 Marcos D. Pinho1*
Marcos D. Pinho1* Geoffrey Foster2
Geoffrey Foster2 Constança Pomba3Miguel P. Machado1
Constança Pomba3Miguel P. Machado1 Johanna L. Baily4†Thijs Kuiken5José Melo-Cristino1
Johanna L. Baily4†Thijs Kuiken5José Melo-Cristino1 Mário Ramirez1the Portuguese Group for the Study of Streptococcal Infections
Mário Ramirez1the Portuguese Group for the Study of Streptococcal Infections- 1Instituto de Microbiologia, Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
- 2SRUC Veterinary Services, Inverness, United Kingdom
- 3Faculdade de Medicina Veterinária, Universidade de Lisboa, Lisbon, Portugal
- 4Moredun Research Institute, Pentlands Science Park, Edinburgh, United Kingdom
- 5Department of Viroscience, Erasmus Medical Center, Rotterdam, Netherlands
Streptococcus canis is an animal pathogen which occasionally causes infections in humans. The S. canis M-like protein (SCM) encoded by the scm gene, is its best characterized virulence factor but previous studies suggested it could be absent in a substantial fraction of isolates. We studied the distribution and variability of the scm gene in 188 S. canis isolates recovered from companion animals (n = 152), wild animal species (n = 20), and humans (n = 14). Multilocus sequence typing, including the first characterization of wildlife isolates, showed that the same lineages are present in all animal hosts, raising the possibility of extensive circulation between species. Whole-genome analysis revealed that emm-like genes found previously in S. canis correspond to divergent scm genes, indicating that what was previously believed to correspond to two genes is in fact the same scm locus. We designed primers allowing for the first time the successful amplification of the scm gene in all isolates. Analysis of the scm sequences identified 12 distinct types, which could be divided into two clusters: group I (76%, n = 142) and group II (24%, n = 46) sharing little sequence similarity. The predicted group I SCM showed extensive similarity with each other outside of the N-terminal hypervariable region and a conserved IgG binding domain. This domain was absent from group II SCM variants found in isolates previously thought to lack the scm gene, which also showed greater amino acid variability. Further studies are necessary to elucidate the possible host interacting partners of the group II SCM variants and their role in virulence.
Introduction
Streptococcus canis is a beta-hemolytic Lancefield group G streptococcus which colonizes the skin, the upper respiratory tract and the reproductive tract of dogs and cats (Devriese et al., 1986; Lyskova et al., 2007; Timoney et al., 2017). S. canis is also an important pathogen of these species, causing skin, and genitourinary tract infections, otitis externa, pneumonia, endocarditis, septic arthritis, septicemia, necrotizing fasciitis, and streptococcal toxic shock syndrome (DeWinter et al., 1999; Lyskova et al., 2007; Morrow et al., 2016). The isolation of S. canis from other animals has been documented, including livestock, in which it is known to cause clinical and subclinical bovine mastitis (Hassan et al., 2005), and various wild animal species, including minks (Chalmers et al., 2015) and feral cats (Hariharan et al., 2011), but also aquatic mammals such as pinnipeds (Castinel et al., 2007; Seguel et al., 2018) and otters (Simpson, 2006). S. canis is a well-recognized zoonotic agent, with a growing number of studies reporting the isolation of S. canis from cases of skin and soft tissue infections, bacteremia and endocarditis in humans (Whatmore et al., 2001; Pinho et al., 2013; Amsallem et al., 2014; Taniyama et al., 2017). Although it has been shown that the same S. canis lineages are found in companion animals, livestock and humans (Richards et al., 2012; Pinho et al., 2013), genotypic characterization of S. canis isolates from wild animals is limited.
Genomic information showed that S. canis is related to beta-hemolytic Streptococcus pyogenes (group A streptococcus [GAS]) and Streptococcus dysgalactiae subsp. equisimilis (SDE), both human pathogens with which S. canis shares many putative virulence factors (Richards et al., 2012). The M protein, encoded by the emm gene, is one of the main virulence factors of both GAS and SDE (Cunningham, 2000; Brandt and Spellerberg, 2009), but application of emm typing – a technique based on amplification and sequencing of a segment of the emm gene encoding the N-terminal hypervariable portion of the protein – revealed that most S. canis isolates were non-typeable (Ahmad et al., 2009; Pinho et al., 2013). Subsequently, an M-like protein was identified in the S. canis genome (termed SCM for S. canis M-like protein; Fulde et al., 2011), encoded by the scm gene, which is now the best characterized of the S. canis virulence factors.
Streptococcus canis M-like protein was first described by Yang et al. (2010) named SPASc by the authors, who showed a protective response in mice after passive immunization and opsonization with specific antiserum. It was shown that SCM binds both plasminogen and IgG from various animal species, including humans, through domains present in the N-terminus and central part of the mature protein, respectively (Fulde et al., 2011; Bergmann et al., 2017). However, these studies also suggested that up to one third of the S. canis isolates lack the scm gene, since the authors correlated failure in amplifying scm by PCR with weak plasminogen and immunoglobulin-binding capacity (Fulde et al., 2011, 2013; Bergmann et al., 2017). The scm gene was detected in S. canis isolates from distinct hosts, including dogs (Fulde et al., 2013), cats (Timoney et al., 2017), cows (Richards et al., 2012) and humans (Fulde et al., 2011; Taniyama et al., 2017), and a recent study showed that allelic variants of scm found among cat isolates in the United States could be classified into four distinct types (Timoney et al., 2017). However, the correspondence between the scm and the emm locus remains uncertain since the scm sequences described to date (Richards et al., 2012; Taniyama et al., 2017;Timoney et al., 2017) are distinct from the emm sequences derived from S. canis isolates (Ahmad et al., 2009; Pinho et al., 2013) and previous studies have applied PCR protocols targeting specifically either scm or emm with no information regarding their genomic context.
The goal of the present study was to determine the distribution of scm in S. canis isolates recovered from different hosts, including wild animals. The draft genomic sequence of an emm-typeable S. canis isolate (Pinho et al., 2013) in which the scm PCR amplification failed was determined, with the aim of clarifying the correspondence between scm and emm and establishing a protocol to allow typing of all S. canis isolates. Multilocus sequence typing (MLST) was used to determine the genetic diversity of the collection and to allow a comparison between S. canis isolates recovered from wild animal species, companion animals and humans.
