 Yu-feng Qiu1,2†
Yu-feng Qiu1,2† Reshma B. Nambiar3†
Reshma B. Nambiar3† Xue-bin Xu4†Shun-tai Weng1,2
Xue-bin Xu4†Shun-tai Weng1,2 Hang Pan3Kui-cheng Zheng1,2,5*
Hang Pan3Kui-cheng Zheng1,2,5* Min Yue3,6,7,8*
Min Yue3,6,7,8*- 1Department of Bacterialogy, Fujian Provincial Center for Disease Control & Prevention, Fuzhou, China
- 2Department of Bacterialogy, Fujian Provincial Key Laboratory of Zoonosis Research, Fuzhou, China
- 3Department of Veterinary Medicine & Institute of Preventive Veterinary Science, Zhejiang University College of Animal Sciences, Hangzhou, China
- 4Department of Microbiology, Shanghai Municipal Center for Disease Control and Prevention, Shanghai, China
- 5School of Public Health, Fujian Medical University, Fuzhou, China
- 6Zhejiang Provincial Key Laboratory of Preventive Veterinary Medicine, Hangzhou, China
- 7State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, National Medical Center for Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 8Hainan Institute of Zhejiang University, Sanya, China
Non-typhoidal Salmonella (NTS) is a common cause for self-limiting gastroenteritis, representing a public health concern globally. NTS is one of the leading causes of foodborne illnesses in China; however, the invasive infection caused by NTS is largely underappreciated. Here, we reported an NTS invasive infection caused by an infrequently reported serovar Telelkebir (13,23:d:e,n,z15) strain FJ001 in China, which carries antimicrobial-resistant genes [fosA7 and aac(6′)-Iaa] and typhoid-toxin genes (cdtB, pltA, and pltB). By conducting the whole genomic sequencing, we also investigated the relatedness of this strain with an additional 120 global contextual Salmonella enterica serovar Telelkebir (S. Telelkebir) isolates, and assessed the antimicrobial-resistant determinants and key virulence factors using the available genomic dataset. Notably, all 121 (100%) of the S. Telelkebir strains possessed the typhoid toxin genes cdtB, pltA, and pltB, and 58.67% (71/121) of S. Telelkebir harbored antimicrobial-resistant gene fosaA7. The study by core genome multilocus sequence typing (cgMLST) and core single-nucleotide polymorphism (SNP)-based phylogenomic analysis demonstrated that the S. Telelkebir isolates from different sources and locations clustered together. This suggests that regular international travels might increase the likelihood of rapid and extensive transmissions of potentially pathogenic bacteria. For the first time, our study revealed the antimicrobial resistance, virulence patterns, and genetic diversity of the serovar S. Telelkebir isolate in humans and similar isolates over the world. The present study also suggests that genomic investigation can facilitate surveillance and could offer added knowledge of a previously unknown threat with the unique combination of virulent and antimicrobial-resistant determinants.
Introduction
Salmonella enterica is a major global foodborne pathogen (Pan et al., 2018; World Health Organization (WHO), 2018; Jiang et al., 2021). S. enterica is divided into six distinct subspecies: enterica, salamae, arizonae, diarizonae, houtenae, and indica (Chand et al., 2020). The S. enterica subsp. enterica consists of more than 1,500 serotypes. A small number of Salmonella serovars (S. Typhi and S. Paratyphi A, B, or C) are human restricted and evoke an invasive, life-threatening systemic disease (Parry et al., 2002; Liu et al., 2021). In contrast, the non-typhoidal Salmonella (NTS) serovars generally cause self-limiting diarrheal illnesses with low case mortality. According to the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD 2017 Causes of Death Collaborators, 2018), it was estimated that NTS accounts for 95 million cases of enterocolitis and 50,771 related deaths.
In sub-Saharan Africa, NTS is the most common cause of bloodstream infection in immunocompromised adults and children with a fatality rate of 20–25% (Feasey et al., 2012). In developed countries, 5% of NTS cases are invasive, focal systemic infections, or extra-intestinal disease leading to bacteremia (Suez et al., 2013). The NTS infections in humans arise through the food chain, but infection can also be contracted through contact with the infected animals, person-to-person transmission, or contaminated water (World Health Organization (WHO), 2018). Various wildlife animals, reptiles, and their contaminated environment could act as a reservoir for various rare and infrequently reported serovars that are normally dissimilar from those strains isolated from the commercial food chain. Currently, most of the research is concentrated on S. Typhimurium and S. Enteritidis, and comparatively lesser consideration has been given to uncommon emerging serovars (Tankson et al., 2006; Mansour et al., 2020). There is an evolving need to investigate the incidence of these uncommon S. enterica serovars as a variety of these have been linked to numerous foodborne outbreaks, acute gastroenteritis, splenic abscesses, etc. (Rodriguez et al., 2006; Berendes et al., 2007; Li et al., 2017), which are largely underappreciated. In China, most of the foodborne bacterial outbreaks (70–80%) are ascribed to Salmonella infections (Wang et al., 2019; Paudyal et al., 2018; Li et al., 2020; Zhou et al., 2020). However, the study on invasive NTS infection remains incomplete in China.
Recently, various reports have indicated the efficiency of whole-genome sequencing (WGS) in the epidemiological investigation of transmittable disease at different geographical locations (Gardy and Loman, 2018). The WGS methods involve either the characterization of core single-nucleotide polymorphisms (SNP) or core-genome multilocus sequence typing (cgMLST) analysis (Mellmann et al., 2016). The cgMLST-based approach involves the direct assessment and comparison of newly determined genotypes with historically available data. However, the SNP-based analysis requires recalculation once there is a change in the data set unless a preliminarily established reference genome is provided (Ruan et al., 2020).
In this study, we have investigated a bloodstream infection case caused by Salmonella Telelkebir in China. To improve our knowledge of the genomic epidemiology of S. Telelkebir, phylogenetic relationship and comparative genomic analysis of additional 120 global S. Telelkebir isolates from various countries were also carried out.
Materials and Methods
Ethics Statements
The FJ001 strain was collected and approved by the Shanghai and Fujian Center for Disease Control and Prevention. Written consent was obtained prior to the study for collecting the samples for surveillance purposes.
Bacterial Isolates
The publicly available isolates with their metadata were collected from Enterobase1 or the sequence read archive2. The S. Telelkebir isolates were used in this study to embody various geographical locations, including the UK (n = 61), United States (n = 23), Germany (n = 18), Ireland (n = 4), France (n = 4), China (n = 1), Mali (n = 1), Netherlands (n = 1), and Turkey (n = 1). The locations of the remaining strains were not available (NA) (Figure 1).
