 Zhen Li1,2,3,4
Zhen Li1,2,3,4 Lishan Zhang1,3Qingli Song1,3Guibin Wang1,5Wenxiao Yang1,3Huamei Tang1,3
Lishan Zhang1,3Qingli Song1,3Guibin Wang1,5Wenxiao Yang1,3Huamei Tang1,3 Ramanathan Srinivasan1,3Ling Lin1,3
Ramanathan Srinivasan1,3Ling Lin1,3 Xiangmin Lin1,3,4*
Xiangmin Lin1,3,4*- 1Fujian Provincial Key Laboratory of Agroecological Processing and Safety Monitoring, School of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, China
- 2Zhangzhou Health Vocational College, Zhangzhou, China
- 3Key Laboratory of Crop Ecology and Molecular Physiology, Fujian Agriculture and Forestry University, Fujian Province University, Fuzhou, China
- 4Key Laboratory of Marine Biotechnology of Fujian Province, Institute of Oceanology, Fujian Agriculture and Forestry University, Fuzhou, China
- 5State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences, Beijing Institute of Lifeomics, Beijing, China
Bacterial antibiotic resistance is a serious global problem; the underlying regulatory mechanisms are largely elusive. The earlier reports states that the vital role of transcriptional regulators (TRs) in bacterial antibiotic resistance. Therefore, we have investigated the role of TRs on enoxacin (ENX) resistance in Aeromonas hydrophila in this study. A label-free quantitative proteomics method was utilized to compare the protein profiles of the ahslyA knockout and wild-type A. hydrophila strains under ENX stress. Bioinformatics analysis showed that the deletion of ahslyA triggers the up-regulated expression of some vital antibiotic resistance proteins in A. hydrophila upon ENX stress and thereby reduce the pressure by preventing the activation of SOS repair system. Moreover, ahslyA directly or indirectly induced at least 11 TRs, which indicates a complicated regulatory network under ENX stress. We also deleted six selected genes in A. hydrophila that altered in proteomics data in order to evaluate their roles in ENX stress. Our results showed that genes such as AHA_0655, narQ, AHA_3721, AHA_2114, and AHA_1239 are regulated by ahslyA and may be involved in ENX resistance. Overall, our data demonstrated the important role of ahslyA in ENX resistance and provided novel insights into the effects of transcriptional regulation on antibiotic resistance in bacteria.
Introduction
Antibiotic-resistant bacterial strains were discovered over 90 years ago. Since then, antibiotic-resistant bacterial strains have been found to be widely distributed in various environments, such as in hospitals, seafood, and aquaculture farms, and as a result, they pose a serious public health problem worldwide (Fritsche et al., 2009; Peng et al., 2015). Although, several mechanisms of antibiotic resistance such as membrane permeability, plasmid transfer, antibiotic modification or degradation, efflux, and biofilm formation have been described in recent years, the underlying mechanism of acquire antibiotic resistance in bacteria is still largely unknown (Peterson and Kaur, 2018). The emergences of drugs resistant bacterial strains have been caused by many complicated characteristics, one of which is the antimicrobial resistance genes (ARGs) transcriptional regulators (TRs). Bacterial TRs play an important role in the transcriptional regulation of functional genes needed to survive environmental stresses, including antibiotic resistance (Zhou and Yang, 2006; Tang et al., 2019a). For example, a mutation in the multiple antibiotic resistance (MarR) TRs in Escherichia coli has been shown to lead to the expression of the marRAB operon. Therefore, it promotes the expression of the transcription factor MarA and activation of the acrAB and tolC efflux pump genes, resulting in multi-drug resistance to tetracycline, quinolones, β-lactams, and phenolic compounds (Pourahmad Jaktaji and Ebadi, 2013; Lankester et al., 2019). Additionally, the TR EmrR is an inhibitor of efflux pump EmrCAB in Chromobacterium violaceum and the mutation of emrRR92H increases the resistance of C. violaceum to nalidixic acid (Barroso et al., 2018). Nonetheless, there are hundreds of bacterial TRs with biological functions that are poorly characterized and most of the direct and indirect effects of TRs on bacterial antibiotic resistance are largely unknown.
Aeromonas hydrophila is a widely distributed environmental bacterium and a well-known fish pathogen. The use of antibiotics in aquaculture industries has resulted in the emergence of multi-drug resistant A. hydrophila strains in aquaculture and even in hospital settings (Li et al., 2020; Zhu et al., 2020). In recent years, many research, including our previous study, have been found that the several metabolic pathways related genes were involved in the drug-resistance on this pathogen by multi omics technologies (Hossain et al., 2018; Sun et al., 2019; Li et al., 2021). In our previous study, we reported that the LysR-type TR YeeY in A. hydrophila plays an important role in the regulation of furazolidone resistance by directly regulating ARGs, including AHA_3222 and AHA_4275. It indicates the crucial role of TRs in the antibiotic resistance of this pathogen. However, the underlying mechanisms of antibiotic resistance regulated by TRs in A. hydrophila are needed to be further investigated.
To better understanding the role and regulatory mechanism of TR on the bacterial physiological function. In this study, we reported on a MarR family TR in A. hydrophila, AhSlyA (gene name ahslyA or AHA_1240). AhSlyA is a winged helix-turn-helix (wHTH) DNA-binding TR. Previous research reported the homologous proteins of this TR in other bacterial species play diverse biological functions such as cell metabolism and virulence, while its biological effect and molecular mechanism are still largely unknown, especially for the bacterial antibiotics resistance in A. hydrophila (Banda et al., 2019; Tian et al., 2021). In this study, we constructed an ahslyA deletion mutant (ΔahslyA) in A. hydrophila and found that it displayed significantly decreased resistance to the quinolone antibiotic, enoxacin (ENX), as compared to the wild-type (WT) parent strain. To further investigate the direct or indirect effect of this TR on the antibiotic resistance of A. hydrophila, a label-free quantitative proteomics method was used to compare the differentially expressed proteins between the ΔahslyA and WT strains under ENX stress. Moreover, a several differentially expressed genes were deleted and their antibiotic susceptibility to ENX was validated. This study will conduce to further understand the complicated antibiotic resistance mechanisms mediated by bacterial TRs.
Materials and Methods
Bacterial Strains and Culture Conditions
The strains used in this study are A. hydrophila ATCC 7966, ΔahslyA, and the ahslyA complemented strain. E. coli MC1061 and S17 were stored in our laboratory previously. The culture temperature of A. hydrophila and E. coli were 30 and 37°C, respectively. Both bacterial strains were cultured in Luria–Bertani (LB) medium with appropriate antibiotics.
Construction of the Gene Deletion Strain
The deletion strain was constructed based on the principle of two-step homologous recombination using the suicide vector pRE112, as previously described (Yu et al., 2005). Briefly, the pRE112 plasmid fused with about 500-bp of the upstream and downstream flanking regions of the target gene was constructed using A. hydrophila ATCC 7966 genomic DNA as template and then transferred into competent E. coli MC1061 cells. Then, the plasmid of a positive clone was extracted and transferred into E. coli S17 competent cells. After verifying by PCR amplification, the E. coli S17 carrying the pRE112 recombined vector was then conjugated with A. hydrophila in a 4:1 ratio to the first step of homologous recombination. Positive A. hydrophila clones were selected on LB agar plates with ampicillin and chloramphenicol (Yeasen Inc., Shanghai, China). The second step of homologous recombination was carried out in LB medium containing 20% sucrose. The ΔahslyA in A. hydrophila was confirmed by plating chloramphenicol, followed by PCR and DNA sequencing. Finally, after about 20 generations of stable inheritance and correct DNA sequencing, the ΔahslyA mutant was stocked and stored in the freezer at −80°C.