Materials and Methods
Bacterial Isolates
A collection of 188 S. canis isolates was analyzed. Ninety-five S. canis isolates were recovered from dogs (n = 75) and wild animal species (n = 20), including seals (n = 11), otters (n = 6), badgers (n = 2) and a fox (n = 1). Seal species included gray seals (Halichoerus grypus, n = 6 isolates), common seals (Phoca vitulina, n = 2) and Mediterranean monk seals (Monachus monachus, n = 2), while in one case the seal species was not recorded. Isolates from wild animals were recovered during necropsies of carcasses found in the wild, while those of companion animals were recovered within the normal workup for the diagnosis of suspected infections. No samples were obtained specifically for this study. All isolates were recovered in Scotland from 1993 to 2014, however, the two isolates from Mediterranean monk seals were cultured in Scotland, following necropsies in the Netherlands. All isolates were presumptively identified by the contributing laboratories using biochemical tests. For comparison purposes and to maximize the diversity of the S. canis isolates on which the distribution of the scm gene was to be analyzed, an additional set of 93 S. canis isolates was included in the study: 7 isolates recovered from human infections in Portugal, from 2011 to 2017, from blood (n = 2), pus (n = 2), sputum, urine and vaginal exudate (n = 1 each); and 86 isolates previously characterized by MLST (Pinho et al., 2013), recovered in Portugal, Germany and Italy, from dogs, cats, a horse and humans. The study was approved by the Institutional Review Board of the Centro Académico de Medicina de Lisboa. These were considered surveillance activities and were exempt from informed consent. All methods were performed in accordance with the relevant guidelines and regulations. The data and isolates were de-identified so that these were irretrievably unlinked to an identifiable person. Detailed information on all isolates included in the study is provided in Supplementary Table 1.
Hemolysis and Lancefield Grouping
Beta-hemolysis and colony size were confirmed in tryptic soy agar (Oxoid, Hampshire, United Kingdom) supplemented with 5% (vol/vol) defibrinated sheep blood, after overnight incubation at 37°C. The Lancefield group was confirmed by a commercial latex agglutination technique (Streptococcal Grouping Kit, Oxoid, Basingstoke, United Kingdom).
MLST Analysis
Streptococcus canis isolates were characterized using the MLST scheme available for S. canis1 (Pinho et al., 2013). The PCR amplification and sequencing for some of the loci were optimized by designing novel primers based on S. canis genomes (Supplementary Table 2). Unique sequences at each locus were assigned allele numbers. The combination of the seven allele numbers for each isolate was used to define sequence types (STs). An expansion of the goeBURST algorithm implemented in PHYLOViZ (Nascimento et al., 2017) was used to generate a minimum-spanning-tree-like reflecting possible relationships between S. canis STs. Clonal complexes were defined at the single-locus variant level (CCSLV).
Whole Genome Sequencing
The genome of S. canis isolate FMV2238.02 was sequenced using Illumina MiSeq. This isolate was recovered from a dog ear exudate in 2002 in Portugal, belongs to MLST ST1 and has the emm type stG1389 (Pinho et al., 2013).
Whole-genome sequencing library was prepared using paired-end Nextera®XT DNA Library Prep Kit, Index Kit v2 (Illumina©, San Diego, CA, United States) and sequenced on Illumina MiSeq®system (Illumina©) using MiSeq®Reagent Kit v2 Kit (500 cycles) at the Genomics Unit of Instituto Gulbenkian de Ciência (Oeiras, Portugal). The quality of the 250 bp paired-end reads obtained was assessed with INNUca pipeline,2 which also assembles and curates the bacterial genomes. INNUca v3.1 was run using Docker image “ummidock/innuca:3.1”3 using a predicted genome size of 2.1 Mb. Briefly, reads quality were checked with FastQC4 and cleaned using Trimmomatic (Bolger et al., 2014). De novo assembly was performed using SPAdes (Bankevich et al., 2012) and subsequently polished using Pilon (Walker et al., 2014). Genomes were annotated using Prokka pipeline v1.12 (Seemann, 2014) using Docker image “ummidock/prokka:1.12”5. SignalP v4.1 (Petersen et al., 2011) was used to find signal peptide features and RNAmmer v1.2 (Lagesen et al., 2007) was used to find ribosomal RNA features, both were externally provided to the Docker container. Prokka was run using the following parameters: –addgenes –usegenus –rfam –rnammer –gram pos –increment 10 –mincontiglen 1 –gcode 11 –kingdom Bacteria –genus Streptococcus –species canis.
The locus corresponding to emm stG1389 was identified and the flanking regions were extracted and aligned with the corresponding regions where scm was found in the 3 publicly available S. canis genomes,6 namely strains FSL Z3-227 (GenBank accession number NZ_AIDX01000001), G361 (GenBank accession number NZ_NMRV01000001) and TA4 (GenBank accession number NZ_BEWZ01000010).
emm Typing and scm Amplification
Characterization of the S. canis isolates by emm typing was carried out with primers (emm1 and emm2) and conditions available at https://www.cdc.gov/streplab/groupa-strep/emm-typing-protocol.html. The scm gene was initially amplified by using the primers all-canis_fwd and all-canis_rev described by Fulde et al. (2011). To enable scm amplification and sequencing of the whole scm gene in all S. canis isolates, new primers (Sc_Mprot_F1 and Sc_Mprot_R1) were designed (based on the whole genome comparisons) targeting the scm/emm flanking genes, resulting in amplification of an 1825bp fragment in S. canis isolate FMV2238.02 (Supplementary Figure 1 and Supplementary Table 2). Briefly, 3 μl of template DNA was added to the PCR mixture containing 1U GoTaq G2 Flexi DNA polymerase (Promega, Madison, WI, United States), 1X Green GoTaq Flexi Buffer (Promega, Madison, WI, United States), 1.5 mM MgCl2 (Promega, Madison, WI, United States), 200 μM deoxynucleoside triphosphates (Thermo Fisher Scientific, Waltham, MA, United States) and 0.4 μM primers, in a final volume of 50 μl. The PCR conditions were adapted from the emm typing protocol, as follows: 94°C for 1 min; 10 cycles of 94°C for 15 s, 46°C for 30 s and 72°C for 1 min 15 s; 25 cycles of 94°C for 15 s, 46°C for 30 s and 72°C for 1 min 15 s with a 10 s increment for each of the subsequent 24 cycles; final extension at 72°C for 10 min. All PCR reactions were conducted in a Biometra T gradient thermocycler (Goettingen, Germany). Sequencing of the PCR products was carried with the primers used for amplification, plus primers Sc_Mprot_F2 and Sc_Mprot_R2 which have target sequences inside the amplified fragment.
