.
Introduction
The common human viruses, human enteroviruses (EV), consist of more than 100 serotypes most classified within four species for enteroviruses (EV-A to -D) and three species for rhinoviruses. Enteroviruses are ubiquitous and resilient in the environment and primarily transmitted fecal-orally. Human enteroviruses have been implicated in a variety of human diseases, including the common cold, hand foot and mouth disease, acute hemorrhagic conjunctivitis, myocarditis, encephalitis, and poliomyelitis.
Enteroviruses are non-enveloped viruses, approximately 7500 nucleotides (nt) in length with a positive, single-stranded RNA genome. There are two untranslated regions (5′ and 3′ -UTR) flanking a large open reading frame encoding a polyprotein that is cleaved to give three precursors P1 to P3. These precursors are subsequently cleaved to give functional proteins: (1) P1 giving rise to four structural capsid proteins (VP1–VP4) and (2) P2 and P3, the non-structural proteins involved in the virus life cycle.
Studies have been conducted to best determine the most accurate way to identify and classify enteroviruses, giving rise to some “gold standards” for detection and typing. Typically, enteroviruses have been isolated on a variety of cell lines (e.g., RD, GMK, Vero, CaCo-2, L20B, HEp2c, and HeLa) based on their ability to propagate and show cytopathic effect (CPE), followed by serotyping using neutralization assays (Silva et al., 2008; Poh and Tan, 2010; Blomqvist and Roivainen, 2016). Despite cell amplification being an appropriate method, it is laborious, time-consuming, and expensive (Nijhuis et al., 2002). It has been shown that there is a good correlation between the sequences of the VP1 nucleotidic and amino acid sequences and enterovirus serotypes (Oberste et al., 1999a; Caro et al., 2001). Thus, VP1 sequences have been used as a gold standard for typing and, if relevant, subtyping enteroviruses (Oberste et al., 1999a,b; Nix et al., 2006; Poh and Tan, 2010).
In addition to typing, studies dedicated to evaluating the enterovirus genomic diversity have shown that besides mutations, intra- and inter-typic genetic recombination is also a frequent mechanism of viral evolution (Lukashev et al., 2003; Simmonds, 2006; Combelas et al., 2011; Joffret et al., 2012). Enterovirus genomes frequently display mosaicism due to genetic exchanges among different enterovirus strains and types. Since this kind of genetic diversification can be at the source of genotypic and phenotypic diversity (Sadeuh-Mba et al., 2013; Bessaud et al., 2016) it is essential to determine the whole genomic sequences of enteroviruses for surveillance and public health purposes, as well as for basic research.
For obtaining sequencing data, the traditional Sanger method is capable of sequencing the whole genome but it is time-consuming and it cannot simultaneously sequence a mixture of viruses; thereby, making a large-scale surveillance project difficult to conduct due to the presence of many viruses. Furthermore, it makes it challenging to specifically target every known enterovirus and impossible to identify any unknown viruses.
Next generation sequencing (NGS) methods offer a new powerful sequencing tool for the identification and characterization of enteroviruses. This has been successfully used to sequence partial or whole genome sequences of poliovirus and of enteroviruses species C (Bessaud et al., 2016; Montmayeur et al., 2017; Sahoo et al., 2017). Additionally, a generic assay for whole genome amplification and deep sequencing of enterovirus A71 was published (Tan et al., 2015). Despite advances in molecular methods, none of these assays were designed to simultaneously amplify the whole genome for all four enterovirus species, and it is known that mixtures of enteroviruses can be found in human stool or sewage. Hence, the goal of this research was to design generic and species-specific primers in order to develop an assay capable of viral typing and sequencing the whole genome for all enteroviruses of species A to D present in samples containing viral mixtures.
Materials and Methods
Viruses
For this study 15-EVA, 40-EVB, 20-EVC, and 2-EVD viruses were used (Table 1) to test and analyze whole-genome sequencing. The prototype strains selected were from the European Virus Archive (EVAg)1. Additionally, we used 20 enterovirus positive supernatants from human rhabdomyosarcoma cells (RD cells) from the Madagascar National Polio Laboratory. These cells were infected with either 10 human stool extracts or 10 sewage concentrates collected during poliomyelitis surveillance in Madagascar and prepared according to WHO protocols (World Health Organization Polio Laboratory manual, 4th edition, 2004). Infections of RD cells with these extracts were followed by full cytopathogenic effect, resembling that induced by enteroviruses.