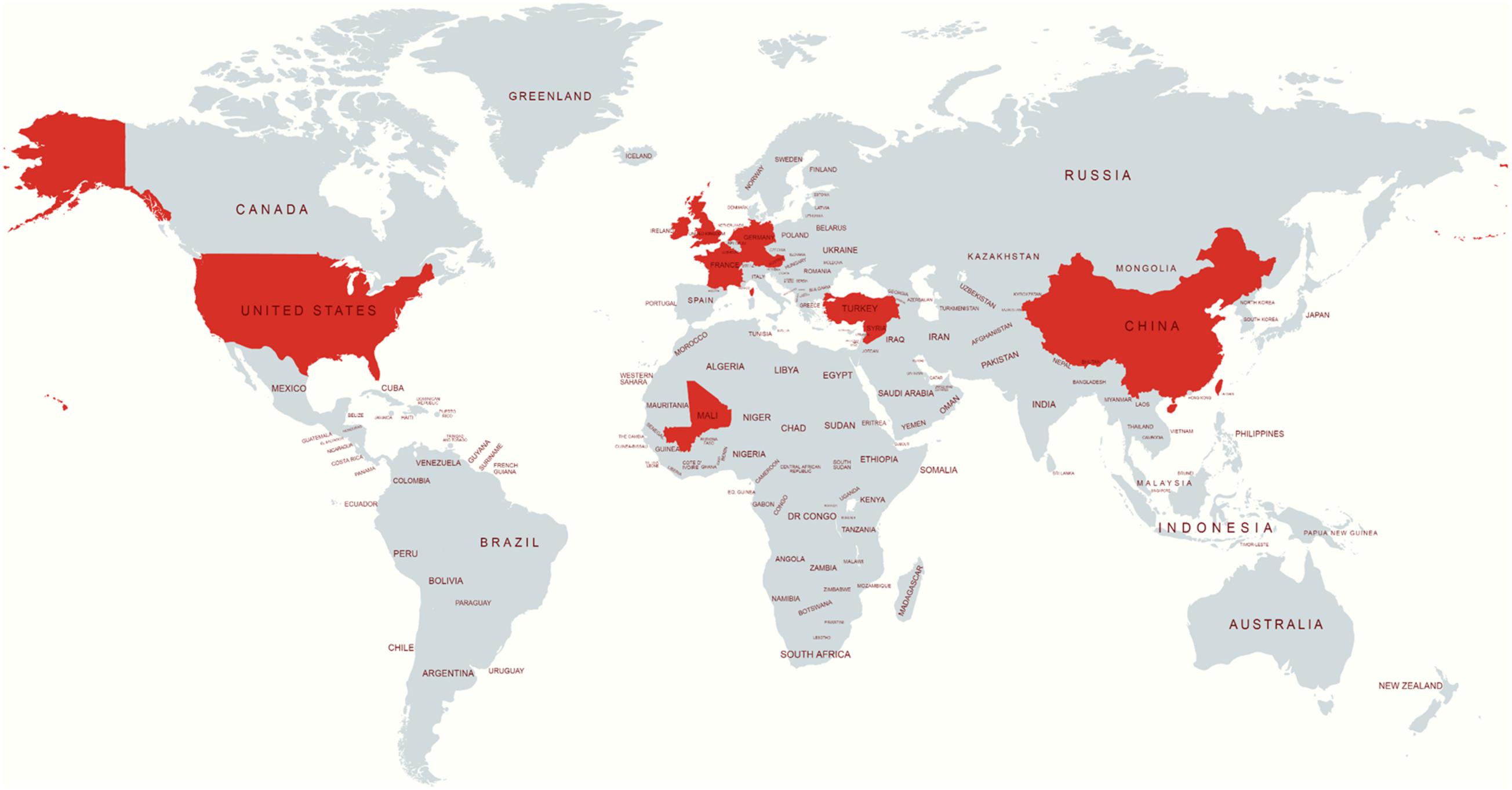
Figure 1. Source location of S. Telelkebir strain genomes used in this study. The red color indicates the presence of this serovar. This map was created using an online service (https://mapchart.net/).
Clinical Study
A patient with no significant medical history was admitted to a tertiary hospital for cardiovascular medicine in Fujian, with fever, abdominal pain, diarrhea, and syncope on December 12, 2018. The patient had no history of travel or contact with any wildlife or domestic animals. The patient had a history of eating outside (roasted meat, the type of meat was unclear) 1 week prior to the onset of the disease. However, other food sources may have been the vehicle of infection. During admission, the patient had a body temperature of 36.6°C, and the blood pressure was 124/60 mmHg. Blood tests showed a normal white blood count (9.4 × 109/L) and a monocyte count (1.12 × 109/L). The stool sample was normal and was negative for fungi and parasites via microscopic examination. The stool sample was negative for Salmonella and Shigella. On the second day of hospital admission, the patient developed diarrhea, abdominal pain, and fever. The injection treatments with Sulperazon (cefoperazone sulbactam) and heparin were ineffective. During the treatment, the patient had intermittent fever and chills. On December 16, a combination of treatments with Sulperazon (cefoperazone sulbactam) and Cravit (levofloxacin) injection also failed. On December 17, the bacterial blood culture indicated the presence of Salmonella Group G, and the patient was diagnosed for sepsis and multiple organ dysfunction syndromes. On December 18, the patient was transferred to the intensive care unit with combined treatment of imipenem–cilastatin and vancomycin, and the body temperature of the patient and chills decreased. After a successive 4-day treatment, the blood culture was negative for Salmonella. The patient completely recovered on December 26 and was discharged from the hospital (Figure 2).
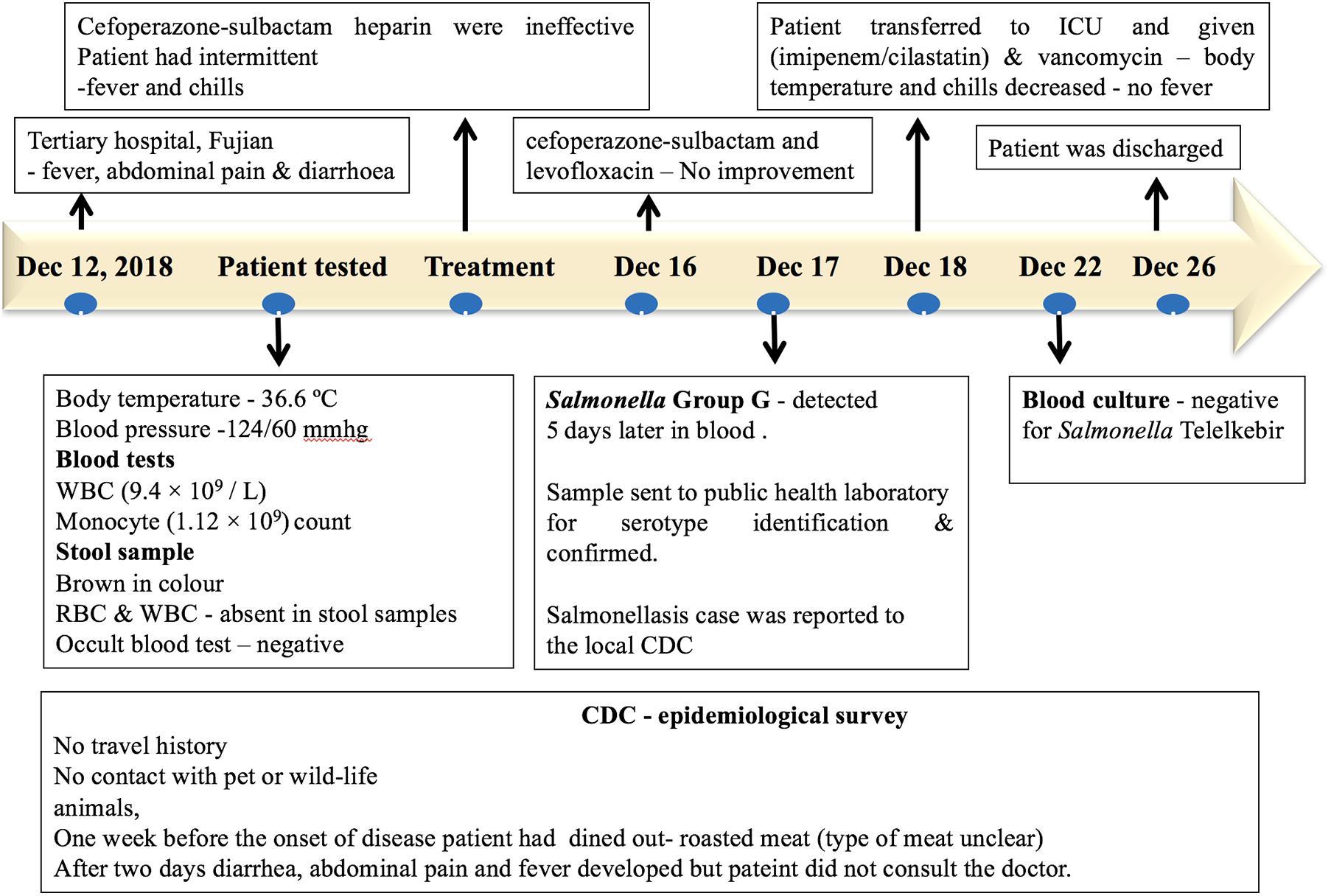
Figure 2. Timeline of the salmonellosis diagnosis, treatment, and investigation since December 12, 2018.
