 Jacqueline Wolf1†
Jacqueline Wolf1† Julia Koblitz1,2†
Julia Koblitz1,2† Andreas Albersmeier3
Andreas Albersmeier3 Jörn Kalinowski3
Jörn Kalinowski3 Bettina Siebers4Dietmar Schomburg1,2
Bettina Siebers4Dietmar Schomburg1,2 Meina Neumann-Schaal2,5*
Meina Neumann-Schaal2,5*- 1Department of Bioinformatics and Biochemistry, Technische Universität Braunschweig, Braunschweig, Germany
- 2Braunschweig Integrated Centre of Systems Biology (BRICS), Braunschweig, Germany
- 3Center for Biotechnology-CeBiTec, Universität Bielefeld, Bielefeld, Germany
- 4Molecular Enzyme Technology and Biochemistry (MEB), Environmental Microbiology and Biotechnology (EMB), Centre for Water and Environmental Research (CWE), University of Duisburg-Essen, Essen, Germany
- 5Junior Research Group Bacterial Metabolomics, Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany
Present in many industrial effluents and as common degradation product of organic matter, phenol is a widespread compound which may cause serious environmental problems, due to its toxicity to animals and humans. Degradation of phenol from the environment by mesophilic bacteria has been studied extensively over the past decades, but only little is known about phenol biodegradation at high temperatures or low pH. In this work we studied phenol degradation in the thermoacidophilic archaeon Saccharolobus solfataricus P2 (basonym: Sulfolobus solfataricus) under extreme conditions (80°C, pH 3.5). We combined metabolomics and transcriptomics together with metabolic modeling to elucidate the organism’s response to growth with phenol as sole carbon source. Although S. solfataricus is able to utilize phenol for biomass production, the carbon source induces profound stress reactions, including genome rearrangement as well as a strong intracellular accumulation of polyamines. Furthermore, computational modeling revealed a 40% higher oxygen demand for substrate oxidation, compared to growth on glucose. However, only 16.5% of oxygen is used for oxidation of phenol to catechol, resulting in a less efficient integration of carbon into the biomass. Finally, our data underlines the importance of the phenol meta-degradation pathway in S. solfataricus and enables us to predict enzyme candidates involved in the degradation processes downstream of 2-hydroxymucconic acid.
Introduction
Phenol is a widespread environmental pollutant, present in the effluents of various industries including coal conversion, production of phenolic resins and the textile industry (Naguib and Badawy, 2020). If it is not removed from ground water, phenol could cause serious health problems to humans, animals and aquatic life (Saha et al., 1999; Michalowicz and Duda, 2007). Additionally, phenolic compounds can result from degradation of natural organic matter in the environment, e.g., from the decomposition of plants and animals (Anku et al., 2017), and are thus ubiquitous in the environment. A number of mesophilic bacteria and yeasts can degrade phenol, e.g., Acinetobacter radioresistens, Glutamicibacter nicotianae, and Candida subhashii (Filipowicz et al., 2017; Duraisamy et al., 2020; Liu et al., 2020). The mechanism of phenol degradation in mesophilic microorganisms has been thoroughly studied because of their application in bioremediation. Although there have been several studies on thermophilic phenol degradation in Bacilli (Gurujeyalakshmi and Oriel, 1989; Antranikian, 1998; Duffner et al., 2000), Saccharolobus solfataricus (basonym: Sulfolobus solfataricus) was the first hyperthermophilic archaeon reported to be able to grow aerobically on phenol (Izzo et al., 2005). Since then this organism has been studied extensively with respect to its phenol degrading capacity and evidence has been found for an active phenol meta-degradation pathway in S. solfataricus (Christen et al., 2011, 2012; Comte et al., 2013). However, only little is known on the participating enzymes in the degradation process, as the homology of archaeal proteins to their bacterial counterparts is often very low. Only the monooxygenase gene cluster and the catechol-2,3-diooxygenase of S. solfataricus have been characterized so far (Izzo et al., 2005; Chae et al., 2007).
In this work we combined data from transcriptome and metabolome analysis together with metabolic modeling, to explore the global changes in S. solfataricus P2 in response to growth on phenol as sole carbon source. Our data strengthen the evidence for the activity of the meta-degradation route and predict gene candidates encoding for the missing proteins of this pathway in this hyperthermophilic archaeon.
Materials and Methods
Strain and Growth Conditions
Saccharolobus solfataricus P2 (Zillig et al., 1980; Sakai and Kurosawa, 2018) was cultivated in long neck flasks at 80°C, pH 3.5 and 160 rpm (Thermotron, Infors AG, Switzerland) on defined minimal medium (Brock et al., 1972), containing either 10 mM phenol or 22.2 mM D-glucose as sole carbon source. For adaption to phenol, pre-cultures, containing 2 mM D-glucose and 5 mM phenol, were inoculated with glucose adapted stock cultures, prepared as described earlier (Zaparty et al., 2010). These pre-cultures were used to inoculate the main cultures to an OD600 of ∼0.05. D-glucose main cultures were directly inoculated, using glucose adapted stock cultures. Cell growth was monitored photometrically, following the increase in optical density at 600 nm. To monitor phenol consumption, 1 ml samples were taken in regular intervals, cells were harvested by centrifugation (20,000 g; 5 min, 20°C) and supernatants were stored at −20°C until further processing. Determination of major biomass fractions was performed as described previously (Wolf et al., 2016). All phenotypic data for growth on D-glucose (growth rate, substrate uptake rate, and biomass composition) have been published previously (Wolf et al., 2016).
Extraction of Intra- and Extracellular Metabolites and Coenzyme A Derivatives
Phenol consumption was measured by GC-MS analysis, following the protocol for extraction, measurement and data processing as described previously (Neumann-Schaal et al., 2015). Briefly, 300 μl phenol-containing supernatant was mixed with 200 μl o-xylene as internal standard. After addition of 20 μl H2SO4 (49–51%), 200 μl methyl-tert-butylether was used to extract phenol. The extract was directly injected into the GC-MS without further derivatization. For calculation of absolute concentrations, an external phenol calibration curve ranging from 2 to 10 mM was measured.
Intracellular metabolites, coenzyme A derivatives and supernatants for analysis of secreted compounds were extracted, measured and analyzed as described earlier (Zaparty et al., 2010; Wolf et al., 2016). Briefly, for intracellular metabolome analysis, cells corresponding to 15 mg cell dry weight were harvested and washed twice with 0.9% NaCl. Afterward 1.5 ml methanol containing 30 μl ribitol (c = 0.2 g⋅l–1) as internal standard were added and cells were lysed in an ultrasonic bath at 70°C for 15 min. Subsequently, cells were chilled on ice and mixed with 1.5 ml deionized water. For extraction of metabolites, 1 ml of chloroform were added and samples were mixed vigorously. After phase separation the upper polar phase was collected and dried in a vacuum concentrator at 15°C overnight.
For extracellular metabolome analysis, supernatants were mixed with 20 μl ribitol (c = 0.2 g⋅l–1) as internal standard and dried in a vacuum concentrator overnight.
For extraction of Coenzyme A (CoA) derivatives, cells corresponding to 10 mg cell dry weight were harvested and resuspended in 1 ml methanol, containing 0.2 mg⋅l–1 biochanin A as internal standard. Cells were lysed using a Precellys 24 homogeniser (Peqlab, Germany) at −10°C with three cycles of homogenization (6,800 rpm, 30 s, with equivalent breaks). Afterward, the lysate was transferred to ice cold ammonium acetate solution (25 mM, pH 6) and cleared by centrifugation. CoA derivatives were extracted by solid phase extraction, using Strata XL-AW solid phase extraction columns (Phenomenex, Germany). Columns were sequentially equilibrated using 1 ml methanol, followed by 1 ml methanol:H2O:formic acid (50:45:5) and 1 ml H2O. After loading the cell lysate onto the column it was washed with 1 ml ammonium acetate (25 mM, pH 7.2) and dried at 700 mbar for 3 min. CoA derivatives were eluted using 1 ml methanol containing 5% (v/v) ammonia. Extracts were dried in a vacuum concentrator at 15°C overnight. For HPLC-MS analysis dried extracts were dissolved in 0.2 ml sample buffer (25 mM ammonium acetate pH 3.5, 2% (v/v) methanol).
GC-MS Measurement of Intra- and Extracellular Metabolites and HPLC-MS Measurement of CoA Derivatives
Metabolites for intra- and extracellular metabolome analysis were derivatized, following the protocol described by Zaparty et al. (2010). Briefly, dried extracts were dissolved in 40 μl 20 mg⋅ml–1 methoxyamine hydrochloride in pyridine and incubated for 90 min at 30°C. Afterward 60 μl N-methyl-N-(trimethylsilyl)-trifluoro-acetamide was added and incubated for 30 min at 37°C, followed by 120 min at 25°C. After centrifugation 60 μl of derivatized extracts were transferred into glass vials for GC-MS analysis.
Metabolites were analyzed using a Leco Pegasus 4D GCxGC TOFMS (Leco, Germany), equipped with a 30 m, 0.25 mm Zebron ZB-5 ms column (Phenomenex, Germany). Ionization of analytes was performed by positive electron ionization (EI+). Gas chromatography was performed using a helium gas flow of 1.2 ml⋅min–1, using the following temperature gradient: 0.02 min at 70°C, increase to 330°C with 12°C⋅s–1, constant temperature period at 330°C for 5 min. Full scan mass spectra were recorded from 45 to 600 m/z.
Phenol consumption was monitored on a Thermo GC Ultra coupled to a DSQ II mass spectrometer Thermo Fisher Scientific, Schwerte, Germany). Chromatography was performed as described previously (Neumann-Schaal et al., 2015) using an Agilent VF-WAX-MS column (30 m, 0.25 mm diameter; Agilent, Santa Clara, United States) with the following temperature gradient: 1 min at 55°C, increase to 245°C with 10°C⋅min–1, constant temperature period at 245°C for 2 min. Full scan mass spectra were recorded from 20 to 400 m/z.
Analysis of CoA derivatives was performed on a Dionex ultimate 3,000 system (Thermo Fisher Scientific Inc., Schwerte, Germany) coupled to a Bruker MicroTOF QII mass spectrometer (Bruker Daltonik GmbH, Bremen Germany). Chromatography was performed based on the protocol described by Peyraud et al. (2009), using a C18 analytical column (Gemini 150 × 2.0 mm, particle size 3 μm; Phenomenex, Aschaffenburg, Germany). At a constant temperature of 35°C and a flow rate of 220 μl⋅min–1 the following gradient was used: 1 min 5% B (methanol): 95% A (50 mM ammonium formiate, pH 8.1); 18 min 30% B: 70% A; 7 min 95% B: 5% A; 4 min 95% B: 5% A. MS analysis was done in positive ESI mode with 3 Hz data acquisition and automated MS2 acquisition. Mass spectra were recorded from 90 to 1178 m/z.
Data Processing and Statistical Analysis
Data analysis was performed as described previously (Wolf et al., 2016). Briefly, GC-MS data was processed using the MetaboliteDetector software for automated peak detection and deconvolution (Hiller et al., 2009; Zech et al., 2013). Compounds were identified by comparison of mass spectra and retention indices to a compound library which was created by merging our in-house library with the Golm metabolome database (Kopka et al., 2005). Peak areas were normalized to the internal standards and peak areas of different derivatives were summarized to the corresponding metabolites. Non-biological and artificial peaks were eliminated by the aid of blank samples. Finally, the data was normalized using a central normalization procedure to the reference D-glucose.