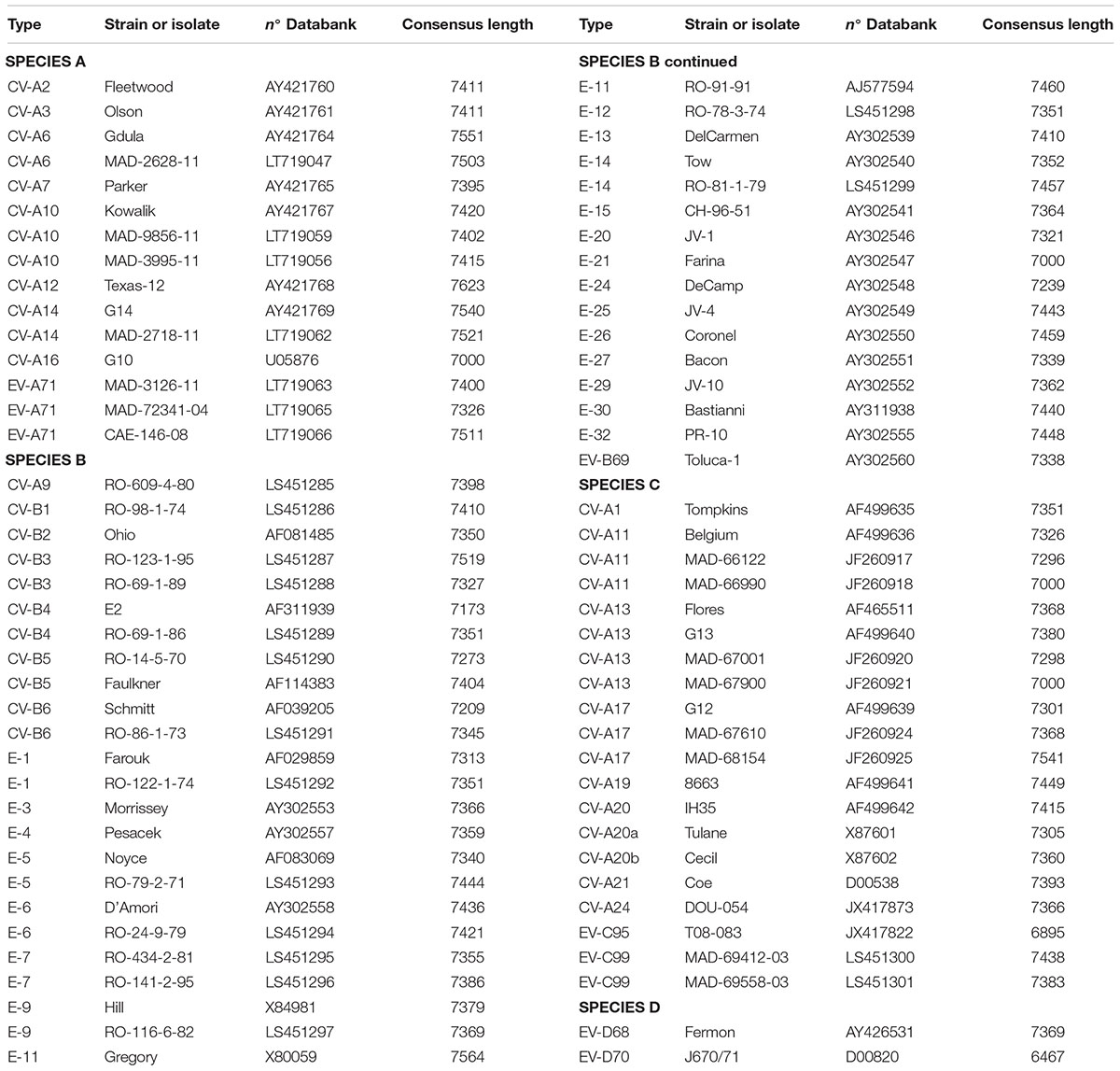
TABLE 1. Viruses used in this study.
For the collection of stool from Madagascar, the protocol was approved by the National Ethics Committee of the Ministry of Public Health in Madagascar (Agreement N° 22-MSANP/CE). Parental written informed consent was given.
Primer Design
For each enterovirus group, complete genome sequences were separately aligned using CLC Main Workbench. To achieve the two overlapping fragments, the first half of the genome was amplified using the previously described C004 primer (Bessaud et al., 2016) in combination with a newly designed degenerated primer targeting the conserved CRE (cis-acting replication element) region for enteroviruses A-D. The second half of the genome was amplified using one degenerated primer set designed to specifically target the conserved region for each enterovirus species. Therefore, we performed five PCR mixes for each isolate and we pooled the PCR products for sequencing. All primers used in this study are described in Table 2 and Figure 1 illustrates the primer locations.
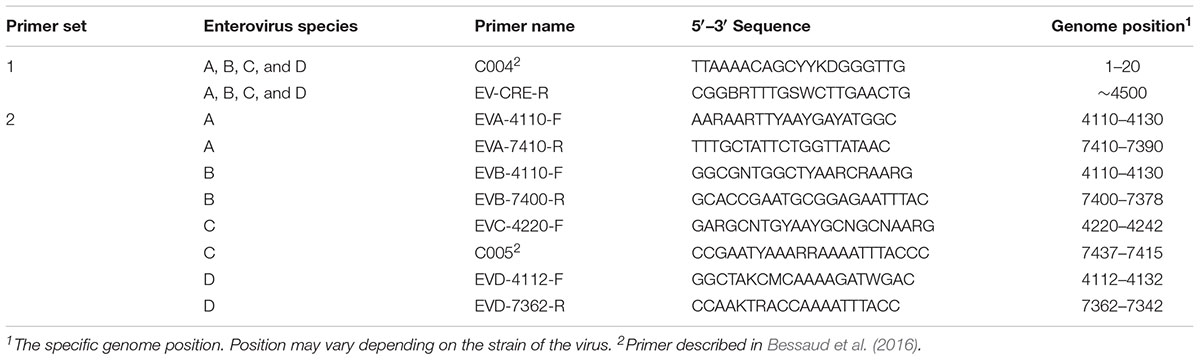
TABLE 2. Primers used for this study.