Protein Sample Preparation
Bacterial strains were inoculated in 5 mL LB medium, cultured for 16 h and then transferred at the ratio of 1:100 into 100 mL of LB medium containing ENX at a final concentration of 0.0078 μg/mL. After culturing for about 3 h (until the culture reached an OD600 of approximately 1.0), the cells were collected by centrifugation at 8,000 × g at 4°C for 20 min and washed twice with PBS. The bacterial samples were then resuspended in 5 mL of PBS buffer containing 1 mM phenylmethanesulfonyl fluoride (PMSF) and then lysed by ultra-sonication. The supernatant was collected by centrifugation at 8,000 × g for 20 min and the total protein concentration was detected via the Bradford method. About 50 μg of each protein sample was reduced with 50 mM dithiothreitol at 56°C, alkylated with 25 mM iodoacetamide in dark and then digested to peptides with a 1:20 ratio of trypsin (Promega Inc., Shanghai, China). The enzymatic peptides were desalted with a C18 column (Waters Inc., Milford, MA, United States) and dried with a centrivap concentrator (Labconco Inc., Kansas City, MO, United States). Each group sample was performed three independent repeats for biological replicates.
Label-Free LC-MS/MS
The desalted peptides were dissolved in liquid chromatography mobile phase buffer A [containing 2% acetonitrile, 0.1% formic acid (FA)], loaded onto the pre-column at a flow rate of 4.5 μL/min on the chromatographic system and then injected into the column at a flow rate of 300 nL/min by an easy-nlc1200 system (Thermo Scientific Inc., Waltham, MA, United States). The liquid gradient setting was as follows: 0–3 min, buffer B (containing 80% acetonitrile, 0.1% FA) increased linearly from 2 to 5%; 3–103 min, solution B increased linearly from 5 to 28%; 103–108 min, solution B increased linearly from 35 to 90%; 110–120 min, the solubility of solution B was maintained at 90%. Mass spectrometry was performed with an Orbitrap Fusion Lumos system (Thermo Scientific Inc., Waltham, MA, United States) nanospray ion source. The spray voltage was 2.0 KV and the ion transfer tube temperature was 300°C. We used the data-dependent acquisition mode to the collect data. The parameters were as follows: the precursors from 350 to 1,600 m/z were scanned at a resolution of 60,000, and the AGC target was set at 4e5. For MS/MS, the HCD collision energy was 30% with a resolution of 15,000. The AGC target was set to 5e4. The cycle time was 3 s. All raw data were searched by Maxquant software v.1.6.3.4 against Uniprot A. hydrophila ATCC7966 database. Proteins with the number of peptides greater than 2, p-value less than 0.05, and protein ratio difference greater than 1.5 times were selected as differential proteins for bioinformatics analysis. The raw MS files were submitted to the iProx (Integrated Proteome resources) database under the accession number IPX0002908000 (Ma et al., 2019).
Bioinformatics Analysis
The GO (gene ontology) analysis of altered proteins were performed using the online software DAVID1 and visualized with GOplot package in R language software (Walter et al., 2015; Zhang L.S. et al., 2020). The protein-protein interaction (PPI) network was predicted using the String2 online database with a confidence score ≥ 0.7 and the network was clustered using the “Markov Cluster Algorithm (MCL)” and the inflation parameter was set as 4 (Szklarczyk et al., 2017). Finally, the visualized network diagram of PPI was drawn using Cyctoscape 3.8.03 (Shannon et al., 2003).
Determination of Minimal Bactericidal Concentrations
The minimal bactericidal concentration (MBC) assay was performed by the agar dilution method, as previously described (Jiang et al., 2020). Briefly, an overnight bacterial culture was passaged into fresh LB medium, incubated at 30°C with shaking until the OD600 reached about 1.0 and then diluted 100 times. Then, 2 μL of each dilution was spotted onto an LB agar plates with twofold dilution gradient concentration of antibiotics (ciprofloxacin, levofloxacin, enoxacin, and enrofloxacin purchased from Yeasen biotech, Ltd., Shanghai, China), respectively, and incubated at 30°C for 16 h. Each experiment was performed at least three times with biological duplicates.
Results
ahslyA Mutant Affects Antibiotic Susceptibility in Aeromonas hydrophila
In order to better understand the characteristics of the TR AhslyA on bacterial antibiotic resistance, we first constructed ΔahslyA mutant strain and then tested its antibiotic susceptibilities against various antibiotics, including several quinolone antibiotics. As shown in Figure 1, loss of ahslyA caused a twofold increase in the MBC to ciprofloxacin (CIP) and levofloxacin (LVX), and increased the MBC of ENX by four times; however, it did not affect the susceptibility to enrofloxacin (ENR). Moreover, complementation of the ΔahslyA strain restored the antibiotic susceptibilities similar level to the WT strain, which is suggesting that the ahslyA gene in A. hydrophila is involved in the regulation of several antibiotic resistances, especially ENX.
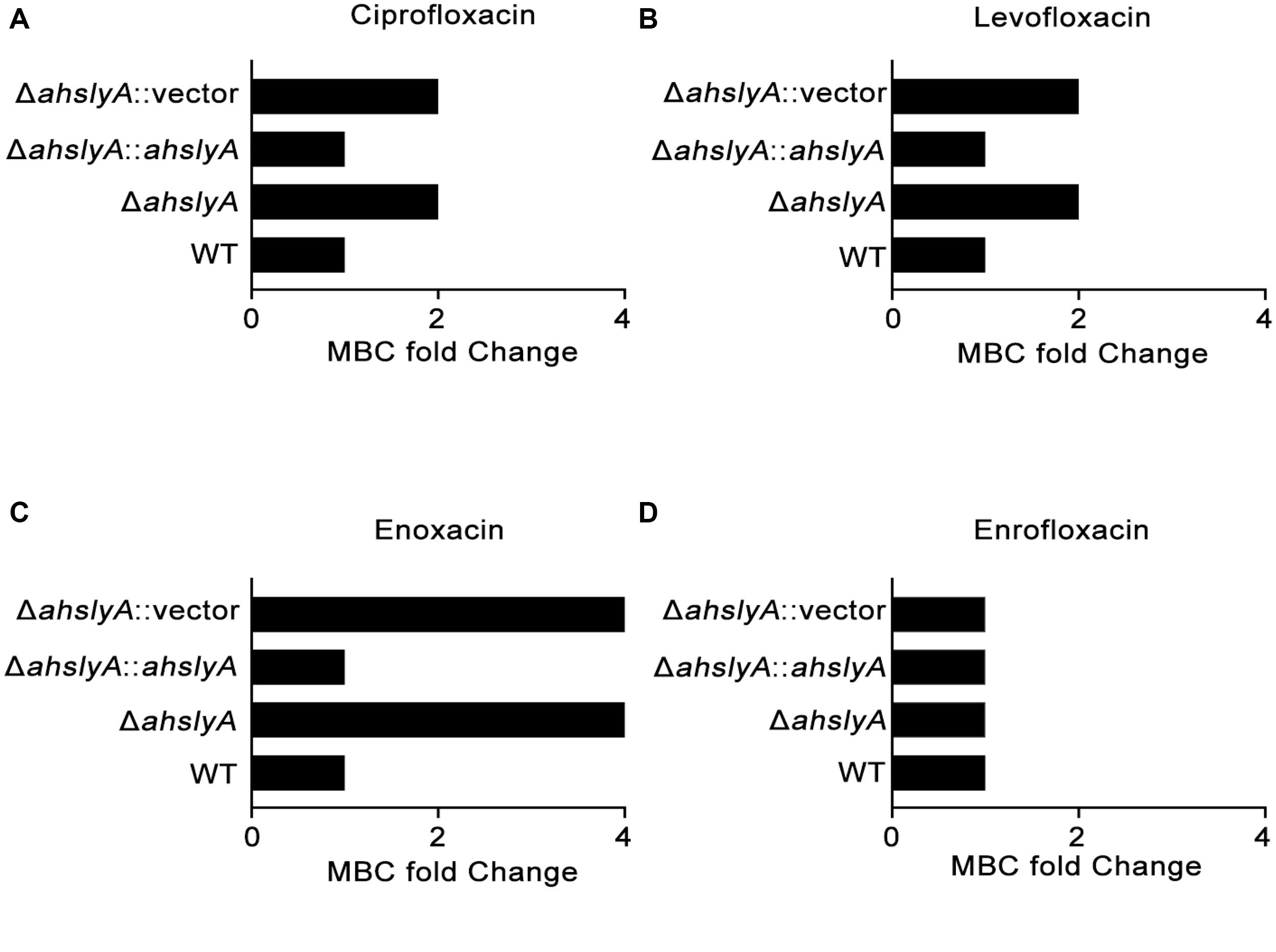
Figure 1. Measurement of the MBCs of ahslyA derivatives in A. hydrophila to four quinolone antibiotics. (A) The MBCs of ΔahslyA and complemented ΔahslyA strains, in addition to WT and ΔahslyA carrying an empty vector as negative controls, were tested for their sensitivity to (A) ciprofloxacin, (B) levofloxacin, (C) enoxacin, and (D) enrofloxacin. The MBCs of each strain to antibiotics were compared with WT as MBC fold change.
