 Qian Li
Qian Li Jun Wang
Jun Wang Mengji Lu
Mengji Lu Yuanwang Qiu
Yuanwang Qiu Hongzhou Lu
Hongzhou Lu- 1Department of Infectious Diseases, Shanghai Public Health Clinical Center, Shanghai, China
- 2Center of Clinical Laboratory, The Fifth People’s Hospital of Wuxi, Jiangnan University, Wuxi, China
- 3Institute of Virology, University Hospital of Essen, University of Duisburg-Essen, Essen, Germany
- 4Department of Hepatology, The Fifth People’s Hospital of Wuxi, Jiangnan University, Wuxi, China
Acute-on-chronic liver failure (ACLF) is an acute syndrome accompanied with decompensation of cirrhosis, organ failure with high 28-day mortality rate. Systemic inflammation is the main feature of ACLF, and poor outcome is closely related with exacerbated systemic inflammatory responses. It is well known that severe systemic inflammation is an important event in chronic hepatitis B (CHB)-ACLF, which eventually leads to liver injury. However, the initial CHB-ACLF events are unclear; moreover, the effect of these events on host immunity as well as that of immune imbalance on CHB-ACLF progression are unknown. Here, we investigate the initial events of ACLF progression, discuss possible mechanisms underlying ACLF progression, and provide a new model for ACLF prediction and treatment. We review the characteristics of ACLF, and consider its plausible immune predictors and alternative treatment strategies.
Introduction
Acute-on-chronic liver failure (ACLF) is defined as the exacerbation of chronic liver disease initiated by a precipitating event, typically resulting in elevated short-term mortality (Alam et al., 2017). Liver complications or decompensation and rapid disease progression lead to high organ failure risk (45%) and high short-term mortality (90%) (Alam et al., 2017). Acute liver failure (ALF) usually occurs following a precipitating event in the context of chronic liver diseases. In Western countries, alcoholism induced cirrhosis is the main reason of ALF (50–70%). However, in Asia countries, hepatitis B virus (HBV) infection is the main cause of ALF (70%) (Sarin et al., 2009). And HBV-related ALF occurs after acute HBV infection or during chronic HBV infection (Oketani et al., 2014; Manka et al., 2016). The primary cause of ALF is an acute exacerbation of chronic hepatitis B (CHB) infection, which accounts for more than 80% of all cases in China (You et al., 2013). ALF caused by CHB is known as chronic hepatitis B-related ACLF (CHB-ACLF).
CHB-ACLF presents with a poor prognosis because antiviral therapy cannot improve short-term survival, but it can control HBV DNA levels. Liver transplantation (LT) is thought to be an effective treatment approach, but it is not always feasible owing to the lack of liver donors and high cost (Cui et al., 2010; Finkenstedt et al., 2013). Therefore, there is a need for early indicators that can predict CHB progression and acute exacerbation to ACLF. The most effective approach to reduce morbidity and mortality may be to elucidate CHB-ACLF progression and to initiate a suitable therapy at an early stage.
In naturally resolved HBV-infected individuals, synergistic aspects of the immune response can function to efficiently repress residual virus. However, the virus typically persists in cases of CHB due to impaired immunity (Hui et al., 2005; Tan et al., 2015). Immune dysregulation is thought to be involved in the progression of CHB to CHB-ACLF, which involves systemic inflammation and exacerbation of the innate immune system resulting in a cytokine storm (Wu et al., 2018). However, relatively less is known about how impaired host immunity triggers the initiation of CHB-ACLF pathogenesis.
In this review, we emphasize on the mechanisms underlying immune pathogenesis in CHB-ACLF, including new insights into virus-induced immune dysfunction and systemic inflammation interaction with immune exhaustion during final liver damage and multiple organ failure. Finally, we will explore strategies for detecting early indicators of CHB-ACLF and immunotherapy as a treatment approach.
What Is the Original Trigger Controlling the Switch From Chronicity to Acute Exacerbation?
CHB is distinguished by 4 stages in clinical—immune tolerant (IT), immune clearance/active (IA), inactive carrier (IC), and Hepatitis-B-Virus e-Antigen (HBeAg)-negative hepatitis (ENH) (EASL Jury, 2003; Fattovich et al., 2008). Acute severe exacerbation occurs either in the IA phase (40–50%) or in the ENH phase (15–30%) (Figure 1; Sheen et al., 1985; Lok et al., 1987). Notably, CHB-ACLF is a dynamic process ranging from the chronically active phases (IA and ENH) to the acute exacerbation phase that culminates in liver failure. Precipitating factors reported in ACLF include sepsis, alcoholism, and relapse of chronic viral hepatitis, but 40–50% of CHB-ACLF cases have no identifiable triggers (Hernaez et al., 2017).
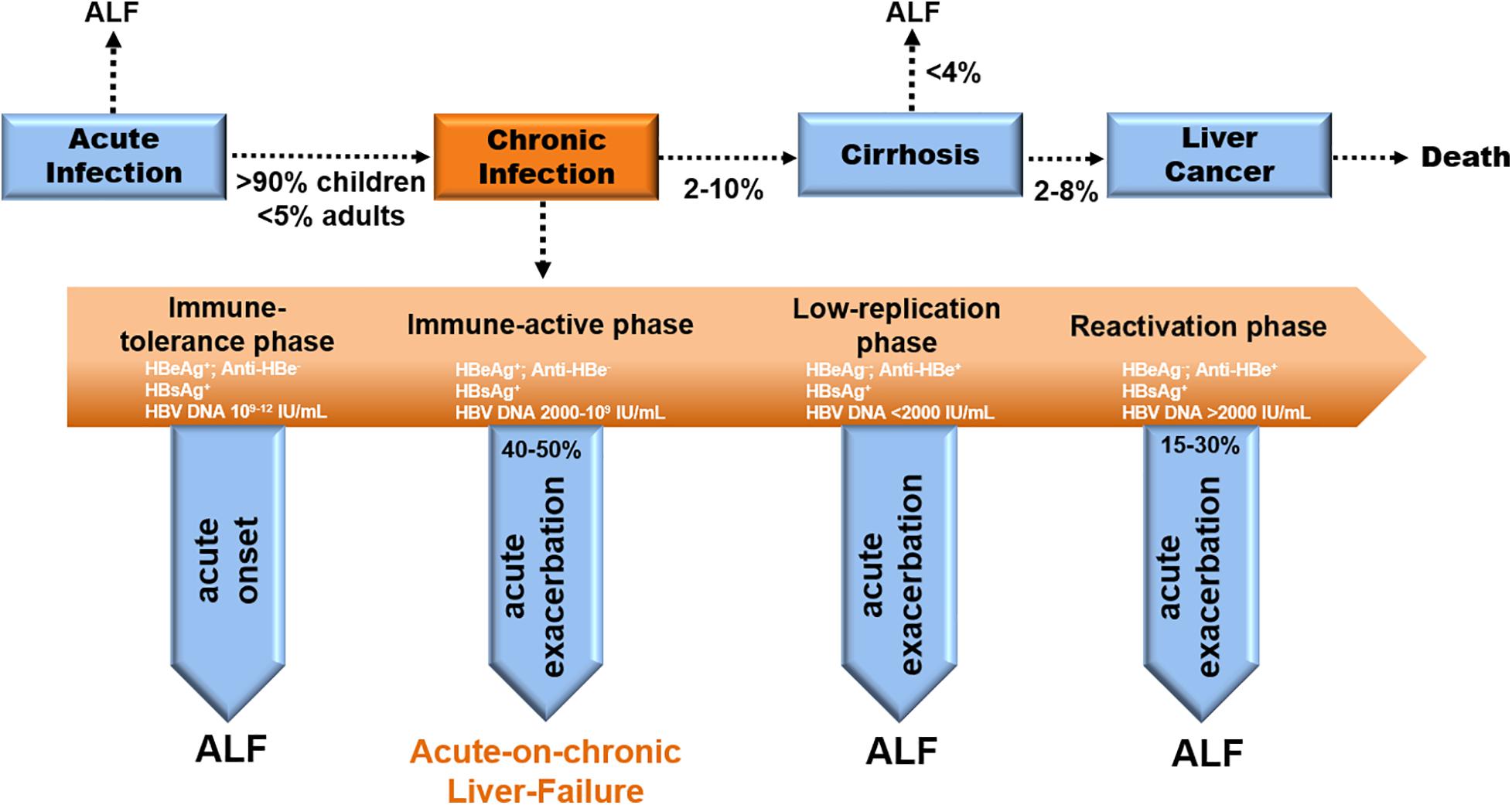
Figure 1. Natural history and disease progression in CHB patients. Evolution to CHB occurs in > 90% of children and < 5% of adults with acute HBV infection. Progression to cirrhosis from CHB appears to be 2–10% per year. The incidence of ALF from cirrhosis is less than 4% yearly. The rate of cirrhosis to liver cancer is 2–8% annually (EASL Jury, 2003). ACLF occurs in the IA phase (40–50%) during chronic infection (Sheen et al., 1985; Lok et al., 1987).
Is Innate Immune Recognition the Original Trigger?
Early pathogen detection generally occurs via the recognition of Pathogen-Associated Molecular Patterns (PAMPs) by Pathogen Recognition Receptors (PRRs). The known PRRs include nucleotide-binding oligomerization domain-containing protein (NOD)-like, retinoic acid-inducible gene I (RIG)-like, toll-like receptor (TLR), DNA-sensing receptors and C-type Lectin (Akira et al., 2006; Kawai and Akira, 2009; Takeuchi and Akira, 2010; Pandey et al., 2014). These immunity sensors are distributed across various liver cell types and intrahepatic liver endothelial cells (Ding et al., 2016; Faure-Dupuy et al., 2018). After PAMP recognition by their corresponding PRRs, downstream signaling pathways are sequentially activated involving the expression of various adaptor molecules [e.g., myeloid differentiation primary response gene 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF)], kinase activation or phosphorylation [e.g., TANK binding kinase 1 (TBK1)/transforming growth factor-β activated kinase 1 (TAK1)]. Thereafter, transcription factors are expressed [e.g., nuclear factor kappa-B (NF-κB), interferon regulatory transcription factors (IRFs), c-fos/c-jun, etc.]. This process ultimately leads to effector gene expression including interferon-stimulated-genes (ISGs), NF-κB-inducible genes, and pro-inflammatory genes (IL-2, IL-12, IL-16, etc.) (Kumar et al., 2009, 2011; Kawai and Akira, 2011; Pandey et al., 2014).
