 Junqing Luo†
Junqing Luo† Wei Guo
Wei Guo- Inner Mongolia Key Laboratory of Environmental Pollution Control and Waste Resource Recycle, Ministry of Education Collaborative Innovation Center for Grassland Ecological Security, Ministry of Education Key Laboratory of Ecology and Resource Use of the Mongolian Plateau, School of Ecology and Environment, Inner Mongolia University, Hohhot, China
Lake littoral zones can also be regarded as another extremely hypersaline environment due to hypersaline properties of salt lakes. In this study, high-throughput sequencing technique was used to analyze bacteria and fungi from different rhizocompartments (rhizosphere and endosphere) of four dominant plants along the salinity gradient in the littoral zones of Ejinur Salt Lake. The study found that microbial α-diversity did not increase with the decrease of salinity, indicating that salinity was not the main factor on the effect of microbial diversity. Distance-based redundancy analysis and regression analysis were used to further reveal the relationship between microorganisms from different rhizocompartments and plant species and soil physicochemical properties. Bacteria and fungi in the rhizosphere and endosphere were the most significantly affected by SO42–, SOC, HCO3–, and SOC, respectively. Correlation network analysis revealed the potential role of microorganisms in different root compartments on the regulation of salt stress through synergistic and antagonistic interactions. LEfSe analysis further indicated that dominant microbial taxa in different rhizocompartments had a positive response to plants, such as Marinobacter, Palleronia, Arthrobacter, and Penicillium. This study was of great significance and practical value for understanding salt environments around salt lakes to excavate the potential microbial resources.
Introduction
Salt lakes belong to extremely hypersaline environments and they are widely distributed all over the world. The salt lakes account for about half of the total inland aquatic ecosystems (Yang et al., 2016). For China, salt lakes are generally distributed in northwest China, such as Tibet, Qinghai, Xinjiang, and Inner Mongolia (Zheng et al., 1993). Lakes in these areas provide water resources and play an important role in the fragile environment (Zhang et al., 2020). In addition, although salt inhibits the diversity of microorganisms, certain microorganisms living in salt lakes still have high activity and involve many geochemical cycles necessary for life, such as carbon, nitrogen, and sulfur (Sorokin et al., 2014). Therefore, salt lakes are often considered as one of the best choices for studying the relationship between microbial diversity and environmental factors in extremely hypersaline environments (Kalwasinska et al., 2019). Importantly, due to hypersaline properties of salt lakes, the microbial communities inhabited in lake littoral zone soils may present particularity. The lake littoral zone is a functional transition zone connecting lake aquatic ecosystem and terrestrial ecosystem (Kou et al., 2020). Thus, the lake littoral zones of salt lakes are similarly regarded as an extremely high salt environment.
There is well-known basic work on the genetic diversity and structure of microbial communities in salt lakes, in which halophilic microorganisms (including bacteria and archaea) isolated and identified from the salt lakes and their sediments can be regarded as an important part of biological community (Guven et al., 2018; Selivanova et al., 2018). At present, most of the sampling points in these studies are from estuaries, salt ponds, and sediments of salt lakes (Wu et al., 2006; Boujelben et al., 2012; Liu et al., 2015). However, few researchers have attended to the bacterial and fungal diversity in the littoral zone soils of salt lakes. Salinity is the primary environmental determinant for microbial communities in different saline ecosystems, salt lakes, and other saline soils (Oren, 2008; Hrynkiewicz et al., 2015; Zhong et al., 2016). The salinity increase had negative impacts on soil microbial activity (Chen et al., 2017). However, it is unknown how soil microbial communities change along the salinity gradients in the littoral zones of salt lakes, because there are different dominant plant species along the salinity gradient in the littoral zones of salt lakes (Williams, 1991; Apaydin et al., 2009). Therefore, it is important to ascertain the dominant factors for the diversity and structure of the microbial community under different salinities in the littoral zone of the salt lakes.
Several studies have shown that soil microorganisms exert important effects on maintaining soil health and helping plants resist adverse environments (Wang et al., 2016; Jiang et al., 2019). Beneficial microorganisms can promote plant growth under salt stress. Rhizosphere bacteria from Salicornia strobilacea has been shown to settle stably on the root surface and promote the growth of its host plants (Yamamoto et al., 2020). Fan et al. (2016) found that salt-tolerant bacteria isolated from the rhizosphere of Suaeda salsa growing on saline soil can be used to alleviate the salt stress of crops. Many studies have revealed that certain bacteria play beneficial effects in plants and regulate a variety of direct or indirect mechanisms by the ways of rhizosphere colonization at the high-salt conditions (Paul and Lade, 2014; Mallick et al., 2018). Ali et al. (2014) found that endophytic bacteria containing 1-Aminocyclopropane-1-carboxylate deaminase (ACC deaminase) can promote plant growth and development under salt stress, but some endophytic fungi from saline–alkaline soils usually stimulate plant growth and give plants tolerance to biotic or abiotic stresses (Khan et al., 2011; Pan et al., 2018). Recent works have successfully used the cooperative relationship among plants and rhizospheric and endophytic bacteria to effectively remediate soil (Fatima et al., 2015; Hussain et al., 2018). Therefore, it is crucial to understand the microbial composition from different rhizocompartments of the plants in hypersaline soil environments, particularly around salt lakes. Exploring the mode of different rhizocompartments of microorganisms of dominant plants in the littoral zones of salt lakes is important to understand how microorganisms help plants resist saline stress and maintain the stability of different saline environments.
In recent years, high-throughput sequencing has been widely used to determine the diversity and structure of microbial communities, understanding the interaction and adaptability of microbes in the environment (Valenzuela-Encinas et al., 2008; Han et al., 2017). In this study, the salt lakes areas of Inner Mongolia Plateau have naturally formed a good soil salinity gradient, and it has become the better experimental platform for studying the microbial diversity in different rhizocompartments of various plants (Du et al., 2007). Through the investigation, dominant plants were discovered to have obvious regional divisions along the salt gradient changes, and the salinization degree of the littoral zone soil environments decreased gradually from the edge of the lake to the periphery. Therefore, we investigated the microbial communities of rhizosphere soil samples and root endophytic samples of four different plants under salinity gradient around Ejinur Salt Lake. Our aim was to (1) study the difference and relationship of rhizosphere soil and endophytic microbial community structure under salinity gradient; (2) explore the dominant microorganisms that can resist salt stress in plants in different soil environments of the littoral zones of salt lakes along the salinity gradient; and (3) reveal the dominant factors affecting microorganisms in different rhizocompartments. This study provided new insights for in-depth understanding of the roles of microorganisms, which was beneficial to excavate the potential microbial resources resistant to saline–alkali stress in the littoral zones of salt lakes.
Materials and Methods
Study Sites and Soil Preparation
The Ejinur Salt Lake is located in the north of Dabusu basin in Inner Mongolia Plateau, China (116°32′44′′E, 45°14′34′′N), which is characterized by a typical temperate semi-arid continental climate. The average annual precipitation is 255 mm, and the soil types primarily belong to the saline–alkali soil of chloride type. Rhizosphere soils were sampled along the salinity gradient in the littoral zones of Ejinur Salt Lake in September 2019. Four sampling points [rhizosphere soil sample points were marked as S. europaea rhizosphere soils (GA), S. salsa rhizosphere soils (GB), Phragmites communis rhizosphere soils (GC), and Achnatherum splendens rhizosphere soils (GD)] were set up along the salinity gradient, and the plant species changed with the distance from the lake shore. Each sample point was composed of three sub-sample points (Rath et al., 2019) (a single sample point uses the four corners of a 10 m × 10 m quadrilateral and a center point to collect plants sample, and the three points on the diagonal are collected rhizosphere soil samples) at four sample points of the salinity gradient. We excavated whole plants including the surrounding soil in blocks. The collected soil and plant samples were stored in an incubator at 4°C and transported them to the laboratory.
The soil close to the roots (0–3 mm from the roots’ surface) was defined as the rhizosphere soil (Zhang and Kong, 2014; Tian and Zhang, 2017). The rhizosphere soil samples in the root zone were gently separated from the roots according to the method described by Hrynkiewicz et al. (2010). Subsequently, the separated rhizosphere soil samples were air-dried and sieved through a 2-mm sieve and used to determine physical and chemical indicators. In the aseptic operating conditions, the plant roots with firmly attached soil were put into a centrifuge tube containing 50 ml of sterile water and shaken using a shaker (4°C) (Simmons et al., 2018; Wang et al., 2020). After the plant roots were removed from the centrifuge tube, the centrifuge tube containing soils was centrifuged (4°C, 8,000 rpm) for 10 min (Edwards et al., 2015). After the supernatants were removed, the remaining sediments were used as rhizosphere soils for microbial determination. The plant roots were soaked in 70% ethanol for 2 min and 2.5% NaClO for 5 min and then transferred to 70% sterile ethanol and soaked for 30 s. Finally, the plant root tissue was washed five times with sterile water. The treated rhizosphere soils and plant roots were stored in a refrigerator at −80°C for subsequent high-throughput sequencing.
Soil Physical and Chemical Properties
The separated soils were placed in an oven and dried to a constant weight at 105°C. The water content of the soil (MC) was determined by the percentage of the weight of the soil lost water to the weight of the dried soil. The previously air-dried and sieved soil pH values were measured at (1:2.5 w/v) soil:water extraction ratio by the pH potentiometer. Soil electrical conductivity (EC) values were measured using an EC meter after shaking the soil:water (1:5 w/v) mixture. Total nitrogen (TN) and soil organic carbon (SOC) were analyzed using an elemental analyzer (Vario MACRO, CHNOS Elemental Analyzer, Elementar Co., Germany). Soils ranging in mass about 0.2–0.3 g were digested with nitric acid (HNO3), hydrofluoric acid (HF), hydrochloric acid (HCl), and perchloric acid (HClO4) (guaranteed reagent) (Sommers and Nelson, 1972). After the digestion, samples were centrifuged, decanted, diluted, and transferred quantitatively to centrifuge tubes, and total phosphorus (TP) was analyzed using an inductively coupled plasma optical emission spectroscopy (ICP-OES, Optima 7000 DV, PerkinElmer, United States). The total salt content of the soil was sent to an external laboratory (Inner Mongolia Hengtai Testing Technology Co., Ltd., China) to complete, including carbonate (CO32–), bicarbonate (HCO3–), sulfate (SO42–), chloride ion (Cl–), calcium ion (Ca2+), magnesium ion (Mg2+), and sodium ion and potassium ion (Na+ and K+). The total salt content was measured at least three times using the standard measurement method, and the measurement standard was referenced to the determination of the total water-soluble salt in the soil (NY/T 1121.16-2006). The measured values of physical and chemical factors are shown in Table 1. For more information on soil property, see Supplementary Table 1.
