 Céline Henry1*†
Céline Henry1*† Lucia Haller1,2†
Lucia Haller1,2† Mélisande Blein-Nicolas3Michel Zivy3
Mélisande Blein-Nicolas3Michel Zivy3 Alexis Canette4
Alexis Canette4 Morgane Verbrugghe2Christine Mézange2Mylène Boulay2Rozenn Gardan2Samantha Samson5
Morgane Verbrugghe2Christine Mézange2Mylène Boulay2Rozenn Gardan2Samantha Samson5 Véronique Martin5
Véronique Martin5 Gwenaëlle André-Leroux5Véronique Monnet1,2*
Gwenaëlle André-Leroux5Véronique Monnet1,2*- 1Micalis Institute, PAPPSO, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France
- 2Micalis Institute, ComBac, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France
- 3PAPPSO, GQE – Le Moulon, INRA, Université Paris-Sud, CNRS, AgroParisTech, Université Paris-Saclay, Gif-sur-Yvette, France
- 4Micalis Institute, MIMA2, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France
- 5MaIAGE, INRA, Université Paris-Saclay, Jouy-en-Josas, France
Protein phosphorylation especially on serine/threonine/tyrosine residues are frequent in many bacteria. This post-translational modification has been associated with pathogenicity and virulence in various species. However, only few data have been produced so far on generally recognized as safe bacteria used in food fermentations. A family of kinases known as Hanks-type kinases is suspected to be responsible for, at least, a part of these phosphorylations in eukaryotes as in bacteria. The objective of our work was to establish the first phosphoproteome of Streptococcus thermophilus, a lactic acid bacterium widely used in dairy fermentations in order to identified the proteins and pathways tagged by Ser/Thr/Tyr phosphorylations. In addition, we have evaluated the role in this process of the only Hanks-type kinase encoded in the S. thermophilus genome. We have constructed a mutant defective for the Hanks type kinase in S. thermophilus and established the proteomes and phosphoproteomes of the wild type and the mutant strains. To do that, we have enriched our samples in phosphopeptides with titane beads and used dimethyl tags to compare phosphopeptide abundances. Peptides and phosphopeptides were analyzed on a last generation LC-MS/MS system. We have identified and quantified 891 proteins representing half of the theoretical proteome. Among these proteins, 106 contained phosphorylated peptides. Various functional groups of proteins (amino acid, carbon and nucleotide metabolism, translation, cell cycle, stress response, …) were found phosphorylated. The phosphoproteome was only weakly reduced in the Hanks-type kinase mutant indicating that this enzyme is only one of the players in the phosphorylation process. The proteins that are modified by the Hanks-type kinase mainly belong to the divisome.
Introduction
Protein post-translational modifications are frequent and diverse in bacteria. These modifications, not genetically encoded, affect protein conformation, activity and function. They allow quick sensing and responses to environmental changes and participate in transduction networks. Among described protein modifications, phosphorylation is the most documented. It targets several amino acids, but serine, threonine and tyrosine phosphorylations are the most studied, as they are stable enough to be analyzed by mass spectrometry. Ser/Thr/Tyr phosphorylation has frequently been associated with bacterial pathogenicity in various species. Kinases, belonging to a main structural family, first described by Hanks et al. (1988) and widespread in both eukaryotes and procaryotes, have emerged as potentially responsible for these phosphorylations (Cousin et al., 2013). Stancik et al. (2018) recently proposed to call these kinases ‘Hanks-type kinases.’
We were, in this work, interested in the role of Ser/Thr/Tyr protein phosphorylation in Streptococcus thermophilus, that is the only GRAS (Generally Recognized As Safe) species in the Streptococcaceae family, which contains mostly pathogenic and commensal species. S. thermophilus is widely and worldwide used as a technological starter bacterium in various milk product fermentations. S. thermophilus also belongs to the so-called group of lactic acid bacteria merging diverse bacterial species characterized by their ability to produce lactic acid during fermentation and associated to plant, meat and dairy products. During technological processes, S. thermophilus has to adapt to various nutrition and physical-chemical stresses that require rapid adjustments. We already demonstrated that streptococci have developed specific cell-cell communication and regulation systems based on peptide pheromones to control specific functions (Fleuchot et al., 2011). In this study, we investigated another way for S. thermophilus to regulate specific pathways namely post-translational protein modifications and more specifically protein serine/threonine/tyrosine phosphorylation. We aimed at identifying the pathways that are modulated by phosphorylation in this bacterium.
Phosphoproteomic studies are very scare in lactic acid bacteria and exclusively concern serine/threonine/tyrosine residues. A pioneer study on the Ser/Thr/Tyr phosphoproteome of Lactococcus lactis, another dairy technological starter identified 73 phosphorylation sites and multiple multi-phosphorylated proteins. The phosphorylated proteins are well represented in house-keeping and glycolytic pathways (Soufi et al., 2008). In Oenococcus oeni, a bacterium used in grape juice fermentation, 39 phosphorylation sites were identified in 19 proteins (Napoli et al., 2014). Finally, a study on the effect of acid stress on protein phosphorylation in Lactobacillus rhamnosus GG identified 15 proteins from central pathways and especially from carbon metabolism and glycolysis that are phosphorylated in response to acid (Koponen et al., 2012).
In streptococci, global Ser/Thr/Tyr phosphoproteomic studies exclusively concern the model pathogenic species, S. pneumoniae. Phosphoproteomics of this bacterium, established in exponentially growing state, allowed to identify 84 phosphorylated proteins. The latter are involved in carbon, nitrogen, nucleic acid metabolisms, cell division but also targets proteins of unknown functions (Sun et al., 2010). The kinases involved in Ser/Thr/Tyr phosphorylation in streptococci and lactic acid bacteria are not well identified. S. pneumoniae, Streptococcus suis as well as Streptococcus agalactiae have one Hanks-type kinase encoding gene in their genomes. The S. pneumoniae Hanks-type kinase is localized at the division site and involved in cell division control (Beilharz et al., 2012). It phosphorylates at least 10 proteins, including cell division proteins, proteins of unknown functions and the Hanks-type kinase itself (Silvestroni et al., 2009). The Hanks-type kinase from S. suis is involved in the phosphorylation of 12 proteins associated with cell cycle, glycolysis and translation and participates in pathogenicity in mice (Zhang et al., 2017). The one of S. agalactiae phosphorylates at least six proteins, including a pyrophosphatase. It also modulates purine biosynthesis and growth (Rajagopal et al., 2003, 2005). From these studies, realized in different conditions and with different experimental protocols, it remains difficult to predict the set of proteins that are targets of Hanks-type kinases. In addition, the relative role of the Hanks-type kinase in the whole Ser/Thr/Tyr process is not well established and the other kinases acting in the process are not identified. Production of new bacterial phosphoproteomics studies will help to identify the actors of phosphorylation and the basis of their specificities. The objectives of the present work were to investigate the Ser/Thr/Tyr phosphorylation process and its role in S. thermophilus, the only streptococcal species used in food fermentation, and to assess the specific role of the only predicted Hanks-type kinase, named PknB, encoded in its genome.
We showed that the S. thermophilus Ser/Thr/Tyr phosphoproteome is of the same order of magnitude than the other streptococci ones as peptides belonging to 106 proteins from various metabolic pathways were found phosphorylated in one bacterial growth condition. We demonstrated that the Hanks-type kinase, named PknB in S. thermophilus, targets the divisome and is only one of the players in the Ser/Thr/Tyr phosphorylation process. Consistent with these results, the pknB deletion mutant exhibits a clear phenotype with longer whimsical chains and affected division process. Finally, we identified two putative kinases that could, with PknB, phosphorylate proteins in S. thermophilus.
Materials and Methods
In silico Screening of the Whole Genome of Streptococcus thermophilus LMD9 and Identification of Structural Homologs to PknB
The whole genome was downloaded from the NCBI1 and each of the 1681 coding genes was split into a single fasta sequence. Then the HHsuite (Söding et al., 2005), dedicated to homology detection and structure prediction, was used to predict sequences that could share the 3D fold of the kinase domain of PknB from Mycobacterium tuberculosis that serves as template. For each fasta sequence of the genome, a multiple sequence analysis (MSA) was performed using HHblits, and a matrix of similarity score using Hidden Markov Model was calculated. Each profile was then compared to the HMM profile of the PknBMtb kinase template. Only were kept for sequence/structure/function analysis the sequences that associated a probability above 95% to share the PknBMtb kinase fold, with a size above 100 amino-acid residues and an e-value below 1.e−15.
Homology Modeling of PknB Catalytic Domain and PASTA Domains
The catalytic domain of PknBSt (residues 3–274) was homology-modeled using the model-building software Modeller (version 9.18) (Sali and Blundell, 1993). The crystal structure of the catalytic domain of PknB from M. tuberculosis (PknBMtb 1O6Y pdb id) in complex with AMP-PCP served as 3D template (Ortiz-Lombardia et al., 2003). Hundred models were generated. A final model of PknBSt was selected upon the lowest score function values (DOPE score) and best stereochemistry, checked by Molprobity (Chen et al., 2010). Subsequently, a molecule of ATP was docked in the binding pocket of this model of PknBSt, with Autodock tools, using a grid box, centered on the ATP-binding pocket, and the genetic algorithm of Lamarck (Morris et al., 2009). To validate this procedure, a crystal molecule of AMP-PCP was previously redocked in the ATP-binding pocket of PknBMtb using similar gridbox and algorithm. Similarly, the three PASTA domains (residues 363–579) of the extracellular domain (ECD) of PknBSt were homology modeled using Modeller, from the PASTA-domain crystal templates of PknBMtb (pdb id 5E12, Prigozhin et al., 2016) and PASTA from Ser/Thr protein kinase (STK1) of Staphylococcus aureus (pdb id 3M9G, Paracuellos et al., 2010) as 3D templates. A final model of PASTA-ECDSt was chosen upon lowest score functions and subsequently checked by Molprobity. Using HHpred2 (Zimmermann et al., 2018) the residues 275–362 that correspond to the juxtamembrane and the transmembrane segments, showed no structural homolog so that this portion could not be modeled. For the same reason, the C-terminal stretch of 44 residues could not be modeled. All the selected models were visually inspected and figures generated are rendered with Pymol 2.0.7. (Schrödinger, LLC).
Construction of a ΔpknB Mutant Strain
We used the overlapping PCR method to delete a part of the PknB encoding gene (STER_1392; STER_RS06845) in the LMD-9 strain (Makarova et al., 2006). Briefly, we generated 3 fragments by PCR using the oligonucleotides described in Table 1. The first fragment (1350 bp) was the kanamycin (aphaA3) cassette, which was amplified with oligonucleotides AphaA3-F and AphaA3-R using the plasmid pKa as template (Débarbouillé et al., 1990). The two other fragments (450 bp each), called F1 and F2, were located upstream and downstream the pknB gene and were amplified with oligonucleotides 1–2 and 3–4, respectively, using chromosome DNA from S. thermophilus LMD-9 as template (Table 1). Thanks to the presence of extensions compatible with the apha3 cassette in oligonucleotides 2 and 3, the three fragments aphaA3, F1 and F2 were fused together in one PCR. The resulting PCR fragment was used to transform S. thermophilus taking advantage of its natural competence in chemically defined medium (Letort and Juillard, 2001). The clones in which a double recombination occurred and which were kanamycin resistant were selected and checked for integration of the aphaA3 cassette at the pknB locus with oligonucleotides 5 and 6 (2431 bp in the mutant) (Table 1). One clone was kept, verified by sequencing and stored under the number TIL1509.