The role of TLRs in CHB is debatable. Due to technical limitations, the entirety of the dynamic innate immune response elicited in vivo is unknown. It was initially believed that HBV activates TLR2 signaling in vitro (Cooper et al., 2005). However, it has been shown that synthesized HBV capsid/core antigen (HBcAg) could be polluted with lipopolysaccharide (LPS)-like ligands in bacteria (Vanlandschoot et al., 2007). In contrast, HBV components are now widely believed to block TLR signaling at various levels. It has been suggested that HBeAg can bind to the co-adaptor of Myd88 and TIRAP, to interfere with TLR2 signaling (Lang et al., 2011). And HBsAg could inhibit TLR2 pathway activation by binding the c-Jun N-terminal protein kinase (JNK) (Wang et al., 2013). Sufficient evidence has demonstrated that HBV is not recognized by TLRs during entry, suggesting that HBV is a “stealth virus” in the early phase of infection (Wieland et al., 2004).
The role of TLRs in CHB-ACLF is also debatable. TLR4 mRNA levels have been reported to be up-regulated in the peripheral blood mononuclear cells (PBMCs) of ACLF patients compared to those in healthy individuals and CHB patients. TLR4 expression in both CD4 + and CD8 + T cells from PBMCs was significantly increased and positively correlated with liver injury severity in ACLF patients, indicating that TLR4 plays a vital role in disease progression (Xu et al., 2015). Xu et al. (2017) found a significant increase in TLR2/4/6/8 expression in the PBMCs of the early stage ACLF patients compared to that in CHB patients. Since TLR2 forms the heterodimers TLR1 and TLR6 to recognize bacterial LPS, it was assumed that TLR up-regulation in the PBMCs of ACLF patients represented the secondary recognition of bacteria from the circulation or gut (Xu et al., 2017). However, Wang et al. (2010) reported that the mRNA levels of TLR3/5/7/9/10 were down-regulated, and the protein level of TLR7 was confirmed to be decreased in CHB-ACLF patients compared to that in CHB patients in the IA phase. Since CHB-ACLF is a dynamic disease, patient data originating from different phases of the disease lead to different TLR recognition. These results suggest that TLR expression varies among different CHB-ACLF stages. However, there is no definite evidence regarding whether TLR recognition triggers the progression from CHB to acute CHB-ACLF exacerbation.
Evidence suggests that strong HBV replication may be sensed by the innate immune system (Durantel and Zoulim, 2009). For example, ex vivo experiments have proven that HBV replication in HepaRG cell lines activates some ISGs (Lucifora et al., 2010). RIG1 and MDA5 are the primary DNA sensors that are also involved in inducing low chronic inflammation (Safari-Arababadi et al., 2019). Additionally, in vivo experiments demonstrated some activation of IFN-α stimulated genes in HBV-infected hepatocytes in chimeric mice (Lutgehetmann et al., 2011). The mechanism by which HBV is sensed in infected cells is likely mediated by the growing family of PRRs capable of discriminating intracellular pathogen DNA from the host’s DNA (Sharma et al., 2015). Interestingly, it has been reported that HBV does not interfere with the innate immune response in the human liver because HBV patient liver specimens fail to induce ISG expression following TLR3 treatment (Suslov et al., 2018). However, subsequent studies have refuted this hypothesis because the model of a pre-existing long-standing infection is not ideal (Vyas et al., 2018). It is generally accepted that the weak IFN response serves as a strategy to enable HBV to escape innate immune recognition (Wieland and Chisari, 2005).
The IFN response is also suppressed in the exacerbation phase of CHB-ACLF as indicated by in vitro experiments demonstrating that SOCS3, a well-known IFN signaling suppressor, was significantly elevated in the liver and PBMCs of a mouse model of CHB-ACLF (Li et al., 2014). This result supports the hypothesis that SOCS3 is involved in immune homeostasis in pathological conditions by negatively regulating cytokine or hormone signaling (Carow and Rottenberg, 2014). However, the newly identified interferon-gamma induced protein (IFI16) is significantly more highly expressed in ACLF patients than in CHB patients and healthy individuals (Kerur et al., 2011; Connolly and Bowie, 2014). In contrast to the weak IFN response observed in CHB patients, the high IFN response in the ACLF patient livers indicates that selective IFI16 recruitment in the liver may initiate a destructive immune response leading to liver injury (Pang et al., 2018). Interestingly, this study was performed over a late time course of liver damage, which is clinically diagnosed as high-grade liver failure (> 2).
The hypothesis that TLR recognition initiates the transition from CHB to ACLF has been refuted due to the lack of a defined turning point from chronicity to the acute exacerbation phase of CHB-ACLF and the limited observations regarding the relationship between TLR recognition, the IFN response, and the prognosis of CHB-ACLF patients.
Serum Viral Load of CHB-ACLF Patients Does Not Differ From That of CHB Patients
In CHB patients, the IT phase is characterized by high HBV DNA levels but minimal liver damage as detected by histological analysis (Andreani et al., 2007). From the data of other patient cohorts, it can be observed that the serum HBV DNA load in ACLF patients is lower than that in CHB patients (Table 1; Gao et al., 2017; Lei et al., 2019). In addition, other serum parameters such as serum HBsAg level are lower in CHB-ACLF patients than in CHB patients. Furthermore, the serum HBeAg level is lower in CHB-ACLF patients than in HBeAg-positive CHB patients (Table 1; Zhang Y.M. et al., 2014).

Table 1. Serum virus load in ACLF and different stage of CHB.
It is reasonable to assume that innate immune recognition like via TLRs cannot induce CHB progression to CHB-ACLF. It is essential to explore and uncover the potential triggers to achieve early ACLF indicators because the serological characteristics of CHB are similar to those of CHB-ACLF.
Potential Triggers of CHB-ACLF
The potential triggers of CHB-ACLF were widely researched. Bacterial infections and alcoholism were found to be the two major identifiable factors in the CANONIC study, while hepatitis B relapse was found to be the predominant factor followed by bacterial infections in studies conducted in China (Moreau et al., 2013; Shi et al., 2015). The CANONIC criteria primarily identified hepatic-ACLF triggers categorized as liver toxins (alcohol, hepatitis) and extrahepatic-ACLF triggers (e.g., infections). Despite exhaustive examination, the trigger for CHB-ACLF remains unknown in 20–45% of cases, particularly for cases in China (Baumert et al., 1996). We speculate that the potential triggers among these CHB-ACLF patients are related to HBV characteristics such as HBV genotype and mutations (Table 2).

Table 2. Potential triggers of HBV-ACLF.
HBV Genotype
HBV is classified into 8 genotypes (A–H) by viral characteristics, geographical distribution, and clinical outcomes. CHB patients with genotype B with basal core promoter/pre-core (BCP/PC) mutations were reported more susceptible to develop ALF compared to patients with genotype C with wild-type BCP/PC regions (Baumert et al., 1996; Tong et al., 2005; Kay and Zoulim, 2007). Another report also indicated that patients infected with HBV/Bj are more frequently to develop ALF than patients with HBV/Ae (Ozasa et al., 2006).
HBV Viral Mutations
Mutations in the core promoter and pre-core (PC) regions have been found to enhance HBV replication in vitro, and the PC mutation inhibits HBeAg translation, which may enable the escape from immune attacks on the infected hepatocytes. Higher G1896A PC mutation and BCP double mutation (A1762T/G1764A) occurred in ALF patients than in patients with acute HBV infection (Kosaka et al., 1991; Liang et al., 1991; Sato et al., 1995; Inoue et al., 1998; Friedt et al., 1999; Ozasa et al., 2006). Additionally, single mutations in the BCP/PC region, including C1766T, T1753V (C/A/G), G1862T, T1768A, and G1899A have been shown to be related to elevated HBV replication in some cases with ALF (Liang et al., 1991; Hou et al., 2002; Parekh et al., 2003; Wai et al., 2005). Xu et al. (2011) found CHB patients with BCP/PC mutant are more likely to have ACLF than patients without such a mutation. Strong evidence demonstrated that the A1846T and C1913A mutations are positively associated with the presentation of severe liver diseases (Zang et al., 2018). However, the traditional technique utilized for HBV virus sequencing is PCR amplification, which targets a limited and select number of clones. Additionally, there is limited evidence regarding how HBV mutations in the BCP and PC induce the immune response. HBeAg, a secreted soluble protein, can suppress seroconversion to anti-HBcAg antibody production in some mouse strains, and it could serve as a regulator (Chen et al., 2004). HBeAg is typically present along with HBcAg in HBV infections, but BCP mutations in HBeAg can cause a loss-of-function phenotype (called a basal-core mutation). HBeAg is not synthesized in these conditions, and the lack of its immunomodulatory functions is thought to induce an uncontrolled and enforced immune response to HBcAg that is associated with ALF in some cases (Fagan et al., 1986; Liang et al., 1991).
Unlike the indirect immune dysregulation caused by HBcAg, amino acid mutations in HBsAg, large HBxAg, and HBcAg have been found to induce immune dysfunction. In large HBsAg, I161 and N177 exhibit higher variation frequencies in ACLF patients than in acute hepatitis B patients of genotype C. Meanwhile, in HBxAg, V131, K130, and C143 have exhibited the highest frequency of variation in ACLF patients compared to that in CHB patients (40% of clones for 2/5 patients) with genotype C (Yang et al., 2015). Zhang et al. also found that A131, T114, E113, and P135 in HBcAg had more variation frequency in ACLF patients with both genotype B/C. Additionally, numerous deletion mutations in different clones from base 1997 to 2279 resulted in frameshift mutations in ACLF patients, which introduced a premature stop codon and produced a truncated HBcAg (Yang et al., 2015). However, the sequence read length (<500 bp) obtained here using next-generation sequencing does not cover the complete HBV genome length. Additionally, patients enrolled for research studies are limited. Since HBV genome quasispecies of all individuals were not longitudinally analyzed, the selective pressure of the immune response caused by mutant viral antibodies is unclear (Yang et al., 2015).
Therefore, there is a need for studies on the immune pressure of mutant viral Ag, especially for HBcAg. Mutations or deletions at L60–I97 of HBcAg have been widely studied as T- or B-cell epitopes in ACLF patients, much more than that in IT and acute hepatitis B patients (Alexopoulou et al., 2009; Chen et al., 2010). Other studies have shown accumulated mutations within HBc141-149 epitope in HBV-infected individuals. HBc141-149 is an HLA-A2-restricted epitope can be naturally processed in HBV-infected individuals, and evoke antiviral T cell response in HLA-A2.1/HBV transgenic mice (Sun et al., 2014). Chen et al. compared genetic and functional difference of the virus and host immunity in the liver tissue from chimpanzees of HBV-associated ALF to those with acute hepatitis B. In contrast to acute hepatitis B, HBV strains displayed highly mutated HBcAg, lacked corresponding somatic mutations in heavy chain variable region (VH) genes, and exhibited low binding affinity to HBcAb, ultimately leading to increased HBcAg expression ex vivo independent of viral replication levels in the liver of ALF (Chen et al., 2018).