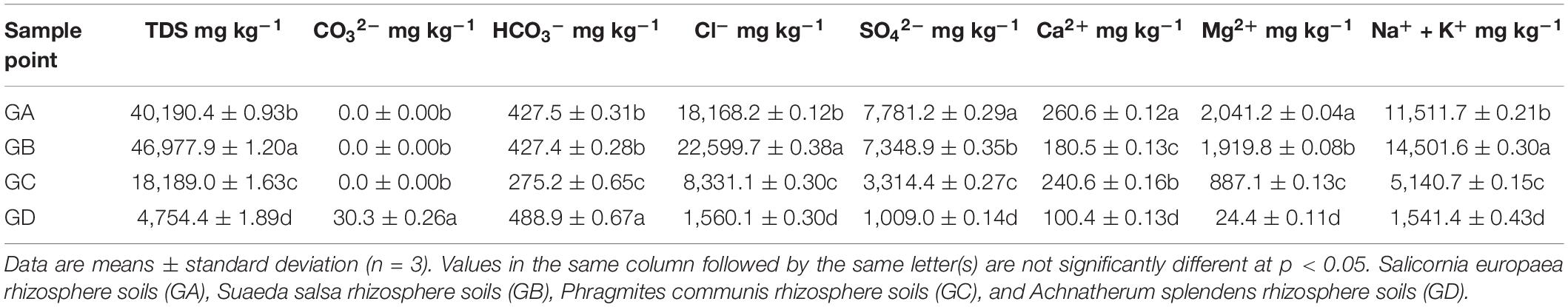
Table 1. The total salt and eight major ions in plant rhizosphere soil around the Ejinur Salt Lake.
DNA Extraction and High-Throughput Sequencing
Microbial community genomic DNA was extracted from rhizosphere soils and root endophytic samples using the E.Z.N.A.®, soil DNA Kit (Omega Bio-tek, Norcross, GA, United States) according to the manufacturer’s instructions. The DNA extract was checked on 1% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, DE, United States). The hypervariable region V3–V4 of the bacterial 16S rRNA gene was amplified with primer pairs 338F (5′-ACT CCT ACG GGA GGC AGC AG-3′) and 806R (5′-ACT CCT ACG GGA GGC AGC AG-3′) (for rhizosphere soil bacterial community), and 799F (5′-AAC MGG ATT AGA TAC CCK G-3′) and 1193R (5′-ACG TCA TCC CCA CCT TCC-3′) (for endophytic bacterial community), respectively. ITS hypervariable regions of the fungal ITS rRNA gene were amplified with primer pairs ITS1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) and ITS2R (5′-GCT GCG TTC TTC ATC GAT GC-3′) (for rhizospheric and endophytic fungal community). Briefly, PCR amplification had the following conditions: initial denaturation at 95°C for 3 min, denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s, a total of 40 cycles, and final extension at 72°C for 10 min; the experiment was repeated three times. The reaction product was detected by 2% agarose gel electrophoresis. The purified amplicons were paired-end sequenced (2 × 300 bp) on an Illumina MiSeq platform according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The raw sequencing data have been deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Accession Number: PRJNA669550).
Sequence Data Processing
The unique sequences among all reads were used to define operational taxonomic units (OTUs) using UPARSE version 7.1 with 97% sequence similarity (Edgar, 2013). In order to compare different samples at the same sequencing level, the samples were flattened according to the minimum number of sample sequences (Fang et al., 2018), and the representative sequences in each OTU of bacteria and fungi were compared with the Silva132 and 16S rRNA bacteria and Unite7.2 and ITS rRNA fungi database (Wang et al., 2007). The sample rarefaction curve was used to evaluate the sequencing depth of the high-throughput sequencing results of bacteria and fungi in eight groups of rhizosphere soil samples and eight groups of plant root samples. The evaluation results are shown in Supplementary Files and Supplementary Figure 1. At the same time, the alpha diversity (Shannon index and Sobs index) of each group of samples was analyzed, and species richness analysis in 16 groups of samples was performed.
Statistical Analyses
All data were analyzed by SPSS 17.0 software, and the data were reflected as the mean ± standard deviation (SD) based on three replicates. The statistical significance of the difference between the means was performed by one-way analysis of variance (ANOVA). Significance was calculated by Duncan’s multiple range test at the p < 0.05 level.
The results of microbial α-diversity were drawn by using Origin 2017 software. Regression analysis was used to show linear correlation between plant species (PBI) and soil environmental factors and β-diversity (PC1 axis value in PCoA results based on Bray–Curtis) of different rhizocompartments of microorganisms (details are in the Supplementary Materials). Distance-based redundancy analysis (db-RDA) was used to analyze the microbial community structure and the relationship between the microbial communities and environmental factors based on Bray–Curtis (McArdle and Anderson, 2001). The environmental factors included SOC, TP, TN, TDS, CO32––, HCO3––, SO42––, Cl––, and Ca2+. Linear discriminant analysis effect size (Lefse) was performed online1. The logarithmic LDA score was set to 4.0 with statistical significance (p < 0.05). LEfSe was conducted to identify taxa that display significant differences from phylum to genus level in the rhizosphere soil and root endophytic samples. Microbial correlation networks were constructed to explore separately the interactions of bacteria and fungi from rhizospheric and root endophytic samples. The bacterial and fungal networks showed different patterns of correlation based on significant correlations (Spearman’s correlation coefficients, | r| > 0.6, p < 0.01). Networks were visualized in Cytoscape 3.4.0 (Shannon et al., 2003). PICRUSt2 was used to predict the bacterial and fungal metabolic pathways based on MetaCyc database (Caspi et al., 2020).
Results
Analysis of Soil Physicochemical Parameters
From the edge of the Ejinur Salt Lake to its periphery, the degree of soil salinization decreased gradually and plants showed obvious zonal distribution characteristics [the dominant plants were S. europaea (GA), S. salsa (GB), P. communis (GC), and A. splendens (GD)]. Rhizosphere soil samples in this study were all alkaline soil (pH was from 7.64 to 8.47) (Supplementary Table 1). Table 1 shows the total salt content and eight major ion contents of rhizosphere soil samples. One-way ANOVA analysis shows that the total salt content and eight major ion contents of rhizosphere soil changed significantly. Soil vegetation covers were closely related to the soil parameters. For instance, S. europaea rhizosphere soils were characterized with relative high concentrations of SO42–, Ca2+, Mg2+ (Table 1), TN, and MC (Supplementary Table 1). S. salsa rhizosphere soils featured relatively high TDS, Cl–, Na+, and K+ (Table 1), SOC, TP, and EC levels (Supplementary Table 1), while A. splendens rhizosphere soils had high levels of CO32–, HCO3–, and pH (Table 1 and Supplementary Table 1).
Microbial Community Composition and Their α-Diversity
Based on 97% sequence similarity, the bacterial and fungal sequences of rhizosphere soil samples were aggregated into 6,497 and 996 OTUs, and the bacterial and fungal sequences of root endophytic samples were aggregated into 1,153 and 279 OTUs. The rarefaction curve for observing the rhizosphere soil and the endophytic bacteria and fungi OTUs reached a plateau, indicating that sequencing effort was sufficient to represent the entire bacterial and fungal diversity (Supplementary Figure 1). In filtered high-throughput sequences, the results demonstrated that the composition of major bacterial phylum or sub-phylum in rhizosphere soil and root endophytic samples was not the same, of which 12 bacterial and 3 fungal major phyla in rhizosphere soil samples accounted for more than 95%. For root endophytic samples, seven bacterial and two fungal major phyla accounted for >95%. Proteobacteria (31.55–40.20%) and Actinobacteria (7.99–35.35%) were the major bacterial groups in rhizosphere soil, representing more than 40.00% of the bacterial sequences. The bacterial groups in the rhizosphere soil were Bacteroidetes, Firmicutes, Chloroflexi, Gemmatimonadetes, Patescibacteria, Acidobacteria, Deinococcus-Thermus, Halanaerobiaeota, Planctomycetes, and Cyanobacteria (Supplementary Figure 2). For the root endophytic samples, Proteobacteria (49.63–64.56%) and Actinobacteria (5.78–32.01%) were mainly the predominant bacterial groups, representing more than 60.00% of the bacterial sequences. Bacteroidetes, Firmicutes, Planctomycetes, Chloroflexi, and Fibrobacteres were bacterial groups in root endophytic samples (Supplementary Figure 2). Ascomycota (94.27–96.77%) was the most abundant fungal phylum in rhizosphere soil followed by Basidiomycota (2.10–5.51%) (Supplementary Figure 2). In contrast, the most abundant fungal phylum of the endosphere was Ascomycota, which accounted for 72.65–99.98%, followed by Basidiomycota, which accounted for 0.00–27.32% (Supplementary Figure 2). The main microbial communities of rhizosphere soil and root endophytic samples are shown in Figure 1, and their composition at the genus level was also not the same. The relative abundances of the main genera are shown in Supplementary Table 2.
