 Manisha Goyal1
Manisha Goyal1 Fabien Javerliat2
Fabien Javerliat2 Mattia Palmieri1Caroline Mirande2Willem van Wamel3Mehri Tavakol3
Mattia Palmieri1Caroline Mirande2Willem van Wamel3Mehri Tavakol3 Nelianne J. Verkaik3
Nelianne J. Verkaik3 Alex van Belkum1*
Alex van Belkum1*- 1Data Analytics Unit, bioMérieux, La Balme-les-Grottes, France
- 2Microbiology R&D, bioMérieux, La Balme-les-Grottes, France
- 3Department of Medical Microbiology and Infectious Diseases, Erasmus University Medical Center, Rotterdam, Netherlands
Staphylococcus aureus can colonize the human vestibulum nasi for many years. It is unknown whether and, how S. aureus adapts to this ecological niche during colonization. We determined the short (1 and 3 months) and mid-term (36 months) genomic evolution of S. aureus in natural carriers and artificially colonized volunteers. Eighty-five S. aureus strains were collected from 6 natural carriers during 3 years and 6 artificially colonized volunteers during 1 month. Multi-locus sequence typing (MLST) and single nucleotide polymorphism (SNP) analysis based on whole-genome sequencing (WGS) were carried out. Mutation frequencies within resident bacterial populations over time were quantified using core genome SNP counts (comparing groups of genomes) and pairwise SNP divergence assessment (comparing two genomes from strains originating from one host and sharing identical MLST). SNP counts (within 1–3 months) in all naturally colonizing strains varied from 0 to 757 (median 4). These strains showed random and independent patterns of pairwise SNP divergence (0 to 44 SNPs, median 7). When the different core genome SNP counts over a period of 3 years were considered, the median SNP count was 4 (range 0–26). Host-specific pairwise SNP divergence for the same period ranged from 9 to 57 SNPs (median 20). During short term artificial colonization the mutation frequency was even lower (0–7 SNPs, median 2) and the pairwise SNP distances were 0 to 5 SNPs (median 2). Quantifying mutation frequencies is important for the longitudinal follow-up of epidemics of infections and outbreak management. Random pattern of pairwise SNP divergence between the strains isolated from single carriers suggested that the WGS of multiple colonies is necessary in this context. Over periods up to 3 years, maximum median core genome SNP counts and SNP divergence for the strains studied were 4 and 20 SNPs or lower. During artificial colonization, where median core genome SNP and pairwise SNP distance scores were 2, there is no early stage selection of different genotypes. Therefore, we suggest an epidemiological cut off value of 20 SNPs as a marker of S. aureus strain identity during studies on nasal colonization and also outbreaks of infection.
Introduction
Extensive use of antibiotics in the environment and the clinical domain contributes toward the emergence of (multi-)drug resistant bacterial pathogens. This has become a global threat (Roca et al., 2015). Staphylococcus aureus (S. aureus) is among the bacterial species associated with increasing drug resistance, morbidity, invasive disease, and mortality in humans as well as animals (Chambers and Deleo, 2009; Schmidt et al., 2015; Li and Webster, 2018). S. aureus is a common opportunistic human pathogen identified most often on the nasal epithelium, About 30–50% of healthy individuals are persistently colonized (Wertheim et al., 2005). S. aureus causes a large variety of community as well as hospital-acquired infections. These include deep abscesses, endocarditis, osteomyelitis, pneumonia, and bloodstream infections (Foster and Höök, 1998; Rasigade and Vandenesch, 2014; Taylor and Unakal, 2018). S. aureus nasal carriage is a risk factor for the development of staphylococcal infections. Adherence to the human nasal epithelial cells is a prerequisite for S. aureus colonization and initiation of infection (Roche et al., 2003). The prevalence of non-symptomatic colonization with methicillin resistant S. aureus strains in the open United States population escalated from 0.8 to 1.5% over recent years (Gorwitz et al., 2008).
Colonization begins with the interaction between nasal epithelial ligands and bacterial receptors often cataloged as microbial surface components recognizing adhesive matrix molecules (MSCRAMMS) (Foster and Höök, 1998; Ghasemian et al., 2015). During colonization S. aureus expresses adherence genes (clf B, isdA, fnbA, eap, sceD, oatA, and atlA) and several immune-modulating genes (e.g., sak, chp, spa, and scn) (Burian et al., 2010; Baur et al., 2014). Host factors and local microbiota can affect the adhesion and colonization properties of S. aureus as well (Emonts et al., 2007; Frank et al., 2010; Ruimy et al., 2010).
During colonization, S. aureus secretes a number of immune-modulating proteins. Staphylococcal complement inhibitor (SCIN), encoded by the scn gene, can efficiently protect S. aureus by inhibiting the innate immune response mediated by human neutrophils. SCIN and other immune modulating proteins are encoded on the immune evasion cluster (IEC) (Goerke et al., 2006). The scn gene was identified as a conserved one being present in all IEC (van Wamel et al., 2006). To test the role and stability of IEC human artificial inoculation was performed using isolates with and without IEC. It was concluded that IEC may not play a significant role in adherence but it did display an essential role in propagation and long term survival (Verkaik et al., 2011).
We have here used whole genome sequencing (WGS) to quantify the mutational changes occurring in S. aureus strains during natural and artificial nasal colonization during periods ranging between 1 and 36 months. The numbers of human volunteers and hence the overall number of S. aureus nasal isolates are limited due to the technical and logistic complexity of the studies involved (Verkaik et al., 2011). In addition, studies involving colonization of human volunteers have to follow extensive ethical procedures and protocols. We applied bio-informatics approaches to assign MLST types and to detect genetic variation at the single nucleotide polymorphism (SNP) level. Moreover, we analyzed selective presence of virulence factors for all strains.
Materials and Methods
Description of the Strain Collection
Staphylococcus aureus strain collection was carried out as described earlier (Verkaik et al., 2011) at Erasmus Medical Center (Rotterdam, Netherlands). Naturally colonizing strains were isolated from nasal swab cultures from healthy persistent carriers who were positive for S. aureus at five culture moments over a time interval of 3 months in both 2007 and 2010. Artificially colonizing strains were collected from the human volunteers inoculated with S. aureus strain NCTC 8325-4 with or without IEC and follow-up cultures were performed in 2008 (days 1, 2, 3, 4, 7, 14, 21, and 28 after inoculation). The latter strains were susceptible to all common antibiotics and were free from staphylococcal toxin genes (Williams et al., 1997; Wertheim et al., 2008). A review of all strains sequenced is provided in Supplementary Table 1.