Quantitative Proteomics Comparison Between WT and ΔahslyA Strains Under ENX Stress
In order to further investigate the regulatory mechanism of ahslyA on ENX antibiotic resistance, we isolated whole protein samples from WT and ΔahslyA strains with or without exposure to 0.0078 μg/mL ENX treatments. After trypsin digestion, each sample was quantified by a label-free quantitative proteomics method to compare the differentially expressed proteins between both groups. As shown in Figure 2A, positive correlations greater than 0.98 were found between the intensities of the MS of each sample with the regression coefficients, which indicated that the quantitative analysis results of MS in this study were stable and reliable. A principal component analysis (PCA) scatter diagram was drawn to cluster the samples. The dots of different colors in Figure 2B represent three groups of repeats of the same sample. It can be seen from the diagram that the three dots of the same color are relatively close; indicating that the gene expression pattern of the three repeats of the same sample had a small difference and suitable data repeatability. The proteomic analysis of the ΔahslyA + ENX group was significantly separated from the WT + ENX group in the direction of PC1, indicating that there may be significant differences between them. These results indicate that the effect of the deletion of ahslyA on bacterial proteome is more than the effect of ENX stress.
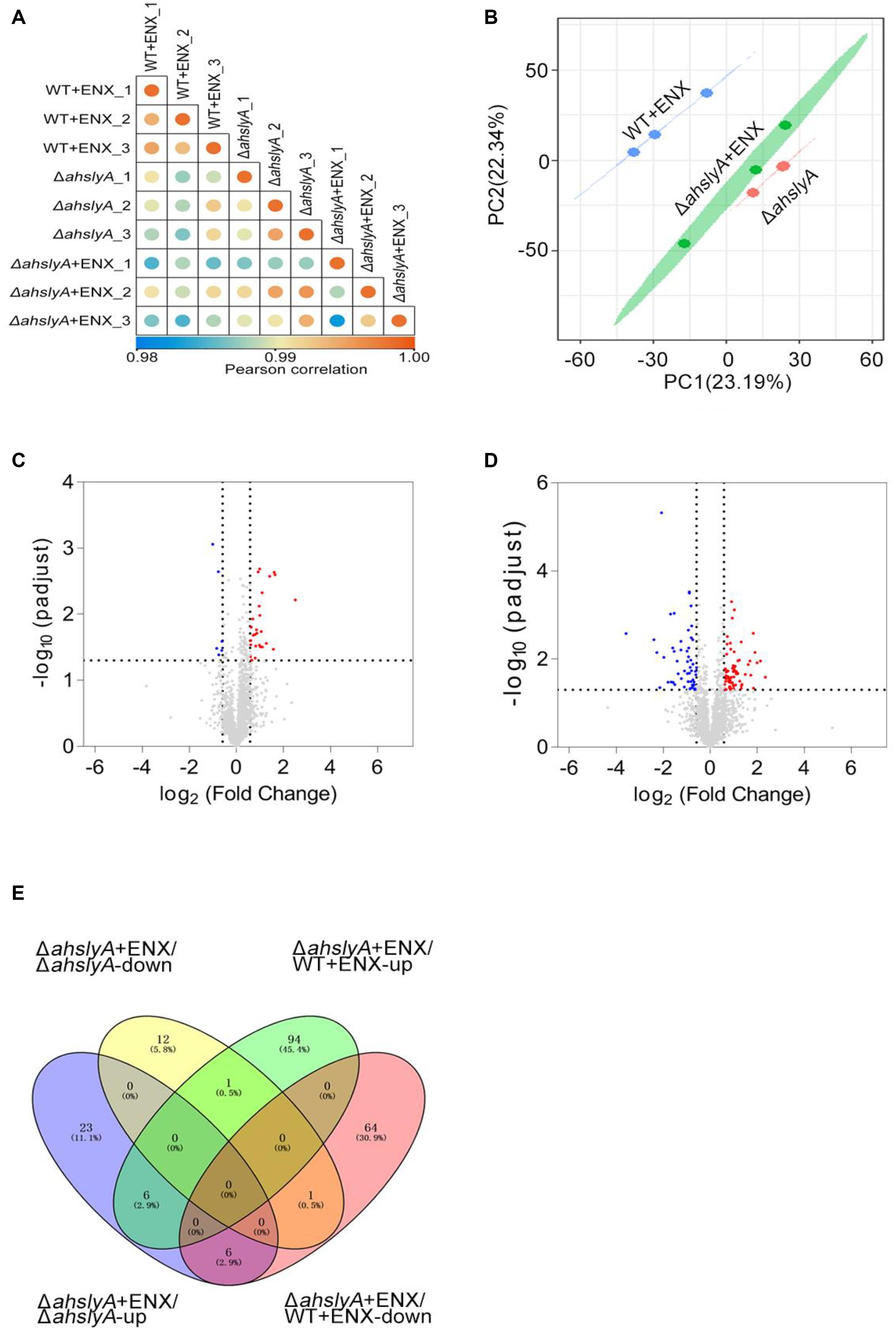
Figure 2. Label-free quantitative proteomics data analysis. (A) Correlation coefficient analysis of protein MS intensity in three biological replications. (B) PCA of WT + ENX, ΔahslyA + ENX, and ΔahslyA. (C,D) Volcano maps comparing the abundance ratios of proteins with significant differences in expression between ΔahslyA + ENX vs. ΔahslyA and ΔahslyA + ENX vs. WT + ENX, respectively. The blue dots represent downregulated proteins, red dots represent upregulated proteins, and the gray color represents non-differentially expressed proteins. (E) Venn-diagram of differentially expressed proteins between two comparisons.