Are T-Cell Independent B Cell Response, the Driver of ACLF?
Since innate immune recognition lacks specificity and the virological parameters fail to change in ACLF patients as mentioned above, the TLR pathway was suppressed in these patients. In this circumstance, it is reasonable to exclude the belief that TLR recognition may trigger immune bias (Akira et al., 2006). Only B cells but not dendritic cells (DC)/phagocytosis (Mφ) were found to effectively present HBcAg and prime naive T cells (Lee et al., 2009). Farci et al. (2010) further utilized phage display technology to uncover a massive intrahepatic antibody response that was essentially directed exclusively against HBcAg in ALF patients, and the inherently enhanced immunogenicity of HBcAg was demonstrated to be linked to its function as a T cell-independent antigen. Moreover, Chen et al. analyzed the miRNA expression profiles in ALF patients. Their data revealed a distinctive B cell signature with enriched intrahepatic IgM and IgG production exclusively targeting to HBcAg with high affinity. The limited expression of T cell-associated genes, the major negative regulators in ALF patients were found (Chen et al., 2018). Naive-like B cells produced immature IgM through somatic mutation deficiency that has lower cross-reactivation of the original viral Ag. The exaggerated IgM response exclusively leads to binding of the germline antigen to further encounter immune complexes on the surface of infected hepatocytes, resulting in the activation and deposition of C1q, C3, and C4d led to massive liver necrosis (Chen et al., 2018).
Above all, these findings supported the hypothesis that CHB-ACLF appears to be driven by an unusual T cell-independent B cell response inherited from a highly mutated core antigen, leading to complement activation and massive liver necrosis.
Systematic Inflammation Following DAMP/PAMP Recognition
Cytokine Storm
Acting in combination, bacterial infection (PAMPs) and liver injury (DAMPs) trigger inflammation to restore tissue homeostasis (Kumar et al., 2011; Kubes and Mehal, 2012). Systemic inflammation that causes severe liver injury through an excessive immune response is a hallmark of ACLF. In contrast to the weak inflammatory response in CHB patients, ACLF patients predominantly secrete inflammatory cytokines or chemokines. Bao et al. (2017) reported that serum IL-6, IL-12, IL-17, IL-23, and TNF-α levels are higher in ACLF patients than in CHB patients. Additionally, ACLF patients have been shown to have high levels of pro-inflammatory molecules such as IL-1β and IL-8 (Claria et al., 2016). The new IL-6/IL-12 family member IL-27 was also higher in ACLF patients than in CHB patients (Zhang G.L. et al., 2014). Colony-stimulating factors that contribute to myelopoiesis, including G-CSF and GM-CSF, were also higher in ACLF patients than in CHB patients and healthy controls. Chemokines such as IP-10 were expressed at a higher level, while others such as RANTS, MCP-1, and MIG were expressed at a lower level in ACLF patients than in CHB patients (Wu et al., 2018). Xin et al. (2016) proposed that six cytokine levels, including hepatocyte growth factor, macrophage inflammatory protein 3α (MIP-3α), carcinoembryonic antigen-related cell adhesion molecule 1, growth differentiation factor 15, E-selectin, and osteopontin, were significantly elevated in CHB-ACLF patients compared to that in CHB patients. This was achieved by detecting cytokine levels in 279 patients with ACLF, 116 patients with CHB, and 20 normal adults. Intrahepatic pro-inflammatory IFN-γ and TNF-α expression was markedly up-regulated in ACLF patients compared to that in CHB and normal controls (Zou et al., 2009). Interestingly, the anti-inflammatory cytokine IL-10 was significantly increased in CHB-ACLF patients, as was the pro-inflammatory IL-6 compared to that in No-ACLF patients. This indicated increased reparative inflammation. Together, these results suggest an excessive host immune response mediated by increased expression of anti-bacterial immunity genes, indicating an increased microbial burden (Wu et al., 2018).
Immune Cell Dysfunction
Immune cells are thought to be dysregulated during the inflammatory response. In ACLF patients, circulating neutrophil and monocyte counts have been reported to be higher and lymphocyte counts to be lower than those of severe exacerbation of chronic hepatitis B (SE−CHB) patients (Wu et al., 2018). Along with the changes in cell subtype proportions, immune cell phenotypes and functions are also altered.
For example, IL-23 was significantly up-regulated in monocyte-derived dendritic cells (MoDCs) from ACLF patients compared to that in MoDCs from CHB patients in the IT phase (Kubes and Mehal, 2012). The up-regulation of TNF-α in ACLF is positively correlated with increased Kupffer Cells (KCs) (Su and Liu, 1991). The population of peripheral CD14 + CD33 + CD11b + HLA-DR–/low myeloid-derived suppressor cells (MDSCs) in ACLF patients was significantly increased compared to that in CHB patients and healthy controls (Yi et al., 2016; Zeng et al., 2019).
It is well known that natural killer (NK) cells play a crucial role in innate immunity. The percentage of CD3-CD56+ NK cells was found to be higher in ACLF patients than in CHB patients. The inhibitory molecule NKG2D was significantly less expressed in NKs in ACLF patients compared to that in CHB patients. In contrast, the activation marker NKG2A was significantly highly expressed in ACLF patients than in CHB and NC patients. These findings indicated an imbalance between NKG2A and NKG2D in NK cells contributing to ACLF progression (Yi et al., 2016). In addition, peripheral NK cells from CHB-ACLF patients expressed higher levels of TNF-related apoptosis-inducing ligand (TRAIL) than those from CHB patients did. TRAIL expression in NK cells was positively correlated with serum IL-6 and IL-8 concentrations in CHB-ACLF patients (Wan et al., 2016). Moreover, expression of other activating receptors such as NKG2D, NKp30, NKp44, and NKp46 was elevated in ACLF patients, but NK cytotoxicity was impaired in patients with ACLF and CHB due to an obvious decrease in cytotoxic CD56dimCD16bright NK cells (Liu et al., 2016).
CD8 + T cells were found to be required to control HBV infection through cytolytic and non-cytolytic effector functions (Thimme et al., 2003; Guidotti and Chisari, 2006; Guidotti et al., 2015). Despite no significant difference in the proportion of CD8+ T cells in total plasma lymphocytes between ACLF and CHB patients, IFN-γ over-expression and its significant positive correlation with intrahepatic CD8 + and CD4 + T cell accumulation was verified in ACLF patients (Zou et al., 2009; Yi et al., 2016). Increased circulating Th22 inversely correlates with HBV-ACLF prognosis. High Th17 levels in ACLF patients were positively correlated with IL-21 secretion (Mo et al., 2017; Zhang G.L. et al., 2014; Zhang et al., 2018). Th9 cells were unlikely to be involved in HBV pathogenesis, but elevated levels of IL-9 and IL-10 may signal poor ACLF prognosis (Yu et al., 2016).
B cells were also found to be dysfunctional during this period based on increased serum IgA, IgM, and IgG in ACLF patients compared to CHB patients and healthy controls. Furthermore, IgG levels were positively related to IL-27 levels in CHB-ACLF patients. Increased serum immunoglobulins were measured in CHB-ACLF patients (Zhang et al., 2019).
Immune Exhaustion Accompanied by Bacterial Co-Infection Following the Systemic Inflammation to Organ Failure
Immune Exhaustion
The host immune system is over-activated during systemic inflammation. However, it is subsequently exhausted at the late ACLF phase. This is strongly supported by the fact that up-regulated inhibitory molecule expression, down-regulated Ag presentation molecules, exhaustion phenotype, and damaged cell function have all been reported in ACLF patients.
The ratio of circulating CD3+ T cells to monocytes decreased in ACLF patients beginning at the early to the intermediate stage. This ratio reached the lowest level at the late stage in ACLF, likely due the drastic up-regulation of programmed death-1 receptor (PD-1) in both CD4+ T and CD8+ T cells, indicating T-cell exhaustion. These CD4+ T and CD8+ T cells in ACLF patients, along with regulatory T cells, inhibit TNF-α secretion by monocytes (Shi et al., 2010). Moreover, additional B7 super family members have been studied in ACLF patients, including PD-L1 (B7-H1), PD-L2 (B7-DC), B7-H3, and B7-H4. These are highly expressed in liver sections from CHB-ACLF patients (Guo et al., 2012; Cao et al., 2013). Other potential negative regulators such as BTLA are also highly expressed in ALCF patients (Xu et al., 2012). Zhang et al. found KCTD9 to be highly expressed in peripheral and hepatic NK cells from CHB-ACLF patients compared to mild CHB patients. KCTD9 is involved in inhibiting CD69 expression, cytotoxicity, IFN-γ secretion, and inducing a significant decrease in NKG2A receptor expression in vitro (Chen T. et al., 2013). Other inhibitory receptors, such as CD158a, on NK cells have also been shown to be increased in ACLF patients (Liu et al., 2016).
Contrary to negative regulators, HLA-DR expression gradually decreased in the monocytes of patients with CHB, liver cirrhosis, and ACLF, especially in the late stage of ACLF compared to that in healthy controls (Xing et al., 2007; Zhang et al., 2016).
Bacterial Co-infection
It is well known that bacterial infections lead to sepsis and increased mortality in ACLF (Moreau et al., 2013). Spontaneous bacteremia, spontaneous bacterial peritonitis, urinary tract infection, pneumonia, cellulitis, bacterial enteritis, and fungal infection are the most frequent among bacterial infections (Cai et al., 2019). If ACLF is truly a B cell-driven disease that leads to complement activation and massive liver necrosis, DAMPs released from the injured hepatocytes may explain the over-active immune system and the worsened liver injury (Kubes and Mehal, 2012). The liver injury, in turn, leads to the pathological translocation of bacteria or bacterial products, typically originating from the gut floor (Wiest et al., 2014).
Microcirculatory changes in the hepatic and splanchnic vasculature occur at the early stage of ACLF due to immune inflammation (Gronbaek et al., 2012; Laleman et al., 2005). With the exhaustion of the “immune army,” the immune system can no longer fight off infection. The exhausted immune system leads to the worsening of bacterial or fungal infections as endotoxin is released and accelerates disease progression from sepsis to multiple organ failure (Figure 2; Garcia-Tsao and Wiest, 2004; Wiest and Garcia-Tsao, 2005).