Characterization of Salmonella Telelkebir
For screening Salmonella and Shigella, blood and stool samples were spread onto xylose lysine deoxycholate agar (XLD) and MacConkey agar, respectively, and were incubated at 36°C for 24 h. The stool cultures were negative for Salmonella, while the blood culture displayed positive colonies on XLD after 24 h. The plate was purple-red in color, and colonies with the typical characteristic of Salmonella, i.e., round, moist, smooth, colorless, translucent, and black in the center were selected and inoculated in triple sugar iron (TSI) plate for 24 h at 36°C. The Salmonella isolate was further subjected to biochemical analysis (VITEK2 COMPACT; bioMérieux, France) and identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS). The serotype agglutination assay was conducted using the anti-serum purchased from Denmark (SSI Diagnostica, Denmark), and was confirmed as a newly reported serovar (13,23:d:e,n,z15 or Telelkebir) in mainland China.
Antimicrobial Susceptibility Testing
The bacteria isolate was used for antimicrobial susceptibility testing (AST) against 17 antimicrobial agents. Broth microdilution minimum inhibitory concentration (MIC) determination was performed according to the Clinical and Laboratory Standards Institute (CLSI) guidelines and interpretation (CLSI, 2018), the European Committee for Antimicrobial Susceptibility Testing (EUCAST, 2018), and where no EUCAST or CLSI interpretative criteria were available, breakpoints were harmonized with those of the National Antimicrobial Resistance Monitoring System (NARMS), United States (FDA, 2017). The following antimicrobials were tested: fosfomycin, levofloxacin, cefoperazone sulbactam, streptomycin, ampicillin, amoxicillin–clavulanic acid, cefoxitin, imipenem, nalidixic acid, ciprofloxacin, chloramphenicol, tetracycline, kanamycin, gentamicin, trimethoprim–sulfamethoxazole, ceftiofur, and azithromycin (Sangon Biotech, China). Escherichia coli ATCC 25922 were used as a control strain. The AST against all antibiotics were carried out in Muller–Hinton broth (MHB) medium in both aerobic and anaerobic conditions and incubated at 37°C. The AST was also performed in Dulbecco’s modified Eagle’s medium (DMEM) (Dulbecco and Freeman, 1959) and incubated in a 5% CO2 incubator.
DNA Extraction, Genomic Sequencing, and Data Analysis
The genomic DNA of FJ001 isolate was extracted from overnight cultures grown at 37°C in Luria–Bertani broth under 180 rpm shaking conditions by using a TIANamp bacteria DNA kit (Tiangen Biotech, China) according to the manufacturer’s protocol. The Qubit Broad Range assay kit (Invitrogen, United States) was used for quantification. The Genomic DNA library was constructed using Nextera XT DNA library construction kit (No. FC-131-1024; Illumina, United States). High-throughput genome sequencing was accomplished by the Illumina NovaSeq 6000 platform, using paired-end sequencing of 2 × 150-bp reads as previously described (Biswas et al., 2019; Paudyal et al., 2019; Yu et al., 2020). For the comparative genomic analysis, an additional 119 isolates (Supplementary Table 1), with assembled contigs in FASTA format, were obtained from the Enterobase (see text footnote 1). One additional strain, with raw reads in SRA format was obtained from the NCBI SRA dataset (see text footnote 2). The quality of sequencing was checked with FastQC toolkit (Rouzeau-Szynalski et al., 2019). The raw reads of the FJ001 genome were trimmed with trimmomatic software prior to genome assembly. The raw reads for each strain were assembled by using SPAdes 4.0.1 (Bolger et al., 2014). QUAST (Gurevich et al., 2013) was used to assess the assembled genomes through basic statistics generation, including the total number of contig, the length of contig, and N50 (Supplementary Table 2). Prokka v.1.14 with default settings under the in-house galaxy platform and Rapid Annotation Subsystem Technology (RAST) server3 were used for annotation of the assembled genomes. The NCBI Basic Local Alignment Search Tool (BLAST) BLASTp4 program was used for similarity alignment. The plasmid types and antimicrobial-resistant genes were determined using the PlasmidFinder 2.05 and ResFinder 3.2,6 respectively (Zankari et al., 2012). The virulence factors in the genome were examined using the Virulence Factors Database (VFDB) (Liu et al., 2019). Salmonella Telelkebir in silico serotyping was conducted by the Salmonella In Silico Typing Resource (SISTR) platform7 and SeqSero2.8 The multilocus sequence typing of the isolates were carried out using MLST.9 The contigs were used for variant calling against reference strain 98-12414 and outgroup control strain Poona ATCC® BAA-1673 by software Snippy v4.4.4 to obtain core single-nucleotide polymorphism (SNP) for determining the population structure of 121 available S. Telelkebir isolates. After being filtered by 95% gap parameter to get the core SNPs, a total of 85,694 SNPs were used to build a maximum-likelihood phylogenetic tree with 1,000 bootstraps using IQ-TREE v.1.6.12 with the best model TVM + F + ASC (Letunic and Bork, 2007).
Core Genome Multilocus Sequence Typing Analysis of Salmonella Telelkebir Isolates
The cgMLST analysis was carried out using the Ridom SeqSphere+ software v6.0.2 (Ridom). An ad hoc core genome MLST (cgMLST) scheme was created for the gene-by-gene analysis with SeqSphere+ (Ridom® GmbH, Münster, Germany). Hence, the S. Typhimurium LT2 (NC_003197.1) genome comprising 4,451 genes was used as annotated reference. The cgMLST target definer tool was applied to 121 Salmonella Telelkebir genomes with the default settings of the software to define the core genome loci (Simon et al., 2018). A cgMLST tree was built using the neighbor-joining method. The cgMLST distance matrix showing pairwise comparison of allelic differences between 121 isolates is given in Supplementary Table 3.
Data Availability
The genome for the strain FJ001 was deposited in the NCBI (BioProject PRJNA666303). Associated metadata and virulence genes can be found in Supplementary Table 1.
Results
Salmonella Telelkebir Clinical Isolate FJ001
In this study, we reported a bloodstream infection caused by Salmonella Telelkebir (FJ001) isolate in mainland China. The Salmonella isolate was serotyped by agglutination assay at both the Fujian and Shanghai Center for Disease Prevention and Control, and two independent groups confirmed this causative isolate as an uncommon serovar S. enterica subsp. enterica Telelkebir (13,23:d:e,n,z15). Here, a retrospective investigation confirmed that the patient had no history of travel and pet contact for 1 month prior to the onset of the disease. Exposure factors are mainly related to the history of unclean diet, i.e., roasted meat, but this cannot make accurate traceability judgments for the source of infection. Also, other food source may have been the vehicle of infection. However, Fujian province, located in the sub-tropic region of Eastern China, has abundant natural species resources, local processing, and consumption habits of wild and farmed snakes. Therefore, we cannot rule out the possibility of infection sources as snakes and other reptiles (Yu-feng et al., 2020), for this particular case.
Antimicrobial Resistance Profile for FJ001
The MIC analysis showed that the strain FJ001 was susceptible against all examined antimicrobial agents in both the tested medium under the tested conditions (Supplementary Table 4). It is interesting to note that earlier treatment with cefoperazone sulbactam alone or combined with levofloxacin failed to inhibit the bacterial replication in the patient.
Antimicrobial Resistance Genes and Plasmids in Salmonella Telelkebir Population
WGS analysis indicated that the FJ001 strain carries two antimicrobial-resistant genes, fosA7 and aac(6′)-Iaa. To understand the global resistance patterns, plasmid profile, and virulence patterns of the S. Telelkebir strains, an additional 120 strains were retrieved from Enterobase and NCBI. The WGS analysis revealed that the global strains contains resistance genes against eight antibiotic families: aminoglycosides including aac(3)-Vla (1/121), aac(6′)-Iaa_1 (121/121), ant(3″)-Ia_1 (1/121), aph(3)-Ib_5 (2/121), and aph(6)-Id_1 (3/121); beta-lactamases, including blaTEM–1B (3/121), blaCTX–M–15 (1/121), and blaOXA–1 (1/121); phenicols, including catB3_1 (1/121) and catA1_1 (1/121); trimethoprims including dfrA14_5 (1/121); quinolones including qnrB19_1 (10/121), qnrB1_1 (2/121), and qnrS1_1 (1/121); sulfonamides, including sul1_5 (12/121) and sul2_2 (2/121); tetracyclines, including tet(A) (3/121), and fosfomycins including fosA7_1 (71/121) (Figure 3). All the S. Telelkebir genomes harbored at least one antimicrobial-resistant gene aac(6′)-Iaa_1 (Supplementary Table 1). A total of 58.67% (71/121) isolates harbored resistance genes for fosfomycin, and 11.57% (14/121) isolates possessed resistance gene for quinolones. Three isolates recovered from human infections from different countries harbored antimicrobial resistance gene blaTEM–1B. Two human isolates from Mali (07-1331) and UK (56980) harbored resistance genes to more than three antibiotic families (Figure 3), which indicates that these strains could be multiple drug-resistant (Elbediwi et al., 2019; Mansour et al., 2020). A total of 35.2% isolates harbored plasmids, mainly IncI1α, IncFII, Col440I, IncHI2, IncN, and IncI2 (Delta) (Figure 3).
