 Brody Barton
Brody Barton Randy M. Morgenstein
Randy M. Morgenstein- Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, OK, United States
The bacterial actin homolog, MreB, is highly conserved among rod-shaped bacteria and essential for growth under normal growth conditions. MreB directs the localization of cell wall synthesis and loss of MreB results in round cells and death. Using the MreB depolymerizing drug, A22, we show that changes to central metabolism through deletion of malate dehydrogenase from the tricarboxylic acid (TCA) cycle results in cells with an increased tolerance to A22. We hypothesize that deletion of malate dehydrogenase leads to the upregulation of gluconeogenesis resulting in an increase in cell wall precursors. Consistent with this idea, metabolite analysis revealed that malate dehydrogenase (mdh) deletion cells possess elevated levels of several glycolysis/gluconeogenesis compounds and the cell wall precursor, uridine diphosphate N-acetylglucosamine (UDP-NAG). In agreement with these results, the increased A22 resistance phenotype can be recapitulated through the addition of glucose to the media. Finally, we show that this increase in antibiotic tolerance is not specific to A22 but also applies to the cell wall-targeting antibiotic, mecillinam.
Introduction
The bacterial actin homolog, MreB, is a highly conserved and conditionally essential protein necessary for shape and growth in many rod-shaped human pathogens, including Escherichia coli, Salmonella enterica, Pseudomonas aeruginosa, and Vibrio cholerae (van den Ent et al., 2001; Alyahya et al., 2009). MreB polymers direct the localization of cell wall synthesis, and disruption of MreB, either through the use of antibioitics such as A22 or genetic modifications, causes cells to become misshapen and lyse. A22 binds adjacent to the ATP binding pocket of MreB and alters MreB’s polymerization dynamics (Gitai et al., 2005; Bean et al., 2009; van den Ent et al., 2014; Awuni et al., 2016; Awuni and Mu, 2019).
Slow growth or overexpression of the cell division genes ftsZAQ can overcome the loss of mreB (Kruse et al., 2005; Bendezú and de Boer, 2008). While overexpression of the cell division genes does not prevent the cell from becoming round upon disruption of MreB, the increased levels of FtsZAQ are thought to help overcome the change in cell diameter, aiding the round cells in dividing. It is currently unknown if over expression or deletion of other genes can also overcome MreB disruption.
There is ample evidence to support a connection between central metabolism and cell shape (Weart et al., 2007; Elbaz and Ben-Yehuda, 2010; Yao et al., 2012; Hill et al., 2013; Beaufay et al., 2015). In addition, a connection between the tricarboxylic acid (TCA) cycle and cell size has been shown in Caulobacter crescentus. In C. crescentus, mutations that result in the accumulation of α-ketoglutarate cause cell shape defects by reducing the synthesis of cell wall precursors (Irnov et al., 2017).
Similar to cell shape regulation, cell size is coordinated with nutrient availability. Cell size is regulated with growth rate so that cells grown in rich medium are larger than those grown in nutrient poor medium (Schaechter et al., 1958; Sargent, 1975; Pierucci, 1978). In Bacillus subtilis, pyruvate has been shown to help coordinate growth and division through FtsZ-ring assembly (Monahan et al., 2014). Furthermore, in both E. coli and B. subtilis, the nucleotide sugar uridine diphosphate (UDP)-glucose is used as a way to link carbon availability with cell size (Weart et al., 2007; Chien et al., 2012; Hill et al., 2013).
Because of the well-known connection between cell shape and size and metabolism, specifically the TCA cycle, we screened deletions of genes involved in each enzymatic reaction of the TCA cycle for changes to the minimal inhibitory concentration (MIC) of A22 (MICA22) in E. coli. We found that three gene deletions lead to an increase in the tolerance of cells to A22 and focused our efforts on understanding how disruption of the TCA cycle protein, malate dehydrogenase (Mdh), leads to a higher MICA22.
We propose the increase in MICA22 works through the activation of gluconeogenesis leading to an increase in the levels of cell wall precursors. While cell shape changes are not suppressed, we did see an increase to the tolerance of cells to A22 and mecillinam (Mec), an inhibitor of the cell wall synthesis enzyme penicillin binding protein 2 (PBP2). Additionally, we found that this higher tolerance for A22 and mecillinam is phenocopied by the addition of glucose to the growth medium. Finally, we found that the cell wall synthesis protein PBP1B is epistatic to Mdh, leading to a reduction of the MICA22 in a PBP1Bmdh double mutant. These results further support cross talk between metabolism, the cell elongation machinery, and the cell division machinery.
Materials and Methods
Bacterial Growth
Bacteria were grown using standard laboratory conditions. Cultures were grown overnight in LB medium (10 g/L NaCl, 10 g/L tryptone, and 5 g/L yeast extract), subcultured 1:1,000, and grown to exponential phase (O.D.600 0.3–0.6) at 37°C in a shaking incubator. Cells were grown in antibiotics at concentrations listed: A22 (made in house), kanamycin (Goldbio K120), ampicillin (Amp; Goldbio A301), mecillinam (Sigma 33,447), cephalexin (Ceph; Goldbio C800), and cefsulodin (Cef; Alfa Aesar J66120).
Bacterial Strains
All gene deletions in MG1655 were made by P1 transduction from the Keio collection. Transductants were selected on kanamycin (30 μg/ml) and confirmed by PCR. To produce double mutants, pCP20 (Supplementary Table S1) was transformed into strain 1 in order to remove the kanamycin resistance cassette. P1 transduction was then used to move the second mutation into the strain. See Supplementary Table S1 for a list of strains used in this study.
Malate dehydrogenase was cloned into pBad33 by PCR and digestion-ligation at the EcoRI and HindIII sites. The ligation mixture was used to transform S17 cells and sequence verified before being transformed into MG1655.
MIC Assay
Optical densities of all cultures used in the MIC were checked at 600 nm (OD600) with a Thermo Scientific Biomate-3S. Overnight cultures were grown in 2 ml test tubes in LB medium in a shaking incubator and normalized to have the same O.D.600 of the slowest growing culture. About 1:100 dilutions were made into 96-well plates filled with 100 μl of LB medium plus indicated antibiotics and/or specified sugars or tricarboxylic acids. Two-fold dilutions of each antibiotic were made. Each plate had one row left uninoculated for a blank and one row drug free as a growth control. The O.D. from the drug free wells were halved and used to determine the MIC values as the concentration of drug that caused half the growth as that in the no drug control. Experiments were performed at minimum in triplicate.
Microscopy
For all imaging, cells were grown at 37°C in indicated medium. Imaging was done on 1% M63-agarose pads at room temperature. Images were collected on a Nikon Ni-E epifluorescent microscope equipped with a 100X/1.45 NA objective (Nikon), Zyla 4.2 plus cooled sCMOS camera (Andor), and NIS Elements software (Nikon).
Intracellular diameter deviation (IDD) was calculated using the Matlab software Morphometrics (Ursell et al., 2017), and custom software as previously described (Morgenstein et al., 2015; Bratton et al., 2018). Briefly, a centerline is drawn through each cell and the diameter is measured across the cell body. The SD of these center lines is divided by the mean width of the cell to calculate a coefficient of variation of IDD. Only non-dividing cells were used for analysis.
Total cell fluorescence was calculated using custom Matlab software. Cell contours were made using Morphometrics to determine the cell boundaries. The total fluorescence within this contour was divided by the amount of pixels to determine the cell brightness.
Metabolomics
Collection
Cells were grown overnight shaking at 37°C in LB medium and then subcultured 1:1,000 into a flask with 10 ml of fresh LB and grown shaking at 37°C for ~4 h until in exponential phase (O.D.600 0.3–0.4). Cells were passed through a 0.2 μm filter via vacuum filtration. The filter was placed in a glass dish with cold acetonitrile:methanol:water (40:40:20) + 0.5% formic acid and placed at −20°C for 15 min to quench metabolism in the cells. This quenching solution was used to wash the cells off of the filter before the solution was neutralized with 1.9 M ammonium bicarbonate. The cells were centrifuged at 15,000 rpm for 5 min to remove debris. Supernatants were collected for mass spectrometry.
Analysis
Supernatants were analyzed via the methods of Su et al. (2020). Specificity was achieved through a combination of chromatographic separations followed by high-resolution MS. This method allows for the identification of ~300 water soluble metabolites. Covariant ion analysis (COVINA) was used to identify peaks. Cells were run in triplicate, and the effect of mdh deletion on metabolites was only determined for metabolites that were identified in all three replicates and showed a consistent ratio between WT:mdh cells.
Results
Deletions in the TCA Cycle Result in an Increase in the MICA22
It is known that mutations that cause an increase in expression of the cell division genes ftsZAQ suppress the growth defects of gene deletions of many cell shape determinants, including MreB (Vinella et al., 1993; Kruse et al., 2005; Bendezú and de Boer, 2008). We wanted to determine if there were gene deletions that result in a cell’s ability to live without the essential bacterial actin homolog, MreB. We used the MreB-depolymerizing drug A22 to mimic the loss of MreB and screened genes from the Keio collection involved in the TCA cycle for mutants that cause an increase in the MIC of A22. For quantification purposes, we report the concentration of A22 that results in less than half the growth (O.D.600) compared to cells grown only in LB medium (henceforth referred to as, MICA22; Iwai et al., 2002; Baba et al., 2006).