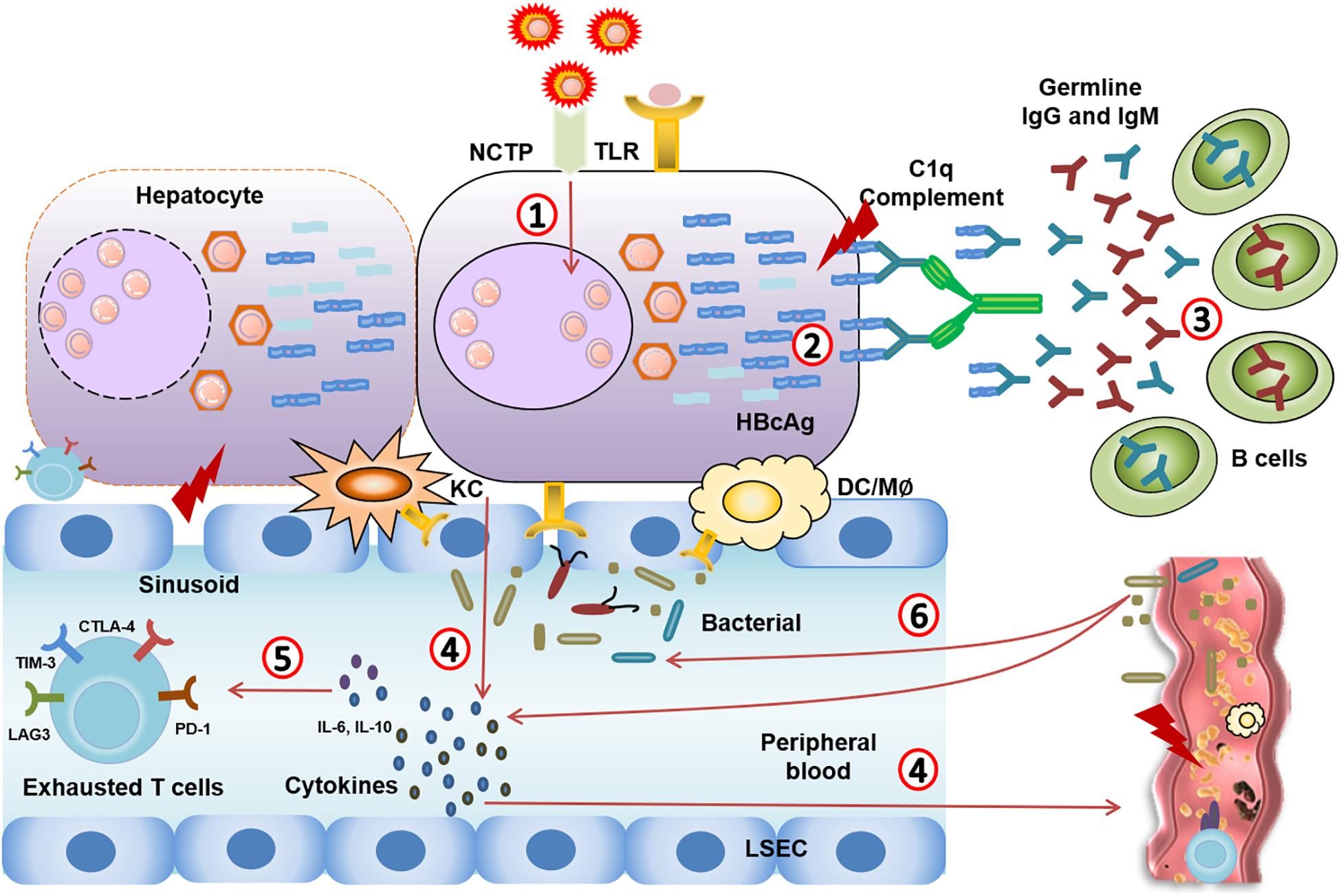
Figure 2. Immune events from CHB to CHB-ACLF. (1) HBV virus mutation leads to highly mutated Ags. (2,3) Highly mutated core antigens induce overwhelming T cell-independent B cell response, leading to complement activation and massive liver necrosis. (4) The damage of hepatocytes induces TLR recognition, immune cells activation, and excessive inflammatory cytokines release in circulating system. (5) Systemic inflammation (IL-10, IL-6…) that causes severe liver injury through an excessive immune response, and leads to immune cell dysfunction and immune exhaustion. (6) The exhausted immune system leads to the worsening of bacterial or fungal infections. Meanwhile, pathological translocation of bacteria or bacterial products aggregates the systemic inflammation.
Emerging Early Indicators of ACLF and Potential Therapy Targets
Early Indicators of ACLF
Even though several biomarkers such as microRNA (hsa-miR-21-5p), the inflammation-mediated protein (N-myc and ATAT interactor, NMI) may be useful novel biomarkers of ACLF (Ding et al., 2015; Xiong et al., 2019), more appropriate, accurate, and early indicators are still needed owing to the rapid progression of ACLF. It is well known that ALCF patients have higher complexity and diversity within the BCP/PC region of HBV, indicating distinct quasispecies characteristics among ALCF patients (Chen L. et al., 2013). ACLF patients infected with HBV show a high frequency of single mutations, including A1762T, T1753V (C/A/G), G1764A, G1896A, and G1899A; double mutations, including G1896A/G1899A and A1762T/G1764A; and triple mutations, including A1762T/C1766T/G1764A and A1762T/T1753V/G1764A (Xiao et al., 2011; Ma et al., 2012). Moreover, HBV mutations such as A1846T is also highly related to the increased risk of ACLF (Nian et al., 2016). These data suggest that BCP/PC mutations play a vital role in ACLF progression and may be useful as early indicators of CHB-ACLF. Beyond viral mutations, more complex factors such as genotypes should also be considered. CHB patients with BCP/PC mutations of genotype B have the high risk of ACLF than patients of genotype C without BCP/PC mutations. In patients of genotype B, A1846T, G1896A, and A1762T/G1764A mutations are more extensive in ACLF patients to CHB patients. Patients of Genotype B with G1896A and A1762T/G1764A are more likely to occur ACLF than patients of genotype C (Ren et al., 2010; Xiao et al., 2011). Thus, on the viral side, specific species and HBV genotypes and variants may be useful early indicators of ACLF. However, clinical data regarding the quantitative and qualitative changes in HBV and the effect of specific genotypes must be collected from ACLF patients and compared to CHB patients in different phases.
On the host side, because many HBcAg mutations have been documented, and their corresponding antibody display is restricted to a VH repertoire lacking somatic mutations, we believe that the detection of HBcAg-targeted unusual humoral immunity may be another potential early indicator for ACLF. Intrahepatic antibodies were targeted to HBcAg exclusively in the germline configuration may present only in ALF individuals (the mutated HBcAg). In chimpanzees with classic acute hepatitis B, no antibodies from the liver were in the germline configuration. In addition, the remarkable affinity of these anti-HBc antibodies is unusual for germline antibodies. And HBcAg is one of the few determined antigens of human germline antibodies, suggesting it to be vital for disease pathogenesis (Chen et al., 2018). Moreover, the significant deposition of complement components C1q, C3, and C4d in the liver tissue of both ALF cases further support a key role for humoral immunity in HBV-associated ALF pathogenesis (Farci et al., 2010). Thus, detecting cell-surface HBcAb and intrahepatic complement components may be a potential early indicator of ACLF. However, these results are from studies on chimpanzee. The dominant B cell signature and exclusive changes of humoral immunity observed in ACLF patients require additional clinical research.
Therapy Strategies
Classical Therapy Strategies
Liver transplantation (LT) is an effective therapy to reduce mortality and increase survival rate (Finkenstedt et al., 2013; Chan and Fan, 2015; Artru et al., 2017). However, the lack of donor organs and high medical cost are major barriers for LT. Non-biological artificial liver support (ALS) devices remove circulating toxins using dialysis-based techniques (Chen et al., 2016; Alshamsi et al., 2020). However, ALS-like MARS provide no significant survival benefit (Banares et al., 2013). Bio-artificial devices that incorporate porcine or human hepatic cells have been developed to replace hepatic detoxification and synthetic functions. But, these devices including extracorporeal liver assist device (ELAD), AMC-BAL bio-reactor, and HepatAssist device have not been tested in ACLF patients and require validation in large studies (Demetriou et al., 2004; Sussman and Kelly, 2014). Anti-viral therapy is required for long-term prognosis. For example, Tenofovir and nucleoside and nucleotide analogs (NUCs) have been reported to improve the transplant-free survival rate in ACLF patients (Garg et al., 2011). However, long-term use of NUCs readily induces acute kidney injury. Peg-IFNα treatment is associated with hepatitis flares. Additionally, glucocorticoids administration in the early phase of HBV-ACLF elevated short-term survival obviously. However, the specifics of GC therapies such as type, dose, and duration should be considered among heterogeneous patients (Fujiwara et al., 2008, 2010; Chen et al., 2014).
Advanced Immunotherapy
Granulocyte-colony stimulating factor (G-CSF) has been introduced as a therapeutic alternative to LT in ACLF (Rolando et al., 2000; Garg et al., 2012; Khanam et al., 2014). G-CSF may reduce short-term mortality and prevent worsening of prognosis in mild adverse events (Duan et al., 2013; Chavez-Tapia et al., 2015). Other cell therapies such as bone hepatocyte transplantation, marrow-derived stem cell therapy, mesenchymal/multipotent mesenchymal stromal cell therapy are emerging therapeutic approaches. The most studied cell therapies are mesenchymal stem cells and stromal cells due to they are easy to obtain and able to differentiate into hepatocyte-like cells (Tsolaki and Yannaki, 2015). They can act as regulators of the immune response and repair hepatocyte injury in the recipient (Shi et al., 2017). MSC transplantation has improved liver function and short-term survival in CHB-ACLF patients. However, these studies are lacking of sufficient sample sizes and follow-up periods (Shi et al., 2012; Lin et al., 2017).
In summary, LT is the advised therapy for CHB-ACLF. Cell therapy is an optional method, but it needs validation and has persisting concerns over the long-term safety. Other methods such as NUCs, artificial liver devices, and glucocorticoids could also improve survival in subgroups. Considering the function of host immunity against HBcAg in the pathogenesis of ACLF, it is advised that therapeutic and prophylactic methods for reducing over-activated anti-HBc immune response may be effective. (Figure 3; Farci et al., 2010).