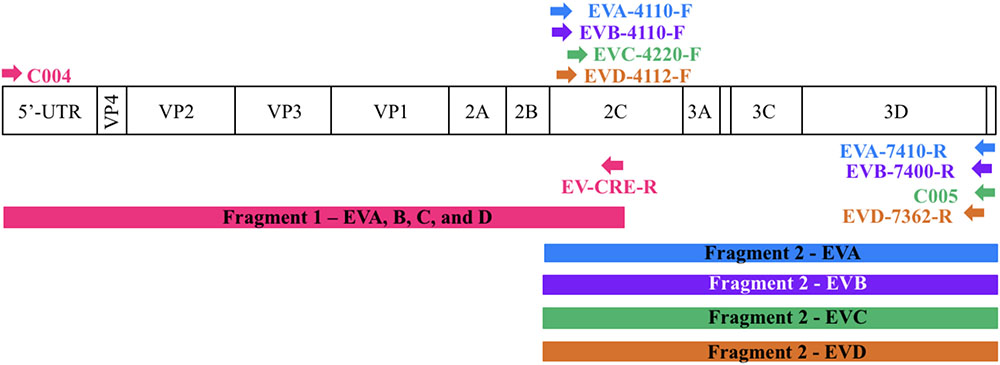
FIGURE 1. Location of primers used in this study. The diagram shows the organization of the enterovirus genome. Arrows indicate the sites targeted by the proposed primers. The RT-PCR products are colored coded to represent the corresponding primer pair. The first half of the genome (fragment 1) is amplified using one primer pair whereas the second half (fragment 2) is amplified using primers targeting each enterovirus species.
RNA Extraction
Viral RNA was extracted from supernatants of infected cells, using the High Pure Viral RNA kit (Roche Diagnostics, Meylan, France) per manufacturer’s protocol. All RNA was either immediately used for PCR amplification or stored at -80°C for further analysis.
One-Step RT-PCR Amplification
For entire length genome amplification, we used two primer sets (Table 2) per enterovirus group (A, B, C, and D). This allowed us to synthesize the two overlapping amplicons by using the One-Step RT-PCR kit (ref G174 Applied Biological Materials Inc.). The reaction mixture contained 1.5 μl of purified RNA, 12.5 μl of 2× One-step RT-PCR buffer, 1 μl of each forward and reverse primer (20 μM), 0.5 μl of EasyScript RTase (200 U/μl), 1 μl of Bestaq DNA Polymerase (5 U/μl), and 9 μl of DNAse free water. PCR amplification was performed using a thermocycler with the following protocol: 42°C for 30 min, 94°C for 3 min, 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 4 min 30 s, and a last step at 72°C for 10 min, ending with 2 min at 4°C. All PCR products were visualized on ethidium bromide-stained agarose gels to ensure appropriate size products.
The sensitivity of this assay was evaluated using serial fourfold dilutions of four viruses (EV-A71 strain MAD-72341, CV-B4 strain E2, CV-A13 strain MAD 67001, and EV-D 68 strain Fermon) representing each species (Table 3). To maintain similar amounts of cellular nucleic acids throughout dilutions, viral stocks were diluted using a supernatant of confluent non-infected HEp-2c cell monolayers frozen and thawed twice and clarified by centrifugation.
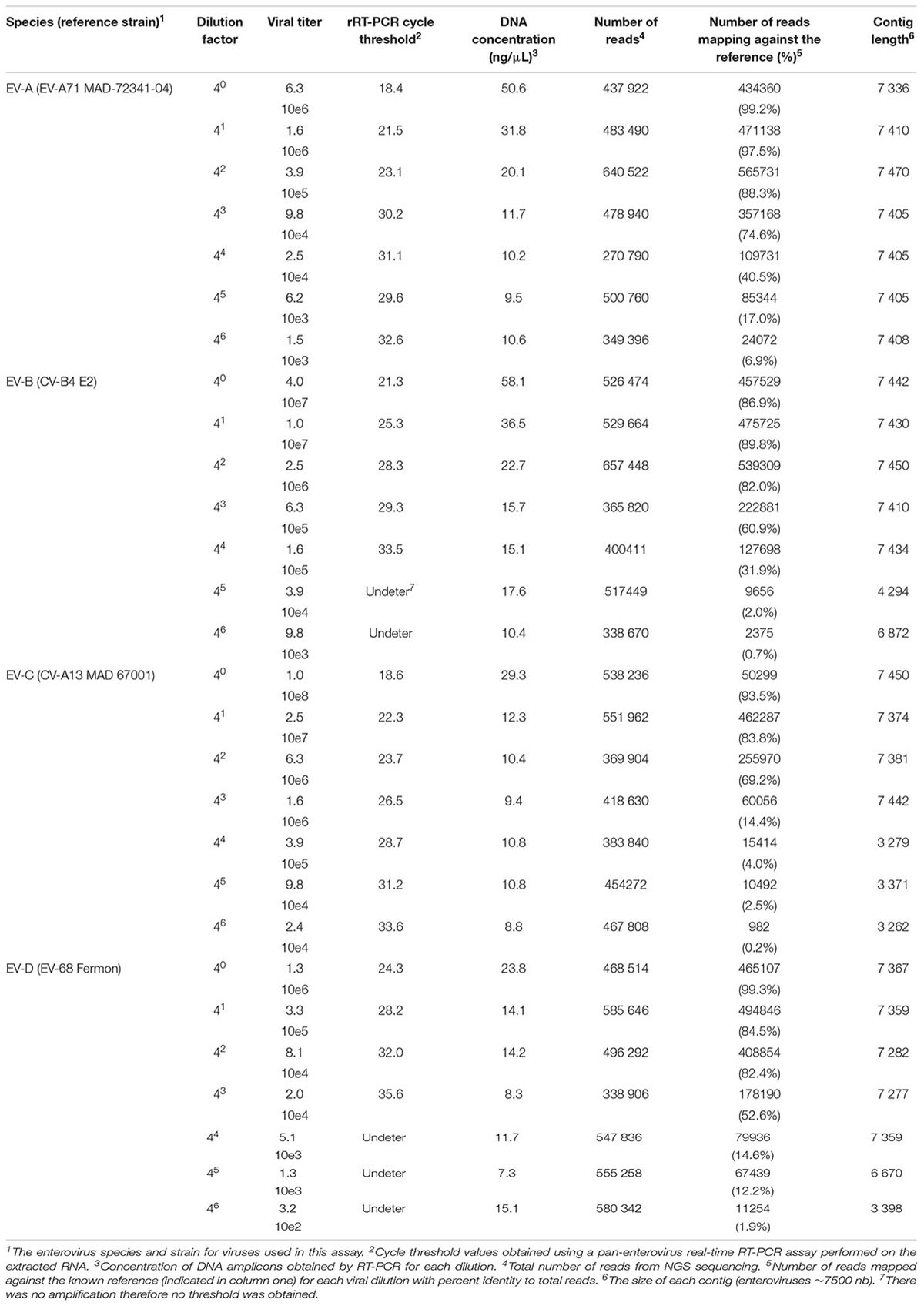
TABLE 3. Sensitivity of the NGS assay.