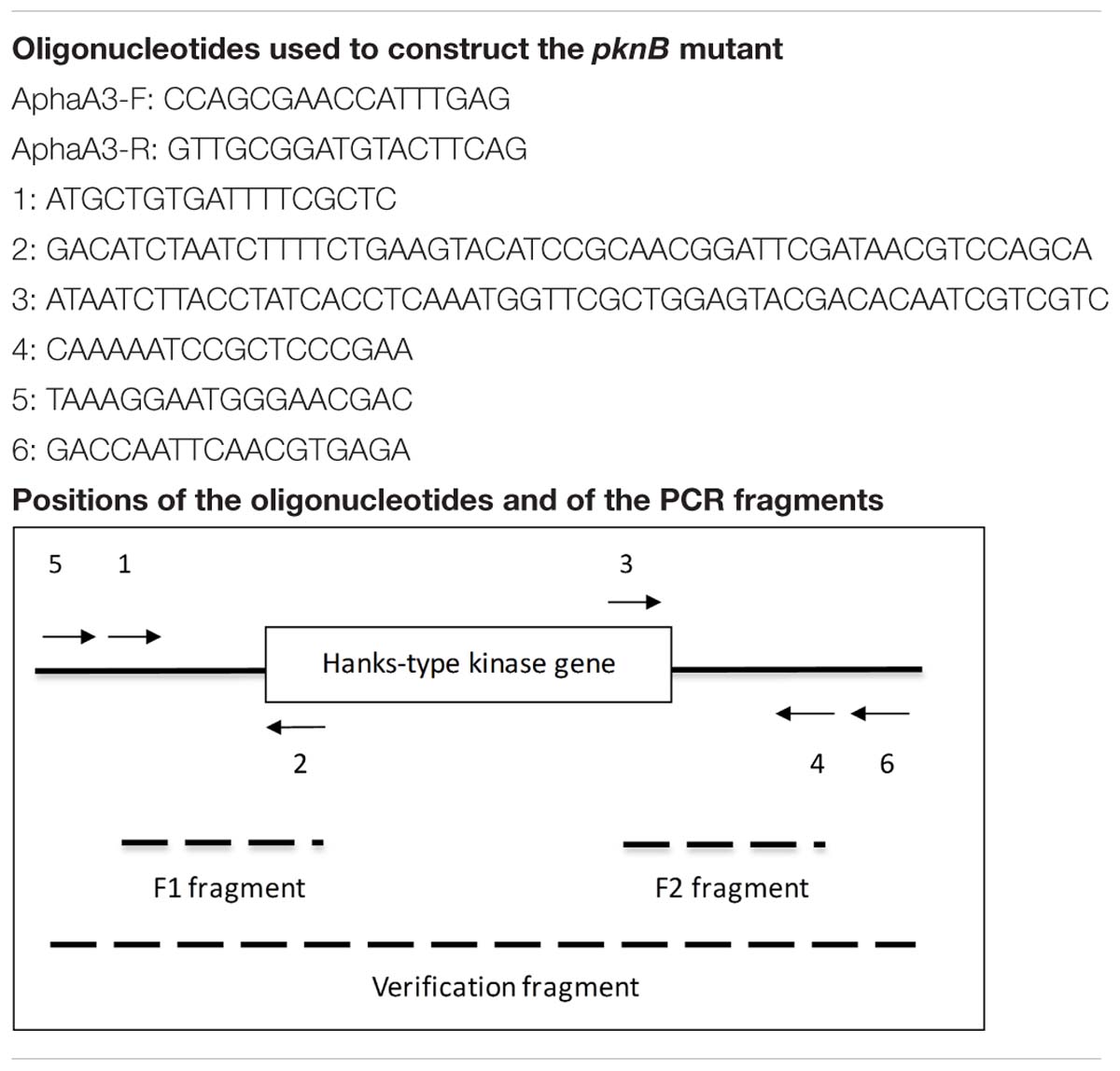
Table 1. Strains and oligonucleotides.
Culture Conditions
S. thermophilus strain LMD-9 (WT) and its ΔpknB mutant (Mut) were grown in M17 medium (Difco) supplemented with 10 g.L−1 lactose (M17Lac) at 42°C and stopped in the exponential growth phase when pH reached 6.20 (after 2,5 h for the wild type and 3 h for the mutant). Agar (1.5%) was added to the medium as appropriate. When required, kanamycin (1 mg.ml−1) was added to the medium. The optical density at 600 nm of cultures was measured with an Uvikon 931 spectrophotometer (Kontron). pH was also followed using a pH-meter. For proteomics experiments, four independent replicates of the culture of each strain (WT 1–4, Mut 1–4) were performed, so that the total number of samples was eight.
A stress test was realized as follows: cultures were grown in triplicates up to stationary phase. Numerations were done at this time, and again after one night freezing at −20°C and defreezing at 25°C. Bacterial counts were compared for each strain, before and after freezing.
Scanning Electron Microscopy Analysis
For two replicates of WT and ΔpknB mutant, one 40 μL drop of fresh M17 liquid culture (collected at exponential phase; OD600nm = 0.7) was deposited onto a sterile aluminum coupon (10 mm diameter, sterilized just before use by sonication in ethanol and dried during UV exposure) placed into one well of a 24-wells polystyrene plate. Sedimentation of bacteria lasted 1.5 h. Samples were fixed via careful immersion in a solution of 2.5% (v/v) glutaraldehyde in 0.1 M sodium cacodylate buffer at pH 7.4 and room temperature (RT) followed by overnight waiting time at 4°C. Samples were then washed three times for 5 min with 0.1 M sodium cacodylate buffer. Samples were dehydrated with increasing concentrations of ethanol at room temperature (50–70–90%-3 × 100%, v/v with ultra-pure water) for 10 min at each step. Samples were critical-point dried (Quorum Technologies K850, Elexience, France) at 70 bar and 37°C with liquid CO2 as the transition fluid and then depressurized slowly (400 cm3 min−1). Each aluminum support carrying the sample was then mounted on an aluminum stub with double-sided carbon tape. Samples were sputter-coated (Polaron SC7640, Elexience, France) in Ar plasma with Pt (approximately 30 nm thick) at 10 mA and 0.8 kV over a duration of 200 s. Observations were performed in a field-emission SEM (Hitachi S4500, Elexience, France) in high vacuum, with a secondary electron low detector, at 2 kV and 16 mm working distance, on the MIMA2 imaging platform (INRA3).
Protein Extraction
At an OD600 of 0.7, bacteria were harvested by centrifugation (10 min at 6,000 g, 4°C). The cell pellet was washed twice with phosphate buffered saline (200 mM, pH 6.4) and resuspended in lysis buffer (Tris-HCl 50 mM pH 8.0; 50 μl/ml Protease Inhibitor Cocktail P8465, Sigma; 10 μl/ml Halt Phosphatase Inhibitor Cocktail 78420, Thermo Scientific). Cells were lysed using a Cell Disruptor (One Shot 0.75 kW, Constant Systems Ltd.) and non-lysed cells as well as cell debris were separated from the protein-containing supernatant by centrifugation (15 min at 20,000 g, 4°C). The protein content was measured using the Bradford assay (Bradford, 1976) and the protein extract was used for the proteome as well as for the phosphoproteome analyses.
Sample Preparation for Proteome Analysis
For the proteome analysis (Figure 1), 10 μg of protein extract were used for a short migration 1D gel electrophoresis (NuPAGE® 4-12% Bis-Tris Gel, novex). Proteins were visualized using Coomassie G-250 (SimplyBlueTM SafeStain, Invitrogen) and the whole colored part of each lane was cut into small pieces. The gel pieces were destained using Solvent A (10% v/v acetic acid, 40% v/v ethanol) and Solvent B (50% v/v 50 mM ammonium bicarbonate, 50% v/v acetonitrile). The proteins contained in the gel were reduced by 10 mM dithiothreitol (Sigma) and alkylated by 55 mM iodoacetamide (Sigma). The proteins were digested with 200 ng trypsin (Promega) and afterwards extracted using a solution of 0.5% v/v trifluoroacetic acid and 50% v/v acetonitrile. The peptides were dried completely using a concentrator (SavantTM SPD121D, Thermo Fisher Scientific) and taken up in 50 μl loading buffer (0.08% v/v trifluoroacetic acid, 2% v/v acetonitrile) for LC-MS/MS proteome analysis (4 μl = 800 ng peptide per injection).