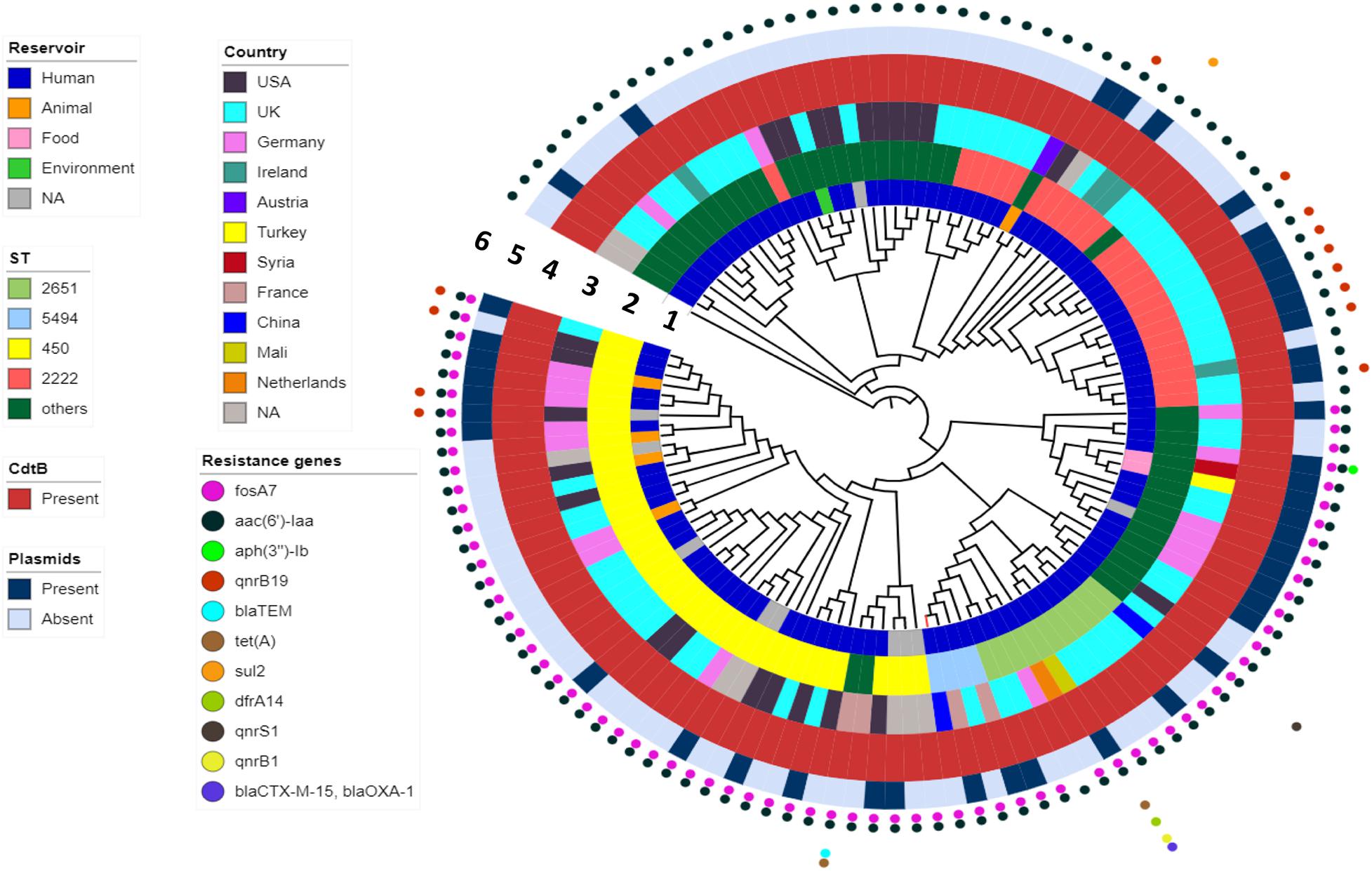
Figure 3. The phylogenomic relationship among 121 Salmonella Telelkebir strains. The tree has been rooted using the serovar Poona ATCC® BAA-1673 as the outgroup. The numbers 1–6 represents the inner to outer rings corresponding to reservoir, ST, country, cdtB, plasmids, and resistance genes, respectively. The strain FJ001 containing leaf node is represented in red. The following plasmids were present in the Telelkebir strains: IncI1α, IncFII, Col440I, IncHI2, IncN, and IncI2 (Delta).
Virulence Genes in Salmonella Telelkebir Population
The virulence gene profile for the 121 strains was carried out using the virulence factor database (VFDB) (Supplementary Table 1). The FJ001 strain harbored cytolethal distending toxin (cdtB) gene and carried fimbrial adherence genes and secretion system genes. A total of 55.81% of the genes (72/129) were conserved among all strains. Twenty-one genes were not present in any of the strains, and the rest of the genes were variable. All the strains carried fimbrial adherence genes fim, csg, and inv, the secretion system genes, sopA, sopB, sopE2, spaOPQRS, spiC, sscAB, sifA, and pipB, and the operons sse and ssa. The secretion system genes pipB2, sopD2, and slrP, and the effector genes, sseK1, and sseK2, and bacteriophage-related genes sodCI, sspH2, and sspH1 genes were variable among the strains. Importantly, all strains (100%) were positive for the cytolethal distending toxin (CDT) genes (cdtB, pltA, and pltB). The cdtB gene is considered as one of the typhoid toxins of Salmonella Typhi, which causes cell arrest due to DNA damage (Chang et al., 2019). The BLASTp analysis of the proteins CdtB, PltA, and PltB of Salmonella Telelkebir showed that all the proteins were highly similar (100–97%) to the CdtB, PltA, and PltB proteins of the available S. enterica strains in NCBI.
Phylogenetic Analysis of Salmonella Telelkebir Population
To determine the phylogenetic relationship, Snippy v.4.4.4 was used to obtain SNP alignment. A total of 18 ST types were identified in 121 isolates from 10 countries across four continents (Figure 3 and Supplementary Table 1). The present study showed that ST450 is the predominant sequence type (33%, 40/121), followed by ST2222 (19%, 23/121), and ST2651 (9%, 11/121) (Figure 3). The FJ001 strain belongs to ST5494, and it clustered with a human isolate isolated from France. The R17.4518 obtained from Taiwan, China, had the ST2651 and clustered with the UK isolates. Interestingly, the two Chinese strains did not cluster together. Among the ST450 isolates, 29 isolates (72.5%) were obtained from humans; four isolates were recovered from animals (10%), whereas the source of the remaining seven isolates could not be obtained. The ST2222 and ST2651 strains were mainly isolated from humans (23 and 11 strains, respectively). The ST450 and ST2249 were associated with strains of animal origin. The phylogenomic analysis reveals that the closely related strains are from very diverse geographical origins. In the ST profiles, the UK region presented a substantial higher diversity among the examined isolates (Figure 3 and Supplementary Table 1). ST2155 was associated with the food products (baklava from Turkey).
