 Carmen Hogendoorn1
Carmen Hogendoorn1 Arjan Pol1
Arjan Pol1 Nunzia Picone1
Nunzia Picone1 Geert Cremers1
Geert Cremers1 Theo A. van Alen1
Theo A. van Alen1 Antonina L. Gagliano2
Antonina L. Gagliano2 Mike S. M. Jetten1
Mike S. M. Jetten1 Walter D’Alessandro2
Walter D’Alessandro2 Paola Quatrini3
Paola Quatrini3 Huub J. M. Op den Camp1*
Huub J. M. Op den Camp1*- 1Department of Microbiology, Institute for Water and Wetland Research, Radboud University, Nijmegen, Netherlands
- 2Istituto Nazionale di Geofisica e Vulcanologia, Palermo, Italy
- 3Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Palermo, Italy
Volcanic and geothermal areas are hot and often acidic environments that emit geothermal gasses, including H2, CO and CO2. Geothermal gasses mix with air, creating conditions where thermoacidophilic aerobic H2- and CO-oxidizing microorganisms could thrive. Here, we describe the isolation of two Kyrpidia spormannii strains, which can grow autotrophically by oxidizing H2 and CO with oxygen. These strains, FAVT5 and COOX1, were isolated from the geothermal soils of the Favara Grande on Pantelleria Island, Italy. Extended physiology studies were performed with K. spormannii FAVT5, and showed that this strain grows optimally at 55°C and pH 5.0. The highest growth rate is obtained using H2 as energy source (μmax 0.19 ± 0.02 h–1, doubling time 3.6 h). K. spormannii FAVT5 can additionally grow on a variety of organic substrates, including some alcohols, volatile fatty acids and amino acids. The genome of each strain encodes for two O2-tolerant hydrogenases belonging to [NiFe] group 2a hydrogenases and transcriptome studies using K. spormannii FAVT5 showed that both hydrogenases are expressed under H2 limiting conditions. So far no Firmicutes except K. spormannii FAVT5 have been reported to exhibit a high affinity for H2, with a Ks of 327 ± 24 nM. The genomes of each strain encode for one putative CO dehydrogenase, belonging to Form II aerobic CO dehydrogenases. The genomic potential and physiological properties of these Kyrpidia strains seem to be quite well adapted to thrive in the harsh environmental volcanic conditions.
Introduction
Volcanic and geothermal areas represent the result of the dynamics involving the deeper layers of the earth and resulting in the emission of several gases from volcanic soils. In general, geothermal gases are mainly composed of H2O and CO2 as the most dominant species, and other minor species as the more reduced gases H2S, CH4, H2, CO, and NH3 (Oppenheimer et al., 2014). The abundance of the latter gases, and consequently their impact to the atmosphere, is strictly related to the energy of the system, the water-rock interactions, gas-gas interactions and also to the gas-biota interactions. In fact, the minor gas species represent the driving forces for the establishment of an active microbial community (Shock et al., 2010; Lindsay et al., 2019). Commonly, geothermal soils are characterized by high temperatures and often a low pH, which may be caused by the presence of H2S and CS2 in the emitted gases and their conversion to sulfuric acid by sulfide-oxidizing microbes (Chiodini et al., 2001; D’Alessandro et al., 2009; Smeulders et al., 2011, 2013; Quatrini and Johnson, 2018). Despite the harsh conditions, these hot and acidic terrestrial areas harbor distinctive microbial communities (Colman et al., 2019a, b; Lindsay et al., 2019). Analyses of soil microbial communities show that at low pH and high temperature, the dominant phyla differ from areas with moderate temperature and neutral pH (Strazzulli et al., 2017). Beside temperature and acidity, the available nutrients determine the microbial composition.
Gagliano et al. (2016) showed that 99% of the sequences obtained at the FAV1 site at the geothermal soils of the Favara Grande (Pantelleria Island) could be assigned to four main phyla: Proteobacteria, Firmicutes, Actinobacteria and Chloroflexi. At the FAV2 site 98% of the sequences were distributed in only three phyla Proteobacteria, Firmicutes and Actinobacteria. Especially Chloroflexi were much less represented than in FAV1. Apparently sites with low NH4+ concentration are dominated by a diverse methanotrophic community, whereas high NH4+ soils harbor thermo-acidophilic chemolithotrophs, despite CH4 being present. Alphaproteobacterial methanotrophs were isolated from the low NH4+ site (Gagliano et al., 2014). Previously, key players in the conversion of H2S, CS2, and CH4 from the Solfatara geothermal area near Naples, Italy, have been isolated and characterized (Pol et al., 2007; Smeulders et al., 2011, 2013; van Teeseling et al., 2014). Besides H2S, CS2 and CH4, H2 and CO are available and suitable electron donors for novel chemolithoautotrophs in geothermal soils. In this study, we focused on H2 and CO-utilizing bacteria from the Favara Grande geothermal site on Pantelleria Island, Italy. These hot and acidic soils are characterized by H2 (5-168,000 ppm) and CO (1.6-26 ppm) emissions (D’Alessandro et al., 2009) and in situ H2 consumption was already observed (Gagliano et al., 2016). Both H2 and CO have a high potential as electron donor for microbial growth.
H2 is an important electron donor in acidic and neutral hot springs (Spear et al., 2005) and can be used in a lithotrophic lifestyle under aerobic or anaerobic conditions. Oxidation of H2 is catalyzed by the enzyme hydrogenase, which can be classified as [NiFe]-, [FeFe]-, or [Fe]-hydrogenase depending on the metal ion in the active site (Lubitz et al., 2014). Hydrogenases are found among many different phyla, such as Proteobacteria, Firmicutes, Cyanobacteria, Chloroflexi, Verrucomicrobia, Aquificae, Euryarchaeota and Crenarchaeota (Greening et al., 2015). Aerobic H2-oxidizing microorganisms are referred to as “Knallgas” bacteria.
CO is a highly reactive gas, and often it is not detected due to field and laboratory equipment detection limits. Despite being a minor species in the hydrothermal gas mixture, CO analysis is very important because any variation in its amount may indicate some variations of the hydrothermal system. CO can be oxidized under aerobic and anaerobic conditions (Robb and Techtmann, 2018). Anaerobic CO-oxidizing bacteria have been detected in geothermal springs of neutral pH (Brady et al., 2015). In addition, anaerobic microbial CO consumption has been observed in acidic hot spring mud of pH 2.9 (Ikeda et al., 2015). In both cases, the microbial population was dominated by Firmicutes. Two distinct classes of CO dehydrogenases (CODH) catalyze CO transformation. The active site of the CODH from anaerobes contains a sulfur-coordinated Ni in a [Ni-4Fe-5S] cluster (Robb and Techtmann, 2018) and can be monofunctional producing CO2 or bifunctional producing acetyl-CoA and H2. Aerobic CO-oxidizers, or carboxydotrophs, possess heterotrimeric CO dehydrogenases with an active site containing a [Mo-Cu] cluster in the CoxL subunit. The other components are a flavoprotein (CoxM) and an iron-sulfur protein (CoxS). These enzyme complexes catalyze the unidirectional conversion of CO to CO2. The aerobic [Mo-Cu]-CO dehydrogenases can be categorized in two groups, Form I and Form II (King and Weber, 2007). Biochemical analysis showed that Form I has highest CO oxidation rates and is responsible for aerobic growth on CO. Form II is phylogenetically closer related to Form I CO dehydrogenase than to other enzymes of the molybdenum hydroxylases family. So far, no firm evidence is reported for CO oxidation by Form II enzymes, suggesting that Form II CO dehydrogenases may also catalyze the oxidation of a different substrate (Cunliffe, 2011).
In the upper layer of geothermal soils air is mixed with the geothermal gasses (Gagliano et al., 2016). Since soil porosity is higher than in the deeper soil horizon and the uprising hydrothermal flux lost part of its energy, this results in conditions where (micro)aerobic H2 and CO-oxidizing bacteria could thrive. Here, we used samples of the Favare grande soil to enrich H2-oxidizing microorganisms. After serial dilution to extinction of active enrichments we isolated and characterized two Kyrpidia spormannii species capable of using H2 and CO as sole energy source. We show that the genomic potential and physiological properties of the two Kyrpidia strains seem very suited to thrive in harsh volcanic conditions.
Materials and Methods
Geological Setting
Pantelleria Island is a quiescent volcano located in the Sicily Channel, and characterized by several hydrothermal manifestations as mofettes, fumaroles and passive degassing from geothermal soils. The main exhalative area is Favara Grande, where soil temperatures can reach 115°C at 5 cm of depth and soil pH may be down to 3. The geothermal field passively degases CO2, CH4, H2 in order of magnitude of percent per volume unit, and minor species as CO in the order of magnitude of ppm per volume unit (see Table 2 in D’Alessandro et al., 2009). Soil samples were taken in June 2017 at Favara Grande from two sites, FAV1 (23°21′80″N; 40°73′170″E) and FAV2 (23°21′77″N; 40°73160E) (Gagliano et al., 2016), using a core sampler (diameter 1.5 cm), divided into subsections of 5 cm and stored in sterile 50 ml tubes at room temperature.
Enrichment and Isolation
The cultivation medium based on geochemical data (Gagliano et al., 2016) was composed of 0.5 mM MgCl2.H2O, 0.5 mM CaCl2.H2O, 1 mM Na2SO4, 2 mM K2SO4, 1 mM (NH4)2SO2 and 1 mM NaH2PO4.H2O. The final trace element concentrations were 1 μM CoCl2.6H2O, NaMoO4.2H2O, Na2SeO3, CeCl3.6H2O and ZnSO4.7H2O, 5 μM MnCl2.4H2O and FeSO4.7H2O and 10 μM CuSO4.5H2O and NiCl2.6H2O, with 50 μM NTA as complexing agent. The pH was set to 3.0 or 5.0 by adding 1M H2SO4 or 1M NaOH.
Within 6 h after taking the samples, the soil was mixed with sterile minimal medium of either pH 3 or pH 5. The mixtures were shaken for 10 min to extract the microorganisms from the soil, after which the suspensions were left settling for a few minutes. One ml of the liquid phase was transferred to a sterile 60 ml bottle, containing 10 ml sterile medium, either pH 3 or pH 5, and 87% (v/v) N2, 10% (v/v) CO2, 1.5% (v/v) air and either 1.5% (v/v) CO or H2. Immediately after this, 1 ml of the first batch is transferred to a new bottle (10x diluted). The bottles were stored at room temperature and after 24 h, the incubations were transferred to a shaking incubator operated at either 50 or 60°C and 50 rpm. Bottles that showed H2 or CO consumption were serial diluted to extinction to obtain pure cultures.