S. aureus Genome Sequences
Isolates were sequenced by WGS (Illumina HiSeq 2000 platform). Raw reads were assembled using the A5 MiSeq-20140604 assembler. Datasets for strains cultured in 2007 and 2010 contained 35 and 22 isolates, respectively, involving natural nasal colonization in 6 persistent carriers. The dataset from 2008 (28 isolates) was collected for strains from 6 different volunteers artificially colonized with S. aureus strain NCTC 8325-4. DNA isolation was performed for up to three colonies from each culture to define their genotypic stability at different point of times during short term as well longer term nasal carriage (see Supplementary Table 1). The sequences obtained from the 1 to 3 independent colonies taken for some of the individual strains were analyzed independently by the bioinformatics tools applied. Using bioinformatics tools as BioNumerics (Applied Maths, bioMérieux, Belgium), kSNP3 (Computations/Global Security, Lawrence Livermore National Laboratory, Livermore, CA, United States and Bellingham Research Institute, Bellingham, WA, United States), and Abricate (Torsten Seemann, University of Melbourne, Australia), all genomes were analyzed extensively.
MLST Typing and SNP Detection
To understand the genetic diversity of all the isolates multi-locus sequence typing (MLST) was performed using BioNumerics1. The MLST method is known to have a higher discriminatory power for S. aureus strains than PFGE (Peacock et al., 2002a). For classical MLST typing seven housekeeping genes and their various alleles were used to define strain relatedness2 (Jolley et al., 2018). A phylogenetic tree was constructed by executing the Linux based stand-alone source code of kSNP3 (Gardner et al., 2015), which identified core genome SNP counts and provided a consensus parsimony phylogenetic tree. The kmer size was set to 19, the optimum size estimated by the kSNP3 utility program Kchooser (Gardner et al., 2015). Pairwise SNP distances between later stage isolates as compared to early stage isolates from each individual were calculated to define mutation over time. The python script kSNPdist was used to calculate the pairwise SNP divergence between all S. aureus isolates. kSNP3 and kSNPdist executables for OS X and Linux are freely available at https://sourceforge.net/projects/ksnp/.
Resistance and Virulence Gene Identification
All the genomes were screened for the presence of 40 known and putative virulence genes3 (Shukla et al., 2010) (enterotoxin genes, exotoxin genes, leucocidin genes, hemolysin genes, surface protein genes, and putative virulence genes) and the S. aureus antibiotic resistance genes available in the ResFinder database4. Those 40 genes are grouped as classical staphylococcal. The Linux-based command line tool known as Abricate was downloaded5 to perform additional mass screening for antimicrobial resistance or virulence genes. All the identified resistance and virulence genes in the dataset were summarized in Supplementary Table 3. Additionally, in silico-based mapping of the scn gene using BioNumerics was carried out to determine the presence of IEC (van Wamel et al., 2006).
Results
Quality Testing of Genome Datasets
Genome sizes varied from 2,647 to 2,827 Kilo base (kb). The average number of contigs generated per genome was 64 contigs (ranging from 40 to 315 contigs). The average N50 contig length was 171778 bp (Supplementary Table 2). Isolates (and hence their genomes) from a single individual are expected to be part of a single clade as predicted by the MLST data and phylogenetic clustering (Figures 1, 2).
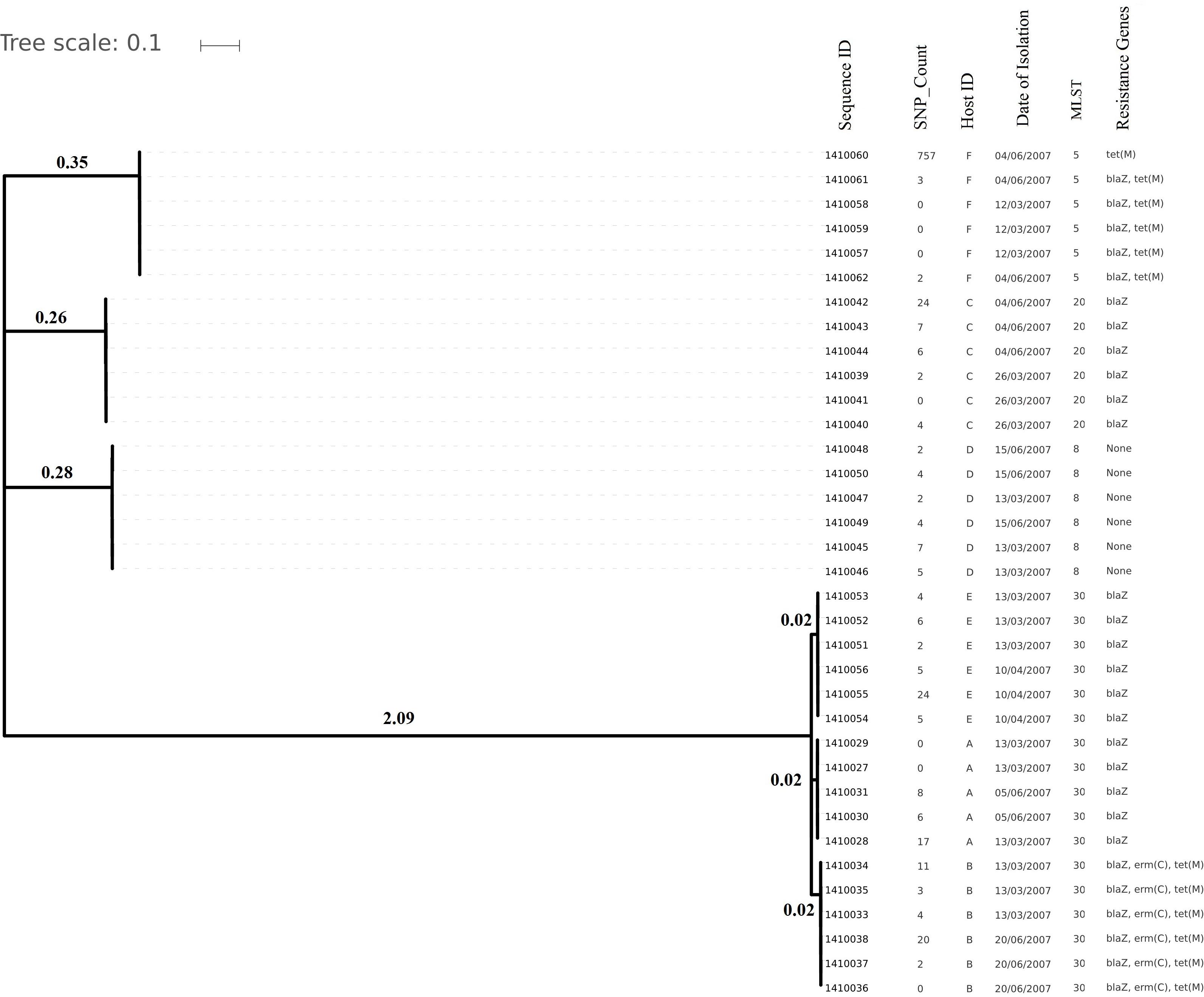
Figure 1. Phylogenetic tree depicting clustering on the basis of core SNP count ranges from 0 to 757 SNPs (median 4 SNPs) in all the Staphylococcus aureus strains colonized during 3 months (2007 subgroup) of follow up along with their date of isolation, persistent carriers from which they have isolated after maximum three cultural moments, their sequence type and resistance genes. Note that all isolates are clustered together on the basis of the original individual they were cultured from.