The Differential Protein Abundances of WT and ΔahslyA in Response to ENX Stress
In this study, a total of 2,534 proteins were identified by mass spectrometry (unique peptide number ≥ 2 and false discovery rate (FDR) < 1%). The protein abundance ratio that was ≥ 1.5 (upregulated expression) and ≤0.667 (downregulated expression) with a p-value < 0.05 of each compared group was regarded as differentially expressed proteins. We analyzed the data by two comparisons, ΔahslyA + ENX vs. ΔahslyA and ΔahslyA + ENX vs. WT + ENX, in order to interpret the ahslyA mediated ENX resistance in this study. When compared with ΔahslyA without antibiotic treatment, there was 49 differentially expressed proteins were found, which accounted for 1.77% of the total identified proteins, including 35 increasing and 14 decreasing in abundance in the ΔahslyA + ENX treatment group (Figure 2C). When compared with WT + ENX, a total of 172 proteins, including 101 increasing and 71 decreasing in abundance, were altered in the ΔahslyA + ENX treatment group (Figure 2D). The following overlap analysis between both group comparisons showed that ΔahslyA + ENX vs. ΔahslyA and ΔahslyA + ENX vs. WT + ENX have 14 common altered proteins, of which seven have the same protein expression trend and another seven have the opposite expression (Supplementary Table 1 and Figure 2E).
Bioinformatics Analysis of Altered Proteins in ΔahslyA + ENX/ΔahslyA and ΔahslyA + ENX/WT + ENX Comparisons
We then used GO terms enrichment to analyze altered proteins in both comparisons. In the biological process (BP) enrichment, the DNA metabolic process, cellular response to stimulus, SOS response, and cell communication were the most enriched terms in the ΔahslyA + ENX vs. ΔahslyA comparison. Additionally, the response to stress, the response to stimulus, the DNA metabolic process, and the cellular response to external stimulus were the most enriched terms in the ΔahslyA + ENX vs. WT + ENX comparison. When comparing both group, the response to stress and the cellular response to stress in the ΔahslyA + ENX vs. WT + ENX comparison changed significantly, while no change were observed in the ΔahslyA + ENX vs. ΔahslyA comparison (Figures 3A,B).
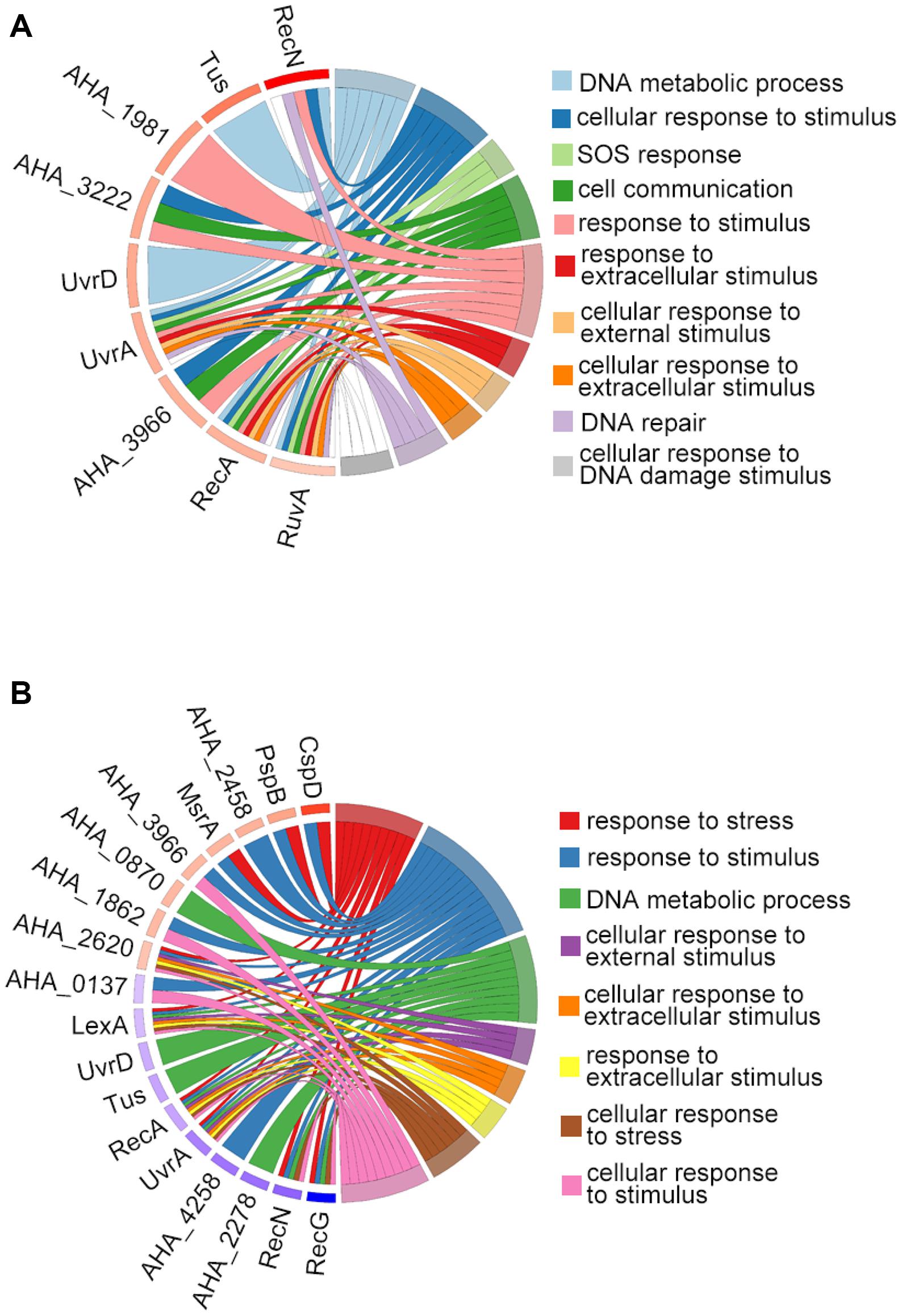
Figure 3. Chord plot showing GO terms BP analysis of differentially expressed proteins in the two comparisons. (A) ΔahslyA + ENX vs. WT + ENX, and (B) ΔahslyA + ENX vs. ΔahslyA.
We further analyzed the predicted PPI network of altered proteins in both comparisons using STRING software and then clustered them using the MCL algorithm. In the ΔahslyA + ENX vs. ΔahslyA comparison, there were five clusters that were enriched (Figure 4). Eight increasing proteins were clustered in cluster 1, and most of them were SOS or DNA repair-related proteins. There were six metabolic pathway-related proteins clustered in cluster 2, including three proteins that might be involved in acyl-CoA metabolism, namely, acyl-CoA thioester hydrolase YciA (gene name AHA_1563), acyl-CoA thioesterase I (AHA_3489), and acyl carrier protein (acpP). Both AHA_3297 and AHA_0044 are a sensor histidine kinase and sensory box/GGDEF family gene, respectively, which were present in clusters 3–5. It indicates that ahslyA may affect a bacterial two-component or cyclic di-GMP signaling system. The top 14 clusters of the ΔahslyA + ENX vs. WT + ENX comparison are shown in Figure 3B. In cluster 1, at least seven proteins, including RecA, UvrA, UvrD, LexA, DinB, RecN, and RecG are involved in DNA repair processes and all seven were decreasing in abundance. AHA_3525 is a response regulator, which was the network hub in cluster 2. Moreover, AHA_3525 interacted with three TRs, namely, AHA_0137 (response regulator, GltR), AHA_1862 (response regulator protein), and AHA_3966 (DNA-binding response regulator), in addition to AHA_3297 (diguanylate cyclase DosC), and most of these genes were increasing in abundance. In cluster 3, most of altered proteins were metal ion-related proteins. For example, SelA, SelD, and the SelT/SelW/SelH domain (AHA_1610) were involved in selenium metabolism in prokaryotes. Additionally, clusters 4–6 were mostly clustered in oxidative respiration, uncharacterized protein and sulfate metabolism.