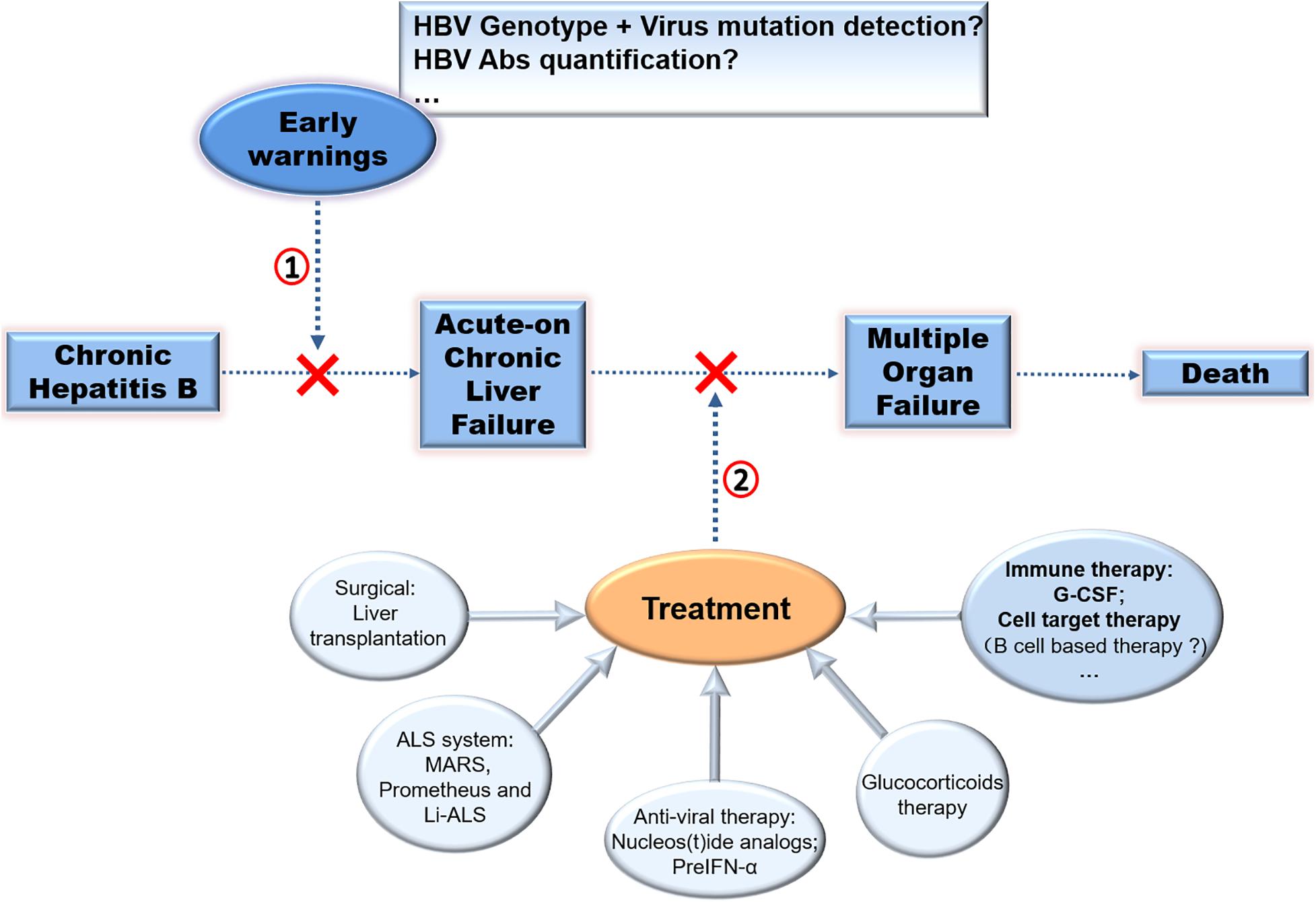
Figure 3. Therapy strategies in CHB-ACLF. Different approaches to prevent disease progression of CHB was exhibited. (1) Early warnings of ACLF by detecting virus mutation or HBV Abs quantification, (2) treatment of ACLF especially by immuno-based therapy for ACLF require further exploration to improve clinical applications.
Conclusion
The imbalanced immune system leads to different HBV progressions, including ACLF. The associated unusual immune events in CHB-ACLF patients have been elucidated. We provide a new understanding of how viral mutations induce immune pressure and how systemic inflammation leads to immune exhaustion culminating in liver damage and multiple organ failure. Additionally, we provide new insights regarding early indicators and immunotherapy for CHB-ACLF. Finally, readily testable early indicators and immuno-based therapy for ACLF require further exploration to improve clinical applications.
Author Contributions
QL and JW drafted and revised the manuscript. YQ, ML, and HL conceived and designed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the foundation of Wuxi Medical Development Discipline (FZXK006) for Infectious Disease, Wuxi Young Medical Talents (Q201743), Healthy and Scientific Bureau of Wuxi (MS201731 and CSE31N1712), and Key Clinical Specialty Construction Project in Shanghai (shslczdzk01102).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to HL and YQ for his guidance. We appreciate for the revision of ML. In addition, thanks to the Editage company for revising our manuscript.
Abbreviations
ACLF, Acute-on-chronic liver failure; ALF, Acute liver failure; CHB, Chronic hepatitis B; BCP, Basal core promoter; CHB-ACLF, Chronic hepatitis B-related ACLF; DCs, Dendritic cells; ELAD, extracorporeal liver assist device; ENH, HBeAg-negative hepatitis; G-CSF, Granulocyte-colony stimulating factor; HBV, Hepatitis B virus; HbeAg, Hepatitis-B-Virus e-Antigen; HBcAg, HBV, capsid/core antigen; IT, Immune tolerant; IC, Immune clearance; IA, Immune active; IRFs, interferon regulatory transcription factors; ISGs, interferon-stimulated-genes; JNK, c-JunN-terminal protein kinase; KCs, Kupffer Cells; LT, Liver transplantation; LPS, lipopolysaccharide; M φ, phagocytosis; MIP-3 α, macrophage inflammatory protein 3 α; MoDCs, monocyte-derived dendritic cells; MDSCs, myeloid-derived suppressor cells; MyD88, myeloid differentiation primary response gene 88; NOD, nucleotide-binding oligomerization domain-containing protein; NK, natural killer; NUCs, nucleotide analogs; PBMCs, peripheral blood mononuclear cells; PAMPs, Pathogen-Associated Molecular Patterns; PRRs, Pathogen Recognition Receptors; PC, pre-core; PD-1, programmed death-1 receptor; SE − CHB, severe exacerbation of chronic hepatitis B; MIP-3 α, macrophage inflammatory protein 3 α; RIG, retinoic acid-inducible gene I; TRAIL, TNF-related apoptosis-inducing ligand; TLR, toll-like receptor; TRIF, TIR-domain-containing adapter-inducing interferon- β; TBK1, TANK binding kinase 1; TAK1, transforming growth factor- β activated kinase 1.
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Alam, A., Chun Suen, K., and Ma, D. (2017). Acute-on-chronic liver failure: recent update. J. Biomed. Res. 31, 283–300. doi: 10.7555/JBR.31.20160060
Alexopoulou, A., Baltayiannis, G., Eroglu, C., Nastos, T., Dourakis, S. P., Archimandritis, A. J., et al. (2009). Core mutations in patients with acute episodes of chronic HBV infection are associated with the emergence of new immune recognition sites and the development of high IgM anti-HBc index values. J. Med. Virol. 81, 34–41. doi: 10.1002/jmv.21337
Alshamsi, F., Alshammari, K., Belley-Cote, E., Dionne, J., Albrahim, T., Albudoor, B., et al. (2020). Extracorporeal liver support in patients with liver failure: a systematic review and meta-analysis of randomized trials. Intensive Care Med. 46, 1–16. doi: 10.1007/s00134-019-05783-y
Andreani, T., Serfaty, L., Mohand, D., Dernaika, S., Wendum, D., Chazouillères, O., et al. (2007). Chronic hepatitis B virus carriers in the immunotolerant phase of infection: histologic findings and outcome. Clin. Gastroenterol. Hepatol. 5, 636–641. doi: 10.1016/j.cgh.2007.01.005
Artru, F., Louvet, A., Ruiz, I., Levesque, E., Labreuche, E., Bedoya, J., et al. (2017). Liver transplantation in the most severely ill cirrhotic patients: a multicenter study in acute-on-chronic liver failure grade 3. J. Hepatol. 67, 708–715. doi: 10.1016/j.jhep.2017.06.009
Banares, R., Nevens, F., Larsen, F. S., Jalan, R., Albillos, A., Dollinger, M., et al. (2013). Extracorporeal albumin dialysis with the molecular adsorbent recirculating system in acute-on-chronic liver failure: the RELIEF trial. Hepatology 57, 1153–1162. doi: 10.1002/hep.26185
Bao, S., Zheng, J., Li, N., Huang, C., Chen, M., Cheng, Q., et al. (2017). Role of interleukin-23 in monocyte-derived dendritic cells of HBV-related acute-on-chronic liver failure and its correlation with the severity of liver damage. Clin. Res. Hepatol. Gastroenterol. 41, 147–155. doi: 10.1016/j.clinre.2016.10.005
Baumert, T. F., Rogers, S. A., Hasegawa, K., and Liang, T. J. (1996). Two core promotor mutations identified in a hepatitis B virus strain associated with fulminant hepatitis result in enhanced viral replication. J. Clin. Invest. 98, 2268–2276. doi: 10.1172/JCI119037
Cai, Q., Liu, W., Zhu, M., and Sheng, J. (2019). Microbial infections as a trigger for acute-on-chronic liver failure: a review. Med. Sci. Monit. 25, 4773–4783. doi: 10.12659/MSM.915637
Cao, D., Xu, H., Guo, G., Ruan, Z., Fei, L., Xie, Z., et al. (2013). Intrahepatic expression of programmed death-1 and its ligands in patients with HBV-related acute-on-chronic liver failure. Inflammation 36, 110–120. doi: 10.1007/s10753-012-9525-7
Carow, B., and Rottenberg, M. E. (2014). SOCS3, a major regulator of infection and inflammation. Front. Immunol. 5:58. doi: 10.3389/fimmu.2014.00058
Chan, A. C., and Fan, S. T. (2015). Criteria for liver transplantation in ACLF and outcome. Hepatol Int. 9, 355–359. doi: 10.1007/s12072-014-9585-x
Chavez-Tapia, N. C., Mendiola-Pastrana, I., Ornelas-Arroyo, V. J., Camilo, N. H., Desiree, V. P., Guadalupe, D. S., et al. (2015). Granulocyte-colony stimulating factor for acute-on-chronic liver failure: systematic review and meta-analysis. Ann. Hepatol. 14, 631–641. doi: 10.1016/S1665-2681(19)30757-4
Chen, C. H., Lee, C. M., Tung, W. C., Wang, J. H., Chao, H. H., Hu, T. H., et al. (2010). Evolution of full-length HBV sequences in chronic hepatitis B patients with sequential lamivudine and adefovir dipivoxil resistance. J. Hepatol. 52, 478–485. doi: 10.1016/j.jhep.2010.01.006
Chen, J. F., Wang, K. W., Zhang, S. Q., Lei, Z. Y., Xie, J. Q., Zhu, J. Y., et al. (2014). Dexamethasone in outcome of patients with hepatitis B virus-related acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 29, 800–806. doi: 10.1111/jgh.12454
Chen, J. J., Huang, J. R., Yang, Q., Xu, X. W., Liu, L. L., Hao, S. R., et al. (2016). Plasma exchange-centered artificial liver support system in hepatitis B virus-related acute-on-chronic liver failure: a nationwide prospective multicenter study in China. Hepatobiliary Pancreat. Dis. Int. 15, 275–281. doi: 10.1016/s1499-3872(16)60084-x
Chen, L., Zheng, C. X., Lin, M. H., Huang, Z. X., Chen, R. H., Li, Q., et al. (2013). Distinct quasispecies characteristics and positive selection within precore/core gene in hepatitis B virus HBV associated acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 28, 1040–1046. doi: 10.1111/jgh.12109
Chen, T., Zhu, L., Zhou, Y., Pi, B., Liu, X., Deng, G., et al. (2013). KCTD9 contributes to liver injury through NK cell activation during hepatitis B virus-induced acute-on-chronic liver failure. Clin. Immunol. 146, 207–216. doi: 10.1016/j.clim.2012.12.013
Chen, M. T., Billaud, J. N., Sallberg, M., Guidotti, L. G., Chisari, F. V., Jones, J., et al. (2004). A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. U.S.A. 101, 14913–14918. doi: 10.1073/pnas.0406282101