Analysis of scm Sequences
Geneious (version 8.1.9, Biomatters, Auckland, New Zealand) was used to identify the putative scm open reading frames (ORFs). Sequences were aligned with MUSCLE and MEGA (version 7.0.18) (Kumar et al., 2016) was used to construct a neighbor-joining (NJ) tree of scm alleles by using the Kimura two-parameter substitution model. Branch support was tested by 1,000 replicate bootstrap tests in each analysis. The sequences of the scm gene extracted from the S. canis genomes and those previously reported by Timoney et al. (2017) were included in the analysis for comparison. The scm types were assigned based on the full scm sequence alignment and named following the designations of the allelic variants identified previously (Timoney et al., 2017). The scm ORFs were translated into amino acid sequence and the SignalP 4.1 server7 was used to identify the signal peptides. The SCM amino acid sequence derived from the genome of isolate G361, a strain included in the studies that described the IgG and plasminogen-binding regions of SCM (Fulde et al., 2011; Bergmann et al., 2017), was used as reference to identify these regions in S. canis isolates. Search for protein domains was carried out with InterPro version 69.08.
Statistical Analysis
The diversity and congruence of MLST STs and scm types were quantitatively evaluated by calculating Simpson’s indices of diversity (SID) (Carriço et al., 2006) and adjusted Wallace (AW) coefficients (Severiano et al., 2011) with 95% confidence intervals (CIs). Calculations were performed using the Comparing Partitions website9. The Fisher exact test was used to explore differences between the distribution of the two scm groups between hosts.
Results
Characteristics and Clonality of S. canis Isolates
All isolates were beta-hemolytic and carried the Lancefield group G polysaccharide, as is characteristic of S. canis. The 188 S. canis isolates belonged to 37 STs (SID ± 95% CI, 0.899 ± 0.030), 13 of which were novel (STs 26 to 38, n = 23 isolates). Eight CCSLV were identified (Figure 1). The 95 isolates from the Scottish collection belonged to 24 STs (SID ± 95% CI, 0.899 ± 0.039), a similar diversity to the one observed for the remaining S. canis isolates included in the analysis (24 STs, SID ± 95% CI, 0.887 ± 0.042). Dog isolates recovered in Scotland and those recovered elsewhere shared 10 STs present in the main CCSLV. Wildlife isolates belonged to 6 different STs, three of which (STs 2, 9, and 29) were identical to those found among dog isolates, while ST26 (two badgers), ST27 (one fox), and ST33 (one otter) were, respectively SLV, double-locus variants and triple-locus variants of dog isolates. Seal isolates belonged to a single CCSLV, presenting either ST9 (n = 9) or ST29 (the two Mediterranean monk seals sampled in the Netherlands). On the other hand, S. canis isolates from otters belonged to three STs: ST9 (n = 4), ST2 and ST33 (one isolate each).
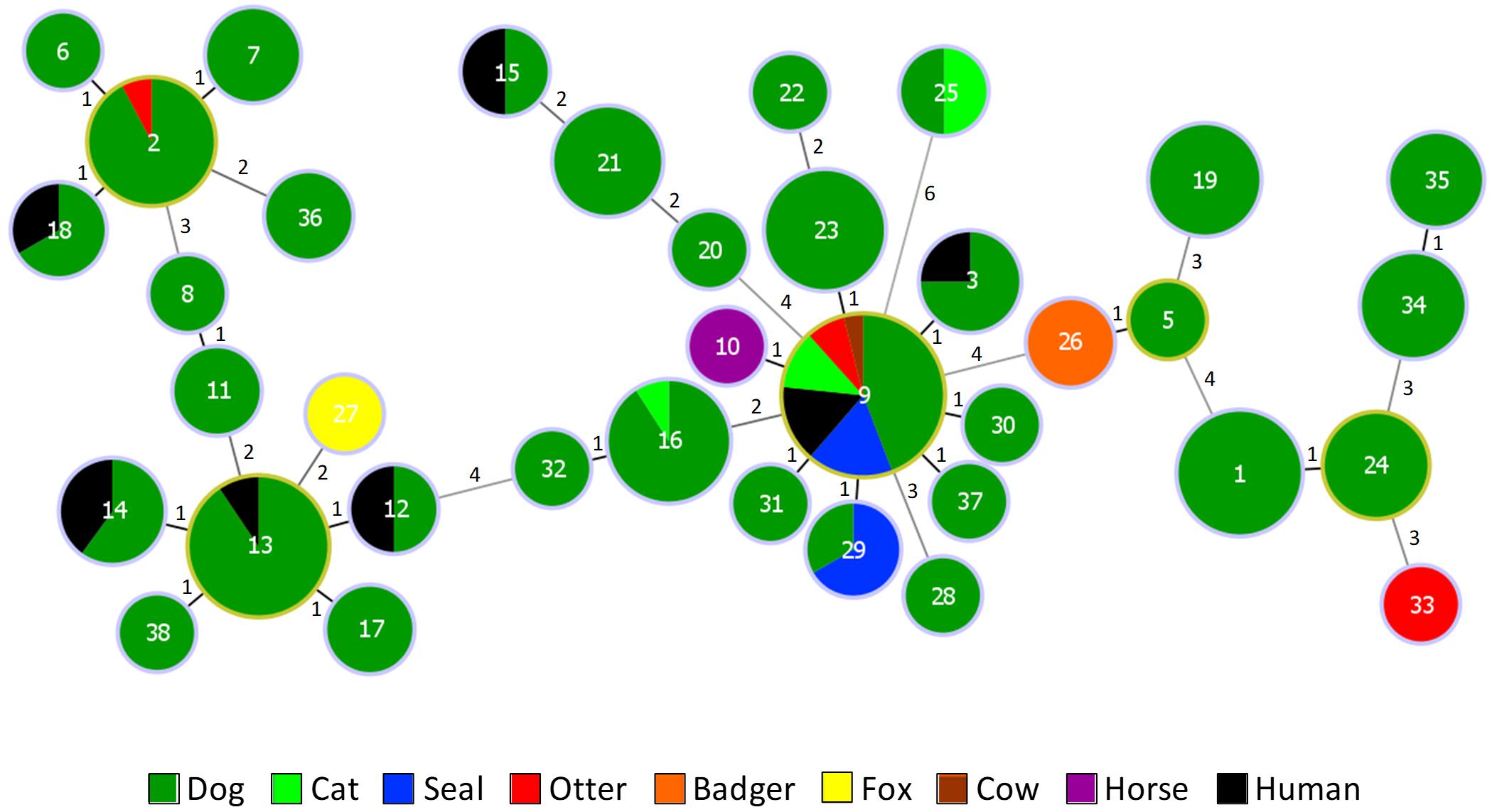
Figure 1. goeBURST diagram of S. canis isolates recovered from animal and human infections. The diagram includes the 188 isolates studied plus the 3 S. canis isolates with publicly available genomes (from which MLST data was extracted). Numbers inside the circles identify the ST and numbers near the lines indicate the number of alleles different between the two connected STs. The size of each circle is proportional to the number of isolates in a logarithmic scale. The number of isolates with the same characteristic is proportional to the respective color. Putative CC founders are identified by an outer light green circle and correspond to the STs with the higher number of SLVs.