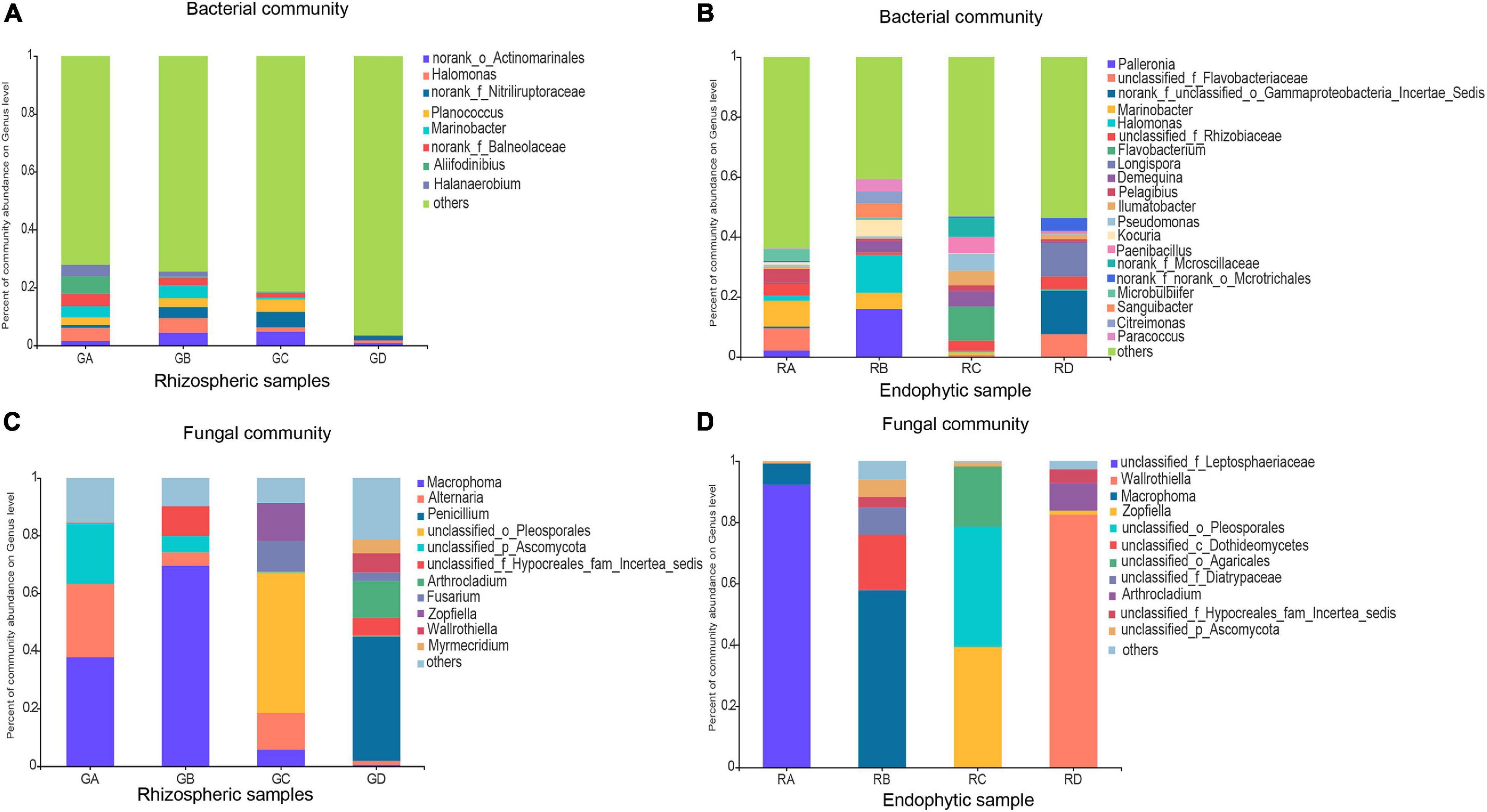
Figure 1. Effects of rhizosphere soil and root endophytic samples on the community composition at the genus level of Bacteria and Fungi in the Ejinur Salt Lake. Genus with relative abundance less than 4% were defined as others. (A) Bacterial rhizospheric samples, (B) bacterial endophytic samples, (C) fungal rhizospheric samples, and (D) fungal endophytic samples. Salicornia europaea (A), Suaeda salsa (B), Phragmites communis (C), and Achnatherum splendens (D).
To compare the significant differences in microbial diversity between samples, α-diversity was represented by statistical methods (Figure 2). The microbial communities belonged to rhizosphere soil and root endophytic samples. Among them, the observed value of bacteria is the highest, followed by fungi. In addition, it was demonstrated that the abundance and diversity of microorganisms in rhizosphere soil were higher than that of root endophytic samples. For bacterial community, the microbial richness of rhizospheric samples A (2319 ± 99.35) and endophytic samples A (389.33 ± 25.42) were the higher. The rhizospheric sample C (6.08 ± 0.01) and endophytic sample A (4.27 ± 0.11) had the higher microbial diversity. For fungal community, the microbial richness (373 ± 30.05) and diversity (2.51 ± 0.14) of rhizospheric sample A were higher. Endophytic sample B had higher microbial richness (85.33 ± 40.02) and diversity (1.28 ± 0.54) (Figure 2 and Supplementary Table 3).
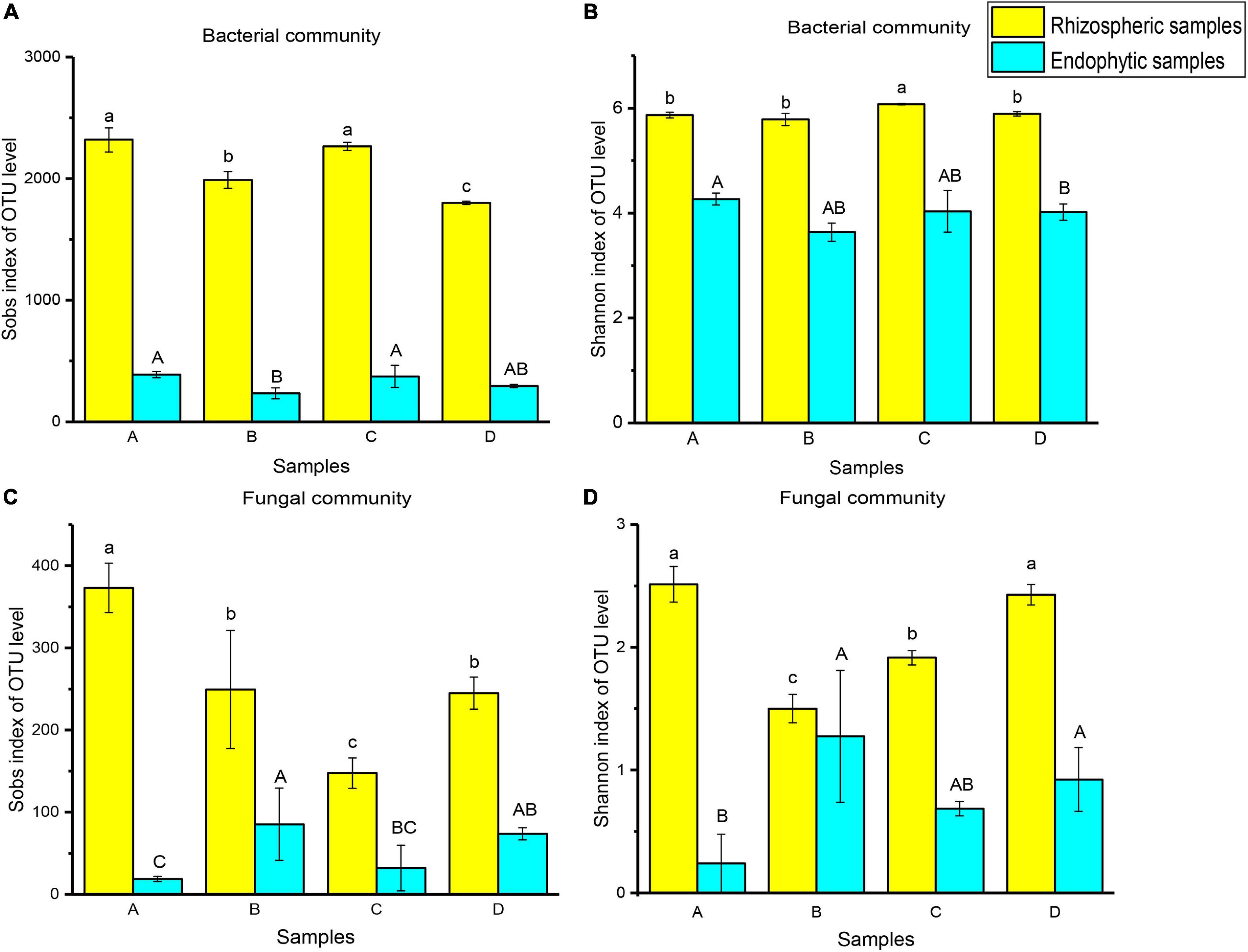
Figure 2. Ejinur Salt Lake different rhizosphere soil and root endophytic samples of microbial α-diversity. Different uppercase (Endophytic samples) and lowercase (Rhizospheric samples) letters indicate significant differences among plant species (p < 0.05). The abscissa letters indicate the plant species [S. europaea (A), S. salsa (B), P. communis (C), and A. splendens (D)].
Distance-Based Redundancy Analysis and Variance Distribution Analysis of Microbial Communities and Soil Characteristics
In both rhizospheric samples and endophytic samples, the plant species were positively correlated with the β-diversity of the microbial communities (Supplementary Figures 3–6). Regression analysis showed that the PC1 axis value in the PCoA results increased with increasing CO32– (r = 0.957, p < 0.0001) in bacterial rhizospheric samples. The PC1 axis value decreased with increasing TP (r = 0.895, p < 0.001), SOC (r = 0.815, p < 0.01), TDS (r = 0.916, p < 0.0001), SO42– (r = 0.933, p < 0.0001), Cl– (r = 0.912, p < 0.0001), and Ca2+ (r = 0.820, p < 0.01) in bacterial rhizospheric samples. For bacterial endophytic samples, TP (r = 0.952, p < 0.0001), SOC (r = 0.947, p < 0.0001), TDS (r = 0.974, p < 0.0001), SO42– (r = 0.942, p < 0.0001), Cl– (r = 0.979, p < 0.0001), and Ca2+ (r = 0.592, p < 0.05) were negatively correlated with microbial β-diversity, while CO32– (r = 0.851, p < 0.001) was positively correlated with microbial β-diversity. For fungal community, the PC1 axis value decreased with increasing TP (r = 0.860, p < 0.001), SOC (r = 0.777, p < 0.01), TDS (r = 0.884, p < 0.001), SO42– (r = 0.906, p < 0.0001), Cl– (r = 0.879, p < 0.001), and Ca2+ (r = 0.856, p < 0.001) in fungal rhizospheric samples. Similarly, the PC1 axis value decreased with increasing TP (r = 0.940, p < 0.0001), SOC (r = 0.789, p < 0.01), TDS (r = 0.938, p < 0.0001), SO42– (r = 0.979, p < 0.0001), Cl– (r = 0.924, p < 0.0001), and Ca2+ (r = 0.736, p < 0.01) in fungal endophytic samples. Moreover, the PC1 axis value increased with increasing CO32– (r = 0.973, p < 0.0001) in fungal rhizospheric samples, while the PC1 axis value was not significantly correlated with CO32– in the fungal endophytic samples.