This RT-PCR amplification method was developed for the analysis of certain amount of viruses present following amplification in infected cells. It could probably be applied to the direct analysis of human or environmental samples, including different compartment-specific human fluids, provided that the sensitivity of the method has been adapted to the amount of virus present in these samples.
Virus Mixture Detection
To confirm the ability to detect viruses in a mixture, different samples containing known viral isolates under the following conditions were prepared: (1) equal amounts of four viral isolates representing enterovirus species A, B, C, and D and (2) four viral isolates belonging to enterovirus species B. The viral isolates used to perform these mixture experiments are listed in Table 4.
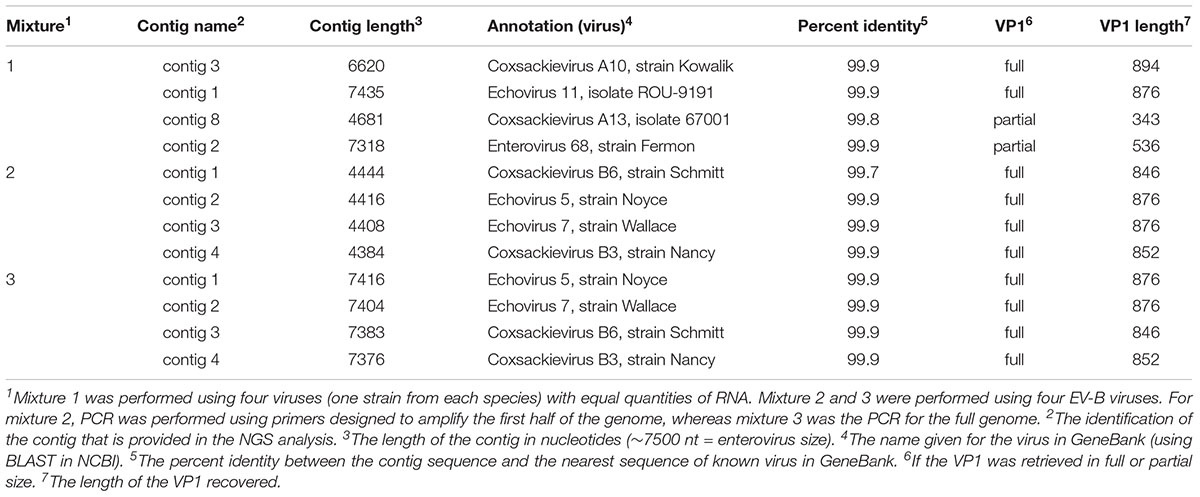
TABLE 4. NGS analysis in three mixtures containing four viruses.
To confirm the ability to detect a mixture of viruses from “real life” conditions, we used 20 supernatants from RD cells which were infected with 10 stool extracts and 10 sewage concentrates, respectively (Table 5). RNA extraction, RT-PCR, and NGS sequencing were performed as described in the related sections.