Distribution of emm and scm
By emm-typing we could only type 22 of the 188 isolates (11.7%). stG1389 (n = 18), stG1451 (n = 2) and stG663 (n = 2) were the three emm types found. On the other hand, 142 isolates (75.5%) were positive for the scm gene in the PCR using the all-canis primers (Fulde et al., 2011). The 46 (24.5%) S. canis isolates that were scm negative in this PCR included the 22 isolates with an assigned emm type and 24 isolates negative in both PCRs.
Identification of scm in the Genome of an stG1389 Isolate
To clarify if the amplification obtained in the emm typing protocol corresponded to the scm locus, the genome of S. canis isolate FMV2238.02 (ST1, emm type stG1389) was sequenced. The stG1389 sequence in this isolate was found to correspond to an emm-like gene flanked by genes encoding a putative trans-acting positive regulator (upstream) and a bifunctional enzyme (downstream). Alignment with the corresponding regions in the three available S. canis genomes, showed that while the stG1389 emm-like gene had less than 58% sequence identity to the scm genes of the other strains, the flanking genes have more than 96% (trans-acting positive regulator) and 97% (bifunctional enzyme) sequence identity in the 4 strains, confirming that the emm-like and scm genes of these strains are present in the same genomic context and therefore correspond to the same locus, henceforth named scm (an example of the alignment with strain FSL Z3-227 is provided in Supplementary Figure 1). PCR amplification and sequencing using primers targeting the extremities of the two flanking genes (primer pair Sc_MProt_F1/R1) confirmed the presence of an scm gene in this location in all 188 S. canis isolates (fragment size ranging from approximately 1.5 to 2 Kb), including those that did not yield a PCR product with either the emm typing protocol or with the all-canis primers. The extensive sequence variation in the scm regions where the emm1/2 and all-canis_fwd/rev primers bind could potentially justify the failure of the amplification with these primers in some isolates (Supplementary Figure 1).
Correlation of scm Types With MLST STs
There were 41 distinct scm alleles among the 188 S. canis isolates studied, which could be divided into 12 scm types (SID ± 95% CI, 0.776 ± 0.042; Table 1). Types 1 (n = 73, 38.8%) and 2 (n = 41, 21.8%) were the dominant scm types, together accounting for around 60% of the isolates. The NJ tree of the scm alleles found (Figure 2) shows two main clusters, one including scm types 1 to 7 (group I scm alleles), present in 75.5% of the isolates (n = 142), and the other including scm types 8 to 12 (group II scm alleles), found in 24.5% of the isolates (n = 46). While group I included all the isolates that were scm-typeable with the all-canis primers previously described (Fulde et al., 2011), group II included the non-typeable isolates, some of which had an emm type assigned in the emm typing protocol (scm types 8, 9, and 10, corresponding to emm types stG1389, stG1451, and stG663, respectively). Group II scm alleles were found among 42 dog isolates, 2 badgers, 1 fox, and 1 human isolate (Table 1), with no differences in the distribution of the two groups between hosts.
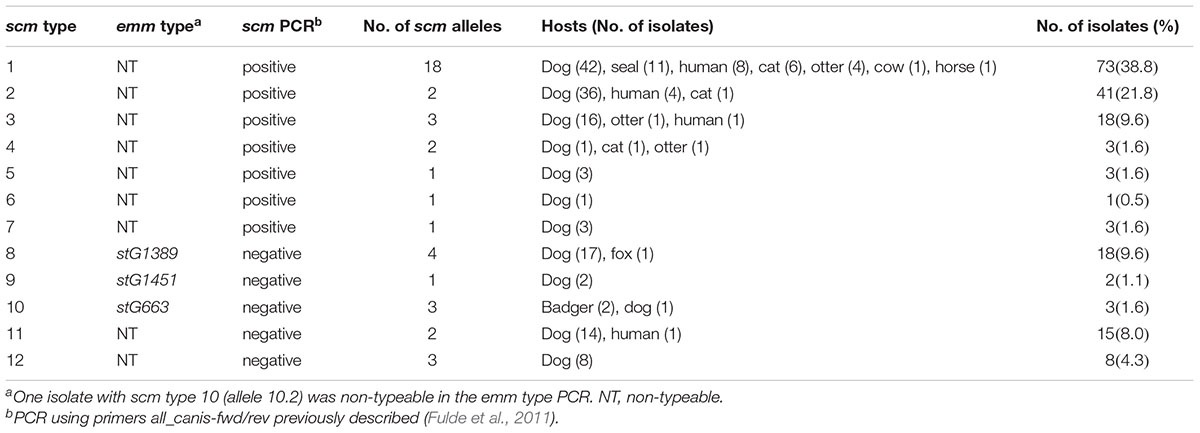
Table 1. Characteristics of the 188 S. canis isolates.

Figure 2. Neighbor-joining tree of scm alleles found among S. canis isolates. The scm alleles found in the current study are shown in bold and the numbers of isolates presenting each allele are indicated in brackets. Sequences from other studies are identified by their GenBank accession number and include those previously described by Timoney et al. (2017), the original scm sequence (FJ594772) described by Yang et al. (2010) and those extracted from S. canis genomes (indicated by an asterisk following the accession number and the name of the strain). scm alleles 1.16 to 1.18 have premature stop codons and were not included in the tree. Branches supported in the bootstrap test (1,000 replicates) by > 75%, have the values shown next to the branches. The tree is drawn to scale, with branch lengths being in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed by the Kimura two-parameter method and are in units of number of base substitutions per site. #indicates scm types that correspond to previously identified emm types, namely stG1389 (scm8), stg1451 (scm9), and stG663 (scm10).
Most isolates of a given ST had the same scm type, resulting in ST being a good predictor of scm type (AWST→scmtype ± 95% CI, 0.966 ± 0.031) (Supplementary Figure 2). In contrast, scm type was a poor predictor of ST (AWscmtype→ST ± 95% CI, 0.374 ± 0.104), although the same scm type was often found among isolates belonging to the same CCSLV (AWCC→scmtype ± 95% CI, 0.949 ± 0.034) with a weaker correspondence in the reverse direction (AWscm type→CC ± 95% CI, 0.793 ± 0.072). The following were the main lineages found ( > 10 isolates):CC9/scm1 (n = 71, 37.8%), CC13/scm2 (n = 28, 14.9%), CC2/scm3 (n = 18, 9.6%), CC1/scm8 (n = 17, 9.0%), and CC16/scm2 (n = 11, 5.9%) (Supplementary Table 3).