The distance-based redundancy analysis (db-RDA) was used to reflect the relationship between microbial structure and soil characteristics (Figure 3). The results showed that the significant differences in microbial community structure at the OTU level and the salinity gradient could influence the rhizospheric and endophytic microbial structure (Figures 3A–D). The microbial structure could be determined according to the size of the angle between bacterial or fungal and environmental factors and the length of the connection in distance-based redundancy analysis. Among all the environmental parameters, the SO42– (R2 = 0.9963, p = 0.001) had a significant influence on the rhizosphere bacterial communities (Figure 3A), while the SOC (R2 = 0.9848, p = 0.001) had the greatest effect on endophytic bacterial communities (Figure 3B). For the fungal communities, the HCO3– (R2 = 0.9906, p = 0.001) and SOC (R2 = 0.6906, p = 0.007) were significantly related to the rhizospheric fungal communities and the endophytic fungal communities, respectively (Figures 3C,D). Overall, the influence of environmental factors on bacterial communities were significantly greater than that of fungal communities, and the results showed that bacteria are more widely affected by soil characteristics.
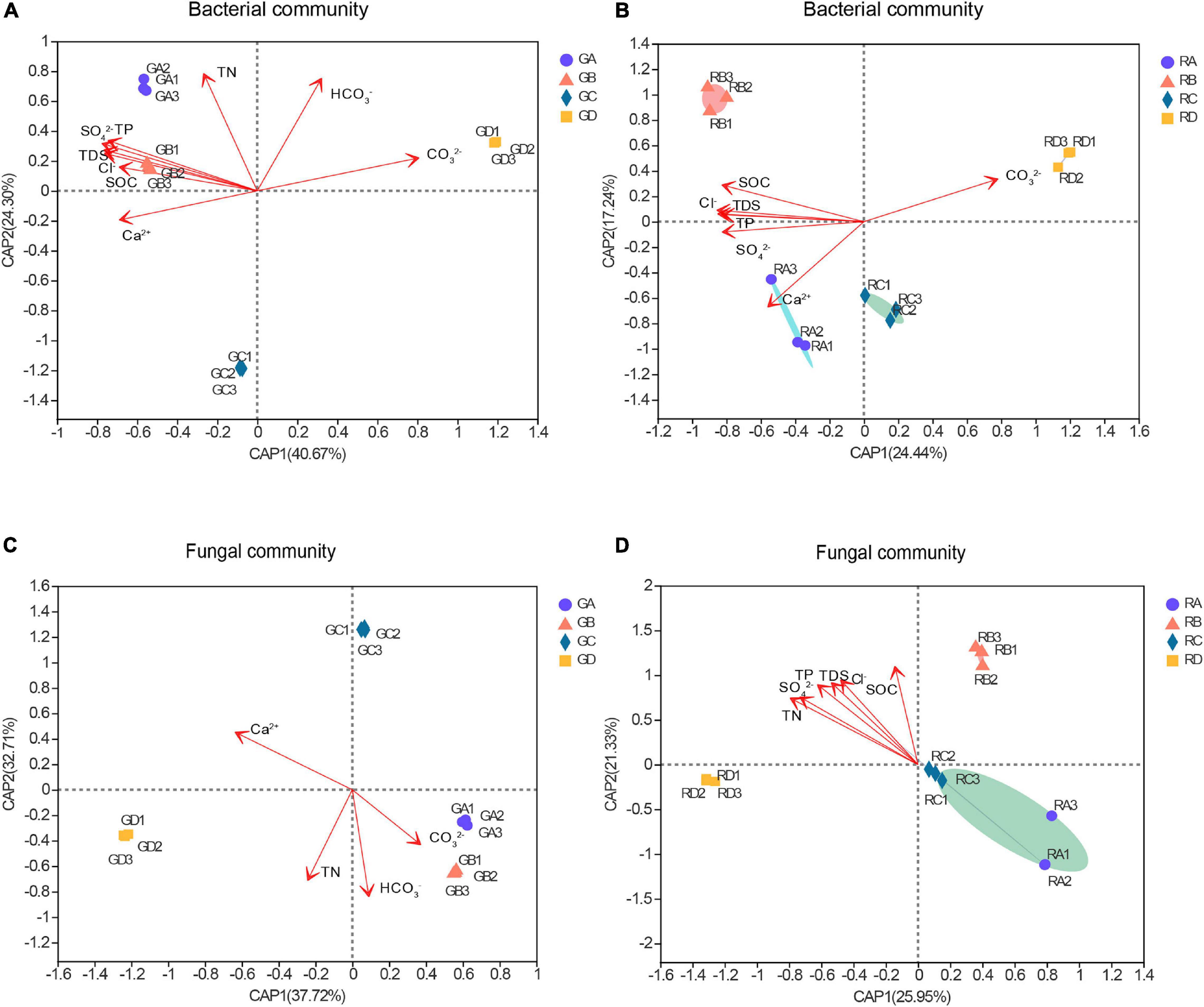
Figure 3. The distance-based redundancy analysis (db-RDA) of these microbial community structure and environmental parameters. Numbers in parentheses in the axis labels was a percentage explanatory variable. (A) Bacterial rhizospheric samples, (B) bacterial endophytic samples, (C) fungal rhizospheric samples, and (D) fungal endophytic samples. S. europaea (A), S. salsa (B), P. communis (C), and A. splendens (D).
LEfSe Analysis to Reveal Dominant Microbial Taxa in Different Rhizocompartments
LEfSe was used to evaluate the different taxa from rhizosphere soil and root endophytic samples. The results showed that the number of dominant bacteria and fungi were 18 and 15 in rhizospheric samples (Figures 4A,C), respectively, while the number of dominant endophytic bacteria and fungi were 16 and 6 in endophytic samples (Figures 4B,D), respectively. This study indicated that whether it was bacterial communities or fungal communities, the rhizosphere soil exhibited a greater number of dominant genera than that in the endophytic environments. Along the salinity gradient decreasing, Aliifodinibius, Alkalibacterium, Halanaerobium (GA), Halomonas, Marinobacter, Paracoccus, Salegentibacter (GB), Kocuria, Planococcus, Truepera (GC), and Pseudomonas and Arthrobacter (GD) were the dominant taxa at the genus level of rhizosphere bacterial communities (Figure 4A). The dominant taxa in the endophytic bacterial communities were Marinobacter, Microbulbifer (RA), Planococcus, Citreimonas, Palleronia, Paracoccus, Halomonas, Nesterenkonia, Arthrobacter, Kocuria (RB), Ilumatobacter (RC), and Pelagibacterium (RD) (Figure 4B). For the fungal communities, Alternaria (GA), Macrophoma (GB), Zopfiella, Fusarium (GC), and Sigarispora, Beauveria, Wallrothiella, Penicillium, and Arthrocladium (GD) were the dominant genus in the rhizosphere fungal communities (Figure 4C), and Macrophoma, Alternaria (RB), Zopfiella (RC), and Fusarium and Wallrothiella (RD) were the dominant taxa in the endophytic fungal communities (Figure 4D). Interestingly, the dominant species in the root were the same as those in rhizosphere soil for the fungal communities.
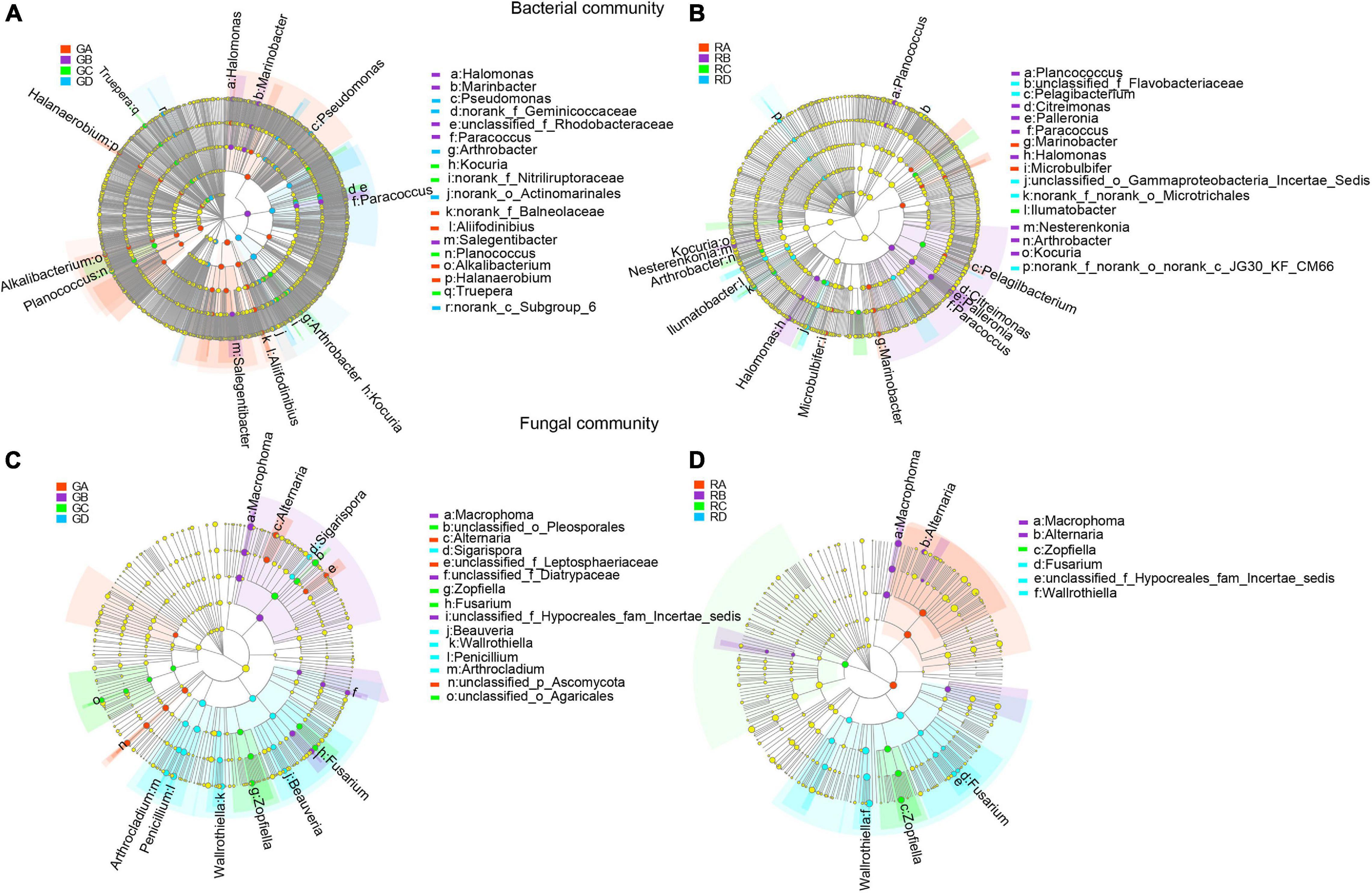
Figure 4. The differential phylogenetic distribution of bacteria (A,B) and fungi (C,D) in Ejinur Salt Lake different rhizosphere soil and root endophytic samples. The linear discriminant analysis scores of >4. The five rings of the cladogram stand for phylum (innermost), class, order, family, and genus. Different colored nodes represent microorganisms that play an important role in the grouping represented by color; yellow represents non-significant. Uppercase “G” represents the rhizospheric sample, and uppercase “R” represents the endophytic samples. S. europaea (A), S. salsa (B), P. communis (C), and A. splendens (D).