Chen, Z., Diaz, G., Pollicino, T., Zhao, H., Engle, R. E., Schuck, P., et al. (2018). Role of humoral immunity against hepatitis B virus core antigen in the pathogenesis of acute liver failure. Proc. Natl. Acad. Sci. U.S.A. 115, E11369–E11378. doi: 10.1073/pnas.1809028115
Claria, J., Stauber, R. E., Coenraad, M. J., Moreau, R., Jalan, R., Pavesi, M., et al. (2016). Systemic inflammation in decompensated cirrhosis: characterization and role in acute-on-chronic liver failure. Hepatology 64, 1249–1264. doi: 10.1002/hep.28740
Connolly, D. J., and Bowie, A. G. (2014). The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem. Pharmacol. 92, 405–414. doi: 10.1016/j.bcp.2014.08.031
Cooper, A., Tal, G., Lider, O., and Shaul, Y. (2005). Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J. Immunol. 175, 3165–3176. doi: 10.4049/jimmunol.175.5.3165
Cui, Y. L., Yan, F., Wang, Y. B., Song, X. Q., Liu, L., Lei, X. Z., et al. (2010). Nucleoside analogue can improve the long-term prognosis of patients with hepatitis B virus infection-associated acute on chronic liver failure. Dig. Dis. Sci. 55, 2373–2380. doi: 10.1007/s10620-010-1257-7
Demetriou, A. A., Brown, R. S. Jr., Busuttil, R. W., Fair, J., McGuire, B. M., Rosenthal, P., et al. (2004). Prospective, randomized, multicenter, controlled trial of a bioartificial liver in treating acute liver failure. Ann. Surg. 239, 660–667. doi: 10.1097/01.sla.0000124298.74199.e5
Ding, C., Li, Y., Guo, F., Jing, Y., Ying, W., Li, D., et al. (2016). A cell-type-resolved liver proteome. Mol. Cell. Proteom. 15, 3190–3202. doi: 10.1074/mcp.M116.060145
Ding, W., Jiang, L., Zhou, Q., Wu, T., Shi, D., Lin, B., et al. (2015). Characterisation of peripheral blood mononuclear cell microRNA in hepatitis B-related acute-on-chronic liver failure. Sci. Rep. 5:13098. doi: 10.1038/srep13098
Duan, X. Z., Liu, F. F., Tong, J. J., Yang, H. Z., Chen, J., Liu, X. Y., et al. (2013). Granulocyte-colony stimulating factor therapy improves survival in patients with hepatitis B virus-associated acute-on-chronic liver failure. World J. Gastroenterol. 19, 1104–1110. doi: 10.3748/wjg.v19.i7.1104
Durantel, D., and Zoulim, F. (2009). Innate response to hepatitis B virus infection: observations challenging the concept of a stealth virus. Hepatology 50, 1692–1695. doi: 10.1002/hep.23361
Fagan, E. A., Smith, P. M., Davison, F., and Williams, R. (1986). Fulminant hepatitis B in successive female sexual partners of two anti-HBe-positive males. Lancet 2, 538–540. doi: 10.1016/s0140-6736(86)90112-1
Farci, P., Diaz, G., Chen, Z., Govindarajan, S., Tice, A., Agulto, L., et al. (2010). B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc. Natl. Acad. Sci. U.S.A. 107, 8766–8771. doi: 10.1073/pnas.1003854107
Fattovich, G., Bortolotti, F., and Donato, F. (2008). Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J. Hepatol. 48, 335–352. doi: 10.1016/j.jhep.2007.11.011
Faure-Dupuy, S., Vegna, S., Aillot, L., Dimier, L., Esser, K., Broxtermann, M., et al. (2018). Characterization of pattern recognition receptor expression and functionality in liver primary cells and derived cell lines. J. Innate. Immun. 10, 339–348. doi: 10.1159/000489966
Finkenstedt, A., Nachbaur, K., Zoller, H., Joannidis, M., Pratschke, J., Graziadei, I. W., et al. (2013). Acute-on-chronic liver failure: excellent outcomes after liver transplantation but high mortality on the wait list. Liver Transpl. 19, 879–886. doi: 10.1002/lt.23678
Friedt, M., Gerner, P., Lausch, E., Trubel, H., Zabel, B., and Wirth, S. (1999). Mutations in the basic core promotor and the precore region of hepatitis B virus and their selection in children with fulminant and chronic hepatitis B. Hepatology 29, 1252–1258. doi: 10.1002/hep.510290418
Fujiwara, K., Yasui, S., Okitsu, K., Yonemitsu, Y., Oda, S., and Yokosuka, O. (2010). The requirement for a sufficient period of corticosteroid treatment in combination with nucleoside analogue for severe acute exacerbation of chronic hepatitis B. J. Gastroenterol. 45, 1255–1262. doi: 10.1007/s00535-010-0280-y
Fujiwara, K., Yasui, S., Yonemitsu, Y., Fukai, K., Arai, M., Imazeki, F., et al. (2008). Efficacy of combination therapy of antiviral and immunosuppressive drugs for the treatment of severe acute exacerbation of chronic hepatitis B. J. Gastroenterol. 43, 711–719. doi: 10.1007/s00535-008-2222-5
Gao, S., Joshi, S. S., Osiowy, C., Chen, Y., Coffin, C. S., and Duan, Z. P. (2017). Chronic hepatitis B carriers with acute on chronic liver failure show increased HBV surface gene mutations, including immune escape variants. Virol. J. 14:203. doi: 10.1186/s12985-017-0870-x
Garcia-Tsao, G., and Wiest, R. (2004). Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract. Res. Clin. Gastroenterol. 18, 353–372. doi: 10.1016/j.bpg.2003.10.005
Garg, H., Sarin, S. K., Kumar, M., Garg, V., Sharma, B. C., and Kumar, A. (2011). Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 53, 774–780. doi: 10.1002/hep.24109
Garg, V., Garg, H., Khan, A., Trehanpati, N., Kumar, A., Chander, B., et al. (2012). Granulocyte colony-stimulating factor mobilizes CD34(+) cells and improves survival of patients with acute-on-chronic liver failure. Gastroenterology 142, 505–512. doi: 10.1053/j.gastro.2011.11.027
Gronbaek, H., Sandahl, T. D., Mortensen, C., Vilstrup, H., Moller, H. J., and Moller, S. (2012). Soluble CD163, a marker of Kupffer cell activation, is related to portal hypertension in patients with liver cirrhosis. Aliment. Pharmacol. Ther. 36, 173–180. doi: 10.1111/j.1365-2036.2012.05134.x
Guidotti, L. G., and Chisari, F. V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1, 23–61. doi: 10.1146/annurev.pathol.1.110304.100230
Guidotti, L. G., Isogawa, M., and Chisari, F. V. (2015). Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 36, 61–66. doi: 10.1016/j.coi.2015.06.016
Guo, G., Cao, D., Xu, H., Ruan, Z., Fei, L., Xie, Z., et al. (2012). The characteristic expression of B7-H3 and B7-H4 in liver biopsies from patients with HBV-related acute-on-chronic liver failure. Pathol. Int. 62, 665–674. doi: 10.1111/j.1440-1827.2012.02856.x
Hernaez, R., Sola, E., Moreau, R., and Gines, P. (2017). Acute-on-chronic liver failure: an update. Gut 66, 541–553. doi: 10.1136/gutjnl-2016-312670
Hou, J., Lin, Y., Waters, J., Wang, Z., Min, J., Liao, H., et al. (2002). Detection and significance of a G1862T variant of hepatitis B virus in Chinese patients with fulminant hepatitis. J. Gen. Virol. 83, 2291–2298. doi: 10.1099/0022-1317-83-9-2291
Hui, C. K., Lie, A., Au, W. Y., Leung, Y. H., Ma, S., Cheung, W., et al. (2005). A long-term follow-up study on hepatitis B surface antigen-positive patients undergoing allogeneic hematopoietic stem cell transplantation. Blood 106, 464–469. doi: 10.1182/blood-2005-02-0698
Inoue, K., Yoshiba, M., Sekiyama, K., Okamoto, H., and Mayumi, M. (1998). Clinical and molecular virological differences between fulminant hepatic failures following acute and chronic infection with hepatitis B virus. J. Med. Virol. 55, 35–41. doi: 10.1002/(SICI)1096-9071(199805)55:1<35::AID-JMV7>3.0.CO;2-P
Jury, E. A. S. L. (2003). EASL international consensus conference on hepatitis B. J. Hepatol. 38, 533–540. doi: 10.1016/s0168-8278(03)00083-7
Kawai, T., and Akira, S. (2009). The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337. doi: 10.1093/intimm/dxp017
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. doi: 10.1016/j.immuni.2011.05.006
Kay, A., and Zoulim, F. (2007). Hepatitis B virus genetic variability and evolution. Virus Res. 127, 164–176. doi: 10.1016/j.virusres.2007.02.021
Kerur, N., Veettil, M. V., Sharma-Walia, N., Bottero, V., Sadagopan, S., Otageri, P., et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 9, 363–375. doi: 10.1016/j.chom.2011.04.008
Khanam, A., Trehanpati, N., Garg, V., Kumar, C., Garg, H., Barjesh, C., et al. (2014). Altered frequencies of dendritic cells and IFN-gamma-secreting T cells with granulocyte colony-stimulating factor (G-CSF) therapy in acute-on-chronic liver failure. Liver Int. 34, 505–513. doi: 10.1111/liv.12415
Kosaka, Y., Takase, K., Kojima, M., Shimizu, M., Inoue, K., Yoshiba, M., et al. (1991). Fulminant hepatitis B: induction by hepatitis B virus mutants defective in the precore region and incapable of encoding e antigen. Gastroenterology 100, 1087–1094. doi: 10.1016/0016-5085(91)90286-t