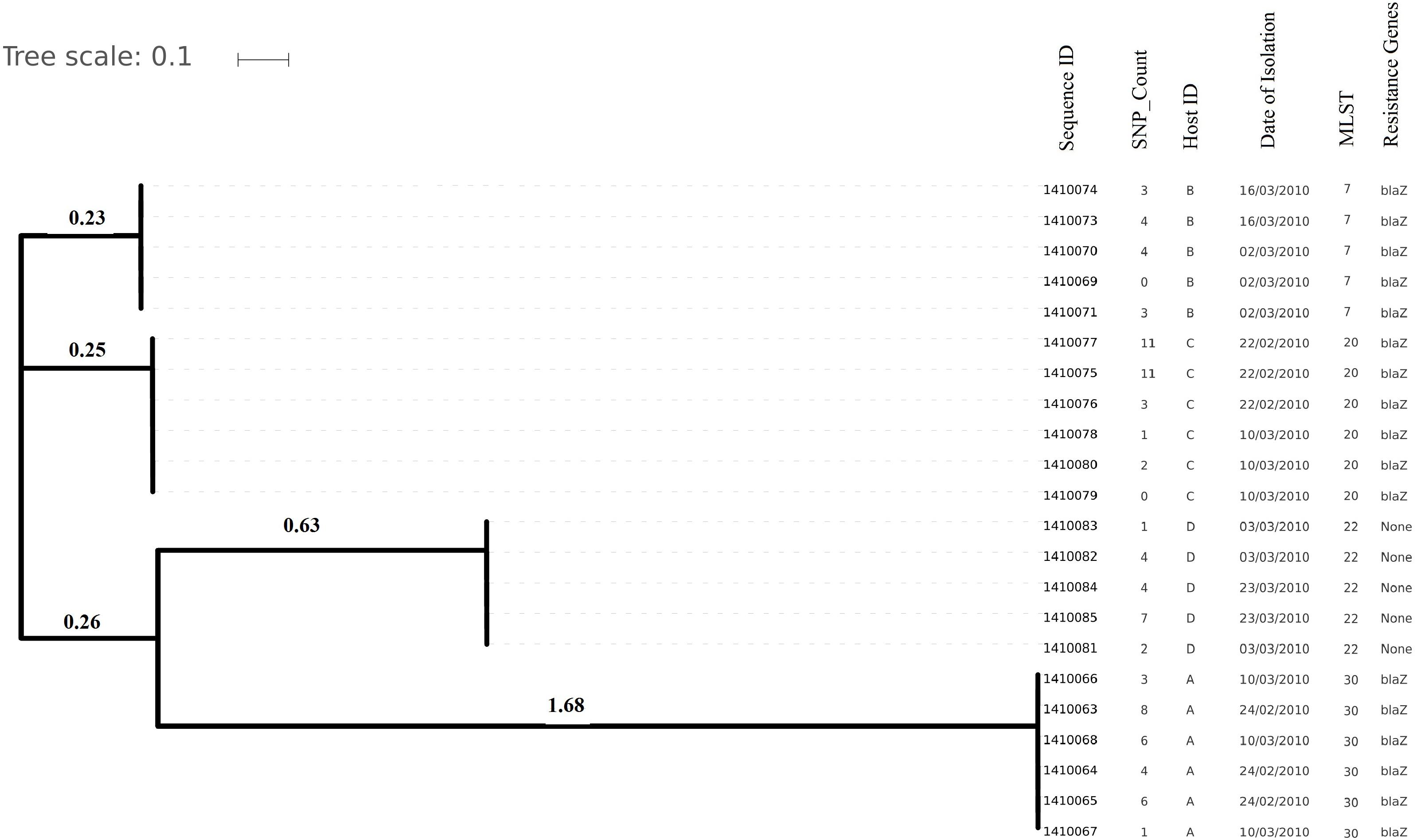
Figure 2. Evolutionary relationship on the basis of core genome SNP counts detected (range 0 to 11 SNPs) in S. aureus strains colonized and isolated during 1 month (2010 subgroup) along with the date of their isolation, the host from which they have isolated, MLST and resistance genotype. Isolates from the same host are clustered together showing their higher strain relatedness.
Short Term Evolution (1–3 Months) in Naturally Colonizing S. aureus Strains
Staphylococcus aureus isolates from 2007 and 2010 (35 and 22 genomes, respectively) from carriers A to F were analyzed for short term genomic changes, over 3 months (in 2007), and 1 month (in 2010). ST30 (2007: 47%; 2010: 30%) and ST20 (2007: 17.60%; 2010: 30%) were found to be the dominant MLST types followed by ST8 and ST5 (18% each) in 2007 and ST22 and ST7 (20% each) in the 2010 subgroups (Table 1). Over the period of 3 years some of the strains were replaced by different sequence type strains within a same carrier. For instance isolates from carrier B and D in 2007 were ST30 and ST8 but in 2010, isolates from the same carries were ST7 and ST22, respectively. These strains were not included for longer term pairwise SNP divergence analysis (Table 1).