Batch Cultivation
Growth experiments were performed in triplicate in 120 ml flasks with 20 ml medium. The headspace contained 10% (v/v) H2 or CO, 5% (v/v) O2, 5% (v/v) CO2 and 80% (v/v) N2. 2-(N-morpholino)ethanesulfonic acid (25 mM) was used to buffer the medium. Bottles were incubated at 55°C, unless stated otherwise, in a shaking incubator operating at 250 rpm. To test the growth on organic substrates, H2 or CO was replaced by the organic substrate (25 mM) or 1 g/l yeast extract. To test nitrogen fixation, medium without ammonium was used. To test for growth on urea, ammonium was replaced by 2 mM urea.
Continuous Culture
Cultivation was performed in a 500 ml bioreactor (Applikon, Delft, Netherlands) with a working volume of 350 ml and the medium described above. The temperature was maintained at 55°C using a Peltier element. The pH was set to 5.0, measured by a pH electrode (Applikon, Delft, Netherlands) and maintained at pH 5.0 ± 0.1 by adding 0.2 M NaOH. The dissolved oxygen (DO) concentration was measured by a DO electrode (Applikon, Delft, Netherlands). The airflow was regulated to maintain a dissolved oxygen concentration of 5% air saturation. Temperature, pH and DO were controlled using the in-Control process controller (Applikon, Delft, Netherlands). The reactor was stirred at 1000 rpm using a stirrer with two Rushton impellers. The reactor was supplied with air (regulated), 9 ml.min–1 CO2-Argon (5%:95%, v/v), 2.5 ml.min–1 H2 and operated at a dilution rate of 0.045 h–1. Growth under this conditions is H2 limited since all other substrates are in excess.
Gas Analysis
H2 and CO2 in the headspace of the bottles and the in- and outflow of the chemostat cultures were measured using a HP 5890 gas chromatograph (Agilent, Santa Clara, California) equipped with a Porapak Q column (1.8 m, ID 2 mm) and a thermal conductivity detector. For this analysis, 50–100 μl gas samples were injected. To determine the CO and O2 consumption, 25–100 μl gas was injected and measured on an Agilent series 6890 GC-MS (Agilent, Santa Clara, CA, United States) and analyzed as described before (Ettwig et al., 2008).
Optical Density and Dry Weight
The optical density was measured using a Cary 50 UV-VIS spectrophotometer at a wavelength of 600 nm (Agilent, Santa Clara, California). Dry weight was determined as described before (Mohammadi et al., 2016).
Membrane-Inlet Mass Spectrometry (MIMS)
Liquid concentrations of H2 were measured by Membrane Inlet Mass Spectrometry (HPR40, Positive Ion Counting detector, Hiden Analytical, Warrington, United Kingdom) in a 10 ml chamber. Briefly, the MIMS probe mounted with a 10 μm thin silicon (Hiden Analytical, Warrington, United Kingdom) or PTFE (Hansatech, Pentney, United Kingdom) membrane (8 mm2) was inserted inside of the bottom part just above the glass stirrer bar (1000 rpm). Liquid was equilibrated with the desired gas by bubbling via a metal capillary. The liquid volume was adjusted with a piston removing gas bubbles (8–9 ml). All additions were done via the metal capillary by gastight syringes (Hamilton, Reno, Nevada). Signal of mass 2 was not only derived from H2 but resulted also from water and gases such as CO2. Therefore, water vapor and other gases that pass the silicon/PTFE membrane were trapped using a coiled part inserted in liquid nitrogen before entering the mass spectrometer. In this way, the background mass 2 signal was minimized. For calibrations, known amounts of gas-saturated water were administered from 50 ml serum bottles containing 10 ml of water. The medium and pH set was identical to the growth medium.
DNA Sequencing and Genome Reconstruction
For DNA isolation 2 ml cell suspension (OD600 0.5–1.0) from the continuous culture was harvested by centrifugation (2 min, 14,000 × g) and resuspended in 100 μl sterile MQ water. DNA was extracted with the PowerSoil DNA isolation kit or the DNeasy Blood and Tissue kit according to the manufacturer’s instructions (Qiagen Benelux B.V, Venlo, The Netherlands). The quality and quantity of the DNA was analyzed using the Qubit (Thermo Fisher Scientific, Waltham, MA, United States) and the Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, Waltham, MA, United States).
The genome was reconstructed using a combination of short-read Illumina sequencing and long-read Nanopore sequencing. For Illumina library preparation, the Nextera XT kit (Illumina, San Diego, CA, United States) was used according to the manufacturer’s instructions. Enzymatic tagmentation was performed starting with 1 ng of DNA, followed by incorporation of the indexed adapters and amplification of the library. After purification of the amplified library using AMPure XP beads (Beckman Coulter, Indianapolis, IN, United States), libraries were checked for quality and size distribution using the Agilent 2100 Bioanalyzer and the High sensitivity DNA kit. Quantitation of the library was performed by Qubit using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States). The libraries were pooled, denatured and sequenced with the Illumina Miseq sequence machine (San Diego, California). Paired end sequencing of 2 × 301 base pairs was performed using the MiSeq Reagent Kit v3 (Illumina, San Diego, CA, United States) according to the manufacturer’s protocol.
For Nanopore library preparation, 1–1.5 μg of DNA was used. The input DNA was checked for high molecular DNA and absence of degradation by agarose (0.5%) gel electrophoresis. For Nanopore sequencing the DNA Library construction was performed using the Ligation Sequencing Kit 1D (SQK-LSK108) in combination the Native barcoding Expansion Kit (EXP-NBD103 or EXP-NBD104) according to the manufacturers protocol (Oxford Nanopore Technologies, Oxford United Kingdom). The libraries were loaded and sequenced on a Flow Cell (R9.4.1) and run on a MinION device (Oxford Nanopore Technologies, Oxford, United Kingdom), according to the manufacturer’s instructions. Base calling after sequencing was done using the guppy_basecaller in combination with guppy_barcoder (Oxford Nanopore Technologies, Limited Version 2.3.7).
The genome was assembled from Nanopore reads using Canu (v1.8) (Koren et al., 2017). Assembled contigs were first polished with Racon (v1.3.1) (Vaser et al., 2017) followed by two iterations of Pilon (v1.23) polishing with Illumina reads (Walker et al., 2014). The genome was annotated using the MicroScope platform (Vallenet et al., 2013) and annotations were checked manually.
Phylogenomic Tree Reconstruction
Bacterial genome-based phylogenetic analyses were performed using the up-to-date core gene (UBCG) set and pipeline for phylogenomic tree reconstruction (Na et al., 2018). Extracted genes of the genomes of the isolates and the reference genomes were aligned and concatenated using UBCG with default parameters. Maximum likelihood phylogenetic trees were made from the concatenated nucleotide alignment using RAxML version 8.2.10 (Stamatakis, 2014) on the CIPRES science gateway (Miller et al., 2012) with the GTR substitution and GAMMA rate heterogeneity models and 100 bootstrap iterations.
RNA Sequencing and RNA-Seq Analysis
For transcriptome analysis, triplicate samples each of 10 ml cell suspension (OD600 0.5) were taken from the continuous culture and harvested by centrifugation. mRNA was isolated using the RiboPureTM-Bacteria kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, United States). The quality and quantity of the RNA was analyzed using the Qubit (Thermo Fisher Scientific, Waltham, MA, United States) and the Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, Waltham, MA, United States). The transcriptome libraries were constructed using the TruSeq® Stranded mRNA Library Prep protocol (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions. Total mRNA was used for library preparation and obtained libraries were checked qualitatively and quantitatively as described above. Pooled libraries were sequenced using the Illumina Miseq sequence machine (Illumina, San Diego, CA, United States). For sequencing the 151 bp sequence chemistry was performed using the MiSeq Reagent Kit v3 (Illumina, San Diego, CA, United States) according to the manufacturers protocol in one direction. CLCBio software (version 10.1.1, Qiagen, Aarhus, Denmark) was used to perform RNA-seq analysis. Gene expression levels were compared by calculating the reads per kilobase per million reads (RPKM) values for the CDSs and calculating the log2-fold to median (Mundinger et al., 2019).
16S rRNA Gene Analysis
The 16S rRNA gene was PCR amplified from isolated DNA using the primers 616F (AGAGTTTGATYMTGGCTCAG) and 1492R (GGTTACCTTGTTACGACTT) using the PCR program 5 min 94°C, 30 cycles 40 s at 96°C, 40 s 55°C, 40 s 72°C and finally 10 min 72°C. The amplicon was cloned into the pGEM-T Easy cloning vector (Promega) and transformed into competent E. coli cells. After growth of the cells, the 16S rRNA amplicon in the vector was PCR amplified, cleaned (GeneJET PCR purification kit, Thermo Fisher Scientific, Waltham, MA, United States) and sequenced using the Sanger sequencing platform (BaseClear B.V., Leiden, the Netherlands).
Deposition of Cultures and Sequences
The two isolated strains were deposited to the DSMZ culture collection as Kyrpidia spormannii FAVT5 (DSM 109470) and Kyrpidia spormannii COOX1 (DSM 109471). The genomes of the two strains are available at the MaGe platform (accession numbers KFAV.1 and KSCOOX1.1)1.
Results and Discussion
Enrichment and Isolation
In this study, aerobic H2 and CO-oxidizing soil bacteria from the geothermal active area of Favara Grande on the island of Pantelleria, Italy were enriched, isolated and characterized. The primary enrichment minimal medium (low nutrient, autotrophic) was based upon geochemical analyses (Gagliano et al., 2016). The incubations were performed at two different pH values, pH 3 and pH 5, and at either 50 or 60°C with H2 or CO as electron donor. All enrichment cultures were checked for H2 or CO consumption, after which the most active cultures (pH 5, 60°C, from FAV1) were diluted to extinction using fresh medium. All bottles containing H2 in the headspace showed H2 consumption. After three transfers, a 16S rRNA gene analysis of these active cultures was performed and revealed that in all cultures the same microorganism was present and dominant (>95%). Therefore, only one culture was used to continue the isolation. After another three consecutive rounds of serial dilutions this resulted in the isolation of strain FAVT5. In contrast, only one of the enrichments with CO as electron donor showed activity, the one incubated at pH 5 and 60°C and inoculated with soil of FAV1. This culture was serial diluted to extinction for five consecutive rounds, resulting in the isolation of the strain COOX1.