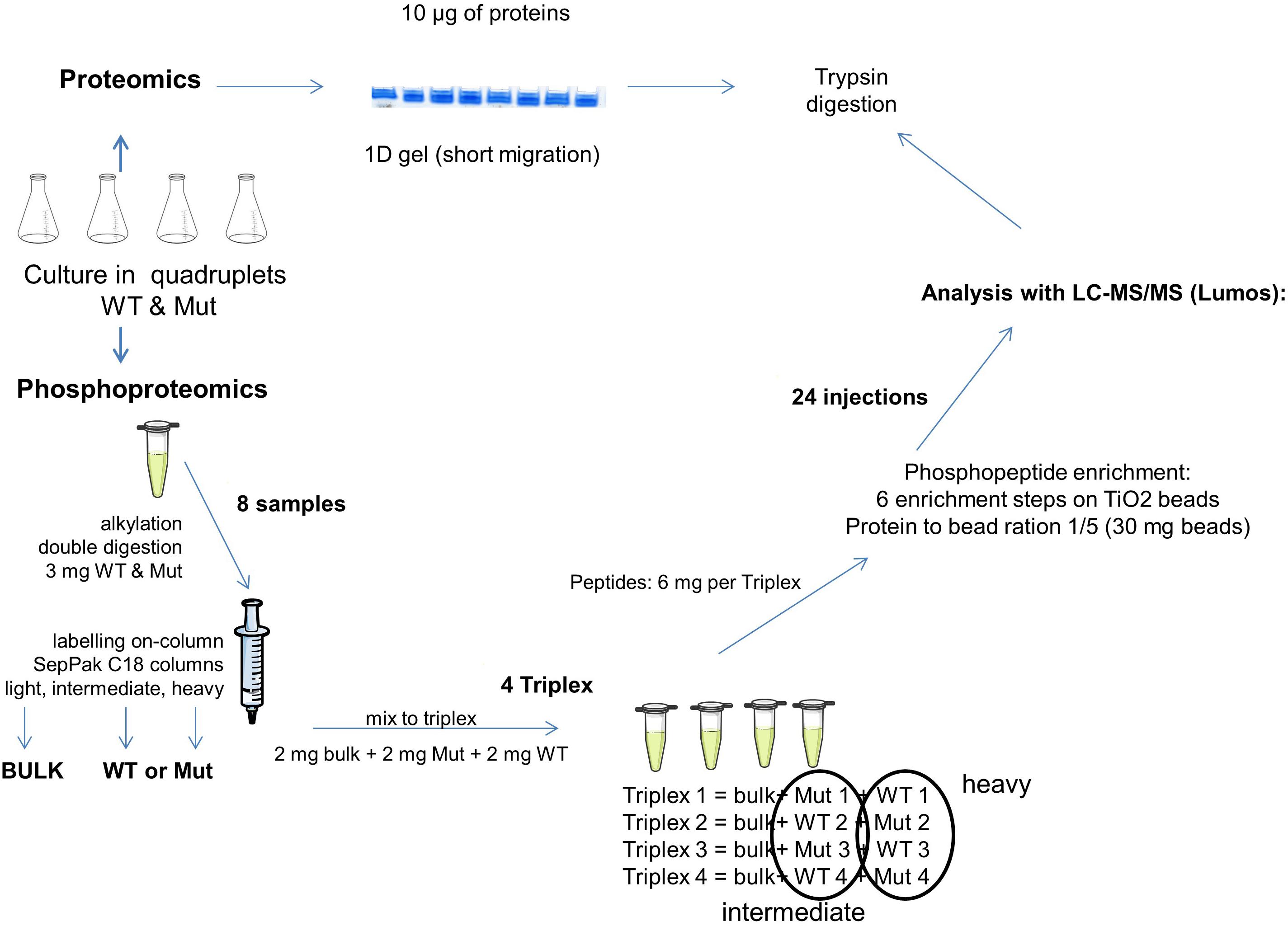
Figure 1. Schematic representation of the proteomics and phosphoproteomics pipeline used in this study. Bulk, mixture of all samples; WT, samples from the wild type strain; Mut, samples from the mutant strain.
Peptide Labeling for Phosphoproteome Analysis
For the phosphoproteome analysis (Figure 1), 3 mg of protein extract were denatured using 6 M urea and 2 M thiourea as well as 0.1% w/v of RapiGest (RapiGest SF Surfactant, Waters). Reduction and alkylation were achieved by 5 mM dithiothreitol and 15 mM iodoacetamide. Proteins were digested by Lys-C (Lysyl Endopeptidase®, Mass Spectrometry Grade, Wako; enzyme/substrate ratio of 1:50 w/w) and, after a dilution to 1 M urea, by trypsin (Sequencing Grade Modified Trypsin, Promega; 1:50 w/w). The reaction was terminated with 0.1% v/v trifluoroacetic acid.
The samples were labeled on SepPak C18 cartridge columns 3 cc (Waters) with 7 times 1 ml of their respective labeling solutions during 10 min (Boersema et al., 2009). The solution for light labeling was composed of 90% v/v 50 mM sodium phosphate buffer pH 7.5, 5% v/v formaldehyde 4%, 5% v/v 0.6 M cyanoborohydride. For the intermediate labeling, formaldehyde was replaced with deuterated formaldehyde and for heavy labeling, formaldehyde was replaced with 13C and deuterium labeled formaldehyde and cyanoborohydride was replaced with cyanoborodeuteride. The labeled peptides were eluted twice with 500 μl elution buffer (0.6% v/v acetic acid, 80% v/v acetonitrile).
Four triplex containing light-, intermediate-, and heavy-labeled samples in equal proportions (2 mg of peptide per labeling) were assembled. The light sample was the technical control. It was prepared by mixing equal quantities of all samples of the experiment. To eliminate any possible bias due to different labeling efficiencies or binding affinities, the labeling of the different replicates was switched, so that in fine two replicates of each strain were labeled “heavy” and the two others were labeled “intermediate” (Figure 1).
Phosphopeptide Enrichment
The experimental protocol for phosphoproteomics used in this study involved a titane beads enrichment step. It was lighter and more rapid than a protocol based on SCX chromatography while preserving the same effectiveness (Misra et al., 2014).
The triplexes were concentrated to a volume of 200 μl before being mixed with 555 μl lactic acid, 1,200 μl acetonitrile, 2 μl trifluoroacetic acid and filled up to 2 ml with purified H2O (Milli-Q®, Merck Millipore). The enrichment was performed in six consecutive steps with 30 mg TiO2 beads (Titansphere TiO, 10 μm, GL Sciences) per step. The TiO2 beads were first washed with loading solution (80% v/v acetonitrile, 6% v/v trifluoroacetic acid) and then transferred to the sample. The sample-bead mixture was incubated at room temperature for 20 min at 7 rpm. After spinning down (3 min at 4,000 g) the beads, the supernatant was transferred to the beads of the next enrichment step and incubation and spinning down were repeated as above. The beads with the bound phosphopeptides were washed once with Wash solution I (30% v/v acetonitrile, 1% v/v trifluoroacetic acid) and once more with Wash solution II (80% v/v acetonitrile, 1% v/v trifluoroacetic acid) before being transferred to a tip containing a 3M EmporeTM phase (Sigma-Aldrich). Elution of the phosphopeptides into a tube containing 20 μl 20% v/v trifluoroacetic acid was completed in 2 steps, first with 2 times 100 μl of the basic Elution solution I (5% v/v ammonium hydroxide, 60% v/v acetonitrile) and then with 50 μl of the acidic Elution solution II (80% v/v acetonitrile, 1% v/v formic acid) under rotation (3 min at 4,000 g). Samples were reduced in a concentrator to a volume of 1 μl and taken up in 30 μl loading buffer (0.08% v/v trifluoroacetic acid, 2% v/v acetonitrile) for LC-MS/MS phosphoproteome analysis. The mass shift caused by light, intermediate and heavy dimethylation was +28, +32, and +36 Da, respectively.
Liquid Chromatography – Mass Spectrometry
Mass spectrometry was performed on the PAPPSO platform (MICALIS, INRA, Jouy-en-Josas, France4). An Orbitrap FusionTM LumosTM TribridTM (Thermo Fisher Scientific) coupled to an UltiMateTM 3000 RSLCnano System (Thermo Fisher Scientific) was used.
A 4 μl sample was loaded at 20 μl/min on a precolumn (μ-Precolumn, 300 μm i.d × 5 mm, C18 PepMap100, 5 μm, 100 Å, Thermo Fisher) and washed with loading buffer. After 3 min, the precolumn cartridge was connected to the separating column (Acclaim PepMap®, 75 μm × 500 mm, C18, 3 μm, 100 Å, Thermo Fisher). Buffer A consisted of 0.1% formic acid in 2% acetonitrile and buffer B of 0.1% formic acid in 80% acetonitrile.
The proteome runs were executed at 300 nl/min with a linear gradient from 0 to 45% buffer B for 118 min. Including regeneration (98% buffer B), one run took 137 min. Ionization (1.6 kV ionization potential) and capillary transfer (275°C) were performed with a liquid junction and a capillary probe. Peptide ions were analyzed using Xcalibur 3.1.66.10 with the following machine setup in CID (Collision Induced Dissociation) mode: (1) full MS scan in Orbitrap (scan range [m/z] = 400–1500) and (2) MS/MS using CID (35% collision energy) in Ion Trap (AGC target = 4.0 × 103, max. injection time = 300 ms, data type = centroid). Analyzed charge states were set to 2–7, the dynamic exclusion to 60 s and the intensity threshold was fixed at 5.0 × 103.
The peptide separation for the phosphoproteome analysis was achieved at 300 nl/min with a linear gradient from 0 to 35% buffer B for 48 min and 35–45% for 5 min. One run took 65 min, including the regeneration step at 98% buffer B. Ionization (1.6 kV ionization potential) and capillary transfer (275°C) were performed with a liquid junction and a capillary probe (SilicaTipTM Emitter, 10 μm, New Objective). Peptide ions were analyzed using Xcalibur 3.1.66.10. In HCD (Higher-energy Collisional Dissociation) mode the machine settings were as follows: (1) full MS scan in Orbitrap (scan range [m/z] = 400–1600) and (2) MS/MS using HCD (30% collision energy) in Orbitrap (AGC target = 5.0 × 104, max. injection time = 55 ms, data type = centroid). Analyzed charge states were set to 2–5, the dynamic exclusion to 40 s and the intensity threshold was fixed at 104.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the dataset identifier PXD011391.
Data Analyses
The S. thermophilus LMD-9 database (NCBI, version 2010, 1710 entries) was searched by using X!TandemPipeline version 3.4.2 (Langella et al., 2017). In the database used, the proteins where initially annotated using a Blast against the genomes of the strains CNRZ1066 and LMG18311 (which were the two first strains sequenced) with variable e-values which are not always very high and which consequently must be considered carefully.
The proteome identification was run with a precursor mass tolerance of 10 ppm and a fragment mass tolerance of 0.5 Da. Enzymatic cleavage rules were set to trypsin digestion (“after Arg and Lys, unless Pro follows directly after”) and no semi-enzymatic cleavage rules were allowed. The fix modification was set to cysteine carbamidomethylation and methionine oxidation was considered as a potential modification. In a second pass, N-terminal acetylation was added as another potential modification, whereas all other previous settings were retained. The identified proteins were filtered as follows: (1) peptide E-value < 0.05 with a minimum of 2 peptides per protein and (2) a protein E-value of <10−4. In routine analyses, protein identification was associated to a function prediction obtained via an elementary Blast toward two S. thermophilus genome sequences (LMG13811 and CNRZ1066). This function prediction need to be carefully considered. A more in-depth Blast was realized against the whole NCBI database for the proteins which were identified as phosphorylated.
For the phosphoproteome identification, the precursor mass tolerance was set at 10 ppm, while the fragment mass tolerance was set at 0.02 Da. The enzymatic cleavage was set to tryptic rules with no semi-enzymatic cleavages being considered. Cysteine carbamidomethylation as well as the mass-increments due to labeling (+28.0313 Da, +32.0564 Da, +36.0756 Da) at the C-terminus and lysine residues were set to static modifications. Methionine oxidation and S/T/Y phosphorylation were considered as potential modifications. A refinement search was performed with the above mentioned parameters and the additional potential modification of N-terminal acetylation. The identified phosphoproteins were filtered as follows: (1) stringent peptide E-value < 0.001 with a minimum of 1 peptide per protein and (2) a protein E-value of <10−2. The labeling efficiency was at 99.7%.
The PAPPSO tool X!TandemPipeline (Langella et al., 2017) works with phosphoislands and identified the best phosphorylation position as well as all possible positions when several phosphorylations are possible within the same peptide.
Proteins and phosphoproteins were quantified by the spectral counting (SC) method. MassChroQ “Black Caiman” version 2.2.1 (Valot et al., 2011) was used for the alignment of retention time. The range for peak detection was set to 10 ppm with a detection threshold ranging from 30,000 to 50,000.