Diversity of Predicted SCM Proteins
Translation of the 41 scm alleles resulted in 40 distinct predicted proteins (alleles scm1.1 and scm1.7 resulted in the same amino acid sequence). These included three scm type 1 isolates with premature stop codons, generating ORFs with 594 bp (scm1.16 allele) and 192 bp (scm1.17 and scm1.18 alleles). In the later two isolates, an alternative reading frame was identified starting 148bp downstream of the usual start-site, resulting in 1164 bp long ORFs (387 amino acids), in which no signal peptide could be identified. This was also the case of one isolate with an scm type 2 allele (scm2.2) which started 48 bp downstream of the start-site of alleles of the same type. In all other SCM variants the presence of a signal peptide was predicted by SignalP 4.1, with three possible cleavage sites found for different proteins, namely between amino acids 34 and 35 (types 1 to 3 and 5 to 7), amino acids 32 and 33 (type 4), and 41 and 42 (types 8 to 12). The SCM proteins of group I (SCM types 1 to 7) and group II (SCM types 8 to 12) had less than 33% amino acid identity to each other, with high sequence identity (> 90%) being observable only in the last 50 amino acids of the C-terminus of the proteins, which corresponds to the LPXTG cell wall anchor domain.
Most of the sequence variability among proteins predicted from group I scm alleles (SCM types 1 to 7) occurred in a region encompassing approximately the first 100 amino acids of the mature SCM protein, corresponding to the hypervariable N-terminal portion of the protein (Supplementary Figure 3). The remaining portion of the sequence was highly conserved, with amino acid sequence identity higher than 98% for most of the group I SCM variants newly identified in our study, although more divergent sequences (75% amino acid sequence identity) were present among those previously reported (Timoney et al., 2017). The IgG-binding region of SCM, corresponding to amino acids 173 to 225 of the mature protein of isolate G361 (Bergmann et al., 2017), is located in this conserved region and showed minimal sequence variation. An amino acid sequence identical to that of isolate G361 was found in 132 of the 142 isolates representing group I SCM, with only 4 alleles (9 isolates), SCM1.11 (L222F), SCM1.17 (L222F), SCM4.1 (E185K), and SCM5.1 (E185A and R195S), presenting amino acid changes in this region of the protein (Figure 3). Up to 4 amino acid differences were observed in the SCM protein derived from sequences described elsewhere (Timoney et al., 2017), but not found in this study. The predicted sequence found in the genome of strain FSL Z3-227 had a deletion of a stretch of 49 amino acids (amino acids 191 to 239 of the G361 protein), resulting in the absence of the IgG-binding region. This SCM protein and the SCM1.16 variant (noted above for possessing a premature stop codon), were the only group I SCM variants in which this region was not present.

Figure 3. Alignment of predicted amino acid sequences of IgG binding regions in SCM group I proteins. The IgG binding region derived from isolate G361 comprising amino acids 173 to 225 of the mature protein (Bergmann et al., 2017) was used as reference and corresponds to the consensus of predicted SCM group I proteins. Representative alleles of sequences found in our study are indicated by their numbers and sequences found elsewhere by their GenBank numbers.
In group II scm alleles (SCM types 8 to 12) the hypervariable portion of the protein was larger (up to 280 amino acids in SCM type 11) and we could find no conserved IgG-binding region (Supplementary Figure 3). Despite the low sequence identity to group I SCM types, the presence of an N-terminal signal peptide, a C-terminal LPXTG cell wall anchor domain (M protein-type) and a two-stranded coiled-coil conformation was predicted in all group II SCM variants, consistent with these two groups being functional homologs.
Discussion
The recognized pathogenic role of S. canis in companion animals, cows and more recently humans, has prompted an increasing number of studies probing the genetic properties of S. canis isolates recovered from these hosts (Richards et al., 2012; Pinho et al., 2013; Taniyama et al., 2017; Timoney et al., 2017). Isolation from wild animal species has been reported sporadically and genotyping of these isolates has been rare, although the range of distinct hosts from which it has been recovered (Castinel et al., 2007; Hariharan et al., 2011; Nikolaisen et al., 2017; Seguel et al., 2018) suggests that a broad host tropism is one of the hallmarks of S. canis.
Our study describes the first MLST characterization of S. canis wildlife isolates. Most of the wildlife isolates were associated with STs or CCSLV found among isolates recovered from companion animals, livestock and humans, indicating that they are not genetically distinct and confirming previous observations that some S. canis lineages are found in many animal species (Richards et al., 2012; Pinho et al., 2013). A prime example is the CC9 lineage, comprising more than one third of the isolates analyzed (37.8%), which has been previously shown to dominate among companion animals (Pinho et al., 2013), humans (Pinho et al., 2013) and cows (Richards et al., 2012) from distinct geographic regions. We found CC9 to predominate in seal and otter isolates and showed that this genetic lineage is associated with scm type 1, also the most common scm type in our study. Taken together, our results indicate that CC9/scm1 is the most important S. canis genetic lineage for many distinct animal hosts.
Although isolation of S. canis from pinnipeds has been documented (Castinel et al., 2007; Seguel et al., 2018), previous studies characterizing beta-hemolytic streptococci from marine mammals from the North and Baltic Seas identified Streptococcus phocae, Streptococcus equi subsp. zooepidemicus and Streptococcus dysgalactiae, but not S. canis (Swenshon et al., 1998; Vossen et al., 2004; Akineden et al., 2007). These species were also recovered from seal carcasses in our study. While S. phocae predominated, similar isolation rates were observed for S. canis and the two other species (data not shown). Despite being collected over a period of 20 years (1993 to 2012), all seal isolates represented CC9. This uniformity seems not to be attributable to the restricted geographical distribution of the seals (most were isolated from seals found along the Scottish Coast), since it was not observed among isolates from otters and the dog isolates, also exclusively recovered in Scotland, which showed a diversity comparable to the one of S. canis isolates recovered in other European countries (Pinho et al., 2013). Our results are similar to the ones reported for S. equi subsp. zooepidemicus (Akineden et al., 2007) and S. dysgalactiae (Swenshon et al., 1998) from marine mammals, in which dominance of a single clone has been noted. It is conceivable that the wide host distribution of CC9 may facilitate transmission, perhaps also through environmental sources, or that this lineage has specific factors allowing infection and persistent in multiple host populations.