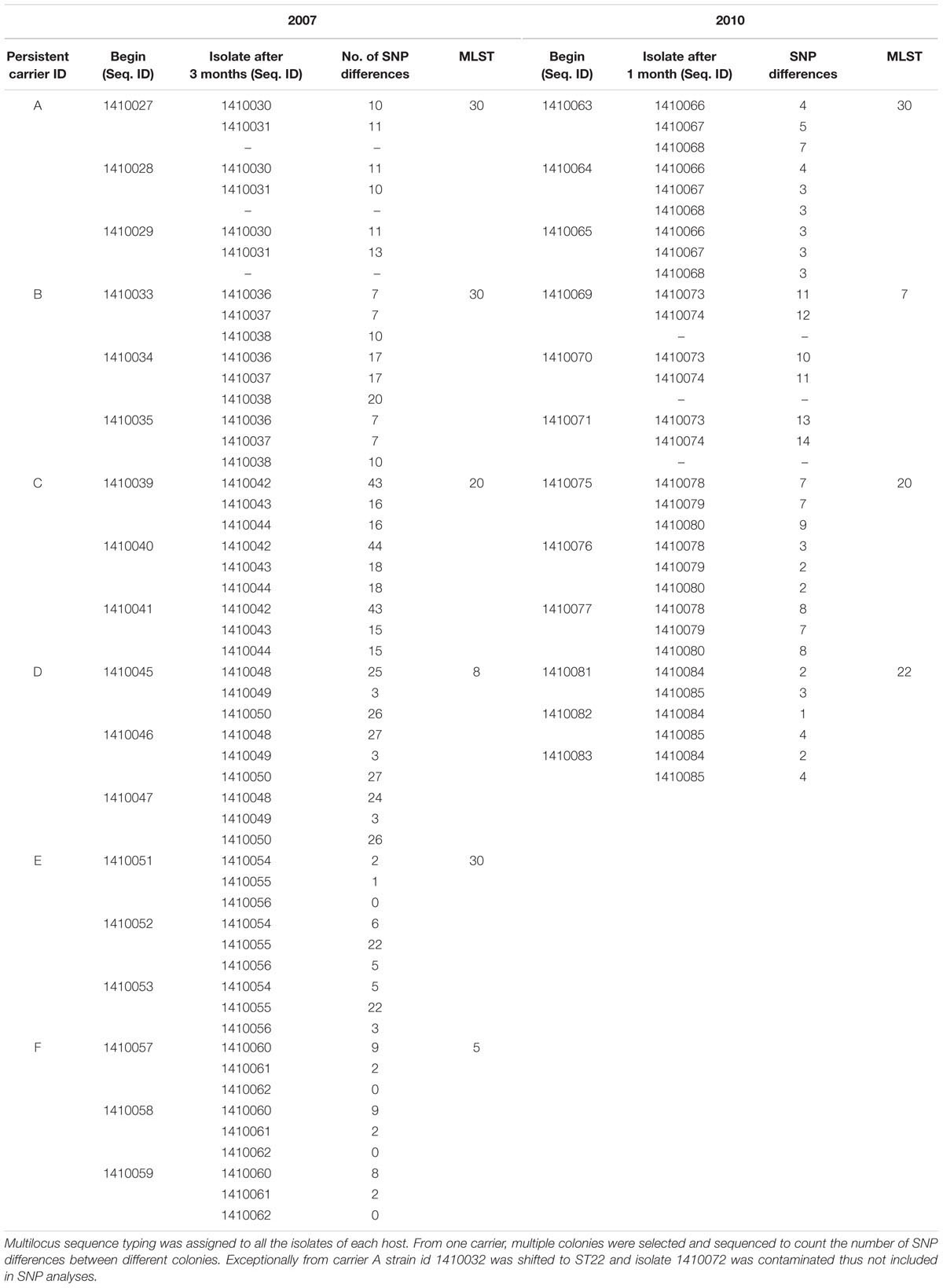
Table 1. Pairwise SNP distances identified between all the early and later stages isolates (according to their isolation date) among the S. aureus strains of subgroup 2007 and 2010 independently from each persistent nasal carrier.
Core genome SNP counts for the genomes of all the strains collected in 2007 and 2010 ranged from 0 to 757 SNPs (median 4 SNPs) and 0 to 11 SNPs (median 3.5 SNPs), respectively (Figures 1, 2). We observed a small pairwise SNP distance between all the early and the later stage isolates within a carrier (all carriers pooled, 2007 median number of SNP divergence was 10 and in 2010 median SNP distance was 4 (Table 1). The maximum number of pairwise SNP differences calculated for the genomes of the isolates of carrier C ranged from 15 to 44 SNPs followed by 3 to 27 SNPs in strains from carrier D, 0 to 22 in strains from E, 7 to 20 in strains from B, 10 to 13 in strains from A and 0 to 9 SNPs in strains from F (Table 1). Paired SNP differences were also calculated for strains from subgroup 2010 illustrating the highest ranges (10–14) among strains from host B followed by 2 to 9 SNPs in strains from C, 3 to 7 in strains from A, and 1 to 4 in strains from individual D (Table 1). On an individual basis, the pattern of pairwise SNP differences is relatively random between the isolates from early and later stages of colonization.
Longer Term Evolution (3 Years) in Naturally Colonizing S. aureus Strains
Evolutionary analysis over a period of 3 years (2007–2010) could only be done for the isolates from two persistent carriers, A, and C. In these carriers the MLST type remained unchanged over time, suggesting persistent colonization with the same strains (Table 1). All isolates of carrier A and C from both 2007 and 2010 were analyzed for the presence of core genome SNPs which ranged from 0 to 26 SNPs (median 4 SNPs) (Figure 3). Host specific pairwise SNP differences between the isolates dating 2007 and 2010 for both carriers A and C individually were 9–33 SNPs (median 19) and 15-57 SNPs (median 24), respectively (Figure 4). All strains from one carrier showed random distribution of SNPs; e.g., SNP distances between strains 1410027 and 1410029 versus the later stage strain 1410066 were 9 and 11 SNPs (Figure 4). This demonstrated that genomic evolution was random and none of the SNPs were fixed genetically over time.