MassChroqR (version 0.3.7), an R package developed by PAPPSO platform5 was used to check the quality of data and practice statistical analysis in proteomic. The abundance in number of spectra was modeled using the following generalized mixed model (GLM) with a Poisson distribution as already described (Millan-Oropeza et al., 2017). Protein abundance change were detected by analysis of variance (ANOVA) using a Chi-square test. The obtained p-values were adjusted for multiple testing by the Benjamini-Hochberg approach (Benjamini and Hochberg, 1995). Adjusted P-values obtained from ANOVA for the proteome was considered significant below a value of 0.05.
For phosphoproteomics, quantification was performed at the peptide level and not at the protein level. In this case, the number of spectra associated to each peptide is generally low (i.e., comprised between 0 and 2), which prevent proper statistical analysis of spectral counting data. We thus focused on the phosphopeptides showing presence/absence variations between the WT and the Mut strains. To ensure robustness of the results, a peptide was considered as present when detected with at least one spectrum in all the four replicates and absent when not detected in none of the four replicates of a strain.
The proteins identified as included in the phosphoproteome have been blasted against the non-redundant NCBI database (update 2018/07/07) for a more specific annotation.
Results
The objectives of this work were, first, to establish the phosphoproteome of S. thermophilus, the only GRAS species in the streptococci genus, second, to identify the pathways targeted by phosphorylation and third, to evaluate the role of the only Hanks-type kinase identified in the genome of this species in this process. To achieve these goals, we performed in parallel a global shotgun proteomics and a specific phosphoproteomics analyses on both the wild type strain and its Hanks-type kinase deletion mutant. These two types of analyses were necessary to be able to identify peptides whose phosphorylation level but not abundance varied.
In silico Screening of the Whole Genome of S. thermophilus LMD9 and Identification of Structural Homologs to PknB
Our in silico screening of the entire genome of LMD9 led to the detection of three sequences with a threshold above 95% probability to share the PknB kinase fold, a size above 100 residues and an e-value below 1.e−15. These are WP_011681404.1 (PknB), WP_011681723.1 (STER_1920; STER_RS09410), and WP_011681158.1 (STER_1047; STER_RS05195) with scores of probability of 100, 99.97, and 99.43%, sequence size of 271, 251, and 138, and e-value of 4.8 e−45, 2.9 e−35, and 2.2 e−17, respectively. Expectedly, the first sequence showing 40% sequence identity with the reference fold is PknB from S. thermophilus, despite being annotated as hypothetical protein. This sequence was homology modeled and the combination sequence/structure/function was thoroughly analyzed (see next section). Interestingly the second, which is annotated aminoglycoside phosphotransferase, shows 19% sequence identity, nevertheless among the 719 residues of this putative kinase-like protein, 251 amino acid residues are aligned onto 278 residues of the PknB kinase domain. Notably, the sequence evidences an additional N-terminal domain of 200 amino acid residues. The HMM detection shows that this protein WP_011681723.1 associates the prediction of kinase domain with a putatively conserved catalytic machinery. Accordingly, it displays functional motifs that are degenerated; LEDx4N aligns with HRDx4N, DLE aligns with DFG, and SQN aligns with SPE, both located at the start and end of the activation loop, respectively. Markedly, the three catalytic residues that belong to HRDx4N and DFG motifs are strictly conserved with PknBSTh Asp331, Asn336, and Asp349 that correspond to PknBMtb Asp138, Asn143, and Asp156, respectively. Eventually, this kinase-like portion of WP_011681723.1 could display an activation loop, which contains several Ser and Thr residues known to accept phosphorylation (Supplementary Figure S1). All these elements strongly suggest that this domain could act as a kinase and should be able to phosphorylate substrates, notably in absence of PknB. The third and last sequence identified is WP_011681158.1, annotated as AarF/ABC1/UbiB kinase family protein, which shows 25% sequence identity with PknBMtb. Similarly, this sequence, despite displaying large insertions in the putative kinase domain, contains nevertheless conserved aspartate and asparagine catalytic residues embedded into the degenerated functional motif HGDx4N instead of HRDx4N and the conserved aspartate of the DFG motif (Supplementary Figure S2). The structural homology and annotation to kinase family as well as the highly conserved catalytic residues suggest that this protein could act as a kinase and could be able to phosphorylate Ser/Thr residues. This result should deserve further assessment.
In silico Structural Characterization Showed That S. thermophilus PknB Kinase Exhibits All the Signatures of a Hanks-Type Kinase
The Hanks-type serine/threonine kinase PknB from S. thermophilus exhibits a classical organization. First, a N-terminal part containing the protein kinase ATP-binding region and the kinase active site. At the C-terminal part, three extracellular PASTA domains interact with peptidoglycan. Between, a transmembrane segment separates the intracellular part from the extracellular part of PknB (Figure 2A). This sequence is well conserved in similar enzymes from different streptococci species (43–58% of identity with PknB from S. agalactiae, Streptococcus mutans, S. pneumoniae, and S. suis), except the number of PASTA domain which is higher (four) in S. pneumoniae and S. suis and only three in the three other species. A gene encoding a serine/threonine phosphatase (STER_1393) is the close neighbor of pknB. The two genes are predicted to belong to the same operon6 (Figure 3).
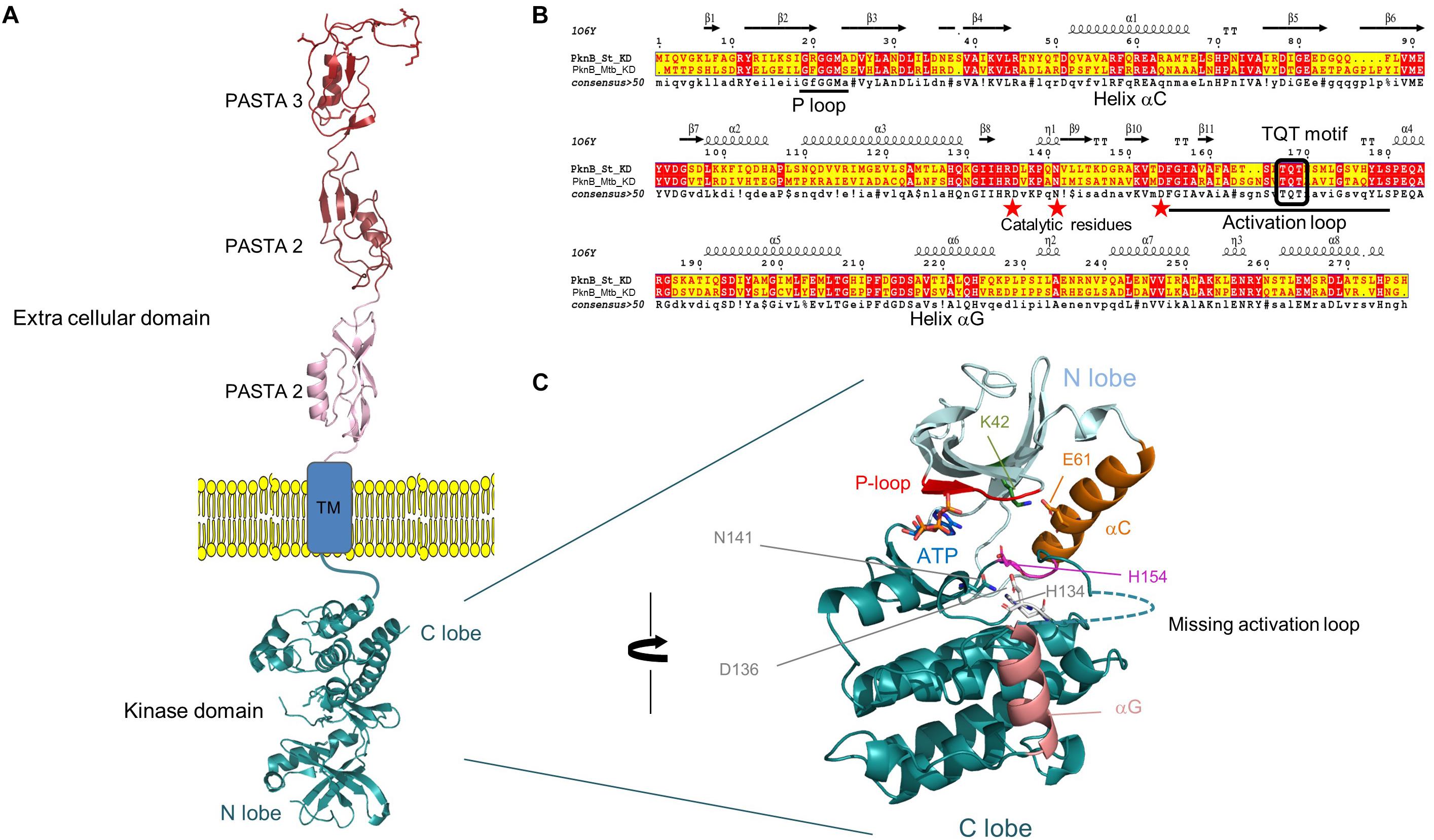
Figure 2. (A) Topology of PknBSt full length, kinase and extra cellular domain (ECD) have been modeled by homology using PknB, and PASTA coordinates from M. tuberculosis and PASTA coordinates of S. aureus. Only the TM domain lacks solved structure and could not be modeled. The PASTA domains are shown extended across the peptidoglycan layer. (B) ESPript alignment of the kinase domain between PknB from S. thermophilus and M. tuberculosis. The structural elements that sign up the hallmarks of Hanks type kinase are conserved and reported in the sequence: helices αC, αG, P- and activation loops. Red stars evidence the catalytic residues. (C) Close view of the kinase domain, with the accommodation of an ATP molecule docked in the active site. The structural and functional elements are highlighted: P-loop in red, αC in orange, its residue E61 forming a salt-bridge with K42 that locks αC in a conformation and participates to create a competent active site are in stick, the active triad composed of H134, H138, and N141 are also shown as sticks. Helix αG is in salmon and the missing activation that could not be modeled because too long and with no reference coordinates is drawn as dotted line. The figure has been prepared using PyMOL2.2 Schrödinger.