Core Genome Multilocus Sequence Typing Analysis of Salmonella Telelkebir Population
The cgMLST based approach was employed to evaluate the genomic epidemiological features of the global S. Telelkebir strains. The cgMLST study was performed on a collection of loci that were shared by all Salmonella isolates, which were then used for gene by gene comparison. The difference in the nucleotide sequences of these loci determines the clustering of isolates (Tang et al., 2017; Pearce et al., 2018). According to cgMLST results, these samples were diverse as depicted by the different number of alleles between the isolates (Figure 4). Nineteen clusters and 68 singletons were identified in the global phylogenetic tree. The FJ001 strain did not form any cluster. The nearest neighbor for the FJ001 strain was 201702574, which were obtained from France with an allelic difference of 53 alleles. The clusters were perfectly coherent with the STs assignment. The ST450 group was subdivided into seven different clusters, with ST450-cluster (C1) being the dominant type. The ST450 strains were isolated from various geographical locations and different years and the reservoirs for these strains where humans (n = 4) and animals (n = 4) (Supplementary Table 1), whereas the cluster C1 mainly included the strains recovered from humans and animals that had an allelic difference of one to six alleles. Two S. Telelkebir isolates from China had resistant genes [fosA7 and aac(6′)-Iaa] but belonged to different types both by MLST and cgMLST. The results suggested that the antimicrobial resistance gene profile varied according to the cgMLST clusters (Supplementary Table 1). The fosfomycin resistance genes were observed in C1, C4, C5, C6, C7, C13, C14, C15, C16, C18, and C19 clusters. The qnrB19 resistance gene was present among clusters C1 (28.57%, 2/7), C8 (100%, 3/3), and C10 (100%, 2/2) and it was mainly associated with plasmid Col440I.
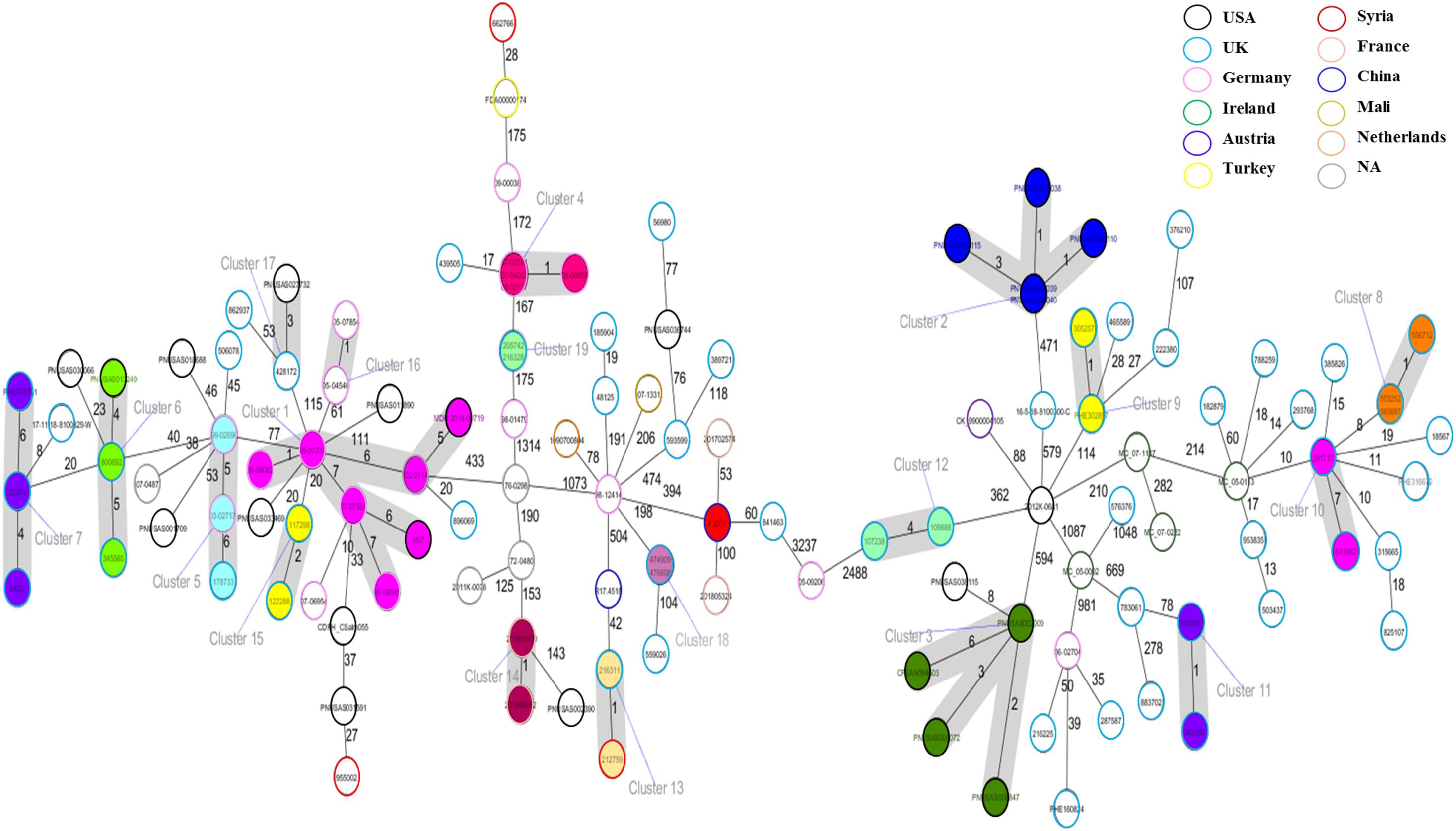
Figure 4. Minimum spanning tree of 121 Salmonella Telelkebir isolates from different origins are depicted here. The numbers on the connecting lines illustrate the numbers of different alleles between connected samples. Closely related samples are represented with shaded circles of closely related genotypes (≤7 different cgMLST alleles) are shaded with gray. The FJ001 strain is shaded in red.
Discussion
There are earlier reports on the isolation of uncommon Salmonella Telelkebir serovar (Tankson et al., 2006; Octavia et al., 2019). However, little is known about its resistance profile, epidemiology, and disease-causing potential. Previous reports suggested that Salmonella serotype Telelkebir is an infrequently reported variant that is mainly associated with exotic animal species, mostly reptiles (Berendes et al., 2007). Between 1995 and 2007, S. Telelkebir strains were obtained from animal-feed ingredients in Poland (Dera-Tomaszewska, 2012). Berendes et al. (2007) reported a case study where a 17-year-old girl had sepsis splenic abscesses caused by Salmonella serovar Telelkebir. The same variant was cultivated from the feces of the reptile pets that were held in the home of the patient. Our study demonstrates that the four global animal isolates had between one and six cgMLST allelic differences to the closest human isolates. In a previous study, three 2017 Bavarian S. Agona feed-origin outbreak strains were compared with a French outbreak isolate and 48 S. Agona isolates collected from 1993 to 2018, out of which 28 were epidemiologically outbreak related. The cgMLST analysis revealed that four most relevant clusters comprised 3 to 15 samples with a maximum within-cluster difference of zero to five alleles (Dangel et al., 2019). The study revealed that cgMLST can be used for reasonable, reproducible, and reliable high-resolution classification to monitor outbreak clusters and relationships among past or international cases, which could also be interpreted using representative public data.
Even though the FJ001 strain was pan-susceptible toward the tested antimicrobial agents, treatment with cefoperazone sulbactam alone or combined with levofloxacin failed when administrated to the patient. Interestingly, the WGS analysis revealed that the strain FJ001 had no antimicrobial resistance genes against quinolones and β-lactam class of antibiotics. One of the possible reason could be that for Salmonella spp. and Shigella spp., aminoglycosides, first- and second-generation cephalosporins and cephamycins may appear active in vitro, but are not effective clinically (CLSI, 2016). Also, according to “the 90–60 rule” coined by Rex and Pfaller (2002), a susceptible result in in vitro is associated with a favorable therapeutic response in 90–95% of patients. Various host or pathogen factors influence the efficiency of antibiotic therapy in vivo, such as the host immune system, site of infection (penetration of antimicrobial agents into the site), and bacterial virulence factors that may increase or hinder the immune response leading to poor clinical outcomes (Stratton, 2006; Ersoy et al., 2017). This could be the possible explanation of the ineffectiveness of these drugs in the patient.