The S. canis M-like protein SCM has been the subject of recent studies focusing on its function (Yang et al., 2010; Fulde et al., 2011, 2013; Bergmann et al., 2017) or using it to type S. canis (Taniyama et al., 2017; Timoney et al., 2017). Although the scm locus was identified in the first S. canis genome available (Richards et al., 2012), the correspondence of scm and the emm types found in S. canis (Ahmad et al., 2009; Pinho et al., 2013) had not been further explored. The percentage of scm-typeable isolates in our collection (75%) using the previously proposed method was comparable to the one previously reported (68%) (Fulde et al., 2011), while an emm type was obtained for a minority of isolates, also in line with previous studies reporting the inability of emm-typing most S. canis isolates (Whatmore et al., 2001; Ahmad et al., 2009; Pinho et al., 2013). The genome arrangement around the stG1389 emm-like gene resembled the genomic context of the emm gene in SDE and was identical to the genomic context of the scm gene in the three previously available S. canis genomes (all scm-typeable isolates). We expanded this observation to all S. canis isolates in our collection by using a novel PCR targeting the scm flanking genes, which showed that an scm gene was present in all S. canis isolates tested. Moreover, identical scm sequences were obtained by both the novel and emm typing PCRs for stG1389, stG1451, and stG663 isolates, confirming that these 3 scm genes are the only ones that have been designated as emm types within the CDC emm type database10 because these are detected using the primer pair that has been most often used for emm typing (Ahmad et al., 2009; Pinho et al., 2013). Taken together, our results confirmed that the S. canis scm locus corresponds to the emm locus present in SDE and GAS and that this locus is universally present among S. canis isolates, contrary to what had been reported.
The new PCR protocol described in this study is the first allowing assignment of an scm/emm type to all S. canis isolates tested. The new method improves emm typing, the most widely used typing method in SDE and GAS, by allowing its universal application in S. canis typing. In emm typing, emm types are defined based on less than 92% identity within the first 30 codons encoding the mature M protein,11 corresponding to the N-terminal hypervariable terminus of the protein. We confirmed that the scm types defined in our study comply with the criteria used in emm typing based on the NJ tree (Figure 2). Since it is not necessary to sequence the entire scm gene to determine the scm type, we propose to use primers Sc_Mprot_F2 and Sc_Mprot_R2 (Supplementary Table 2) for amplification (fragment size ranging from approximately 1 to 1.4 kb) and of Sc_Mprot_F2 for sequencing to define scm types, similarly to the emm typing protocol.
The current study expands the known diversity of SCM proteins from 4 (Timoney et al., 2017) to 12 different types and shows that SCM proteins of S. canis can be divided into two major groups. SCM types belonging to group I dominate among S. canis from all hosts and what is known about SCM function concerns this group of proteins since it corresponds to the previously scm-typeable isolates. SCM is thought to have an anti-phagocytic role by interacting with the conserved Fc domain of IgG in a non-opsonic manner (Bergmann et al., 2017). We found the IgG-binding region, consisting of 52 amino acids in the central part of the mature SCM (Bergmann et al., 2017), to be highly conserved among group I SCM types, in agreement with previous observations (Bergmann et al., 2017). Plasminogen has also been identified as an SCM ligand (Fulde et al., 2011, 2013) and the plasminogen-binding region in SCM has been identified within the first 214 amino acids of the mature protein (Fulde et al., 2011). This region encompasses the hypervariable portion of SCM and we found substantial amino acid variation between each of the individual SCM types. Thus, inferences on plasminogen binding ability of the different SCM protein variants based solely on their sequences may be unreliable, and it is conceivable that differences in plasminogen-binding ability may occur even within group I SCM types.
Our study extended previous observations by showing that S. canis isolates from wildlife were not genetically distinct from those found among animals with close contact with humans and humans themselves. This is compatible with frequent transmission between species which could have important consequences when considering the zoonotic potential of S. canis. We found that the scm gene is universally present in S. canis and identified divergent SCM proteins among isolates previously reported as scm-negative. The function of these group II SCM proteins is unclear since they present limited sequence similarity with those of group I along almost the full protein length and the IgG binding domain, typical of group I SCM types, is absent. Nevertheless, group II SCM proteins present structural features typical of M-like proteins indicating these may also interact with host factors and have a role in pathogenesis. Since there were no apparent differences in host distribution or infection type between isolates carrying either group I or group II SCM variants, we have no clues as to the potential phenotypic differences conferred by the presence of each of these variants. It was recently suggested a novel emm-cluster-based system to classify the M-proteins of GAS (Sanderson-Smith et al., 2014). The M-proteins encoded by each of the two major clusters identified among GAS emm (clades X and Y), shared few interactions with host factors (Sanderson-Smith et al., 2014) and this seems to be also the case with SCM’s two groups. Despite these similarities, both clades X and Y of GAS are outgroups of the S. canis scm gene (data not shown), not indicating a recent relationship of any of the S. canis groups with any of the GAS clades. Further studies are necessary to identify the possible host interaction partners of group II SCM to clarify the role of these proteins in pathogenesis.
Data Availability
The datasets generated for this study can be found in S. canis PubMLST database, GenBank and European Nucleotide Archive (ENA). All MLST data, including novel alleles and STs, were submitted to the S. canis PubMLST database (http://pubmlst.org/scanis/). scm sequences were submitted to GenBank under accession numbers MH996642 to MH996682. The raw sequencing reads and the annotated assembled genome of S. canis isolate FMV2238.02 are available through the European Nucleotide Archive (ENA) under the study accession number PRJEB29117.
Portuguese Group for the Study of Streptococcal Infections
Members of the Portuguese Group for the Study of Streptococcal Infections are: Teresa Vaz, Marília Gião, Rui Ferreira (Centro Hospitalar do Barlavento Algarvio); Ana Cristina Silva, Hermínia Costa, Maria Fátima Silva, Maria Amélia Afonso (Centro Hospitalar de Entre Douro e Vouga); Ana Domingos, Gina Marrão, José Grossinho (Centro Hospitalar de Leiria); Paulo Lopes, Angelina Lameirão, Gabriela Abreu; Aurélia Selaru (Centro Hospitalar de Vila Nova de Gaia/Espinho); Hermínia Marques, Margarida Tomaz, Paula Mota (Centro Hospitalar do Alto Ave); Maria Helena Ramos, Ana Paula Castro (Centro Hospitalar do Porto); Fernando Fonseca (Centro Hospitalar da Póvoa do Varzim/Vila do Conde); Nuno Canhoto, Teresa Afonso (Hospital Central do Funchal); Teresa Pina, Helena Peres, Odete Chantre, João Marques, Cristina Marcelo, Isabel Peres, Isabel Lourenço, Margarida Pinto (Centro Hospitalar de Lisboa Central); Lurdes Monteiro, Luís Marques Lito (Centro Hospitalar Lisboa Norte); Cristina Toscano, Maria Ana Pessanha (Centro Hospitalar Lisboa Ocidental); Elmano Ramalheira, Raquel Diaz, Sónia Ferreira, Inês Cravo Roxo (Centro Hospitalar do Baixo Vouga); Ana Paula Castro (Hospital de Vila Real); Graça Ribeiro, Rui Tomé, Celeste Pontes, Luísa Boaventura, Catarina Chaves, Teresa Reis (Hospitais da Universidade de Coimbra); Ana Buschy Fonseca (Hospital de Cascais); Manuela Ribeiro, Helena Gonçalves (Hospital de São João, Porto); Alberta Faustino, Adelaide Alves, Maria Cármen Iglesias (Hospital de Braga); Ilse Fontes, Paulo Martinho (Hospital de Santa Luzia, Elvas); Maria Luísa Gonçalves, Olga Neto (Hospital dos SAMS, Lisboa); Luísa Sancho (Hospital Dr. Fernando da Fonseca, Amadora/Sintra); Adriana Coutinho (Hospital do Espírito Santo, Évora); José Diogo, Ana Rodrigues (Hospital Garcia de Orta, Almada); Maria Antónia Read, Valquíria Alves, Margarida Monteiro (Hospital Pedro Hispano, Matosinhos); Rosa Bento (Unidade Local de Saúde do Baixo Alentejo, Beja).