We broke each enzymatic step of the TCA cycle by deleting genes for citrate synthase (gltA), aconitate hydratase (acnB), isocitrate dehydrogenase (icd), 2-oxoglutarate dehydrogenase (sucA), succinyl-CoA synthetase (sucC), succinate:quinone oxidoreductase (sdhA), the major anaerobic fumarase (fumA), and malate dehydrogenase (mdh; Figure 1A). Of these, only deletion of acnB, sucC, and mdh shows a higher MICA22 than WT cells (Figure 1B).
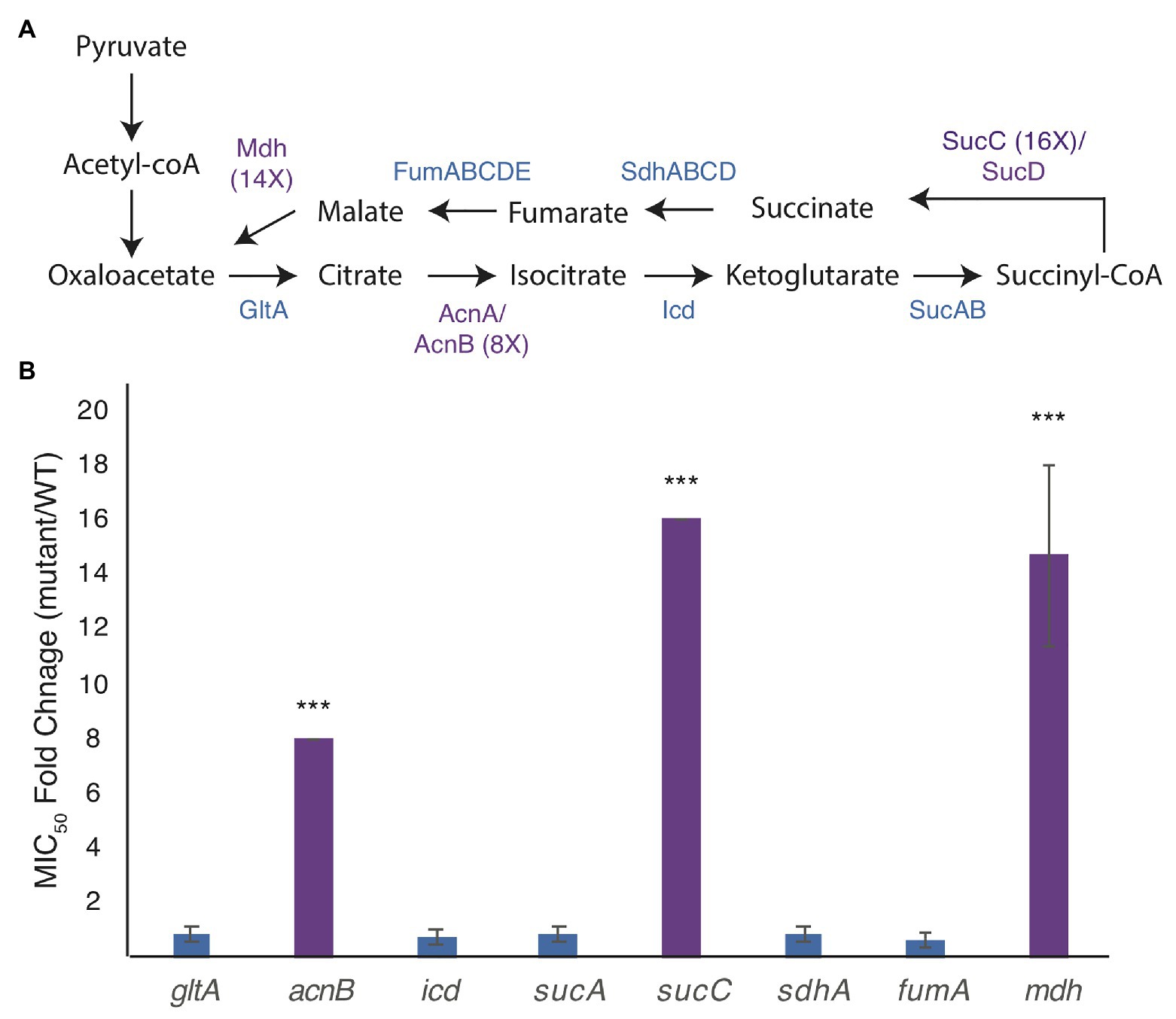
Figure 1. Mutations in the genes of the tricarboxylic acid (TCA) cycle increase the minimal inhibitory concentration (MIC) of A22 (MICA22). (A) Model of the TCA cycle. Proteins in purple have higher MICA22 when the associated gene is deleted. (B) MICA22 fold change of mutants in the TCA cycle. Only malate dehydrogenase (mdh), aconitate hydratase (acnB), and succinyl-CoA synthetase (sucC) mutants show an increased MICA22. ***p < 0.001 comparing each strain to 1 (no change).
Deletion of acnB has been shown to increase antibiotic tolerance by decreasing ATP levels in the cell, but to our knowledge, neither mutations in mdh nor sucC have been reported to cause a change in antibiotic susceptibility (Rowe et al., 2020). In this paper, we focus on understanding the effects of the mdh mutation on A22 tolerance. We do not expect the increased tolerance to A22 in the mdh mutant to function through ATP synthesis, like acnB, because the action of Mdh occurs after the steps in the TCA cycle where reducing power is created.
Mdh Is Responsible for the Increased Tolerance to A22
To ensure that there were not any second site mutations in the Keio mdh deletion strain, we transfered the mutation into an MG1655 background using P1 transduction. The MG1655 mdh mutant retained the increased tolerance to A22 and was used for all subsequent experiments (Supplementary Table S1). It cannot be ruled out that a second site mutation is genetically linked to mdh.
Although mdh is not in an operon, it is encoded just downstream of the degQS operon on the opposite strand. To confirm that the increase in A22 tolerance was from deletion of mdh and not disrupted transcriptional termination of degS, we complemented mdh on an arabinose-inducible plasmid. When expression is induced with 0.2% arabinose WT cells with the empty vector have an MICA22 of 5.63 ± 1.25 μg/ml, which is slightly higher than WT cells without the vector (1.5 ± 0.55). However, mdh deletion cells with the empty vector control show a much higher MICA22 than the complemented strain (35.94 ± 14.5 vs. 7.81 ± 2.71), or WT cells, suggesting that ectopic expression of mdh can complement the observed A22 tolerance. As was seen with WT cells, the mdh cells with the empty vector have a slightly higher MIC. This decrease in the MICA22 when mdh is expressed ectopically supports the idea that there is not a second linked mutation to mdh and that deletion of mdh results in a higher MICA22. We note that in these conditions, the MIC values are slightly higher which may be due to either the addition of arabinose or the empty vector (Supplementary Table S2).
Slow growth is known to help cells grow without MreB (Bendezú and de Boer, 2008; Figure 1B). Growth curves of WT and mdh cells show that in our test conditions (LB medium) there is no appreciable change in growth rates, indicating that slow growth is not contributing to the increase in MICA22 (Figure 2A) and that ATP production is not severely affected in this strain. We did however observe that the mdh mutant reaches a maximum optical density at a lower point than WT cells. Because the growth rate is the same between WT and mdh cells and increased tolerance to A22 is not a universal feature of TCA cycle gene deletions (Figure 1B), we hypothesize that the lack of the Mdh enzyme or changes in malate levels (the substrate of Mdh) is the most likely cause of the increased MICA22 seen in the mdh mutant; however, it is also possible the lack of oxaloacetate, the product of Mdh is responsible.