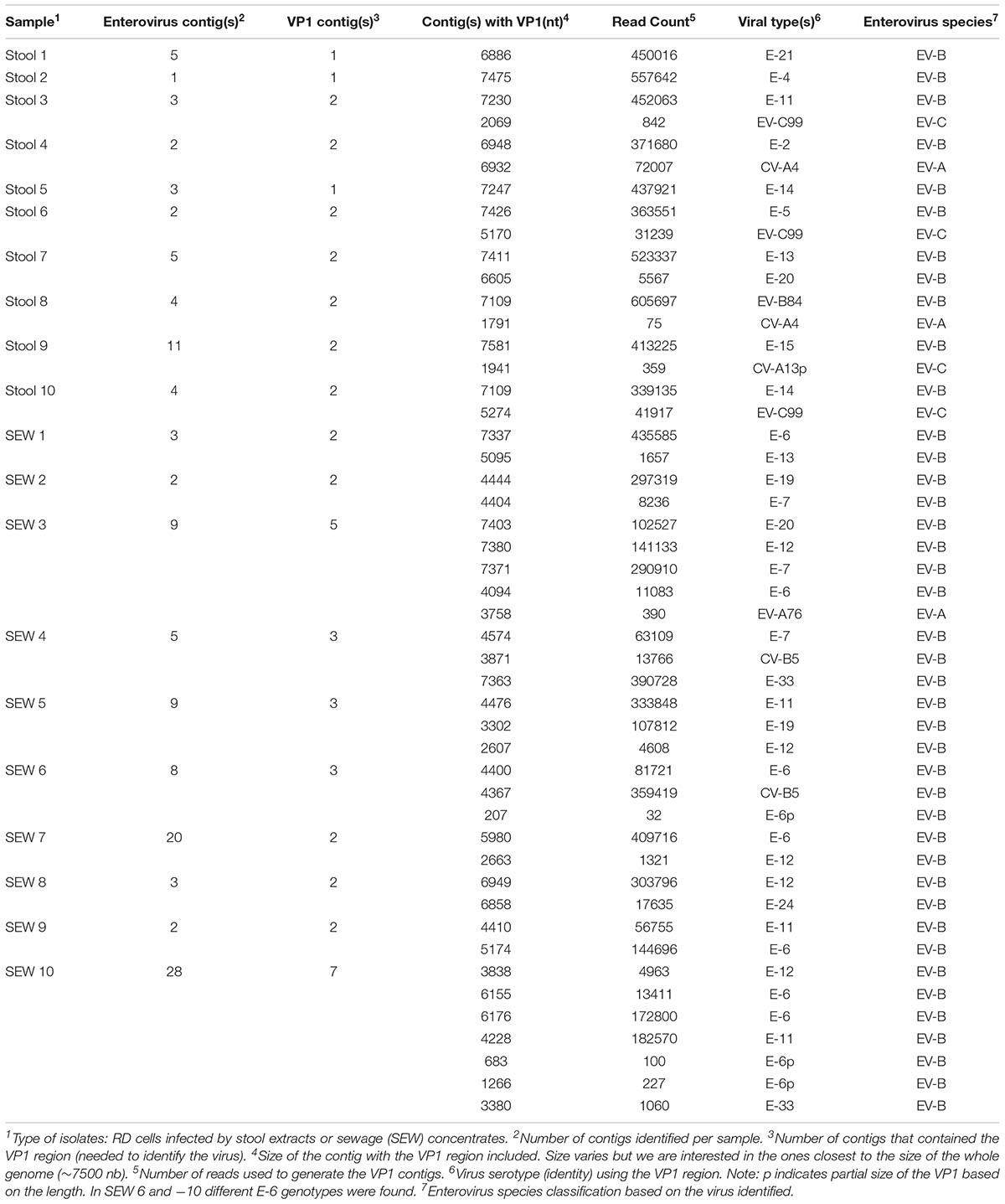
TABLE 5. Detection of viral sequences in supernatants of RD cells infected with stool and sewage samples, using de novo assembly.
PCR Purification and Next Generation Sequencing (NGS)
For each sample, the five PCR products were pooled and purified using a vacuum method and then sent to the sequencing platform PIBNET (Pasteur International Bioresources Network, Institute Pasteur Paris). The libraries were created using 1 ng of DNA with the Nextera XT DNA Library Preparation Kit in a SureCycler 8800 thermocycler (Agilent). Following purification on AMPure beads (Beckman), the libraries were controlled using the High Sensitivity D1000 assay (Agilent) on a TapeStation 2200. The products were sequenced using Illumina NextSeq HiSeq. All kits were used following manufacturer’s instructions.
Data Analysis
Using CLC Genomics Workbench 8.5 (CLCbio), we paired and assembled contigs from the raw reads. Next, de novo assembly was performed using CLC Main Workbench (CLCbio) with the following parameters: Mismatch cost = 2; Insertion cost = 2; Deletion cost = 2; Length Fraction = 0.5; Similarity Fraction = 0.95. All contigs longer than 200 nt were submitted to National Center for Biotechnology Information (NCBI) for BLAST analysis.
Results
Sequence Analysis and Primer Design
To sequence all EVs, we designed primers targeting conserved genomic regions that allowed the synthesis of overlapping amplicons. To amplify the first half of the genome primer C004 (Bessaud et al., 2016) was used in combination with a newly designed generic primer (EV-CRE-R), targeting the CRE region. This primer set was used to amplify the first half of the genome for the four EV species (EV A-D). To ensure the best amplification of the second half of the genome, we designed primers specifically targeting species A, B, C, and D. The combination of the two primer sets (Table 2), allowed for the amplification of the 5′ and 3′ parts of the genomes, which led to the synthesis of two overlapping DNA fragments per virus (approximately 400-nt long see Figure 1). This method resulted in the amplification of the whole genome for samples containing a single virus or mixture and the amplicon products were used for NGS.
To validate the proposed primer sets capability of generating whole genome sequencing data, we tested 15-EVA, 40-EVB, 20-EVC, and 2-EVD viruses (Table 1). We obtained the full genome sequencing data for all viruses except two (EV-C95 T08-083 and EV-D70), for which we obtained 93.6 and 87.5% of the genomic sequences, respectively. In all cases the sequences of the VP1 capsid protein could be used to confirm the type of virus. These results indicated the effectiveness of the proposed primers in conjunction with NGS sequencing to correctly identify the viruses and to reconstruct the whole genome using de novo assembly.