Genome Sequencing and Phylogeny
The genomes of both isolates, FAVT5 and COOX1, were sequenced and assembled using a combination of short read Illumina sequencing and long read Nanopore MinION sequencing. This resulted in closed genomes and the general features of both genomes are compiled in Table 1. The genomes are both 3.3 Mb in size and have a GC content of 59% and both contain five complete rRNA operons (16S, 23S, 5S) and 59 tRNAs with 1–5 copies per tRNA type. The 16S rRNA gene sequences in the genomes were identical to the sequences obtained by PCR analysis of the enrichments (see above).
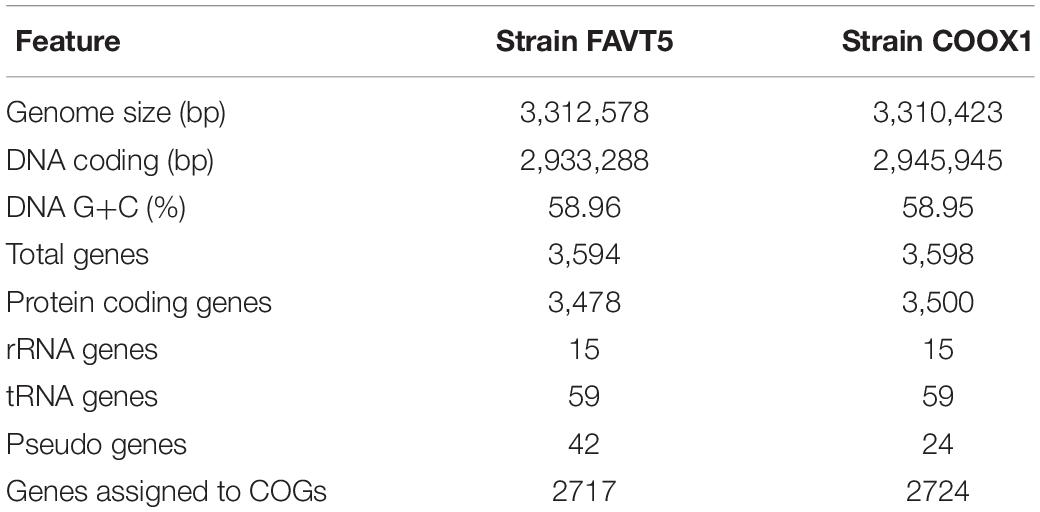
Table 1. Genomic features of K. spormannii strains FAVT5 and COOX1.
The 16S rRNA gene sequences (5 copies in each isolate) showed a high identity with each other (98.7–100%) (Supplementary Table S1 and Supplementary Figure S1), indicating that both strains belong to the same species. Some studies claim that 16S rRNA gene identity is not suitable for phylogeny and that better results can be obtained using genome-based phylogeny (Mahato et al., 2017). Therefore, an up-to-date bacterial core gene (UBCG) phylogenetic tree, based on 92 concatenated core gene alignments, was constructed. Our two isolates clustered within the genus Kyrpidia (Figure 1). The genus Kyrpidia, with thus far two cultured representatives, is the second genus in the parent family Alicyclobacillaceae (Klenk et al., 2011). Kyrpidia species, including our isolates, are Gram-stain-positive, aerobic, endospore-forming, non-motile rods (0.7–1.2 × 4–8 μm) (Reiner et al., 2018). To distinguish between species level, the average nucleotide identity (ANI) was calculated (Goris et al., 2007). These two isolates showed an ANI of 98.9% with each other and 97.4 and 97.5% with Kyrpidia spormannii EA-1 (Supplementary Table S2). ANI uses a species boundary of 95–96% identity (Chun et al., 2018), indicating that our two isolates are two novel strains of the species Kyrpidia spormannii, which we named K. spormannii strain FAVT5 and K. spormannii strain COOX1.
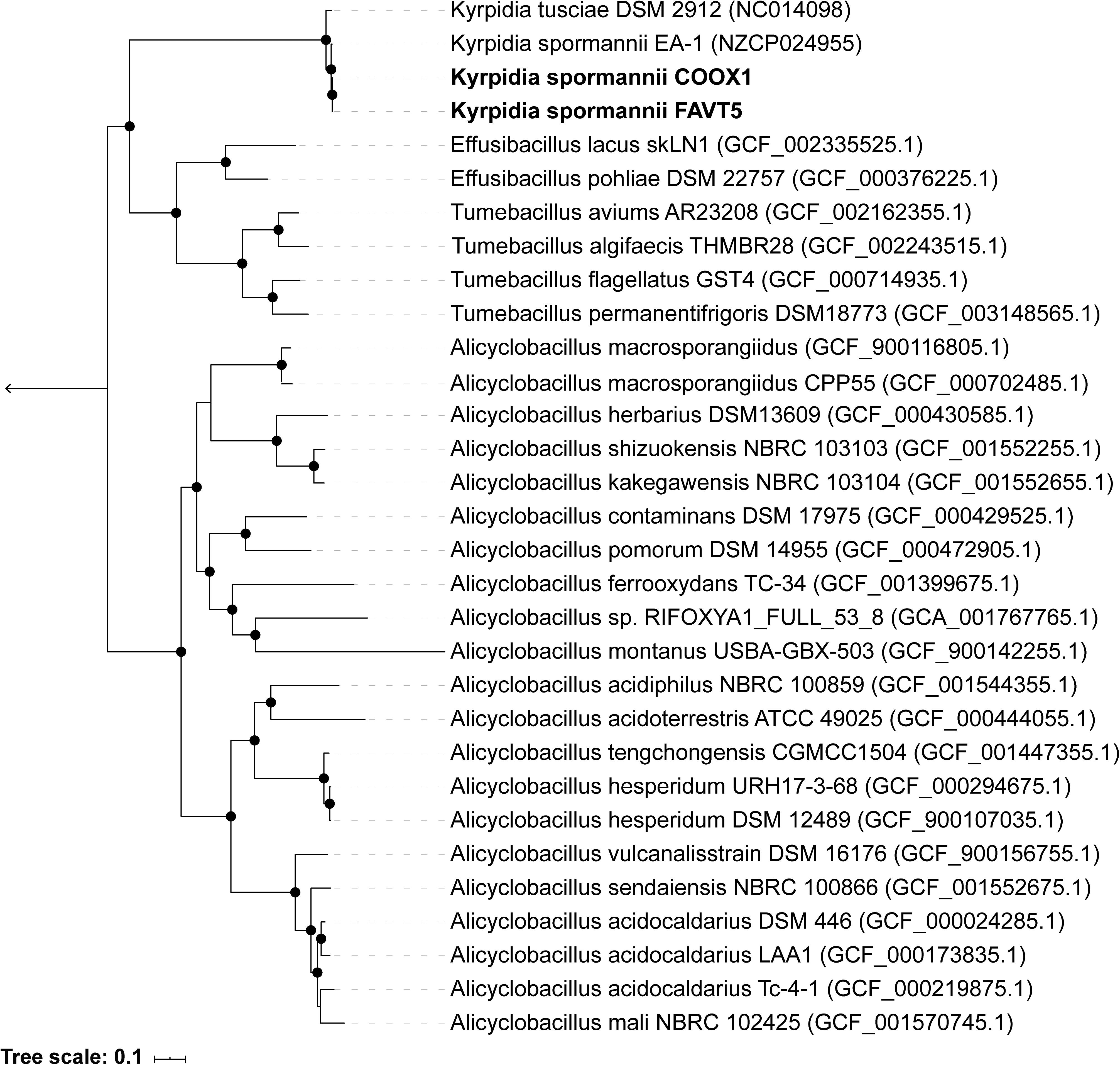
Figure 1. Up-to-date Bacterial Core Gene (UBCG) phylogenetic tree of the two isolates and members of the family Alicyclobacillaceae. Hyphomicrobium denitrificans was used to root the tree, but removed from the tree for clarity. The tree was constructed using RAxML. Bootstrap analysis was carried using 100 replications and percentage bootstrap values > 95% are indicated by a black dot at the nodes.
So far, two other Kyrpidia species have been isolated from geothermal areas. K. spormannii EA-1 was isolated from geothermal soils on the Azores, São Miguel, Portugal (Reiner et al., 2018) and K. tusciae was isolated from a geothermal pond in Tuscany, Italy (Bonjour and Aragno, 1984). 16S rRNA gene sequencing of environmental samples showed that members of the genus Kyrpidia were also found in sugarcane bagasse feedstock piles which are slightly acidic with a temperature of 49–52°C (Rattanachomsri et al., 2011). Other sequence matches were with biofilms of microbial fuel cells operated at 55°C and inoculated with biomass from a methanogenic anaerobic digester (Wrighton et al., 2008). Gagliano et al. (2016) performed 16S rRNA amplicon sequencing of the geothermal soils of our sampling site and this revealed the presence of three amplicon sequences related to K. spormannii within these soils (Supplementary Figure S1).
Autotrophic Growth on H2
Since the two strains are phylogenetically very closely related and both grew on H2 and CO as sole source of energy, all physiological studies were performed with K. spormannii FAVT5. The optimal pH and temperature were determined during batch experiments with H2 as electron donor and CO2 as carbon source. The highest growth rate was achieved at pH 5, but growth occurred as low as pH 3. The strain has an optimal growth temperature of 55°C at which a maximal growth rate of 0.19 ± 0.02 h–1 was calculated (doubling time of 3.6 h) (Supplementary Figure S2). Spores were formed during the stationary phase (Supplementary Figure S3). To determine the stoichiometry of K. spormannii FAVT5, a H2-limited chemostat (D = 0.045 h–1) was started. Based on dry weight, 4.3 g DW was produced per mole H2 consumed and the following stoichiometry was obtained:
The yield of biomass on H2 is slightly lower compared to other “Knallgas” bacteria, such as Ralstonia eutropha (4.6 g DW/mol H2) (Morinaga et al., 1978) or Hydrogenomonas eutropha (5 g DW/mol H2) (Bongers, 1970), but higher than reported for Methylacidiphilum fumariolicum SolV (3.4 g DW/mol H2) (Mohammadi et al., 2016).