Author Contributions
MP performed the experiments. PGSSI, GF, CP, JB and TK collected data. MP, MM and MR analyzed and interpreted the data. MP, GF, JM-C and MR were involved in the conception and design of the study. MP, GF, CP, MM, JM-C and MR were involved in drafting the manuscript and revising it critically for important intellectual content.
Funding
This work, including the high throughput sequencing at the Genomics Unit of Instituto Gulbenkian de Ciencia (Oeiras, Portugal), was partially supported by the ONEIDA project (LISBOA-01-0145-FEDER-016417) co-funded by FEEI – “Fundos Europeus Estruturais e de Investimento” from “Programa Operacional Regional Lisboa 2020”, by national funds from FCT – “Fundação para a Ciência e a Tecnologia”, and by project UID/BIM/50005/2019, funded by Fundação para a Ciência e a Tecnologia (FCT)/Ministério da Ciência, Tecnologia e Ensino Superior (MCTES) through Fundos do Orçamento de Estado”. SRUC Veterinary Services receives funding from the Scottish Government as part of its public good Veterinary Service Programme and the Scottish Marine Animal Stranding Scheme receives financial support from the Scottish Government Marine Directorate and the UK Department of Environment, Farming and Rural Affairs (Defra).
Conflict of Interest Statement
JM-C has received research grants administered through his university and received honoraria for serving on the speakers bureaus of Pfizer and Merck Sharp, and Dohme. MR has received honoraria for serving on the speakers bureau of Pfizer and for consulting for GlaxoSmithKline and Merck Sharp and Dohme.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00631/full#supplementary-material
Footnotes
- ^ http://pubmlst.org/scanis/
- ^ https://github.com/B-UMMI/INNUca
- ^ https://hub.docker.com/r/ummidock/innuca/
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://hub.docker.com/r/ummidock/prokka/
- ^ https://www.ncbi.nlm.nih.gov/genome/11108
- ^ http://www.cbs.dtu.dk/services/SignalP/
- ^ https://www.ebi.ac.uk/interpro/
- ^ http://www.comparingpartitions.info
- ^ https://www.cdc.gov/streplab/groupa-strep/emm-typing-protocol.html
- ^ https://www.cdc.gov/streplab/groupa-strep/emm-background.html
References
Ahmad, Y., Gertz, R. E., Li, Z., Sakota, V., Broyles, L. N., Van Beneden, C., et al. (2009). Genetic relationships deduced from emm and multilocus sequence typing of invasive Streptococcus dysgalactiae subsp. equisimilis and S. canis recovered from isolates collected in the United States. J. Clin. Microbiol. 47, 2046–2054. doi: 10.1128/JCM.00246-09
Akineden, O., Alber, J., Lämmler, C., Weiss, R., Siebert, U., Foster, G., et al. (2007). Relatedness of Streptococcus equi subsp. zooepidemicus strains isolated from harbour seals (Phoca Vitulina) and grey seals (Halichoerus grypus) of various origins of the North Sea during 1988-2005. Vet. Microbiol. 121, 158–162. doi: 10.1016/j.vetmic.2006.11.015
Amsallem, M., Iung, B., Bouleti, C., Armand-Lefevre, L., Eme, A. L., Touati, A., et al. (2014). First reported human case of native mitral infective endocarditis caused by Streptococcus canis. Can. J. Cardiol. 30, 1462.e1–1462.e2. doi: 10.1016/j.cjca.2014.07.013
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and ats applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bergmann, S., Eichhorn, I., Kohler, T. P., Hammerschmidt, S., Goldmann, O., Rohde, M., et al. (2017). SCM, the M protein of Streptococcus canis binds immunoglobulin G. Front. Cell. Infect. Microbiol. 7:80. doi: 10.3389/fcimb.2017.00080
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brandt, C. M., and Spellerberg, B. (2009). Human infections due to Streptococcus dysgalactiae subspecies equisimilis. Clin. Infect. Dis. 49, 766–772. doi: 10.1086/605085
Carriço, J. A., Silva-Costa, C., Melo-Cristino, J., Pinto, F. R., Lencastre, H., Almeida, J. S., et al. (2006). Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44, 2524–2532. doi: 10.1128/JCM.02536-05
Castinel, A., Duignan, P. J., Pomroy, W. E., López-Villalobos, N., Gibbs, N. J., Chilvers, B. L., et al. (2007). Neonatal mortality in New Zealand sea lions (Phocarctos Hookeri) at Sandy Bay, Enderby Island, Auckland Islands from 1998 to 2005. J. Wild Dis. 43, 461–474. doi: 10.7589/0090-3558-43.3.461
Chalmers, G., McLean, J., Hunter, D. B., Brash, M., Slavic, D., Pearl, D. L., et al. (2015). Staphylococcus spp., Streptococcus canis, and Arcanobacterium phocae of healthy Canadian farmed mink and mink with pododermatitis. Can. J. Vet. Res. 79, 129–135.