Internal mass calibration with the sodium formiate cluster and export of HPLC data to the mzXML format was performed using the Bruker DataAnalysis software (Bruker Daltonik GmbH, Bremen, Germany). Raw data was processed using the XCMS package for R (Smith et al., 2006; Benton et al., 2008; Tautenhahn et al., 2008). Peak identification was performed using the accurate masses of the [M+2H]2+ ions and retention times of synthetic CoA standards. If there was no synthetic standard available for a certain compound, both the calculation of the molecular mass from [M+2H]2+ and [M+H]+ ions and sum formula predictions using the accurate masses and the isotopic pattern were used for identification. MS2 fragmentation was used to confirm the presence of the CoA moiety. Peak areas were normalized by central normalization to the reference condition.
Statistical significant differences for all metabolites were determined by non-parametric Kruskal-Wallis test (Kruskal and Wallis, 1952), using the Benjamini–Yekutieli correction to control the false discovery rate (Benjamini and Yekutieli, 2001). Changes with a p < 0.01 were considered to be significant.
RNA Isolation, Sequencing, and Data Analysis
RNA was extracted from frozen cell pellets using Trizol (Thermo Fisher Scientific, United States) as described earlier (Hottes et al., 2004). Afterward, ribosomal RNA was depleted using the RiboZero magnetic kit for bacteria (Epicenter, United States) according to the manufacturer’s protocol with slight modifications: only 90 μl of magnetic beads were used. For the rRNA removal reaction 1 μg was mixed with 4 μl removal solution in a total volume of 20 μl. Sequencing libraries for all samples and replicates were prepared with the TruSeq® Stranded mRNA HT kit (Illumina) starting with the RNA fragmentation step after elution of precipitated RNA in 19 μl of the Fragment-Prime-Mix. Sequencing was performed on a HiSeq1500 instrument (Illumina) in rapid mode with a read length of 2 × 25 nt. Sequencing reads were mapped with Bowtie2 (Langmead and Salzberg, 2012) against the reference genome (Saccharolobus solfataricus P2, genome size: 2,992,245 nt, RefSeq ID: NC_002754.1) and calculation of mapped reads per gene were done using ReadXplorer (Hilker et al., 2014). Subsequently, RPKM values were computed for each gene (Mortazavi et al., 2008). To reduce influence of global and systemic changes on transcript abundance the data was additionally normalized by a central normalization procedure. To distinguish between background activity and active gene expression a mean log intensity (A-value, Dudoit et al., 2002) cut-off value of 2 was applied. Furthermore, only genes with an at least fourfold difference between both conditions and a RPKM value higher than the median RPKM value of the whole dataset (median RPKM = 69 ± 5.2) were included into the analysis. Statistical analysis was performed using DeSeq (Anders and Huber, 2010) and changes with a p < 0.01 were considered to be significant.
Metabolic Modeling
The published metabolic model of S. solfataricus (Wolf et al., 2016; Stark et al., 2017) was used to elucidate changes in metabolic fluxes during growth on phenol. A glucose standard scenario was chosen as reference condition. Phenol degradation pathways were revised. Because of their importance in this study, the protons pumped by the two terminal oxidases SoxABCD and SoxM were also revised by the use of primary literature (Gleißner et al., 1997; Komorowski et al., 2002). The biomass composition for growth on phenol was added according to experimental data. The amount of polyamines in S. solfataricus grown on glucose was calculated from Giles and Graham (2008). The relative changes in metabolic polyamine levels were used to calculate the polyamine content for the growth on phenol. Transcriptomic and metabolomic measurements were included by limiting the metabolic fluxes within the model.
Metabolic modeling was performed using the metabolic modeling toolbox MMTB1. The metabolic model used during this study as well as glucose and phenol scenarios were uploaded to the MMTB platform to enhance further usage by the community. Data were visualized using MetaboMAPS (Helmecke et al., 2020). Swiss-Model was used to model the 3D-structure of proteins.
Results
Phenotypic Characterization of S. solfataricus P2 Growing on Phenol as Sole Carbon Source
Basic physiological parameters of S. solfataricus P2 were determined using Brock minimal medium (Brock et al., 1972) containing 10 mM phenol as sole carbon source. No growth was observed at phenol concentrations above 14 mM (Supplementary Material S1), indicating toxicity of the substrate at higher concentrations. Data for comparison with glucose as substrate (growth curve, substrate uptake rate and biomass composition) were published previously (Wolf et al., 2016). Overall, S. solfataricus showed a reduced growth rate and a lower biomass yield on 10 mM phenol compared to growth on 22 mM D-glucose (Supplementary Material S2).
Although the maximum substrate uptake rate of S. solfataricus P2 grown on phenol is comparable to growth on glucose, phenol has shown to be only a poor source of carbon and energy as a considerably decreased growth rate and less efficient integration of carbon into the biomass was observed (Table 1).
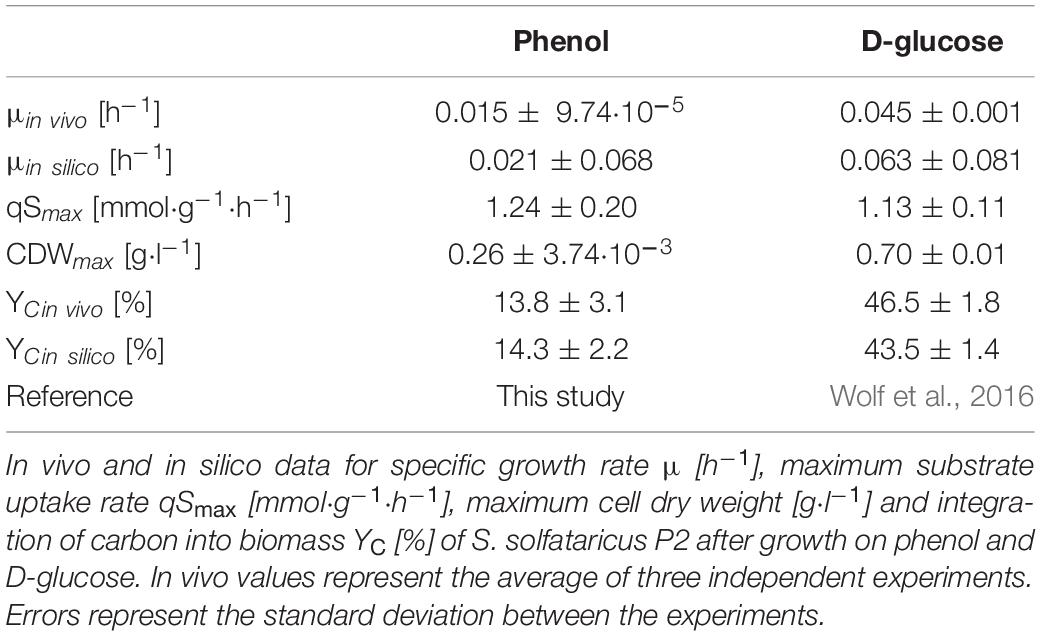
Table 1. Comparison of in vivo and in silico growth parameters.
To improve our metabolic model of S. solfataricus P2 for modeling phenol metabolism, we further analyzed the macromolecular composition of the organism after growth on phenol (Supplementary Material S3), as it has been shown that biomass composition could strongly depend on the environmental conditions (Neidhardt, 1963). Phenol grown cells show a similar composition of the nucleic acid fractions as compared to cells grown on D-glucose (RNA: 5.4 ± 0.5%, DNA: 1.1 ± 0.1%; Wolf et al., 2016). However, protein content of phenol grown cells is strongly decreased to only 47 ± 2.3% (D-glucose protein content: 64.3 ± 3.4%).
To investigate if the secretion of carbon containing compounds contributes to the poor conversion of carbon into biomass, we analyzed supernatants of S. solfataricus P2 after growth on phenol by GC-MS. Only small amounts of hydroquinone and oxalate were identified in the supernatants during the exponential growth phase (Supplementary Material S4). The oxalate amount in the media increases slightly in the stationary phase, whereas the hydroquinone concentration decreases. In contrast, none of these compounds could be detected under glucose growth conditions.
Estimating the conversion of carbon into biomass during phenol degradation using the metabolic model, results in an incorporation value of 14.3% (Table 1), which is consistent with the experimental findings. The model predicted twice as much oxygen consumption in simulations with phenol (15.0 mmol/gCDW/h) compared to glucose (7.2 mmol/gCDW/h). The respiratory activity increased in a similar manner.
The Metabolic Response of S. solfataricus P2 to Phenol
To monitor changes in the metabolic activity of S. solfataricus P2 after growth on phenol as sole carbon source, we compared the intracellular metabolite proportions in the late exponential growth phase to cells grown on D-glucose as reference (Figure 1, Table 2, and Supplementary Material S5).
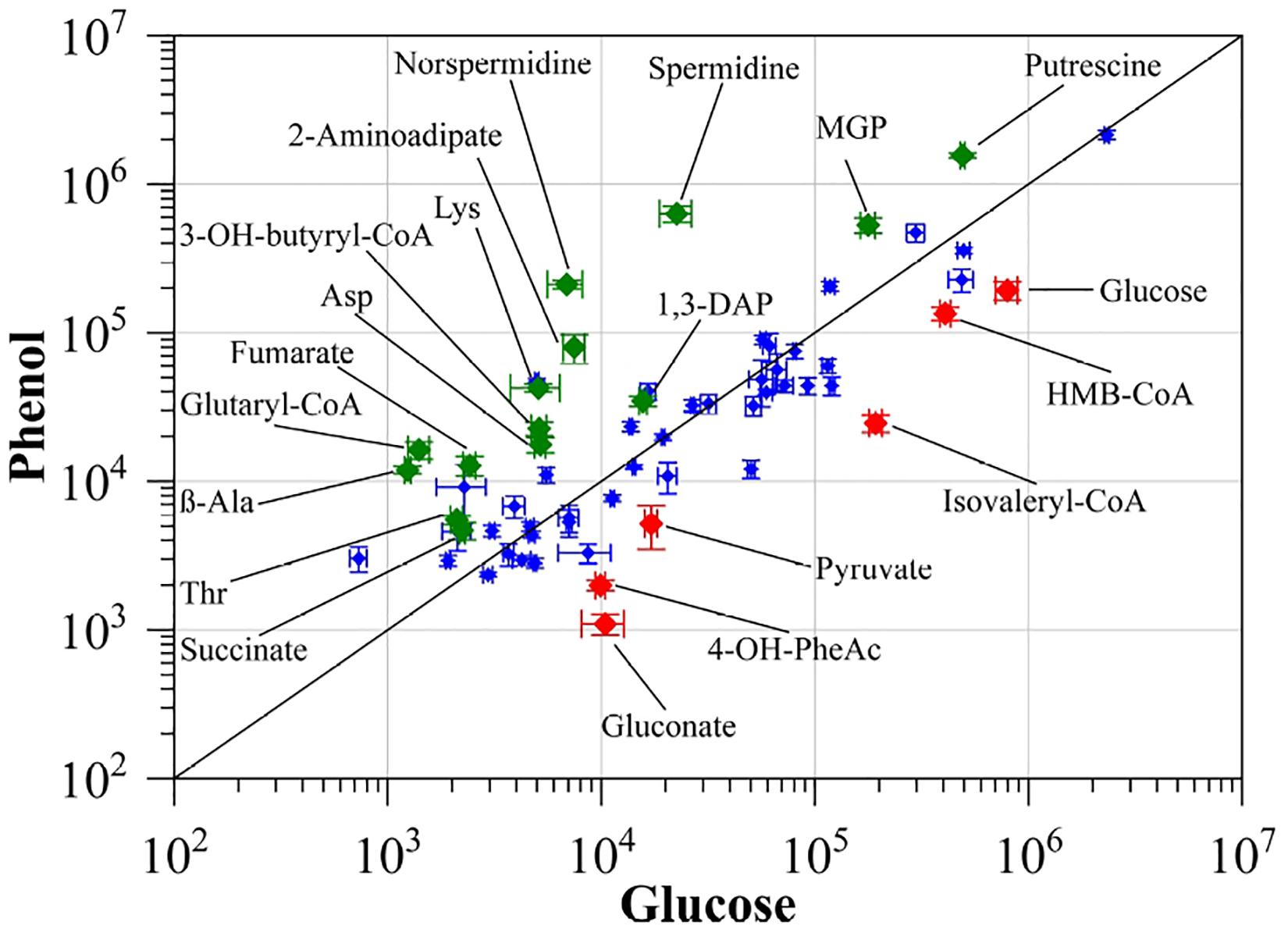
Figure 1. Comparison of intracellularly detected metabolites of S. solfataricus P2 grown on either phenol or D-glucose as sole carbon source. Normalized peak areas were plotted on a double logarithmic scale. Green diamonds: more abundant in cells grown on phenol, red diamonds: more abundant in cells grown on D-glucose. Only identified metabolites with significantly altered concentrations (p < 0.01) and at least twofold differences between both conditions were labeled. Values represent the average of three independent cultivations. Errors represent the standard error between the experiments. 1,3-DAP, 1,3-diaminopropane; MGP, methyl-α-D-glucopyranoside; HMB-CoA, 3-hydroxy-2-methylbutanoyl-CoA; 3-OH-butanoyl-CoA, 3-hydroxybutanoyl-CoA; 4-OH-PheAc, 4-hydroxy-phenylacetate.