Growth on Other Substrates
The K. spormannii FAVT5 repertoire of alternative electron donors besides H2 was tested. Compared to H2 the growth rate on CO was reduced to a doubling time of 15.7 h (μmax = 0.04 ± 0.01 h–1). Furthermore, K. spormannii FAVT5 grew on the alcohols ethanol, propanol and butanol, but not on methanol (Table 2). In addition, the volatile fatty acids acetate, propionate and butyrate supported growth, but not formate. Growth on yeast extract and succinate was observed (Table 2). No growth was observed on the alkanes, methane, ethane, propane or butane. No growth occurred on oxaloacetate, citric acid, α-ketoglutarate, pyruvate, fumaric acid or malic acid. No growth was observed on the following sugars: glucose, galactose, fructose, maltose, ribose and lactose. This is in contrast to K. spormannii EA-1, since this strain could utilize pyruvate and sugars (Reiner et al., 2018). However, K. tusciae did not show growth on sugars and pyruvate either (Bonjour and Aragno, 1984). As for K. tusciae, the highest growth rate was obtained during growth on H2.

Table 2. Growth rates of K. spormannii strain FAVT5 on different electron donors, different nitrogen sources and different amino acids.
K. spormannii FAVT5 can use the following amino acids as energy and nitrogen source: alanine, valine, isoleucine, and phenylalanine. As nitrogen source, ammonium, urea and nitrogen gas can be used, but no growth was observed with nitrate or nitrite. Nitrogen fixation was tested under low O2 concentration (max. 5%, v/v). There is a difference in utilization of nitrogen sources between the different Kyrpidia species. K. spormannii EA-1 does not grow using urea or N2 as nitrogen source. The genome of this strain does not contain a urease gene, but the nif-genes could be identified (Reiner et al., 2018). The genome of K. tusciae contains an urease and growth on urea has been reported, but no N2 fixation has been observed (Bonjour and Aragno, 1984; Klenk et al., 2011).
Kinetics of H2 Consumption
K. spormannii FAVT5 was isolated from a volcanic soil, and soil microorganisms can have an extremely high affinity for H2 and oxidize atmospheric H2 (Novelli et al., 1999). Therefore, we tested the affinity for H2 of K. spormannii FAVT5. The kinetics were measured using Membrane-Inlet Mass Spectrometry (MIMS). For these experiments, the cells were grown in batch and during exponential phase transferred to the MIMS chamber. After addition of H2 to the chamber, H2 consumption started immediately and the maximal H2 oxidation rate was achieved within 1 min (Figure 2). The H2 depletion followed Michaelis-Menten kinetics and, resulted in a Vmax of 60 ± 3 nmol H2/min/mg DW and the strain has a high affinity for H2, since a Ks of 327 ± 24 nM was found. These high affinities are also observed for different Actinobacteria, Acidobacteria, Chloroflexi and Verrucomicrobia (Constant et al., 2010; Greening et al., 2014; Myers and King, 2016; Mohammadi et al., 2016; Islam et al., 2019), however such a high affinity for H2 was not yet reported for any strain belonging to the phylum of Firmicutes.
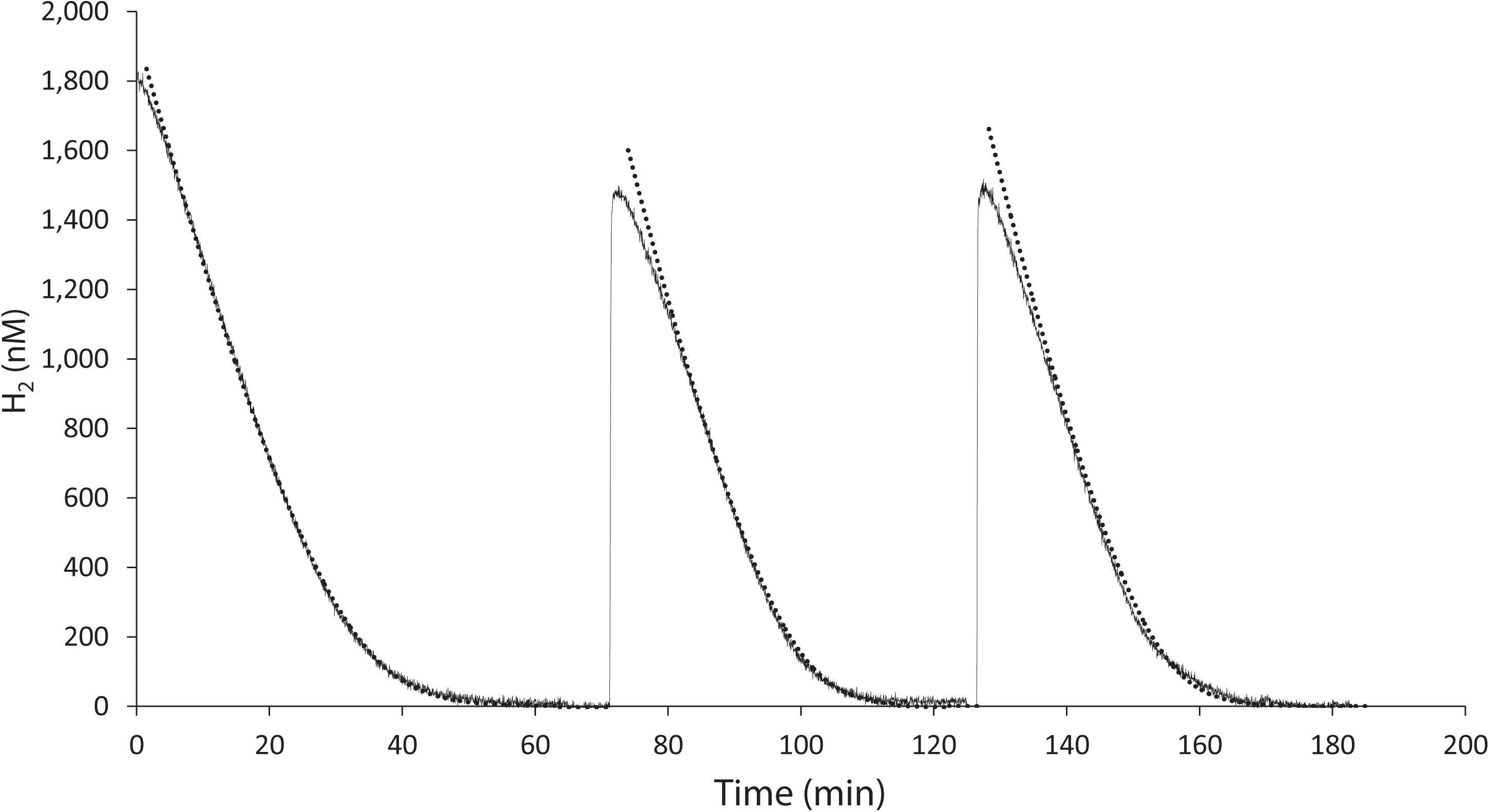
Figure 2. H2 uptake by K. spormannii strain FAVT in the MIMS-system (black line) and calculated hydrogen concentration based on Michaelis-Menten kinetics (dashed line).
Hydrogenases in the Genomes
High affinity hydrogenases belong to either the group 1h or group 2a [NiFe]-hydrogenases (Constant et al., 2010; Greening et al., 2014; Myers and King, 2016; Mohammadi et al., 2016; Islam et al., 2019). The genomes of K. spormannii FAVT5 and K. spormannii COOX1 were checked for the presence of all known hydrogenase genes. Both strains possess two hydrogenases, all belonging to the oxygen tolerant group 2a [NiFe] hydrogenases (hydDB classification) (Greening et al., 2015). Group 2a hydrogenases are typically found in Actinobacteria (Greening et al., 2015) or Cyanobacteria to recycle H2 derived from nitrogen fixation (Tamagnini et al., 2007). Group 2a [NiFe] hydrogenases are classified as H2-uptake hydrogenases and some have a high affinity for H2. For example, the group 2a hydrogenase of the Actinobacteria Mycobacterium smegmatis mc2 has a Km(app) of 180 nM for H2 (Greening et al., 2014), showing a bit higher affinity for H2 compared to K. spormannii FAVT5.
The hydrogenase large subunits (HucL) of all Kyrpidia sp. contain the L1 and L2 motifs typical for group 2a [NiFe] hydrogenases. The two cysteine residues, that bind the metal ions, are well conserved. However in all Kyrpidia sp. the phenylalanine in the L2 motif is replaced by a tyrosine (Figure 3). Changes in neighboring residues are assumed to affect the catalytic behavior of the hydrogenases (Greening et al., 2015).