Correlation Molecular Network Analysis of Microbial Communities
The correlation microbial networks were explored microbial interactions in rhizosphere soil and root endophytic samples (Figure 5 and Supplementary Figure 7). The results showed that there were differences in the interaction between bacterial and fungal taxa under a salinity gradient (Supplementary Tables 4, 5). For endophytic samples of bacteria, the number of negative links from RA to RC was lower than RD (Supplementary Table 4). Except for the A. splendens sample (RD), whether bacteria or fungi, the positive links between endophytes were greater than those between rhizosphere soil microorganisms (Supplementary Tables 4, 5). The number of positive links in fungal taxa was far greater than the number of negative links (Supplementary Table 5). Interestingly, the same genus (e.g., Kocuria in the A. splendens sample) was observed in rhizosphere soil and root endophytic samples. It implied that fungal taxa might cooperate more to adapt to high salinity niches. In addition, Proteobacteria were defined as the core microbial taxa of GA–GC samples based on its abundance and close connections with other taxa. It accounted for 40.20, 36.03, and 31.55% of GB, GA, and GC samples, respectively. The core bacterial taxa of GD were Actinobacteria (35.55%) (Figures 5A–D). The core bacterial taxa of RA–RD were primarily Proteobacteria. It accounted for 64.56, 58.50, 52.10, and 49.63% of RA, RB, RD, and RC, respectively (Figures 5E–H). Whether in the rhizosphere fungal communities or the endophytic fungal communities (Supplementary Figure 7), Ascomycota were core fungal taxa (>72%). Moreover, the bacterial and fungal 10 metabolic pathways in rhizospheric and endophytic samples were predicted by PICRUSt2. The differences in the average relative abundance of different metabolic pathways are shown in Supplementary Tables 8, 9.
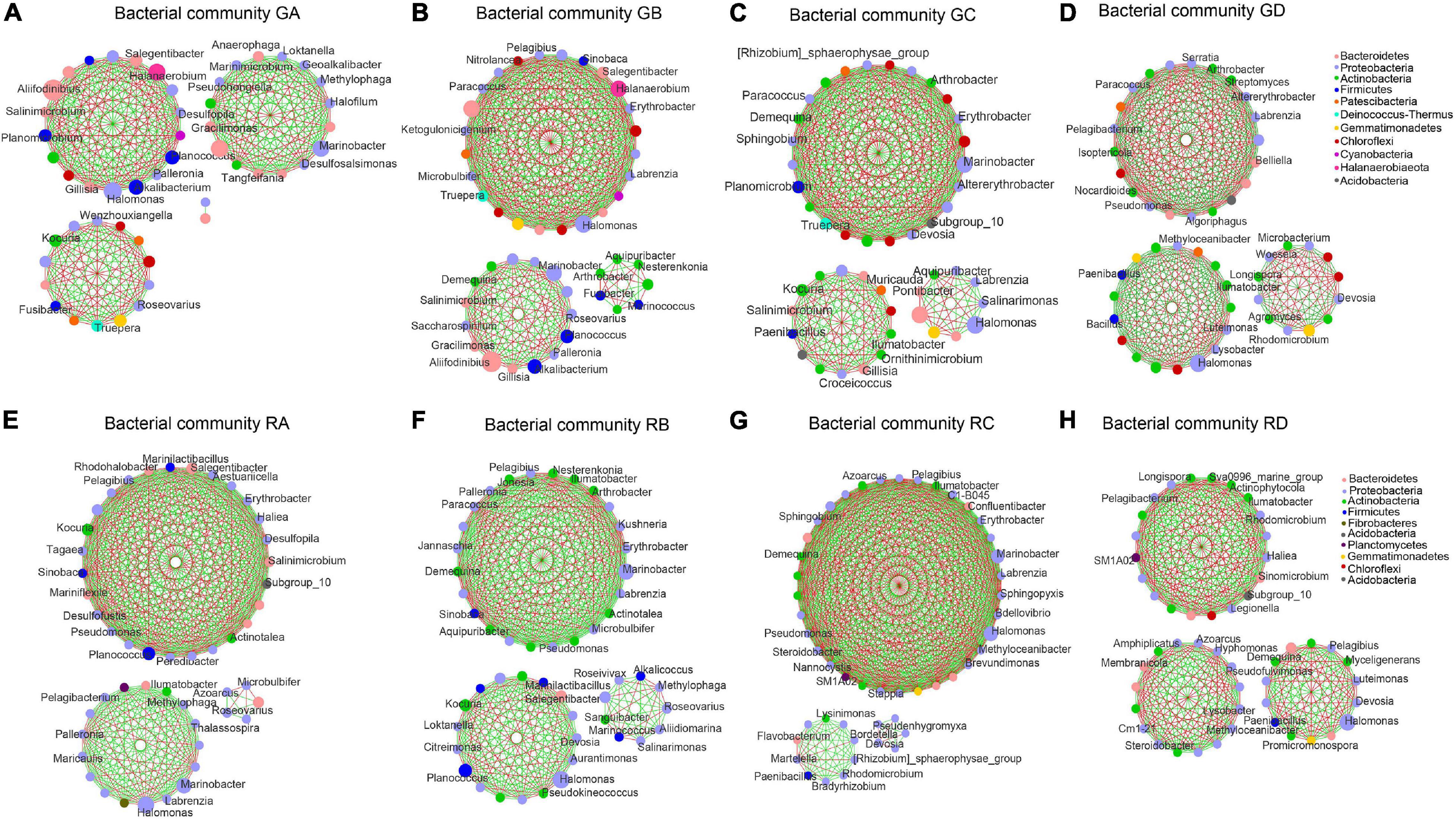
Figure 5. An overview of the bacterial network interactions in Ejinur Salt Lake different rhizosphere soil and root endophytic samples. The connections in the network represent a strong (| r| > 0.6) and significant (p < 0.01) correlation. Colors of nodes represent different major phyla; the node representing genus is written inside. A green link indicates a negative interaction, whereas a red link implies a positive interaction between two individual nodes. Uppercase “G” represents the rhizospheric sample, and lowercase “R” represents the endophytic samples. (A–D) S. europaea rhizosphere soil (GA), S. salsa rhizosphere soil (GB), P. communis rhizosphere soil (GC) and A. splendens rhizosphere soil (GD). (E–H) Salicornia europaea endophytic samples (RA), Suaeda salsa endophytic samples (RB), Phragmites communis endophytic samples (RC) and Achnatherum splendens endophytic samples (RD).
Discussion
Drivers of Microbiome Niche Differentiation
Soil salinity can affect not only the distribution, composition, and diversity of plant communities, but also the diversity and community structure of microbial in soil (Xi et al., 2016; Zhang et al., 2019). In the study, the high-throughput sequencing technique was used to reveal the diversity and abundance of bacterial and fungal communities in rhizosphere soil samples and root endophytic samples under different salinity. The study showed that the bacterial and fungal communities in rhizosphere soil were more abundant than root endophytic microbial communities, which is consistent with previous studies (Lu et al., 2016; Li et al., 2018). Based on the significant difference of microbial composition at the phylum level (Supplementary Tables S6 and S7), microbial α-diversity (Figure 2) and other results (Figures 3–5), it indicates that a strong habitat filtering mechanism that may affect the microbial composition from different rhizocompartments of plants (Qin et al., 2016). These filtering effects have previously been reported in the different rhizocompartments of soya and alfalfa (Xiao et al., 2017) and the rhizosphere and endosphere of Arabidopsis thaliana (Lundberg et al., 2012). This filtration was related to the physical and chemical characteristics of the rhizosphere soil and the change from the soil to the endosphere environment (Gottel et al., 2011). Moreover, previous works have found that the richness and evenness of microbial communities decreased with the increase of salt-rich soil and sediment (Wang et al., 2010; Zhao et al., 2018). However, some studies have shown that bacterial and fungal communities are positively correlated with salinity in a saline–alkali environment, and the existence of plants will increase the diversity of microorganisms independently of salinity (Foti et al., 2008; Vanegas et al., 2019). In this study, our investigation found that the degree of soil salinization decreased gradually and dominant plants showed obvious zonal distribution characteristics from the edge of the lake to the periphery. Moreover, there were also significant differences in soluble salt ion concentrations and soil nutrients under different salinity in the littoral zones of Ejinur Salt Lake (Table 1). The difference in soil properties and plant species may result in the difference of microbial community composition and structure. Although salinity plays an important role in the construction of bacterial and fungal communities, plant species and other soil parameters (e.g., soil pH value or soil nutrient content) may also affect the bacterial and fungal diversity and community composition from different rhizocompartments in soil environments of salt lakes littoral zones.
An additional analysis of significant difference in community structure were identified among different rhizocompartments (Figure 3). This finding was consistent with previous studies on the microbial communities in different root chambers of tomato in different geographical locations. For endophytic microbial communities, the high difference of community structure may depend on the inherent immune system of plants, while rhizosphere soil microbes are less affected by host-dependent selection (Lee et al., 2019). The growth of host plants may be affected by the soil environments. Therefore, the high difference of endophytic microorganisms depends on the plant and other soil physicochemical properties. In addition, the microbial composition related to salinity, soluble salt ion concentration, and soil nutrients (Lozupone and Knight, 2007; Barin et al., 2015). Our regression analysis results confirmed the inference that rhizosphere soil microorganisms and root endophytic microorganisms are affected by plants, soil nutrients, and different soluble salt ions (Supplementary Figures 3–6). This was also consistent with our db-RDA results that show that rhizosphere soil and root endophytic microbial communities have significant relationships with soil salinity, different soluble salt ions, and soil nutrients (Figure 3). The SOC in soil nutrients was overall an important driving factor for the microbial community structure in the whole range of the salinity gradient, because SOC can mitigate the salinity pressure in microorganisms. Microorganisms may produce some compounds such as osmotic regulators (e.g., glutamine and proline) to cope with salt stress (Barin et al., 2015). The synthesis of these compounds depletes a lot of energy, which is obtained by the decomposition of organic compounds (Oren, 1999).