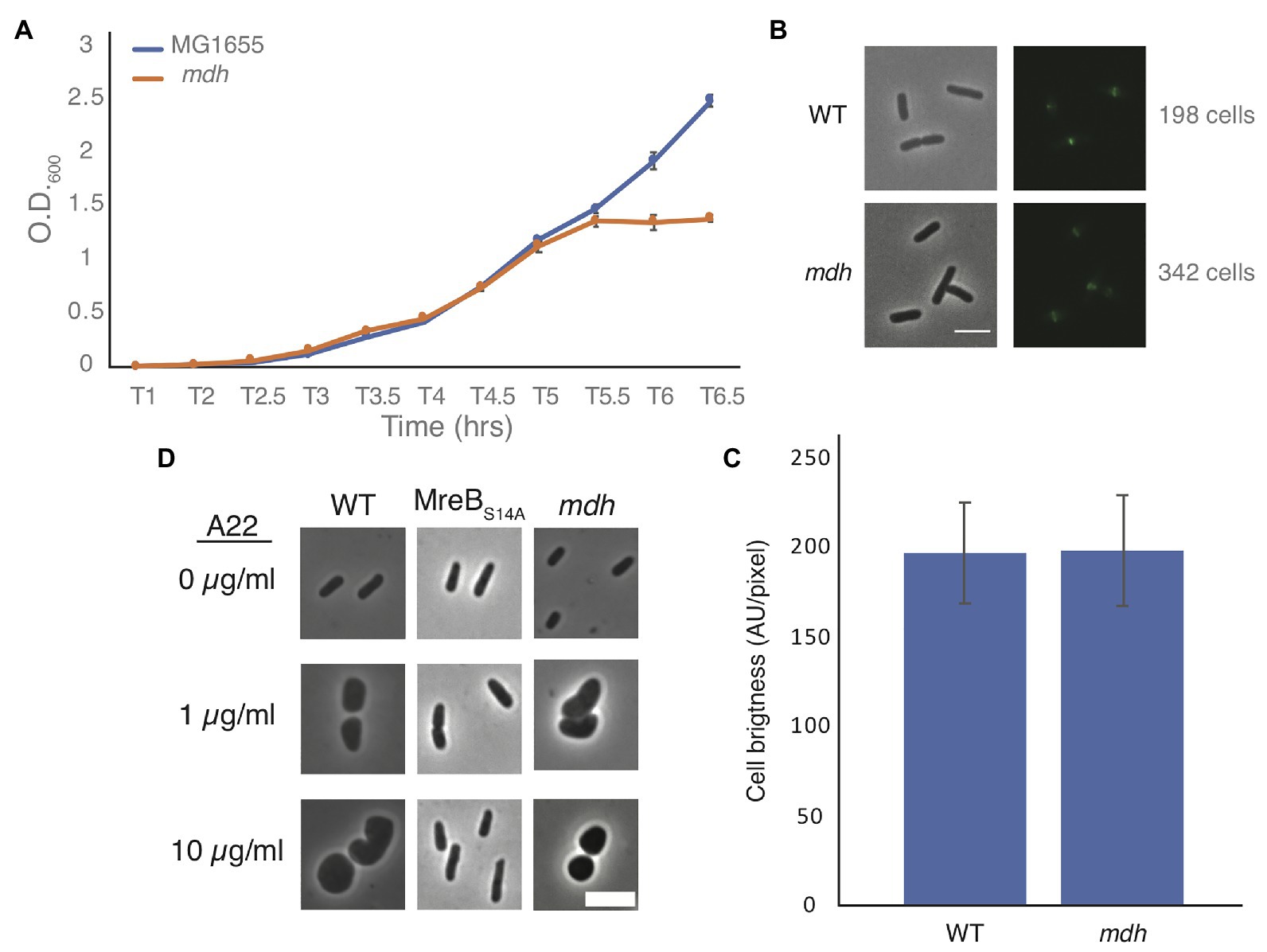
Figure 2. Malate dehydrogenase deletion cells grow at the same rate as WT cells but lose shape under A22 treatment. (A) Representative growth curve of WT and mdh deletion cells grown in triplicate. Cells were grown overnight in LB and set to the same O.D. before a 1:1,000 dilution was made into fresh LB. Error bars are SD. (B) Phase-contrast and GFP images of WT or mdh cells with a functional fluorescent FtsZ-GFP fusion. (C) Quantification of cell brightness from cells expressing FtsZ-GFP in exponential phase. (D) Phase-contrast images of cells grown with different A22 concentrations. Cells were grown for 4 h in LB or A22 1 μg/ml. Cells were grown for 2 h in LB before 10 μg/ml, A22 was added for an additional 2 h. Scale bars are 4 μm.
Additionally, it is possible that changes in FtsZ levels are responsible for the increased survival of mdh cells. The fact that both the Keio and MG1655 strains display an increase in MICA22 suggests that there is not a second site mutation increasing ftsZ levels. It is possible that the metabolic changes caused by the deletion of mdh results in changes to ftsZ levels. We modified a functional native site FtsZ-GFP fusion by using monomeric superfolder GFP at an internal site of FtsZ previously determined to accommodate a fusion protein (Landgraf et al., 2012; Moore et al., 2017). This new fusion was used to measure FtsZ levels in both WT and mdh cells as it is the sole copy of FtsZ in the cell (Supplementary Figure S1). There was no difference in fluorescence intensity between the strains suggesting that FtsZ levels are not affected by deletion of mdh (Figures 2B,C). If FtsZ is overexpressed to a high enough level, minicells can form (Belhumeur and Drapeau, 1984; Ward and Lutkenhaus, 1985). To further support the idea that FtsZ is not upregulated in mdh cells, we measured the cell length of non-dividing WT and mdh cells and while mdh cells are slightly shorter than WT cells, minicells were not produced, suggesting that there are not large changes to FtsZ levels, although the shorter cells do suggest a possible change in divisome activity (Supplementary Figure S2A). The lack of minicells can be observed by the lack of small cell outliers (red plus) in the box plot as well as the fact that the whisker representing the most extreme data points is actually lower for the WT cells than the mdh mutant, indicating that while the mdh mutant produces smaller cells on average the distribution of those cells is also smaller. We also measured the cell length of the mdh complementation strain induced with arabinose and found that along with sensitivity to A22, this strain complemented the shorter cell shape phenotype (Supplementary Figure S2B). The loss of mdh also leads to a smaller width and cell area, both of which are also complemented when mdh is expressed ectopically (Supplementary Figures S2C–F).
Cells could also show an increased MICA22 due to a mechanism that prevents A22 from depolymerizing MreB, as is seen with MreB resistant point mutations. When MreB is disrupted cells become round. To determine if MreB is still being affected by the addition of A22 when mdh is deleted, we imaged WT cells, a previously described MreB point mutant resistant to A22 (MreBS14A), and mdh deletion cells after growth in LB alone, LB with a sublethal concentration of A22 (1 μg/ml) for 4 h, or LB spiked with 10 μg/ml of A22 for 2 h after 2 h of growth (Morgenstein et al., 2015). As expected, both A22 treatments cause WT cells to become round, whereas the MreB point mutant remains a rod regardless of A22 treatment. Interestingly, mdh cells became round, like WT cells, under both A22 conditions, confirming that MreB is still susceptible to A22 in the mdh mutant (Figure 2D). These data further support the hypothesis that the loss of mdh is responsible for the increased MICA22.
mdh Mutants Have an Altered Metabolic Profile
We performed metabolomic analysis to identify changes in metabolites in mdh mutant cells to determine if these changes might be the cause of the increased MICA22. Both WT and mdh cells were grown in LB (without glucose) to exponential phase before metabolite extraction. These experiments were done three independent times and reported results are the ion counts from each of these experiments in order to establish trends in metabolite changes. Only metabolites that show a consistent trend were considered for further analysis. As expected, malate levels were higher in the mdh mutant than in WT cells in all three experiments, although the amount varies (Figure 3A). These increased levels of malate could increase metabolic flux through the alternative malate dehydrogenases MaeA or MaeB, into pyruvate. An increase in pyruvate levels was seen in the mdh mutant (Figure 3B). Pyruvate is involved in many cellular reactions, including glycolysis/gluconeogenesis; therefore, this increased pyruvate could flow into gluconeogenesis to produce sugars (Sauer and Eikmanns, 2005). While the levels of glucose-6-phosphate (G6P), the end product of gluconeogenesis, are very low in cells grown in LB and are not seen in one of the trials at all, there was a consistent increase seen in the mdh mutant, which could only come from increased gluconeogenesis because the cells were grown in LB medium without any sugar (Figure 3C). The increased levels of G6P suggest that the elevated levels of malate seen in the mdh mutant led to an increase in gluconeogenesis. G6P can be converted into fructose-6-phosphate which can be used to form N-acetylglucosamine (NAG), while pyruvate can be converted to phosphoenolpyruvate (PEP), another metabolite used in the synthesis of cell wall precursors (Barreteau et al., 2008).