Table 2. Overview of detected metabolites of S. solfataricus P2 after growth on phenol.
The most obvious metabolic changes after growth on phenol were observed for the components of the glycolytic Entner-Doudoroff pathway and gluconeogenesis (Figure 2 and Table 3). We observed strongly decreased amounts not only for glucose, but also for gluconate, pyruvate and 2-keto-3-deoxygluconate, with the latter being only detected under glucose reference conditions (Table 3). Furthermore, the phosphorylated sugars, fructose-6-phosphate and glucose-6-phosphate, which belong to the gluconeogenic Embden-Meyerhof-Parnas pathway, as well as several other sugars (e.g., ribulose, xylulose, fructose) were also detected only with glucose as sole carbon source (Figure 2 and Table 3).
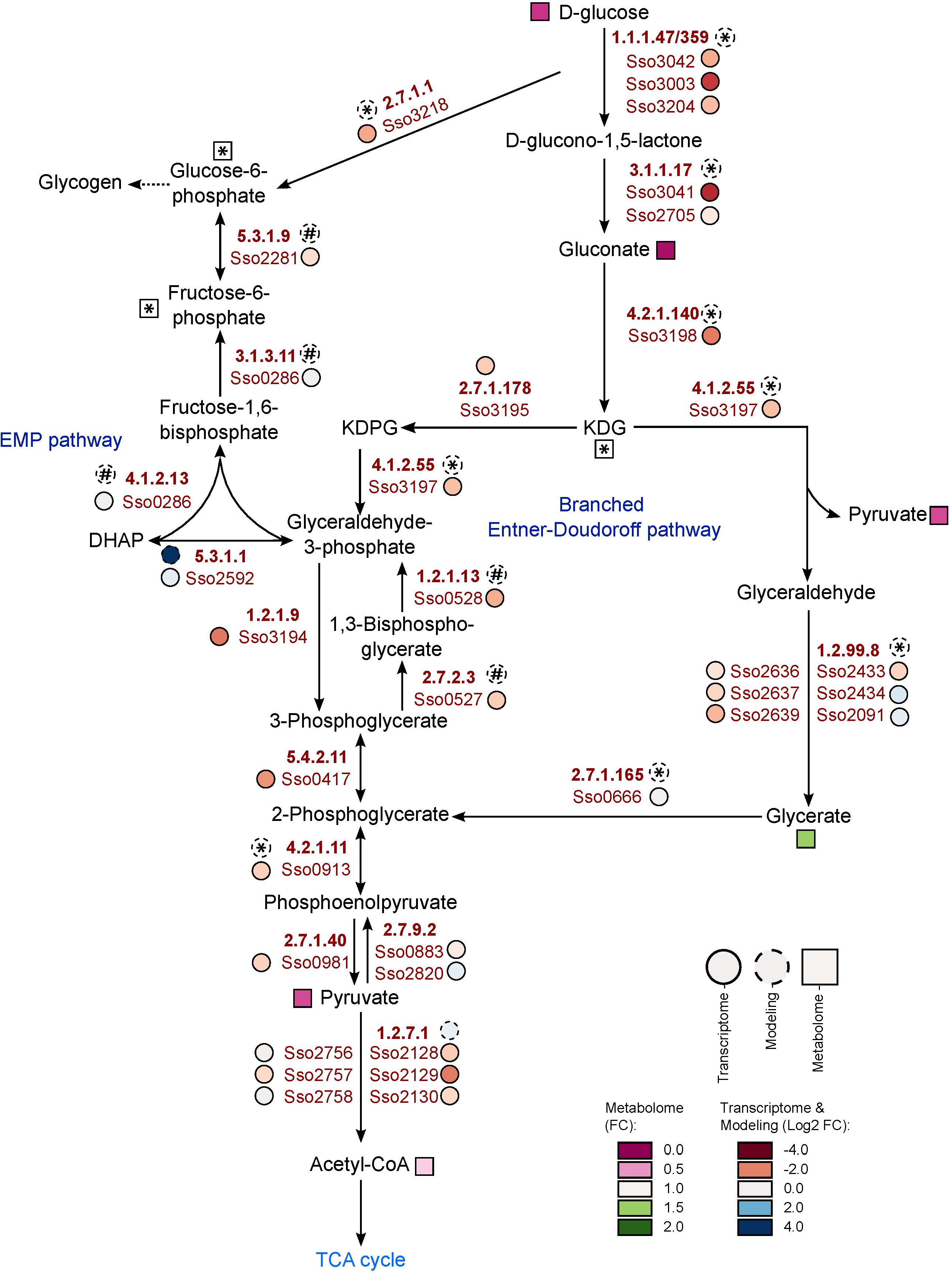
Figure 2. Metabolic and transcriptomic changes within the Entner-Doudoroff pathway and the gluconeogenesis during growth of S. solfataricus P2 on phenol Relative metabolite concentrations (squares) were expressed as fold-changes compared to the reference D-glucose. Transcript abundance (solid circles) and flux distributions (dashed circles) were expressed as Log2 fold-changes compared to the reference D-glucose. Asterisks: Data are only available under glucose conditions. Hash: Data are only available under phenol conditions. Dotted arrows indicate multiple enzymatic reactions. KDG, 2-keto-3-deoxygluconate. This pathway is available at MetaboMAPS (accession: P101).
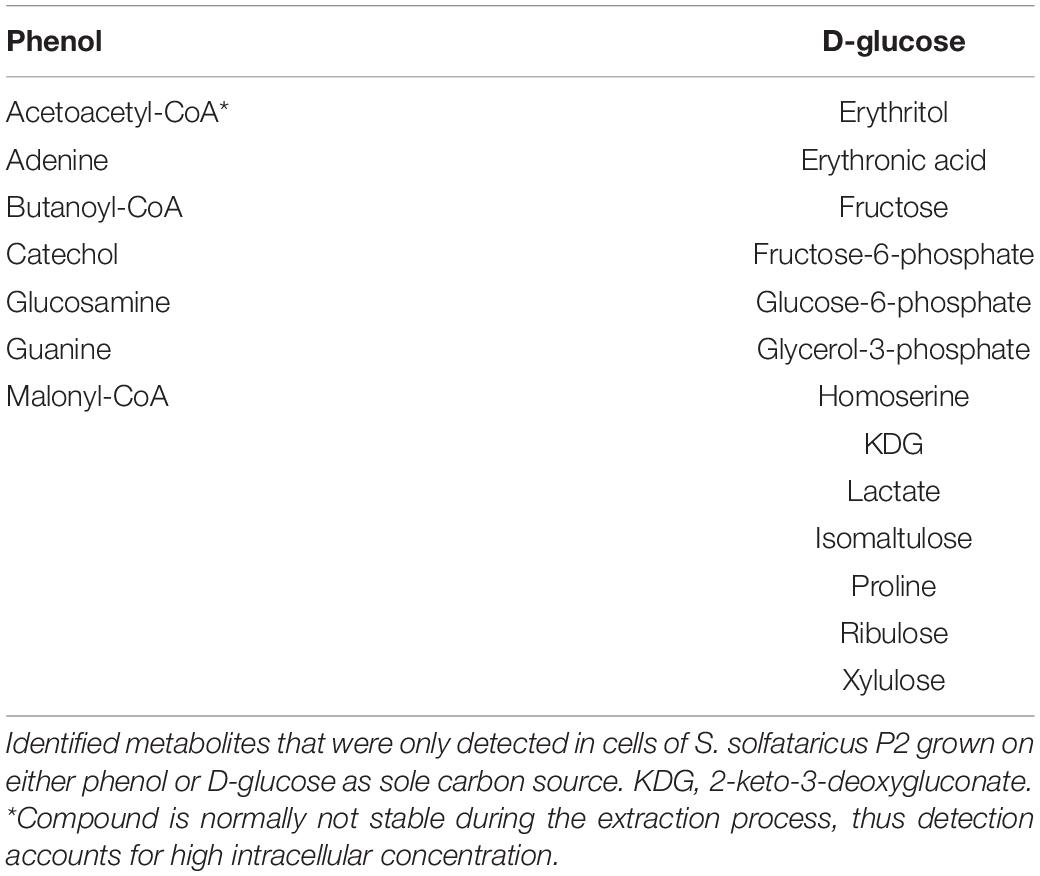
Table 3. Phenol or D-glucose specific metabolites.
As degradation of phenol finally leads to formation of pyruvate and acetyl-CoA (Shingler et al., 1992; Sridevi et al., 2012), the TCA cycle represents the entry point to central carbon metabolism for this carbon source. From the metabolites of the TCA cycle we observed significantly increased amounts only for succinate (2.09-fold) and fumarate (5.27-fold). Malate and citrate show increased concentrations by more than twofold with respect to the glucose reference, but these changes were not statistically significant (Figure 3).