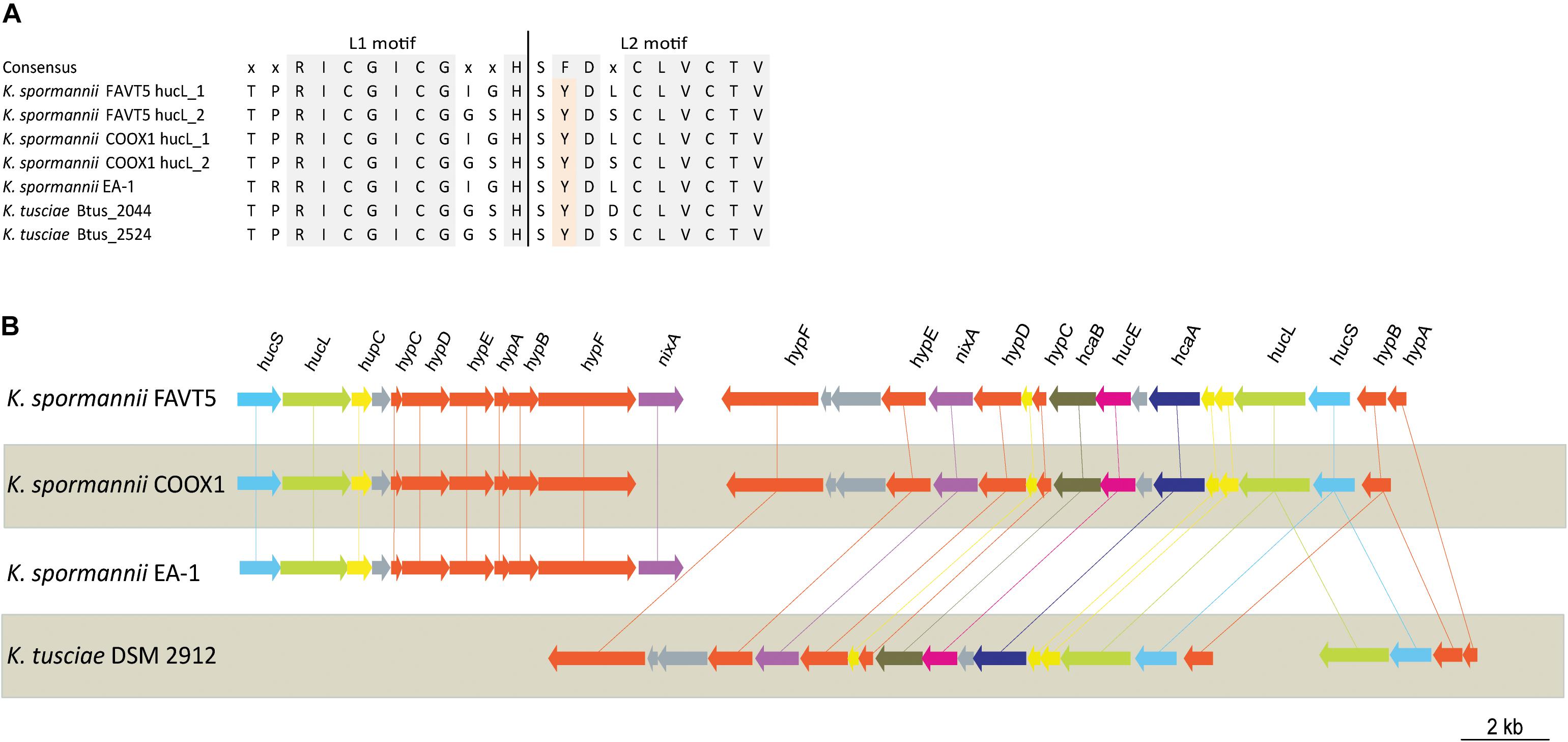
Figure 3. (A) Multiple sequence alignment of L1 and L2 motif of the [NiFe] hydrogenase large subunit of the different Kyrpidia species and strains. (B) Gene arrangement of the hydrogenases in different Kyrpidia isolates. Encoded proteins are colored as follows: green = large subunit, blue = small subunits, yellow = (putative) maturation proteins, orange = accessory proteins, purple = nickel transporter, pink = FeS cluster protein, dark purple = TTP repeat domain protein and dark green = NHL repeat domain protein.
Phylogenetic analysis of the large subunits of our strains reveals that one of the goup 2a [NiFe] hydrogenases clustered together with the [NiFe] hydrogenase of K. tusciae, whereas the other hydrogenases clustered outside the group 2a [NiFe] hydrogenase phylogenetic tree, together with the hydrogenase of K. spormannnii strain EA-1 (Figure 4). Based on amino acids, these latter hydrogenases showed little identity (55%) with the other [NiFe] hydrogenases belonging to class 2a. Also, the identity of the small subunit of this hydrogenase to other group 2a types was low (41%).
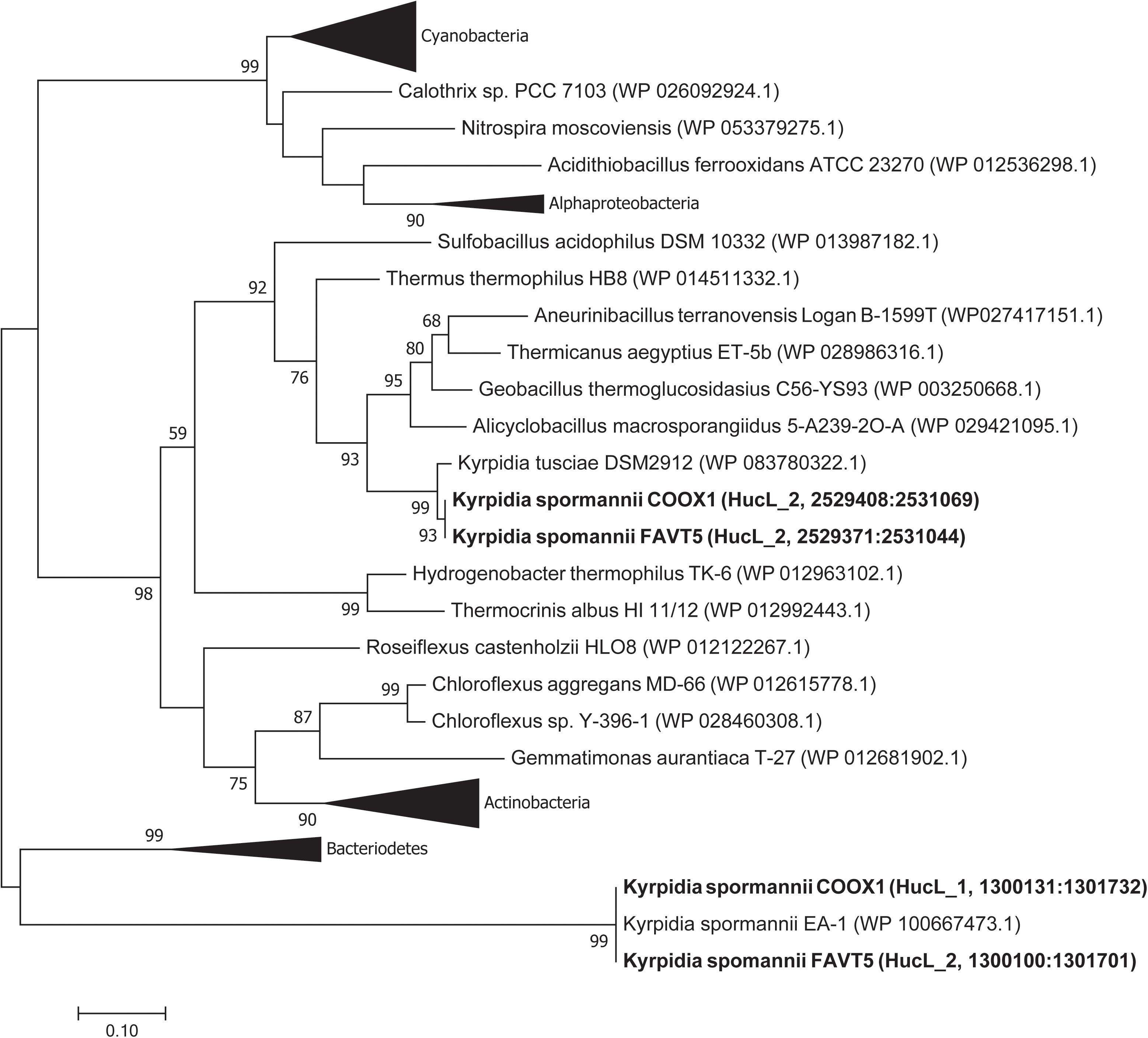
Figure 4. Phylogenetic tree of the group 2a [NiFe] hydrogenases. The tree was constructed using the Maximum Likelihood method (Saitou and Nei, 1987). Bootstrap percentage values above 50% (1000 replicates) are given at each node. Evolutionary distances were calculated using the Poisson correction method (Zuckerkandl and Pauling, 1965). The analysis involved 74 amino acid sequences and was performed using MEGA7 (Kumar et al., 2016).
The two group 2a [NiFe] hydrogenase gene clusters are differently arranged. Figure 3 shows the hydrogenase gene arrangement of the K. spormannnii strains FAVT5, COOX1, EA-1 and K. tusciae, whereby the first gene cluster encodes for the distinct Kyrpidia group 2a [NiFe] hydrogenase and the second operon encodes a more conventional group 2a [NiFe] hydrogenase. The hypABCDEF genes coding for accessory proteins were found in both operons, although they are differently arranged. The gene encoding for HypD was only found in the first operon. This protein is needed to cleave off the C-terminus from the large subunit to ensure that the small subunit can bind. However, it is expected that KFAV_v1_2744 fulfills this job in the hydrogenases encoded by the second operon, since a Pfam motif search annotated the encoded protein as HycI maturation protein. In K. spormannii COOX1, the high affinity nickel transporter was only found in the conventional hydrogenase operon, whereas in K. spormannii FAVT5, both operons contain a high affinity nickel transporter. The FeS cluster was only found in the second/conventional operon. Hydrogenase type 2a operons often contain genes with a TTP repeat domain (hcaA) and a NHL repeat domain (hcaB) (Greening et al., 2015). The functions of these domains remain unclear, and these genes are only found in the second, conventional hydrogenase operon.
CO Dehydrogenases in the Genome
K. spormannii COOX1 was isolated from a CO enrichment culture. Although originally isolated on H2, K. spormannii FAVT5 also grows on CO as sole energy source. A maximal growth rate of 0.04 ± 0.01 h–1 (Table 2) was observed, which is much lower compared to growth on H2. The genomes of both K. spormannii FAVT5 and K. spormannii COOX1 were analyzed for CO dehydrogenase genes. Both genomes encode for one candidate CO dehydrogenase gene cluster. The large subunits of all Kyrpidia strains contain the active site with the amino acid motive AYRGAGR, which groups them as a Form II CO dehydrogenases (Figure 5). BlastP searches with the Oligotropha carboxidovorans Type I CoxL (AEI08106) did not give a hit in the high quality closed genomes of strains FAVT5 and COOX1 and the very closely related K. tusciae. The K. spormannii EA-1genome encodes for both types of CoxL. The corresponding gene order in our strains, coxSLM, is characteristic for Form II CO dehydrogenases (King and Weber, 2007). This gene cluster is complemented with coxG (Figure 5), which encodes the pleckstrin homology (PH) domain protein. This domain binds to lipids, resulting in the hypothesis that CoxG is involved in anchoring the CO dehydrogenase to the cytoplasmic membrane to improve electron transfer to the respiratory chain (Pelzmann et al., 2014).