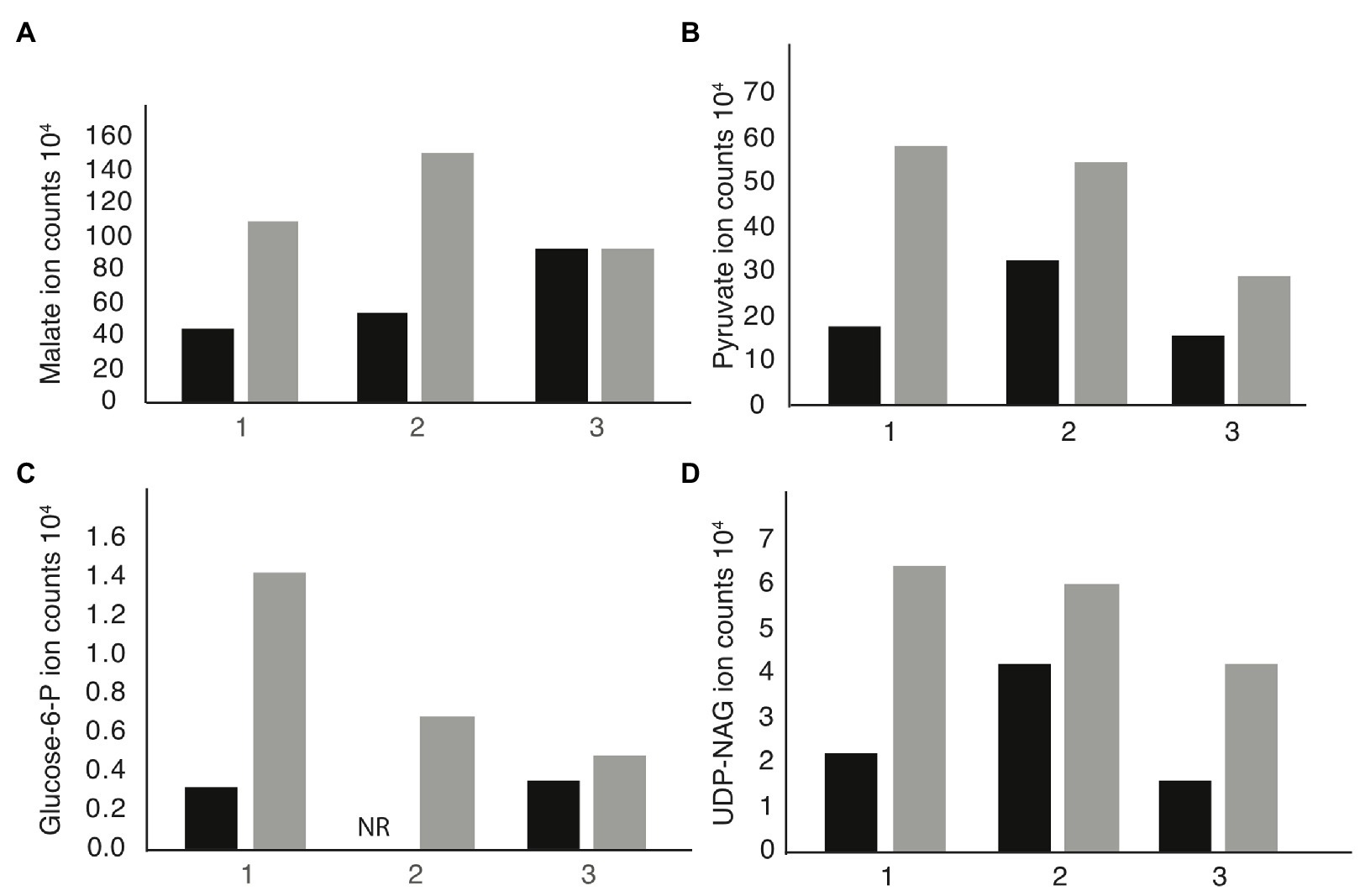
Figure 3. Metabolomics analysis reveals upregulation of gluconeogenesis in mdh cells. Cells were grown in LB medium without the addition of sugar on three independent cultures. The individual ion counts from each day were analyzed to ensure a consistent pattern. (A) Ion counts of malate levels in WT and mdh cells. (B) Ion counts of pyruvate levels in WT and mdh cells. (C) Ion counts of glucose-6P levels in WT and mdh cells. NR, not recorded. (D) Ion counts of uridine diphosphate N-acetylglucosamine (UDP-NAG) levels in WT and mdh cells. Black bars – WT cells, gray bars – mdh cells.
Thus, the increased levels of both G6P and pyruvate led us to hypothesize that the mdh deletion causes an increase in gluconeogenesis resulting in increased levels of cell wall precursors, which may cause an increase in the activity of cell wall synthesis systems not disrupted during A22 treatment (Mengin-Lecreulx et al., 1983). To this end, we looked for changes in the level of cell wall precursors in the metabolomics data and saw an increase in UDP-NAG, supporting our hypothesis (Figure 3D). These results suggest that increasing the amount of cell wall precursors can suppress the lethal effects of MreB depolymerization.
The Addition of Glucose Mimics Deletion of mdh
Glycolysis and gluconeogenesis are the reverse reactions of each other; thus, because G6P levels increased in mdh cells, we reasoned that the addition of glucose to the growth medium would have a similar effect on the MICA22 to deletion of mdh: providing additional substrate for the synthesis of cell wall precursors. In Staphylococcus aureus, it was shown that ~50% of exogenously added glucose ended up as part of the cell wall, supporting the hypothesis that exogenously added glucose can be easily converted into UDP-NAG and incorporated into the cell wall (Komatsuzawa et al., 2004). Additionally, glucose has been shown to compensate for mutations in aconitase to restore antibiotic susceptibility (Rowe et al., 2020).
Minimal inhibitory concentration assays were performed on WT cells grown in LB medium with increasing amounts of glucose or α-methyl-D-glucoside (αMG), a nonhydrolyzable form of glucose (Rogers and Yu, 1962; Hernandez-Asensio et al., 1975). While very low levels (<0.125%) of glucose had no effect on the MICA22 in WT cells, even glucose levels as low as 0.125% resulted in a statistically significant increase in the fold change of the MICA22 in cells grown with glucose vs. LB alone compared with cells grown with αMG vs. LB (Figure 4A). An increase in the glucose levels results in even higher MICA22 fold changes. The significant increase in MICA22 during glucose vs. αMG treatment suggests that the changes are not due to osmotic protection caused by the addition of sugar. These results further support our hypothesis that increasing the levels of cell wall precursors leads to an increase in MICA22.
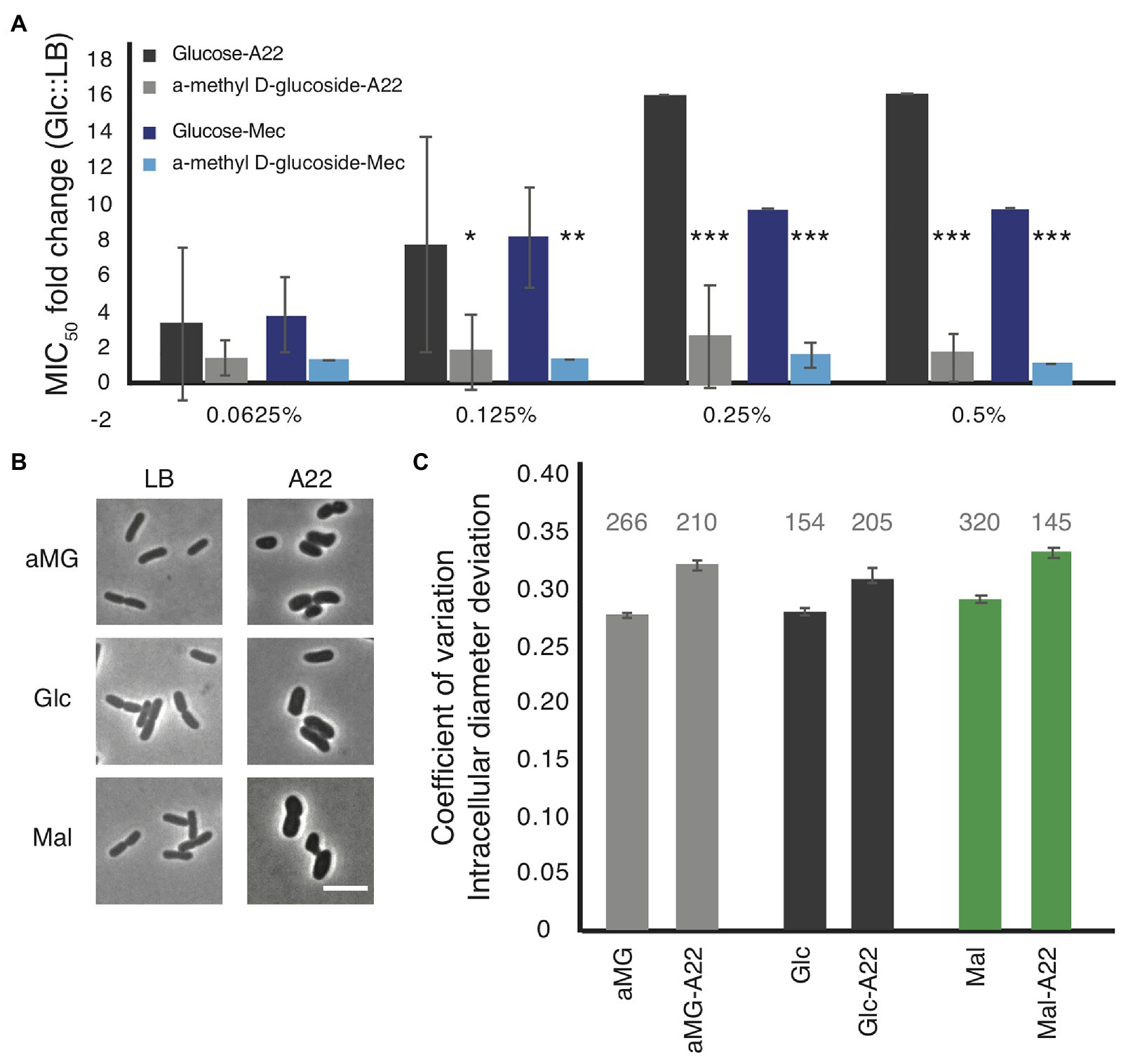
Figure 4. The addition of glucose to the medium phenocopies, the antibiotic tolerant phenotypes of the mdh deletion. (A) WT cells were grown in LB medium supplemented with the indicated amount of either glucose or a non-hydrolizable glucose mimic, α-methyl D-glucoside (αMG). Cells were treated with either A22 (black bars) or mecillinam (blue bars). Error shown is SD from three independent experiments. *p < 0.05, ***p < 0.001 comparing glucose to αMG for each condition. (B,C) Cells were grown with 0.2% αMG, glucose (glc), or malate (mal) for 4 h before imaging (LB) or for 2 h before a spike of 10 μg/ml, A22 for another 2 h. (B) Images of cells. (C) Quantification of how rod-like cells are. Coefficient of variation of the intracellular diameter deviation (IDD) measures rodness. The higher the number the less rod-like cells appear. A22 causes cells to round up in all conditions but the effect is less when cells are grown in glucose. Number above each bar represents the total number of cells analyzed.