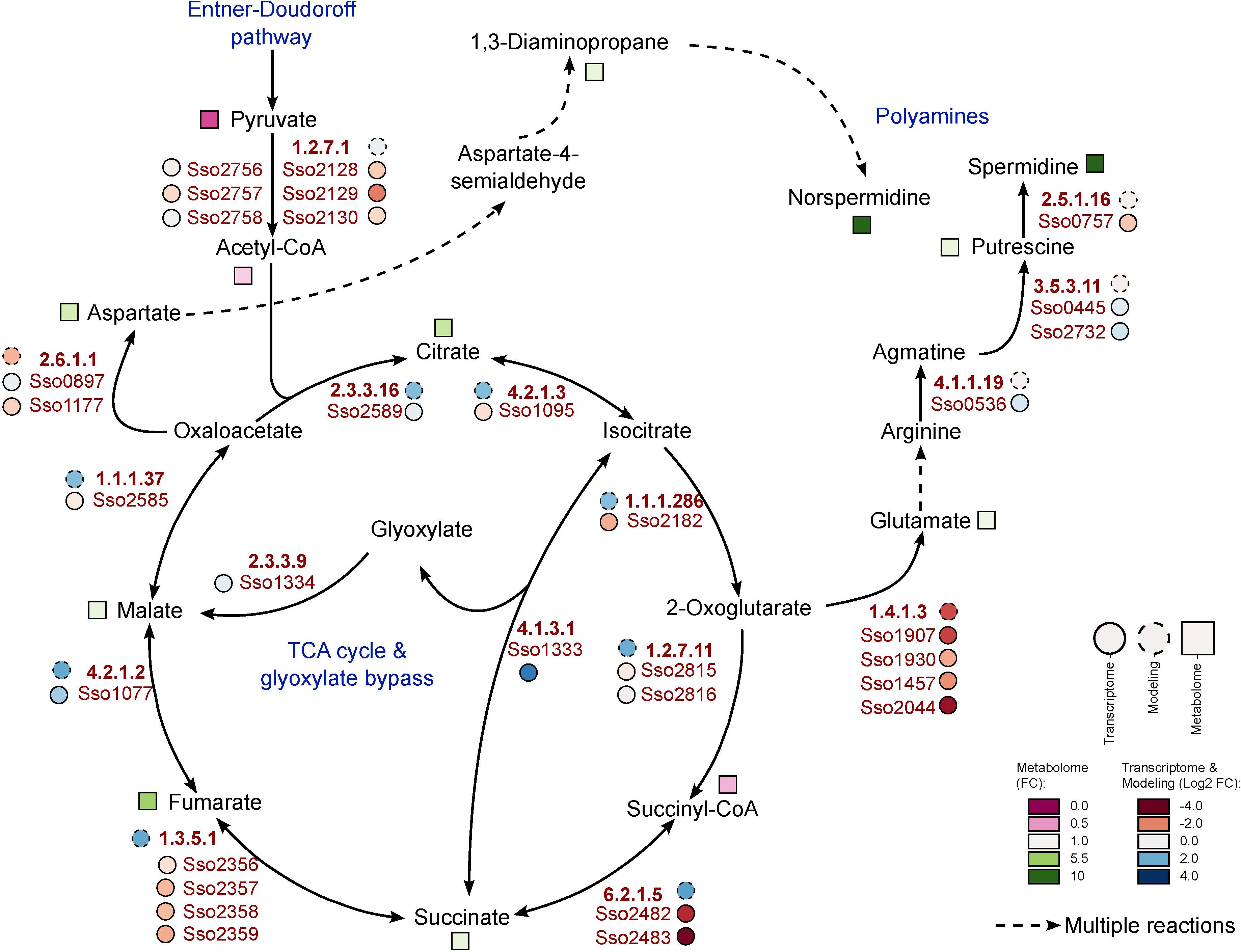
Figure 3. Metabolic changes within the TCA cycle during growth of S. solfataricus P2 on phenol. Relative metabolite concentrations (squares) were expressed as fold-changes compared to the reference D-glucose. transcript abundance (solid circles) and flux distributions (dashed circles) were expressed as Log2 fold-changes compared to the reference D-glucose. Dotted arrows indicate multiple enzymatic reactions. Note that the fold-changes of spermidine and norspermidine exceeds the color scale with values of 28 and 30, respectively. This pathway is available at MetaboMAPS (accession: P103).
In addition to the changes in the central carbon metabolism, we observed noticeable changes in polyamine formation if S. solfataricus P2 is cultivated with phenol as sole carbon source. Under these conditions, the relative concentrations of spermidine and norspermidine were increased by approximately 28- and 31-fold, as compared to the reference condition. Furthermore, elevated, but less pronounced levels were also found for 1,3-diaminopropane and putrescine (Figure 3).
In addition to small polar metabolites, we also monitored changes in the coenzyme A content of the cells between the different growth conditions. If S. solfataricus P2 grows on phenol as carbon source, we found a strong accumulation of glutaryl-CoA (11.57-fold) and 3-hydroxy-butanoyl-CoA (4.4-fold). Furthermore, butanoyl-CoA, malonyl-CoA and acetoacetyl-CoA could only be detected under phenol growth conditions (Table 3). It should be noted, that acetoacetyl-CoA is normally not stable during the extraction process, thus the detection of this compound in phenol cell extracts accounts for a high intracellular concentration. In conjunction with an increased coenzyme A biosynthesis, we also found the precursor beta-alanine (Genschel, 2004; Tomita et al., 2014) accumulating during growth of S. solfataricus P2 on phenol (Figure 1).
Finally, decreased concentrations were found for 3-hydroxy-2-methylbutanoyl-CoA (0.33-fold) and isovaleryl-CoA (0.13-fold) (Figure 1), which are both connected to amino acid degradation in S. solfataricus P2 (Stark et al., 2017).
Global Transcriptomic Changes of S. solfataricus P2 During Growth on Phenol
Amongst others, phenol is formed during lignin degradation and toxic to most organisms. Its utilization as a carbon source is less common and requires a complex metabolic adaption. To investigate the cellular changes of this adaption process, we analyzed the global transcriptomic response of S. solfataricus P2 to growth on phenol, using RNASeq analysis and growth on D-glucose as reference condition. From the 3,213 genes encoded on the S. solfataricus P2 genome, we could detect 2,760 genes to be actively transcribed under both growth conditions. Out of these, 274 genes showed significantly altered abundance with changes of at least fourfold (log2FC > 2, p <0.01) (Table 4 and Supplementary Material S6).
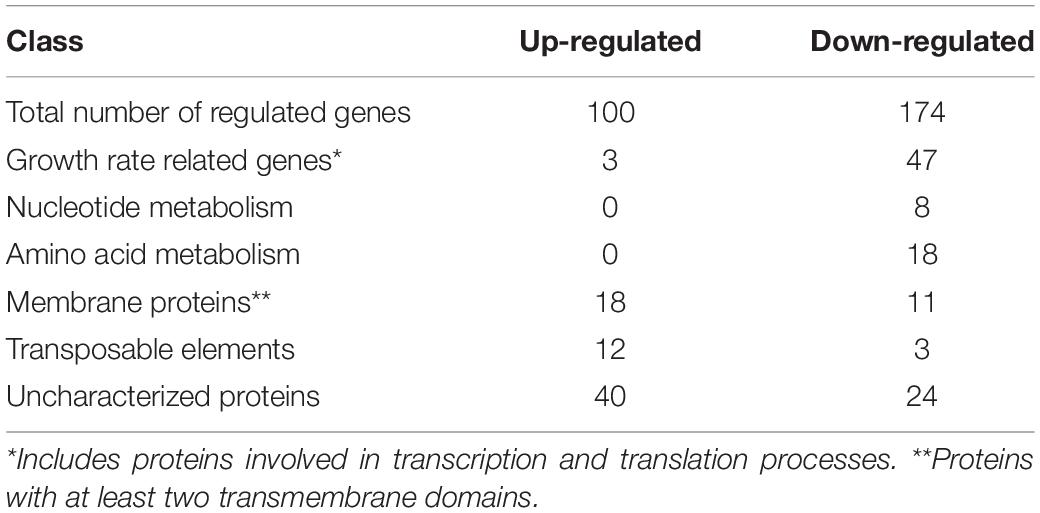
Table 4. Overview of differential expressed protein classes in S. solfataricus P2 after growth on phenol.
The majority of the 100 up-regulated genes found under phenol growth conditions belong either to membrane proteins, mobile elements or uncharacterized proteins (Table 4), In contrast, the majority of the 174 down-regulated transcripts are associated with growth rate related genes, including ribosomal proteins, tRNA synthetases and enzymes that are involved in replication, translation and transcription processes (Table 4). Further a strong down regulation of genes belonging to amino acid and nucleotide biosynthesis was observed.
Among the strongly induced transcripts observed under phenol growth conditions, we found four of the seven subunits of a monooxygenase gene cluster (Sso1225–Sso1233) showing significant differential expression with fold-changes ranging from 59 up to 384-fold (Table 5). The remaining three subunits of these gene cluster also show high transcription levels compared to growth on D-glucose (fold-change 10 up to 73-fold), but due to higher standard errors, these changes were not statistically significant (Table 5). Interestingly, together with the catechol-2,3-dioxygenase (Sso1223), this gene cluster has already been shown to be involved in phenol degradation in S. solfataricus (Izzo et al., 2005). Furthermore, several genes located upstream of the monooxygenase gene cluster also show strong induction under phenol growth conditions, including an alcohol dehydrogenase (Sso1220), two acetoacetate decarboxylase related enzymes (Sso1221 and Sso1222) and methylmalonate-semialdehyde dehydrogenase (Sso1218).
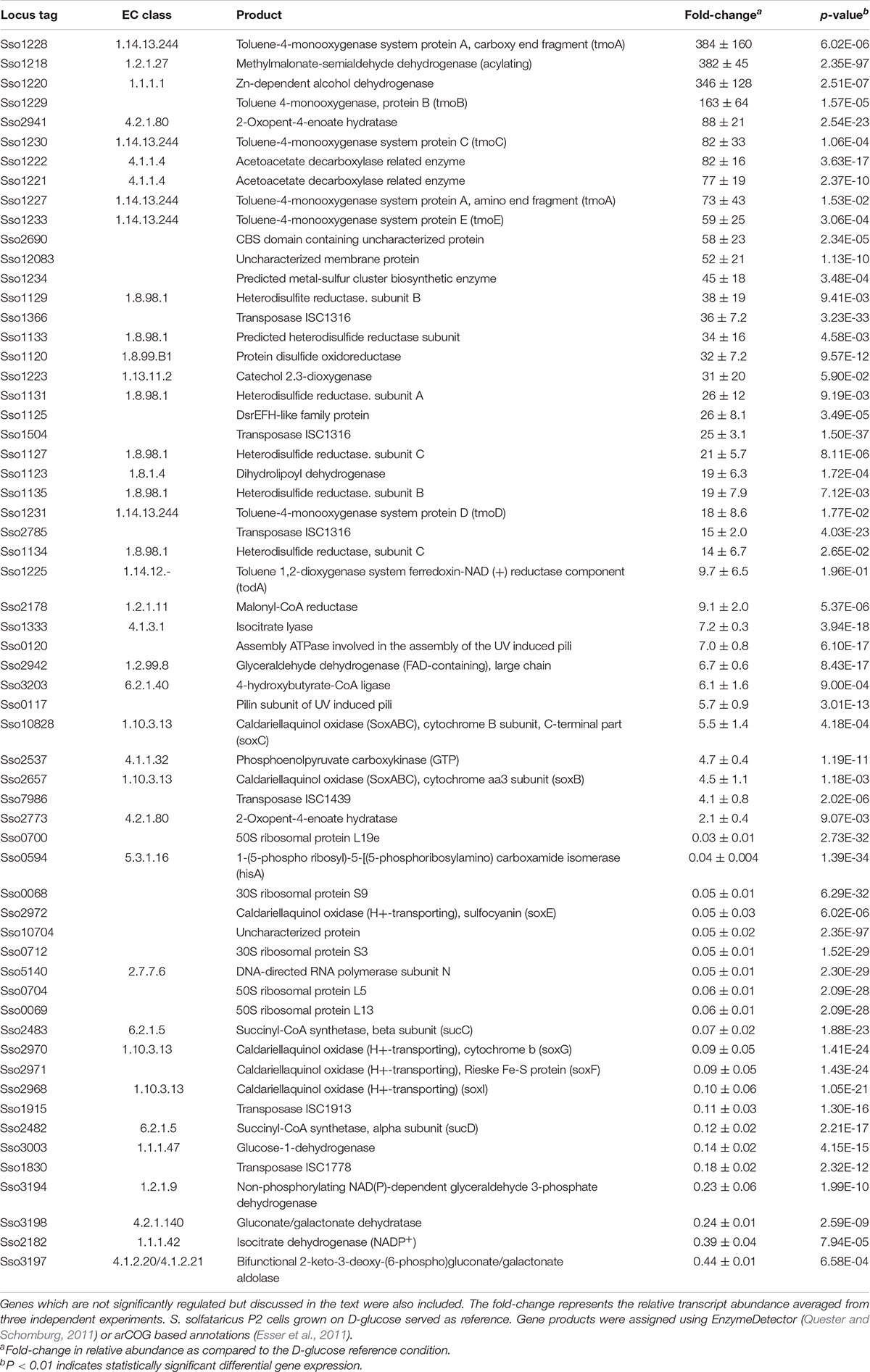
Table 5. Overview of the most differentially expressed genes in S. solfataricus P2 cells after growth on phenol.