Figure 5. (A) Multiple sequence alignment of the active site of Form II CO dehydrogenase of the different Kyrpidia species. (B) Gene arrangement of the CO dehydrogenases in K. spormannnii FAVT5 and K. spormannii COOX1. Genes are color coded as follows: green = large subunit, blue = small subunit, yellow = medium subunit, orange = accessory protein.
Phylogenetic analysis of the CoxL proteins of K. spormannii FAVT5 and K. spormannii COOX1 shows that these proteins cluster with Form II CO dehydrogenases (Figure 6). Most biochemical evidence for Form II CODH is obtained from members of the BMS (Burkholderia, Mesorhizobium, Sinorhizobium) clade, a group of Form II CODH found in α-Proteobacteria (Nunoura et al., 2005; Wu et al., 2017). It remains puzzling if the Form II enzymes are real CO dehydrogenases since the large subunit of type II-CODH does not possess the canonical motif bridging the active site of CODH (a cysteine residue is essential to accommodate the copper and molybdenum atoms of the active site). Recently, the CODH hydrogenase activity was observed for a putative Form II CODH from the thermophilic Archaeon Aeropyrum pernix TB5, which clusters outside the BMS clade (Nishimura et al., 2010). However, the enzyme was purified but no direct coupling of the purified protein with its encoding gene could be made. These strains oxidizes CO at heterotrophic and aerobic conditions and at high CO concentrations (>25%, v/v), however, growth on CO as sole electron source is impossible by this Archaeon, since it lacks a CO2 fixation pathway. This CO dehydrogenase could be purified and shows highest activity at 95°C and is oxygen stable (Nishimura et al., 2010). The CODH of the different K. spormannii isolates cluster outside the BMS clade as well and are phylogenetically closer to the archaeal CODH (Figure 6).
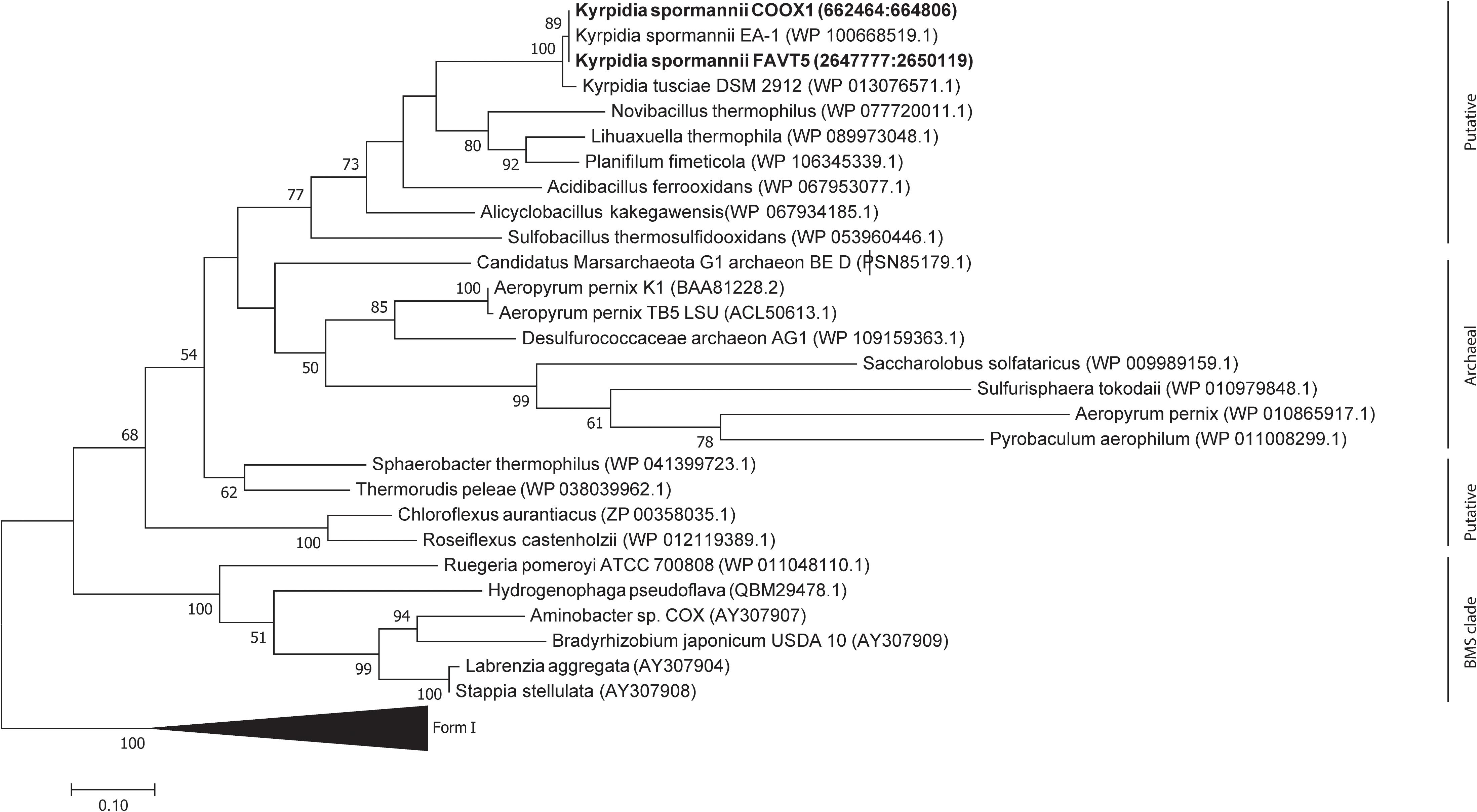
Figure 6. Phylogenetic tree of the CO dehydrogenase large subunit, form I and II. The tree was constructed using the Maximum Likelihood method. Bootstrap percentage values above 50% (1000 replicates) are given at each node. Evolutionary distances were calculated using the Poisson correction method (Zuckerkandl and Pauling, 1965) and the analysis was performed in MEGA7 (Kumar et al., 2016).
Transcriptomics
The genomes of both K. spormannii FAVT5 and K. spormannii COOX1 contain 2 group 2a [NiFe] hydrogenase gene clusters, one of which is phylogenetically distinct from other group 2a hydrogenases (Figure 4). To test whether both hydrogenases are being expressed, transcriptome analysis (RNAseq) was performed on strain FAVT5 cells from the H2-limited chemostat culture. The results of the transcriptome analysis were calculated as log2fold change of the RPKM-value compared to the median expression level (Mundinger et al., 2019). In this way, the genes that show expression values above the median get positive values and genes with RPKM values below the median will give a negative log2fold change. The results of the transcriptome studies revealed that K. spormannii FAVT5 expressed its two hydrogenases under H2-limiting conditions and their expression values largely exceeded the median RPKM values (Figure 7). The expression values of the large and small hydrogenase subunits were amongst the highest expressed genes (Supplementary Table S4). The expression values (RPKM) were between 25 and 52 times higher than the median values. The other genes in the hydrogenase gene clusters showed high expression values too (Supplementary Table S4). The reason for having two hydrogenases remains unclear, but it is likely that the enzymes function under different conditions (Sanchez-Perez et al., 2008) as in Methylacidiphilum fumariolicum SolV. In fact, this methanotroph can grow as “Knallgas”-bacterium and possesses two different hydrogenases, an oxygen-sensitive (group 1d) and an oxygen-insensitive hydrogenase (group 1h), giving it the possibility to oxidize H2 at higher oxygen concentrations (Mohammadi et al., 2016). Detailed transcriptomic studies combined with biochemical studies are needed to elucidate under which conditions the two different hydrogenases in the novel Kyrpidia isolates function.
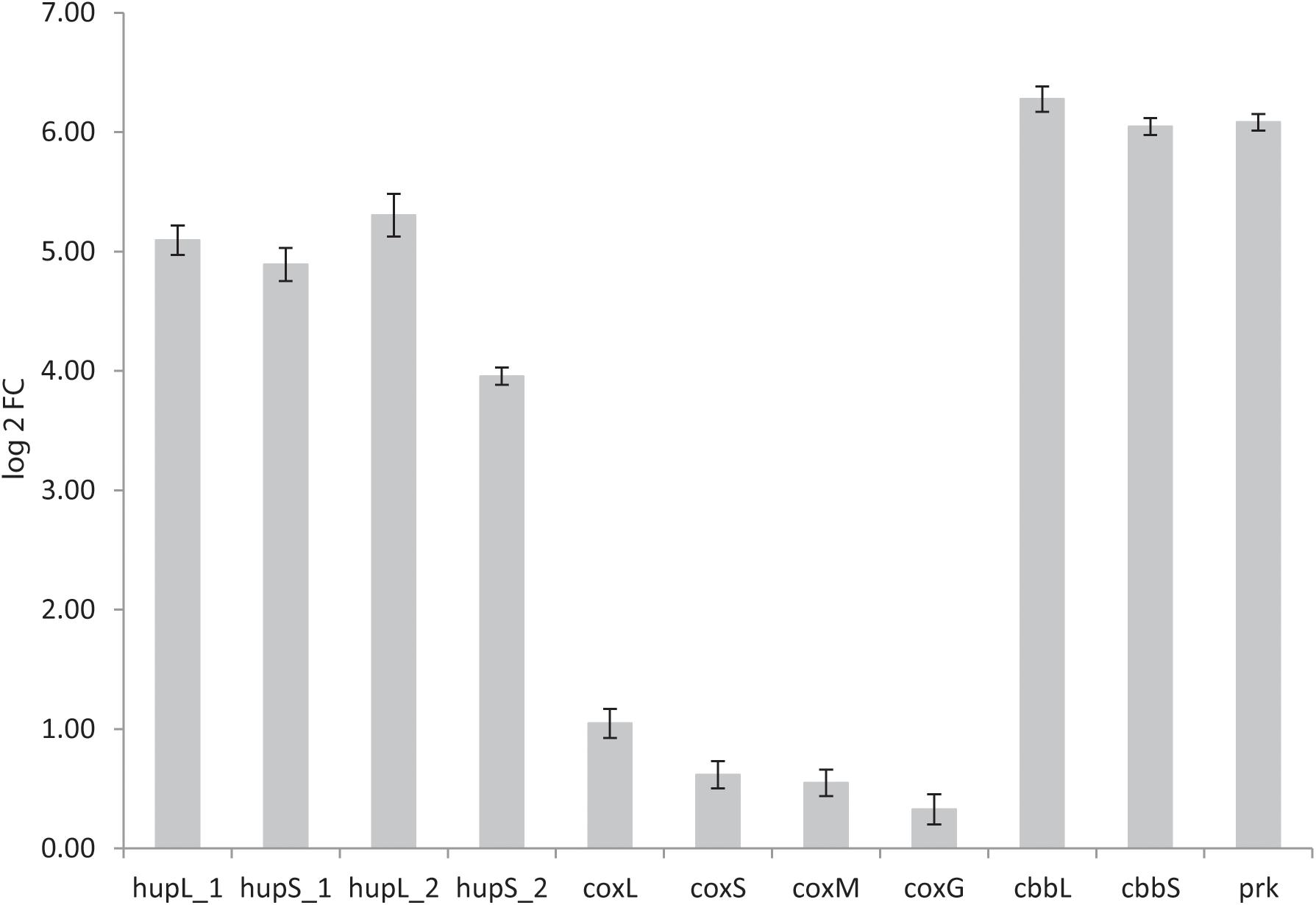
Figure 7. Log 2fold change of reads per kilobase per million reads (RPKM) values over the median RPKM. The values are the average of three biological replicates (chemostat samples taken on three different days). hupL_1, hydrogenase large subunit 1 (FAVT5_v1_1432); hupS_1, hydrogenase small subunit 1 (FAVT5_v1_1431); hupL_2, hydrogenase large subunit 2 (FAVT5_v1_2745); hupS_2, hydrogenase small subunit 2 (FAVT5_v1_2746); coxL, CO dehydrogenase large subunit; coxS, CO dehydrogenase small subunit; coxM, CO dehydrogenase medium subunit; coxG, pleckstrin homology (PH) domain protein; cbbL, ribulose bisphosphate carboxylase large chain; cbbS, ribulose bisphosphate carboxylase small chain; prk, phosphoribulokinase.