Because glucose was able to provide WT cells some protection from A22, we wanted to determine how it affected cell shape. WT cells were grown in LB or A22-spiked media supplemented with 0.2% glucose, αMG, or malate. In all conditions, A22 caused a loss of rod shape measured by increases in the IDD (Figures 4B,C; Morgenstein et al., 2015). We observed that the change in IDD was smaller (although still statistically significant) when cells were treated with glucose than either αMG or malate. Using a two-way ANOVA analysis, we are able to show that the addition of glucose has a significant effect on reducing the change in IDD when cells are treated with A22 (p < 0.05).
Deletion of mdh Leads to an Increased MIC of Mecillinam
The primary role of MreB is to direct the location of cell wall synthesis enzymes. It has been suggested that MreB forms a complex with the SEDS family of cell wall synthesis enzymes (Cho et al., 2016; Meeske et al., 2016; Leclercq et al., 2017). Specifically, recent work has suggested that MreB interacts with RodA and PBP2 to regulate cell elongation, while PBP1A/1B work independently of MreB (Cho et al., 2016). Because the mdh deletion results in cells with a higher MICA22, we hypothesized that cells might also have a higher MIC against other cell wall targeting antibiotics. To that end, we performed MIC assays with other antibiotics that target cell wall synthesis proteins to determine how general the effects of the mdh deletion are on the MICs of cell wall-targeting drugs.
We performed MIC assays with cell wall-targeting antibiotics that inhibit either cell elongation (mecillinam, cefsulodin), cell division (cephalexin), or both (ampicillin) by blocking the activity of different PBPs involved in cell wall synthesis. An MIC fold change >1 indicates that the mdh deletion causes cells to be more resistant and a fold change <1 indicates that mdh cells are more sensitive to the specified antibiotic. The only other cell wall-targeting antibiotic to which the mdh deletion shows an increased MIC is mecillinam, which targets the MreB complex partner, PBP2 (Spratt, 1977; Figure 5A). The fact that the mdh deletion strain does not have a higher MICcef supports the idea that PBP1A/B is not part of the MreB complex because cefsulodin specifically targets PBP1A and PBP1B. Cephalexin targets PBP3 (FtsI), which is part of the division machinery, a different complex from the MreB-elongasome. We did not see an increase in the MICceph in the mdh deletion, suggesting that an increase in gluconeogenesis cannot overcome cell division inhibition. Ampicillin targets multiple PBPs, with the highest affinity for PBP4, followed by PBP3, and PBP2 (Curtis et al., 1979; Preston et al., 1990). Because ampicillin binds to PBP3 tightly and there was no increase in the MICceph, it stands to reason that deletion of mdh would not have an effect on the MICamp. These results suggest that the upregulation of gluconeogenesis caused by the mdh deletion specifically causes an increase in the MIC of drugs that target the MreB elongation synthesis complex (A22 and mecillinam), but not to other cell wall-targeting antibiotics.
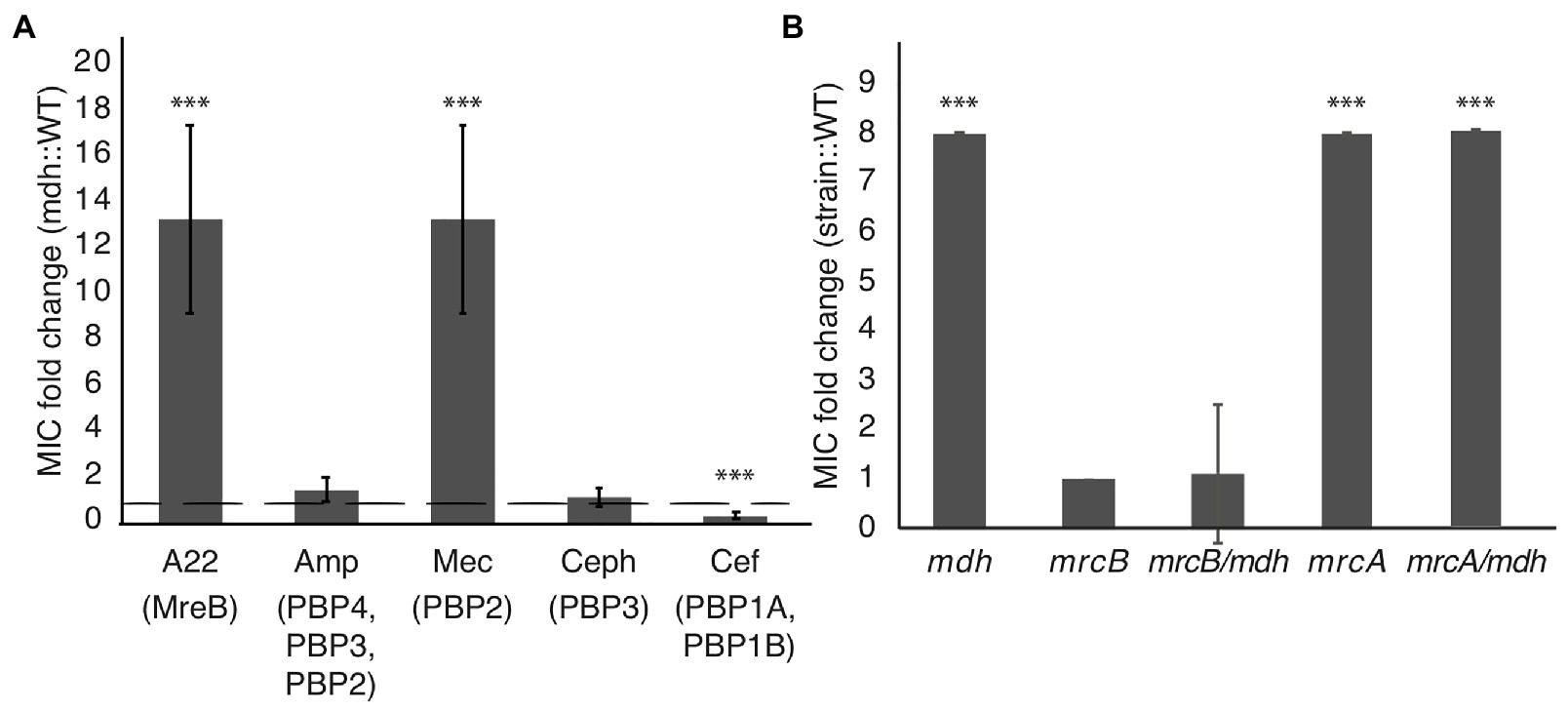
Figure 5. Malate dehydrogenase mutation provides increased MIC specifically of A22 and mecillinam. (A) MIC assays were performed for the indicated antibiotics. Amp, ampicillin; Mec, mecillinam; Ceph, cephalexin; and Cef, cefsulodin. A fold change of 1 (dotted line) indicates no change. (B) A22 MIC comparing WT cells with the indicated strains. ***p < 0.001 comparing each strain to 1.
Inactivation of aconitase (acnB) or sucC leads to an increase in the MICA22 (Figure 1B); therefore, we tested whether deletions of these genes also lead to an increase in the MICmec. The acnB mutation displays an increased MICmec, while deletion of sucC does not (Supplementary Figure S2). This suggests that the role of SucC in A22 resistance is different from that of Mdh. However, it is unclear if AcnB acts through a general antibiotic resistance mechanism via ATP depletion or through a specific MreB-elongasome mechanism.
Glucose Phenocopies the mdh Mutation Leading to an Increased MIC of Mecillinam
The addition of glucose to the media was able to phenocopy the effects of deleting mdh on the MICA22 (Figure 4A). Because we saw a similar increase in the MICmec as with the MICA22 in the mdh deletion, we wanted to test whether the addition of glucose would also lead to an increased MICmec in WT cells. The addition of glucose leads to a statistically significant increase in the MICmec but not αMG (Figure 4). Taken together, the fact that there is only a change in the MIC for antibiotics that target MreB and PBP2 (the major elongasome components), but not other PBPs suggest that an increase in the levels of cell wall precursors, either through an increase in gluconeogenesis or the addition of glucose to the medium, induces a mechanism that specifically targets the MreB elongasome, but not other cell wall synthesis enzymes.