Cunningham, M. W. (2000). Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511. doi: 10.1128/CMR.13.3.470
Devriese, L. A., Hommez, J., Kilpper-Balz, R., and Schleifer, K. H. (1986). Streptococcus canis sp. nov.: a species of group G streptococci from animals. Int. J. Syst. Bacteriol. 36, 422–425. doi: 10.1099/00207713-36-3-422
DeWinter, L. M., Low, D. E., and Prescott, J. F. (1999). Virulence of Streptococcus canis from canine streptococcal toxic shock syndrome and necrotizing fasciitis. Vet. Microbiol. 70, 95–110. doi: 10.1016/S0378-1135(99)00128-5
Fulde, M., Rohde, M., Hitzmann, A., Preissner, K. T., Nitsche-Schmitz, D. P., Nerlich, A., et al. (2011). SCM, a novel M-like protein from Streptococcus canis, binds (mini)-plasminogen with high affinity and facilitates bacterial transmigration. Biochem. J. 434, 523–535. doi: 10.1042/BJ20101121
Fulde, M., Rohde, M., Polok, A., Preissner, K. T., Chhatwal, G. S., and Bergmann, S. (2013). Cooperative plasminogen recruitment to the surface of Streptococcus canis via M protein and enolase enhances bacterial survival. mBio 4:e00629-12. doi: 10.1128/mBio.00629-12
Hariharan, H., Matthew, V., Fountain, J., Snell, A., Doherty, D., King, B., et al. (2011). Aerobic bacteria from mucous membranes, ear canals, and skin wounds of feral cats in Grenada, and the antimicrobial drug susceptibility of major isolates. Comp. Immunol. Microbiol. Infect. Dis. 34, 129–134. doi: 10.1016/j.cimid.2010.05.001
Hassan, A. A., Akineden, O., and Usleber, E. (2005). Identification of Streptococcus canis isolated from milk of dairy cows with subclinical mastitis. J. Clin. Microbiol. 43, 1234–1238. doi: 10.1128/JCM.43.3.1234-1238.2005
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lyskova, P., Vydrzalova, M., and Mazurova, J. (2007). Identification and antimicrobial susceptibility of bacteria and yeasts isolated from healthy dogs and dogs with otitis externa. J. Vet. Med. A Physiol. Pathol. Clin. Med. 54, 559–563. doi: 10.1111/j.1439-0442.2007.00996.x
Morrow, B. L., McNatt, R., Joyce, L., McBride, S., Morgan, D., Tressler, C., et al. (2016). Highly pathogenic beta-hemolytic streptococcal infections in cats from an institutionalized hoarding facility and a multi-species comparison. J. Feline Med. Surg. 18, 318–327. doi: 10.1177/1098612X15582233
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carriço, J. A., and Vaz, C. (2017). PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129. doi: 10.1093/bioinformatics/btw582
Nikolaisen, N. K., Lassen, D. C. K., Chriél, M., Larsen, G., Jensen, V. F., and Pedersen, K. (2017). Antimicrobial resistance among pathogenic bacteria from mink (Neovison vison) in Denmark. Acta Vet. Scand. 59:60. doi: 10.1186/s13028-017-0328-6
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pinho, M. D., Matos, S. C., Pomba, C., Lübke-Becker, A., Wieler, L. H., Preziuso, S., et al. (2013). Multilocus sequence analysis of Streptococcus canis confirms the zoonotic origin of human infections and reveals genetic exchange with Streptococcus dysgalactiae subsp. equisimilis. J. Clin. Microbiol. 51, 1099–1109. doi: 10.1128/JCM.02912-12
Richards, V. P., Zadoks, R. N., Bitar, P. D. P., Lefébure, T., Lang, P., Werner, B., et al. (2012). Genome characterization and population genetic structure of the zoonotic pathogen. Streptococcus canis. BMC Microbiol. 12:293. doi: 10.1186/1471-2180-12-293
Sanderson-Smith, M., De Oliveira, D. M., Guglielmini, J., McMillan, D. J., Vu, T., Holien, J. K., et al. (2014). A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J. Infect. Dis. 210, 1325–1338. doi: 10.1093/infdis/jiu260
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seguel, M., Gutiérrez, J., Hernández, C., Montalva, F., and Verdugo, C. (2018). Respiratory mites (Orthohalarachne diminuata) and β-hemolytic streptococci-associated bronchopneumonia outbreak in South American fur seal pups (Arctocephalus australis). J. Wild Dis. 54, 380–385. doi: 10.7589/2017-09-214
Severiano, A., Pinto, F. R., Ramirez, M., and Carriço, J. A. (2011). Adjusted Wallace coefficient as a measure of congruence between typing methods. J. Clin. Microbiol. 49, 3997–4000. doi: 10.1128/JCM.00624-11
Simpson, V. R. (2006). Patterns and significance of bite wounds in Eurasian otters (Lutra lutra) in southern and south-west England. Vet. Rec. 158, 113–119. doi: 10.1136/vr.158.4.113
Swenshon, M., Lämmler, C., and Siebert, U. (1998). Identification and molecular characterization of beta-hemolytic streptococci isolated from harbor porpoises (Phocoena phocoena) of the North and Baltic Seas. J. Clin. Microbiol. 36, 1902–1906.
Taniyama, D., Abe, Y., Sakai, T., Kikuchi, T., and Takahashi, T. (2017). Human case of bacteremia caused by Streptococcus canis sequence type 9 harboring the scm gene. IDCases 7, 48–52. doi: 10.1016/j.idcr.2017.01.002
Timoney, J. F., Velineni, S., Ulrich, B., and Blanchard, P. (2017). Biotypes and ScM types of isolates of Streptococcus canis from diseased and healthy cats. Vet. Rec. 180:358. doi: 10.1136/vr.103868
Vossen, A., Abdulmawjood, A., Lämmler, C., Weiss, R., and Siebert, U. (2004). Identification and molecular characterization of beta-hemolytic streptococci isolated from harbor seals (Phoca vitulina) and grey seals (Halichoerus grypus) of the German North and Baltic Seas. J. Clin. Microbiol. 42, 469–473. doi: 10.1128/JCM.42.1.469-473.2004
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., and Sakthikumar, S. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Whatmore, A. M., Engler, K. H., Gudmundsdottir, G., and Efstratiou, A. (2001). Identification of isolates of Streptococcus canis infecting humans. J. Clin. Microbiol. 39, 4196–4199. doi: 10.1128/JCM.39.1.4196-4199.2001
Keywords: Streptococcus canis, multilocus sequence typing, M-like protein (SCM) gene, wildlife, genome
Citation: Pinho MD, Foster G, Pomba C, Machado MP, Baily JL, Kuiken T, Melo-Cristino J, Ramirez M and the Portuguese Group for the Study of Streptococcal Infections (2019) Streptococcus canis Are a Single Population Infecting Multiple Animal Hosts Despite the Diversity of the Universally Present M-Like Protein SCM. Front. Microbiol. 10:631. doi: 10.3389/fmicb.2019.00631
Received: 12 October 2018; Accepted: 13 March 2019;
Published: 29 March 2019.
Edited by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Bernard Beall, Centers for Disease Control and Prevention (CDC), United StatesSilvia Preziuso, University of Camerino, Italy
Claire Turner, The University of Sheffield, United Kingdom
Copyright © 2019 Pinho, Foster, Pomba, Machado, Baily, Kuiken, Melo-Cristino, Ramirez and the Portuguese Group for the Study of Streptococcal Infections. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcos D. Pinho, bWRwaW5ob0BtZWRpY2luYS51bGlzYm9hLnB0
†Present address: Johanna L. Baily, Institute of Aquaculture, University of Stirling, Stirling, United Kingdom