Oxygen availability is a crucial factor for phenol metabolism (Comte et al., 2013), as it is the co-substrate of the monooxygenase, catalyzing the first step in phenol degradation. Interestingly, we found differential regulation in the expression of two different quinol oxidase complexes in S. solfataricus P2. Whereas the quinol oxidase complex SoxABC (Sso2656–Sso2662 and Sso10828) was significantly induced under phenol growth conditions (fold-changes: 3.1–7.1), the second quinol oxidase complex SoxM (Sso2968–Sso2973) showed a strong decrease in relative abundance, compared to the D-glucose reference condition (Table 5 and Supplementary Material S6). In addition, transcription of genes involved in the glycolytic Entner-Doudoroff pathway like the glucose-1-dehydrongease (Sso3003) or the GAP-dehydrogenase (Sso3194; Table 5) was significantly decreased.
We further found a gene cluster comprising the loci Sso1120–Sso1135 (fold-change 12–34) among the upregulated transcripts. Next to some proteins with unknown function, this gene cluster encodes for a putative heterodisulfide reductase (Sso1127 -Sso1135), a protein-disulfide oxidoreductase (Sso1120; Limauro et al., 2014) and a putative dihydrolipoyl dehydrogenase (Sso1123).
Furthermore, the data indicates a high plasticity of the genome, if S. solfataricus P2 is grown on phenol as carbon source. In total twenty-two transposable elements showed increased transcription under these conditions (Tables 4, 5 and Supplementary Material S6), although only 12 show expression levels above the median RPKM of the complete dataset. In contrast, during growth on D-glucose only three mobile elements show significantly increased expression (Table 4 and Supplementary Material S6). Finally, we found an increased transcription of the UV-inducible pili operon (Sso0117- Sso0121; fold-change 2.8–7.0) during growth on phenol, although two of the five subunits still show low relative expression levels (Supplementary Material S6).
Discussion
Global Response of Saccharolobus solfataricus P2 to Phenol and Metabolic Modeling
Compared to growth on glucose, phenol represents a poor source of carbon and energy for Saccharolobus solfataricus P2, as growth rate and biomass production were strongly decreased on this substrate (Table 1). A similar growth rate for batch cultures of S. solfataricus grown on phenol has been reported previously (Izzo et al., 2005). Furthermore, it has been shown from bioreactor cultivations that the growth rate of the organism on phenol is strongly dependent on the concentration of dissolved oxygen and decreases with reduced oxygen supply (Comte et al., 2013). Considering the fact that oxygen is the co-substrate of the mono- and dioxygenase catalyzing the first steps in phenol breakdown, it is evident that a sufficient oxygen supply is a crucial factor for efficient phenol degradation.
The strongly reduced growth rate during growth with phenol as carbon source is also reflected in the transcription of several growth rate related genes, as we found strongest down regulation of different ribosomal genes, tRNAs synthetases and genes involved in purine and amino acid metabolism (Tables 4, 5 and Supplementary Material S6). Interestingly, nearly all down regulated genes belonging to purine biosynthesis are involved in ATP consuming reactions. This has also been reported for Bacillus subtilis and P. putida during phenol exposure (Santos et al., 2004; Tam et al., 2006), suggesting that growth rate reduction prevents the organisms of consuming ATP for nucleotide biosynthesis and cell division and rather channeling the available energy to processes crucial for phenol adaptation.
Next to the reduced growth rate, we also observed a less efficient conversion of carbon into biomass if S. solfataricus is grown on phenol (13.8 ± 3.1%, Table 1). The carbon distribution is in concordance with that reported from Christen et al. (2011), if glucose adapted cells were grown with phenol as sole carbon source, indicating a higher respiratory activity on this substrate.
In conjunction with an increased oxygen demand, we found that phenol influences the expression of different quinol oxidase complexes in S. solfataricus as the SoxABC complex is induced, whereas a second quinol oxidase complex (SoxM) showed a strongly decreased abundance (Table 5 and Supplementary Material S6). A similar expression pattern has been previously reported when S. solfataricus is exposed to low oxygen concentrations (Simon et al., 2009). This indicates a higher oxygen affinity of the SoxABC complex, which is linked to a smaller number of protons pumped via the membrane per electron transferred (Gleißner et al., 1997; Komorowski et al., 2002). Thus, increased expression of this complex under phenol growth conditions emphasizes the importance of sufficient oxygen supply for efficient phenol utilization.
The metabolic model of S. solfataricus considers transcriptomic and metabolic changes that are determined in this study, meaning it takes the different quinol oxidases into account. The model is in consent with in vivo studies by showing a higher oxygen consumption in simulations with phenol. 16.5% of the oxygen is already used for the oxidation of phenol to catechol. The model showed also a higher respiratory activity leading to ∼40% more oxygen consumption than in glucose simulations and a lower incorporation of carbon into the biomass. The low carbon yield and growth rate on phenol results from a higher demand of oxygen which leads to the usage of a less efficient terminal oxidase and further decreases the ATP yield per substrate.
Reconstruction of Phenol Degradation in S. solfataricus P2
Previous studies already predicted the phenol meta-cleavage pathway to be active in S. solfataricus growing on phenol (Izzo et al., 2005; Comte et al., 2013). On the metabolic level clear evidence for the activity of this pathway suffers from the missing commercial availability of intermediates that could serve as standards for mass spectra libraries. Only catechol, cis, cis-muconate and 3-oxoadipate are available, the latter two compounds representing intermediates of the ortho-cleavage pathway (Ornston, 1966; Feist and Hegeman, 1969). With the exception of catechol none of these intermediates could be detected in our experiments, indicating a minor importance of the ortho-route in this thermophilic archaeon.
Independent of which route for phenol breakdown is used, the initial step is the transformation to catechol, which requires the presence of an active monooxygenase. A putative monooxygenase gene cluster (Sso1225–Sso1233) involved in this reaction has already been identified by Izzo et al. (2005). Accordingly, we found that the expression of all genes of this cluster was strongly induced under phenol growth conditions (Figure 4, reaction 1).
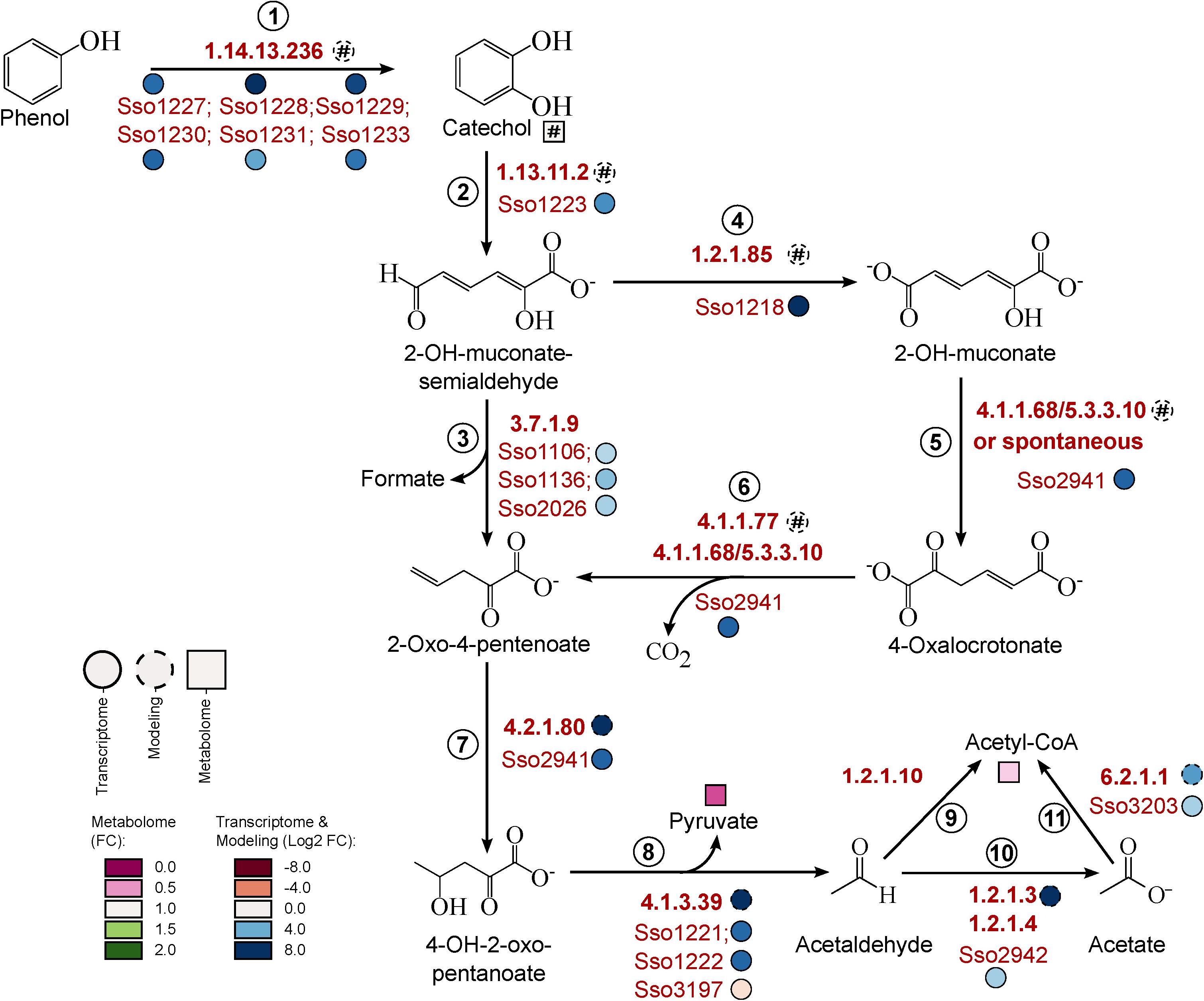
Figure 4. Refinement of phenol meta-degradation pathway in S. solfataricus P2. For simplicity, additional Co-substrates and Co-factors were not shown. Proteins predicted to be involved in phenol degradation were indicated by the corresponding gene loci. Relative metabolite concentrations (squares) were expressed as fold-changes compared to the reference D-glucose. Transcript abundance (solid circles) and flux distributions (dashed circles) were expressed as Log2 fold-changes compared to the reference D-glucose. Hash: Data are only available under phenol conditions. 2-OH-muconate-semialdehyde, 2-hydroxymuconate-semialdehyde; 2-OH-muconate, 2-hydroxymuconate; 4-OH-2-oxo-pentanoate, 4-hydroxy-2-oxo-pentanoate. This pathway is available at MetaboMAPS (accession: P100).