PBP1B Is Epistatic to mdh for A22 Tolerance
If there are two cell wall synthesis machinery systems at work, it stands to reason that inhibition of one could be compensated by activation of the other. Cefsulodin specifically targets the bifunctional PBP1A and PBP1B proteins, which have been suggested to work separately from MreB-PBP2 (Cho et al., 2016; Meeske et al., 2016). The mdh mutation does not increase the MICcef; therefore, to test if either PBP1A or PBP1B can compensate for disrupted MreB, we performed MIC assays on mdh cells with either mrcA (PBP1A) or mrcB (PBP1B) deleted. While the mrcA deletion had no effect on A22 sensitivity, the mrcB deletion, either alone or with mdh, made cells highly sensitive to A22 (Figure 5B). Interestingly, while WT cells display reduced growth above 1.25 μg/ml A22, cells lacking PBP1B show no growth above this concentration but did not have increased sensitivity at lower concentrations resulting in the same MIC when measuring 50% of growth of cells vs. growth in LB. We hypothesize that the increased levels of cell wall precursors observed in the mdh mutant can be used by PBP1B to keep cells alive in the absence of the MreB-PBP2 complex.
Discussion
While studying novel mechanisms of A22 resistance, we found that loss of Mdh, an enzyme in the TCA cycle, leads to an increase in the MICA22 (Figure 1), which we hypothesize is through the induction of gluconeogenesis, leading to an increase in cell wall precursors. We propose that this accumulation of cell wall precursors results in increased MICs of both the MreB-targeting drug A22 and the PBP2-targeting drug, mecillinam through the activation of PBP1B. This effect appears to be specific for the MreB cell wall synthesis complex, as we do not see an increase in MIC of antibiotics that target other enzymes in the cell wall synthesis pathway.
Mutations in the TCA cycle have been implicated in resistance to antibiotics. Deletion of icd, which encodes isocitrate dehydrogenase, results in the accumulation of toxic intermediates leading to the activation of the AcrAB-TolC efflux pump which can pump nalidixic acid out of the cell (Helling et al., 2002). A similar mechanism is most likely not at work in the mdh mutant, as the cells still become round upon A22 treatment, suggesting that A22 is in the cell at a high enough concentration to depolymerize MreB. More recently, it was shown that antibiotic tolerance can be induced by inhibiting ATP production (Conlon et al., 2016; Shan et al., 2017). One way this could be achieved is by blocking enzymes in the TCA cycle such as aconitase (acnB) or succinate dehydrogenase (sdhA), which would lower the reducing power available for oxidative respiration (Rowe et al., 2020).
Multiple Systems Involved in Rod Shape Maintenance
The bacterial cell wall is a macromolecule made up of sugars crosslinked by proteins to provide shape and support to the cell. In rod-shaped bacteria, such as E. coli, B. subtilis, and C. crescentus, MreB is thought to form the main protein of the elongasome complex consisting of multiple PBPs, MreBCD, RodA, and RodZ. Work in the above species led to the hypothesis that MreB interacts with the cytoplasmic cell wall synthesis components and helps to direct the localization of the periplasmic-acting enzymes (Figge et al., 2004; Divakaruni et al., 2005, 2007; Kruse et al., 2005; Kawai et al., 2009; White et al., 2010; Ursell et al., 2014; Morgenstein et al., 2015).
Recent work has questioned the model that the MreB elongosome is a large complex composed of both the bifunctional class A PBPs (aPBPs) and monofunctional class B PBPs (bPBPs). The aPBPs contain both transglycosylation and transpeptidation activities, and thus should be functional alone, while the bPBPs, such as PBP2, possess only transpeptidation activity. However, PBP2 is essential in E. coli, and in both B. subtilis and E. coli, polymerization of glycan strands has been shown to proceed without the aPBPs (Cho et al., 2016; Meeske et al., 2016). If elongation of glycan strands occurs without aPBPs, then there must be another enzyme capable of transglycosylation reactions. RodA has been suggested to be the transglycosylase that works with PBP2 (bPBP; Cho et al., 2016; Meeske et al., 2016). These authors also showed that the RodA-PBP2 complex works with the MreB cytoskeleton while the aPBPs act separately from this complex. Our previous work studying MreB dynamics suggested that RodZ helps modulate MreB motion through interactions with RodA/PBP2, further supporting the idea that they are in a complex together (Morgenstein et al., 2015). Here we show that antibiotics that target MreB (A22) or PBP2 (mecillinam), but not aPBPs (cefsulodin) are less effective when mdh is deleted and cell wall precursor synthesis levels are proposed to be upregulated (Figure 5A), suggesting that a second PG synthesis system is dominating.
These results reinforce the model that MreB forms a cell wall synthesis complex with PBP2 but not PBP1A/B and suggest that cell wall synthesis deriving from the activity of aPBPs may be upregulated when there is an abundance of precursor molecules in the cell and the MreB elongation machinery is disrupted. Upregulation of an aPBP-specific synthesis mechanism would explain how cells can grow when MreB or PBP2 are inhibited. To test this idea, we made an mdh double deletion with either mrcA (PBP1A) or mrcB (PBP1B). Surprisingly, loss of PBP1B either alone or with mdh results in cells more sensitive to A22, but loss of PBP1A actually lead to increased tolerance to A22 (Figure 5B). PBP1A and PBP1B can compensate for each other as a single deletion is not lethal but a double deletion is lethal. When PBP1A is deleted, PBP1B may become hyperactivated in order to compensate for the loss of PBP1A, which could lead to increased resistance to A22. Our model for the effects of the mdh deletion relies on the activation of PBP1B; therefore, it would be expected that there is not an additive effect on deleting both mrcA and mdh as we propose both work through the activation of PBP1B.
To the best of our knowledge, the rate-limiting step of cell wall synthesis is currently unknown as it is very difficult to measure in vitro cell wall synthesis, which is further complicated by the rapid turnover of the PG. Possibilities include substrate availability (precursor synthesis), enzyme (PBPs) kinetics, or enzyme availability. Our results suggest that substrate availability is rate limiting, as increasing precursor synthesis provides a mechanism to bypass the need for PBP2 or MreB. Experiments that reduce substrate levels, either by modulating of the activity or cellular levels of the synthesis enzymes, will be needed in the future.
It is possible that the observed increase in UDP-NAG actually results from a decrease in cell wall synthesis. This model would also be consistent with cells becoming more sensitive to the inactivation of PBP1B; if cell wall synthesis is slowed in the mdh mutant, loss of another cell wall synthesis enzyme (PBP1B) would be more deleterious than in WT cells. However, it is unclear why the addition of glucose to the medium would result in increased tolerance. How aPBP activity is modulated when other cell wall synthesis mechanisms are inhibited is currently not known but would be an interesting topic for further study. If reduced wall synthesis was happening, one would expect that deletion of PBP1A would also reduce the MICA22 in the mdh deletion background, but this was not observed.
While MreB and PBP2 may act in the same complex, inhibition of each protein results in different effects on cell wall synthesis. MreB acts upstream of cell wall synthesis to organize the complex; therefore, A22 treatment breaks the ability of the cell to build an organized cell wall. PBP2 is an enzyme that actually builds the cell wall acting upstream of MreB and A22 inhibition. Inhibition by mecillinam and other PBP inhibitors, causes a futile cycle where not only is synthesis or crosslinking inhibited but also cell wall recycling is increased (Uehara and Park, 2008; Cho et al., 2014). While most antibiotics that target cell wall synthesis enzymes result in this futile cycle, we only see an increase in resistance to mecillinam, suggesting a specific phenotype for inhibition of the MreB-PBP2 elongasome. We propose that if precursor synthesis is elevated and MreB or PBP2 is inhibited, then the increased activity of PBP1B can break the futile cycle by using the excess precursors.
Drug Efficacy Is Affected by the Metabolic State of Cells
Antibiotics resistance normally comes from genetic changes in a cell. However, in addition to genetic changes, the efficacy of antibiotics changes with the growth state of cells. Slow growing cells show an increased tolerance to many antibiotics (Tuomanen et al., 1986; Pontes and Groisman, 2019). When only a subfraction of the population is tolerant to antibiotic exposure, the surviving cells are termed “persisters” (Brauner et al., 2016). In addition, changes in metabolic state can affect the efficacy of antibiotics. Previous studies have shown changes to antibiotic sensitivity when mutations are made in the genes involved in the TCA cycle (Helling et al., 2002; Irnov et al., 2017; Rowe et al., 2020). Mutations in icd can activate efflux pumps rendering cells more tolerant to antibiotics that can be targeted by those efflux pumps (Helling et al., 2002). Additionally, acnB mutations are thought to alter antibiotic susceptibility through decreases in cellular ATP levels (Conlon et al., 2016).
Here, we show that mutations in the metabolic genes mdh, sucC, and acnB increase a cell’s tolerance to A22. Both mdh and acnB mutations also lead to increases in mecillinam tolerance, while sucC deletion does not affect mecillinam tolerance (Supplementary Table S2). This suggests that the changes in A22 tolerance in sucC and mdh mutants work through different mechanisms. It will be interesting in the future to perform metabolite analysis on the sucC deletion strain to better understand the changes in metabolites. Do mutations in other steps of the TCA cycle increase gluconeogenesis? It would seem that they do not as only three of the eight reactions of the TCA lead to increased A22 tolerance when broken. Metabolite analysis of the sucC and acnB mutants could also help to unravel the differences in mecillinam resistance. Perhaps, the changes in ATP levels in the acnB mutant provide a general antibiotic resistance mechanism. However, SucC and Mdh occur after the formation of reducing power in the TCA cycle. While the substrate of Mdh (malate), can be directly converted into gluconeogenesis substrates, succinyl-CoA, the substrate of SucC, is more likely to be consumed in the modification of amino acids; arginine degradation, and methionine and lysine biosynthesis. It is possible that the levels of these specific amino acids relate to A22 resistance. Unfortunately, we were unable to record PEP or oxaloacetate levels in our metabolomic dataset. This leaves open the possibility that other metabolic changes have occurred that account for the increase in the antibiotic tolerance in the mdh mutant.