The fosA7 is a new antimicrobial resistance gene against fosfomycin that was recently identified in S. Heidelberg from broiler chickens in Canada (Rehman et al., 2017). The gene fosA7 confers resistance to broad-spectrum antibiotic fosfomycin, which is extensively used to treat drug-resistant Gram-negative bacteria (Balbin et al., 2020). A total of 58.67% of the tested S. Telelkebir strains (71 strains) harbored the fosA7 gene. The fluoroquinolones are the drugs of choice for the treatment of iNTS due to their broad-spectrum antimicrobial activity. Nevertheless, the extensive use of these drugs has led to the appearance of resistant strains globally, mainly in Gram-negative bacterial species. The qnrB19 gene is one the most frequent variants of qnr genes globally, and out of the 121 strains, 12 isolates are positive for qnrB19 gene (Supplementary Table 1; Ciesielczuk et al., 2013). The qnr is often found in association with genes that impart resistance to other antibiotics classes, e.g., β-lactams and aminoglycosides (Robicsek et al., 2006). In the present study, the qnr genes were mostly associated with aminoglycosides which suggests that Enterobacteriaceae strains harboring qnr resistance may denote a serious threat to public health. Also, the presence of blaTEM–1B gene was observed in three human S. Telelkebir isolates from the United Kingdom, Ireland, and Mali. The broad-spectrum β-lactamase enzymes can hydrolyze almost all β-lactams and are commonly linked with genes conferring resistance to several other classes of antibiotics (Bush and Bradford, 2016).
The screening of the virulence gene profile demonstrated that all S. Telelkebir isolates harbored various fimbrial genes (bcf, fim, inv, and csg) and secretion systems involved in cell invasion and bacterial viability in phagocytes. Previous results suggest that fimbriae are involved in differential intestinal colonization of animal species (Weening et al., 2005; Yue et al., 2012). The cytolethal distending toxin (CDT) or typhoid toxin is a bacterial genotoxin, which are encoded by several Gram-negative bacteria, including S. enterica. Our comparative genome analysis, for the first time, revealed that FJ001 strain and all the Salmonella Telelkebir strains harbored the gene cdtB, pltA, and pltB typhoid toxin gene, which highlights particular concern in public health. Typhoid toxin is recognized as a major virulence factor of S. Typhi, and it is reported to play a central role in the pathogenicity of S. Typhi. It has been observed that typhoid toxin is involved in the establishment of S. Typhi persistent infection most likely by altering the immune cell functions to its favor (Song et al., 2010; Chong et al., 2017). The genes encoding Salmonella-CDT (i.e., genes pltA, pltB, and cdtB) have been characterized in around 40 NTS serovars (den Bakker et al., 2011; Mezal et al., 2014a). Previous reports suggest that the amino acid alignments of CdtB, PltA, and PltB are extremely conserved among S. enterica serotypes (Rodriguez-Rivera et al., 2015). The BLASTp analysis of all the proteins CdtB, PltA, and PltB of Salmonella Telelkebir strains showed high sequence similarity (100–97%) to S. enterica strains. This suggests that the CdtB, PltA, and PltB are highly conserved among S. enterica serotypes. Also, cellular-level Salmonella CDT significantly alters the outcome of infection by inducing DNA damage, which is associated with a cell cycle arrest and activation of the host cell’s DNA damage response (Chang et al., 2019). Earlier findings revealed that S. Javiana isolates harboring cdtB, pltA, and pltB caused cytoplasmic and nuclear enlargement together with cell cycle arrest in G2/M phase, increased invasion and cytotoxicity toward HeLa cells that are characteristic of CDT activity (Mezal et al., 2014a). Recently, Xu et al. (2020) reported the presence of the cdtB, pltA, and pltB genes in iNTS serovar S. Uzaramo, which showed a higher killing rate than the classic gastrointestinal infectious agent S. Typhimurium and suggested it to be a possible explanation of the invasiveness of these serovars. Ahn et al. (2021) demonstrated that even when host cells infected with S. Typhi are treated with antibiotics, typhoid toxin is continuously secreted by antibiotic-resistant S. Typhi. These NTS serovars might have acquired these unique virulence factors through horizontal gene transfer during homologous recombination between S. Typhi and non-typhoidal serovars, by a prophage that integrates into the bacterial DNA chromosome (Bakker et al., 2011), and it might have led to a higher capacity of invasion in NTS (Mezal et al., 2013; Mezal et al., 2014b).
The SNP-based phylogenomic analysis also revealed that the strains clustered together irrespective of the time of isolation, source, and geographical location. This could be the result of global dissemination, due to the convenient and regular global travels, there is an increase in the possibility for fast and extensive transmissions of bacterial pathogens, which permits these isolates to effortlessly cross geographical barriers. Regardless of the improved surveillance followed today in response to the outbreak of infectious diseases, transmission connections cannot be identified in the event of an infectious disease outbreak due to missed screening, antimicrobial therapy suppression, and difficulties recognizing contacts (Jia et al., 2019).
Conclusion
In summary, we reported a bloodstream infection caused by an uncommon NTS serovar Telelkebir in mainland China. These infrequently reported Salmonella serovars can possibly be misidentified in clinics, and the actual threat possessed by these serovars may be underestimated. Although the real threat of human diseases caused by this infrequently reported Salmonella serovar remains unknown, the combination of unique virulence factors and antimicrobial resistance genes carried by these minority NTS may lead to adverse clinical outcomes, demonstrating the necessity for an enhanced surveillance for the clinically important typhoid-toxin-containing serovars.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA666303.
Author Contributions
YQ, RN, XX, and MY conceptualized the study, performed the data curation, and formulated the methodology. YQ, RN, and XX were in charge of the resources and performed the investigation. YQ, RN, XX, SW, and HP performed the formal analysis and validation. MY was in charge of the software and visualization. KZ and MY supervised the study, were in charge of project administration, and acquisition of the funding. RN wrote the original draft of the study. RN and MY reviewed and edited the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Program on Key Research Project of China (2017YFC1600103 and 2019YFE0103900) as well as the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 861917–SAFFI, the Zhejiang Provincial Natural Science Foundation of China (LR19C180001), the Zhejiang Provincial Key R&D Program of China (2020C02032 and 2021C02008), the Opening Fund of Key Laboratory of Microorganism Technology and Bioinformatics Research of Zhejiang Province (2017E10010), Construction of Fujian Provincial Scientific and Technological Innovation Platform (2019Y2001), and National Major Science and Technology Program of China (2017ZX10103008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the staff from the Fujian and Shanghai CDCs for providing technical assistance during the clinical investigation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.704152/full#supplementary-material
Supplementary Table 1 | A complete analytic information for 121 the examined Salmonella Telelkebir isolates in this study.
Supplementary Table 2 | A summary table of the genomic assembly information for 121 the examined Salmonella Telelkebir isolates.
Supplementary Table 3 | cgMLST distance matrix table with a pairwise comparison of allelic differences among each two of 121 S. Telelkebir isolates.
Supplementary Table 4 | Minimum inhibitory concentration values for S. Telelkebir strain FJ001 under various different conditions.