Additionally, to evaluate the sensitivity of this assay, we tested a fourfold serial dilution of one representative of each enterovirus species (Table 3). For undiluted viruses, the number of reads that mapped against the reference strains ranged between 86.9 to 99.3%, depending on the species. The full-length genome was reconstructed, using de novo assembly, for the majority of viruses and dilutions (ranging from 1.0 10e8 to 1.5 10e3 TCID50/ml). For some strains, long contigs overlapping almost the whole genomic sequences were recovered when high amounts of viruses were used (1.6 10e5 for CV-B4 and 1.6 10e6 for CV-A13).
Mixture Detection
To assess the capacity of the method to detect and identify different enteroviruses in samples containing mixtures, we performed two types of experiments: (1) mixed samples containing different mixtures of known viral isolates under controlled conditions (spiked) and (2) supernatants of cells infected with stool or sewage extracts in which the mixture of viruses was unknown and the conditions were not controlled (unspiked).
Spiked Experiments
To simulate true clinical and/or environmental conditions, four representative viral isolates for each enterovirus species were used to make mixture 1 (similar quantities or viral RNA); and four viral isolates of species B were used for mixtures 2 and 3. For all mixtures, RNA was extracted and DNA amplification was performed using the newly designed primer sets.
For mixture 1, we were successful in generating one contig per virus with 99.9% identity with the corresponding sequences in GeneBank using BLAST algorithm (Table 4). We were able to recover data about the four viral isolates and to consider the VP1 region used to identify the virus(es) present, either in its full or partial length.
For mixtures 2 and 3 we were able to correctly isolate and identify all four serotypes from species B with 99.7–99.9% identity and we recovered the full length of the VP1 region for the four B viral isolate strains under both conditions (half and full genome amplification). Additionally, for all three experiments, we confirmed that the parameters used by de novo could assemble the reads into separate contigs, resulting in the expected viruses.
Unspiked Experiments
To validate this method for field studies, we tested unknown mixtures of viral isolates present in the supernatant of RD cells infected with extracts from stools and sewage samples. Ten samples from stools and ten samples from sewage were used, following the protocols described in the materials and methods section. Full genomic sequences were identified for 8 viruses present in RD supernatants from stool samples and 5 viruses present in those from sewage samples. However, for certain viruses, contigs covering only partial genomic sequences could be obtained. In these cases, we could use the VP1 contigs to identify the viruses present. In cell supernatants of infected cells, we were able to recover 1 to 2 viruses per stool sample and 2 to 7 viruses per sewage sample (Table 5). Among twenty samples, we were able to find 48 enteroviruses based on the VP1 region. For 21 viruses, more than 6100 nt per genome could be determined, whereas, for 27 other viruses, genomic contigs were not as long. The genome of 18 viruses was higher than 90% in length. Overall, 41 EV-Bs, 3 EV-As, 3 EV-Cs, and no EV-Ds were identified.
Discussion
The goal of this research was to genetically sequence the entire genome for all enteroviruses (EV-A, -B, -C, and –D) present in a given sample, using a simple method for detection and genetic characterization. To accomplish this, we developed an RT-PCR assay where we designed degenerate primers targetting conserved regions of the genome, which allowed the DNA fragments to be amplified, followed by NGS. This paper described our approach and demonstrated the feasibility of our methods to successfully identify EV in mixtures.
The effectiveness of this assay was achieved by selecting two primer sets per enterovirus species that generated two overlapping fragments decreasing labor, time, and cost per sample tested. The first generic primer set (C004-F and CRE-R) corresponds to two conserved regions for all enteroviruses (-A through -D) and was capable of successfully amplifying the first half of the genome (∼4500 nt). The second half of the genome was amplified using generic primers that were designed to specifically target all viruses within the respective species (EV-A, -B, -C, and -D). We were able to amplify nearly the entire genome for all enteroviruses, using only five PCR mixtures. The sensitivity of the primers was determined and the EV-A primers appeared to be more sensitive than the others (Table 3). However, the primers selected for this project were sensitive enough to detect all enteroviruses and specific enough to detect only one species type in the presence of the other species.
To validate our sequencing method, we performed it using twenty RD isolate samples from human stools and sewage concentrates. The consensus lengths after de novo assembly were obtained for the ten isolate samples from stool tested, indicating that the method was sensitive enough to determine the entire genome. The results were different for sewage because mixtures of strains appeared to be more complex (up to 7 different strains) than those present in stools (two strains maximum). Only three of the ten isolate samples resulted in sequencing data for the whole genome. However, the sequencing data for the other seven provided long genomic contigs and the coverage necessary to identify the type of virus(es) present using VP1 sequences (Nix et al., 2006). Additionally, we effectively recovered the four viruses used to produce the known mixtures described in Table 4 and the enterovirus field mixtures from Table 5. Being able to differentiate viruses in mixtures is important and in this study, and in a previous study (Bessaud et al., 2016), we have found that de novo assembly is able to properly construct contigs when mixed viruses are genetically divergent (i.e., when they do not share identical genetic sequences). However, recombination can create genomes that, together, are clearly divergent in some genomic regions and completely identical in others. In this case, de novo assembly can fail to build full-length contigs because it is impossible for the software to determine which reads are from virus 1 or from virus 2. This limitation is not related to our method used to generate DNA from RNA genomes, but it is due to the fact that most NGS methods (i.e., Illumina) cannot deal with long DNA fragments, requiring the shearing of the DNA amplicons prior to sequencing. Other NGS methods (such as PacBio or MinION) can sequence long DNA fragments and could be used to overcome the assembly problems but these methods have a high error rate compared to Illumina. Therefore, there is a balance between the accuracy of the sequences and the length of the contigs that can be generated in cases of mixed recombinant genomes. Nonetheless, this challenge did not compromise the main objective of our method, which was to allow the identification of the enterovirus lineages found in a given sample. The typing of EVs relies on their capsid sequence and intertypic recombination is very uncommon inside this genomic region, the contigs generated through de novo assembly generally span all the capsid-encoding region.