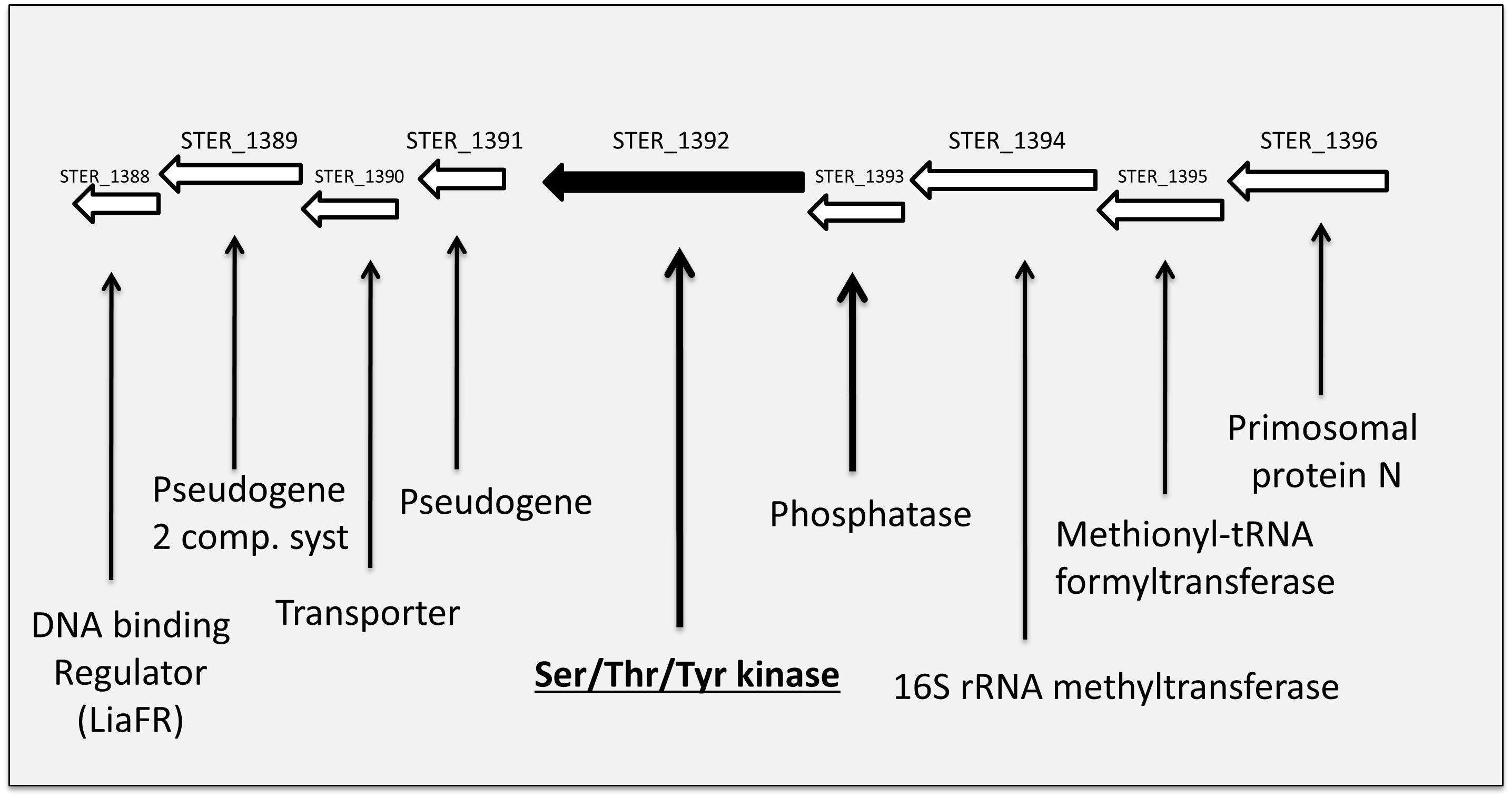
Figure 3. Schematic view of the genetic environment of the PknB encoding gene in the S. thermophilus LMD-9 genome.
The homology modeling of PknB-KDSt catalytic domain displays the extremely conserved protein kinase fold with a N-terminal lobe mainly composed of β-sheet plus the regulatory α-C helix, and the C-terminal lobe that contains almost exclusively helices. The model shows an active and closed conformation that is able to accommodate the cognate ATP molecule in the dedicated pocket, located at the interface between the N and C-lobes. It includes the conserved glycine residues of the phosphate loop (P-loop), the strictly conserved salt-bridge between Lys42 and helix α-C Glu61 that locks the kinase into an active conformation, the DFG motif and the catalytic triad composed of His134, Asp136, and Asn141 (Figures 2B,C). Notably, PknB-KDSt contains two main features very close to the bacterial prototypical Ser/Thr kinase PknB-KDMtb that both deal with the activation loop. The first trait is a similarly long activation loop of 23 residues, starting right after the conserved DFG motif and ending with the conserved SPE motif. As this activation is highly floppy and is hardly seen in crystal structure of template, it could not be modeled. Notably, such flexibility is expected to correlate to versatility of the kinase to tune its binding to the structurally unrelated substrates. This is in total agreement with the 10 highly dissimilar substrates identified in our study. The second feature is a double phosphorylation site contained in the TQT motif located in the middle of the activation loop. PknB-KDMtb has been shown to require the two threonine to be phosphorylated for a fully active kinase. Possibly, this could be also a regulation trait of PknBSt shared with PknBMtb, but this need to be documented. The homology modeling of the extra cellular domain of PknBSt displays three PASTA repeats of roughly 70 residues each. In S. pneumoniae, these PASTA123 repeats have been shown to behave as interchangeable modules that are nevertheless required to trigger the kinase activity and to control the septal cell wall thickness (Zucchini et al., 2018). In contrast, the distal PASTA moiety drives PknB localization at the division septum and displays a specific motif critical for cell separation. Clearly, this fourth PASTA is missing in S. thermophilus and the third one does not display the specific motif expected to interact with the cell wall hydrolase LytB (Zucchini et al., 2018). Markedly, while the shorter C-terminal extremity (44 residues) shares no sequence homology with any PASTA module, this segment shows a very low complexity of amino-acids, with 19 and 10 serine and threonine residues, respectively. Possibly, due to this low complexity, this portion could be intrinsically disordered. Its role in cell division and position at the distal extremity of the extracellular domain remain to be deciphered.
The S. thermophilus Phosphoproteome Concerns Diverse Central and Essential Functions
Our phosphoproteome analysis identified 106 proteins and 410 phosphopeptides corresponding to 161 peptide sequences with different phosphosite positions (Supplementary Table S1 for complete phosphoproteomic data and Table 2 for phosphorylated protein identification and functional categories). The phosphorylation occurred for 43% on serine, 33% on threonine and 23% on tyrosine. In the phosphoproteome, we identified only a few multi-phosphorylated peptides. Those carrying three phosphorylations were found in four proteins: the tyrosine kinase (STER_1393, WP_011681174.1), the carboxyl carrier protein (STER_0435, WP_002946351.1), the UDP-glucose 4-epimerase (STER_1369, WP_011681394.1) and a protein of unknown function (STER_0283, WP_002945999.1) in both strains. No peptide phosphorylation was confidently detectable before the enrichment procedure indicating that the level of peptide phosphorylation is low.
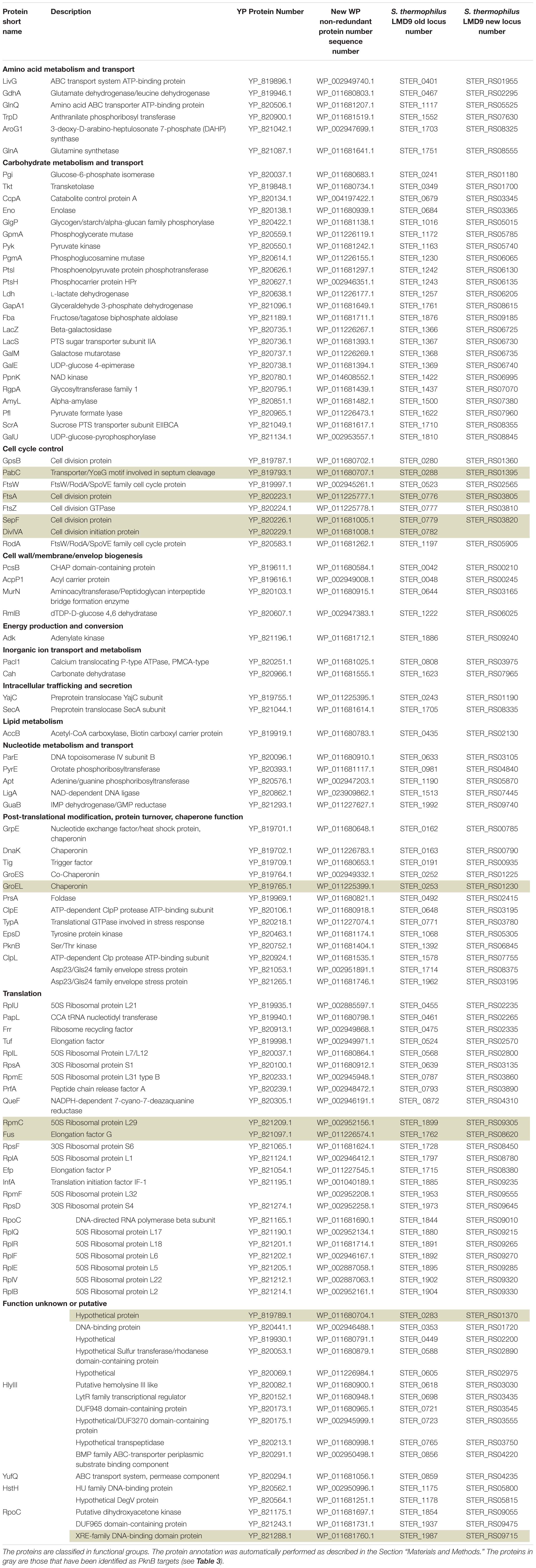
Table 2. List of S. thermophilus proteins in which phosphorylated peptides have been identified.
The phosphorylation especially targeted proteins involved in essential pathways (Table 2). In order of diminishing importance, we found, first, many phosphorylated peptides belonging to proteins necessary for translation including 15 ribosomal proteins. The second main group concerns proteins involved in carbon metabolism including glycolysis enzymes, PTS carbohydrate transporters and the catabolite control regulator. We merged 16 proteins with unknown or putative functions in a third group that contain a few putative regulators or DNA-binding proteins. A group of proteins involved in cell cycle control, especially cell division proteins, was also found phosphorylated. Chaperonin and stress response proteins constituted another important group of phosphorylated proteins. Finally, we identified proteins involved cell wall/membrane/envelope biogenesis, intracellular trafficking and secretion, inorganic ion, lipid and amino acids metabolisms and energy production and conversion. Both the serine/threonine (PknB) and the putative tyrosine kinases contained phosphorylated peptides. Only one PknB amino acid (threonine 291) was found phosphorylated in the four WT replicates. However, because the modeling step identified a TQT motif that needs to be phosphorylated for a fully active kinase in M. tuberculosis and that is conserved in S. thermophilus, we specifically looked at this part of the protein. In our dataset, we identified one peptide including this TQT motif and that was found phosphorylated but only in one replicate and was therefore not retained with our stringent parameters. It, however, indicates that this position might be also phosphorylated in S. thermophilus. Moreover, it has to be noticed that 6 proteins in which phosphorylation sites have been detected were not identified in the proteome data set probably because of their low abundance.