Previous research has shown that while persister cells are more tolerant of aminoglycoside antibiotics, the addition of different sugars specifically potentiated the effects of gentamycin and other aminoglycosides, resulting in a reduction of persister cell survival after treatment (Allison et al., 2011). The work presented here suggests that the addition of glucose has the opposite effect on the efficacy of mecillinam and decreases its effectiveness. Additionally, inhibiting ATP production can lead to increases in antibiotic tolerance which can be bypassed through the addition of glucose (Conlon et al., 2016; Shan et al., 2017; Rowe et al., 2020). While the addition of glucose may help to treat cells when using aminoglycosides by killing both actively growing cells and persister cells, the opposite is true when using mecillinam, as the addition of glucose actually increases the required dose (MIC).
Furthermore, gluconeogenic metabolism has been shown to promote infection. Colonization by E. coli of both cows and mice has been shown to increase during gluconeogenic growth, especially when undergoing competition in the gut (Miranda et al., 2004; Bertin et al., 2014). Moreover, a gluconeogenic environment leads to an increase in virulence factor expression of enterohemorrhagic E. coli (Njoroge et al., 2012). Therefore, the use of metabolites to potentially help eliminate a subset of cells, such as persister cells, may have the unintended consequences of both increasing colonization and virulence of other cells and diminishing the efficacy of other antibiotics.
The properties that make MreB an attractive drug target – conservation across many pathogens and essentiality – are true for other members of the MreB complex, including RodA and RodZ. Our results suggest that drugs that target these proteins would be less effective if gluconeogenesis is activated. As new antibiotics are developed against novel targets, these results show it is important to think about the broad function of the target protein because the efficacy of a drug may be affected in seemingly unknown ways, as we have shown that antibiotic tolerance against MreB-targeting drugs can be increased through a mutation in the TCA cycle gene mdh.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
BB performed and designed the experiments and interpreted the data. AG performed the experiments. RM designed the experiments, interpreted the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by NIGMS grant R15GM129636-01A1 awarded to RM for research related to this project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Elizabeth Ohneck, PhD and Tyrrell Conway, PhD for editing of this manuscript. We would also like to thank the Rutgers Cancer Institute of New Jersey for help with metabolomics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.664281/full#supplementary-material
References
Allison, K. R., Brynildsen, M. P., and Collins, J. J. (2011). Metabolite-enabled rradication of bacterial persisters by aminoglycosides. Nature 473, 216–220. doi: 10.1038/nature10069
Alyahya, S. A., Alexander, R., Costa, T., Henriques, A. O., Emonet, T., and Jacobs-Wagner, C. (2009). RodZ, a component of the bacterial core morphogenic apparatus. Proc. Natl. Acad. Sci. 106, 1239–1244. doi: 10.1073/pnas.0810794106
Awuni, Y., Jiang, S., Robinson, R. C., and Mu, Y. (2016). Exploring the A22-bacterial actin MreB interaction through molecular dynamics simulations. J. Phys. Chem. B 120, 9867–9874. doi: 10.1021/acs.jpcb.6b05199
Awuni, E., and Mu, Y. (2019). Effect of A22 on the conformation of bacterial actin MreB. Int. J. Mol. Sci. 20:1304. doi: 10.3390/ijms20061304
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the keio collection. Mol. Syst. Biol. 2:2006.0008. doi: 10.1038/msb4100050
Barreteau, H., Kovač, A., Boniface, A., Sova, M., Gobec, S., and Blanot, D. (2008). Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 168–207. doi: 10.1111/j.1574-6976.2008.00104.x
Bean, G. J., Flickinger, S. T., Westler, W. M., McCully, M. E., Sept, D., Weibel, D. B., et al. (2009). A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry 48, 4852–4857. doi: 10.1021/bi900014d
Beaufay, F., Coppine, J., Mayard, A., Laloux, G., De Bolle, X., and Hallez, R. (2015). A NAD-dependent glutamate dehydrogenase coordinates metabolism with cell division in Caulobacter crescentus. EMBO J. 34, 1786–1800. doi: 10.15252/embj.201490730
Belhumeur, P., and Drapeau, G. R. (1984). Regulation of cell division in Escherichia coli: properties of new FtsZ mutants. Mol. Gen. Genet. 197, 254–260. doi: 10.1007/BF00330971
Bendezú, F. O., and de Boer, P. A. J. (2008). Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 190, 1792–1811. doi: 10.1128/JB.01322-07
Bertin, Y., Deval, C., de la Foye, A., Masson, L., Gannon, V., Harel, J., et al. (2014). The gluconeogenesis pathway is involved in maintenance of enterohaemorrhagic Escherichia coli O157:H7 in bovine intestinal content. PLoS One 9:e98367. doi: 10.1371/journal.pone.0098367
Bratton, B. P., Shaevitz, J. W., Gitai, Z., and Morgenstein, R. M. (2018). MreB polymers and curvature localization are enhanced by RodZ and predict E. coli’s cylindrical uniformity. Nat. Commun. 9:2797. doi: 10.1038/s41467-018-05186-5
Brauner, A., Fridman, O., Gefen, O., and Balaban, N. Q. (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330. doi: 10.1038/nrmicro.2016.34
Chien, A. C., Zareh, S. K., Wang, Y. M., and Levin, P. A. (2012). Changes in the oligomerization potential of the division inhibitor UgtP co-ordinate Bacillus subtilis cell size with nutrient availability. Mol. Microbiol. 86, 594–610. doi: 10.1111/mmi.12007
Cho, H., Uehara, T., and Bernhardt, T. G. (2014). Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159, 1300–1311. doi: 10.1016/j.cell.2014.11.017
Cho, H., Wivagg, C. N., Kapoor, M., Barry, Z., Rohs, P. D. A., Suh, H., et al. (2016). Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol. 1:16172. doi: 10.1038/nmicrobiol.2016.172
Conlon, B. P., Rowe, S. E., Gandt, A. B., Nuxoll, A. S., Donegan, N. P., Zalis, E. A., et al. (2016). Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 1:16051. doi: 10.1038/nmicrobiol.2016.51
Curtis, N. A., Orr, D., Ross, G. W., and Boulton, M. G. (1979). Affinities of penicillins and cephalosporins for the penicillin-binding proteins of Escherichia coli K-12 and their antibacterial activity. Antimicrob. Agents Chemother. 16, 533–539. doi: 10.1128/AAC.16.5.533
Divakaruni, A. V., Baida, C., White, C. L., and Gober, J. W. (2007). The cell shape proteins MreB and MreC control cell morphogenesis by positioning cell wall synthetic complexes. Mol. Microbiol. 66, 174–188. doi: 10.1111/j.1365-2958.2007.05910.x
Divakaruni, A. V., Loo, R. R. O., Xie, Y., Loo, J. A., and Gober, J. W. (2005). The cell-shape protein MreC interacts with extracytoplasmic proteins including cell wall assembly complexes in Caulobacter crescentus. Proc. Natl. Acad. Sci. U. S. A. 102, 18602–18607. doi: 10.1073/pnas.0507937102
Elbaz, M., and Ben-Yehuda, S. (2010). The metabolic enzyme ManA reveals a link between cell wall integrity and chromosome morphology. PLoS Genet. 6:e1001119. doi: 10.1371/journal.pgen.1001119
Figge, R. M., Divakaruni, A. V., and Gober, J. W. (2004). MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol. Microbiol. 51, 1321–1332. doi: 10.1111/j.1365-2958.2003.03936.x
Gitai, Z., Dye, N. A., Reisenauer, A., Wachi, M., and Shapiro, L. (2005). MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120, 329–341. doi: 10.1016/j.cell.2005.01.007
Helling, R. B., Janes, B. K., Kimball, H., Tran, T., Bundesmann, M., Check, P., et al. (2002). Toxic waste disposal in Escherichia coli. J. Bacteriol. 184, 3699–3703. doi: 10.1128/JB.184.13.3699-3703.2002
Hernandez-Asensio, M., Ramirez, J. M., and Del Campo, F. F. (1975). The control by respiration of the uptake of α-methyl-glucoside in Escherichia coli K12. Arch. Microbiol. 103, 155–162. doi: 10.1007/BF00436343