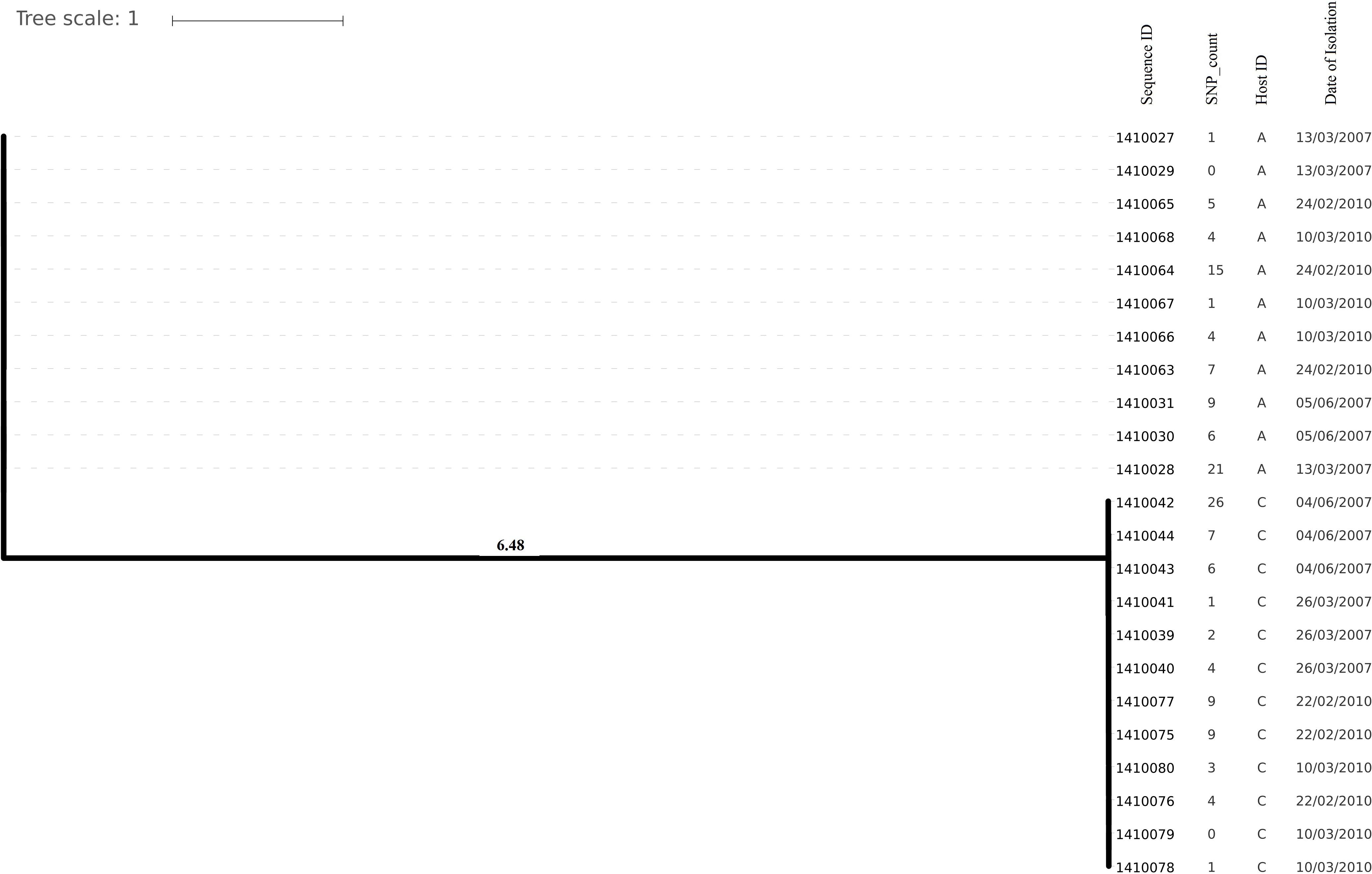
Figure 3. Phylogenetic tree showing longer term (3 years) diversity and relatedness of S. aureus strains on the bases of core genome SNP counts ranged from 0 to 26 SNPs in all the isolates from two nasal carriage individuals (A and C) for both the years 2007 and 2010.
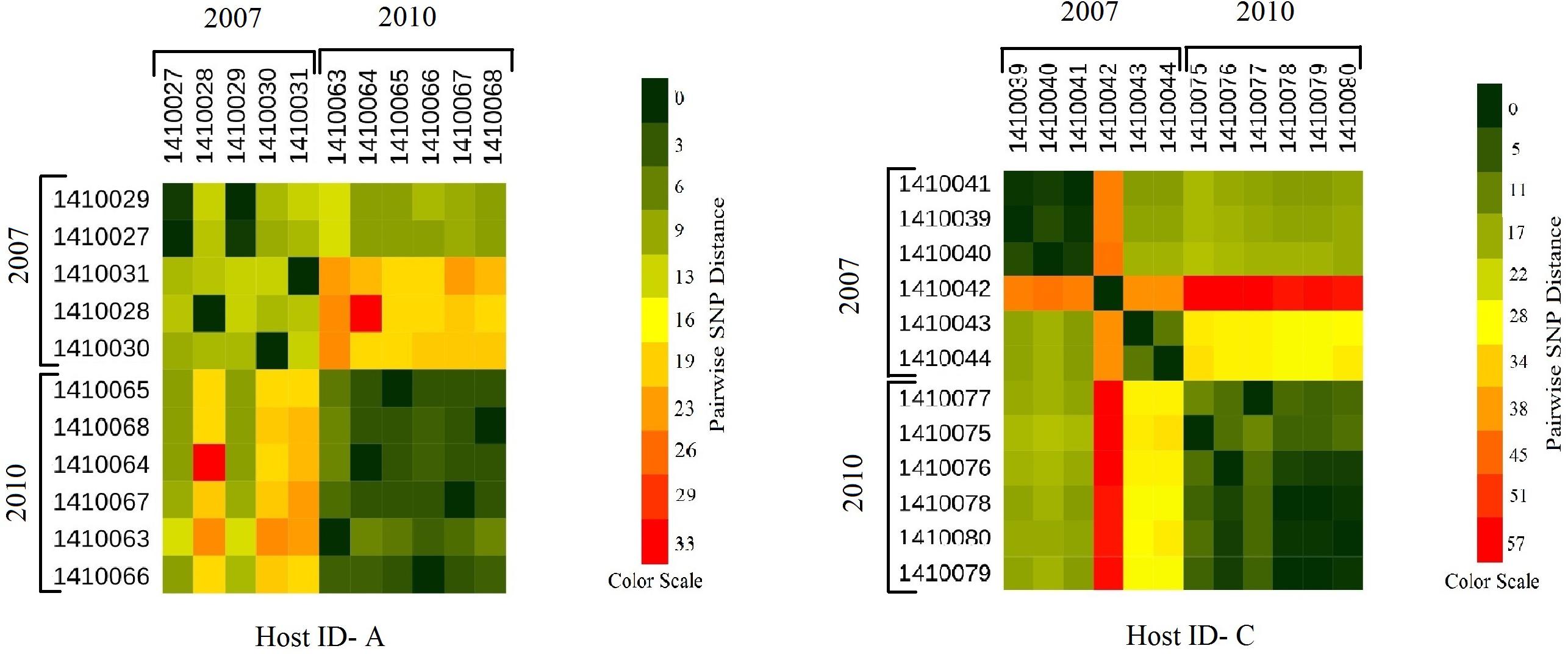
Figure 4. Heat maps showing the host specific pairwise SNP divergence (longer term) among all the early (2007) and later stage (2010) isolates of carrier A and carrier C individually with the color range of dark green (least SNP divergence) to red (higher SNP divergence). In both the hosts A and C pairwise SNP distances between the isolates of 2007 and 2010 datasets are visibly higher (from yellow to red boxes) than that of within the dataset itself (from dark green to light green boxes) with one exceptional isolate 1410042 in carrier C which showed higher pairwise SNP divergence within its dataset (orange boxes) as well as with the isolates of 2010 dataset (red boxes).
Mutational Analyses of Strains From Artificially Colonized Humans
All strains were of ST8. No considerable identity was observed with resistance genes (Supplementary Table 3) from the database which was in agreement with the pan-susceptibility of the isolates. The overall core genome SNP counts among the isolates ranged from 0 to 7 SNPs (average 2) (Figure 5). The maximum range of pairwise SNP distances between the isolates within a host was 0 to 5 SNPs (median SNP distance 2) after 28 days of colonization in S. aureus nasal carriers (Table 2).
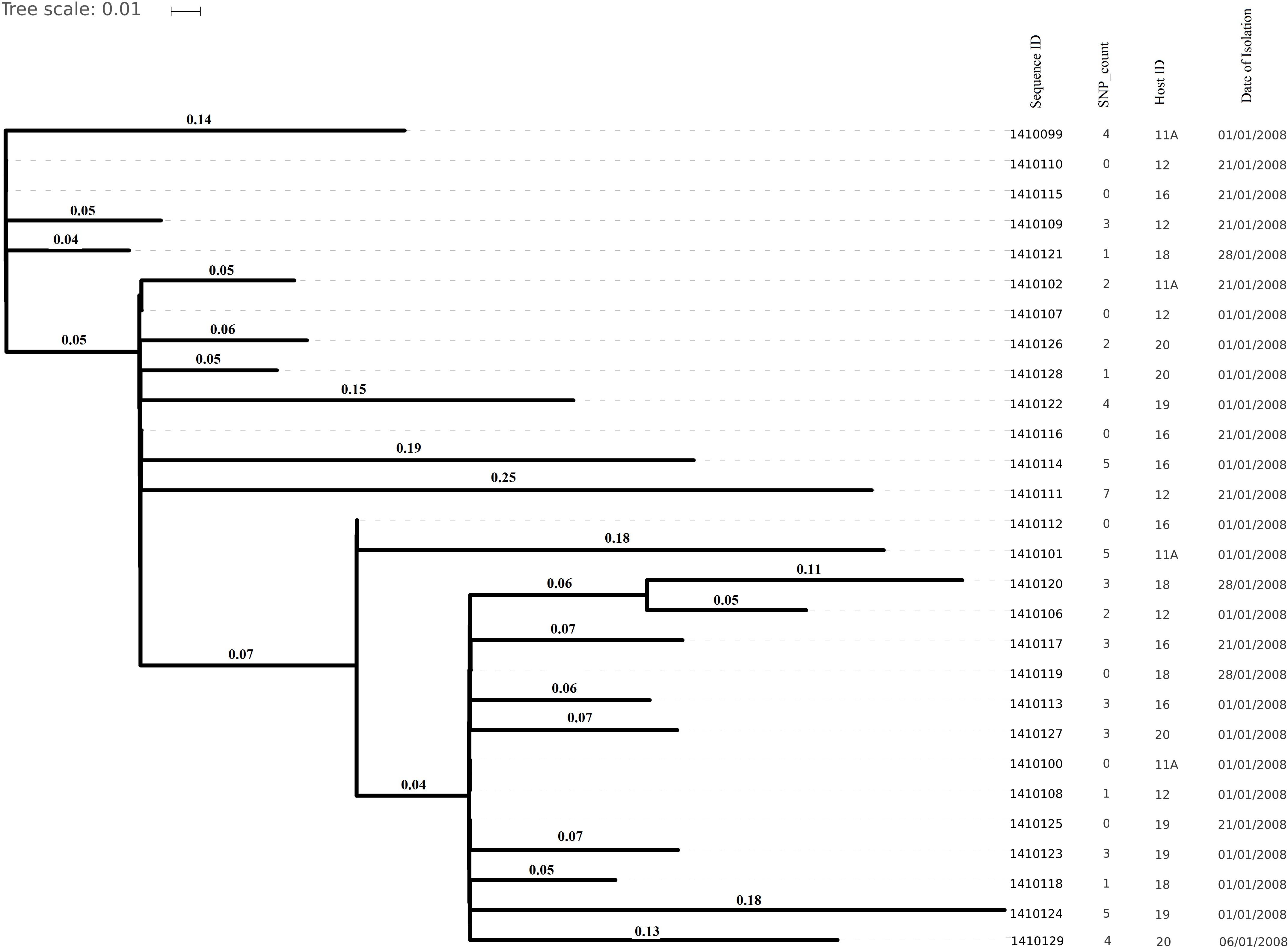
Figure 5. Core genome SNP counts based phylogenetic tree illustrating the close resemblance among the genomes isolated from artificially inoculated S. aureus nasal carriers in 2008. Core genome SNP counts here ranged from 0 to 7 core SNPs and each cluster is showing random collection of the strains irrespective of their specific host depicted very less genomic evolution (in 1 month) in artificially colonizing strains.

Table 2. Pairwise SNP distances found in artificially colonizing strains isolated during short term colonization in different individuals.
Fourteen virulence genes (sea, hla, hlb, hld, hlgB, clfA, clfB, fnbA, fnbB, icaA, sdrC, sdrD, sdrE, and tsst-1) were identified in the current sequence dataset (Supplementary Table 3). The virulence factor fnbA was not found in isolates from host B and was also missing in one of the isolates from carrier F (1410060). Two strains (1410054 and 1410055) were shown to have acquired the cna gene during colonization of host E (Supplementary Table 3). Absence of the scn gene corroborating the complete lack of IEC in artificially colonized strains (Supplementary Table 3).
Discussion
In the present work, we have studied the evolutionary patterns in nasal S. aureus strains to better understand their local adaptive behavior and mutational frequency. Low core genome SNP values among all the genomes defines the significant strain relatedness witnessed in this study. This is experimentally supported by the outcomes of previous research (Ankrum and Hall, 2017) where S. aureus strains with <71 SNP differences were considered as non-discriminate. Similar findings by Golubchik et al. (2013) suggested that SNP divergence with in a host varied from ∼ 0 to 27 SNPs among host specific isolates. In our study, one isolate from host C (1410042) was showing an exceptionally high SNP divergence value for which we have no clear explanation (Figure 5 and Table 1). Phylogenetic trees (Figures 1, 2, 3, 5) were constructed on the basis of core genome SNPs identified within strains from all individual hosts showing different numbers of mutations as compared to their pairwise frequency of SNP divergence. The level of diversity (SNP divergence) within the hosts was consistently lower than that detected between different hosts and of same MLST type (Golubchik et al., 2013).
Prior studies tried to assess the number of SNPs accumulating over time, but mostly under selective conditions. Rouard et al. (2018) calculated that during selection for linezolid resistance an expected 17–93 mutations should accumulate per genome per year. A more global calculation using a significantly larger strain collection resulted in average number of less than 10 SNPs per genome per year (Harris et al., 2010). Ankrum and Hall (2017) came up with figures around 70 SNPs per year. Obviously, the discussion on epidemiological SNP cut off values defining identity (or not) or close relatedness between clinical isolates have not been finalized yet. On the basis of this study, we suggest a median SNP cut off 20 SNPs. Although in our study limited numbers of individuals are included, a high number of strains per individual were included to thoroughly study mutation frequency over time. Our suggested cut off can be used to identify S. aureus strains as identical or not in outbreak management.
Nasal colonization with strains carrying virulence determinants such as fibronectin (fnb) and collagen adhesions (cna) may represent risk for subsequent invasive infections in carriers (Peacock et al., 2002b; Nashev et al., 2004).
We acknowledge that our study is likely to be underpowered: limited numbers of individuals were able to take part in these long-lasting and logistically complicated studies. Still, the artificial inoculation model is a unique feature of this study. So far and except for our own work, very few studies have been done using artificial inoculation in humans (Cole et al., 2018). On the other hand, epidemiological studies usually take place in similar time frames as used here. The mutation frequency we observe here during weeks and months will be well aligned with those occurring during active outbreaks since these mostly also span weeks rather than months.
Conclusion
Median core genome SNP counts and pairwise SNP divergence for all the strains studied here were always lower than 20 over periods up to 3 years of evolution in individual carriers. During artificial colonization, where median core genome SNP, and pairwise SNP distance scores were 2, there is no early stage selection of different genotypes. In addition, during stable long(er) term colonization (up to 3 years) the number of accumulating SNPs was low as well. We here suggest an epidemiological median cut off value of 20 SNPs as a marker of S. aureus strain identity during outbreaks of infection. Random pattern of pairwise SNP divergence between the strains isolated from single carrier suggested that the WGS of multiple colonies is necessary for outbreak infection analysis.
Data Availability
The datasets for this manuscript are not publicly available because we are still in the process of submitting data on NCBI. Requests to access the datasets should be directed tobWFuaXNoYS5nb3lhbEBiaW9tZXJpZXV4LmNvbQ==.
Author Contributions
WvW, NV, and AvB conceived the study. MT conducted the microbiological experimentation for S. aureus strains. MG, FJ, MP, and CM carried out the whole genome sequencing studies. MG interpreted the sequence data and wrote the first version of the manuscript. All authors discussed the results and edited the manuscript.
Funding
This work was funded by bioMérieux, France, the Erasmus University Medical Center, Netherlands, and the ViBrANT (ITN project Marie Skłodowska-Curie Grant Agreement No. 765042 funded by the European Union).
Conflict of Interest Statement
AvB, MP, CM, FJ, and MG were employees of bioMérieux, a company designing, developing, and marketing tests in the domain of infectious diseases. The company was not involved in the design of the current review and the opinions expressed are those of the authors and may be different from formal company opinions and policies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the volunteers who were inoculated with S. aureus strains to carry out the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01525/full#supplementary-material
Footnotes
- ^ http://www.applied-maths.com/
- ^ https://pubmlst.org/saureus/
- ^ http://www.mgc.ac.cn/VFs/main.htm
- ^ http://www.genomicepidemiology.org/
- ^ https://github.com/tseemann/abricate
References
Ankrum, A., and Hall, B. G. (2017). Population dynamics of staphylococcus aureus in cystic fibrosis patients to determine transmission events by use of whole-genome sequencing. J. Clin. Microbiol. 55, 2143–2152. doi: 10.1128/JCM.00164-17
Baur, S., Rautenberg, M., Faulstich, M., Grau, T., Severin, Y., Unger, C., et al. (2014). A nasal epithelial receptor for Staphylococcus aureus WTA governs adhesion to epithelial cells and modulates nasal colonization. PLoS Pathog 10:e1004089. doi: 10.1371/journal.ppat.1004089
Burian, M., Wolz, C., and Goerke, C. (2010). Regulatory adaptation of Staphylococcus aureus during nasal colonization of humans. PLoS One 5:e10040. doi: 10.1371/journal.pone.0010040
Chambers, H. F., and Deleo, F. R. (2009). Waves of resistance: staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641. doi: 10.1038/nrmicro2200
Cole, A. L., Schmidt-Owens, M., Beavis, A. C., Chong, C. F., Tarwater, P. M., Schaus, J., et al. (2018). Cessation from smoking improves innate host defense and clearance of experimentally inoculated nasal Staphylococcus aureus. Infect. Immun. 86, e912–17. doi: 10.1128/IAI.00912-917
Emonts, M., Uitterlinden, A. G., Nouwen, J. L., Kardys, I., de Maat, M. P. M., Melles, D. C., et al. (2007). Host polymorphisms in interleukin 4, complement factor H, and C-reactive protein associated with nasal carriage of staphylococcus aureus and occurrence of boils. J. Infect. Dis. 197, 1244–1253. doi: 10.1086/533501
Foster, T. M., and Höök, M. (1998). Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 6, 484–488. doi: 10.1016/s0966-842x(98)01400-0
Frank, D. N., Feazel, L. M., Bessesen, M. T., Price, C. S., Janoff, E. N., and Pace, N. R. (2010). The human nasal microbiota and Staphylococcus aureus carriage. PLoS One 5:e10598. doi: 10.1371/journal.pone.0010598
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Ghasemian, A., Peerayeh, S. N., Bakhshi, B., and Mirzaee, M. (2015). The microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) genes among clinical isolates of staphylococcus aureus from hospitalized children. Iran J. Pathol. 10, 258–264.