The pknB Gene Disruption Clearly Affects the Phenotype and the Phosphoproteome of S. thermophilus but Not Its Proteome
Taking advantage of the natural competence of the S. thermophilus strain used, we easily replaced the pknB gene by an antibiotic cassette. The phenotype of the mutant during growth in liquid medium was visually different from the one of the wild type. A higher sedimentation of the bacteria was observed with the mutant (Figure 4A). Microscopy observation revealed that the PknB bacteria form aggregates in addition to longer chains (Figure 4B). We also observed clear cell division defects. After growth on plates, mutant colonies appear bigger that the wild type ones, which is in agreement with longer chains in the mutant (Figure 4C).The two strains grew well in rich medium, but it was difficult to precisely compare the growth rates of the two strains using absorbance measurements due to culture heterogeneity of the mutant. We therefore followed pH evolution of the two strains during growth in M17 lactose medium (Figure 5). pH decrease was delayed in the mutant but reached the same value than the wild type, i.e., 5,3 at the end of exponential phase.
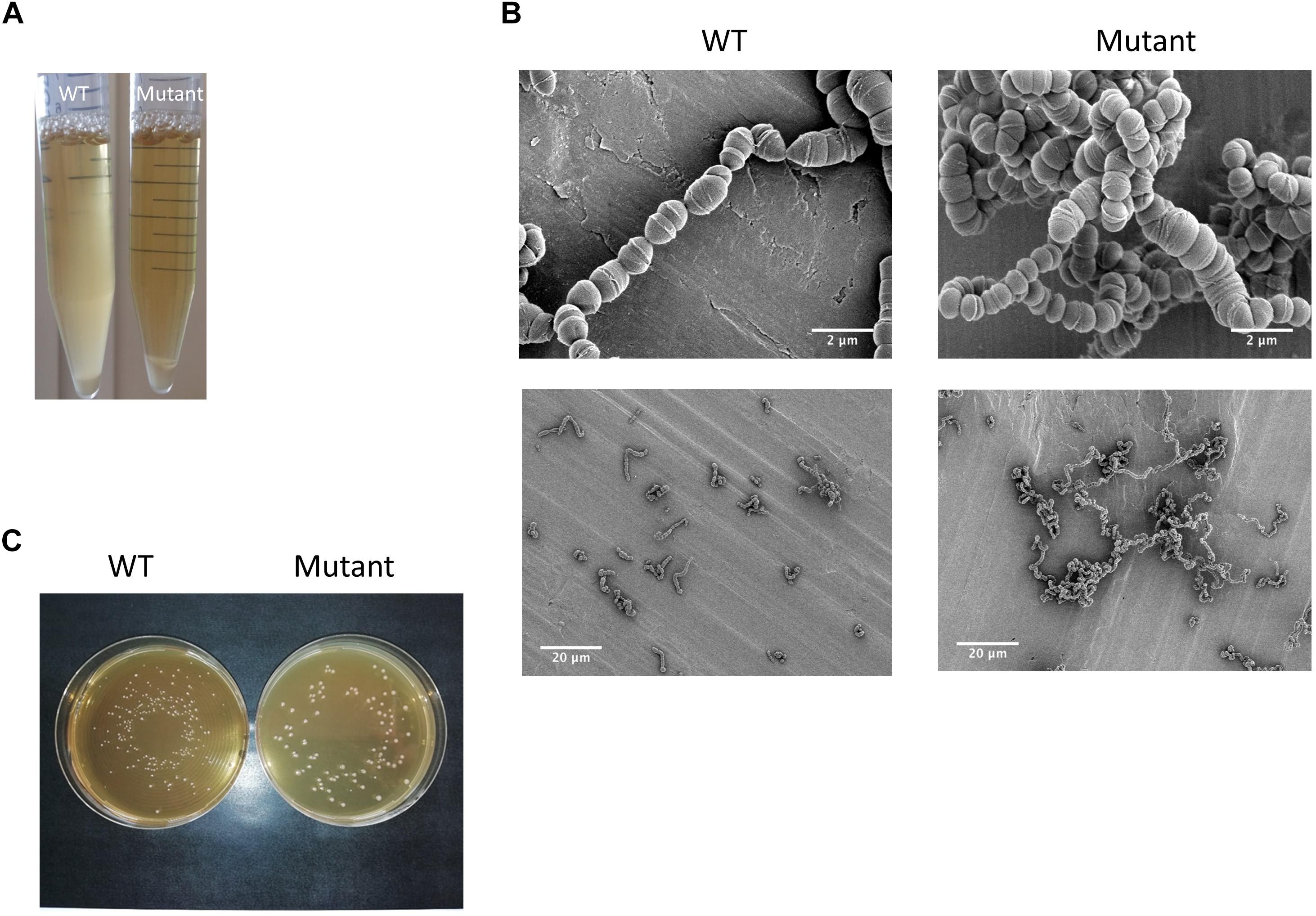
Figure 4. (A) Visual and (B) microscopic observation at two degrees of magnification of the wild type strain and the ΔpknB mutant grown in M17 lactose medium; (C) colonies phenotypes of the wild type and the mutant on M17 lactose agar medium.
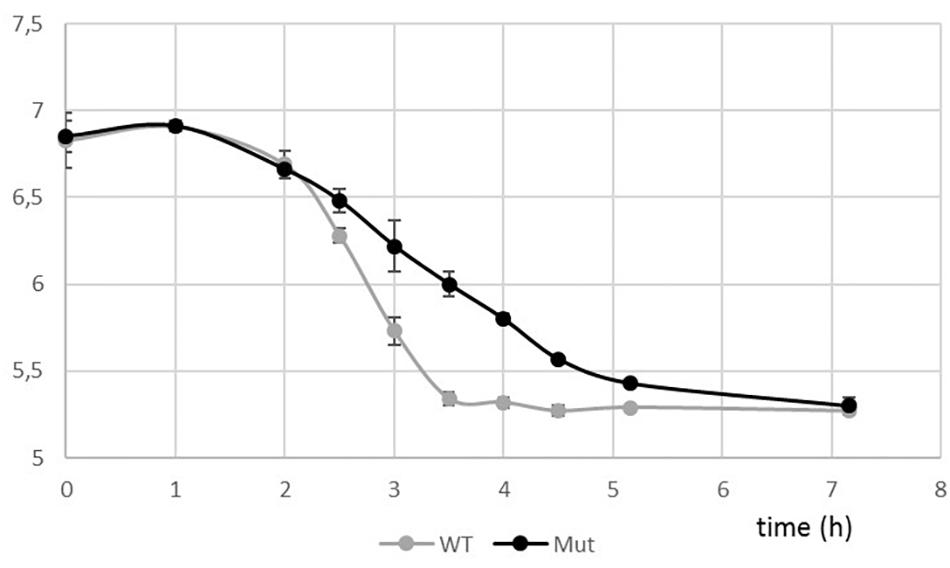
Figure 5. pH evolution curves during growth of the wild type and the ΔpknB strains in M17 lactose medium.
We evaluated the survival of the two strains after a −20°C freezing stress. It was not possible to compare the bacterial counts of the two strains since their chain lengths were different. We therefore calculated for each strain the percentage of cells that growth on plate after freezing without any additive. Unexpectedly, the mutant exhibited a higher survival rate (73%) than the wild type strain (9%). Longer chains could have a protective effect against a freezing stress.
The proteome analysis, realized in one culture condition (exponential phase in rich M17 lac medium) on a total bacterial extract, allowed to identify 891 proteins on the basis of at least two peptides per protein (listed in Supplementary Table S2) representing half of the theoretical proteome. Based on spectral counts, no statistically significant difference in protein abundance was found in our data set except for PknB that was no more detected, as expected, in the mutant. Among the proteins identified, 82% are predicted to be cytoplasmic (LocateP, Zhou et al., 2008) while the others are associated in different manners to bacterial envelopes. The PknB encoding gene is predicted to be in operon with a gene encoding a phosphatase (STER_1393, WP_011681405.1)7 (Figure 3) which is present in the proteomes of the two strains. Another putative protein kinase, annotated tyrosine kinase (WP_011681174.1) is also present in the proteomes. Although they were clearly identified and quantified, these two kinases and the phosphatase were not at the top of the most abundant proteins identified.
Proteome and phosphoproteome were realized in parallel with mutant and wild type strains. Differences in protein phosphorylation, which were not imputable to protein abundance differences, were highlighted. Concerning the phosphorylation, we identified phosphorylated peptides from 9 proteins that showed presence/absence variations between the wild type and mutant strains in the four replicates (Table 3 and Supplementary Figure S3). We can conclude that at least these 9 proteins plus probably PknB itself, are, direct or indirect, targets of PknB. However, many other peptides remain fully or partially phosphorylated in the mutant (Supplementary Table S3) indicating that other kinase(s) than PknB participate in the protein Ser/Thr/Tyr phosphorylation process.
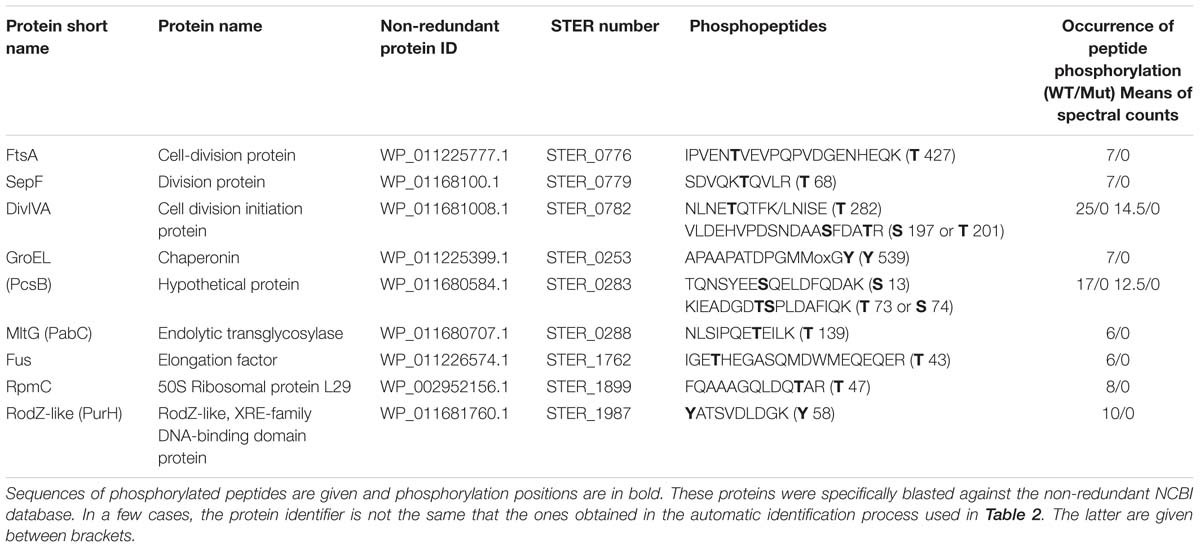
Table 3. List of phosphorylated proteins that are no more phosphorylated in the ΔpknB mutant in the four replicates bases on spectral counts.