The microbial interaction is also very important for exploring the structure and dynamics of soil bacterial and fungal communities (Faust and Raes, 2012; Xue et al., 2017). It is possible to determine the positive and negative co-occurrence correlation between bacterial and fungal members and further reflect the functional relationship between synergy and antagonism (Benidire et al., 2020). Our results showed that bacterial hubs were mainly members of Proteobacteria and Actinomycota, such as Labrenzia and Kocuria. PGPR strains belonging to genera Kocuria have been proven to indirectly improve the salt tolerance of maize (Li et al., 2020). A strain isolated from the genus Labrenzia in the rhizosphere of the halophyte Arthrocnemum macrostachyum has been verified to have halophilic properties (Camacho et al., 2016). Moreover, the root endophytic fungi played a certain role in assisting plants to adapt to the salt environments (Chadha et al., 2015). The composition of the genera among different modules were not the same, which further reflects the unique ecological characteristics of these combinations. The aggregation patterns of these microorganisms showed that the interaction between species and environmental filtration plays a core role in the formation of community structure. In the results, except for the A. splendens sample, whether bacteria or fungi, the positive links between endophytes were greater than those between rhizosphere soil microorganisms (Supplementary Tables 4, 5). Generally speaking, positive connections mean cross-feeding, while negative relationships represent competition in the network (Zheng et al., 2017). The pMENs analysis of Wang et al. (2019) found that salinity and alkalinity significantly increased microbial interactions from different cadmium-contaminated soils. Therefore, it can be inferred that the regulation of microbial interaction may be a strategy for microorganisms to deal with strong salt stress. Previous study showed that the hubs involved in positive interactions under certain conditions may behave differently under stress conditions and mainly establish antagonistic interactions. The results may be related to the expression of genes involved in stress tolerance (Benidire et al., 2020). The study found that in the network structure, some members of the hubs belonged to Neocamarosporium and Aspergillus (Supplementary Figures 5C,D). They mainly participated in the collaborative interaction only under salt stress, while they showed antagonistic interaction under non-salt stress. Some genera in the endophyte network structure were found to occur in the network structure of rhizosphere soil. The results also found that the number of OTUs and microbial diversity of rhizosphere soil was much larger than that of root endophytic samples. These results suggested that endophytes may be a part of rhizosphere microbes. Tian et al. (2015) also indicated that tomato endophytic microbiota was obtained from rhizosphere microbes.
Dominant Microbiome Within Different Rhizocompartments
According to the LEfSe analysis (LDA > 4.0), the microbial taxa shown in the results (Figure 4) were defined as the dominant taxa (Richa et al., 2017; Zhao et al., 2020; Zhang et al., 2021). These dominant taxa can be used as indicator species for different plants around the salt lake and may assist plants to adapt to unfavorable environments. For example, Marinobacter, Palleronia, Ilumatobacter, and Arthrobacter were the dominant genera in the samples of S. europaea, S. salsa, P. communis, and A. splendens, respectively. Previous works have shown that Marinobacter and Palleronia have good endurance in a broad range of salinity with exceptional adaptation capacities (Martinez-Checa et al., 2005; Li et al., 2019). Ilumatobacter and Arthrobacter are beneficial taxa, and PGPR strains belonging to genera Arthrobacter can promote plant growth, increase plant salt tolerance, and effectively improve agricultural productivity (Kumar et al., 2019; Zhao et al., 2019; Stassinos et al., 2021). Alternaria, Macrophoma, Zopfiella, and Penicillium were the dominant fungal taxa with high abundance in plant samples of S. europaea, S. salsa, P. communis, and A. splendens, respectively. In previous studies, Alternaria and Macrophoma have been found in the salt crusts and roots of halophytes (You et al., 2012; Liu et al., 2014). These genera are known plant pathogens that can cause leaf spots and fusarium wilt (Chaerani and Voorrips, 2006; Jeyaraman and Robert, 2017). Penicillium has been shown to confer soybean resistance to salt stress (Khan et al., 2011). Moreover, microbial metabolic functions also had a certain positive effect on salt stress. Yu et al. (2021) found that Maize bHLH55 functions regulated the biosynthesis of ascorbic acid by directly regulating GDP-mannose pathway genes and thus play a positive role in salt tolerance. Therefore, these dominant microbial taxa had been proven to improve abiotic stress, promote plant growth, and confer plants the ability to resist salt stress.
Previous studies also found that the bacterial communities were more sensitive to the soil environments. The research results confirmed that dominant taxa of bacterial communities in rhizosphere soil or the root endosphere are significantly more than those of fungal communities. Bacteria generally is smaller and has a shorter life cycle compared with fungi (Chen et al., 2020), making them more susceptible to external interference and causing them to be more sensitive. The rhizosphere soil microbial communities showed higher sensitivity than the root endophytic microbial communities, containing more biomarkers. The rhizosphere has the most direct resource exchange interface between the soil environments and the root systems (Yu and Hochholdinger, 2018) and is more easily affected by the soil environments than the root endosphere. These results indicated that improving plant tolerance to salt stress depends on not only the interaction of the entire microbial communities but also some specific microbial consortia (de Zelicourt et al., 2013). Exploring the interaction between microorganisms and the dominant taxa may be beneficial to understand the salt tolerance of plants and the importance of related microorganisms. In short, exploring the interaction between microorganisms and excavating the potential functional roles in dominant taxa are beneficial for understanding the interaction mechanism between plants and related microorganisms in the littoral zones of salt lakes. Although the findings of this study expand our understanding of the difference in bacterial and fungal community and their interactions from rhizosphere and endosphere of four dominant plants in the littoral zones along the salinity gradient, there is still much to be learned about the special roles of functional microorganisms and their relationships. Therefore, it should be further explored by using advanced sequencing technology (e.g., metagenomics and multiomics) in the future.
Conclusion
This study demonstrated the microbial community composition in different rhizocompartments of four dominant plants around the Ejinur Salt Lake and revealed the main driving factors affecting the microbial community composition. The bacterial and fungal richness and phylogenetic diversity did not increase with the decrease of salinity, indicating that salinity may not be the main driving factor for microorganisms from different rhizocompartments of plants. Regression analysis indicated that rhizospheric and endophytic microorganisms were affected by soil nutrients and different soil soluble salt ions and plant species. The db-RDA future revealed that bacteria from rhizospheric samples were the most significantly affected by SO42–, while fungi were the most significantly affected by HCO3–. For endophytic samples, both bacteria and fungi were the most significantly affected by SOC. In addition, the correlation network analysis indicated that the interaction mode between microorganisms has potential beneficial effects in regulating salt stress. LEfSe analysis further revealed that there were dominant microbial taxa under soil environments of salinity gradient, and those microbial taxa had a positive response to plant species. This study provided new insights for in-depth understanding of the roles of microorganisms from different rhizocompartments (rhizosphere and endosphere), and it was of great significance for excavating the potential microbial resources resistant to saline–alkali stress in the littoral zones of salt lakes.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA669550.
Author Contributions
JL: writing—original draft, investigation, methodology, validation, visualization, data analysis, and software. ZZ: writing—review and editing, data curation, visualization, formal analysis, and software. YH: investigation, resources, and methodology. FD: software and data curation. BH and LW: methodology. ZB: conceptualization. WG: writing—review and editing, supervision, funding acquisition, and project. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (31860170 and 41977113), the Major Science and Technology Projects in Inner Mongolia Autonomous Region (ZDZX2018054 and 2020ZD0020), and the Natural Science Foundation of Inner Mongolia (2018MS04003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.698479/full#supplementary-material
Footnotes
References
Ali, S., Charles, T. C., and Glick, B. R. (2014). Amelioration of high salinity stress damage by plant growth-promoting bacterial endophytes that contain ACC deaminase. Plant Physiol. Biochem. 80, 160–167. doi: 10.1016/j.plaphy.2014.04.003
Apaydin, Z., Kutbay, H. G., Ozbucak, T., Yalcin, E., and Bilgin, A. (2009). Relationships between vegetation zonation and edaphic factors in a salt-marsh community (black sea coast). Polish J. Ecol. 57, 99–112.
Barin, M., Aliasgharzad, N., Olsson, P. A., and Rasouli-Sadaghiani, M. (2015). Salinity-induced differences in soil microbial communities around the hypersaline Lake Urmia. Soil Res. 53, 494–504. doi: 10.1071/sr14090
Benidire, L., El Khalloufi, F., Oufdou, K., Barakat, M., Tulumello, J., Ortet, P., et al. (2020). Phytobeneficial bacteria improve saline stress tolerance in Vicia faba and modulate microbial interaction network. Sci. Total Environ. 729:139020. doi: 10.1016/j.scitotenv.2020.139020
Boujelben, I., Gomariz, M., Martinez-Garcia, M., Santos, F., Pena, A., Lopez, C., et al. (2012). Spatial and seasonal prokaryotic community dynamics in ponds of increasing salinity of Sfax solar saltern in Tunisia. Antonie Van Leeuwenhoek 101, 845–857. doi: 10.1007/s10482-012-9701-7
Camacho, M., Redondo-Gomez, S., Rodriguez-Llorente, I., Rohde, M., Sproer, C., Schumann, P., et al. (2016). Labrenzia salina sp nov., isolated from the rhizosphere of the halophyte Arthrocnemum macrostachyum. Int. J. Syst. Evol. Microbiol. 66, 5173–5180. doi: 10.1099/ijsem.0.001492
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., et al. (2020). The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 48, D445–D453.