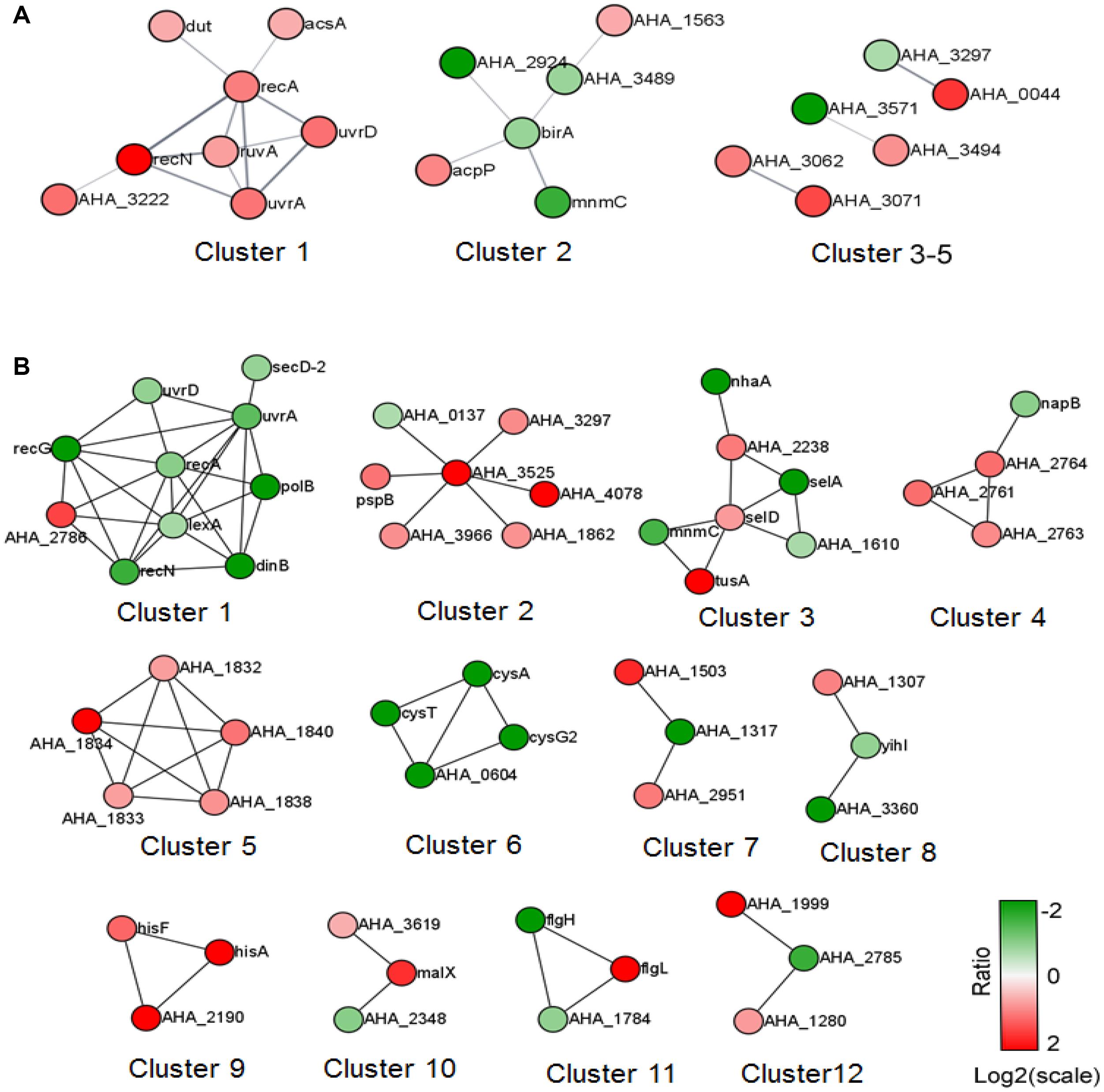
Figure 4. The PPI prediction network of altered proteins in the two comparisons. (A) ΔahslyA + ENX vs. WT + ENX, and (B) ΔahslyA + ENX vs. ΔahslyA. The green dots represent downregulated proteins and red dots represent upregulated proteins.
The ENX-Resistance Capabilities of Proteins Regulated by ahslyA
In order to better understand the ENX resistance mechanism mediated by the TR ahslyA, we further assessed the effect of several gene mutants on the resistance to ENX, which were shown to be regulated directly or indirectly by ahslyA in our proteomics results. Selected gene deletion strains, including three genes related to decreasing in protein abundance (AHA_0655, AHA_1195, and AHA_3721) and three genes related to increasing in protein abundance (AHA_1239, AHA_2114, and narQ) in the ΔahslyA + ENX vs. WT + ENX proteomic comparison, were successfully constructed via a two-step homologous recombination method using the primer pairs listed in Supplementary Table 2. Each mutant was assessed for sensitivity to ENX using an antibiotic susceptibility assay. As showed in Figure 5, the ΔAHA_0655 exhibited a slightly decreased resistance while other mutants showed no significant difference to 0.0078 μg/mL of ENX. The ΔAHA_2114 and ΔnarQ showed a slight decrease in resistance to 0.01 μg/mL of ENX, and the ΔAHA_1239 and ΔAHA_3721 demonstrated significantly increased resistance to 0.01 μg/mL of ENX. These results suggested that ahslyA may regulate the transcription of AHA_0655, narQ, AHA_3721, AHA_2114, or AHA_1239 to against ENX stress.