The 9 proteins containing phosphorylated peptides that were no more phosphorylated in the ΔpknB mutant are mainly strongly linked to the division process and located, as PknB, close or within the cytoplasmic membrane, probably at the division site. Four of them (PknB, RodZ, MltG, and Ster_0283) are predicted to contain a transmembrane segment. In addition, RodZ contains a HTH_XRE intracellular motif indicating a possible transcriptional regulatory role for this protein. FtsA, SepF, DivIVA, and RodZ are proteins well-known to be involved, as PknB, in the division process in streptococci as well as in many bacteria (Massidda et al., 2013). The cases of the chaperonin GroEL and the elongation factor Fus are, in a less obvious way, probably linked to PknB and the cell division in S. thermophilus. They are indeed known to be phosphorylated and identified as substrates of the Hanks-type kinase PrkC in Bacillus anthracis. In this bacterium, the phosphorylation of GroEL is critical for biofilm formation (Arora et al., 2017). GroEL was also found co-localized with FtsZ in Escherichia coli suggesting a role in cell division (Ogino et al., 2004). MltG is a protein containing a transmembrane segment. It is described as an endolytic transglycosylase from the YceG family, well-conserved within bacteria and cleaves nascent peptidoglycan polymers and terminates their elongation (Yunck et al., 2016). Finally, the presence of RpmC in the list of PknB targets remains unexplained at this step.
The sites of phosphorylation of the nine proteins targets of PknB have been identified (Table 3). Phosphorylated amino acids were, as expected mainly serines and threonines. Our results also indicate that two phosphopeptides (in GroEL and RodZ proteins), impacted by the absence of PknB, were phosphorylated on tyrosines. Tyrosine phosphorylation by Hanks-type kinases was already observed in M. tuberculosis (Kusebauch et al., 2014). We cannot discount the possibility of an indirect effect of PknB via the modification of the tyrosine kinase phosphorylation state. Evidences of cross-phosphorylation processes between Hanks-type and tyrosine kinases have been given by Shi et al. (2014). However, in our data set, we cannot confirm this hypothesis since we did not identify significant modification of the tyrosine kinase phosphorylation state between the two strains.
Discussion
Several research groups, working with pathogen bacteria, have hypothesized that a phosphoproteome composed of a large number of proteins and a high proportion of multiple phosphorylation sites may be closely related to bacterial virulence (Sun et al., 2010). We demonstrated in this work done on S. thermophilus, a bacterium recognized as GRAS and massively used in the dairy industry, that protein phosphorylation is also an important process in non-pathogenic bacteria. Although not directly comparable, the phosphoproteome of this bacterium, established in one culture condition is of the same order of magnitude (around 100 phosphorylated proteins) than the one of S. pneumoniae (Sun et al., 2010). Our results are also coherent with those obtained with another main starter bacterium: L. lactis (Soufi et al., 2008). The level of phosphorylation is too low to be detected without an enrichment step although a very sensitive last generation mass spectrometer was used. We did not identified a clear recognition motif for phosphorylation and it therefore remains impossible to predict which protein will be phosphorylated and where. In the S. pneumoniae phosphoproteome, Sun et al. (2010) did not identify a specific sequence for phosphorylation either and only found that phosphorylation prefers hydrophobic sequences to occur.
We demonstrated that, in S. thermophilus as in other bacteria, phosphorylation target central functions sustaining growth such as translation, division and carbon metabolism. Even if these functional categories are globally well conserved, the specific proteins phosphorylated can differ from one set to another one. The sites of phosphorylation of a same protein in different data sets can also vary as observed in M. tuberculosis (Calder et al., 2016). Elsewhere, even if their proportion is lower compared to what was noticed in S. pneumoniae, we also observed proteins harboring multi phosphorylation sites (up to four in our study). The level of phosphorylated tyrosine observed in S. thermophilus is rather high compared to other phosphoproteomes (S. pneumoniae, L. lactis, and Bacillus subtilis) in which it hardly reaches 10% (Macek et al., 2007; Soufi et al., 2008; Sun et al., 2010).
The impact of pknB gene disruption is very clear on cell division and morphogenesis as observed with the naked eye and by microscopy. Longer chain and division defects were observed in the ΔpknB mutant. Longer chains were also observed in S. suis and S. agalactiae ΔpknB mutants (Rajagopal et al., 2003; Zhang et al., 2017). In S. thermophilus, these observations correlate well with phosphoproteome modifications between the two strains, which affect, in an important manner, proteins involved in cell division process.
Unexpectedly, the proteomes obtained from the S. thermophilus wild type strain and the ΔpknB mutant did not differ. No protein abundance variation, based on spectral counts, was significant apart from PknB which was completely absent from the mutant data set consequently to the deletion of its encoding gene. We cannot exclude that abundance of some proteins, not visible in our preparation, varied. We indeed worked with a total extract, which do not allow an optimal visualization of all membrane, and cell wall proteins, that are less abundant, compared to cytoplasmic ones in a global extract. A prior fractionation of cell into cytoplasm and envelopes would allow the quantification of more proteins and confirm or not the present result.
Via this work, we brought new data confirming that bacterial Ser/Thr Hanks-type kinase targets bacterial division. We can hypothesize that all proteins, substrates of PknB, are involved in the cell division process and located near the cytoplasmic membrane of PknB. As we did identify neither conserved sequences nor FHA motif in the proteins that are PknB targets, we cannot explain PknB specificity based on primary sequence analysis. We hypothesized that the co-localization of PknB with its substrates could be the main factor driving its specificity. A possible role of the hypothetical protein (STER_0283) and of RpmC in cell division, as well as the identification of genes possibly regulated by RodZ need therefore to be investigated. The phosphorylation of other proteins is also affected by the absence of PknB but do not fully fulfill the stringent criteria fixed. We can mention, as example, the protein FtsW, involved in division and whose peptides were no more phosphorylated in the mutant and phosphorylated in the wild type but not in all replicates.
The protein DivIVA, involved in cell division initiation, is the most emblematic substrate of streptococcal Hanks-type kinases. Several phosphopeptides have been identified within streptococcal DivIVA proteins. One containing the threonine 201 (in S. thermophilus) seems conserved among the four streptococcal species while others have been identified only in one species and could be more specific (Figure 6). The phosphorylation of the threonine 282 in the C-terminal peptide in S. thermophilus was not yet reported and this amino acid is not conserved in S. pneumoniae.

Figure 6. Alignment of DivIVA proteins from Streptococcus thermophilus (St), pneumoniae (Sp), agalactiae (Sa), and suis (Ss) and identification of phosphopeptides (framed sequences, this work) and others identified phosphorylated amino acids (highlighted in yellow).
As already reported, transcriptional regulators and histidine kinases from two-component systems (TCS), can be regulated via Ser/Thr/Tyr phosphorylation (Kalantari et al., 2015; Khara et al., 2018). In our data set, we identified components of three TCS among the eight present in the strain: the orphan response regulator CovR-like (STER_0354), the whole WalR-like system (STER_1115 and STER_1116), the Lia-like response regulator. None of these proteins was found phosphorylated in our experiment.
As already observed in pathogen streptococci, PknB is only responsible for a minor part of the Ser/Thr/Tyr protein phosphorylation observed. We identified, with a stringent process, 9 proteins substrates of PknB while 7 and 12 proteins were found substrates of homolog kinases in S. pneumoniae (Novakova et al., 2010) and S. suis (Zhang et al., 2017), respectively. These observations indicate PknB is probably specialized in the phosphorylation of co-localized proteins involved in cell division and that other enzymes are involved in the global phosphorylation process. Of course, these PknB substrates should be confirmed by targeted proteomics approaches. A more specific analysis of an extract enriched in envelope-associated proteins could help to identify additional PknB targets.
It appears clearly from our work that PknB is not responsible for the whole phosphorylation process. We identified two potential candidates showing a kinase structural homology with PknB and whose activity and targets need to be demonstrated and identified, respectively.
Author Contributions
CH and LH set up and realized proteomics and phosphoproteomics experiments. CH, LH, MZ, and MB-N participated in proteomics data analysis and manuscript. AC realized microscopic observations. MV, RG, CM, and MB were in charge of molecular biology and microbiology experiments, especially the construction of the mutant and its characterization. SS, VMa, and GA-L characterized PknB from a structural point of view, modeled it, and identified other possible kinases. VMo initiated the study, coordinated it, participated in data analysis and interpretation, and wrote the main part of the manuscript.
Funding
Proteomics analyses were performed on the PAPPSO platform (http://pappso.inra.fr) which was supported by the INRA (http://www.inra.fr), the Ile-de-France Regional Council especially via the DIM ASTREA (https://www.iledefrance.fr/education-recherche), the IBiSA (https://www.ibisa.net), and the CNRS (http://www.cnrs.fr).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank F. Rul, V. Juillard, and A. Gruss for their critical and constructive reading of the article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01329/full#supplementary-material
FIGURE S1 | The figure shows a clustalW alignement using Espript (http://espript.ibcp.fr/ESPript/ESPript/) which evidences the secondary structure elements of PknB kinase domain M. tuberculosis (pdb id code 1O6Y) along with the sequences between PknB kinase domain from S. thermophilus, and putative kinase named WP_0116817231.
FIGURE S2 | The figure shows a clustalW alignement using Espript (http://espript.ibcp.fr/ESPript/ESPript/) which evidences the secondary structure elements of PknB kinase domain M. tuberculosis (pdb id code 1O6Y) along with the sequences between PknB kinase domain from S. thermophilus, and putative kinase named WP_0116811581.
FIGURE S3 | Phosphopeptides spectra of the protein targets of PknB listed in Table 3.
TABLE S1 | Identification of 891 proteins found by LC-MS/MS and present in the 8 samples (4 WT and 4 mutants). This table gives details about peptides and proteins.
TABLE S2 | Identification by LC-MS/MS of 161 phosphoproteins present in the triplex with light, medium and heavy dimethyl labeling (48 Samples).