The bioreactor was fed with CO2 as carbon source. The transcriptomics data also show high expression levels of the genes encoding the small (cbbS) and large (cbbL) subunits of the RuBisCO enzyme and the phosphoribulokinase gene (prk), key enzymes of the Calvin–Benson–Bassham (CBB) cycle.
Although the bioreactor was not fed with CO, the CO dehydrogenase gene cluster showed expression values higher than the mean. It is possible that K. spormannii FAVT5 transcribed the CO dehydrogenases constitutively or that the expression increases under nutrient limitation, as was observed with the Chloroflexi Thermomicrobium roseum. This strain upregulates the expression of its CO dehydrogenase under nutrient starvation to oxidize atmospheric CO for microbial persistence (Islam et al., 2019).
Genome Data Supporting Alternative Substrate Utilization
The genome of K. spormannii strain FAVT5 contains a urease gene and the nif-genes for growth on urea and performing nitrogen fixation. The genomic analysis revealed multiple alcohol dehydrogenases and two acetaldehyde dehydrogenases (Supplementary Table S3) for the heterotrophic growth on alcohols. Growth on sugars was not observed, despite all genes for the glycolysis pathway being present. Since the K. spormannii FAVT5 genome does not encode any sugar transport proteins, it is more likely that the gluconeogenesis and glycolysis will be used for the production and consumption of storage material. During H2-limited growth, the genes for the glycolysis pathway were expressed, and especially the genes for the fructose 1,6-bisphosphatase class II, fructose 1,6-bisphosphate aldolase and glyceraldehyde-3-phosphate dehydrogenase showed high expression levels (Supplementary Table S4). Furthermore, all genes for the TCA cycle, the glyoxylate cycle and the non-oxidative part of pentose phosphate pathway were found in the genome (Supplementary Table S3). All of these pathways were expressed under H2-limited growth and high expression levels were observed for ribose 5-phosphate isomerase and transketolase genes (Supplementary Table S4). Despite any organic substrates being present, these pathways were expressed for the synthesis of different biomolecules, such as nucleotides, amino acids and lipids.
Conclusion
This study shows that two different strains of the species Kyrpidia spormannii were isolated from a geothermal site using CO and H2 as energy source. The two isolates were genetically very similar and strain FAVT5, isolated on H2 as sole energy source, was also able to grow on CO. Therefore, all physiology experiment were performed with K. spormannii FAVT5. This strain grows on H2, CO and a variety of organic substrates with an optimum at 55 °C and pH 5.0. The affinity for H2 is high, namely 327 ± 24 nM, the first high H2 affinity reported for Firmicutes so far. The genome encodes for two group 2a [NiFe] hydrogenases of which one is distantly related to other group 2a hydrogenases. Both encoded hydrogenases are expressed under H2-limiting conditions in a chemostat. The genome encodes for a candidate CO dehydrogenase gene cluster and these genes are transcribed during H2 limited growth. The presence of Kyrpidia spormannii strains at Favara Grande and their capability to grow on CO are highly important.
Data Availability Statement
The genomes of strains FAVT5 and COOX1 are available under accession numbers GCA_902829265 and GCA_902829275. The transcriptome sequencing data are available under Project number PRJNA616194.
Author Contributions
CH, AP, MJ, and HO designed the projects and experiments. CH, AP, NP, AG, WD’A, PQ, and HO sampled the geothermal soils. CH and NP performed the enrichment and isolation experiments. CH and AP conducted the experiments. GC and TA sequenced the genome and transcriptome and analyzed the reconstructed the genome. CH, AP, and HO carried out the data analysis. CH and HO wrote the manuscript. All authors contributed to revision of the manuscript, and read and approved the submitted version.
Funding
CH, NP, and HO were supported by the European Research Council (ERC Advanced Grant project VOLCANO 669371). MJ was supported by the European Research Council (ERC Advanced Grant project Eco_MoM 339880).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00951/full#supplementary-material
Footnotes
References
Bongers, L. (1970). Yields of Hydrogenomonas eutropha from growth on succinate and fumarate. J. Bacteriol. 102, 598–599.
Bonjour, F., and Aragno, M. (1984). Bacillus tusciae, a new species of thermoacidophilic, facultatively chemolithoautotrophic hydrogen oxidizing sporeformer from a geothermal area. Arch. Microbiol. 139, 397–401. doi: 10.1007/BF00408386
Brady, A., Sharp, C., Grasby, S., and Dunfield, P. (2015). Anaerobic carboxydotrophic bacteria in geothermal springs identified using stable isotope probing. Front. Microbiol. 6:897. doi: 10.3389/fmicb.2015.00897
Chiodini, G., Frondini, F., Cardellini, C., Granieri, D., Marini, L., and Ventura, G. (2001). CO2 degassing and energy release at Solfatara volcano, Campi Flegrei, Italy. J. Geophys. Res. Solid Earth 106, 16213–16221. doi: 10.1029/2001JB000246
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Colman, D. R., Lindsay, M. R., and Boyd, E. S. (2019a). Mixing of meteoric and geothermal fluids supports hyperdiverse chemosynthetic hydrothermal communities. Nat. Commun. 10:681. doi: 10.1038/s41467-019-08499-1
Colman, D. R., Lindsay, M. R., Maximiliano, J. A., and Boyd, E. S. (2019b). The intersection of geology, geochemistry, and microbiology in continental hydrothermal systems. Astrobiology 19, 1505–1522. doi: 10.1089/ast.2018.2016
Constant, P., Chowdhury, S. P., Pratscher, J., and Conrad, R. (2010). Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high-affinity [NiFe]-hydrogenase. Environ. Microbiol. 12, 821–829. doi: 10.1111/j.1462-2920.2009.02130.x
Cunliffe, M. (2011). Correlating carbon monoxide oxidation with cox genes in the abundant marine Roseobacter clade. ISME J. 5, 685–691. doi: 10.1038/ismej.2010.170
D’Alessandro, W., Bellomo, S., Brusca, L., Fiebig, J., Longo, M., Martelli, M., et al. (2009). Hydrothermal methane fluxes from the soil at Pantelleria Island (Italy). J. Volcanol. Geotherm. Res. 187, 147–157. doi: 10.1016/j.jvolgeores.2009.08.018
Ettwig, K. F., Shima, S., van der Pas−Schoonen, K. T., Kahnt, J., Medema, M. H., Op den Camp, H. J. M., et al. (2008). Denitrifying bacteria anaerobically oxidize methane in the absence of archaea. Environ. Microbiol. 10, 3164–3173. doi: 10.1111/j.1462-2920.2008.01724.x
Gagliano, A. L., D’Alessandro, W., Tagliavia, M., Parello, F., and Quatrini, P. (2014). Methanotrophic activity and diversity of methanotrophs in volcanic geothermal soils at Pantelleria (Italy). Biogeosciences 11, 5865–5875. doi: 10.5194/bg-11-5865-2014
Gagliano, A. L., Tagliavia, M., D’Alessandro, W., Franzetti, A., Parello, F., and Quatrini, P. (2016). So close, so different: Geothermal flux shapes divergent soil microbial communities at neighbouring sites. Geobiology 14, 150–162. doi: 10.1111/gbi.12167
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Greening, C., Berney, M., Hards, K., Cook, G. M., and Conrad, R. (2014). A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. PNAS 111, 4257–4261. doi: 10.1073/pnas.1320586111
Greening, C., Biswas, A., Carere, C. R., Jackson, C. J., Taylor, M. C., Stott, M. B., et al. (2015). Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 10, 761–777. doi: 10.1038/ismej.2015.153
Ikeda, E., Omae, K., Kano, S. I., Kimura-Sakai, S., Daifuku, T., Yoshida, T., et al. (2015). Detection of anaerobic carbon monoxide-oxidizing thermophiles in hydrothermal environments. FEMS Microbiol. Ecol. 91, 1–9. doi: 10.1093/femsec/fiv093
Islam, Z. F., Cordero, P. R. F., Feng, J., Chen, Y.-J., Bay, S. K., Jirapanjawat, T., et al. (2019). Two chloroflexi classes independently evolved the ability to persist on atmospheric hydrogen and carbon monoxide. ISME J. 13, 1801–1813. doi: 10.1038/s41396-019-0393-0
King, G. M., and Weber, C. F. (2007). Distribution, diversity and ecology of aerobic co-oxidizing bacteria. Nat. Rev. Microbiol. 5, 107–118. doi: 10.1038/nrmicro1595
Klenk, H. P., Lapidus, A., Chertkov, O., Copeland, A., Del Rio, T. G., Nolan, J. A., et al. (2011). Complete genome sequence of the thermophilic, hydrogen-oxidizing Bacillus Tusciae type strain (T2) and reclassification in the new genus, Kyrpidia gen. nov. as Kyrpidia tusciae comb. nov. and emendation of the family Alicyclobacillaceae da Costa and Rainey, 2010. Stand. Genomic. Sci. 5, 121–134. doi: 10.4056/sigs.2144922
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., and Tamura, K. (2016). Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lindsay, M. R., Colman, D. R., Amenabar, M. J., Fristad, K. E., Fecteau, K. M., Debes, I. R. V., et al. (2019). Probing the geological source and biological fate of hydrogen in Yellowstone hot springs. Environ. Microbiol. 21, 3816–3830. doi: 10.1111/1462-2920.14730
Lubitz, W., Ogata, H., Rüdiger, O., and Reijerse, E. (2014). Hydrogenases. Chem. Rev. 114, 4081–4148. doi: 10.1021/cr4005814
Mahato, N. K., Gupta, V., Singh, P., Kumari, R., Verma, H., Tripathi, C., et al. (2017). Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques. Antonie Van Leeuwenhoek 110, 1357–1371. doi: 10.1007/s10482-017-0928-1
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2012). “The cipres science gateway: enabling high-impact science for phylogenetics researchers with limited resources,” in Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond, (Chicago, IL: ACM).