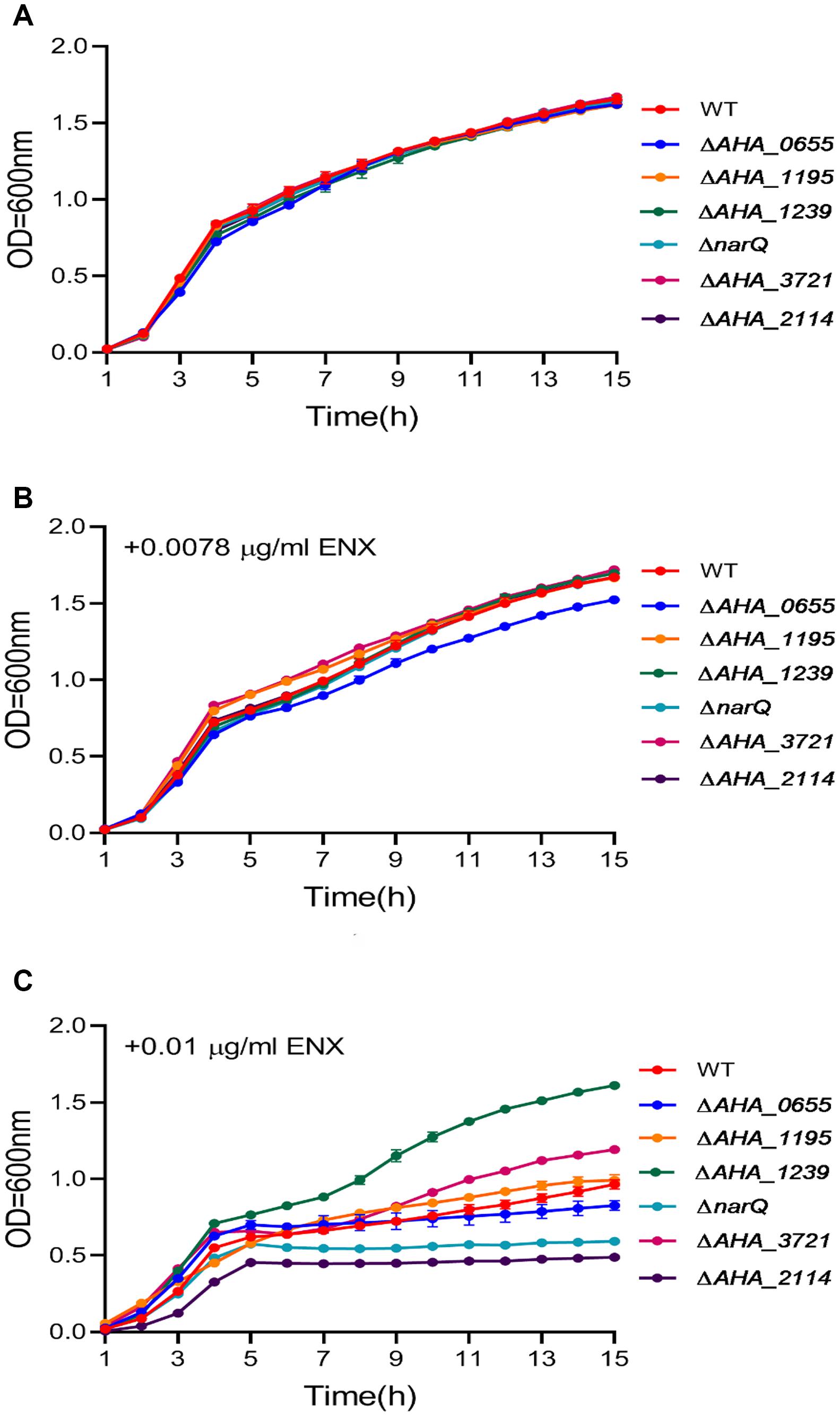
Figure 5. The Growth curves of A. hydrophila WT and a total six gene deletion strains with or without ENX treatment. (A) LB medium alone; (B) LB medium + 0.0078 μg/mL ENX; (C) LB medium + 0.01 μg/mL ENX.
Discussion
It is well known that bacterial TRs play a crucial regulatory role in diverse physiological and pathological functions. However, as there are hundreds of TRs are identified in prokaryotes and only a few of them have been well described, the intrinsic regulatory mechanisms of prokaryotic TRs are largely unknown (Brown et al., 2003). SlyA belongs to the TR MarR family that possesses a winged-helix DNA binding domain (Wilkinson and Grove, 2006). It was first reported to regulate virulence in many pathogens, such as Salmonella typhimurium, Enterococcus faecalis, and Dickeya dadantii (Libby et al., 1994; Haque et al., 2009; Michaux et al., 2011b). It has also been shown to play important roles in oxidative stress, bile salt stress, antimicrobial peptide resistance, heat, and acid stress (Buchmeier et al., 1997; Spory et al., 2002; Shi et al., 2004; Michaux et al., 2011a). Therefore, it suggests that this TR plays an important role in numerous physiological functions. However, the mechanism underlying these biological functions remains elusive.
Since SlyA plays an important role in the resistance to environmental stresses, we speculated that it may contribute to bacterial antibiotic resistance, because antibiotics are toxic small molecules like bile salts. To test our hypothesis, the antibiotic susceptibilities of the slyA gene deletion mutant and its complemented strains were analyzed in this study. We found that the ΔahslyA had increased resistance against several quinolone antibiotics, suggesting that this TR may negatively regulate resistance to certain antibiotics, especially ENX. We then performed label-free quantitative MS to characterize the effect of ahslyA gene deletion on the proteome of A. hydrophila cells with or without ENX treatment. Here, we analyzed two group comparisons in order to understand the ENX resistance mediated by ahslyA. In the ΔahslyA + ENX vs. ΔahslyA, we identified 49 altered proteins, and when ΔahslyA + ENX was compared to WT + ENX, there were 172 differentially expressed proteins. GO bioinformatics analysis showed that both comparisons were related to a stress response or stimulus, which indicated that ahslyA may play an important role in stress resistance to environmental factors, including antibiotics. Although, we have not obtained a significant KEGG (Kyoto Encyclopedia of genes and genes) pathway enriched altered proteins in either of the comparisons in this study. The PPI prediction plus the MCL algorithm showed that these altered proteins can be classified into several clusters. In the ΔahslyA + ENX vs. ΔahslyA comparison, five DNA repair-related proteins (RecA, RecN, RuvA, UvrA, and UrvD) were significantly increased in abundance. It is well known that quinolone antibiotics inhibit DNA synthesis and cause DNA strand cleavage or cell death. The underlying mechanism of quinolone antibiotic resistance in bacteria is due to the upregulation of DNA repair related proteins. In our previous study, 11 SOS responses or DNA repair-related proteins of A. hydrophila were reported to be increased under ENX stress (Zhang L.S. et al., 2020). Moreover, the deletion of uvrA decreased the ENX tolerance in A. hydrophila, which suggests the important role of the DNA repair process in protecting the DNA from quinolone-induced damage. Interestingly, we found that many of the DNA repair-related proteins decreased in abundance in the ΔahslyA + ENX vs. WT + ENX comparison. This could have happened for a few reasons. First, the loss of ahslyA likely slowed down the toxic effect of ENX, so that bacteria did not trigger the DNA repair system. Second, the DNA repair response should be a last resort against ENX, as it may cause genetic mutations that influence bacterial survival. Third, ahslyA may regulate other drug resistance genes, aside from just DNA repair processes.
In both proteomic comparisons, the ΔahslyA + ENX vs. WT + ENX comparison may be better than the ΔahslyA + ENX vs. ΔahslyA comparison to interpret ahslyA-mediated ENX resistance, because the first comparison is on the same ENX background. Therefore, we focused more on the properties of altered proteins between ΔahslyA + ENX compared to ΔahslyA. Of these altered proteins, we found ahslyA could directly or indirectly regulate at least 11 TRs (gltR, yidZ, ycaN, citA, AHA_3297, AHA_0117, AHA_3721, AHA_1240, AHA_4233, AHA_1862, and AHA_3966), which indicating that these TRs may construct a complicated gene regulatory network to maintain the intracellular homeostasis during ENX stress. Of these TRs, AHA_3966 is homologous with E. coli ompR, with an identity of 49%. The ompR gene belongs to a well-known two-component regulatory system and plays important roles in multiple physiological functions, including antibiotic resistance (Lin et al., 2012; Zhang M.M. et al., 2020). Moreover, both AHA_1862 and AHA_3297 coded diguanylate cyclase and AHA_3525 coded phosphodiesterase that govern the cellular level of c-di-GMP, which acts as a unique bacterial second messenger to trigger various cellular responses, such as in motility, biofilm formation, and antibiotics resistance (De et al., 2008; Gupta et al., 2014). However, the relationship between ahslyA and c-di-GMP is not clear.