Footnotes
- ^ http://enterobase.warwick.ac.uk
- ^ https://www.ncbi.nlm.nih.gov/sra
- ^ http://rast.nmpdr.org/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
- ^ https://cge.cbs.dtu.dk/services/PlasmidFinder/
- ^ https://cge.cbs.dtu.dk/services/ResFinder/
- ^ https://lfz.corefacility.ca/sistr-app/
- ^ https://cge.cbs.dtu.dk/services/SeqSero/
- ^ https://cge.cbs.dtu.dk/services/MLST/
References
Ahn, C., Yang, Y.-A., Neupane, D. P., Nguyen, T., Richards, A. F., Sim, J. H., et al. (2021). Mechanisms of typhoid toxin neutralization by antibodies targeting glycan receptor binding and nuclease subunits. iScience 24:102454. doi: 10.1016/j.isci.2021.102454
Bakker, H. C., Switt, A. I. M., Cummings, C. A., Hoelzer, K., Degoricija, L., Rodriguez-Rivera, L. D., et al. (2011). A whole-genome single nucleotide polymorphism-based approach to trace and identify outbreaks linked to a common Salmonella enterica subsp. enterica serovar Montevideo pulsed-field gel electrophoresis type. Appl. Environ. Microbiol. 77, 8648–8655. doi: 10.1128/AEM.06538-11
Balbin, M. M., Hull, D., Guest, C., Nichols, L., Dunn, R., and Thakur, S. (2020). Antimicrobial resistance and virulence factors profile of Salmonella spp. and Escherichia coli isolated from different environments exposed to anthropogenic activity. J. Glob. Antimicrob. Resist. 22, 578–583. doi: 10.1016/j.jgar.2020.05.016
Berendes, T. D., Keijman, J. M. G., te Velde, L. F., and Oostenbroek, R. J. (2007). Splenic abscesses caused by a reptile-associated Salmonella infection. Dig. Surg. 24, 397–399. doi: 10.1159/000107718
Biswas, S., Li, Y., Elbediwi, M., and Yue, M. (2019). Emergence and dissemination of mcr-carrying clinically relevant Salmonella Typhimurium Monophasic Clone ST34. Microorganisms 7:298. doi: 10.3390/microorganisms7090298
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bush, K., and Bradford, P. A. (2016). β-Lactams and β-Lactamase inhibitors: an overview. Cold Spring Harb. Perspect. Med. 6:a025247. doi: 10.1101/cshperspect.a025247
Chand, Y., Alam, M. A., and Singh, S. (2020). Pan-genomic analysis of the species Salmonella enterica: identification of core essential and putative essential genes. Gene Rep. 20:100669. doi: 10.1016/j.genrep.2020.100669
Chang, S.-J., Jin, S. C., Jiao, X., and Galán, J. E. (2019). Unique features in the intracellular transport of typhoid toxin revealed by a genome-wide screen. PLoS Pathog. 15:e1007704. doi: 10.1371/journal.ppat.1007704
Chong, A., Lee, S., Yang, Y.-A., and Song, J. (2017). The role of typhoid Toxin in Salmonella Typhi Virulence. Yale J. Biol. Med. 90, 283–290.
Ciesielczuk, H., Hornsey, M., Choi, V., Woodford, N., and Wareham, D. W. (2013). Development and evaluation of a multiplex PCR for eight plasmid-mediated quinolone-resistance determinants. J. Med. Microbiol. 62, 1823–1827. doi: 10.1099/jmm.0.064428-0
CLSI (2016). Performance Standards for Antimicrobial Susceptibility Testing Twenty-Sixth Informational Supplement M100-S26. Wayne, PA: CLSI.
CLSI (2018). Performance Standards for Antimicrobial Susceptibility Testing, 28th Edn. Wayne, PA: CLSI, 608–608.
Dangel, A., Berger, A., Messelhäußer, U., Konrad, R., Hörmansdorfer, S., Ackermann, N., et al. (2019). Genetic diversity and delineation of Salmonella Agona outbreak strains by next generation sequencing, Bavaria, Germany, 1993 to 2018. Euro Surveill. 24:1800303. doi: 10.2807/1560-7917.ES.2019.24.18.1800303
den Bakker, H. C., Moreno Switt, A. I., Govoni, G., Cummings, C. A., Ranieri, M. L., Degoricija, L., et al. (2011). Genome sequencing reveals diversification of virulence factor content and possible host adaptation in distinct subpopulations of Salmonella enterica. BMC Genomics 12:425. doi: 10.1186/1471-2164-12-425
Dera-Tomaszewska, B. (2012). Salmonella serovars isolated for the first time in Poland, 1995-2007. Int. J. Occup. Med. Environ. Health 25, 294–303. doi: 10.2478/S13382-012-0038-2
Dulbecco, R., and Freeman, G. (1959). Plaque production by the polyoma virus. Virology 8, 396–397. doi: 10.1016/0042-6822(59)90043-1
Elbediwi, M., Li, Y., Paudyal, N., Pan, H., Li, X., Xie, S., et al. (2019). Global burden of colistin resistant bacteria: mobilized colistin resistant genes study 1980-2018. Microorganisms 7:461. doi: 10.3390/microorganisms7100461
Ersoy, S. C., Heithoff, D. M., Barnes, L., Tripp, G. K., House, J. K., Marth, J. D., et al. (2017). Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine 20, 173–181. doi: 10.1016/j.ebiom.2017.05.026
EUCAST (2018). EUCAST Breakpoint Tables for Interpretation of MICs and Zone Diameters, Version, 8th Edn. Växjö: The European Committee on Antimicrobial Susceptibility Testing.
FDA (2017). The National Antimicrobial Resistance Monitoring System: NARMS Integrated Report, 2015. Laurel, MD: U.S. Department of Health and Human Services.
Feasey, N. A., Dougan, G., Kingsley, R. A., Heyderman, R. S., and Gordon, M. A. (2012). Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379, 2489–2499. doi: 10.1016/S0140-6736(11)61752-2
Gardy, J. L., and Loman, N. J. (2018). Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20. doi: 10.1038/nrg.2017.88
GBD 2017 Causes of Death Collaborators (2018). Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788. doi: 10.1016/S0140-6736(18)32203-7
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Jia, H., Chen, Y., Wang, J., Xie, X., and Ruan, Z. (2019). Emerging challenges of whole-genome-sequencing–powered epidemiological surveillance of globally distributed clonal groups of bacterial infections, giving Acinetobacter baumannii ST195 as an example. Int. J. Med. Microbiol. 309:151339. doi: 10.1016/j.ijmm.2019.151339
Jiang, Z., Anwar, T., Peng, X., Biswas, S., Elbediwi, M., Li, Y., et al. (2021). Prevalence and antimicrobial resistance of Salmonella recovered from pig-borne food products in Henan, China. Food Control 121:107535. doi: 10.1016/j.foodcont.2020.107535
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li, S., Zhou, Y., and Miao, Z. (2017). Prevalence and antibiotic resistance of non-typhoidal Salmonella isolated from raw chicken carcasses of commercial broilers and spent Hens in Tai’an, China. Front. Microbiol. 8:2106. doi: 10.3389/fmicb.2017.02106
Li, Y., Yang, Q., Cao, C., Cui, S., Wu, Y., Yang, H., et al. (2020). Prevalence and characteristics of Salmonella isolates recovered from retail raw chickens in Shaanxi Province, China. Poult. Sci. 99, 6031–6044. doi: 10.1016/j.psj.2020.07.038
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Liu, Y., Jiang, J., -Dra, A., Li, X., Peng, X., Xia, L., et al. (2021). Prevalence and genomic investigation of Salmonella isolates recovered from animal food-chain in Xinjiang, China. Food Res. Int. 142:110198. doi: 10.1016/j.foodres.2021.110198
Mansour, M. N., Yaghi, J., El Khoury, A., Felten, A., Mistou, M.-Y., Atoui, A., et al. (2020). Prediction of Salmonella serovars isolated from clinical and food matrices in Lebanon and genomic-based investigation focusing on Enteritidis serovar. Int. J. Food Microbiol. 333:108831. doi: 10.1016/j.ijfoodmicro.2020.108831
Mellmann, A., Bletz, S., Böking, T., Kipp, F., Becker, K., Schultes, A., et al. (2016). Real-time genome sequencing of resistant bacteria provides precision infection control in an institutional setting. J. Clin. Microbiol. 54, 2874–2881. doi: 10.1128/JCM.00790-16