Chadha, N., Mishra, M., Rajpal, K., Bajaj, R., Choudhary, D. K., and Varma, A. (2015). An ecological role of fungal endophytes to ameliorate plants under biotic stress. Arch. Microbiol. 197, 869–881. doi: 10.1007/s00203-015-1130-3
Chaerani, R., and Voorrips, R. E. (2006). Tomato early blight (Alternaria solani): the pathogen, genetics, and breeding for resistance. J. Gen. Plant Pathol. 72, 335–347. doi: 10.1007/s10327-006-0299-3
Chen, J., Xu, D. L., Chao, L. M., Liu, H. J., and Bao, Y. Y. (2020). Microbial assemblages associated with the rhizosphere and endosphere of an herbage, Leymus chinensis. Microb. Biotechnol. 13, 1390–1402. doi: 10.1111/1751-7915.13558
Chen, L. J., Li, C. S., Feng, Q., Wei, Y. P., Zheng, H., Zhao, Y., et al. (2017). Shifts in soil microbial metabolic activities and community structures along a salinity gradient of irrigation water in a typical arid region of China. Sci. Total Environ. 598, 64–70. doi: 10.1016/j.scitotenv.2017.04.105
de Zelicourt, A., Al-Yousif, M., and Hirt, H. (2013). Rhizosphere microbes as essential partners for plant stress tolerance. Mol. Plant 6, 242–245. doi: 10.1093/mp/sst028
Du, Q. Y., Li, Z., Liu, S. R., and Liu, H. Y. (2007). Dynamics of species diversity and characteristics of halophytic plant communities around saline lakes in and semi-arid regions of Inner Mongolia. Biodivers. Sci. 15, 271–281. doi: 10.1360/biodiv.060214
Edgar, R. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10:996. doi: 10.1038/nmeth.2604
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, 911–920.
Fan, P., Chen, D., He, Y., Zhou, Q., Tian, Y., and Gao, L. (2016). Alleviating salt stress in tomato seedlings using Arthrobacter and Bacillus megaterium isolated from the rhizosphere of wild plants grown on saline-alkaline lands. Int. J. Phytoremediation 18, 1113–1121. doi: 10.1080/15226514.2016.1183583
Fang, D., Zhao, G., Xu, X., Zhang, Q., Shen, Q., Fang, Z., et al. (2018). Microbial community structures and functions of wastewater treatment systems in plateau and cold regions. Bioresour. Technol. 249, 684–693. doi: 10.1016/j.biortech.2017.10.063
Fatima, K., Afzal, M., Imran, A., and Khan, Q. M. (2015). Bacterial rhizosphere and endosphere populations associated with grasses and trees to be used for phytoremediation of crude oil contaminated soil. Bull. Environ. Contam. Toxicol. 94, 314–320. doi: 10.1007/s00128-015-1489-5
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Foti, M. J., Sorokin, D. Y., Zacharova, E. E., Pimenov, N. V., Kuenen, J. G., and Muyzer, G. (2008). Bacterial diversity and activity along a salinity gradient in soda lakes of the Kulunda Steppe (Altai, Russia). Extremophiles 12, 133–145. doi: 10.1007/s00792-007-0117-7
Gottel, N. R., Castro, H. F., Kerley, M., Yang, Z. M., Pelletier, D. A., Podar, M., et al. (2011). Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl. Environ. Microbiol. 77, 5934–5944. doi: 10.1128/aem.05255-11
Guven, K., Matpan Bekler, F., and Gul Guven, R. (2018). “Thermophilic and halophilic microorganisms isolated from extreme environments of turkey, with potential biotechnological applications,” in Extremophiles in Eurasian Ecosystems: Ecology, Diversity, and Applications, eds D. Egamberdieva, N.-K. Birkeland, H. Panosyan, and W.-J. Li (Singapore: Springer), 219–264. doi: 10.1007/978-981-13-0329-6_8
Han, R., Zhang, X., Liu, J., Long, Q. F., Chen, L. S., Liu, D. L., et al. (2017). Microbial community structure and diversity within hypersaline keke salt lake environments. Can. J. Microbiol. 63, 895–908. doi: 10.1139/cjm-2016-0773
Hrynkiewicz, K., Baum, C., and Leinweber, P. (2010). Density, metabolic activity, and identity of cultivable rhizosphere bacteria on Salix viminalis in disturbed arable and landfill soils. J. Plant Nutr. Soil Sci. 173, 747–756. doi: 10.1002/jpln.200900286
Hrynkiewicz, K., Szymanska, S., Piernik, A., and Thiem, D. (2015). Ectomycorrhizal community structure of Salix and Betula spp. at a saline site in central poland in relation to the seasons and soil parameters. Water Air Soil Pollut. 226:99.
Hussain, I., Aleti, G., Naidu, R., Puschenreiter, M., Mahmood, Q., Rahman, M. M., et al. (2018). Microbe and plant assisted-remediation of organic xenobiotics and its enhancement by genetically modified organisms and recombinant technology: a review. Sci. Total Environ. 628-629, 1582–1599. doi: 10.1016/j.scitotenv.2018.02.037
Jeyaraman, M., and Robert, P. S. A. (2017). Integrated management of branch canker disease (Macrophoma sp.) in tea under field level. J. Plant Dis. Prot. 124, 115–119. doi: 10.1007/s41348-017-0072-1
Jiang, S. Q., Yu, Y. N., Gao, R. W., Wang, H., Zhang, J., Li, R., et al. (2019). High-throughput absolute quantification sequencing reveals the effect of different fertilizer applications on bacterial community in a tomato cultivated coastal saline soil. Sci. Total Environ. 687, 601–609. doi: 10.1016/j.scitotenv.2019.06.105
Kalwasinska, A., Deja-Sikora, E., Szabo, A., Felfoldi, T., Kosobucki, P., Brzezinska, M. S., et al. (2019). Salino-alkaline lime of anthropogenic origin a reservoir of diverse microbial communities. Sci. Total Environ. 655, 842–854. doi: 10.1016/j.scitotenv.2018.11.246
Khan, A. L., Hamayun, M., Kim, Y. H., Kang, S. M., and Lee, I. J. (2011). Ameliorative symbiosis of endophyte (Penicillium funiculosum LHL06) under salt stress elevated plant growth of Glycine max L. Plant Physiol. Biochem. 49, 852–861. doi: 10.1016/j.plaphy.2011.03.005
Kou, X., Li, J. Q., Liu, H. M., Li, B. Q., Yu, X. W., Cao, X. A., et al. (2020). Characteristics of bacterial biodiversity and community structure in non-rhizosphere soils along zonal distribution of plants within littoral wetlands in inner Mongolia, China. Glob. Ecol. Conserv. 24:e01310. doi: 10.1016/j.gecco.2020.e01310
Kumar, A., Patel, J. S., Meena, V. S., and Ramteke, P. W. (2019). Plant growth-promoting rhizobacteria: strategies to improve abiotic stresses under sustainable agriculture. J. Plant Nutr. 42, 1402–1415. doi: 10.1080/01904167.2019.1616757
Lee, S. A., Kim, Y., Kim, J. M., Chu, B., Joa, J. H., Sang, M. K., et al. (2019). A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 9:9300.
Li, H., Chi, Z. F., Li, J. L., Wu, H. T., and Yan, B. X. (2019). Bacterial community structure and function in soils from tidal freshwater wetlands in a Chinese delta: potential impacts of salinity and nutrient. Sci. Total Environ. 696:134029. doi: 10.1016/j.scitotenv.2019.134029
Li, X., Sun, P., Zhang, Y., Jin, C., and Guan, C. (2020). A novel PGPR strain Kocuria rhizophila Y1 enhances salt stress tolerance in maize by regulating phytohormone levels, nutrient acquisition, redox potential, ion homeostasis, photosynthetic capacity and stress-responsive genes expression. Environ. Exp. Bot. 174:104023. doi: 10.1016/j.envexpbot.2020.104023
Li, Y., Kong, Y., Teng, D. X., Zhang, X. N., He, X. M., Zhang, Y., et al. (2018). Rhizobacterial communities of five co-occurring desert halophytes. PeerJ 6:e5508. doi: 10.7717/peerj.5508
Liu, J. W., Fu, B. B., Yang, H. M., Zhao, M. X., He, B. Y., and Zhang, X. H. (2015). Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: the potential impact of hypoxia and nutrients. Front. Microbiol. 6:64. doi: 10.3389/fmicb.2015.00064
Liu, K. H., Ding, X. W., Wang, H. F., Zhang, X. M., Hozzein, W. N., Wadaan, M. A. M., et al. (2014). Eukaryotic microbial communities in hypersaline soils and sediments from the alkaline hypersaline Huama Lake as revealed by 454 pyrosequencing. Antonie Van Leeuwenhoek 105, 871–880. doi: 10.1007/s10482-014-0141-4
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U.S.A. 104, 11436–11440. doi: 10.1073/pnas.0611525104
Lu, D. X., Jin, H., Yang, X. Y., Zhang, D. H., Yan, Z. Q., Li, X. Z., et al. (2016). Characterization of rhizosphere and endophytic fungal communities from roots of Stipa purpurea in alpine steppe around qinghai lake. Can. J. Microbiol. 62, 643–656. doi: 10.1139/cjm-2015-0857
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Mallick, I., Bhattacharyya, C., Mukherji, S., Dey, D., Sarkar, S. C., Mukhopadhyay, U. K., et al. (2018). Effective rhizoinoculation and biofilm formation by arsenic immobilizing halophilic plant growth promoting bacteria (PGPB) isolated from mangrove rhizosphere: a step towards arsenic rhizoremediation. Sci. Total Environ. 610-611, 1239–1250. doi: 10.1016/j.scitotenv.2017.07.234
Martinez-Checa, F., Quesada, E., Martinez-Canovas, M. J., Llamas, I., and Bejar, V. (2005). Palleronia marisminoris gen. nov., sp. nov., a moderately halophilic, exopolysaccharide-producing bacterium belonging to the ‘Alphaproteobacteria’, isolated from a saline soil. Int. J. Syst. Evol. Microbiol. 55(Pt. 6), 2525–2530. doi: 10.1099/ijs.0.63906-0
McArdle, B. H., and Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297. doi: 10.1890/0012-9658(2001)082[0290:fmmtcd]2.0.co;2
Oren, A. (1999). Bioenergetic aspects of halophilism. Microbiol. Mol. Biol. Rev. 63, 334–348. doi: 10.1128/mmbr.63.2.334-348.1999
Oren, A. (2008). Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst. 4:2. doi: 10.1186/1746-1448-4-2
Pan, X., Qin, Y., and Yuan, Z. (2018). Potential of a halophyte-associated endophytic fungus for sustaining Chinese white poplar growth under salinity. Symbiosis 76, 109–116. doi: 10.1007/s13199-018-0541-8
Paul, D., and Lade, H. (2014). Plant-growth-promoting rhizobacteria to improve crop growth in saline soils: a review. Agron. Sustain. Dev. 34, 737–752. doi: 10.1007/s13593-014-0233-6
Qin, Y., Druzhinina, I. S., Pan, X. Y., and Yuan, Z. L. (2016). Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 34, 1245–1259. doi: 10.1016/j.biotechadv.2016.08.005
Rath, K. M., Fierer, N., Murphy, D. V., and Rousk, J. (2019). Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 13, 836–846. doi: 10.1038/s41396-018-0313-8
Richa, K., Balestra, C., Piredda, R., Benes, V., Borra, M., Passarelli, A., et al. (2017). Distribution, community composition, and potential metabolic activity of bacterioplankton in an urbanized mediterranean sea coastal zone. Appl. Environ. Microbiol. 83, e00494–17.