Hill, N. S., Buske, P. J., Shi, Y., and Levin, P. A. (2013). A moonlighting enzyme links Escherichia coli cell size with central metabolism. PLoS Genet. 9:e1003663. doi: 10.1371/journal.pgen.1003663
Irnov, I., Wang, Z., Jannetty, N. D., Bustamante, J. A., Rhee, K. Y., and Jacobs-Wagner, C. (2017). Crosstalk between the tricarboxylic acid cycle and peptidoglycan synthesis in Caulobacter crescentus through the homeostatic control of α-ketoglutarate. PLoS Genet. 13:e1006978. doi: 10.1371/journal.pgen.1006978
Iwai, N., Nagai, K., and Wachi, M. (2002). Novel S-benzylisothiourea compound that induces spherical cells in Escherichia coli probably by acting on a rod-shape-determining protein(s) other than penicillin-binding protein 2. Biosci. Biotechnol. Biochem. 66, 2658–2662. doi: 10.1271/bbb.66.2658
Kawai, Y., Daniel, R. A., and Errington, J. (2009). Regulation of cell wall morphogenesis in Bacillus subtilis by recruitment of PBP1 to the MreB helix. Mol. Microbiol. 71, 1131–1144. doi: 10.1111/j.1365-2958.2009.06601.x
Komatsuzawa, H., Fujiwara, T., Nishi, H., Yamada, S., Ohara, M., McCallum, N., et al. (2004). The gate controlling cell wall synthesis in Staphylococcus aureus. Mol. Microbiol. 53, 1221–1231. doi: 10.1111/j.1365-2958.2004.04200.x
Kruse, T., Bork-Jensen, J., and Gerdes, K. (2005). The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 55, 78–89. doi: 10.1111/j.1365-2958.2004.04367.x
Landgraf, D., Okumus, B., Chien, P., Baker, T. A., and Paulsson, J. (2012). Segregation of molecules at cell division reveals native protein localization. Nat. Methods 9, 480–482. doi: 10.1038/nmeth.1955
Leclercq, S., Derouaux, A., Olatunji, S., Fraipont, C., Egan, A. J. F., Vollmer, W., et al. (2017). Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Sci. Rep. 7:43306. doi: 10.1038/srep43306
Meeske, A. J., Riley, E. P., Robins, W. P., Uehara, T., Mekalanos, J. J., Kahne, D., et al. (2016). SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638. doi: 10.1038/nature19331
Mengin-Lecreulx, D., Flouret, B., and van Heijenoort, J. (1983). Pool levels of UDP N-acetylglucosamine and UDP N-acetylglucosamine-enolpyruvate in Escherichia coli and correlation with peptidoglycan synthesis. J. Bacteriol. 154, 1284–1290. doi: 10.1128/JB.154.3.1284-1290.1983
Miranda, R. L., Conway, T., Leatham, M. P., Chang, D. E., Norris, W. E., Allen, J. H., et al. (2004). Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect. Immun. 72, 1666–1676. doi: 10.1128/IAI.72.3.1666-1676.2004
Monahan, L. G., Hajduk, I. V., Blaber, S. P., Charles, I. G., and Harry, E. J. (2014). Coordinating bacterial cell division with nutrient availability: a role for glycolysis. MBio 5:e00935–14. doi: 10.1128/mBio.00935-14
Moore, D. A., Whatley, Z. N., Joshi, C. P., Osawa, M., and Erickson, H. P. (2017). Probing for binding regions of the FtsZ protein surface through site-directed insertions: discovery of fully functional FtsZ-fluorescent proteins. J. Bacteriol. 199:e00553–16. doi: 10.1128/JB.00553-16
Morgenstein, R. M., Bratton, B. P., Ouzounov, N., Nguyen, J. P., Shaevitz, J. W., and Gitai, Z. (2015). RodZ links MreB to cell wall synthesis to mediate MreB rotation and robust morphogenesis. Proc. Natl. Acad. Sci. 112, 12510–12515. doi: 10.1073/pnas.1509610112
Njoroge, J. W., Nguyen, Y., Curtis, M. M., Moreira, C. G., and Sperandio, V. (2012). Virulence meets metabolism: Cra and KdpE gene regulation in enterohemorrhagic Escherichia coli. MBio 3:e00280–12. doi: 10.1128/mBio.00280-12
Pierucci, O. (1978). Dimensions of Escherichia coli at various growth rates: model for envelope growth. J. Bacteriol. 135, 559–574. doi: 10.1128/JB.135.2.559-574.1978
Pontes, M. H., and Groisman, E. A. (2019). Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci. Signal. 12:eaax3938. doi: 10.1126/scisignal.aax3938
Preston, D. A., Wu, C. Y., Blaszczak, L. C., Seitz, D. E., and Halligan, N. G. (1990). Biological characterization of a new radioactive labeling reagent for bacterial penicillin-binding proteins. Antimicrob. Agents Chemother. 34, 718–721. doi: 10.1128/AAC.34.5.718
Rogers, D., and Yu, S.-H. (1962). Substrate specificity of a glucose permease of Escherichia coli. J. Bacteriol. 84, 877–881. doi: 10.1128/JB.84.5.877-881.1962
Rowe, S. E., Wagner, N. J., Li, L., Beam, J. E., Wilkinson, A. D., Radlinski, L. C., et al. (2020). Reactive oxygen species induce antibiotic tolerance during systemic Staphylococcus aureus infection. Nat. Microbiol. 5, 282–290. doi: 10.1038/s41564-019-0627-y
Sargent, M. G. (1975). Control of cell length in Bacillus subtilis. J. Bacteriol. 123, 7–19. doi: 10.1128/JB.123.1.7-19.1975
Sauer, U., and Eikmanns, B. J. (2005). The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 29, 765–794. doi: 10.1016/j.femsre.2004.11.002
Schaechter, M., Maaloe, O., and Kjeldgaard, N. O. (1958). Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J. Gen. Microbiol. 19, 592–606. doi: 10.1099/00221287-19-3-592
Shan, Y., Brown Gandt, A., Rowe, S. E., Deisinger, J. P., Conlon, B. P., and Lewis, K. (2017). ATP-dependent persister formation in Escherichia coli. MBio 8:e02267–16. doi: 10.1128/mBio.02267-16
Spratt, B. G. (1977). The mechanism of action of mecillinam. J. Antimicrob. Chemother. 3(Suppl. B), 13–19. doi: 10.1093/jac/3.suppl_b.13
Su, X., Chiles, E., Maimouni, S., Wondisford, F. E., Zong, W. X., and Song, C. (2020). In-source CID ramping and covariant ion analysis of hydrophilic interaction chromatography metabolomics. Anal. Chem. 92, 4829–4837. doi: 10.1021/acs.analchem.9b04181
Tuomanen, E., Cozens, R., Tosch, W., Zak, O., and Tomasz, A. (1986). The rate of killing of Escherichia coli by β-lactam antibiotics is strictly proportional to the rate of bacterial growth free. J. Gen. Microbiol. 132, 1297–1304. doi: 10.1099/00221287-132-5-1297
Uehara, T., and Park, J. T. (2008). Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and eeptation. J. Bacteriol. 190, 3914–3922. doi: 10.1128/JB.00207-08
Ursell, T., Lee, T. K., Shiomi, D., Shi, H., Tropini, C., Monds, R. D., et al. (2017). Rapid, precise quantification of bacterial cellular dimensions across a genomic-scale knockout library. BMC Biol. 15:17. doi: 10.1186/s12915-017-0348-8
Ursell, T. S., Nguyen, J., Monds, R. D., Colavin, A., Billings, G., Ouzounov, N., et al. (2014). Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc. Natl. Acad. Sci. 111, E1025–E1034. doi: 10.1073/pnas.1317174111
van den Ent, F., Amos, L. A., and Lowe, J. (2001). Prokaryotic origin of the actin cytoskeleton. Nature 413, 39–44. doi: 10.1038/35092500
van den Ent, F., Izoré, T., Bharat, T. B., Johnson, C. M., and Lowe, J. (2014). Bacterial actin MreB forms antiparallel double filaments. elife 3:e02634. doi: 10.7554/eLife.02634
Vinella, D., Joseleau-Petit, D., Thévenet, D., Bouloc, P., and D’Ari, R. (1993). Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J. Bacteriol. 175, 6704–6710. doi: 10.1128/JB.175.20.6704-6710.1993
Ward, J. E. Jr., and Lutkenhaus, J. (1985). Overproduction of FtsZ induces minicell formation in E. coli. Cell 42, 941–949. doi: 10.1016/0092-8674(85)90290-9
Weart, R. B., Lee, A. H., Chien, A.-C., Haeusser, D. P., Hill, N. S., and Levin, P. A. (2007). A metabolic sensor governing cell size in bacteria. Cell 130, 335–347. doi: 10.1016/j.cell.2007.05.043
White, C. L., Kitich, A., and Gober, J. W. (2010). Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol. Microbiol. 76, 616–633. doi: 10.1111/j.1365-2958.2010.07108.x
Keywords: MreB cytoskeleton, antibiotic resistance, gluconeogenesis, tricarboxylic acid cycle, elongasome
Citation: Barton B, Grinnell A and Morgenstein RM (2021) Disruption of the MreB Elongasome Is Overcome by Mutations in the Tricarboxylic Acid Cycle. Front. Microbiol. 12:664281. doi: 10.3389/fmicb.2021.664281
Edited by:
Peter Graumann, University of Marburg, GermanyReviewed by:
William Margolin, University of Texas Health Science Center at Houston, United StatesAkira Nakamura, University of Tsukuba, Japan
Copyright © 2021 Barton, Grinnell and Morgenstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Randy M. Morgenstein, cmFuZHkubW9yZ2Vuc3RlaW5Ab2tzdGF0ZS5lZHU=