TABLE S3 | Spectral counting of phosphopeptides: present/absent in WT/Mutant by sample and genotype.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/genome/420?genome_assembly_id=300120
- ^ https://toolkit.tuebingen.mgp.de/
- ^ http://www6.jouy.inra.fr/mima2/
- ^ http://pappso.inra.fr/
- ^ http://pappso.inra.fr
- ^ www.microbesonline.org/operons/gnc322159.html
- ^ www.microbesonline.org/operons/gnc322159.html
References
Arora, G., Sajid, A., Virmani, R., Singhal, A., Kumar, C. M. S., Dhasmana, N., et al. (2017). Ser/Thr protein kinase PrkC-mediated regulation of GroEL is critical for biofilm formation in Bacillus anthracis. NPJ Biofilms Microbiomes 3:7. doi: 10.1038/s41522-017-0015-4
Beilharz, K., Novakova, L., Fadda, D., Branny, P., Massidda, O., and Veening, J. W. (2012). Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase. Proc. Natl. Acad. Sci. U.S.A. 19, E905–913. doi: 10.1073/pnas.1119172109
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Royal Stat. Soc. SeriesB- Method. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Boersema, P. J., Mohammed, S., and Heck, A. J. R. (2009). Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spec. 44, 861–878. doi: 10.1002/jms.1599
Bradford, M. M. (1976). Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1006/abio.1976.9999
Calder, B., Albeldas, C., Blacckburn, J. M., and Soares, N. C. (2016). Mass spectrometry offers insight into the role of Ser/Thr/Tyr phosphorylation in the Mycobacteria. Front. Microbiol. 7:141. doi: 10.3589/fmicb.2016.00141
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atomstructure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
Cousin, C., Derouiche, A., Shi, L., Pagot, Y., Poncet, S., and Mijakovic, I. (2013). Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol. Lett. 346, 11–19. doi: 10.1111/1574-6968.12189
Débarbouillé, M., Arnaud, M., Fouet, A., Klier, A., and Rapoport, G. (1990). The sacT gene regulating the sacPA operon in Bacillus subtilis shares strong homology with transcriptional antiterminators. J. Bacteriol. 172, 3966–3973. doi: 10.1128/jb.172.7.3966.3973
Fleuchot, B., Gitton, C., Guillot, A., Vidic, J., Nicolas, P., Besset, C., et al. (2011). Rgg proteins associated with internalized small hydrophobic peptides: a new quorum sensing mechanism in streptococci. Mol. Microbiol. 80, 1102–1119. doi: 10.1111/j.1365-2958.2011.07633.x
Hanks, S. K., Quinn, A. M., and Hunter, T. (1988). The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241, 42–52. doi: 10.1126/science.3291115
Kalantari, A., Derouiche, A., Shi, L., and Mijakovic, I. (2015). Serine/threonine/tyrosine phosphorylation regulates DNA binding of bacterial transcriptional regulators. Microbiology 161, 1720–1729. doi: 10.1099/mic.0.000148
Khara, P., Mohapatra, S. S., and Biswas, I. (2018). Role of CovR phosphorylation in gene transcription in Streptococcus mutans. Microbiology 164, 704–715. doi: 10.1099/mic.0.000641
Koponen, J., Laakso, K., Koskenniemi, K., Kankainen, M., Savijoki, K., Nyman, T. A., et al. (2012). Effect of acid stress on protein expression and phosphorylation in Lactobacillus rhamnosus GG. J. Proteomics 75, 1357–1374. doi: 10.1016/j.jprot.2011.11.009
Kusebauch, U., Ortega, C., Ollodart, A., Rogers, R. S., Sherman, D. R., Moritz, R. L., et al. (2014). Mycobacterium tuberculosis supports protein tyrosine phosphorylation. Proc. Nat. Acad. Sci. U.S.A. 111, 9265–9270. doi: 10.1073/pnas.1323894111
Langella, O., Valot, B., Balliau, T., Blein-Nicolas, M., Bonhonarne, L., and Zivy, M. (2017). X !tandempipeline : a tool to manage sequence redundancy for protein inference and phosphosite identification. J. Prot. Res. 16, 494–503. doi: 10.1021/acs.jproteome.6b00632
Letort, C., and Juillard, V. (2001). Development of a minimal chemically-defined medium for the exponantial growth of Streptococcus thermophilus. J. Appl. Microbiol. 91, 1023–1029. doi: 10.1046/j.1365-2672.2001.01469.x
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., et al. (2007). The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Protomics 6, 697–707. doi: 10.1074/mcp.M600464-MCP200
Makarova, K., Slesarev, A., Wolf, Y., Sorokin, A., Mirkin, B., Koonin, E., et al. (2006). Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 15611–15616. doi: 10.1073/pnas.0607117103
Massidda, O., Novakova, L., and Vollmer, W. (2013). From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ. Microbiol. 15, 3133–3157. doi: 10.1111/1462-2920.12189
Millan-Oropeza, A., Henry, C., Blein-Nicolas, M., Aubert-Frambourg, A., Moussa, F., Bleton, J., et al. (2017). Quantitative proteomics analysis confirmed oxidative metabolism predominates in Streptomyces coelicolor versus glycolytic metabolism in Streptomyces lividans. J. Prot. Res. 16, 2597–2613. doi: 10.1021/acs.jproteome.7b00163
Misra, S. K., Moussan Désirée Aké, F., Wu, Z., Milohanic, E., Cao, T. N., Cossart, P., et al. (2014). Quantitative proteome analyses identify PrfA-responsive proteins and phosphoproteins in Listeria monocytogenes. J. Prot. Res. 13, 6046–6057. doi: 10.1021/pr500929u
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and autodocktools4: automated docking with selective receptor flexibility. J. Comput. Chem. 16, 2785–2791. doi: 10.1002/jcc.21256
Napoli, A., Aiello, D., Cappello, M. S., Di Donna, L., Mazzotti, F., Materazzi, S., et al. (2014). Mass spectrometry-based proteomic approach in Oenococcus oeni enological starter. J. Prot. Res. 13, 2856–2866. doi: 10.1021/pr4012798
Novakova, L., Bezouskova, S., Pompach, P., Spidlova, P., Saskova, L., Weiser, J., et al. (2010). Identification of multiple substrates of the StkP Ser/Thr protein kinase in Streptococcus penumoniae. J. Bacteriol. 192, 3629–3638. doi: 10.1128/JB.01564-09
Ogino, H., Wachi, A., Iwai, N., Nishida, T., Yamada, S., Nagai, K., et al. (2004). FtsZ-dependant localization of GroEL protein at possible division sites. Genes Cells 9, 765–771. doi: 10.1111/j.1365-2443.2004.00770.x
Ortiz-Lombardia, M., Pompeo, F., Boitel, B., and Alzari, P. M. (2003). Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 278, 13094–13100. doi: 10.1074/jbc.M300660200
Paracuellos, P., Ballandras, A., Robert, X., Khan, R., Hervé, M., Mengin-Lecreuls, D., et al. (2010). The extended conformation of the 2.9A crystal structure of the three-PASTA domain of a Ser/Thr kinase from the human pathogen Staphylococcus aureus. J. Mol. Biol. 404, 847–858. doi: 10.1016/j.jmb.2010.10.012
Prigozhin, D. M., Papavinasaundaram, K. G., Baer, C. E., Murphy, K. C., Moskaleva, A., Chen, T. Y., et al. (2016). Structural and genetic analyses of the Mycobacterium tuberculosis protein kinase B sensor domain identify a potential ligand-binding site. J. Biol. Chem. 291, 22961–22969. doi: 10.1074/jbc.M116.731760
Rajagopal, L., Clancy, A., and Rubens, C. E. (2003). A eukaryotic type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation and virulence. J. Biol. Chem. 278, 14429–14441. doi: 10.1074/jbc.M212747200
Rajagopal, L., Vo, A., Silvestroni, A., and Rubens, C. E. (2005). Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol. Mic. 56, 1329–1346. doi: 10.1111/j.1365-2958.2005.04620.x
Sali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–8015. doi: 10.1006/jmbi.1993.1626
Shi, L., Pigeonneau, N., Ravikumar, V., Dobrinic, P., Macek, B., Franjevic, D., et al. (2014). Cross-phosphorylation of bacterial serine/threonine and tyrosine protein kinase on key regulatory residues. Front. Microbiol. 5:495. doi: 10.3389/fmicb.2014.00495
Silvestroni, A., Jewell, K., Lin, W. J., Connelly, J. E., Ivancic, M. M., Tao, W. A., et al. (2009). Identification of serine/threonine kinase substrates in the human pathogen group B streptococcus. J. Prot. Res. 8, 2563–2574. doi: 10.1021/pr900069n
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33(Suppl. 2), W244–W248. doi: 10.1093/nar/gki408
Soufi, B., Gnad, F., Jensen, P. R., Petranovic, D., Mann, M., Mijakovic, I., et al. (2008). The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493. doi: 10.1002/pmic.200800069
Stancik, I. A., Sestak, M. S., Ji, B., Axelson-Fisk, M., Franjevic, D., Jers, C., et al. (2018). Serine/threonine protein kinases from bacteria, archae and eukarya share a common evolutionary origin deeply rooted in the tree of life. J. Mol. Biol. 430, 27–32. doi: 10.1016/j.jmb.2017.11.004
Sun, X., Ge, F., Xia, C. L., Yin, X. F., Ge, R., Zhang, L. H., et al. (2010). Phosphoproteomics analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Prot. Res. 9, 275–282. doi: 10.1021/pr900612v
Valot, B., Langella, O., Nano, E., and Zivy, M. (2011). MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 11, 3572–3577. doi: 10.1002/pmic.201100120
Vizcaino, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and related tools. Nucl. Acids Res. 44, D447–456. doi: 10.1093/nar/gkw880
Yunck, R., Cho, H., and Bernhardt, T. (2016). Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol. Mic 99, 700–718. doi: 10.1111/mmi.13258
Zhang, C., Sun, W., Tan, M., Dong, M., Liu, W., Gao, T., et al. (2017). The eukaryote-like serine/threonine kinase STK regulates the growth and metabolism of zoonotic Streptococcus suis. Front. Cell. Infect. Microbiol. 7:66. doi: 10.3389/fcimb.2017.00066
Zhou, M., Boekhorst, J., Francke, C., and Siezen, R. (2008). LocateP: genome-scale subcellular-location predictor for bacterial proteins. BMC Bioinformatics 9:173. doi: 10.1186/1471-2105-9-173
Zimmermann, L., Stephens, A., Nam, S. Z., Rau, D., Kübler, J., Lozajic, M., et al. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243. doi: 10.1016/j.jmb.2017.12.007
Keywords: protein phosphorylation, Streptococcus thermophilus, Hanks-type kinase, proteomics, cellular division
Citation: Henry C, Haller L, Blein-Nicolas M, Zivy M, Canette A, Verbrugghe M, Mézange C, Boulay M, Gardan R, Samson S, Martin V, André-Leroux G and Monnet V (2019) Identification of Hanks-Type Kinase PknB-Specific Targets in the Streptococcus thermophilus Phosphoproteome. Front. Microbiol. 10:1329. doi: 10.3389/fmicb.2019.01329
Received: 01 February 2019; Accepted: 28 May 2019;
Published: 19 June 2019.
Edited by:
Christophe Grangeasse, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Vinay Kumar Nandicoori, National Institute of Immunology (NII), IndiaNelson da Cruz Soares, University of Cape Town, South Africa
Copyright © 2019 Henry, Haller, Blein-Nicolas, Zivy, Canette, Verbrugghe, Mézange, Boulay, Gardan, Samson, Martin, André-Leroux and Monnet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Céline Henry, Y2VsaW5lLmhlbnJ5QGlucmEuZnI=; Véronique Monnet, dmVyb25pcXVlLm1vbm5ldEBpbnJhLmZy
†These authors have contributed equally to this work