Selivanova, E. A., Poshvina, D. V., Khlopko, Y. A., Gogoleva, N. E., and Plotnikov, A. O. (2018). Diversity of prokaryotes in planktonic communities of saline sol-iletsk lakes (Orenburg Oblast. Russia). Microbiology 87, 569–582. doi: 10.1134/s0026261718040161
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Simmons, T., Caddell, D. F., Deng, S., and Coleman-Derr, D. (2018). Exploring the root microbiome: extracting bacterial community data from the soil, rhizosphere, and root endosphere. J. Vis. Exp. 135:57561. doi: 10.3791/57561
Sommers, L. E., and Nelson, D. W. (1972). Determination of total phosphorus in soils a rapid per chloric-acid digestion procedure. Soil Sci. Soc. Am. Proc. 36, 902–904. doi: 10.2136/sssaj1972.03615995003600060020x
Sorokin, D. Y., Berben, T., Melton, E. D., Overmars, L., Vavourakis, C. D., and Muyzer, G. (2014). Microbial diversity and biogeochemical cycling in soda lakes. Extremophiles 18, 791–809. doi: 10.1007/s00792-014-0670-9
Stassinos, P. M., Rossi, M., Borromeo, I., Capo, C., Beninati, S., and Forni, C. (2021). Amelioration of salt stress tolerance in rapeseed (Brassica napus) cultivars by seed inoculation with Arthrobacter globiformis. Plant Biosyst. 1–14. doi: 10.1080/11263504.2020.1857872
Tian, B. Y., Cao, Y., and Zhang, K. Q. (2015). Metagenomic insights into communities, functions of endophytes, and their associates with infection by root-knot nematode, Meloidogyne incognita, in tomato roots. Sci. Rep. 5:17087.
Tian, X. Y., and Zhang, C. S. (2017). Illumina-based analysis of endophytic and rhizosphere bacterial diversity of the coastal halophyte Messerschmidia sibirica. Front. Microbiol. 8:2288. doi: 10.3389/fmicb.2017.02288
Valenzuela-Encinas, C., Neria-Gonzalez, I., Alcantara-Hernandez, R. J., Enriquez-Aragon, J. A., Estrada-Alvarado, I., Hernandez-Rodriguez, C., et al. (2008). Phylogenetic analysis of the archaeal community in an alkaline-saline soil of the former lake Texcoco (Mexico). Extremophiles 12, 247–254. doi: 10.1007/s00792-007-0121-y
Vanegas, J., Muñoz-García, A., Pérez-Parra, K. A., Figueroa-Galvis, I., Mestanza, O., and Polanía, J. (2019). Effect of salinity on fungal diversity in the rhizosphere of the halophyte Avicennia germinans from a semi-arid mangrove. Fungal Ecol. 42:100855. doi: 10.1016/j.funeco.2019.07.009
Wang, M., Chen, S., Chen, L., and Wang, D. (2019). Responses of soil microbial communities and their network interactions to saline-alkaline stress in Cd-contaminated soils. Environ. Pollut. 252, 1609–1621. doi: 10.1016/j.envpol.2019.06.082
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Wang, W. H., Wang, H., Feng, Y. Z., Wang, L., Xiao, X. J., Xi, Y. G., et al. (2016). Consistent responses of the microbial community structure to organic farming along the middle and lower reaches of the Yangtze River. Sci. Rep. 6:35046.
Wang, X., Sun, R., Tian, Y., Guo, K., Sun, H., Liu, X., et al. (2020). Long-term phytoremediation of coastal saline soil reveals plant species-specific patterns of microbial community recruitment. mSystems 5, e00741–19.
Wang, Z. Y., Xin, Y. Z., Gao, D. M., Li, F. M., Morgan, J., and Xing, B. S. (2010). Microbial community characteristics in a degraded wetland of the yellow river delta. Pedosphere 20, 466–478. doi: 10.1016/s1002-0160(10)60036-7
Williams, W. D. (1991). Chinese and mongolian saline lakes a limnological overview. Hydrobiologia 210, 39–66. doi: 10.1007/bf00014322
Wu, Q. L., Zwart, G., Schauer, M., Kamst-van Agterveld, M. P., and Hahn, M. W. (2006). Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 72, 5478–5485. doi: 10.1128/aem.00767-06
Xi, H. Y., Feng, Q., Zhang, L., Si, J. H., Chang, Z. Q., Yu, T. F., et al. (2016). Effects of water and salinity on plant species composition and community succession in Ejina Desert Oasis, northwest China. Environ. Earth Sci. 75:138.
Xiao, X., Chen, W., Zong, L., Yang, J., Jiao, S., Lin, Y., et al. (2017). Two cultivated legume plants reveal the enrichment process of the microbiome in the rhizocompartments. Mol. Ecol. 26, 1641–1651. doi: 10.1111/mec.14027
Xue, L., Ren, H. D., Li, S., Leng, X. H., and Yao, X. H. (2017). Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front. Microbiol. 8:2377. doi: 10.3389/fmicb.2017.02377
Yamamoto, K., Matsutani, M., Shiwa, Y., Ishige, T., Sakamoto, H., Saitoh, H., et al. (2020). Comparative analysis of bacterial diversity and community structure in the rhizosphere and root endosphere of two halophytes, Salicornia europaea and Glaux maritima, collected from two brackish lakes in Japan. Microbes Environ. 35:ME20072.
Yang, J., Ma, L., Jiang, H., Wu, G., and Dong, H. (2016). Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 6:25078.
You, Y. H., Yoon, H. J., Woo, J. R., Seo, Y. G., Kim, M. A., Lee, G. M., et al. (2012). Diversity of endophytic fungi from the roots of halophytes growing in go-chang salt marsh. Korean J. Mycol. 40, 86–92. doi: 10.4489/kjm.2012.40.2.86
Yu, C., Yan, M., Dong, H., Luo, J., Ke, Y., Guo, A., et al. (2021). Maize bHLH55 functions positively in salt tolerance through modulation of AsA biosynthesis by directly regulating GDP-mannose pathway genes. Plant Sci. 302:110676. doi: 10.1016/j.plantsci.2020.110676
Yu, P., and Hochholdinger, F. (2018). The role of host genetic signatures on root-microbe interactions in the rhizosphere and endosphere. Front. Plant Sci. 9:1896. doi: 10.3389/fpls.2018.01896
Zhang, C., and Kong, F. (2014). Isolation and identification of potassium-solubilizing bacteria from tobacco rhizospheric soil and their effect on tobacco plants. Appl. Soil Ecol. 82, 18–25. doi: 10.1016/j.apsoil.2014.05.002
Zhang, L., Shen, T., Cheng, Y., Zhao, T., Li, L., and Qi, P. (2020). Temporal and spatial variations in the bacterial community composition in Lake Bosten, a large, brackish lake in China. Sci. Rep. 10:304.
Zhang, W. W., Wang, C., Xue, R., and Wang, L. J. (2019). Effects of salinity on the soil microbial community and soil fertility. J. Integr. Agric. 18, 1360–1368. doi: 10.1016/s2095-3119(18)62077-5
Zhang, Z., Feng, S., Luo, J., Hao, B., Diao, F., Li, X., et al. (2021). Evaluation of microbial assemblages in various saline-alkaline soils driven by soluble salt ion components. J. Agric. Food Chem. 69, 3390–3400. doi: 10.1021/acs.jafc.1c00210
Zhao, F. Y., Zhang, Y. Y., Dong, W. G., Zhang, Y. Q., Zhang, G. X., Sun, Z. P., et al. (2019). Vermicompost can suppress Fusarium oxysporum f. sp. lycopersici via generation of beneficial bacteria in a long-term tomato monoculture soil. Plant Soil 440, 491–505. doi: 10.1007/s11104-019-04104-y
Zhao, Q., Bai, J., Gao, Y., Zhao, H., Zhang, G., and Cui, B. (2020). Shifts in the soil bacterial community along a salinity gradient in the Yellow River Delta. Land Degrad. Dev. 31, 2255–2267. doi: 10.1002/ldr.3594
Zhao, S., Liu, J. J., Banerjee, S., Zhou, N., Zhao, Z. Y., Zhang, K., et al. (2018). Soil pH is equally important as salinity in shaping bacterial communities in saline soils under halophytic vegetation. Sci. Rep. 8:4550.
Zheng, M. P., Tang, J. Y., Liu, J. Y., and Zhang, F. S. (1993). Chinese saline lakes. Hydrobiologia 267, 23–36. doi: 10.1007/978-94-011-2076-0_3
Zheng, W., Xue, D. M., Li, X. Z., Deng, Y., Rui, J. P., Feng, K., et al. (2017). The responses and adaptations of microbial communities to salinity in farmland soils: a molecular ecological network analysis. Appl. Soil Ecol. 120, 239–246. doi: 10.1016/j.apsoil.2017.08.019
Keywords: extreme environments, salt stress, soil properties, rhizosphere soil microorganisms, root endophytic microorganisms
Citation: Luo J, Zhang Z, Hou Y, Diao F, Hao B, Bao Z, Wang L and Guo W (2021) Exploring Microbial Resource of Different Rhizocompartments of Dominant Plants Along the Salinity Gradient Around the Hypersaline Lake Ejinur. Front. Microbiol. 12:698479. doi: 10.3389/fmicb.2021.698479
Received: 21 April 2021; Accepted: 14 June 2021;
Published: 12 July 2021.
Edited by:
Richard Allen White III, The University of North Carolina at Charlotte, United StatesReviewed by:
Candice Lumibao, The University of Tennessee, Knoxville, United StatesAyansina Segun Ayangbenro, North-West University, South Africa
Copyright © 2021 Luo, Zhang, Hou, Diao, Hao, Bao, Wang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Guo, bmRndW93ZWlAMTYzLmNvbQ==
†These authors have contributed equally to this work