To further understand the effect of ahslyA on the regulation of ENX resistance-related proteins, six target genes (AHA_0655, AHA_1195, AHA_3721, AHA_1239, AHA_2114, and narQ) that encode altered proteins in the proteomics data were selected to construct targeted gene deletion strains to determine their roles in susceptibility against ENX. Among these selected genes or proteins, AHA_0655 (A0KG12) encodes an ATP-binding cassette transporter that belongs to a multi-drug efflux transporter family and plays a crucial role in the uptake of nutritional or toxic substrates from the environment, including antibiotics (Pletzer et al., 2015; Tang et al., 2019b). The decreased expression of A0KG12 in this study suggests that ahslyA may negatively regulate this protein to uptake ENX into cells. AHA_1239 encodes a HlyD family secretion protein, was the protein that increased most in abundance in our MS data. AHA_1239 increased 3,899 folds in the ΔahslyA + ENX vs. WT + ENX comparison. Interestingly, AHA_1239 is the downstream gene of ahslyA (AHA_1240), and its amino acid sequence has 30% identity to multi-drug resistance protein MdtN in E. coli. Moreover, the deletion of ahslyA and exposure to ENX stress also caused an upregulation of A0KK37 (AHA_2114), which is the multi-drug resistance protein MdtK. Since both proteins are members of the MATE (multi-drug and toxic compound extrusion) protein family. Further, the overexpression of MdtK increases resistance to norfloxacin, doxorubicin, and acriflavine in Salmonella enterica serovar Typhimurium. More, the ahslyA may negatively regulate both MdtN and MdtK to obtain ENX resistance in this study (Nishino et al., 2006). Additionally, the TRs A0KHI5 (AHA_1195, YcaN) and A0KPG8 (AHA_3721), nitrate sensor protein, A0KIL7 (narQ), belong to a two-component regulatory system play important roles on diverse biological functions in other bacterial species (Oshima et al., 2002). In this study, A0KHI5 and A0KPG8 were down-regulated, while NarQ increased in the ΔahslyA + ENX vs. WT + ENX proteomics data, suggesting that they may affect ENX resistance regulated by ahslyA.
To test our hypothesis, the antibiotic susceptibilities of these six gene deletion mutants were determined. Our results showed that the deletion of AHA_0655 slightly decreased the growth of A. hydrophila under low dose of ENX stress. Further the deletion of AHA_2114 and narQ significantly decreased bacterial growth under high dose of ENX stress, while the deletion of AHA_3721 increased antibiotic susceptibility in A. hydrophila. Which suggested that ahslyA may regulate AHA_0655, narQ, AHA_3721, and AHA_2114 for ENX resistance. Although AHA_1239 may act as a multi-drug resistance protein, the deletion of AHA_1239 caused significant resistance to ENX in this study. The inherent reason is unknown, but based on the fact that AHA_1239 is the downstream neighbor of ahslyA, the deletion of AHA_1239 may affect the expression of ahslyA and then trigger other ARGs or systems against ENX stress. Overall, our data demonstrated the important role of ahslyA in the multiple ARGs regulation during ENX resistance and provided novel insights into the effects of TRs on the antibiotic resistance of bacteria.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.proteomexchange.org/, PXD024843.
Author Contributions
XL and LZ conceptualized and validated the study. ZL, LL, and LZ performed the methodology and the data curation and wrote the manuscript for the final draft. ZL, GW, and QS performed the formal analysis. XL was responsible for the resources, supervised the study, and performed the funding acquisition. RS, WY, HT, and XL wrote, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was sponsored by grants from Key projects of Natural Science Foundation of Fujian Province (No. 2020J02023), NSFC projects (Nos. 31670129 and 31802343), China Post-doctoral Science Foundation (Grant No. 2019M662214), and Program for Innovative Research Team in Fujian Agricultural and Forestry University (No. 712018009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the support of the program for Key Laboratory of Marine Biotechnology of Fujian Province (2020MB04) and the Fujian-Taiwan Joint Innovative Center for Germplasm Resources and Cultivation of Crop (FJ 2011 Program, No. 2015-75, China). We would also like to thank the help from Central Laboratory, Fujian Medical University Union Hospital, China.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.699415/full#supplementary-material
Supplementary Figure 1 | Construction and confirmation of the six gene deletion strains.
Supplementary Table 1 | The identification of 14 common altered proteins between both group comparisons using label-free analysis.
Supplementary Table 2 | The primer pairs used in this study.
Footnotes
References
Banda, M. M., Zavala-Alvarado, C., Perez-Morales, D., and Bustamante, V. H. (2019). SlyA and HilD counteract H-NS-mediated repression on the ssrAB virulence operon of Salmonella enterica Serovar typhimurium and thus promote its activation by OmpR. J. Bacteriol. 201:e00530-18.
Barroso, K. C. M., Previato-Mello, M., Batista, B. B., Batista, J. H., and da Silva, and Neto, J. F. (2018). EmrR-dependent upregulation of the efflux pump EmrCAB contributes to antibiotic resistance in Chromobacterium violaceum. Front. Microbiol. 9:2756.
Brown, N. L., Stoyanov, J. V., Kidd, S. P., and Hobman, J. L. (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163. doi: 10.1016/s0168-6445(03)00051-2
Buchmeier, N., Bossie, S., Chen, C. Y., Fang, F. C., Guiney, D. G., and Libby, S. J. (1997). SlyA, a transcriptional regulator of Salmonella typhimurium, is required for resistance to oxidative stress and is expressed in the intracellular environment of macrophages. Infect. Immun. 65, 3725–3730. doi: 10.1128/iai.65.9.3725-3730.1997
De, N., Pirruccello, M., Krasteva, P. V., Bae, N., Raghavan, R. V., and Sondermann, H. (2008). Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6:e67. doi: 10.1371/journal.pbio.0060067
Fritsche, T. R., Biedenbach, D. J., and Jones, R. N. (2009). Antimicrobial activity of prulifloxacin tested against a worldwide collection of gastroenteritis-producing pathogens, including those causing traveler’s diarrhea. Antimicrob. Agents Chemother. 53, 1221–1224. doi: 10.1128/aac.01260-08
Gupta, K., Liao, J., Petrova, O. E., Cherny, K. E., and Sauer, K. (2014). Elevated levels of the second messenger c-di-GMP contribute to antimicrobial resistance of Pseudomonas aeruginosa. Mol. Microbiol. 92, 488–506. doi: 10.1111/mmi.12587
Haque, M. M., Kabir, M. S., Aini, L. Q., Hirata, H., and Tsuyumu, S. (2009). SlyA, a MarR family transcriptional regulator, is essential for virulence in Dickeya dadantii 3937. J. Bacteriol. 191, 5409–5418. doi: 10.1128/jb.00240-09
Hossain, S., De Silva, B. C. J., Wimalasena, S., Pathirana, H., Dahanayake, P. S., and Heo, G. J. (2018). Distribution of antimicrobial resistance genes and class 1 integron gene cassette arrays in motile Aeromonas spp. isolated from goldfish (Carassius auratus). Microb. Drug Resist. 24, 1217–1225. doi: 10.1089/mdr.2017.0388
Jiang, M., Kuang, S. F., Lai, S. S., Zhang, S., Yang, J., Peng, B., et al. (2020). Na+-NQR confers aminoglycoside resistance via the regulation of l-alanine metabolism. mBio 11:e002086-20.
Lankester, A., Ahmed, S., Lamberte, L. E., Kettles, R. A., and Grainger, D. C. (2019). The Escherichia coli multiple antibiotic resistance activator protein represses transcription of the lac operon. Biochem. Soc. Trans. 47, 671–677. doi: 10.1042/bst20180498
Li, L., Peng, B., Peng, X. X., and Li, H. (2020). Reprogramming metabolomics reveals membrane potential-dependent colistin resistance in Vibrio alginolyticus. Environ. Microbiol. 22, 4295–4313. doi: 10.1111/1462-2920.15021
Li, W. X., Zhao, Y. Y., Yu, J., Lin, L., Ramanathan, S., Wang, G. B., et al. (2021). TonB-dependent receptors affect the spontaneous oxytetracycline resistance evolution in Aeromonas hydrophila. J. Proteome Res. 20, 154–163. doi: 10.1021/acs.jproteome.9b00708
Libby, S. J., Goebel, W., Ludwig, A., Buchmeier, N., Bowe, F., Fang, F. C., et al. (1994). A cytolysin encoded by Salmonella is required for survival within macrophages. Proc. Natl. Acad. Sci. USA 91, 489–493. doi: 10.1073/pnas.91.2.489
Lin, X. M., Wang, C., Guo, C., Tian, Y. M., Li, H., and Peng, X. X. (2012). Differential regulation of OmpC and OmpF by AtpB in Escherichia coli exposed to nalidixic acid and chlortetracycline. J. Proteomics 75, 5898–5910. doi: 10.1016/j.jprot.2012.08.016
Ma, J., Chen, T., Wu, S. F., Yang, C. Y., Bai, M. Z., Shu, K. X., et al. (2019). iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217. doi: 10.1093/nar/gky869
Michaux, C., Martini, C., Hanin, A., Auffray, Y., Hartke, A., and Giard, J.-C. (2011a). SlyA regulator is involved in bile salts stress response of Enterococcus faecalis. FEMS Microbiol. Lett. 324, 142–146. doi: 10.1111/j.1574-6968.2011.02390.x
Michaux, C., Sanguinetti, M., Reffuveille, F., Auffray, Y., Posteraro, B., Gilmore, M. S., et al. (2011b). SlyA is a transcriptional regulator involved in the virulence of Enterococcus faecalis. Infect. Immun. 79, 2638–2645. doi: 10.1128/iai.01132-10
Nishino, K., Latifi, T., and Groisman, E. A. (2006). Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica Serovar Typhimurium. Mol. Microbiol. 59, 126–141. doi: 10.1111/j.1365-2958.2005.04940.x
Oshima, T., Aiba, H., Masuda, Y., Kanaya, S., Sugiura, M., Wanner, B. L., et al. (2002). Transcriptome analysis of all two-component regulatory system mutants of Escherichia coli K-12. Mol. Microbiol. 46, 281–291. doi: 10.1046/j.1365-2958.2002.03170.x
Peng, B., Su, Y. B., Li, H., Han, Y., Guo, C., Tian, Y. M., et al. (2015). Exogenous Alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab. 21, 249–261. doi: 10.1016/j.cmet.2015.01.008
Peterson, E., and Kaur, P. (2018). Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 9:2928. doi: 10.3389/fmicb.2018.02928
Pletzer, D., Braun, Y., Dubiley, S., Lafon, C., Köhler, T., Page, M. G. P., et al. (2015). The Pseudomonas aeruginosa PA14 ABC transporter NppA1A2BCD is required for uptake of peptidyl nucleoside antibiotics. J. Bacteriol. 197, 2217–2228. doi: 10.1128/jb.00234-15
Pourahmad Jaktaji, R., and Ebadi, R. (2013). Study the expression of marA gene in ciprofloxacin and tetracycline resistant mutants of Esherichia coli. Iran. J. Pharm. Res. 12, 923–928.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shi, Y., Latifi, T., Cromie, M. J., and Groisman, E. A. (2004). Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J. Biol. Chem. 279, 38618–38625. doi: 10.1074/jbc.m406149200
Spory, A., Bosserhoff, A., von Rhein, C., Goebel, W., and Ludwig, A. (2002). Differential regulation of multiple proteins of Escherichia coli and Salmonella enterica serovar Typhimurium by the transcriptional regulator SlyA. J. Bacteriol. 184, 3549–3559. doi: 10.1128/jb.184.13.3549-3559.2002
Sun, L. N., Yao, Z. J., Guo, Z., Zhang, L. S., Wang, Y. Q., Mao, R. R., et al. (2019). Comprehensive analysis of the lysine acetylome in Aeromonas hydrophila reveals cross-talk between lysine acetylation and succinylation in LuxS. Emerg. Microbes Infec. 8, 1229–1239. doi: 10.1080/22221751.2019.1656549
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368.
Tang, R. Q., Luo, G., Zhao, L. M., Huang, L. X., Qin, Y. X., Xu, X. J., et al. (2019a). The effect of a LysR-type transcriptional regulator gene of Pseudomonas plecoglossicida on the immune responses of Epinephelus coioides. Fish Shellfish Immunol. 89, 420–427. doi: 10.1016/j.fsi.2019.03.051
Tang, R. Q., Zhao, L. M., Xu, X. J., Huang, L. X., Qin, Y. X., Su, Y. Q., et al. (2019b). Dual RNA-Seq uncovers the function of an ABC transporter gene in the host-pathogen interaction between Epinephelus coioides and Pseudomonas plecoglossicida. Fish Shellfish Immunol. 92, 45–53. doi: 10.1016/j.fsi.2019.05.046
Tian, S. C., Wang, C., Li, Y. Y., Bao, X. M., Zhang, Y. W., and Tang, T. (2021). The impact of SlyA on cell metabolism of Salmonella typhimurium: a joint study of transcriptomics and metabolomics. J. Proteome Res. 20, 184–190. doi: 10.1021/acs.jproteome.0c00281
Walter, W., Sanchez-Cabo, F., and Ricote, M. (2015). GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. doi: 10.1093/bioinformatics/btv300
Wilkinson, S. P., and Grove, A. (2006). Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 8, 51–62.
Yu, H. B., Zhang, Y. L., Lau, Y. L., Yao, F., Vilches, S., Merino, S., et al. (2005). Identification and characterization of putative virulence genes and gene clusters in Aeromonas hydrophila PPD134/91. Appl. Environ. Microbiol. 71, 4469–4477. doi: 10.1128/aem.71.8.4469-4477.2005
Zhang, L. S., Li, W. X., Sun, L. N., Wang, Y. Q., Lin, Y. X., and Lin, X. M. (2020). Quantitative proteomics reveals the molecular mechanism of Aeromonas hydrophila in enoxacin stress. J. Proteomics 211:103561. doi: 10.1016/j.jprot.2019.103561
Zhang, M. M., Kang, J. P., Wu, B., Qin, Y. X., Huang, L. X., Zhao, L. M., et al. (2020). Comparative transcriptome and phenotype analysis revealed the role and mechanism of ompR in the virulence of fish pathogenic Aeromonas hydrophila. Microbiologyopen 9:e1041.
Zhou, D., and Yang, R. (2006). Global analysis of gene transcription regulation in prokaryotes. Cell Mol. Life Sci. 63, 2260–2290. doi: 10.1007/s00018-006-6184-6
Keywords: antibiotics resistance, AhSlyA, enoxacin, quantitative proteomics, Aeromonas hydrophila
Citation: Li Z, Zhang L, Song Q, Wang G, Yang W, Tang H, Srinivasan R, Lin L and Lin X (2021) Proteomics Analysis Reveals Bacterial Antibiotics Resistance Mechanism Mediated by ahslyA Against Enoxacin in Aeromonas hydrophila. Front. Microbiol. 12:699415. doi: 10.3389/fmicb.2021.699415
Received: 23 April 2021; Accepted: 17 May 2021;
Published: 08 June 2021.
Edited by:
Bo Peng, Sun Yat-sen University, ChinaReviewed by:
Lixing Huang, Jimei University, ChinaLewis Oscar Felix Raj Lucas, Rhode Island Hospital, United States
Copyright © 2021 Li, Zhang, Song, Wang, Yang, Tang, Srinivasan, Lin and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangmin Lin, eGlhbmdtaW5AZmFmdS5lZHUuY24=