The following transformation of catechol to 2-hydroxymuconate-semialdehyde requires a ring opening reaction by catechol-2,3-dioxygenase. The corresponding homolog in S. solfataricus (Sso1223) has also been characterized before (Izzo et al., 2005; Chae et al., 2007) and is located in the same gene cluster as the monooxygenase. Concordantly, we observed an increased expression of this gene under phenol growth conditions (Figure 4, reaction 2, Table 5). In addition to the characterized protein a second homolog of the dioxygenase exists on the S. solfataricus genome (Sso2054, Izzo et al., 2005), but this gene showed very low expression levels under both, phenol and glucose growth conditions (Supplementary Material S6), suggesting no specific importance of this protein for phenol degradation.
2-Hydroxymuconate-semialdehyde is then degraded to 2-oxo-4-pentenoate, which could be achieved by two different metabolic routes (Figure 4, reactions 3–6). For none of these reactions the participating enzymes in S. solfataricus are known, because no homologous to known bacterial or eukaryotic enzymes could be identified in the genome. The direct conversion requires hydrolase activity (Figure 4, reaction 3). We found three putative hydrolases to be induced under phenol growth conditions compared to the glucose reference (Sso1106, Sso1136, Sso2026). However, the expression level for these genes were very low under both growth conditions (Supplementary Material S6), indicating a minor importance of the hydrolase cleavage of 2-hydroxymuconate-semialdeyhde. An alternative pathway for conversion of the semialdehyde involves the oxidation to 2-hydroxymuconate (Figure 4, reaction 4). Previously, Comte et al. (2013) reported the formation of small amounts of 2-hydroxymuconate and 4-oxalocrotonate in batch cultures of S. solfataricus, providing evidence for the activity of this branch. The genome encodes for five members of the aldehyde dehydrogenase superfamily: Two of them, Sso3117 and Sso3194, have been extensively studied and proven to be involved in different sugar degradation pathways (Brouns et al., 2006; Ettema et al., 2008). The remaining candidates (Sso1218, Sso1629, and Sso1842) show approximately 30% identity to characterized 2-hydroxymuconate-semialdehyde dehydrogenases of Pseudomonas sp. (Inoue et al., 1995). For Sso1629 and the previously characterized succinate-semialdehyde dehydrogenase (Sso1842) (Esser et al., 2013) no change in transcript abundance was observed in response to carbon source (Supplementary Material S6).
The most promising candidate is Sso1218 based on transcriptomic data, which showed a 382-fold increase in expression in cells grown on phenol (Table 5). Moreover, this gene is located in the same cluster as the mono- and dioxygenase. Interestingly, Sso1218 has previously been characterized as methylmalonate-semialdehyde dehydrogenase and is supposed to be strictly coenzyme A-dependent and involved in degradation of branched-chain amino acids. However, the enzyme showed an unusual low activity for its physiological substrate (Esser et al., 2013). Recently, it has been shown that branched-chain amino acids were not primary degraded via the classical routes in S. solfataricus. In the same study Sso1218 did not show differential expression during growth on amino acids (Stark et al., 2017). This indicates that not methylmalonate-semialdehyde but rather 2-hydroxymuconate-semialdehyde is the physiological substrate of this enzyme.
In the next step of the phenol meta-cleavage pathway, 2-hydroxymucoante is isomerised to 4-oxalocrotonate (Figure 4, reaction 5). This reaction has been shown to occur spontaneously in cell extracts of Azotobacter sp. and Comamonas sp. (Sala-trepat and Evans, 1971; Liu et al., 2007), so it is likely that this might also be true for S. solfataricus. However, an involvement of the strongly induced protein Sso2941 in the isomerisation reaction cannot be excluded (fold-change 88 ± 21, Table 5). This gene is currently annotated as 2-oxo-4-pentanoate hydratase and would therefore be involved in the formation of 4-hydroxy-2-oxo-pentanoate (Figure 4, reaction 7). However, Sso2941 also shows similarity to a bifunctional isomerase/decarboxylase from Escherichia coli (32% identity, e-value: 8e-22) which catalyses the isomerisation of 5-carboxymethyl-2-hydroxymuconate as well as the subsequent decarboxylation to 2-oxo-hetp-4-enedioate (Jervis et al., 1996). In E. coli these reactions are part of the 4-hydroxyphenylacetate meta-degradation pathway, which is closely related to the catechol pathway. Furthermore, Sso2941 belongs to the fumarylacetoacetate hydrolase family, which is also responsible for the decarboxylation of 4-oxalocrotonate (Figure 4, reaction 6). Thus, suggesting that Sso2941 may be involved in different steps of the phenol meta-degradation pathway.
Finally, the cleavage of 4-hydroxy-2-oxo-pentanoate into pyruvate and acetaldehyde requires the action of an aldolase (Figure 4, reaction 8). The 2-keto-3-deoxy-(6-phospho)gluconate/galactonate aldolase (Sso3197) of S. solfataricus has been already shown to possess a broad substrate specificity (Ahmed et al., 2005; Nunn et al., 2010), and thus might be able to catalyze the cleavage of 4-hydroxy-2-oxo-pentanoate. Additionally, we observed a strong induction of two proteins of yet unknown function (Sso1221 and Sso1222, Table 5). Both proteins contain conserved domains assigning them to the acetoacetate decarboxylase superfamily. This superfamily also includes the enturacidine biosynthesis protein MppR, which acts as an aldolase, catalyzing the condensation of pyruvate and 4-imidazolecarboxaldehyde (Burroughs et al., 2013). Considering the reversible nature of aldolase reactions, we analyzed the sequences of Sso1221 and Sso1222 for their similarity to known MppR proteins and found an identity of 21–29% (E-values: 2e–14–6e–34) to the bacterial enzymes. In view of the structural diversity, archaeal proteins often differ strongly from their bacterial counterparts, thus we suggest a possible involvement of the two acetoacetate decarboxylase-related enzymes in phenol degradation. This was supported further by structural modeling using Swiss-Model: the characterized MppR protein from Streptomyces hygroscopicus [PDB:4JM3; Burroughs et al. (2013)] was selected as model template with 94% coverage and a GMQE (Global Model Quality Estimation score) of 0.64 for Sso1221 and 86% coverage and a GMQE of 0.57 for Sso1222. Both proteins showed high structural conservation especially in the center of the homo-tetramer (Supplementary Material S7).
In the last step of phenol degradation acetaldehyde has to be converted to acetyl-CoA. Based on our data, this includes the conversion of acetaldehyde to acetate with subsequent formation of acetyl-CoA (Figure 4, reactions 10 and 11). This mechanism is already proposed to be active in S. solfataricus during growth on ethanol (Chong et al., 2007). To date no acetaldehyde dehydrogenase is known in S. solfataricus, but we observed induction of a currently annotated glyceraldehyde dehydrogenase (Sso2942). The enzyme shares 34% identity to the glyceraldehyde oxidoreductase of S. tokodaii which has shown to be active on acetaldehyde (Wakagi et al., 2016). Together with the also induced 4-hydroxybutyrate-CoA ligase (Sso3203, Table 5) it appears likely, that acetaldehyde is first oxidized to acetate, before it is channeled into the TCA cycle via formation of acetyl-CoA. Finally, excess acetaldehyde could probably also be reduced by the action of an alcohol dehydrogenase, as we found a member of this enzyme amongst the most upregulated transcripts (Sso1220, fold-change 346 ± 128 Table 5). A similar up-regulation of an alcohol dehydrogenase has been previously reported for Pseudomonas sp. during growth on phenol (Collinsworth et al., 1973; Shingler et al., 1992). Produced ethanol would partially evaporate at growth temperatures of 80 °C. In addition to higher CO2 production levels, this may also contribute to the very low integration of carbon into the biomass we observed for growth of S. solfataricus on phenol (Table 1).
Phenol Induces Multiple Stress Responses in S. solfataricus P2
It is well known that benzene and its derivatives cause an increased formation of reactive oxygen species in pro- and eukaryotic cells, which could lead to oxidative damage of the DNA (Winn, 2003; Santos et al., 2004). Microorganisms which are able to use phenol as carbon source possess defense mechanisms against oxidative stress, often including the up-regulation of antioxidant proteins (Santos et al., 2004). In S. solfataricus growth on phenol induces the transcription of a gene cluster encoding for a putative heterodisulfide reductase, as well as for a protein-disulfide oxidoreductase and a putative dihydrolipoyl dehydrogenase (Table 5). The protein-disulfide oxidoreductase (Sso1120) has been characterized previously and does not to interact with peroxiredoxins from S. solfataricus (Limauro et al., 2014). However, this protein interacts with a putative dihydrolipoyl dehydrogenase (Sso1123), which has been shown to take part in an alternative antioxidant system in Mycobacterium tuberculosis (Koshkin et al., 2004). Thus it is suggested that these proteins, together with the heterodisulfide reductase may represent a system against oxidative stress in S. solfataricus in presence of elevated phenol concentrations.
In addition to antioxidant proteins, polyamines have been shown to function as radical scavengers, protecting cells from oxidative stress (Ha et al., 1998; Jung et al., 2003). We found a strong accumulation of different polyamines during growth on phenol, which was absent under the glucose reference condition (Figure 3). Thus, our results indicate that S. solfataricus P2 uses both, an antioxidant system and the accumulation of polyamines to overcome the increased oxidative stress the organism is exposed to during growth on phenol.
When S. solfataricus P2 is grown on phenol, a high plasticity of the genome is observed, as we found 22 transposable elements upregulated between 4- and 34-fold (Table 5 and Supplementary Material S6). Mobile elements normally enable rearrangement of the genome by homologous recombination which leads to loss or induction of gene activity (Syvanen, 1994). In S. solfataricus, these mobile elements are further responsible for the relatively high spontaneous mutation rate (Martusewitsch et al., 2000). S. solfataricus contains several classes of predicted mobile elements, some of them have been shown to be mobile on the experimental level while others were shown to be non-mobile (Brügger et al., 2002; Filee et al., 2007). Among the upregulated transposable elements, we found 14 sequences belonging to the ISC1316 class, which has no known function so far and which was predicted to be non-mobile (Filee et al., 2007). However, we also found members of the ISC1439 class strongly induced under phenol growth conditions. For this class mobility causing mutations has already been reported and there is evidence of mobility for other ISC classes (Brügger et al., 2002). Such a rearrangement of the genome could serve as another mechanism to overcome phenol induced stresses and toxicity, probably by inducing expression of the required proteins. A similar increase in expression of transposable elements has previously been reported for S. solfataricus after exposure to heat shock (Tachdjian and Kelly, 2006). Furthermore, genome rearrangement may also mediate DNA exchange or repair. We found an increased transcription of the gene cluster encoding for the UV-induced pili system. The formation of these pili in S. solfataricus mediates cell-cell contact and formation of cell aggregates which would allow for DNA exchange between cells, thus enabling DNA repair by homologous recombination (Fröls et al., 2007).