Mohammadi, S., Pol, A., van Alen, T. A., Jetten, M. S. M., and Op den Camp, H. J. M. (2016). Methylacidiphilum fumariolicum solv, a thermoacidophilic ‘knallgas’ methanotroph with both an oxygen-sensitive and -insensitive hydrogenase. ISME J. 11, 945–958. doi: 10.1038/ismej.2016.171
Morinaga, Y., Yamanaka, S., Ishizaki, A., and Hirose, Y. (1978). Growth characteristics and cell composition of alcaligenes eutrophus in chemostat culture. Agric. Biol. Chem. 42, 439–444. doi: 10.1080/00021369.1978.10862993
Mundinger, A. B., Lawson, C. E., Jetten, M. S. M., Koch, H., and Lücker, S. (2019). Cultivation and transcriptional analysis of a canonical Nitrospira under stable growth conditions. Front. Microbiol. 10:1935. doi: 10.3389/fmicb.2019.01325
Myers, M. R., and King, G. M. (2016). Isolation and characterization of Acidobacterium ailaaui sp. nov., a novel member of Acidobacteria subdivision 1, from a geothermally heated hawaiian microbial mat. Int. J. Syst. Evol. Microbiol. 66, 5328–5335. doi: 10.1099/ijsem.0.001516
Na, S.-I., Kim, Y. O., Yoon, S.-H., Ha, S.-M., Baek, I., and Chun, J. (2018). UbCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Nishimura, H., Nomura, Y., Iwata, E., Sato, N., and Sako, Y. (2010). Purification and characterization of carbon monoxide dehydrogenase from the aerobic hyperthermophilic archaeon Aeropyrum pernix. Fish. Sci. 76, 999–1006. doi: 10.1007/s12562-010-0277-8
Novelli, P. C., Lang, P. M., Masarie, K. A., Hurst, D. F., Myers, R., and Elkins, J. W. (1999). Molecular hydrogen in the troposphere: global distribution and budget. J. Geophys Res. Atmos. 104, 30427–30444. doi: 10.1029/1999JD900788
Nunoura, T., Hirayama, H., Takami, H., Oida, H., Nishi, S., Shimamura, S., et al. (2005). Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ. Microbiol. 7, 1967–1984. doi: 10.1111/j.1462-2920.2005.00881.x
Oppenheimer, C., Fischer, T. P., and Scaillet, B. (2014). “Volcanic degassing: Process and impact,” in Treatise on Geochemistry, The Crust, 2nd Edn, Vol. 4, eds H. D. Holland and K. K. Turekian (Amsterdam: Elsevier), 111–179. doi: 10.1016/B978-0-08-095975-7.00304-1
Pelzmann, A. M., Mickoleit, F., and Meyer, O. (2014). Insights into the posttranslational assembly of the Mo-, S- and Cu-containing cluster in the active site of CO dehydrogenase of Oligotropha carboxidovorans. J. Biol. Inorg. Chem. 19, 1399–1414. doi: 10.1007/s00775-014-1201-y
Pol, A., Heijmans, K., Harhangi, H. R., Tedesco, D., Jetten, M. S. M., and Op den Camp, H. J. M. (2007). Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature 450, 874–878. doi: 10.1038/nature06222
Quatrini, R., and Johnson, D. B. (2018). Microbiomes in extremely acidic environments: functionalities and interactions that allow survival and growth of prokaryotes at low pH. Curr. Opin. Microbiol. 43, 139–147. doi: 10.1016/j.mib.2018.01.011
Rattanachomsri, U., Kanokratana, P., Eurwilaichitr, L., Igarashi, Y., and Champreda, V. (2011). Culture-independent phylogenetic analysis of the microbial community in industrial sugarcane bagasse feedstock piles. Biosci. Biotechnol. Biochem. 75, 232–239. doi: 10.1271/bbb.100429
Reiner, J. E., Jung, T., Lapp, C. J., Siedler, M., Bunk, B., Overmann, J., et al. (2018). Kyrpidia spormannii sp. nov., a thermophilic, hydrogen-oxidizing, facultative autotroph, isolated from hydrothermal systems at São Miguel Island, and emended description of the genus Kyrpidia. Int. J. Syst. Evol. Microbiol. 68, 3735–3740. doi: 10.1099/ijsem.0.003037
Robb, F., and Techtmann, S. (2018). Life on the fringe: Microbial adaptation to growth on carbon monoxide. F1000Res. 7:1981. doi: 10.12688/f1000research.16059.1
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sanchez-Perez, G., Mira, A., Nyirõ, G., Pašić, L., and Rodriguez-Valera, F. (2008). Adapting to environmental changes using specialized paralogs. Trends Genet. 24, 154–158. doi: 10.1016/j.tig.2008.01.002
Shock, E. L., Holland, M., Meyer-Dombard, D. A., Amend, J. P., Osburn, G. R., and Fischer, T. P. (2010). Quantifying inorganic sources of geochemical energy in hydrothermal ecosystems, Yellowstone National Park, USA. Geochim. Cosmochim. Acta 74, 4005–4043. doi: 10.1016/j.gca.2009.08.036
Smeulders, M. J., Barends, T. R. M., Pol, A., Scherer, A., Zandvoort, M. H., Udvarhelyi, A., et al. (2011). Evolution of a new enzyme for carbon disulphide conversion by an acidothermophilic archaeon. Nature 478, 412–416. doi: 10.1038/nature10464
Smeulders, M. J., Pol, A., Zandvoort, M. H., Jetten, M. S. M., and Op den Camp, H. J. M. (2013). Diversity and ecophysiology of new isolates of extremely acidophilic CS2-converting Acidithiobacillus strains. Appl. Environ. Microbiol. 79, 6784–6794. doi: 10.1128/AEM.02167-13
Spear, J. R., Walker, J. J., McCollom, T. M., and Pace, N. R. (2005). Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. PNAS 102, 2555–2560. doi: 10.1073/pnas.0409574102
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Strazzulli, A., Fusco, S., Cobucci-Ponzano, B., Moracci, M., and Contursi, P. (2017). Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev. Environ. Sci. Biotechnol. 16, 425–454. doi: 10.1007/s11157-017-9435-0
Tamagnini, P., Leitão, E., Oliveira, P., Ferreira, D., Pinto, F., Harris, D. J., et al. (2007). Cyanobacterial hydrogenases: diversity, regulation and applications. FEMS Microbiol. Rev. 31, 692–720. doi: 10.1111/j.1574-6976.2007.00085.x
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013). Microscope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647. doi: 10.1093/nar/gks1194
van Teeseling, M. C., Pol, A., Harhangi, H. R., van der Zwart, S., Jetten, M. S., Op den Camp, H. J. M., et al. (2014). Expanding the verrucomicrobial methanotrophic world: description of three novel species of Methylacidimicrobium gen. nov. Appl. Environ. Microbiol. 80, 6782–6791. doi: 10.1128/AEM.01838-14
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9:e112963. doi: 10.1371/journal.pone.0112963
Wrighton, K. C., Agbo, P., Warnecke, F., Weber, K. A., Brodie, E. L., DeSantis, T. Z., et al. (2008). A novel ecological role of the Firmicutes identified in thermophilic microbial fuel cells. ISME J. 2, 1146–1156. doi: 10.1038/ismej.2008.48
Wu, X., Ge, T., Hu, Y., Wei, X., Chen, L., Whiteley, A. S., et al. (2017). Abundance and diversity of carbon monoxide dehydrogenase genes from bms clade bacteria in different vegetated soils. Eur. J. Soil Biol. 81, 94–99. doi: 10.1016/j.ejsobi.2017.06.012
Keywords: Kyrpidia spormannii, H2, [NiFe]-hydrogenases, CO, thermoacidophilic, phylogeny
Citation: Hogendoorn C, Pol A, Picone N, Cremers G, van Alen TA, Gagliano AL, Jetten MSM, D’Alessandro W, Quatrini P and Op den Camp HJM (2020) Hydrogen and Carbon Monoxide-Utilizing Kyrpidia spormannii Species From Pantelleria Island, Italy. Front. Microbiol. 11:951. doi: 10.3389/fmicb.2020.00951
Received: 21 January 2020; Accepted: 21 April 2020;
Published: 19 May 2020.
Edited by:
Axel Schippers, Federal Institute for Geosciences and Natural Resources, GermanyReviewed by:
Philippe Constant, Institut National de la Recherche Scientifique (INRS), CanadaMarianne Quéméneur, UMR7294 Institut Méditerranéen d’Océanographie (MIO), France
Copyright © 2020 Hogendoorn, Pol, Picone, Cremers, van Alen, Gagliano, Jetten, D’Alessandro, Quatrini and Op den Camp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huub J. M. Op den Camp, aC5vcGRlbmNhbXBAc2NpZW5jZS5ydS5ubA==