Research has shown the importance of analyzing the complete genome for enteroviruses (Oberste et al., 2004; Joffret et al., 2012) because nucleotidic differences and inter and intra-typic recombination events in non-structural regions differentiate types and lineages (Lukashev et al., 2003; Simmonds, 2006; Combelas et al., 2011). The amplification and sequencing of whole genomic sequences of strains belonging to EV-A, -B, and -C species using generic and specific primers were successfully performed (Tan et al., 2015; Bessaud et al., 2016; Montmayeur et al., 2017; Sahoo et al., 2017). One particular study aimed to isolate polioviruses using random amplification and NGS to better optimize current protocols for whole genome sequencing and identification of a variety of vaccine-derived polioviruses (Montmayeur et al., 2017). Although these methods were able to recover the entire genome for a given type or species, they were not able to sequence mixtures of several human enteroviruses in a single run like our described research. However, a recent study focused on detecting polioviruses and non-polio enteroviruses in cellular supernatants infected with sewage concentrates using NGS and random primers or specific primers targeting polioviruses (Majumdar et al., 2018).
Contrary to these previous studies, our assay was designed to specifically amplify enterovirus of species A to D, capturing the diversity of EVs. Because the assay includes species-specific primers, we can increase the amplification of the respective targeted species. This is not the case with random primers that cannot be ensured to capture all enteroviruses within a given species. In addition, the assay can be simplified to target one particular species. Although strategies based on random amplification have the advantage of being able to amplify sequences of viruses that are unexpected or unknown, they have the disadvantage of decreasing the number of relevant reads since the sequence of non-viral origins is also amplified and sequenced. Our strategy is more suitable to specifically amplify the relevant sequences, limiting the requested depth of coverage, which favors multiplexing and thus reducing the cost per genome and per sample.
In conclusion, the method described in this study enables the specific identification of all enteroviruses present in samples during a single sequencing run. This type of assay would be useful when analyzing human and environmental field samples as indicated in our results.
This contributes to the study of EV diversity and ecosystem within given populations. In addition, this method provided a way to collect full genome sequencing data from mixtures of enteroviruses within the four species A to D that were present in cellular supernatants. These data become a necessity for surveillance purposes and when studying the relationships between the genetic characteristics, including the mosaic features of their genomes acquired through frequent intra- and intertypic recombination and their biological properties. Indeed, mutations and recombination events can be implicated in reintroducing virulence factors and have been involved in the evolution of enteroviruses (Riquet et al., 2008; Jegouic et al., 2009; Joffret et al., 2012; Holmblat et al., 2014).
Author Contributions
M-LJ, PP, and FD conceived and designed the experiments and analyzed the data. M-LJ and PP performed the experiments. RR and J-MH contributed to samples and experiments in Madagascar. PP and M-LJ wrote the manuscript. MB, RR, J-MH, and FD critically revised.
Funding
The collection and shipment of samples were supported by the Centers for Disease Control and Prevention through “Intensive Virologic Monitoring of the tOPV/bOPV switch” Project. This work was supported by the Institut Pasteur (PTR 484), and by the Foundation Total grant S-CM15010-05B (http://fondation.total/fr). Patsy Polston is a postdoc sponsored in part by a grant from the Pasteur Foundation and the Dennis and Mireille Gillings Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are indebted to Maud Vanpeene, Andrea Alexandru, Sobhy Wilhame, and Vincent Enouf [Institut Pasteur, Pasteur International Bioresources network (PIBNET), Plateforme de microbiologie mutualisée (P2M), Paris, France] for performing the sequencing experiments. Thanks to Deborah Delaune and Anne-Lou Pinon for their assistance with performing some experiments. We would like to thank the staff involved in the sampling collection and isolation from Institut Pasteur de Madagascar (Seta Andriamamonjy, Fitahiana Michael Rakotoarison, and Sandratana Raharinantoanina). In addition, we are grateful for the isolates used for this study from Anda Baicus (Romania) and Serge Sadeuh-Mba (Cameroon).