Goerke, C., Wirtz, C., Fluckiger, U., and Wolz, C. (2006). Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 61, 1673–1685. doi: 10.1111/j.1365-2958.2006.05354.x
Golubchik, T., Batty, E. M., Miller, R. R., Farr, H., Young, B. C., Larner-Svensson, H., et al. (2013). Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS One 8:e61319. doi: 10.1371/journal.pone.0061319
Gorwitz, R. J., Kruszon-Moran, D., McAllister, S. K., McQuillan, G., McDougal, L. K., Fosheim, G. E., et al. (2008). Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J. Infect. Dis. 197, 1226–1234. doi: 10.1086/533494
Harris, S. R., Feil, E. J., Holden, M. T., Quail, M. A., Nickerson, E. K., Chantratita, N., et al. (2010). Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474. doi: 10.1126/science.1182395
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Li, B., and Webster, T. J. (2018). Bacteria antibiotic resistance: new challenges and opportunities for implant-associated orthopedic infections. J. Orthop. Res. 36, 22–32. doi: 10.1002/jor.23656
Nashev, D., Toshkova, K., Salasia, S. I., Hassan, A. A., Lammler, C., and Zschock, M. (2004). Distribution of virulence genes of Staphylococcus aureus isolated from stable nasal carriers. FEMS Microbiol. Lett. 233, 45–52. doi: 10.1016/j.femsle.2004.01.032
Peacock, S. J., de Silva, G. D., Justice, A., Cowland, A., Moore, C. E., Winearls, C. G., et al. (2002a). Comparison of multilocus sequence typing and pulsed-field gel electrophoresis as tools for typing Staphylococcus aureus isolates in a microepidemiological setting. J. Clin. Microbiol. 40, 3764–3770. doi: 10.1128/jcm.40.10.3764-3770.2002
Peacock, S. J., Moore, C. E., Justice, A., Kantzanou, M., Story, L., Mackie, K., et al. (2002b). Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect. Immun. 70, 4987–4996. doi: 10.1128/iai.70.9.4987-4996.2002
Rasigade, J. P., and Vandenesch, F. (2014). Staphylococcus aureus: a pathogen with still unresolved issues. Infect. Genet. Evol. 21, 510–514. doi: 10.1016/j.meegid.2013.08.018
Roca, I., Akova, M., Baquero, F., Carlet, J., Cavaleri, M., Coenen, S., et al. (2015). The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 6, 22–29. doi: 10.1016/j.nmni.2015.02.007
Roche, F. M., Meehan, M., and Foster, T. J. (2003). The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells. Microbiology 149(Pt 10), 2759–2767. doi: 10.1099/mic.0.26412-26410
Rouard, C., Garnier, F., Leraut, J., Lepainteur, M., Rahajamananav, L., Languepin, J., et al. (2018). Emergence and within-host genetic evolution of methicillinresistant staphylococcus aureus resistant to linezolid in a cystic fibrosis patient. Antimicrob. Agents Chemother. 62, e720–18.
Ruimy, R., Angebault, C., Djossou, F., Dupont, C., Epelboin, L., Jarraud, S., et al. (2010). Are the Host Genetics Predominant Determinant of Persistent Nasal Staphylococcus aureus Carriage In Humans? J. Infect. Dis. 202, 924–934. doi: 10.1086/655901
Schmidt, A., Benard, S., and Cyr, S. (2015). Hospital cost of staphylococcal infection after cardiothoracic or orthopedic operations in France: a retrospective database analysis. Surg. Infect. 16, 428–435. doi: 10.1089/sur.2014.045
Shukla, S. K., Karow, M. E., Brady, J. M., Stemper, M. E., Kislow, J., Moore, N., et al. (2010). Virulence genes and genotypic associations in nasal carriage, community-associated methicillin-susceptible and methicillin-resistant USA400 Staphylococcus aureus isolates. J. Clin. Microbiol. 48, 3582–3592. doi: 10.1128/JCM.00657-610
Taylor, T. A., and Unakal, C. G. (2018). Staphylococcus Aureus. Treasure Island, FL: StatPearls Publishing.
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P., and van Strijp, J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006
Verkaik, N. J., Benard, M., Boelens, H. A., de Vogel, C. P., Nouwen, J. L., Verbrugh, H. A., et al. (2011). Immune evasion cluster-positive bacteriophages are highly prevalent among human Staphylococcus aureus strains, but they are not essential in the first stages of nasal colonization. Clin. Microbiol. Infect. 17, 343–348. doi: 10.1111/j.1469-0691.2010.03227.x
Wertheim, H. F. L., Melles, D. C., Vos, M. C., van Leeuwen, W., van Belkum, A., Verbrugh, H. A., et al. (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762. doi: 10.1016/s1473-3099(05)70295-70294
Wertheim, H. F. L., Walsh, E., Choudhurry, R., Melles, D. C., Boelens, H. A. M., Miajlovic, H., et al. (2008). Key Role for Clumping Factor B in Staphylococcus aureus nasal colonization of humans. PLoS Med. 5:e17. doi: 10.1371/journal.pmed.0050017
Keywords: Staphylococcus aureus, nasal carriage, epidemiology, resistance, MLST, SNP, strain relatedness, genomic evolution
Citation: Goyal M, Javerliat F, Palmieri M, Mirande C, van Wamel W, Tavakol M, Verkaik NJ and van Belkum A (2019) Genomic Evolution of Staphylococcus aureus During Artificial and Natural Colonization of the Human Nose. Front. Microbiol. 10:1525. doi: 10.3389/fmicb.2019.01525
Received: 26 March 2019; Accepted: 18 June 2019;
Published: 05 July 2019.
Edited by:
Baolei Jia, Chung-Ang University, South KoreaReviewed by:
Dominique Missiakas, The University of Chicago, United StatesSima Sadat Seyedjavadi, Pasteur Institute of Iran (PII), Iran
Copyright © 2019 Goyal, Javerliat, Palmieri, Mirande, van Wamel, Tavakol, Verkaik and van Belkum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex van Belkum, YWxleC52YW5iZWxrdW1AYmlvbWVyaWV1eC5jb20=