Mezal, E. H., Bae, D., and Khan, A. A. (2014a). Detection and functionality of the CdtB, PltA, and PltB from Salmonella enterica serovar Javiana. Pathog. Dis. 72, 95–103. doi: 10.1111/2049-632X.12191
Mezal, E. H., Sabol, A., Khan, M. A., Ali, N., Stefanova, R., and Khan, A. A. (2014b). Isolation and molecular characterization of Salmonella enterica serovar Enteritidis from poultry house and clinical samples during 2010. Food Microbiol. 38, 67–74. doi: 10.1016/j.fm.2013.08.003
Mezal, E. H., Stefanova, R., and Khan, A. A. (2013). Isolation and molecular characterization of Salmonella enterica serovar Javiana from food, environmental and clinical samples. Int. J. Food Microbiol. 164, 113–118. doi: 10.1016/j.ijfoodmicro.2013.03.021
Octavia, S., Zulaina, S., Seet, S. K., Tien, W. S., Thu, M., Ooi, P. L., et al. (2019). Whole-genome sequencing of the rare Salmonella enterica serovar Anfo isolated from food handlers. J. Med. Microbiol. 68, 429–431. doi: 10.1099/jmm.0.000934
Pan, H., Paudyal, N., Li, X., Fang, W., and Yue, M. (2018). Multiple food-animal-borne route in transmission of antibiotic-resistant Salmonella newport to humans. Front. Microbiol. 9:23. doi: 10.3389/fmicb.2018.00023
Parry, C. M., Hien, T. T., Dougan, G., White, N. J., and Farrar, J. J. (2002). Typhoid fever. N. Engl. J. Med. 347, 1770–1782. doi: 10.1056/NEJMra020201
Paudyal, N., Pan, H., Elbediwi, M., Zhou, X., Peng, X., Li, X., et al. (2019). Characterization of Salmonella Dublin isolated from bovine and human hosts. BMC Microbiol. 19:226. doi: 10.1186/s12866-019-1598-0
Paudyal, N., Pan, H., Liao, X., Zhang, X., Li, X., Fang, W., et al. (2018). A meta-analysis of major foodborne pathogens in Chinese food commodities between 2006 and 2016. Foodborne Pathog. Dis. 15, 187–197. doi: 10.1089/fpd.2017.2417
Pearce, M. E., Alikhan, N.-F., Dallman, T. J., Zhou, Z., Grant, K., and Maiden, M. C. J. (2018). Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 274, 1–11. doi: 10.1016/j.ijfoodmicro.2018.02.023
Rehman, M. A., Yin, X., Persaud-Lachhman, M. G., and Diarra, M. S. (2017). First detection of a fosfomycin resistance Gene, fosA7, in Salmonella enterica Serovar Heidelberg isolated from broiler chickens. Antimicrob. Agents Chemother. 61, e00410-17. doi: 10.1128/aac.00410-17
Rex, J. H., and Pfaller, M. A. (2002). Has antifungal susceptibility testing come of age? Clin. Infect. Dis. 35, 982–989. doi: 10.1086/342384
Robicsek, A., Strahilevitz, J., Jacoby, G. A., Macielag, M., Abbanat, D., Hye Park, C., et al. (2006). Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 12, 83–88. doi: 10.1038/nm1347
Rodriguez, A., Pangloli, P., Richards, H. A., Mount, J. R., and Draughon, F. A. (2006). Prevalence of Salmonella in diverse environmental farm samples. J. Food Protect. 69, 2576–2580. doi: 10.4315/0362-028X-69.11.2576
Rodriguez-Rivera, L. D., Bowen, B. M., den Bakker, H. C., Duhamel, G. E., and Wiedmann, M. (2015). Characterization of the cytolethal distending toxin (typhoid toxin) in non-typhoidal Salmonella serovars. Gut Pathog. 7:19. doi: 10.1186/s13099-015-0065-1
Rouzeau-Szynalski, K., Barretto, C., Fournier, C., Moine, D., Gimonet, J., and Baert, L. (2019). Whole genome sequencing used in an industrial context reveals a Salmonella laboratory cross-contamination. Int. J. Food Microbiol. 298, 39–43. doi: 10.1016/j.ijfoodmicro.2019.03.007
Ruan, Z., Yu, Y., and Feng, Y. (2020). The global dissemination of bacterial infections necessitates the study of reverse genomic epidemiology. Brief. Bioinform. 21, 741–750. doi: 10.1093/bib/bbz010
Simon, S., Trost, E., Bender, J., Fuchs, S., Malorny, B., Rabsch, W., et al. (2018). Evaluation of WGS based approaches for investigating a food-borne outbreak caused by Salmonella enterica serovar Derby in Germany. Food Microbiol. 71, 46–54. doi: 10.1016/j.fm.2017.08.017
Song, J., Willinger, T., Rongvaux, A., Eynon, E. E., Stevens, S., Manz, M. G., et al. (2010). A mouse model for the human pathogen Salmonella Typhi. Cell Host Microbe 8, 369–376. doi: 10.1016/j.chom.2010.09.003
Stratton, C. W. (2006). In vitro susceptibility testing versus in vivo effectiveness. Med. Clin. North Am. 90, 1077–1088. doi: 10.1016/j.mcna.2006.07.003
Suez, J., Porwollik, S., Dagan, A., Marzel, A., Schorr, Y. I., Desai, P. T., et al. (2013). Virulence gene profiling and pathogenicity characterization of non-typhoidal Salmonella accounted for invasive disease in humans. PLoS One 8:e58449. doi: 10.1371/journal.pone.0058449
Tang, P., Croxen, M. A., Hasan, M. R., Hsiao, W. W. L., and Hoang, L. M. (2017). Infection control in the new age of genomic epidemiology. Am. J. Infect. Control 45, 170–179. doi: 10.1016/j.ajic.2016.05.015
Tankson, J. D., Fedorka-Cray, P. J., Jackson, C. R., and Headrick, M. (2006). Genetic relatedness of a rarely isolated Salmonella: Salmonella enterica serotype Niakhar from NARMS animal isolates. J. Antimicrob. Chemother. 57, 190–198. doi: 10.1093/jac/dki439
Wang, X., Biswas, S., Paudyal, N., Pan, H., Li, X., Fang, W., et al. (2019). Antibiotic resistance in Salmonella typhimurium isolates recovered from the food Chain through National Antimicrobial Resistance Monitoring System between 1996 and 2016. Front. Microbiol. 10:985. doi: 10.3389/fmicb.2019.00985
Weening, E. H., Barker, J. D., Laarakker, M. C., Humphries, A. D., Tsolis, R. M., and Bäumler, A. J. (2005). The Salmonella enterica serotype Typhimurium lpf, bcf, stb, stc, std, and sth fimbrial operons are required for intestinal persistence in mice. Infect. Immun. 73, 3358–3366. doi: 10.1128/IAI.73.6.3358-3366.2005
World Health Organization (WHO) (2018). Salmonella (Non-typhoidal). Available online at: https://www.who.int/news-room/fact-sheets/detail/salmonella-(non-typhoidal) (accessed March 10, 2021).
Xu, X., Chen, Y., Pan, H., Pang, Z., Li, F., Peng, X., et al. (2020). Genomic characterization of Salmonella uzaramo for human invasive infection. Microb. Genom. 6, 1–8. doi: 10.1099/mgen.0.000401
Yu, H., Elbediwi, M., Zhou, X., Shuai, H., Lou, X., Wang, H., et al. (2020). Epidemiological and genomic characterization of Campylobacter jejuni isolates from a foodborne outbreak at Hangzhou, China. Int. J. Mol. Sci. 21:3001. doi: 10.3390/ijms21083001
Yue, M., Rankin, S. C., Blanchet, R. T., Nulton, J. D., Edwards, R. A., and Schifferli, D. M. (2012). Diversification of the Salmonella fimbriae: a model of macro- and microevolution. PLoS One 7:e38596. doi: 10.1371/journal.pone.0038596
Yu-feng, Q., Shun-tai, W., Xin-lan, H., Jian-hui, C., Jin-song, Y., Meng-yin, H., et al. (2020). Salmonella Telelkebir was first detected from clinical bloodstream infection in Fujian Province, China. Chin. J. Zoonoses 36, 643–645.
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Salmonella Telelkebir, invasive infection, antimicrobial resistance gene, typhoid toxin, virulence gene
Citation: Qiu Y-f, Nambiar RB, Xu X-b, Weng S-t, Pan H, Zheng K-c and Yue M (2021) Global Genomic Characterization of Salmonella enterica Serovar Telelkebir. Front. Microbiol. 12:704152. doi: 10.3389/fmicb.2021.704152
Received: 01 May 2021; Accepted: 25 June 2021;
Published: 29 July 2021.
Edited by:
Michael Kemp, University of Southern Denmark, DenmarkCopyright © 2021 Qiu, Nambiar, Xu, Weng, Pan, Zheng and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Yue, bXl1ZUB6anUuZWR1LmNu; Kui-cheng Zheng, a2luZ2RhZGk5OTA5QDEyNi5jb20=
†These authors have contributed equally to this work