Footnote
References
Bessaud, M., Sadeuh-Mba, S. A., Joffret, M.-L., Razafindratsimandresy, R., Polston, P., Volle, R., et al. (2016). Whole genome sequencing of Enterovirus species C isolates by high-throughput sequencing: development of generic primers. Front. Microbiol. 7:1294. doi: 10.3389/fmicb.2016.01294
Blomqvist, S., and Roivainen, M. (2016). Isolation and characterization of enteroviruses from clinical samples. Methods Mol. Biol. 1387, 19–28. doi: 10.1007/978-1-4939-3292-4_3
Caro, V., Guillot, S., Delpeyroux, F., and Crainic, R. (2001). Molecular strategy for ‘serotyping’of human enteroviruses. J. Gen. Virol. 82, 79–91. doi: 10.1099/0022-1317-82-1-79
Combelas, N., Holmblat, B., Joffret, M. L., Colbere-Garapin, F., and Delpeyroux, F. (2011). Recombination between poliovirus and coxsackie A viruses of species C: a model of viral genetic plasticity and emergence. Viruses 3, 1460–1484. doi: 10.3390/v3081460
Holmblat, B., Jégouic, S., Muslin, C., Blondel, B., Joffret, M.-L., and Delpeyroux, F. (2014). Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. mBio 5:e1119-14. doi: 10.1128/mBio.01119-14
Jegouic, S., Joffret, M. L., Blanchard, C., Riquet, F. B., Perret, C., Pelletier, I., et al. (2009). Recombination between polioviruses and co-circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 5:e1000412. doi: 10.1371/journal.ppat.1000412
Joffret, M. L., Jegouic, S., Bessaud, M., Balanant, J., Tran, C., Caro, V., et al. (2012). Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J. Infect. Dis. 205, 1363–1373. doi: 10.1093/infdis/jis204
Lukashev, A. N., Lashkevich, V. A., Ivanova, O. E., Koroleva, G. A., Hinkkanen, A. E., and Ilonen, J. (2003). Recombination in circulating enteroviruses. J. Virol. 77, 10423–10431. doi: 10.1128/jvi.77.19.10423-10431.2003
Majumdar, M., Klapsa, D., Wilton, T., Akello, J., Anscombe, C., Allen, D., et al. (2018). Isolation of vaccine-like poliovirus strains in sewage samples from the United Kingdom. J. Infect. Dis. 217, 1222–1230. doi: 10.1093/infdis/jix667
Montmayeur, A. M., Ng, T. F., Schmidt, A., Zhao, K., Magana, L., Iber, J., et al. (2017). High-throughput next-generation sequencing of polioviruses. J. Clin. Microbiol. 55, 606–615. doi: 10.1128/JCM.02121-16
Nijhuis, M., van Maarseveen, N., Schuurman, R., Verkuijlen, S., de Vos, M., Hendriksen, K., et al. (2002). Rapid and sensitive routine detection of all members of the genus enterovirus in different clinical specimens by real-time PCR. J. Clin. Microbiol. 40, 3666–3670. doi: 10.1128/JCM.40.10.3666-3670.2002
Nix, W. A., Oberste, M. S., and Pallansch, M. A. (2006). Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 44, 2698–2704. doi: 10.1128/JCM.00542-06
Oberste, M. S., Maher, K., Kilpatrick, D. R., Flemister, M. R., Brown, B. A., and Pallansch, M. A. (1999a). Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 37, 1288–1293.
Oberste, M. S., Maher, K., Kilpatrick, D. R., and Pallansch, M. A. (1999b). Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 73, 1941–1948.
Oberste, M. S., Maher, K., and Pallansch, M. A. (2004). Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J. Virol. 78, 855–867. doi: 10.1128/JVI.78.2.855-867.2004
Poh, C. L., and Tan, E. L. (2010). “Detection of enteroviruses from clinical specimens,” in Diagnostic Virology Protocols, eds J. R. Stephenson and A. Warnes (Berlin: Springer), 65–77. doi: 10.1007/978-1-60761-817-1_5
Riquet, F. B., Blanchard, C., Jegouic, S., Balanant, J., Guillot, S., Vibet, M. A., et al. (2008). Impact of exogenous sequences on the characteristics of an epidemic type 2 recombinant vaccine-derived poliovirus. J. Virol. 82, 8927–8932. doi: 10.1128/JVI.00239-08
Sadeuh-Mba, S. A., Bessaud, M., Massenet, D., Joffret, M. L., Endegue, M. C., Njouom, R., et al. (2013). High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J. Clin. Microbiol. 51, 759–770. doi: 10.1128/JCM.02119-12
Sahoo, M. K., Holubar, M., Huang, C., Mohamed-Hadley, A., Liu, Y., Waggoner, J. J., et al. (2017). Detection of emerging vaccine-related polioviruses by deep sequencing. J. Clin. Microbiol. 55, 2162–2171. doi: 10.1128/JCM.00144-17
Silva, P. A., Diedrich, S., de Paula Cardoso, D. D., and Schreier, E. (2008). Identification of enterovirus serotypes by pyrosequencing using multiple sequencing primers. J. Virol. Methods 148, 260–264. doi: 10.1016/j.jviromet.2007.10.008
Simmonds, P. (2006). Recombination and selection in the evolution of picornaviruses and other mammalian positive-stranded RNA viruses. J. Virol. 80, 11124–11140. doi: 10.1128/JVI.01076-06
Keywords: human enteroviruses, whole-genome sequences, high-throughput sequencing, viral mixtures, enterovirus identification
Citation: Joffret M-L, Polston PM, Razafindratsimandresy R, Bessaud M, Heraud J-M and Delpeyroux F (2018) Whole Genome Sequencing of Enteroviruses Species A to D by High-Throughput Sequencing: Application for Viral Mixtures. Front. Microbiol. 9:2339. doi: 10.3389/fmicb.2018.02339
Received: 02 July 2018; Accepted: 12 September 2018;
Published: 28 September 2018.
Edited by:
Hirokazu Kimura, Gunma Paz University, JapanReviewed by:
Flore Rozenberg, Université Paris Descartes, FranceKomei Shirabe, Yamaguchi Prefectural Institute of Public Health and Environment, Japan
Copyright © 2018 Joffret, Polston, Razafindratsimandresy, Bessaud, Heraud and Delpeyroux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Line Joffret, bWFyaWUtbGluZS5qb2ZmcmV0QHBhc3RldXIuZnI= Patsy M. Polston, cGF0c3kucG9sc3RvbkBnbWFpbC5jb20=
†These authors have contributed equally to this work