Conclusion
In this work we describe the global metabolic and transcriptional response of Saccharolobus solfataricus P2 during growth with phenol as sole carbon source. Our data underline the importance of the phenol meta-degradation pathway in S. solfataricus, as intermediates of the ortho-route were not detected. Based on our data, we provide enzyme candidates for the degradation of phenol by the meta-degradation pathway downstream of 2-hydroxymucconic acid. Furthermore, our data clearly showed that the already characterized enzymes of phenol degradation acting upstream of 2-hydroxymucconic acid are strongly up-regulated during growth on phenol, compared to the reference condition. Although S. solfataricus uses phenol for biomass production, it causes several stress reactions, including genome rearrangement, polyamine accumulation and expression of antioxidant proteins. A higher oxygen demand for substrate oxidation leads to the usage of a less efficient terminal oxidase and an increased respiratory activity to meet the energy demand of the cell. This further contributes to an even lower ATP yield per substrate and to the very poor incorporation of carbon into biomass which was furthermore confirmed by the metabolic model.
Data Availability Statement
The datasets presented in this study can be found online at the FAIRDOMHub platform via: https://fairdomhub.org/investigations/401. The metabolic model is available at the MMTB platform via: https://mmtb.brenda-enzymes.org/models/view/3.
Author Contributions
JW, JKa, BS, DS, and MN-S conceived the idea and designed the experiments. JW, AA, and MN-S performed the experiments. JKo performed the computational modeling. JW, JKo, AA, and MN-S analyzed the data. JW, JKo, and MN-S wrote the manuscript with input of all authors. All authors edited the manuscript and approved the final manuscript.
Funding
This work was performed within the e:Bio initiative of the Federal Ministry of Education and Research (BMBF), Germany. The following authors acknowledge the BMBF for financial support: JW (0316188B), AA (0316188D), and JKo (031L0078E). The publication of this article was funded by the Open Access Fund of the Leibniz Association.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sabine Kaltenhäuser and Steffen Lippold for excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.587032/full#supplementary-material
Footnotes
References
Ahmed, H., Ettema, T. J. G., Tjaden, B., Geerling, A. C. M., van der Oost, J., and Siebers, B. (2005). The semi-phosphorylative Entner–Doudoroff pathway in hyperthermophilic archaea: a re-evaluation. Biochem. J. 390, 529–540. doi: 10.1042/bj20041711
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106.
Anku, W. W., Mamo, M. A., and Govender, P. P. (2017). “Phenolic Compounds in Water: Sources, Reactivity, Toxicity and Treatment Methods,” in Phenolic Compounds - Natural Sources, Importance and Applications, eds M. Soto-Hernández, M. Palma-Tenango, and M. R. del Garcia-Mateos (London: InTech).
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 29, 1165–1188.
Benton, H. P., Wong, D. M., Trauger, S. A., and Siuzdak, G. (2008). XCMS 2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem. 80, 6382–6389. doi: 10.1021/ac800795f
Brock, T. D., Brock, K. M., Belly, R. T., and Weiss, R. L. (1972). Sulfolobus: A new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Archiv. Mikrobiol. 84, 54–68. doi: 10.1007/bf00408082
Brouns, S. J. J., Walther, J., Snijders, A. P. L., van de Werken, H. J. G., Willemen, H. L. D. M., Worm, P., et al. (2006). Identification of the missing links in prokaryotic pentose oxidation pathways: evidence for enzyme recruitment. J. Biol. Chem. 281, 27378–27388. doi: 10.1074/jbc.m605549200
Brügger, K., Redder, P., She, Q., Confalonieri, F., Zivanovic, Y., and Garrett, R. A. (2002). Mobile elements in archaeal genomes. FEMS Microbiol. Lett. 206, 131–141. doi: 10.1016/s0378-1097(01)00504-3
Burroughs, A. M., Hoppe, R. W., Goebel, N. C., Sayyed, B. H., Voegtline, T. J., Schwabacher, A. W., et al. (2013). Structural and Functional Characterization of MppR, an Enduracididine Biosynthetic Enzyme from Streptomyces hygroscopicus: Functional Diversity in the Acetoacetate Decarboxylase-like Superfamily. Biochemistry 52, 4492–4506. doi: 10.1021/bi400397k
Chae, J.-C., Kim, E., Bini, E., and Zylstra, G. J. (2007). Comparative analysis of the catechol 2,3-dioxygenase gene locus in thermoacidophilic archaeon Sulfolobus solfataricus strain 98/2. Biochem. Biophys. Res. Commun. 357, 815–819. doi: 10.1016/j.bbrc.2007.04.027
Chong, P. K., Burja, A. M., Radianingtyas, H., Fazeli, A., and Wright, P. C. (2007). Translational and transcriptional analysis of Sulfolobus solfataricus P2 to provide insights into alcohol and ketone utilisation. Proteomics 7, 424–435. doi: 10.1002/pmic.200600746
Christen, P., Davidson, S., Combet-Blanc, Y., and Auria, R. (2011). Phenol biodegradation by the thermoacidophilic archaeon Sulfolobus solfataricus 98/2 in a fed-batch bioreactor. Biodegradation 22, 475–484. doi: 10.1007/s10532-010-9420-6
Christen, P., Vega, A., Casalot, L., Simon, G., and Auria, R. (2012). Kinetics of aerobic phenol biodegradation by the acidophilic and hyperthermophilic archaeon Sulfolobus solfataricus 98/2. Biochem. Eng. J. 62, 56–61. doi: 10.1016/j.bej.2011.12.012
Collinsworth, W. L., Chapman, P. J., and Dagley, S. (1973). Stereospecific enzymes in the degradation of aromatic compounds by Pseudomonas putida. J. Bacteriol. 113, 922–931. doi: 10.1128/jb.113.2.922-931.1973
Comte, A., Christen, P., Davidson, S., Pophillat, M., Lorquin, J., Auria, R., et al. (2013). Biochemical, transcriptional and translational evidences of the Phenol-meta-Degradation Pathway by the Hyperthermophilic Sulfolobus solfataricus 98/2. PLoS One 8:e82397. doi: 10.1371/journal.pone.0082397
Dudoit, S., Yang, Y. H., Callow, M. J., and Speed, T. P. (2002). Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statist. Sin. 12, 111–139.
Duffner, F. M., Kirchner, U., Bauer, M. P., and Müller, R. (2000). Phenol/cresol degradation by the thermophilic Bacillus thermoglucosidasius A7: cloning and sequence analysis of five genes involved in the pathway. Gene 256, 215–221. doi: 10.1016/s0378-1119(00)00352-8
Duraisamy, P., Sekar, J., Arunkumar, A. D., and Ramalingam, P. V. (2020). Kinetics of Phenol Biodegradation by Heavy Metal Tolerant Rhizobacteria Glutamicibacter nicotianae MSSRFPD35 From Distillery Effluent Contaminated Soils. Front. Microbiol. 11:1573. doi: 10.3389/fmicb.2020.01573
Esser, D., Kouril, T., Talfournier, F., Polkowska, J., Schrader, T., Bräsen, C., et al. (2013). Unraveling the function of paralogs of the aldehyde dehydrogenase super family from Sulfolobus solfataricus. Extremophiles 17, 205–216. doi: 10.1007/s00792-012-0507-3
Esser, D., Kouril, T., Zaparty, M., Sierocinski, P., Chan, P. P., Lowe, T., et al. (2011). Functional curation of the Sulfolobus solfataricus P2 and S. acidocaldarius 98-3 complete genome sequences. Extremophiles 15, 711–712. doi: 10.1007/s00792-011-0392-1
Ettema, T. J. G., Ahmed, H., Geerling, A. C. M., van der Oost, J., and Siebers, B. (2008). The non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) of Sulfolobus solfataricus: a key-enzyme of the semi-phosphorylative branch of the Entner–Doudoroff pathway. Extremophiles 12, 75–88. doi: 10.1007/s00792-007-0082-1
Feist, C. F., and Hegeman, G. D. (1969). Phenol and benzoate metabolism by Pseudomonas putida: regulation of tangential pathways. J. Bacteriol. 100, 869–877. doi: 10.1128/jb.100.2.869-877.1969
Filee, J., Siguier, P., and Chandler, M. (2007). Insertion Sequence Diversity in Archaea. Microbiol. Mol. Biol. Rev. 71, 121–157. doi: 10.1128/mmbr.00031-06
Filipowicz, N., Momotko, M., Boczkaj, G., Pawlikowski, T., Wanarska, M., and Cieśliński, H. (2017). Isolation and characterization of phenol-degrading psychrotolerant yeasts. Water Air Soil Pollut. 228:210.
Fröls, S., Gordon, P. M. K., Panlilio, M. A., Duggin, I. G., Bell, S. D., Sensen, C. W., et al. (2007). Response of the Hyperthermophilic Archaeon Sulfolobus solfataricus to UV Damage. Genome Biol. 189, 8708–8718. doi: 10.1128/jb.01016-07
Genschel, U. (2004). Coenzyme A biosynthesis: reconstruction of the pathway in archaea and an evolutionary scenario based on comparative genomics. Mol. Biol. Evol. 21, 1242–1251. doi: 10.1093/molbev/msh119
Giles, T. N., and Graham, D. E. (2008). Crenarchaeal arginine decarboxylase evolved from an S -adenosylmethionine decarboxylase enzyme. J. Biol. Chem. 283, 25829–25838. doi: 10.1074/jbc.m802674200
Gleißner, M., Kaiser, U., Antonopoulos, E., and Schäfer, G. (1997). The Archaeal SoxABCD Complex Is a Proton Pump in Sulfolobus acidocaldarius. J. Biol. Chem. 272, 8417–8426. doi: 10.1074/jbc.272.13.8417
Gurujeyalakshmi, G., and Oriel, P. (1989). Isolation of phenol-degrading Bacillus stearothermophilus and partial characterization of the phenol hydroxylase. Appl. Environ. Microbiol. 55, 500–502. doi: 10.1128/aem.55.2.500-502.1989
Ha, H. C., Sirisoma, N. S., Kuppusamy, P., Zweier, J. L., Woster, P. M., and Casero, R. A. (1998). The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. U.S.A. 95, 11140–11145. doi: 10.1073/pnas.95.19.11140
Helmecke, J., Schomburg, D., and Neumann-Schaal, M. (2020). MetaboMAPS: pathway sharing and multi-omics data visualization in metabolic context. F1000Res 9:288. doi: 10.12688/f1000research.23427.1
Hilker, R., Stadermann, K. B., Doppmeier, D., Kalinowski, J., Stoye, J., Straube, J., et al. (2014). ReadXplorer—visualization and analysis of mapped sequences. Bioinformatics 30, 2247–2254. doi: 10.1093/bioinformatics/btu205
Hiller, K., Hangebrauk, J., Jäger, C., Spura, J., Schreiber, K., and Schomburg, D. (2009). MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem. 81, 3429–3439. doi: 10.1021/ac802689c
Hottes, A. K., Meewan, M., Yang, D., Arana, N., Romero, P., McAdams, H. H., et al. (2004). Transcriptional Profiling of Caulobacter crescentus during Growth on Complex and Minimal Media. J. Bacteriol. 186, 1448–1461. doi: 10.1128/jb.186.5.1448-1461.2004
Inoue, J., Shaw, J. P., Rekik, M., and Harayama, S. (1995). Overlapping substrate specificities of benzaldehyde dehydrogenase (the xylC gene product) and 2-hydroxymuconic semialdehyde dehydrogenase (the xylG gene product) encoded by TOL plasmid pWW0 of Pseudomonas putida. J. Bacteriol. 177, 1196–1201. doi: 10.1128/jb.177.5.1196-1201.1995
Izzo, V., Notomista, E., Picardi, A., Pennacchio, F., and Di Donato, A. (2005). The thermophilic archaeon Sulfolobus solfataricus is able to grow on phenol. Res. Microbiol. 156, 677–689. doi: 10.1016/j.resmic.2005.04.001
Jervis, T. J., Roper, D. I., and Glover, I. D. (1996). Crystallization and preliminary X-ray analysis of a bifunctional enzyme: HHDD isomerase/OPET decarboxylase from Escherichia coli. Acta Crystallogr D Biol. Crystallogr. 52, 1036–1038. doi: 10.1107/s0907444996003381
Jung, I. L., Oh, T. J., and Kim, I. G. (2003). Abnormal growth of polyamine-deficient Escherichia colimutant is partially caused by oxidative stress-induced damage. Arch. Biochem. Biophys. 418, 125–132. doi: 10.1016/j.abb.2003.08.003
Komorowski, L., Verheyen, W., and Schäfer, G. (2002). The archaeal respiratory supercomplex soxm from s. acidocaldarius combines features of quinole and cytochrome c oxidases. Biol. Chem. 383, 1791–1799.