Kubes, P., and Mehal, W. Z. (2012). Sterile inflammation in the liver. Gastroenterology 143, 1158–1172. doi: 10.1053/j.gastro.2012.09.008
Kumar, H., Kawai, T., and Akira, S. (2009). Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388, 621–625. doi: 10.1016/j.bbrc.2009.08.062
Kumar, H., Kawai, T., and Akira, S. (2011). Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34. doi: 10.3109/08830185.2010.529976
Laleman, W., Landeghem, L., Wilmer, A., Fevery, J., and Nevens, F. (2005). Portal hypertension: from pathophysiology to clinical practice. Liver Int. 25, 1079–1090. doi: 10.1111/j.1478-3231.2005.01163.x
Lang, T., Lo, C., Skinner, N., Locarnini, S., Visvanathan, K., and Mansell, A. (2011). The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 55, 762–769. doi: 10.1016/j.jhep.2010.12.042
Lee, B. O., Tucker, A., Frelin, L., Sallberg, M., Jones, J., Peters, C., et al. (2009). Interaction of the hepatitis B core antigen and the innate immune system. J. Immunol. 182, 6670–6681. doi: 10.4049/jimmunol.0803683
Lei, J. H., Peng, F., Chen, Z., and Xiao, X. Q. (2019). Is HBV viral load at admission associated with development of acute-on-chronic liver failure in patients with acute decompensation of chronic hepatitis B related cirrhosis? BMC Infect. Dis. 19:363. doi: 10.1186/s12879-019-3988-1
Li, Y., Han, M. F., Li, W. N., Shi, A. C., Zhang, Y. Y., Wang, H. Y., et al. (2014). SOCS3 expression correlates with severity of inflammation in mouse hepatitis virus strain 3-induced acute liver failure and HBV-ACLF. J. Huazhong Univ. Sci. Technol. 34, 348–353. doi: 10.1007/s11596-014-1281-5
Liang, T. J., Hasegawa, K., Rimon, N., Wands, J. R., and Ben-Porath, E. (1991). A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. N Engl J Med. 324, 1705–1709. doi: 10.1056/NEJM199106133242405
Lin, B. L., Chen, J. F., Qiu, W. H., Wang, K. W., Xie, D., Chen, X., et al. (2017). Allogeneic bone marrow-derived mesenchymal stromal cells for hepatitis B virus-related acute-on-chronic liver failure: a randomized controlled trial. Hepatology 66, 209–219. doi: 10.1002/hep.29189
Liu, F., Duan, X., Wan, Z., Zang, H., You, S., Yang, R., et al. (2016). Lower number and decreased function of natural killer cells in hepatitis B virus related acute-on-chronic liver failure. Clin. Res. Hepatol. Gastroenterol. 40, 605–613. doi: 10.1016/j.clinre.2016.01.004
Lok, A. S., Lai, C. L., Wu, P. C., Leung, E. K., and Lam, T. S. (1987). Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 92, 1839–1843. doi: 10.1016/0016-5085(87)90613-5
Lucifora, J., Durantel, D., Testoni, B., Hantz, O., Levrero, M., and Zoulim, F. (2010). Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 51, 63–72. doi: 10.1002/hep.23230
Lutgehetmann, M., Bornscheuer, T., Volz, T., Allweiss, L., Bockmann, J. H., Pollok, J. M., et al. (2011). Hepatitis B virus limits response of human hepatocytes to interferon-alpha in chimeric mice. Gastroenterology 140, 2074–2083. doi: 10.1053/j.gastro.2011.02.057
Ma, X. Y., Han, T., Pei, Y. Z., Zhao, Z. G., Gao, Y. T., Li, Y., et al. (2012). [Analysis of the relationship between hepatitis B virus precore and basal core promoter mutations and acute-on-chronic liver failure]. Zhonghua gan zang bing za zhi. 20, 644–648. doi: 10.1186/s12985-015-0313-5
Manka, P., Verheyen, J., Gerken, G., and Canbay, A. (2016). Liver failure due to acute viral hepatitis (A-E). Visc. Med. 32, 80–85. doi: 10.1159/000444915
Mo, R., Wang, P., Lai, R., Li, F., Liu, Y., Jiang, S., et al. (2017). Persistently elevated circulating Th22 reversely correlates with prognosis in HBV-related acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 32, 677–686. doi: 10.1111/jgh.13537
Moreau, R., Jalan, R., Gines, P., Marco, P., Paolo, A., Juan, C., et al. (2013). Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 144, 1426–1437. doi: 10.1053/j.gastro.2013.02.042
Nian, X., Xu, Z., Liu, Y., Chen, J., Li, X., and Xu, D. (2016). Association between hepatitis B virus basal core promoter/precore region mutations and the risk of hepatitis B-related acute-on-chronic liver failure in the Chinese population: an updated meta-analysis. Hepatol. Int. 10, 606–615. doi: 10.1007/s12072-016-9716-7
Oketani, M., Uto, H., Ido, A., and Tsubouchi, H. (2014). Management of hepatitis B virus-related acute liver failure. Clin. J. Gastroenterol. 7, 19–26. doi: 10.1007/s12328-013-0447-1
Ozasa, A., Tanaka, Y., Orito, E., Sugiyama, M., Kang, J. H., Hige, S., et al. (2006). Influence of genotypes and precore mutations on fulminant or chronic outcome of acute hepatitis B virus infection. Hepatology 44, 326–334. doi: 10.1002/hep.21249
Pandey, S., Kawai, T., and Akira, S. (2014). Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 7:a016246. doi: 10.1101/cshperspect.a016246
Pang, X., Li, X., Mo, Z., Huang, J., Deng, H., Lei, Z., et al. (2018). IFI16 is involved in HBV-associated acute-on-chronic liver failure inflammation. BMC Gastroenterol. 18:61. doi: 10.1186/s12876-018-0791-1
Parekh, S., Zoulim, F., Ahn, S. H., Tsai, A., Li, J., Kawai, S., et al. (2003). Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J. Virol. 77, 6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003
Ren, X., Xu, Z., Liu, Y., Li, X., Bai, S., Ding, N., et al. (2010). Hepatitis B virus genotype and basal core promoter/precore mutations are associated with hepatitis B-related acute-on-chronic liver failure without pre-existing liver cirrhosis. J. Viral. Hepat. 17, 887–895. doi: 10.1111/j.1365-2893.2009.01254.x
Rolando, N., Clapperton, M., Wade, J., Panetsos, G., Mufti, G., Williams, R., et al. (2000). Granulocyte colony-stimulating factor improves function of neutrophils from patients with acute liver failure. Eur. J. Gastroenterol. Hepatol. 12, 1135–1140. doi: 10.1097/00042737-200012100-00011
Safari-Arababadi, M., Modarressi, M. H., and Arababadi, M. K. (2019). Up-regulation of RIP1 and IPS-1 in chronic HBV infected patients. Genet. Mol. Biol. 42, 337–343. doi: 10.1590/1678-4685-GMB-2018-0071
Sarin, S. K., Kumar, A., Almeida, J. A., Chawla, Y. K., Fan, S. T., Garg, H., et al. (2009). Acute-on-chronic liver failure: consensus recommendations of the Asian Pacific Association for the study of the liver (APASL). Hepatol Int. 3, 269–282. doi: 10.1007/s12072-008-9106-x
Sato, S., Suzuki, K., Akahane, Y., Akamatsu, K., Akiyama, K., Yunomura, K., et al. (1995). Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann. Intern. Med. 122, 241–248. doi: 10.7326/0003-4819-122-4-199502150-00001
Sharma, S., Fitzgerald, K. A., Cancro, M. P., and Marshak-Rothstein, A. (2015). Nucleic acid-sensing receptors: rheostats of autoimmunity and autoinflammation. J. Immunol. 195, 3507–3512. doi: 10.4049/jimmunol.1500964
Sheen, I. S., Liaw, Y. F., Tai, D. I., and Chu, C. M. (1985). Hepatic decompensation associated with hepatitis B e antigen clearance in chronic type B hepatitis. Gastroenterology 89, 732–735. doi: 10.1016/0016-5085(85)90566-9
Shi, D., Zhang, J., Zhou, Q., Xin, J. J., Jiang, J., Jiang, L., et al. (2017). Quantitative evaluation of human bone mesenchymal stem cells rescuing fulminant hepatic failure in pigs. Gut 66, 955–964. doi: 10.1136/gutjnl-2015-311146
Shi, F., Zhang, J. Y., Zeng, Z., Tien, P., and Wang, F. S. (2010). Skewed ratios between CD3(+) T cells and monocytes are associated with poor prognosis in patients with HBV-related acute-on-chronic liver failure. Biochem. Biophys. Res. Commun. 402, 30–36. doi: 10.1016/j.bbrc.2010.09.096
Shi, M., Zhang, Z., Xu, R., Lin, H., Fu, J., Zou, Z., et al. (2012). Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients. Stem Cells Transl. Med. 1, 725–731. doi: 10.5966/sctm.2012-0034
Shi, Y., Yang, Y., Hu, Y., Wu, W., Yang, Q., Zheng, M., et al. (2015). Acute-on-chronic liver failure precipitated by hepatic injury is distinct from that precipitated by extrahepatic insults. Hepatology 62, 232–242. doi: 10.1002/hep.27795
Su, X., and Liu, G. (1991). [Quantitative determination of sugars in radix Aconiti and its processed products by two kinds of colorimetry]. Zhongguo Zhong yao za zhi. 16, 215–217.
Sun, L., Zhang, Y., Zhao, B., Deng, M., Liu, J., Li, X., et al. (2014). A new unconventional HLA-A2-restricted epitope from HBV core protein elicits antiviral cytotoxic T lymphocytes. Protein Cell 5, 317–327. doi: 10.1007/s13238-014-0041-4
Suslov, A., Boldanova, T., Wang, X., Wieland, S., and Heim, M. H. (2018). Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology 154, 1778–1790. doi: 10.1053/j.gastro.2018.01.034
Sussman, N. L., and Kelly, J. H. (2014). Artificial liver. Clin. Gastroenterol. Hepatol. 12, 1439–1442. doi: 10.1016/j.cgh.2014.06.002
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tan, A., Koh, S., and Bertoletti, A. (2015). Immune response in hepatitis b virus infection. Cold Spring Harb. Perspect. Med. 5, a021428. doi: 10.1101/cshperspect.a021428
Thimme, R., Wieland, S., Steiger, C., Ghrayeb, J., Reimann, K. A., Purcell, R. H., et al. (2003). CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 77, 68–76. doi: 10.1128/jvi.77.1.68-76.2003
Tong, S., Kim, K. H., Chante, C., Wands, J., and Li, J. (2005). Hepatitis B virus e antigen variants. Int. J. Med. Sci. 2, 2–7. doi: 10.7150/ijms.2.2
Tsolaki, E., and Yannaki, E. (2015). Stem cell-based regenerative opportunities for the liver: state of the art and beyond. World J. Gastroenterol. 21, 12334–12350. doi: 10.3748/wjg.v21.i43.12334
Vanlandschoot, P., Van Houtte, F., Serruys, B., and Leroux-Roels, G. (2007). Contamination of a recombinant hepatitis B virus nucleocapsid preparation with a human B-cell activator. J. Virol. 81, 2535–2536. doi: 10.1128/JVI.02507-06
Vyas, A. K., Jindal, A., and Trehanpati, N. (2018). Ponder the innate immunity in hepatitis B infection. Gastroenterology 155:1277. doi: 10.1053/j.gastro.2018.05.052
Wai, C. T., Fontana, R. J., Polson, J., Hussain, M., Shakil, A. O., Han, S. H., et al. (2005). Clinical outcome and virological characteristics of hepatitis B-related acute liver failure in the United States. J. Viral. Hepat. 12, 192–198. doi: 10.1111/j.1365-2893.2005.00581.x
Wan, Z., Xie, G., Wu, Y., Liu, F., Xin, S., You, S., et al. (2016). Cytokines elevated in patients with HBV-related acute-on-chronic liver failure promote NK cell mediated cytotoxicity through TRAIL. Dig. Liver Dis. 48, 528–535. doi: 10.1016/j.dld.2016.01.008
Wang, K., Liu, H., He, Y., Chen, T., Yang, Y., Niu, Y., et al. (2010). Correlation of TLR1-10 expression in peripheral blood mononuclear cells with chronic hepatitis B and chronic hepatitis B-related liver failure. Hum. Immunol. 71, 950–956. doi: 10.1016/j.humimm.2010.07.013
Wang, S., Chen, Z., Hu, C., Qian, F., Cheng, Y., Wu, M., et al. (2013). Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 190, 5142–5151. doi: 10.4049/jimmunol.1201625
Wieland, S., Thimme, R., Purcell, R. H., and Chisari, F. V. (2004). Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 6669–6674. doi: 10.1073/pnas.0401771101
Wieland, S. F., and Chisari, F. V. (2005). Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 79, 9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005
Wiest, R., and Garcia-Tsao, G. (2005). Bacterial translocation (BT) in cirrhosis. Hepatology 41, 422–433. doi: 10.1002/hep.20632
Wiest, R., Lawson, M., and Geuking, M. (2014). Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 60, 197–209. doi: 10.1016/j.jhep.2013.07.044
Wu, W., Yan, H., Zhao, H., Sun, W., Yang, Q., Sheng, J., et al. (2018). Characteristics of systemic inflammation in hepatitis B-precipitated ACLF: differentiate it from No-ACLF. Liver Int. 38, 248–257. doi: 10.1111/liv.13504
Xiao, L., Zhou, B., Gao, H., Ma, S., Yang, G. F., Wu, M., et al. (2011). Hepatitis B virus genotype B with G1896A and A1762T/G1764A mutations is associated with hepatitis B related acute-on-chronic liver failure. J. Med. Virol. 83, 1544–1550. doi: 10.1002/jmv.22159
Xin, J., Ding, W., Hao, S., Chen, X., Zhang, J., Jiang, L., et al. (2016). Serum macrophage inflammatory protein 3alpha levels predict the severity of HBV-related acute-on-chronic liver failure. Gut 65, 355–357. doi: 10.1136/gutjnl-2015-309916
Xing, T., Li, L., Cao, H., and Huang, J. (2007). Altered immune function of monocytes in different stages of patients with acute on chronic liver failure. Clin. Exp. Immunol. 147, 184–188. doi: 10.1111/j.1365-2249.2006.03259.x
Xiong, L., Du, Y., Zhou, T., Du, B., Visalath, P., Lin, L., et al. (2019). N-myc and STAT interactor correlates with severity and prognosis in acute-on-chronic liver failure of hepatitis B virus. J. Gastroenterol. Hepatol. 34, 1800–1808. doi: 10.1111/jgh.14634
Xu, C., Lu, Y., Zheng, X., Feng, X., Yang, X. C., Timm, J., et al. (2017). TLR2 expression in peripheral CD4+ T cells promotes Th17 response and is associated with disease aggravation of hepatitis B virus-related acute-on-chronic liver failure. Front. Immunol. 8:1609. doi: 10.3389/fimmu.2017.01609
Xu, C. L., Hao, Y. H., Lu, Y. P., Tang, Z. S., Yang, X. C., Wu, J., et al. (2015). Upregulation of toll-like receptor 4 on T cells in PBMCs is associated with disease aggravation of HBV-related acute-on-chronic liver failure. J. Huazhong Univ. Sci. Technolog. Med. Sci. 35, 910–915. doi: 10.1007/s11596-015-1527-x
Xu, H., Cao, D., Guo, G., Ruan, Z., Wu, Y., and Chen, Y. (2012). The intrahepatic expression and distribution of BTLA and its ligand HVEM in patients with HBV-related acute-on-chronic liver failure. Diagn. Pathol. 7:142. doi: 10.1186/1746-1596-7-142
Xu, Z., Ren, X., Liu, Y., Li, X. D., Bai, S., Zhong, Y., et al. (2011). Association of hepatitis B virus mutations in basal core promoter and precore regions with severity of liver disease: an investigation of 793 Chinese patients with mild and severe chronic hepatitis B and acute-on-chronic liver failure. J. Gastroenterol. 46, 391–400. doi: 10.1007/s00535-010-0315-4
Yang, Z. T., Huang, S. Y., Chen, L., Liu, F., Cai, X. H., Guo, Y. F., et al. (2015). Characterization of full-length genomes of hepatitis B virus quasispecies in sera of patients at different phases of infection. J. Clin. Microbiol. 53, 2203–2214. doi: 10.1128/JCM.00068-15
Yi, R. T., Niu, Y. H., Liu, H. L., Zhang, T. Y., Yang, Y. C., Zhang, Y., et al. (2016). Natural killer group 2A expressed on both peripheral CD3(-)CD56(+)NK cells and CD3(+)CD8(+)T cells plays a pivotal negative regulatory role in the progression of hepatitis B virus-related acute-on-chronic liver failure. J. Interferon Cytokine Res. 36, 689–697. doi: 10.1089/jir.2015.0166
You, S., Rong, Y., Zhu, B., Zhang, A., Zang, H., Liu, H., et al. (2013). Changing etiology of liver failure in 3,916 patients from northern China: a 10-year survey. Hepatol Int. 7, 714–720. doi: 10.1007/s12072-013-9424-5
Yu, X., Zheng, Y., Deng, Y., Li, L. J., Guo, R., Su, M., et al. (2016). Serum interleukin (IL)-9 and IL-10, but not T-Helper 9 (Th9) cells, are associated with survival of patients with acute-on-chronic hepatitis B liver failure. Medicine 95:e3405. doi: 10.1097/MD.0000000000003405
Zang, H., Xu, Z., Liu, Y., Li, X., Rong, Y., Jiang, L., et al. (2018). Clinical and virological implications of A1846T and C1913A/G mutations of hepatitis B virus genome in severe liver diseases. Scand. J. Gastroenterol. 53, 319–328. doi: 10.1080/00365521.2018.1424934
Zeng, Y., Li, Y., Xu, Z., Gan, W., Lu, L., Huang, X., et al. (2019). Myeloid-derived suppressor cells expansion is closely associated with disease severity and progression in HBV-related acute-on-chronic liver failure. J. Med. Virol. 91, 1510–1518. doi: 10.1002/jmv.25466
Zhang, G. L., Xie, D. Y., Ye, Y. N., Lin, C. S., Zhang, X. H., Zheng, Y. B., et al. (2014). High level of IL-27 positively correlated with Th17 cells may indicate liver injury in patients infected with HBV. Liver Int. 34, 266–273. doi: 10.1111/liv.12268
Zhang, Y. M., Yang, Y. D., Jia, H. Y., Zeng, L. Y., Yu, W., Zhou, N., et al. (2014). HBsAg levels in HBeAg-positive chronic hepatitis B patients with different immune conditions. World J. Gastroenterol. 20, 4407–4413. doi: 10.3748/wjg.v20.i15.4407
Zhang, G. L., Zhang, T., Ye, Y. N., Liu, J., Zhang, X., Xie, C., et al. (2016). Complement factor 3 could be an independent risk factor for mortality in patients with HBV related acute-on-chronic liver failure. Biomed. Res. Int. 2016:3524842. doi: 10.1155/2016/3524842
Zhang, G. L., Zhang, T., Zhao, Q. Y., Lin, C. S., and Gao, Z. L. (2018). Th17 cells over 5.9% at admission indicate poor prognosis in patients with HBV-related acute-on-chronic liver failure. Medicine 97:e12656. doi: 10.1097/MD.0000000000012656
Zhang, G. L., Zhao, Q. Y., Xie, C., Peng, L., Zhang, T., and Gao, Z. L. (2019). Elevated serum IgG levels positively correlated with IL-27 may indicate poor outcome in patients with HBV-related acute-on-chronic liver failure. J Immunol Res. 2019:1538439. doi: 10.1155/2019/1538439
Zou, Z., Li, B., Xu, D., Zhang, Z., Zhao, J. M., Zhou, G., et al. (2009). Imbalanced intrahepatic cytokine expression of interferon-gamma, tumor necrosis factor-alpha, and interleukin-10 in patients with acute-on-chronic liver failure associated with hepatitis B virus infection. J. Clin. Gastroenterol. 43, 182–190. doi: 10.1097/MCG.0b013e3181624464
Keywords: ACLF, HBV—hepatitis B virus, B cells, inflammation, cytokines storm
Citation: Li Q, Wang J, Lu M, Qiu Y and Lu H (2020) Acute-on-Chronic Liver Failure From Chronic-Hepatitis-B, Who Is the Behind Scenes. Front. Microbiol. 11:583423. doi: 10.3389/fmicb.2020.583423
Received: 20 July 2020; Accepted: 13 November 2020;
Published: 07 December 2020.
Edited by:
Jorge Quarleri, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Manuel Romero-Gomez, Seville University, SpainYu Shi, Zhejiang University, China
Yu-Chen Fan, Shandong University, China
Copyright © 2020 Li, Wang, Lu, Qiu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongzhou Lu, bHVob25nemhvdUBmdWRhbi5lZHUuY24=; Yuanwang Qiu, cXl3YW5nODM5QDEyNi5jb20=
†These authors have contributed equally to this work