Kopka, J., Schauer, N., Krueger, S., Birkemeyer, C., Usadel, B., Bergmuller, E., et al. (2005). GMD@CSB.DB: the Golm Metabolome Database. Bioinformatics 21, 1635–1638. doi: 10.1093/bioinformatics/bti236
Koshkin, A., Knudsen, G. M., and Ortiz de Montellano, P. R. (2004). Intermolecular interactions in the AhpC/AhpD antioxidant defense system of Mycobacterium tuberculosis. Arch. Biochem. Biophys. 427, 41–47. doi: 10.1016/j.abb.2004.04.017
Kruskal, W. H., and Wallis, W. A. (1952). Use of ranks in one-criterion variance analysis. J. Am. Statist. Assoc. 47, 583–621. doi: 10.1080/01621459.1952.10483441
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Limauro, D., De Simone, G., Pirone, L., Bartolucci, S., D’Ambrosio, K., and Pedone, E. (2014). Sulfolobus solfataricus thiol redox puzzle: characterization of an atypical protein disulfide oxidoreductase. Extremophiles 18, 219–228. doi: 10.1007/s00792-013-0607-8
Liu, L., Wu, J.-F., Ma, Y.-F., Wang, S.-Y., Zhao, G.-P., and Liu, S.-J. (2007). A Novel Deaminase Involved in Chloronitrobenzene and Nitrobenzene Degradation with Comamonas sp. Strain CNB-1. J. Bacteriol. 189, 2677–2682. doi: 10.1128/jb.01762-06
Liu, Y., Wang, W., Shah, S. B., Zanaroli, G., Xu, P., and Tang, H. (2020). Phenol biodegradation by Acinetobacter radioresistens APH1 and its application in soil bioremediation. Appl. Microbiol. Biotechnol. 104, 427–437. doi: 10.1007/s00253-019-10271-w
Martusewitsch, E., Sensen, C. W., and Schleper, C. (2000). High spontaneous mutation rate in the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by transposable elements. J. Bacteriol. 182, 2574–2581. doi: 10.1128/jb.182.9.2574-2581.2000
Michalowicz, J., and Duda, W. (2007). Phenols – Sources and Toxicity. Polish J. Environ. Stud. 16, 347–362.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Naguib, D. M., and Badawy, N. M. (2020). Phenol removal from wastewater using waste products. J. Environ. Chem. Eng. 8:103592. doi: 10.1016/j.jece.2019.103592
Neidhardt, F. C. (1963). Effects of environment on the composition of bacterial cells. Annu. Rev. Microbiol. 17, 61–86. doi: 10.1146/annurev.mi.17.100163.000425
Neumann-Schaal, M., Hofmann, J. D., Will, S. E., and Schomburg, D. (2015). Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiol. 15:281. doi: 10.1186/s12866-015-0614-2
Nunn, C. E. M., Johnsen, U., Schönheit, P., Fuhrer, T., Sauer, U., Hough, D. W., et al. (2010). Metabolism of Pentose Sugars in the Hyperthermophilic Archaea Sulfolobus solfataricus and Sulfolobus acidocaldarius. J. Biol. Chem. 285, 33701–33709.
Ornston, L. N. (1966). The conversion of catechol and protocatechuate to beta-ketoadipate by Pseudomonas putida. J. Biol. Chem. 241, 3800–3810.
Peyraud, R., Kiefer, P., Christen, P., Massou, S., Portais, J.-C., and Vorholt, J. A. (2009). Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Natl. Acad. Sci. U.S.A. 106, 4846–4851. doi: 10.1073/pnas.0810932106
Quester, S., and Schomburg, D. (2011). EnzymeDetector: an integrated enzyme function prediction tool and database. BMC Bioinformatics 12:376. doi: 10.1186/1471-2105-12-376
Saha, N. C., Bhunia, F., and Kaviraj, A. (1999). Toxicity of Phenol to Fish and Aquatic Ecosystems. Bull. Environ. Contam Toxicol. 63, 195–202. doi: 10.1007/s001289900966
Sakai, H. D., and Kurosawa, N. (2018). Saccharolobus caldissimus gen. nov., sp. nov., a facultatively anaerobic iron-reducing hyperthermophilic archaeon isolated from an acidic terrestrial hot spring, and reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. nov. and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Microbiol. 68, 1271–1278. doi: 10.1099/ijsem.0.002665
Sala-trepat, J. M., and Evans, W. C. (1971). The meta Cleavage of Catechol by Azotobacter Species. 4-Oxalocrotonate Pathway. Eur. J. Biochem. 20, 400–413. doi: 10.1111/j.1432-1033.1971.tb01406.x
Santos, P. M., Benndorf, D., and Sá-Correia, I. (2004). Insights into Pseudomonas putida KT2440 response to phenol-induced stress by quantitative proteomics. Proteomics 4, 2640–2652. doi: 10.1002/pmic.200300793
Shingler, V., Powlowski, J., and Marklund, U. (1992). Nucleotide sequence and functional analysis of the complete phenol/3,4-dimethylphenol catabolic pathway of Pseudomonas sp. strain CF600. J. Bacteriol. 174, 711–724. doi: 10.1128/jb.174.3.711-724.1992
Simon, G., Walther, J., Zabeti, N., Combet-Blanc, Y., Auria, R., Oost, J. V., et al. (2009). Effect of O 2 concentrations on Sulfolobus solfataricus P2. FEMS Microbiol. Lett. 299, 255–260.
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R., and Siuzdak, G. (2006). XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment. matching, and identification. Anal. Chem. 78, 779–787. doi: 10.1021/ac051437y
Sridevi, V., Lakshmi, M., Manasa, M., and Sravani, M. (2012). Metabolic pathways for the biodegradation of phenol. Int. J. Eng. Sci. Adv. Technol. 2, 695–705.
Stark, H., Wolf, J., Albersmeier, A., Pham, T. K., Hofmann, J. D., Siebers, B., et al. (2017). Oxidative Stickland reactions in an obligate aerobic organism - amino acid catabolism in the Crenarchaeon Sulfolobus solfataricus. FEBS J. 284, 2078–2095. doi: 10.1111/febs.14105
Syvanen, M. (1994). Horizontal gene transfer: evidence and possible consequences. Annu. Rev. Genet. 28, 237–261. doi: 10.1146/annurev.ge.28.120194.001321
Tachdjian, S., and Kelly, R. M. (2006). Dynamic metabolic adjustments and genome plasticity are implicated in the heat shock response of the extremely thermoacidophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 188, 4553–4559. doi: 10.1128/jb.00080-06
Tam, L. T., Eymann, C., Albrecht, D., Sietmann, R., Schauer, F., Hecker, M., et al. (2006). Differential gene expression in response to phenol and catechol reveals different metabolic activities for the degradation of aromatic compounds in Bacillus subtilis. Environ. Microbiol. 8, 1408–1427. doi: 10.1111/j.1462-2920.2006.01034.x
Tautenhahn, R., Böttcher, C., and Neumann, S. (2008). Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 9:504. doi: 10.1186/1471-2105-9-504
Tomita, H., Yokooji, Y., Ishibashi, T., Imanaka, T., and Atomi, H. (2014). An Archaeal Glutamate Decarboxylase Homolog Functions as an Aspartate Decarboxylase and Is Involved in -Alanine and Coenzyme A Biosynthesis. J. Bacteriol. 196, 1222–1230. doi: 10.1128/jb.01327-13
Wakagi, T., Nishimasu, H., Miyake, M., and Fushinobu, S. (2016). Archaeal Mo-Containing Glyceraldehyde Oxidoreductase Isozymes Exhibit Diverse Substrate Specificities through Unique Subunit Assemblies. PLoS one 11:e0147333. doi: 10.1371/journal.pone.0147333
Winn, L. M. (2003). Homologous recombination initiated by benzene metabolites: a potential role of oxidative stress. Toxicol. Sci. 72, 143–149. doi: 10.1093/toxsci/kfg008
Wolf, J., Stark, H., Fafenrot, K., Albersmeier, A., Pham, T. K., Müller, K. B., et al. (2016). A systems biology approach reveals major metabolic changes in the thermoacidophilic archaeon Sulfolobus solfataricus in response to the carbon source L-fucose versus D-glucose. Mol. Microbiol. 102, 882–908. doi: 10.1111/mmi.13498
Zaparty, M., Esser, D., Gertig, S., Haferkamp, P., Kouril, T., Manica, A., et al. (2010). “Hot standards” for the thermoacidophilic archaeon Sulfolobus solfataricus. Extremophiles 14, 119–142.
Zech, H., Hensler, M., Koßmehl, S., Drüppel, K., Wöhlbrand, L., Trautwein, K., et al. (2013). Adaptation of Phaeobacter inhibens DSM 17395 to growth with complex nutrients. Proteomics 13, 2851–2868. doi: 10.1002/pmic.201200513
Keywords: phenol biodegradation, Sulfolobales, stress response, metabolic modeling, systems biology
Citation: Wolf J, Koblitz J, Albersmeier A, Kalinowski J, Siebers B, Schomburg D and Neumann-Schaal M (2021) Utilization of Phenol as Carbon Source by the Thermoacidophilic Archaeon Saccharolobus solfataricus P2 Is Limited by Oxygen Supply and the Cellular Stress Response. Front. Microbiol. 11:587032. doi: 10.3389/fmicb.2020.587032
Received: 24 July 2020; Accepted: 08 December 2020;
Published: 08 January 2021.
Edited by:
Christa Schleper, University of Vienna, AustriaCopyright © 2021 Wolf, Koblitz, Albersmeier, Kalinowski, Siebers, Schomburg and Neumann-Schaal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meina Neumann-Schaal, bWVpbmEubmV1bWFubi1zY2hhYWxAZHNtei5kZQ==
†Present address: Jacqueline Wolf, Institute of Laboratory Medicine and Pathobiochemistry, Molecular Diagnostics, Philipps-Universität Marburg, Marburg, Germany; Julia Koblitz, Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany