 Chien-Jui Huang
Chien-Jui Huang Ting-Li Wu
Ting-Li Wu Po-Xing Zheng
Po-Xing Zheng Jheng-Yang Ou
Jheng-Yang Ou Hui-Fang Ni3
Hui-Fang Ni3 Yao-Cheng Lin
Yao-Cheng Lin- 1Department of Plant Medicine, National Chiayi University, Chiayi, Taiwan
- 2Biotechnology Center in Southern Taiwan, Agricultural Biotechnology Research Center, Academia Sinica, Tainan, Taiwan
- 3Department of Plant Protection, Chiayi Agricultural Experiment Station, Taiwan Agricultural Research Institute, Chiayi, Taiwan
Background: Worldwide citrus production is severely threatened by Asiatic citrus canker which is caused by the proteobacterium Xanthomonas citri subsp. citri. Foliar sprays of copper-based bactericides are frequently used to control plant bacterial diseases. Despite the sequencing of many X. citri strains, the genome diversity and distribution of genes responsible for metal resistance in X. citri subsp. citri strains from orchards with different management practices in Taiwan are not well understood.
Results: The genomes of three X. citri subsp. citri strains including one copper-resistant strain collected from farms with different management regimes in Taiwan were sequenced by Illumina and Nanopore sequencing and assembled into complete circular chromosomes and plasmids. CRISPR spoligotyping and phylogenomic analysis indicated that the three strains were located in the same phylogenetic lineages and shared ∼3,000 core-genes with published X. citri subsp. citri strains. These strains differed mainly in the CRISPR repeats and pathogenicity-related plasmid-borne transcription activator-like effector (TALE)-encoding pthA genes. The copper-resistant strain has a unique, large copper resistance plasmid due to an unusual ∼40 kbp inverted repeat. Each repeat contains a complete set of the gene cluster responsible for copper and heavy metal resistance. Conversely, the copper sensitive strains carry no metal resistance genes in the plasmid. Through comparative analysis, the origin and evolution of the metal resistance clusters was resolved.
Conclusion: Chromosomes remained constant among three strains collected in Taiwan, but plasmids likely played an important role in maintaining pathogenicity and developing bacterial fitness in the field. The evolution of pathogenicity factors and horizontal gene transfer events were observed in the three strains. These data suggest that agricultural management practices could be a potential trigger for the evolution of citrus canker pathogens. The decrease in the number of CRISPR repeats and pthA genes might be the result of adaptation to a less stressful environment. The metal resistance genes in the copper resistant X. citri strain likely originated from the Mauritian strain not the local copper-resistant X. euvesicatoria strain. This study highlights the importance of plasmids as ‘vehicles’ for exchanging genetic elements between plant pathogenic bacteria and contributing to bacterial adaptation to the environment.
Introduction
Xanthomonas spp. are a large group of Gram-negative bacteria that cause disease in more than 400 different plant hosts (Timilsina et al., 2020); however, the host range of the individual species is often restricted to a single or a handful of plants in the same botanical family. To aid explanation of this phenomenon, the term “pathovar” was coined and is defined as an intra-subspecific group of strains causing the same disease with host and tissue specificity (Timilsina et al., 2020). Most Xanthomonas species infect plants by first colonizing the surface of aerial organs then entering through stomata or wounds; the host may show symptoms. Asiatic citrus canker caused by Xanthomonas citri subsp. citri is a serious threat to citrus production in most citrus-growing regions in the world (Brunings and Gabriel, 2003; Ference et al., 2018). X. citri subsp. citri is a genetically monomorphic bacterium (Leduc et al., 2015; Richard et al., 2017b) and has spread geographically from its Asiatic origin to many citrus-growing regions including Taiwan (Pruvost et al., 2014; Leduc et al., 2015; Huang and Ni, 2017). Recent advances in high throughput sequencing have made it possible to sequence the whole genomes of groups within the microbial community rather than a handful of loci (multilocus sequence typing, MLST) and the reconstruction of the repetitive sequence regions such as the complete CRISPR unit (CRISPR spoligotyping) (Jeong et al., 2019). These advances have allowed the detailed study of the evolution, ecology and dissemination of bacterial pathogens (Vinatzer et al., 2014; Timilsina et al., 2019). Genetically monomorphic bacteria have been considered to have low adaptive potential because of low genetic variability (Achtman, 2008). Yet, how these bacteria adapted to diverse environmental conditions and evolved resistance to antibacterial compounds remains unclear.
Citrus canker pathogens are commonly classified into the three pathotypes: A, B and C. Pathotype A was first reported in Asia in the early nineteenth century (Fawcett and Jenkins, 1933) and later spread to all citrus producing regions worldwide. The first genome of X. citri subsp. citri strain 306 was completely sequenced in 2002 (da Silva et al., 2002). Two variant forms of pathotype A, namely A∗ and AW, have been found in Southeast Asia and Southern Florida in the past 30 years (Timilsina et al., 2020). These variants of the pathotype A showed apparent intraspecific diversity and host specialization. After the reference genome of the pathotype A strain 306 was sequenced, a combination of whole genome sequencing and comparative analysis has contributed to the discovery of polymorphisms associated with potential mechanisms of adaptation in genetically monomorphic bacterium X. citri subsp. citri (Zhang et al., 2015; Richard et al., 2017b; Gochez et al., 2018). To date, multiple factors including transcription activator-like effectors (TALEs), plasmid-mediated horizontal gene transfers and transposons have been found to play important roles in adaptation, evolution and spread of pathogenicity determinants of X. citri subsp. citri (Ferreira et al., 2015; Richard et al., 2017b, 2021; Gochez et al., 2018).
TALEs belonging to the PthA family of type III secretion system effector proteins (T3SEs) are the main pathogenicity factor of X. citri subsp. citri (Brunings and Gabriel, 2003; Abe and Benedetti, 2016; Roeschlin et al., 2019). When injected into host cells, PthA proteins activate expression of disease susceptibility or resistance genes (Brunings and Gabriel, 2003; Abe and Benedetti, 2016; Roeschlin et al., 2019). PthA proteins consist of an N-terminal region for secretion, a central DNA-binding domain and a C-terminal region containing nuclear localization signals and an acidic transcriptional activation domain (Boch and Bonas, 2010). The central DNA-binding domain of the PthA family is composed of almost identical tandem repeats of 33 to 34 amino acids (Boch and Bonas, 2010). Each repeat contains a repeat-variable diresidue (RVD) at the 12th and 13th positions (Boch and Bonas, 2010). In addition, the number of tandem repeats are variable among PthA proteins (Brunings and Gabriel, 2003; Abe and Benedetti, 2016; Roeschlin et al., 2019). The reference X. citri subsp. citri strain 306 carries four TALE-encoding pthA genes located on the plasmids pXAC33 (pthA1 and pthA2) and pXAC64 (pthA3 and pthA4) (da Silva et al., 2002). The pthA1, pthA2, pthA3 and pthA4 genes of strain 306 harbor 16.5, 15.5, 15.5, and 17.5 copies of repeats, respectively (da Silva et al., 2002). Recently, comparative analysis of completely sequenced plasmids from X. citri subsp. citri revealed clues to rearrangements of plasmids and reshuffling of TALEs among citrus canker strains (Gochez et al., 2018). Furthermore, an experimental evolution study showed that in less than 30 cycles of repeated infections, X. citri subsp. citri could accumulate sufficient mutations and rearrangements of repeats of TALEs to cause pathogenicity in incompatible hosts (Teper and Wang, 2021).
Copper-based bactericides have been widely used for control of plant bacterial diseases throughout the world. However, frequent applications of copper-based bactericides induce the evolution and development of bacterial strains that are either resistant or tolerant to copper (Hseu and Hsu, 1991; Wu et al., 1995; Canteros et al., 2008; Behlau et al., 2012b, 2013). Copper resistant (CuR) strains of X. citri subsp. citri have been found across the world (Behlau et al., 2011, 2013). Large-sized plasmids carrying copper resistance genes (cop genes) are predominantly present in CuR xanthomonads (Behlau et al., 2011, 2012a). The cop genes in xanthomonads associated with citrus and solanaceous hosts have been identified and organized in a cluster (Behlau et al., 2011; Timilsina et al., 2019). In CuR Xanthomonas strains, copL, copA, and copB genes in the cop cluster play a major role in copper resistance (Behlau et al., 2011). In addition to the copLAB cluster, the plasmid-borne cluster of copABCD genes has been also identified in CuR strains of X. arboricola pv. juglandis (Lee et al., 1994) and X. citri subsp. citri (Richard et al., 2017b). Copper resistance plasmids of xanthomonads can be mobilized from a donor cell to a copper sensitive (CuS) recipient cell through conjugation (Behlau et al., 2011, 2012a). Recently, copper tolerant (CuT) X. ctri subsp. citri strains whose chromosomal genes cohA and cohB (homologous to copA and copB) were increasingly expressed in the presence of copper, were found in Brazil, but they are not precursors of CuR strains (Marin et al., 2019).
Asiatic citrus canker is an important epidemic disease of citrus production worldwide. Previously we used phylogenetic analysis of copper resistance genes copLAB in combination with polymorphism analysis of complete copB genes to track the possible origin of CuR X. citri subsp. citri strains from Taiwan (Lai et al., 2021). However, hitherto nothing has been known about the detailed genome composition of X. citri subsp. citri in orchards located in different regions managed under distinct agricultural practices. Thus, the aim of this study was to explore the genetic basis of X. citri subsp. citri by comparative analysis of complete genomes of three strains collected from two regions in Taiwan, including one CuR strain. The high quality genome assembly and annotation of the three strains were compared with published X. citri genomes by CRISPR spoligotyping and core genome analysis to reveal the phylogenetic positions of these three strains. By analyzing the structure and variation of the pthA genes, we provided evidence of plasmid fusion in the sequenced genomes. Furthermore, comparative analysis of the copper and arsenate gene cluster helped decipher the origin of citrus canker pathogens with resistance to either copper or heavy metals.
Materials and Methods
Collection of Strains
Two X. citri subsp. citri strains B2 and T4 were isolated from the leaves of Citrus reticulata cvs. “Shiranui” and “Tainung Giant,” respectively, from a commercial citrus orchard in Taichung, Taiwan (Geolocation: 24.27 N, 120.78 E), where copper-based bactericides and fungicides have been routinely applied during the citrus growing season, on October 7, 2016. The other strain, SN3-3, was collected from the leaf of Citrus sinensis cv. “Suenaga” in an orchard with minimal management that has not applied bactericides and fungicides in Chiayi, Taiwan, (Geolocation: 243.48 N, 120.46 E, Chiayi Agricultural Experiment Branch, Taiwan Agricultural Research Institute) on April 14, 2016.
Phenotypic Characterization
The pathogenicity of the three strains was tested based on the previously published method (Huang and Ni, 2017). Briefly, leaves of citrus cv. Murcott were infiltrated with bacterial suspensions (1 × 106 CFU/ml). Symptom development was observed 14 days after the inoculation.
Copper sensitivity test was performed according to the method in Lai et al. (2021). The three strains, cultured overnight on NA (Nutrient agar, Difco) plates, were streaked on NA plates supplemented with 0, 0.1, 0.2, 0.4, 0.6, 0.7, 0.8, 1.6, and 3.2 mM CuSO4. Stains sensitive, tolerant, or resistant to copper were differentiated by their ability to grow on NA plates with maximum concentrations of ≤0.6, 0.6–0.8 and ≥0.8 mM CuSO4, respectively, as rated by Behlau et al. (2013) and Marin et al. (2019).
Illumina Data Generation
Genomic DNA from the three strains was prepared using Bacteria Genomic DNA kit (Geneaid, Taiwan). Nuclei were isolated according to the manufacturer’s instructions. Purity and quantity of DNA samples were estimated using the Qubit dsDNA HS Assay Kit (Thermo-Fisher Scientific) and Agilent BioAnalyzer 2100 High Sensitivity DNA Kit (Agilent). Sequencing libraries were prepared using Nextera DNA Flex Library Prep Kit (Illumina). Whole genome shotgun sequencing was performed on an Illumina MiSeq instrument using MiSeq Reagent Kit v.3 to generate 2 × 300 bp paired-end reads with an average of 2.8–3.1 million paired-end Illumina reads per genome. On average, the final coverage of the assembled genomes exceeded 200 × of the Illumina reads (Supplementary Table 1).
Nanopore Data Generation
High molecular weight DNA was prepared using a modified phenol/chloroform protocol as previously described (Chiang-Ni et al., 2012). The gDNA was sheared using the Covaris g-TUBE (Covaris) to select fragment sizes ranging from 6 to 20 kb. The sheared gDNA was further selected using BluePippin with a 0.75% agarose gel cassette (Sage Science) to select gDNA fragment sizes ranging from 6 to 20 kb. Nanopore sequencing libraries were prepared using the PCR free, ligation-based sequencing kit (SQK-LSK109) with the native barcoding expansion (EXP-NBD104) for sample multiplexing. Nanopore sequencing was performed on an Oxford Nanopore MinION device (R9.4 flow cell FLO-MIN106D). In total, we obtained 161K–183K Nanopore reads for each strain and the average Nanopore read length was 8,875 bp and the L50 was 48,138 bp long.
Genome Assembly and Annotation
The quality of the reads was evaluated using FastQC (v.0.11.9) (Andrews, 2010) and the low quality reads were subsequently removed by Trimmomatic (v.0.36) (Bolger et al., 2014) for the Illumina reads, and Nanofilt (v.2.6.0) (De Coster et al., 2018) for the Nanopore reads. The base quality of the Nanopore reads of greater than 1k bp was further improved by the corresponding high quality Illumina reads using FMLRC (v.1.0.0) (Wang et al., 2018). Taking advantage of the strengths of different algorithms in dealing with the repetitive regions, each strain was assembled by Canu (v.1.8) (Nurk et al., 2020), Flye (v.2.5) (Kolmogorov et al., 2019) and wtdbg2 (v.2.5) (Ruan and Li, 2020) individually to produce multiple versions of the draft genomes. These draft assemblies of each strain were then compared and merged by BLASTN (v.2.10.1) (Altschul et al., 1990) with manual inspection to produce a consensus assembly. The per-base accuracy was improved by Pilon (Walker et al., 2014) using the trimmed Illumina reads. On average, the final coverage of the assembled genomes exceeded 200 ×. The exact sequences of the duplicated TALE repeats and the large inverted repeats of plasmids were based on a second round of local de novo assembly using soft-clipped Illumina and Nanopore reads surrounding the draft pthA genes or the copper resistance gene cluster. The duplicated blocks were identified based on the proviso that the Illumina read coverage was more than two times higher than the background coverage of the corresponding plasmid sequence. The exact duplicated junction sites were then determined by high quality Nanopore long reads. Ambiguous regions where the sequencing depth was lower or higher than two times the standard deviation of the mean coverage were identified (Wang et al., 2021) and were locally reassembled and/or experimentally verified.
The genome sequences of the three strains were individually resolved into a single circular chromosome. All plasmid sequences were completely circularized as well (Supplementary Table 1). The genome annotation was performed by a local NCBI prokaryotic genome annotation pipeline (PGAP) (Lomsadze et al., 2018) with manual curation. In brief, the PGAP pipeline of the Docker image (v.2019-08-22.build3958) was used to perform the initial genome annotation. The assigned annotations were manually checked [Artemis (Carver et al., 2012)], e.g., correct start and stop codon), by comparative analysis with the published X. citri genomes (BLASTP (Altschul et al., 1990)).
Whole Genome Comparison
Two complementary approaches based on the diversity of nucleotide sequences and protein coding genes were used for the genome-wide analysis to understand the overall genome variations. Assembled chromosome and plasmid sequences were compared with published X. citri genomes (Supplementary Table 2) to identify conserved and novel sequence elements. Whole genome sequence comparison was conducted by a series of bioinformatics tools including BLAST (v.2.10.1) (Altschul et al., 1990), fastANI (v.1.20) (Jain et al., 2018), Harvest (Treangen et al., 2014) and MUMmer (v.4.0.0rc1) (Kurtz et al., 2004). The whole genome similarity metrics of the three strains were estimated using the alignment-free approximate sequence mapping algorithm of fast Average Nucleotide Identity (fastANI) (Jain et al., 2018) with default settings. To further understand the origin of these three strains, whole genome sequences of 33 completely assembled X. citri strains (Supplementary Table 2) were included in the phylogeny analysis. The Parsnp algorithm in the Harvest suite (Treangen et al., 2014) used the maximum unique matches (MUMs) from the suffix graph data structure of the whole genome sequences to identify regions of the core-genome. The algorithm then performed multiple sequence alignments of multiple MUMs for subsequent variant calling and produced a SNP tree.
Synteny Analysis, Pan-Genome, and Core Genes
The orthologous relationships between B2, T4 and SN3-3 was investigated. The protein coding genes of these three strains were searched using all-against-all BLASTP (Altschul et al., 1990). The BLASTP result was analyzed by a scalable unsupervised cluster algorithm TribeMCL (Enright et al., 2002) and the orthologous and paralogous relationships were presented as networks based on the e-values. The genome collinearity analysis was based on the genome synteny information calculated by i-ADHoRe 3.0 (Proost et al., 2012).
To further expand the analysis of gene repositories of X. citri, we compared protein coding genes of 79 Xanthomonas genomes (two X. albilineans, two X. oryza and 75 X. citri) (Supplementary Table 2) by BLASTP (Altschul et al., 1990) and TribeMCL (Enright et al., 2002) clustering. The homolog of the gene matrix in each genome was used as an input to calculate the average number of genes added with each additional genome using a method modified from Meric et al. (2014). Genes that were presented in more than one genome were considered as pan-genome and genes that were shared in at least two genomes were considered as core-genome. To obtain sufficient resolution of the phylogenetic positions, a total of 1,512 orthologous genes with a strict one-to-one relationship in 75 X. citri strains were used to construct the phylogenetic tree. In brief, protein sequences of single copy core genes were individually aligned by MUSCLE (Edgar, 2004) and gaps in the multiple sequence alignments were removed by Trimal (Capella-Gutierrez et al., 2009). Individual sequence alignments were concatenated to create a 478,302 amino acid-long sequence of each strain. Maximum likelihood phylogenetic trees were inferred by RAXML-NG (Kozlov et al., 2019) with 1000 bootstrap replicates (v.1.0.1, -model LG+G8+F –seed 2 –bs-trees 1000) and visualized by Figtree1.
TALE Identification and CRISPR Analysis
The location and classification of TALEs were manually searched by BLASTN (Altschul et al., 1990) based on collected TALE sequences including short inverted repeats (IR), mobile insertion cassettes (MICs) and the Tn3 transposon TnXax1 (Ferreira et al., 2015). Two short palindromic sequences and passenger genes were manually inspected as well. The structure of CRISPR and the Cas proteins were detected by CRISPRCasFinder (Couvin et al., 2018). CRISPRCasFinder integrated multiple tools to determine the hidden Markov model (HMM) profile of Cas proteins, the maximal repeat structure, the entropy of repeats, the sequence similarity and the size of spacers, and the sequence similarity with known CRISPR, and eventually assigned different levels of evidence code. The spacer sequences were further identified by BLASTN following the spoligotypes classification in Jeong et al. (2019). Phages and their integration sites were identified using PHASTER (Arndt et al., 2016) where published redundant phage/prophage sequences had been curated and the completeness scores were assigned to the identified phage regions.
Copper and Arsenate Gene Cluster Analysis
The copper resistance gene clusters in the assembled plasmids were identified by comparison with known copper resistance genes in the plasmid pCuR (Gochez et al., 2018) and pLH201.1 (Richard et al., 2017a). Initial BLASTN hits were manually inspected and only regions with >60% sequence identity and >70% coverage of the query sequence were considered as the candidate area. Protein coding genes were then predicted using the PGAP pipeline (Lomsadze et al., 2018) and manually curated using Artemis (Carver et al., 2012). Comparative analysis results of gene clusters in pT4p2, pCuR and pLH201.1 were visualized by Artemis and Circos (Krzywinski et al., 2009).
Horizontal Transfer of Copper Resistance Genes
Horizontal transfer of cop genes between different X. citri subsp. citri strains was tested according to the method in Behlau et al. (2012a). The strain T4 with copper resistance and rifampicin sensitivity was used as a donor. Spontaneous rifampicin resistant mutants of CuS strains B2Rif and SN3-3Rif were used as recipients. Bacterial strains were mated on NA plates at 28°C for 24 h. After mating, bacterial cells were scraped, suspended, and plated at dilutions on NA amended with rifampicin (50 mg/L) to estimate the population of the recipient. To select transconjugants, bacterial suspensions were plated at dilutions on NA supplemented with rifampicin (50 mg/L) and 0.8 mM CuSO4. The conjugation frequency was calculated as the ratio between the number of transconjugants and the population of the recipient (Behlau et al., 2012a).
Results
Complete Genome Sequence of Three X. citri Strains
The three strains of X. citri subsp. citri were able to cause similar levels of canker symptoms on Murcott leaves. No difference among the three strains was observed with regard to induction of symptoms. Furthermore, the strain T4 was characterized as a CuR strain which was able to grow on NA plates supplemented with 0.8 mM CuSO4 (Lai et al., 2021). The other two strains B2 and SN3-3, which could not grow on NA plates supplemented with more than 0.4 mM CuSO4, both showed sensitivity to copper.
To advance understanding of the genome structure and the molecular makeup of the Taiwan X. citri subsp. citri, we applied hybrid assembly by combining short read (Illumina) and long read (Nanopore MinION) sequencing technologies in an integrated bioinformatics workflow. Three type A strains, including two strains from one conventional commercial orchard (B2, copper sensitive and T4, copper resistant), and one strain from an orchard with minimal management (SN3-3, copper sensitive) were assembled into gap-free chromosome sequences (Supplementary Table 1).
The chromosome sequences of the three strains were resolved into a single circular chromosome with length 5,120,747 bp (B2, accession number CP059999), 5,194,482 bp (T4, CP059992) and 5,192,310 bp (SN3-3, CP060002), respectively. To facilitate the comparative analysis, DnaA was organized in the beginning of the completely assembled genomes. The three conserved DnaA boxes of the oriC region could be identified between DnaA and DnaN (da Silva et al., 2002; Yen et al., 2002; Qian et al., 2005). The terminator of replication of each strain was located at around 2.5 Mb of the chromosome sequence. The replichore of the three strains could be identified based on the GC skew plot (Grigoriev, 1998). The assembled chromosome sequence length and GC content (64.76∼64.84%) (Figure 1) were in the same range as the published X. citri subsp. citri genomes (Timilsina et al., 2020). Through the genome assemblies, we also identified two plasmids in B2 (pB2_V1, 137,420 bp, CP060000; pB2_V3, 94,653 bp, CP060001), two plasmids in T4 (pT4p1, 99,130 bp, CP059993; pT4p2, 312,426 bp, CP059994) and one plasmid in SN3-3 (pSN3-3, 63,921 bp, CP060003). All plasmid sequences were completely circularized as well (Supplementary Table 1).
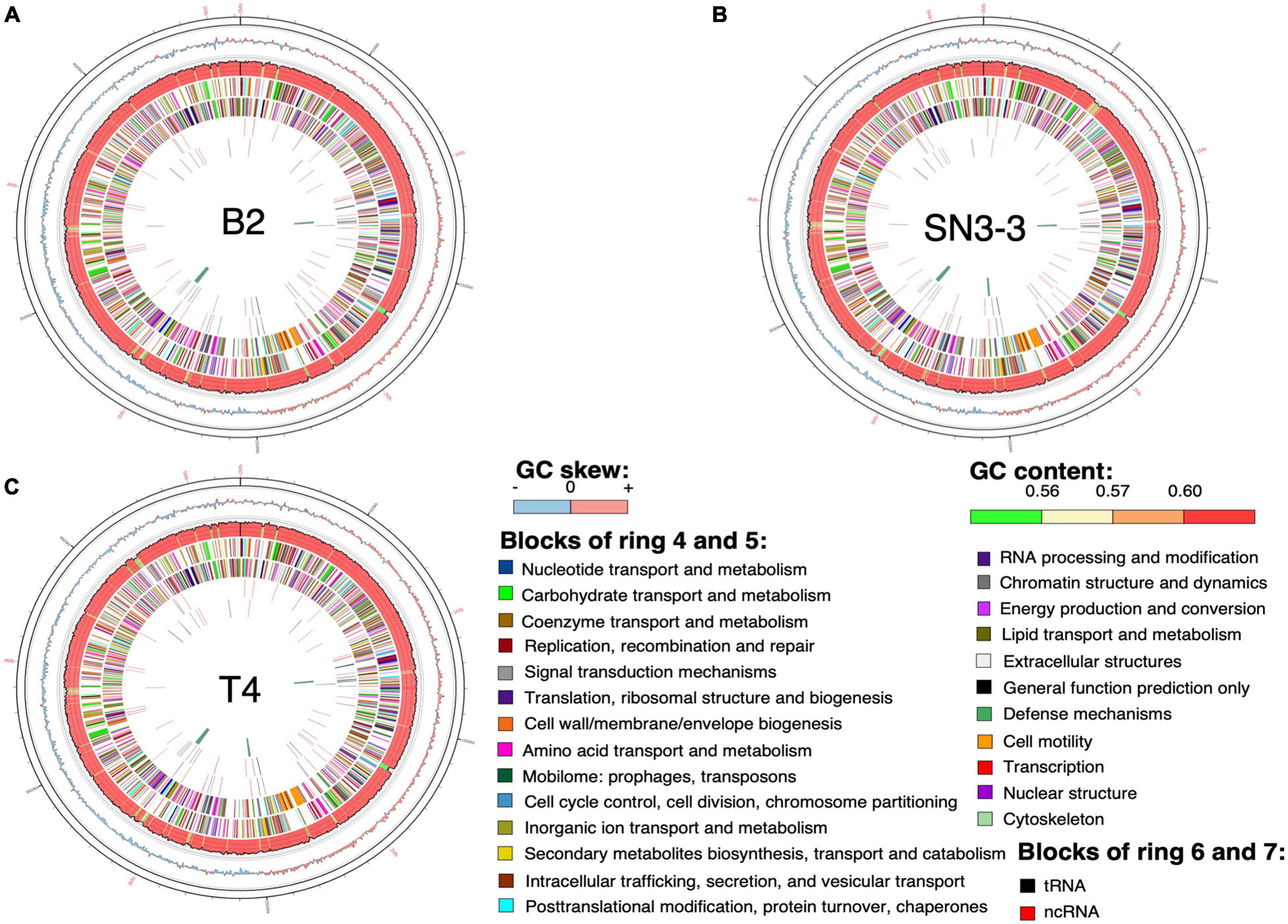
Figure 1. Chromosome organization and genome annotation. (A–C) Overview of three assembled chromosomes and the distribution of genetic elements. Circles from outermost to innermost represent: (1) position, the grid is 100 kb; (2) GC skew with 2500-bp sliding widow and 2500-bp step; (3) GC content in the same sliding window and step size as GC skew; predicted protein-coding sequence (CDS) with known function (4) of the plus strand and (5) the minus strand; predicted RNA of (6) the plus strand and (7) the minus strand; (8) phage sequence.
Using the NCBI prokaryotic genome annotation pipeline (PGAP) (Lomsadze et al., 2018) with manual curation, we identified 4,489 (B2), 4,557 (T4) and 4,539 (SN3-3) protein coding genes in the chromosomes with average coding capacity of 87.2, 87.1, and 87.1%, respectively. The leading strand and the lagging strand harbored a similar number of coding genes. Despite the X. citri reference genome 306 being published in 2002 and many genome sequences having been completed afterwards, gene function of a large part of the genome remains unknown. By comparison with the UniProt protein database and the manually collected Xanthomonas protein sequences from NCBI, ∼20% of the protein coding genes were predicted to be hypothetical genes. In addition to protein coding genes, all three strains contain 106 non-coding genes including 6 rRNA genes, 54 tRNA genes and 46 ncRNA genes (Supplementary Table 1).
Low Nucleotide Sequence Diversity of Chromosomes
To further understand the similarity and possible origin of these three strains, we applied multiple complementary approaches to analyze the genome sequences. We first compared the chromosome sequence of the three strains to investigate the possible large scale chromosomal changes. The three strains shared high sequence identity and collinearity (Supplementary Figure 1). We then examined the nucleotide sequence divergence at the whole genome level. Instead of using the conventional approach for calculating average nucleotide identity (ANI) of orthologous genes, we applied an alignment-free, whole-genome average nucleotide identity (FastANI) analysis (Jain et al., 2018) to determine the pairwise ANI values. The three strains sequenced in this study were found to share very high levels of sequence identity (>99.96%) and there were no significant differences among them (Supplementary Table 3). By focusing on the orthologous chromosome sequences conserved in these three strains and 33 assembled X. citri strains in the same pathovars (Supplementary Table 2), we used Parsnp in the Harvest package to perform the core-genome alignment (Treangen et al., 2014). X. citri strains of the same pathotype were grouped in the same phylogenetic clade (Supplementary Figure 2). Three strains sequenced in this study, B2, T4 and SN3-3, were all classified into the pathotype A. However, these strains were respectively grouped with strains from different geographic origins (Supplementary Figure 2). In particular, T4 shared higher sequence similarity with the reference strain A306 from Brazil and the other strains from Argentina whereas B2 was closer to the strains from Jiangxi, China. SN3-3 was closer to strains from Jiangxi and Guangdong, China.
Spoligotype Classification by CRISPR Spacer Array
We analyzed the compositions and orders of CRISPR and spacer array of our strains by CRISPRCasFinder (Couvin et al., 2018). Following the presence and absence of spacer patterns (spoligotypes, spacer oligonucleotide) (Jeong et al., 2019), B2 contained 19 spacer/repeat units, T4 contained 18 units and SN3-3 contained 17 units, corresponding to spacer Xcc_01 to Xcc_23; spacer Xcc_8, Xcc_10, Xcc_11 and Xcc_14 were absent in these strains (Jeong et al., 2019). In particular, B2 and SN3-3 shared a common Xcc_3 whereas Xcc_9 and Xcc_12 were only seen in B2 and T4 (Figure 2). According to the spoligotype classification by Jeong et al. (2019), B2 belongs to spoligotype 8 (China, Florida, Reunion islands), T4 belongs to spoligotype 14 (Brazil, Mali) and SN3-3 belongs to the spoligotype 21 (Japan). The CRISPR spacer arrays demonstrated that our strains of the same pathotype A belonged to the different spoligotypes originally from East Asia, the Indian Ocean, and South America.
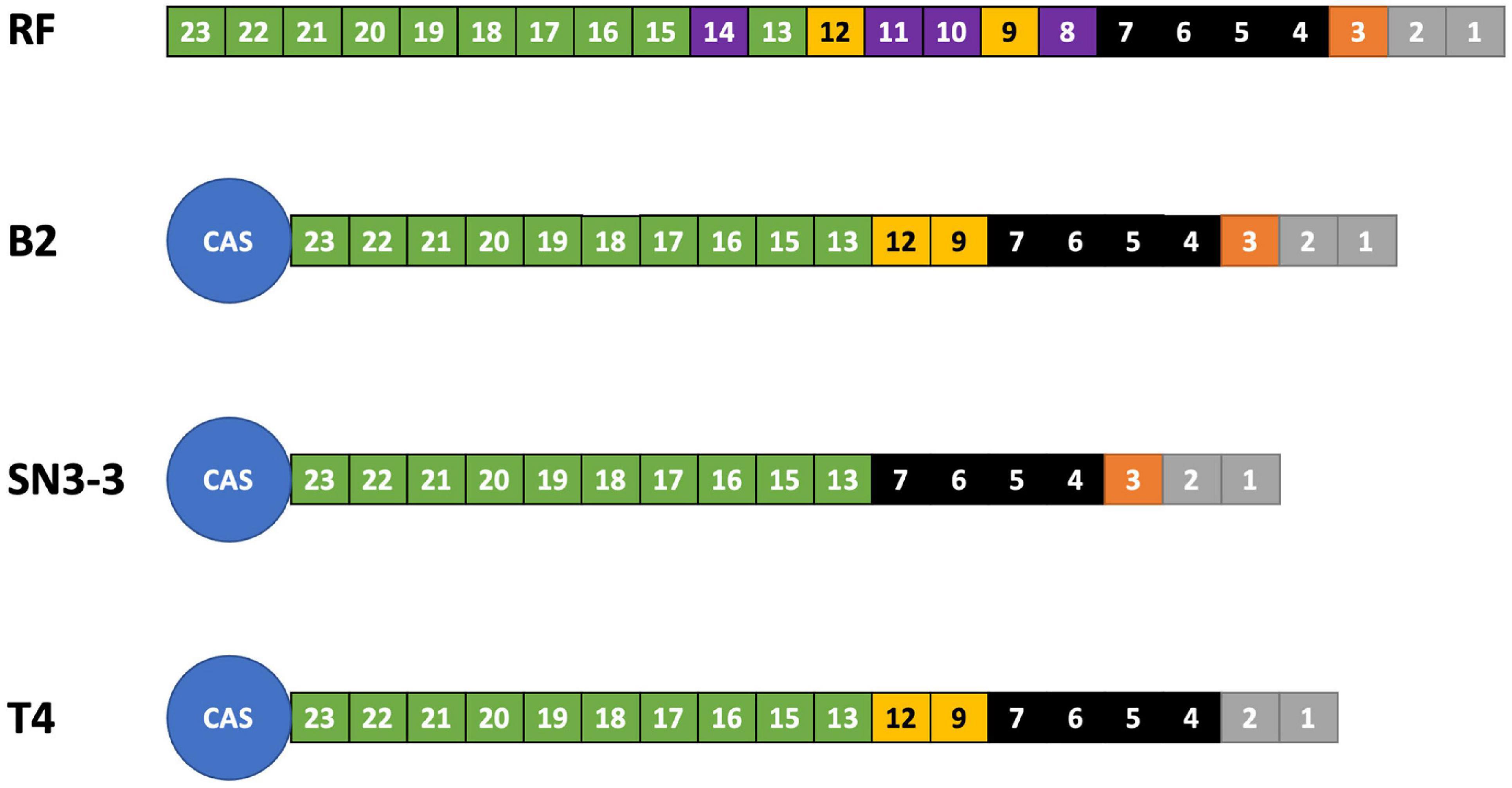
Figure 2. CRISPR spoligotyping. Overview of the CRISPR/Cas locus. The reference (RF) array annotation is based on X. citri 306 (CP009016). The colors of the boxes indicate spacer sequences that correspond to the reference. Purple: absent in the sequenced genomes of this study.
Core Genome Phylogeny
Orthologous genes were first identified by an all-against-all BLASTP search and the BLASTP result was subsequently analyzed by the Markov chain based TribeMCL (Enright et al., 2002) to divide them into orthologous groups. The orthologous information was then used by the synteny based gene family analysis (Proost et al., 2012) to determine genes in the collinearity region. In total, 4,090 orthologous genes were shared between the three strains and did not show chromosome rearrangements (Supplementary Figure 1). Around 144–237 genes are strain specific and were found in only one of the genomes (Supplementary Figure 3). Detailed functional analysis by GO enrichment of these strain specific genes indicated that most of them were enriched with transposon activity-related genes (Supplementary Table 4). This result indicated that the chromosome variations between the three strains, though minimal, were likely associated with the transposon activities.
To better understand the chromosome dynamics of X. citri strains, we conducted pan-genome analysis with protein coding genes of our three sequenced strains in this study and 76 published Xanthomonas genomes. Orthologous genes of 79 strains (including two X. albilineans, two X. oryza and 75 X. citri) (Supplementary Table 2) were determined by the combination of an all-against-all BLASTP search and TribeMCL clustering. We identified 6,342 ortholog clusters that were shared between more than two strains. That is, 99.7% of the genes in one strain were shared in at least one of the 78 sequenced strains. Within the 79 Xanthomonas genomes used for the comparison, the pan-genome size was around 5,000 genes and the core-genome size was less than 2,000 genes (Timilsina et al., 2019; Supplementary Figure 4). Nevertheless, when four outgroup species (2 X. albilineans, 2 X. oryza) were removed from the analysis, the pan-genome size remained around 5,000 genes but the core-genome size increased to ∼3,000 genes (Figure 3A). The number of core genes identified in our study is comparable with those obtained from 58 X. perforans strains (Timilsina et al., 2019).
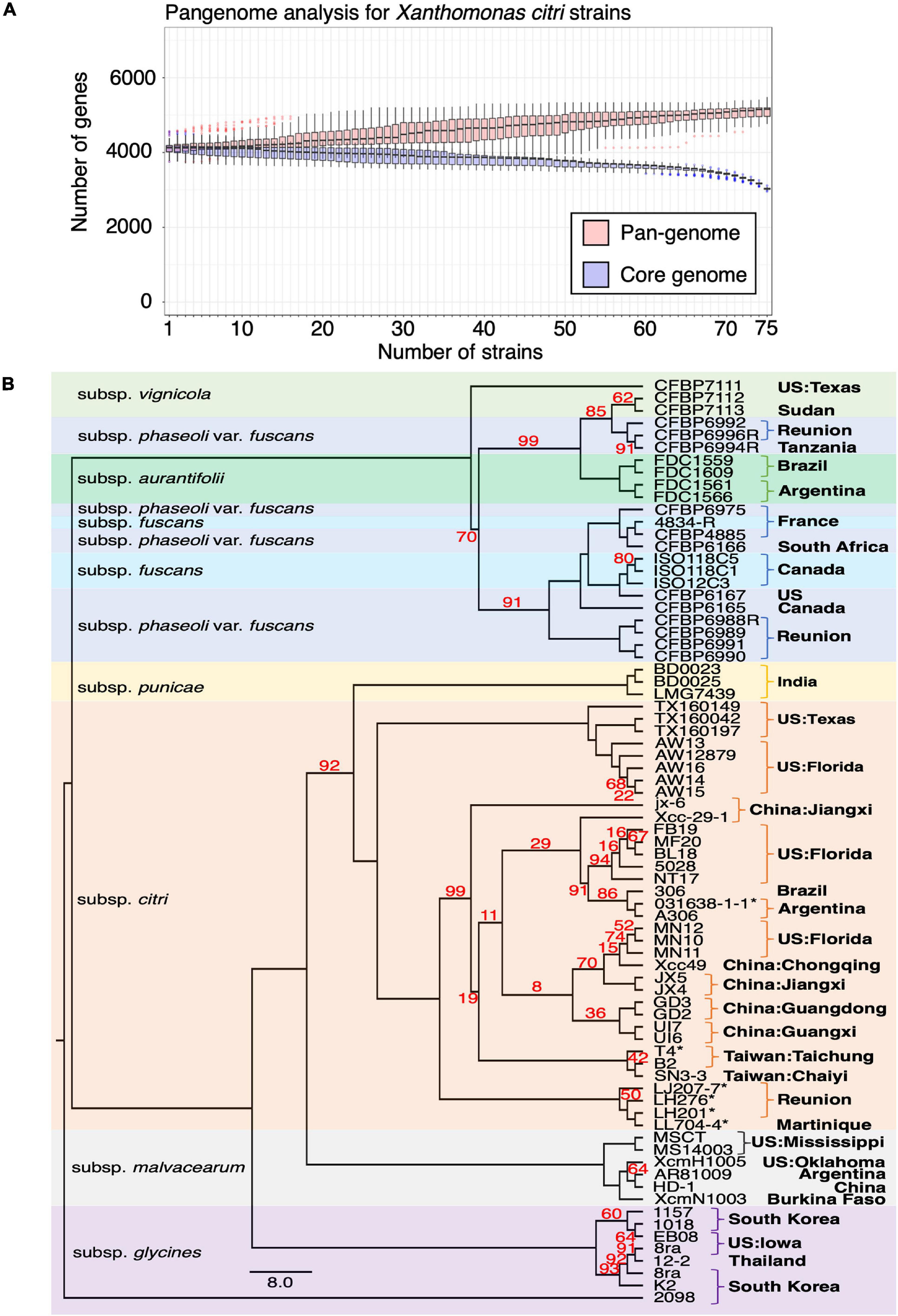
Figure 3. Pangenome analysis of Xanthomonas genomes. (A) Pangenome analysis of 75 X. citri genomes. (B) The core genomes of 75 X. citri sp. strains showed a phylogenetic relationship and shared geographic origins. Maximum likelihood phylogeny based on concatenated sequences of 1,512 core genes. The asterisks (*) indicate copper-resistant strains, and branches with bootstrap value < 100 support are labeled with red numbers.
We used the phylogenomics approach to further understand the phylogenetic positions of three strains in this study and other published genomes. Based on the core genome analysis, we selected 1,512 orthologous genes that had a strict one-to-one single copy relationship to construct the phylogenetic tree. A tree inference tool based on the maximum likelihood (ML) method (Kozlov et al., 2019) was used to construct the phylogenetic tree. As expected, the strains of different pathotypes were clustered on different clades of the phylogenetic tree with high bootstrap value support for each lineage (Figure 3B) and correspondence to the origin of the collection. The pathotype A strains including the reference strain 306 and our three strains were placed together in the same clade together with strains from China, Florida, Argentina and Brazil. On the other hand, four strains from Reunion were placed in an independent clade (Richard et al., 2021).
Type II and Type III Secretion Systems
In Xanthomonas, the type II secretion system (T2SS) is used to translocate folded proteins from the periplasm into the extracellular milieu (Korotkov et al., 2012) and the type III secretion system (T3SS) is essential to pathogenicity through modulating host plant physiology and enabling evasion of host immune responses (Timilsina et al., 2020). In the T2SS, we identified two operons, each containing eleven genes of the general secretory pathway (Gsp) (Korotkov et al., 2012) as was previously reported in the reference strain 306 genome (da Silva et al., 2002; Supplementary Figure 5A and Supplementary Table 5). GspO could be identified in the chromosome but is not located in the two Gsp operons. Gene clusters encoding for hrp (hypersensitive response and pathogenicity), hrc (hrp conserved) and hpa (hrp associated) of the T3SS (Alegria et al., 2004) were identified in the chromosome (Supplementary Figure 5B and Supplementary Table 5). Overall, the T2SS and T3SS were highly conserved in the three sequenced strains.
TALE Variation and Plasmid Fusion
Compared with the chromosome sequences, the plasmid sequences showed higher diversity in plasmid length and number of genes (Supplementary Figure 6; Timilsina et al., 2020). In total, we identified five plasmids in three strains. The plasmid size ranged from 64 to 312 kb and the GC content was around 60%, which was lower than the average of 64% of the chromosomes (Supplementary Table 1). We tried to determine the plasmid copy number in each genome based on the relative whole genome sequencing coverage with the chromosome (Pena-Gonzalez et al., 2018). On average, we estimated the genomes contained 2.6, 3.6, 4.4, 3.5, and 1.6 copies of pB2_V1, pB2_V3, pSN3-3, pT4p1, and pT4p2, respectively (Supplementary Figure 7).
A sequence similarity search of plasmid sequences by BLASTN indicated pT4p2 contained the copper resistance gene cluster as indicated in Gochez et al. (2018). The other four plasmids (pB2_V1, pB2_V3, pSN3-3, and pT4p1) sharing high similarity with known plasmid sequences showed a high level of sequence rearrangement (Figure 4 and Supplementary Figures 6, 8). For instance, mobile genetic elements including Tn3-like transposon and the subclass of insertion sequence (IS) elements were highly variable between the four pathogenicity-related plasmids. Analysis of repeat-variable diresidues (RVDs; Ferreira et al., 2015) indicated that TALE repeats were seen in these four plasmids which contained different copy numbers of pthA encoding genes with various sizes of TALEs (Supplementary Table 6). Three copies of pthA gene were found in strains SN3-3, four copies in T4 and eight copies in strain B2.
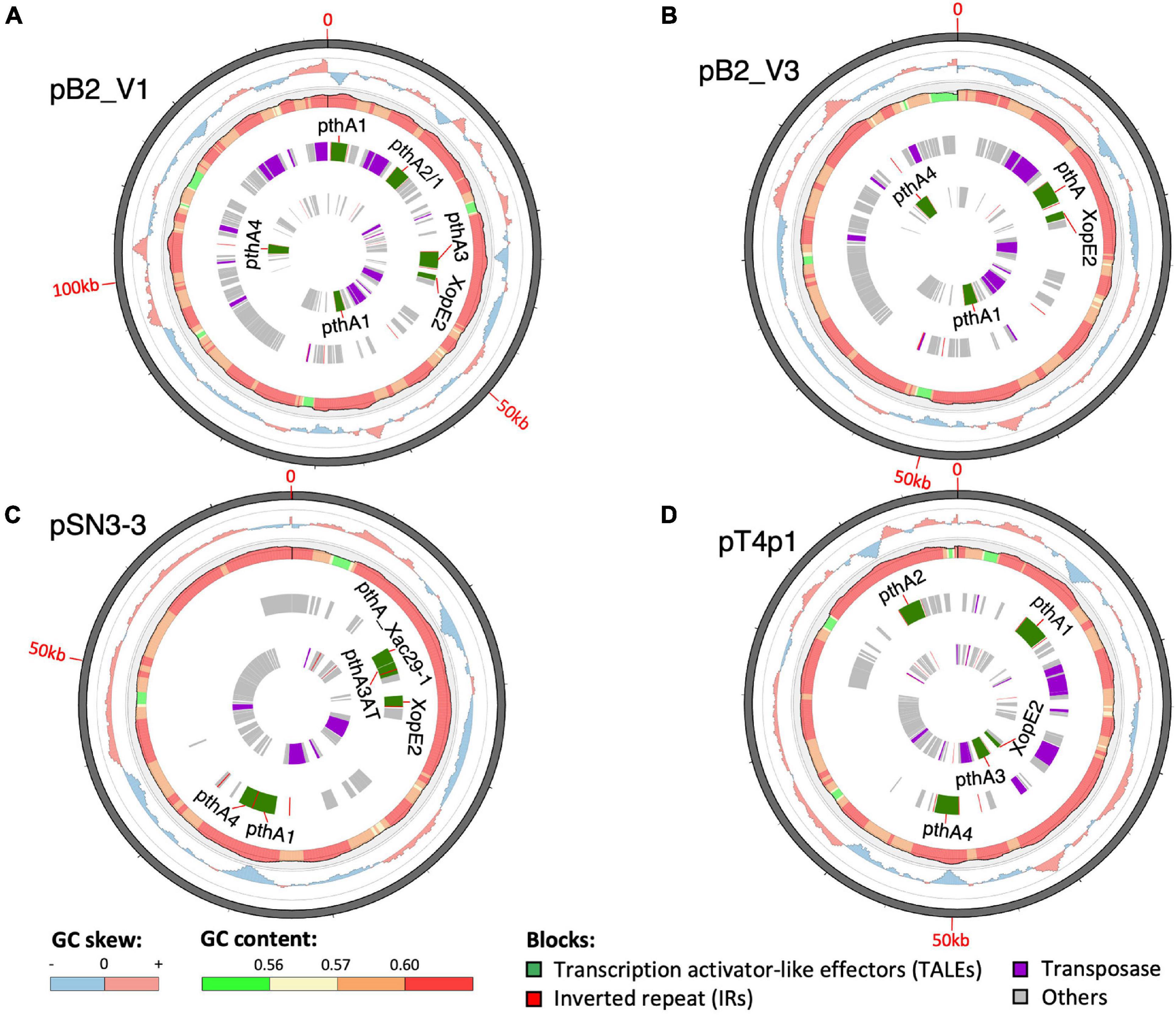
Figure 4. Plasmid organization. (A) pB2_V1, (B) pB2_V3, (C) pSN3-3, and (D) pT4p1. Circles from outermost to innermost represent: (1) GC skew, (2) GC content, (3) reverse, and (4) forward direction of repetitive sequences. Green: TALEs. Red: inverted repeats. Purple: transposase. Gray: other CDS/genes: gray.
Based on the TALE classification, Class I contained pthA2 and pthA3 genes, Class II contained pthA4 and Class III contained pthA1 (Gochez et al., 2018). These three Classes were seen together in plasmids pSN3-3, pB2_V1, and pT4p1 (Figure 4) and represented the signature of plasmid fusion events (Figures 5, Supplementary Figures 6, 8, and Supplementary Table 6). The size of pSN3-3 is similar to that of pXAC64 of the reference strain 306 but the pthA genes of strain SN3-3 carry different copies of repeats (Supplementary Table 6), which are shorter and not comparable with those of the reference strain 306 (da Silva et al., 2002). On the other hand, pthA2 and pthA4 on pSN3-3 partially overlapped with each other (Figure 5 and Supplementary Table 6).
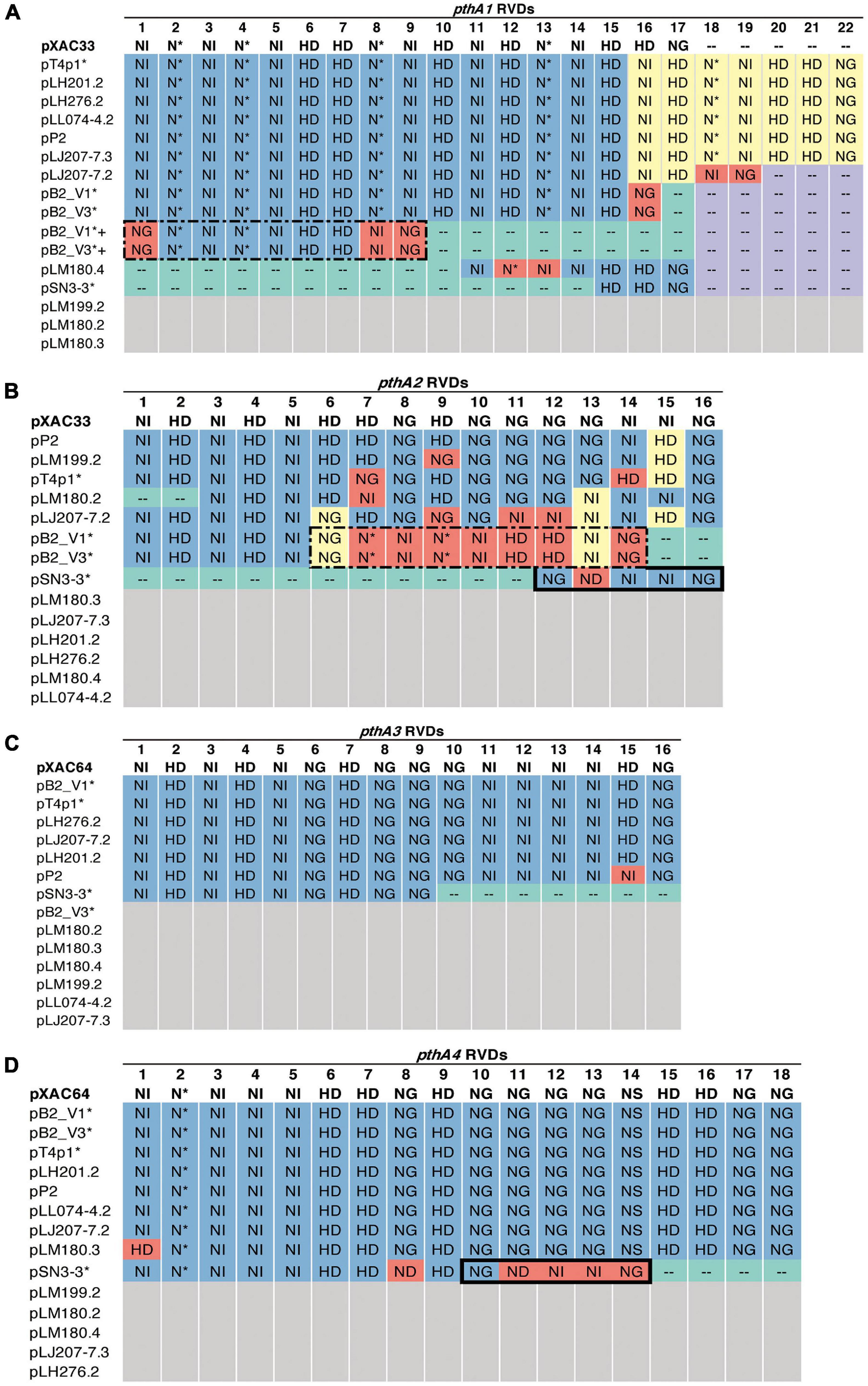
Figure 5. RVD profile of pthA genes. The asterisks (*) indicate plasmids sequenced in this study. NI (Asn-Ile) recognizes A; HD (His-Asp) recognizes C (but not 5′-methyl-C); NG (Asn-Gly) recognizes T and 5′-methyl-C; NN (Asn-Asn) recognizes G or A; NS (Asn-Ser) recognizes A,T,C or G; N* (Asn-*) recognizes C or T [47]. Blue: identical to the reference. Purple: absent, the same as the reference. Green: absent, different from the reference. Yellow: > 50% sequence similarity with the reference. Red: unique. White: no such pthA genes. (A) Plus sign (+): merged pthA genes. (A,B) Dashed box: nine pthA1 repeats were merged with pthA2 repeats. (B,D) Solid box: five pthA2 repeats were merged with pthA4 repeats.
Plasmid pT4p1 was highly similar to plasmid pP2 of the CuR strain Xc-03-1638-1-1 (Supplementary Figure 6 and Supplementary Table 6; Gochez et al., 2018). The two plasmids were almost identical with only 23 bp difference and contained the exact copy number of all four classes of pthA1 genes. Plasmid pT4p1 had 4 pthA genes of 3 classes including two genes with 15.5 (Class I: pthA2 and pthA3), one with 17.5 (Class II: pthA4) and one with 21.5 (Class III: pthA1) repeats comparable with the second largest plasmids of CuR strains pLH276.2 and pLL074-4.2 (Gochez et al., 2018; Supplementary Table 6). The plasmids of strain B2 had 8 pthA genes of 3 classes including two genes with 13.5 (unclassified class: pthA2/1), three with 15.5 (Class III) and two with 17.5 (Class II) repeats partially comparable to the pathogenicity plasmids of CuR strains (Gochez et al., 2018; Supplementary Table 6). The effector genes xopE2 (Ferreira et al., 2015) and pthA4 (Roeschlin et al., 2019) that are involved in the suppression of the hypersensitive response of the host were also identified in these four pathogenicity-associated plasmids.
Structures and Origins of the Copper and Arsenate Resistance Gene Clusters
The CuR strain T4 had the largest plasmid pT4p2 among the three sequenced strains (Supplementary Table 1) for which the sequence is similar to the copper resistance plasmids pLH201.1 and pCuR (Figure 6). Interestingly, the sequence of pT4p2 is larger than those two plasmids due to an unusual ∼40 Kbp inverted repeat. We further confirmed the junction size of the inverted repeat by PCR (Supplementary Figure 9). On the other hand, pT4p2 shared the same inverted repeat pattern as observed in pLH3.1 of X. perforans LH3 (CP018472) (Supplementary Figure 10), where the strain LH3 was collected from a tomato orchard in Mauritius in 2010 (Richard et al., 2017b). Each inverted repeat contains a complete set of the copper resistance gene cluster, except copCDG are missing (Figures 6, 7). Analysis of cop gene clusters revealed that three groups of copper resistance gene clusters could be identified across a diverse set of bacterial species. pLH201.1 and pCuR belonged to Group I. The pT4p2 and copper resistance plasmids from X. perforans, Stenotrophomonas, and Pseudoxanthomonas sharing the same arrangement of the cop gene cluster belonged to Group II (Figure 7 and Supplementary Table 7). In particular, in the NCBI nucleotide database, a unique region between the copper-arsenate clusters was only identified in pT4p2, pLH3.1 and the plasmids of six other Xanthomonas strains that were tomato pathogens, with the exception of pXAC219 that was isolated from the citrus leaf. Plasmids of these strains shared 99% sequence coverage and >96% sequence identity (Supplementary Table 8). The third group, Group III, contained a more diverse set of cop gene clusters where the plasmid backbone was extremely similar to pCuR (Richard et al., 2017b) but only copF and copA shared 70% nucleotide sequence identity with those in pCuR.

Figure 6. Comparison of plasmids containing copper resistance genes in pT4p2, pCur and pLH201.1. Syntenic regions are presented in red color and inverted regions are in blue. Genes involved in copper/arsenate resistance and conjugation are highlighted along the plasmid sequences.
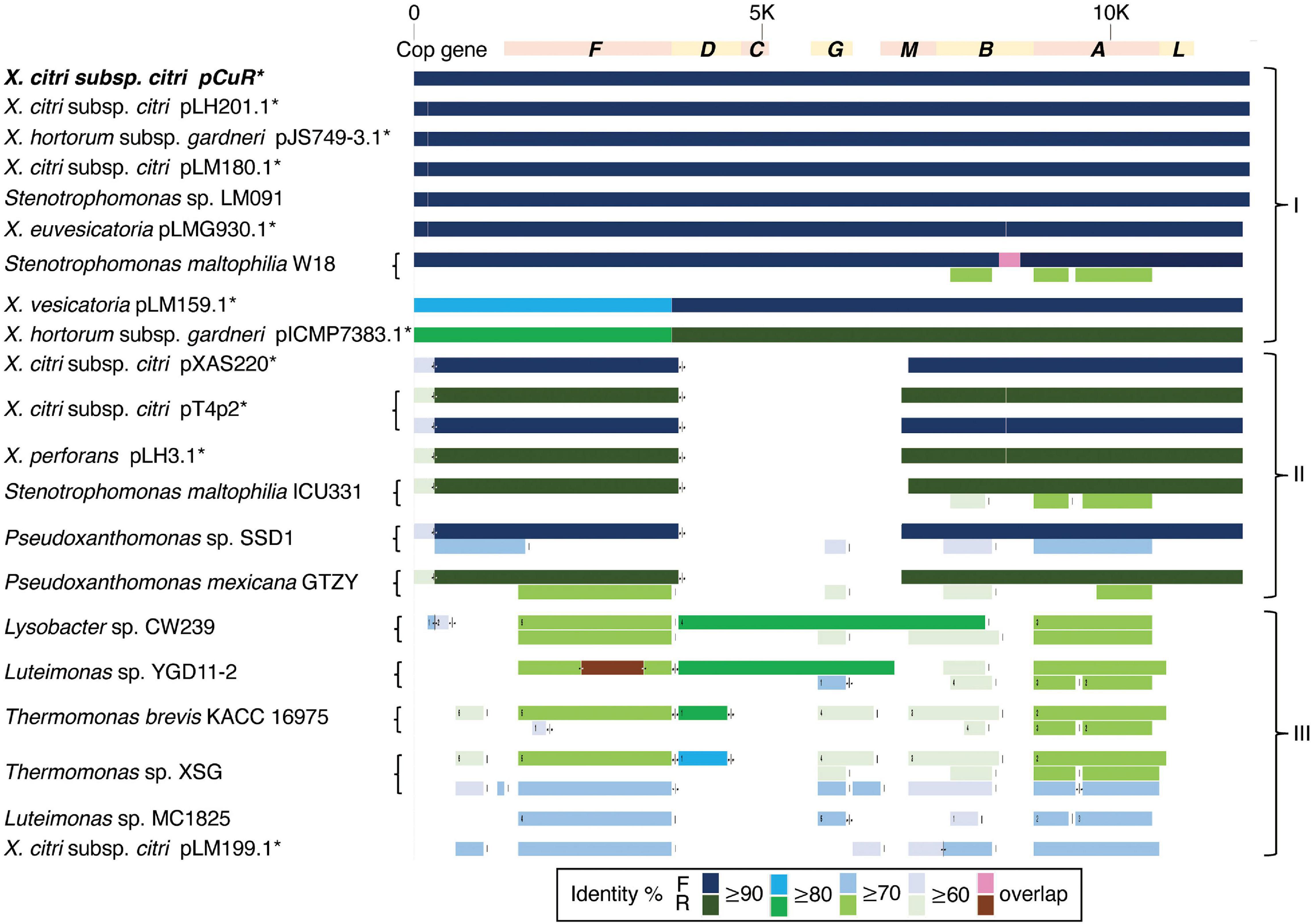
Figure 7. Comparison of gene gain and loss in the copper resistance gene cluster.
To further support our idea about the origin of the copper resistance cluster, we then compared the structure of the arsenate gene cluster. The arsenate cluster was mostly composed of either three (arsRBC) or five (arsRDABC) genes and expressed in a single transcriptional unit in soil bacteria (Achour et al., 2007). Interestingly, the X. citri arsenate gene cluster which was grouped together with that of Stenotrophomonas was composed of four (arsRCHB) genes (Figure 8). Furthermore, the transposases between the arsenate and copper clusters showed higher sequence similarity to those of X. perforans whereas two endonucleases corresponding to pCuR and pLH3.1 were adjacent to each other (Supplementary Figure 10).

Figure 8. Comparison of gene gain and loss in the arsenate resistance gene cluster.
Moreover, the ars and cop gene clusters responsible for resistance to heavy metals including copper and arsenate are adjacent to each other with higher GC content (62.3%) compared with other genomic regions (58.6%) and flanked by two Tn3-like transposons (Figure 6 and Supplementary Figure 11). In addition to the abnormal GC content and transposon insertions, merR and cusAB associated with heavy metal resistance in bacteria were found in the pT4p2. The MerR transcriptional regulator family controlled the expression of copA in Escherichia coli and responded to environmental stimuli, such as heavy metals or antibiotics (Brown et al., 2003). In pT4p2, MerR is 7 Kbp away from the ars cluster but the heavy metal efflux pump cusAB is next to the ars cluster (300 bp). The close proximity of MerR and cusAB to the heavy metal cluster was also observed in pLH201.1 (Richard et al., 2017b). In addition, the heavy metal resistance gene clusters in pT4p2 were surrounded by two Tra gene clusters. The T4 strain showed the conjugation ability and the pT4p2 encoded 16 Tra proteins that are essential for conjugation. Similar to the pCuR and pLH201.1 arrangement, Tra genes of pT4p2 located in two different regions of the plasmid with TraID in one side and other 14 Tra genes grouped into another cluster. Overall, the complete set of Tra genes in the pT4p2 and the high sequence similarity of heavy metal resistance gene clusters with other published plasmids indicated that the heavy metal resistance gene was likely acquired from other microbes.
Conjugative Transfer of Copper Resistance Genes
The copper resistance genes can be transferred from the CuR strain T4 to the CuS strains B2Rif and SN3-3Rif through bacterial conjugation (Supplementary Table 9). The transconjugants showed the same level of copper resistance as the donor strain T4. The frequency of conjugative gene transfer ranged from 10–8 to 10–5 transconjugants per recipient (Supplementary Table 9). B2Rif received the CuR genes from T4 more frequently than SN3-3Rif (Supplementary Table 9).
Discussion
In this study, the genome and plasmid sequences of three X. citri subsp. citri strains from Taiwan were completely sequenced and assembled using the combination of Illumina short-reads and Oxford Nanopore long-reads. Whole genome sequencing has been widely used to understand the pathogenic, taxonomic and phylogenetic status of the xanthomonads (Zhang et al., 2015; Bansal et al., 2017; Patane et al., 2019). Previous studies lacked the resolution of repetitive or duplicate regions as they could not be precisely resolved by the short-read sequencing method and the bioinformatic algorithm (Triplett et al., 2011; Fonseca et al., 2019). Recently, with the availability of the long-read sequencing method and the advances in genome assembly (Kolmogorov et al., 2019), complex genome structures can be fully resolved. Correspondingly, the genome and plasmid sequence dynamics of X. citri. from different geographic locations have gradually emerged (Gochez et al., 2018; Roach et al., 2020; Richard et al., 2021).
The assembled chromosome sequence length, GC content (64.76∼64.84%) and number of genes (Figures 1A–C) are in the same range as the published complete X. citri subsp. citri (Timilsina et al., 2020). The 75 X. citri genomes showed a high level of genome conservation where 2,529 ortholog genes had a strict one-to-one single copy relationship within X. citri strains. The large core-genome size (Figure 3A) and the pangenome analysis (Loiseau et al., 2018) indicated that the X. citri strains showed a ‘close’ genome signature where strains shared a large common gene repertoire. In contrast, the Xanthomonas genus was an ‘open’ genome where gene gain and loss events frequently occurred within different Xanthomonas lineages (Timilsina et al., 2020).
Comparing chromosome sequences and gene contents did not reveal many differences between X. citri subsp. citri strains. In fact, the pangenome analysis confirmed that strains in the X. citri subsp. citri were rather conserved and the core genome was composed of 2,529 one-to-one ortholog genes and ∼3,000 genes (i.e., detected in all 75 strains) (Figure 3A). Nevertheless, the core genome size of this study is smaller than the 4,347 genes of 221 strains in the previous study (Richard et al., 2021). The differences in the three pan-genome analysis strategies likely explain the inconsistency between the studies. First, only protein coding genes in the chromosomes were considered for the orthologous relationships in this study (Timilsina et al., 2019); however, the study by Richard et al. included the plasmid genes in the analysis (Richard et al., 2021). A high gene turnover rate (gain or loss of genes) was observed due to uneven frequencies of acquiring or losing a single large plasmid in different lineages (Richard et al., 2021). The ease of horizontal gene transfer of the entire plasmid between strains has made plasmids the main driver of host adaptation (Ruh et al., 2017). To focus on changes in the chromosome sequences, we decided to focus only on genes of the chromosomes.
Secondly, we used orthology-based analysis where predicted protein coding genes of each of the de novo assembled genomes were used to identify orthologous genes among sequenced strains. The advantage of this strategy is that it did not restrict the analysis in one particular reference strain. That is, unique genome fragments and gene contents that existed in one particular strain could be identified. The main drawback was that each sequencing project applied different gene prediction methods and this might introduce biases in gene models due to technical artifacts. An alternative approach would be based on the read coverage of the whole genome resequencing data (Richard et al., 2021). Using this approach, one strain was selected as the reference genome where resequencing reads were mapped to the reference genome sequence. The presence and absence of the gene was then evaluated based on the pre-defined cut-off of read depth and coverage of the gene length. This method could avoid the fragmented genomes and truncated gene models due to the short read assemblies. Nevertheless, variations due to paralogs, gene copy number or pseudogenization could not be identified. Despite the differences in the exact number of genes of the core genome, our result agrees with previous analysis (Timilsina et al., 2019) in concluding that X. citri subsp. citri is a monomorphic bacterium and the gene numbers are conservative in the lineage (Timilsina et al., 2019).
The pathotype classification is in agreement with the core-genome phylogenetic analysis in showing that these three strains clustered with the pathotype A strains from different geographic origins (Jeong et al., 2019). However, the exact phylogenetic relationship between the three strains sequenced in this study could not be clearly resolved and we did not date the time scale of the evolutionary history (Figure 3B). The three strains were placed in an independent clade with 100% bootstrap value support. The long branch length with strains from other geographic origins suggest that three strains diverged from a common ancestor.
The gap-free complete genome sequences offered a unique opportunity to study the structure of the repetitive sequences such as CRISPR repeats and spacers in detail since their order and the exact copy number have often been mis-assembled in the draft genome assemblies (Wang et al., 2021). In particular, spoligotyping provided an alternative method to infer the evolutionary history and the common ancestor of the microbial strains. Among sequenced X. citri subsp. citri strains, the CRISPR array contains a conserved array of 23 spacers and could be used to infer the evolutionary trajectory among the observed spoligotypes (Jeong et al., 2019). This method was based on the principle associated with the order of the CRISPR array, where the cas genes tend to be associated with more recently acquired CRISPR repeats. For instance, spacer Xcc_23 is likely to be a newly acquired spacer/repeat unit where spacer Xcc_1 might be the oldest spacer (Jeong et al., 2019). The results of the CRISPR array are consistent with the notion that X. citri originated from a common ancestor and formed a monophyletic clade (Bansal et al., 2018). Furthermore, strains B2 and T4 from the commercial orchard under conventional management have more CRISPR repeats than the strain SN3-3 which was from the orchard with minimal management. This result implied that the number of CRISPR repeats could be affected by types of agricultural management. Furthermore, B2 and T4 originated from the same orchard but differed in composition of CRISPR repeats. Xcc_3 was found in B2 but not in T4 (Figure 2). Thus, X. citri subsp. citri in an orchard could evolve into various strains with diverse compositions of CRISPR repeats. The spoligotyping data indicated the rapid evolution and diversity of X. citri subsp. citri pathotype A in Asia.
The function of CRISPR/Cas systems has been proven to be an adaptive bacterial defense system against phage infections. New CRISPR spacers are introduced in the bacterial chromosome near the leader sequence (Datsenko et al., 2012). Yet, frame-shift mutation of csd1/cas8c genes caused by a short tandem repeat of two base pairs (AG) was found in the majority of X. citri strains, suggesting that the CRISPR defense system was mutationally inactivated to further acquire new spacers (Jeong et al., 2019). The results also suggest that most of the 25 spoligotype patterns found in X. citri could evolve by either random deletion of a single spacer/repeat unit or simultaneous deletion of adjacent spacer/repeat units (Jeong et al., 2019). Accordingly, deletion events could occur more often in SN3-3 than in the other 2 strains since SN3-3 had fewer CRISPR repeats than B2 and T4. An extra AG is also present in the csd1/cas8c genes of our X. citri subsp. citri strains (Supplementary Figure 12) reflecting inactivation of the CRISPR system due to frame-shift mutation. Moreover, the CRISPR repeat profile data implied that B2 and T4 could acquire more resistance to phage infections than SN3-3. It will be intriguing to explore whether minimal agricultural management could induce more deletion of CRISPR repeats in citrus canker pathogens or reduce diversity of phages in an orchard.
Plasmids are highly dynamic and present another source of the X. citri genome diversity. The number and size of plasmids vary between X. citri strains and are important contributors to pathogenicity due to their ease of acquisition of foreign genomic elements through recombination and horizontal gene transfer (Timilsina et al., 2020; Rodriguez-Beltran et al., 2021). Mobile genetic elements are known to facilitate the exchange or transfer of genomic fragments between chromosomes and plasmids (Blair et al., 2015). Metagenomic analysis of wastewater identified that antibiotic selection pressure significantly increased the abundance of antibiotic resistance genes, reduced the diversity of the microbial community and in particular, increased the occurrence and abundance of mobile genetic elements (Zhao et al., 2021). Despite the high sequence variability, genes in several important functional categories such as conjugative transfer (traY, traD and mobD/A/L), the type IV secretion system (vir2/3/4/6/8/9/11), toxin/antitoxin system (family vapC or pemK/mazF) and core genes (repA, parA, XRE family transcriptional regulator) were found in all of our plasmids (Gruber et al., 2016).
Multiple types of insertion sequences and transposons including IS3, IS4, ISxac3, ISxac2, TnpA and Tn3 were identified in the plasmids (Figure 4). The xanthomonad Tn3 family transposons were located near different TALEs. The TnXax1 transposable element of Tn3 family in xanthomonadaceae were flanked by short inverted repeat (IR) sequences forming a generic structure of mobile insertion cassettes (MICs; Gochez et al., 2018). In particular, the genetic content of TnXax1 was organized in the following order from left IR (IRL) to right IR (IRR): mlt + TnpA (transposase) + TnpR (resolvase)/TnpS/TnpT (recombinase) + passenger gene as observed in other Xanthomonas strains (Lima-Mendez et al., 2020). The structure of the MICs in the plasmids of this study was similar to TnXax1 containing the same passenger genes, Tn3 transposons and inverted repeats (IRs; Ferreira et al., 2015). The pthA genes in the four pathogenicity-related plasmids were surrounded by the same IRR and IRL as in the pXAC64 plasmid of X. citri 306.
TALEs are located on plasmids in X. citri strains with high variability in order to adapt to different hosts (Ferreira et al., 2015; Timilsina et al., 2020; Figures 4, 5 and Supplementary Table 6). The variability of the TALE repeats has been considered to be a strategy to diversify selection pressure to escape the detection of the host R genes (Doucoure et al., 2018). The changes in repeat arrays in X. oryzea TALEs were mainly associated with repeat deletion, and recombination with other TALEs (Teper and Wang, 2021). Furthermore, Teper and Wang (2021) demonstrated that two to five mismatched TALE repeats of X. citri subsp. citri was sufficient to escape the host NB-LRR recognition and promote disease symptoms in sweet orange. Our data revealed that most pthA4 genes from different X. citri subsp. citri strains have the same number and RVD of tandem repeats (Figure 5 and Supplementary Table 6). Surprisingly, the pthA4 of strain SN3-3 was not only short (13.5 repeats) but also merged with pthA2.
It has been estimated that the optimal functional length of TALEs contained 15.5–19.5 RVD repeats whereas TALEs with fewer than 6.5 repeats did not perform gene activation and could be by-products of recombination events (Boch and Bonas, 2010; Gochez et al., 2018). Nevertheless, some ‘non-classical’ TALEs with unusually short lengths of RVD repeats but that maintain their biological function, have been reported recently (Roeschlin et al., 2019). We also identified a fusion of pthA1 and pthA4 in pSN3-3 and pthA1 that was surrounded by a solo IR sequence containing only 2.5 RVD repeats (Figure 5 and Supplementary Table 6). The pthA4 was indispensable for canker elicitation but pthA1 and pthA3 could contribute to additive roles in developing disease symptoms (Abe and Benedetti, 2016). Since PthA4 is the TALE required for pathogenicity and SN3-3 was collected from a citrus farm with minimal management, we speculated that the PthA4 with 13.5 RVD repeats was sufficient to develop pathogenicity and maintain bacterial fitness under the agroecosystem with low selection pressure. Our data is in full agreement with the notion that the presence of the Tn3-like transposons around TALEs likely contributed to the generation of diverse TALEs of X. citri (Ferreira et al., 2015).
The genomic signatures of the three strains supported the occurrence of independent events of plasmid fusion. Compared with the reference genome of strain 306 (da Silva et al., 2002), we were able to identify the recombination event that caused the plasmid co-integration and the fusion of pthA genes (Figure 5). Unlike different pthA classes that were separated in two plasmids in pXAC33 and pXAC64 (da Silva et al., 2002), pB2_V1 and pT4p1 each contained three pthA classes in one plasmid (Supplementary Table 6). Furthermore, pB2_V1 contained two copies of pthA1 class with high sequence identity and pB2_V1 and pB2_V3 both contained a unique class of pthA2/pthA1 fusion with 13.5 RVD repeats (Figures 5A,B and Supplementary Table 6). The plasmid size of pSN3-3 was similar to pXAC64 of strain 306 (da Silva et al., 2002) but also had three classes of pthA and the fusion of pthA1/pthA4 in one plasmid (Figures 5B,D).
The whole genome sequence of T4 clarified that the cop gene cluster is borne on the big plasmid pT4p2. Though the copper resistance in different Xanthomonas strains might be acquired through independent events, cop genes in the plasmids were genetically related (Richard et al., 2017b) and the backbones of the plasmids are highly similar. Based on the nucleotide sequence identity and presence and absence of cop genes, we could divide plasmids, based on the copper cluster, into three groups (Figure 7). Group I, including the reference plasmid pCuR and pLM091 from Stenotrophomonas (Richard et al., 2017b), contained the complete set of copLABCDMGF genes without gaps and shared high sequence identity (>90%) with plasmids in this group. On the other hand, the most dissimilar group (Group III), including pLM199 from Argentina, had sequence identity with pCuR that was ∼70% and failed to produce PCR amplicons using primers of the copLAB system (Richard et al., 2017b). A distinct copper transposon region (TnpLM199) containing an alternative copper resistance system copABCD was identified in the Argentinian strain LM199 genome (Richard et al., 2017b).
The pT4p2 was classified in Group II together with pLH3.1 of X. perforans and chromosome sequences from other microbials. In particular, a unique nucleotide region between the copper and arsenate cluster was only identified in pT4p2 and pLH3.1. A detailed analysis of the 1,857 bp spacer nucleotide sequence between the copper and arsenate cluster in the NCBI Nucleotide database revealed that only seven Xanthomonas strains contained this fragment. Among these seven strains, the spacer sequence was not located on the same plasmid as that containing copper resistance genes in the X. euvesicatoria strain LMG930. This result is in agreement with a previous analysis that revealed that the copB genes carried by pT4p2 and LMG930 were grouped in the ‘Variant IV’ group and shared a combination of 3, 300 and 36 bp gaps in the complete copB sequences (Lai et al., 2021). On the other hand, the other six Xanthomonas strains showed high levels of sequence coverage (99%) and nucleotide identity (>96%) (Supplementary Table 8). In particular, pLH3.1 of X. euvesicatoria did not only show high identity of the spacer sequence but also the cop genes (Supplementary Figure 10). Accordingly, the comparative genome analysis data provided evidence that pT4p2 could originate from pLH3.1 carried by Mauritian X. perforans LMG930. Although CuR X. euvesicatoria pv. perforans populations have prevalently occurred in domestic tomato orchards in Taiwan since 1989 (Burlakoti et al., 2018), only the variant V group of the copB gene was carried by X. euvesicatoria pv. perforans strains in our recent survey (Lai et al., 2021). Thus, pT4p2 might not originate from local CuR strains of X. euvesicatoria pv. perforans. However, we cannot exclude the possibility that other xanthomonads in other production areas in Taiwan, on other citrus or solanaceous cultivars which carry pLH3.1/pT4p2-like plasmids, could be found by increasing the number of CuR xanthomonad populations studied. A further survey of CuR xanthomonad populations on various hosts in combination with comparative genomic analysis will help to decipher the distribution and spread of CuR plasmids in xanthomonads.
Our whole genome sequencing data clarified that the copLAB cluster is located in the plasmid pT4p2 associated with heavy metal resistance. Horizontal transfer of copper resistance between bacteria occurs more frequently when the cop genes are located in the mobile plasmids (Behlau et al., 2012a). The Tra cluster responsible for the mobility of the plasmid was found in the CuR plasmid pT4p2. The data of conjugative transfer proved that pT4p2 is a mobile plasmid and able to horizontally transfer between different strains of X. citri subsp. citri (Supplementary Table 9). The CuS recipient cells became as resistant to copper as the donor cells while receiving cop genes via conjugation. Accordingly, the horizontal transfer of plasmid borne cop genes between citrus canker xanthomonads may potentially increase CuR xanthomonad populations to reduce the disease control efficacy of copper bactericides.
It is worth noting that the copper resistance gene clusters were located on the plasmid of Xanthomonas strains but were present in the chromosomes of Stenotrophomonas, Pseudoxanthomonas, Lysobacter, Luteimonas and Thermomonas (Figure 7). These Xanthomonadaceae microbes were either human pathogens (Crossman et al., 2008) or presented in the agricultural environment (Turrini et al., 2021) and some are known for carrying heavy metal resistance genes and mobile genetic elements. These environmental microbes could serve as a reservoir for the transfer of heavy metal resistance genes to microbes living in the surrounding environment.
We speculated that one copper resistance cluster in pT4p2 was inserted next to the arsenate cluster due to the Tn3-like transposon activity. Moreover, the additional copy of the arsenate and copper cluster might be the byproduct of conjugation where plasmids were transferred as a single-strand DNA during the conjugation and subsequently activated the bacterial SOS stress response. The recombination or mutagenesis frequencies of genomic fragments were then induced and increased the bacterial evolution rate (Rodriguez-Beltran et al., 2021). The high density of transposable elements around the heavy metal resistance clusters (Supplementary Figure 10) increased the probability that the duplicated fragment was inserted around the same location.
The whole genome sequences of the three strains in this study reflected the potential effect of agricultural practices or agroecosystems on diversification of citrus bacterial canker pathogens. A large sampling of microbes from the same environment over a long period of time would likely improve our understanding of how agricultural practices impact the microbial community.
Conclusion
Complete genome sequencing of three X. citri subsp. citri pathotype A strains from two distant orchards and a comparison with the published genomes in this study clearly illustrated plasticity in chromosomes and plasmids. Our results revealed the evolution of pathogenicity factors and horizontal gene transfer events in the three strains. Type of agricultural management could be a potential trigger for evolution of pathotype A of X. citri subsp. citri. Surprisingly, conventional management might induce less deletion of CRISPR repeats in X. citri subsp. citri or increase the diversity of phages in the orchard. Thus, the X. citri subsp. citri strains under conventional management might have more CRIPSR repeats for immunity to phage infections compared with the strain from the orchard with minimal management. Moreover, the cop gene cluster together with the arsenate resistance gene cluster were only carried by the huge plasmid in the CuR strain of X. citri subsp. citri. Collectively, plasmids represented a hotspot for exchanging foreign genomic elements and accelerated the adaptation of X. citri subsp. citri to the agroecosystem.
Data Availability Statement
The dataset generated for this study including sequencing reads, genome assemblies and genome annotation have been deposited at NCBI under BioProject PRJNA644481. The X. citri strains that support the findings of this study are available on request from C-JH.
Author Contributions
C-JH, H-FN, and Y-CL conceived and designed the experiments. T-LW, P-XZ, and J-YO performed next-generation sequencing and bioinformatics analysis. C-JH, T-LW, and Y-CL interpreted the data. C-JH, T-LW, and Y-CL wrote the manuscript with input from all co-authors. All authors read and approved the final manuscript.
Funding
This research was funded by the Ministry of Science and Technology (MOST, grant numbers 104-2313-B-415-013-MY2, 107-2311-B-415-004, and 108-2313-B-415-006-MY2) to C-JH, the Innovative Translational Agricultural Research Program (AS-KPQ-108-ITAR-10) and Academia Sinica Institutional funding to Y-CL. These funding bodies played no role in the design of the study, collection, analysis or interpretation of data, or in writing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the AS-BCST Bioinformatics Core for the computational support, and Miranda Loney for English editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.731711/full#supplementary-material
Supplementary Figure 1 | Highly conserved chromosome sequences of the three sequenced strains. Each collinearity block represents >5000 bp alignment and >99% sequence identity by BLASTN.
Supplementary Figure 2 | Phylogenetic analysis based on whole genome comparison. The asterisks indicate copper-resistant strains.
Supplementary Figure 3 | Shared and unique protein coding genes in three sequenced strains.
Supplementary Figure 4 | Pangenome analysis of 79 X. citri genomes.
Supplementary Figure 5 | Type II and III secretion systems. (A) Two operons of the type II secretion system. (B) Gene cluster of the type III secretion system.
Supplementary Figure 6 | Pairwise comparison of plasmids. Upper panel: copper resistance plasmids. Lower panel: pathogenicity related plasmids. Blue line: syntenic region in the same direction. Green: syntenic region in the reverse direction. Orange: secondary alignment. a. plasmids sequenced in this study.
Supplementary Figure 7 | Estimated plasmid copy number based on the Nanopore sequencing coverage.
Supplementary Figure 8 | Multiple plasmid alignment of pXAC33, pXAC64 and non-CuR plasmids sequenced in this study. (A) Mauve alignment including pXAC33, pxAC64 and plasmids in this study. (B) Mauve alignment of plasmids in this study. Syntenic regions share the same color.
Supplementary Figure 9 | Genome assembly confirmation of the copper-resistance plasmid pT4p2. (A) Genome structure of the 40 kbp inverted repeat. (B) PCR product of the PF1-CF1 primer pair and (C) PCR product of the PF1-CR3 primer pair.
Supplementary Figure 10 | Inverted duplication of metal-resistance clusters between pT4p2 and pLH3.1. Orange box (Cop): location of copper resistance gene clusters. Pink box (Ars): arsenate-resistant gene clusters. Tns: transposase.
Supplementary Figure 11 | Comparison of plasmids containing copper resistance genes in pT4p2, pCur and pLH201.1. Circles from outermost to innermost represent: (1) position and genes, the grid is 10 kbp [pink, pT4p2 of strain T4; yellow, pLH201.1 (NZ_CP018859.1) of strain LH201; white, pCuR (NZ_CP023286.1) of strain 03-1638-1-1]; (2) GC content with 2000-bp sliding window and 200-bp step; (3) regions with similar gene content and nucleotide sequence identity >90%, and alignment length >1 kb between plasmids.
Supplementary Figure 12 | Multiple sequence alignment of csd1 and cas8c genes. Black box, around 530 nt, indicates the AG frameshift.
Supplementary Table 1 | Summary of assembly and annotation statistics.
Supplementary Table 2 | Strains used for pan-genome analysis.
Supplementary Table 3 | Pairwise nucleotide sequences identified by fastANI.
Supplementary Table 4 | GO enrichment of strain specific genes.
Supplementary Table 5 | Gene clsters of the type II and type III secretion systems.
Supplementary Table 6 | TALEs and repeat number of RVDs in plasmids.
Supplementary Table 7 | Strains used for copper and arsenate resistance gene cluster alignment.
Supplementary Table 8 | Strains containing the unique spacer repeat between the copper and arsenate resistance gene clusters.
Supplementary Table 9 | Conjugation frequency of plasmid-borne copper resistance genes transferred between different Xanthomonas citri subsp. citri strains in vitro.
Footnotes
References
Abe, V. K., and Benedetti, C. E. (2016). Additive roles of PthAs in bacterial growth and pathogenicity associated with nucleotide polymorphisms in effector-binding elements of citrus canker susceptibility genes. Mol. Plant Pathol. 17, 1223–1236. doi: 10.1111/mpp.12359
Achour, A. R., Bauda, P., and Billard, P. (2007). Diversity of arsenite transporter genes from arsenic-resistant soil bacteria. Res. Microbiol. 158, 128–137. doi: 10.1016/j.resmic.2006.11.006
Achtman, M. (2008). Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62, 53–70. doi: 10.1146/annurev.micro.62.081307.162832
Alegria, M. C., Docena, C., Khater, L., Ramos, C. H., Da Silva, A. C., and Farah, C. S. (2004). New protein-protein interactions identified for the regulatory and structural components and substrates of the type III Secretion system of the phytopathogen Xanthomonas axonopodis Pathovar citri. J. Bacteriol. 186, 6186–6197. doi: 10.1128/JB.186.18.6186-6197.2004
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2010). FastQC: a Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics. Cambridge: Babraham Institute.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bansal, K., Kumar, S., and Patil, P. B. (2018). Complete genome sequence reveals evolutionary dynamics of an emerging and variant pathovar of Xanthomonas euvesicatoria. Genome Biol. Evol. 10, 3104–3109. doi: 10.1093/gbe/evy238
Bansal, K., Midha, S., Kumar, S., and Patil, P. B. (2017). Ecological and evolutionary insights into Xanthomonas citri pathovar diversity. Appl. Environ. Microbiol. 83:e02993-16. doi: 10.1128/AEM.02993-16
Behlau, F., Canteros, B. I., Jones, J. B., and Graham, J. H. (2012a). Copper resistance genes from different xanthomonads and citrus epiphytic bacteria confer resistance to Xanthomonas citri subsp. citri. Eur. J. Plant Pathol. 133, 949–963. doi: 10.1007/s10658-012-9966-8
Behlau, F., Canteros, B. I., Minsavage, G. V., Jones, J. B., and Graham, J. H. (2011). Molecular characterization of copper resistance genes from Xanthomonas citri subsp. citri and Xanthomonas alfalfae subsp. citrumelonis. Appl. Environ. Microbiol. 77, 4089–4096. doi: 10.1128/AEM.03043-10
Behlau, F., Hong, J. C., Jones, J. B., and Graham, J. H. (2013). Evidence for acquisition of copper resistance genes from different sources in citrus-associated xanthomonads. Phytopathology 103, 409–418. doi: 10.1094/PHYTO-06-12-0134-R
Behlau, F., Jones, J. B., Myers, M. E., and Graham, J. H. (2012b). Monitoring for resistant populations of Xanthomonas citri subsp. citri and epiphytic bacteria on citrus trees treated with copper or streptomycin using a new semi-selective medium. Eur. J. Plant Pathol. 132, 259–270. doi: 10.1007/s10658-011-9870-7
Blair, J. M., Webber, M. A., Baylay, A. J., Ogbolu, D. O., and Piddock, L. J. (2015). Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51. doi: 10.1038/nrmicro3380
Boch, J., and Bonas, U. (2010). Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 48, 419–436. doi: 10.1146/annurev-phyto-080508-081936
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, N. L., Stoyanov, J. V., Kidd, S. P., and Hobman, J. L. (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163. doi: 10.1016/S0168-6445(03)00051-2
Brunings, A. M., and Gabriel, D. W. (2003). Xanthomonas citri: breaking the surface. Mol. Plant Pathol. 4, 141–157. doi: 10.1046/j.1364-3703.2003.00163.x
Burlakoti, R. R., Hsu, C. F., Chen, J. R., and Wang, J. F. (2018). Population dynamics of Xanthomonads associated with bacterial spot of tomato and pepper during 27 years across Taiwan. Plant Dis. 102, 1348–1356. doi: 10.1094/PDIS-04-17-0465-RE
Canteros, B. I., Rybak, M., Gochez, A., Velazquez, P., Rivadeneira, M., Mitidieri, M., et al. (2008). Occurrence of copper resistance in Xanthomonas axonopodis pv. citri in Argentina. Phytopathology 98:S30.
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and Mcquillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Chiang-Ni, C., Zheng, P. X., Tsai, P. J., Chuang, W. J., Lin, Y. S., Liu, C. C., et al. (2012). Environmental pH changes, but not the LuxS signalling pathway, regulate SpeB expression in M1 group A streptococci. J. Med. Microbiol. 61, 16–22. doi: 10.1099/jmm.0.036012-0
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Crossman, L. C., Gould, V. C., Dow, J. M., Vernikos, G. S., Okazaki, A., Sebaihia, M., et al. (2008). The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 9:R74. doi: 10.1186/gb-2008-9-4-r74
da Silva, A. C., Ferro, J. A., Reinach, F. C., Farah, C. S., Furlan, L. R., Quaggio, R. B., et al. (2002). Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417, 459–463. doi: 10.1038/417459a
Datsenko, K. A., Pougach, K., Tikhonov, A., Wanner, B. L., Severinov, K., and Semenova, E. (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3:945. doi: 10.1038/ncomms1937
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Doucoure, H., Perez-Quintero, A. L., Reshetnyak, G., Tekete, C., Auguy, F., Thomas, E., et al. (2018). Functional and genome sequence-driven characterization of tal effector gene repertoires reveals novel variants with altered specificities in closely related Malian Xanthomonas oryzae pv. oryzae strains. Front. Microbiol. 9:1657. doi: 10.3389/fmicb.2018.01657
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Fawcett, H., and Jenkins, A. (1933). Records of citrus canker from herbarium specimens of the genus Citrus in England and the United States. Phytopathology 23, 820–824.
Ference, C. M., Gochez, A. M., Behlau, F., Wang, N., Graham, J. H., and Jones, J. B. (2018). Recent advances in the understanding of Xanthomonas citri ssp. citri pathogenesis and citrus canker disease management. Mol. Plant Pathol. 19, 1302–1318. doi: 10.1111/mpp.12638
Ferreira, R. M., De Oliveira, A. C., Moreira, L. M., Belasque, J. Jr., Gourbeyre, E., Siguier, P., et al. (2015). A TALE of transposition: Tn3-like transposons play a major role in the spread of pathogenicity determinants of Xanthomonas citri and other xanthomonads. mBio 6, e02505–e02514. doi: 10.1128/mBio.02505-14
Fonseca, N. P., Patane, J. S. L., Varani, A. M., Felestrino, E. B., Caneschi, W. L., Sanchez, A. B., et al. (2019). Analyses of seven new genomes of Xanthomonas citri pv. aurantifolii Strains, causative agents of citrus canker B and C, Show a reduced repertoire of pathogenicity-related genes. Front. Microbiol. 10:2361. doi: 10.3389/fmicb.2019.02361
Gochez, A. M., Huguet-Tapia, J. C., Minsavage, G. V., Shantaraj, D., Jalan, N., Strauss, A., et al. (2018). Pacbio sequencing of copper-tolerant Xanthomonas citri reveals presence of a chimeric plasmid structure and provides insights into reassortment and shuffling of transcription activator-like effectors among X. citri strains. BMC Genomics 19:16. doi: 10.1186/s12864-017-4408-9
Grigoriev, A. (1998). Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 26, 2286–2290. doi: 10.1093/nar/26.10.2286
Gruber, C. J., Lang, S., Rajendra, V. K., Nuk, M., Raffl, S., Schildbach, J. F., et al. (2016). Conjugative DNA transfer is enhanced by plasmid R1 partitioning proteins. Front. Mol. Biosci. 3:32. doi: 10.3389/fmolb.2016.00032
Hseu, S. H., and Hsu, S. T. (1991). Sensitivity of strains of Xanthomonas campestris pv. vesicatoria from Taiwan to copper and other agrochemicals. Plant Prot. Bull. 33, 410–419.
Huang, C. J., and Ni, H. F. (2017). First report of Citrus depressa as a new natural host of Xanthomonas citri subsp. citri pathotype A in Taiwan. J. Plant Pathol. 99:289.
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jeong, K., Muñoz-Bodnar, A., Rojas, N. A., Poulin, L., Rodriguez-R, L. M., Gagnevin, L., et al. (2019). CRISPR elements provide a new framework for the genealogy of the citrus canker pathogen Xanthomonas citri pv. citri. BMC genomics 20:917. doi: 10.1186/s12864-019-6267-z
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Korotkov, K. V., Sandkvist, M., and Hol, W. G. (2012). The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10, 336–351. doi: 10.1038/nrmicro2762
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lai, Y.-R., Lin, C.-H., Chang, C.-P., Ni, H.-F., Tsai, W.-S., and Huang, C.-J. (2021). Distribution of copper resistance gene variants of Xanthomonas citri subsp. citri and Xanthomonas euvesicatoria pv. perforans. Plant Prot. Sci. 57, 206–216. doi: 10.17221/160/2020-PPS
Leduc, A., Traore, Y. N., Boyer, K., Magne, M., Grygiel, P., Juhasz, C. C., et al. (2015). Bridgehead invasion of a monomorphic plant pathogenic bacterium: Xanthomonas citri pv. citri, an emerging citrus pathogen in Mali and Burkina Faso. Environ. Microbiol. 17, 4429–4442. doi: 10.1111/1462-2920.12876
Lee, Y. A., Hendson, M., Panopoulos, N. J., and Schroth, M. N. (1994). Molecular cloning, chromosomal mapping, and sequence analysis of copper resistance genes from Xanthomonas campestris pv. juglandis: homology with small blue copper proteins and multicopper oxidase. J. Bacteriol. 176, 173–188. doi: 10.1128/jb.176.1.173-188.1994
Lima-Mendez, G., Oliveira Alvarenga, D., Ross, K., Hallet, B., Van Melderen, L., Varani, A. M., et al. (2020). Toxin-antitoxin gene pairs found in Tn3 family transposons appear to be an integral part of the transposition module. mBio 11:e00452-20. doi: 10.1128/mBio.00452-20
Loiseau, C., Hatte, V., Andrieu, C., Barlet, L., Cologne, A., De Oliveira, R., et al. (2018). PanGeneHome : a web interface to analyze microbial pangenomes. J. Bioinform. Comput. Syst. Biol. 1:108.
Lomsadze, A., Gemayel, K., Tang, S., and Borodovsky, M. (2018). Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 28, 1079–1089. doi: 10.1101/gr.230615.117
Marin, T. G. S., Galvanin, A. L., Lanza, F. E., and Behlau, F. (2019). Description of copper tolerant Xanthomonas citri subsp. citri and genotypic comparison with sensitive and resistant strains. Plant Pathol. 68, 1088–1098. doi: 10.1111/ppa.13026
Meric, G., Yahara, K., Mageiros, L., Pascoe, B., Maiden, M. C., Jolley, K. A., et al. (2014). A reference pan-genome approach to comparative bacterial genomics: identification of novel epidemiological markers in pathogenic Campylobacter. PLoS One 9:e92798. doi: 10.1371/journal.pone.0092798
Nurk, S., Walenz, B. P., Rhie, A., Vollger, M. R., Logsdon, G. A., Grothe, R., et al. (2020). HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305. doi: 10.1101/gr.263566.120
Patane, J. S. L., Martins, J. Jr., Rangel, L. T., Belasque, J., Digiampietri, L. A., Facincani, A. P., et al. (2019). Origin and diversification of Xanthomonas citri subsp. citri pathotypes revealed by inclusive phylogenomic, dating, and biogeographic analyses. BMC Genomics 20:700. doi: 10.1186/s12864-019-6007-4
Pena-Gonzalez, A., Rodriguez, R. L., Marston, C. K., Gee, J. E., Gulvik, C. A., Kolton, C. B., et al. (2018). Genomic characterization and copy number variation of Bacillus anthracis plasmids pXO1 and pXO2 in a historical collection of 412 strains. mSystems 3:e00065-18. doi: 10.1128/mSystems.00065-18
Proost, S., Fostier, J., De Witte, D., Dhoedt, B., Demeester, P., Van De Peer, Y., et al. (2012). i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 40:e11. doi: 10.1093/nar/gkr955
Pruvost, O., Magne, M., Boyer, K., Leduc, A., Tourterel, C., Drevet, C., et al. (2014). A MLVA genotyping scheme for global surveillance of the citrus pathogen Xanthomonas citri pv. citri suggests a worldwide geographical expansion of a single genetic lineage. PLoS One 9:e98129. doi: 10.1371/journal.pone.0098129
Qian, W., Jia, Y., Ren, S. X., He, Y. Q., Feng, J. X., Lu, L. F., et al. (2005). Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15, 757–767. doi: 10.1101/gr.3378705
Richard, D., Boyer, C., Verniere, C., Canteros, B. I., Lefeuvre, P., and Pruvost, O. (2017a). Complete genome sequences of six copper-resistant Xanthomonas citri pv. citri strains causing asiatic citrus canker, obtained using long-read technology. Genome Announc. 5:e00010-e17. doi: 10.1128/genomeA.00010-17
Richard, D., Pruvost, O., Balloux, F., Boyer, C., Rieux, A., and Lefeuvre, P. (2021). Time-calibrated genomic evolution of a monomorphic bacterium during its establishment as an endemic crop pathogen. Mol. Ecol. 30, 1823–1835. doi: 10.1111/mec.15770
Richard, D., Ravigne, V., Rieux, A., Facon, B., Boyer, C., Boyer, K., et al. (2017b). Adaptation of genetically monomorphic bacteria: evolution of copper resistance through multiple horizontal gene transfers of complex and versatile mobile genetic elements. Mol. Ecol. 26, 2131–2149. doi: 10.1111/mec.14007
Roach, R., Mann, R., Gambley, C., Shivas, R., Chapman, T., and Rodoni, B. (2020). Pathogenicity and copper tolerance in Australian Xanthomonas species associated with bacterial leaf spot. Crop Prot. 127:104923. doi: 10.1016/j.cropro.2019.104923
Rodriguez-Beltran, J., Delafuente, J., Leon-Sampedro, R., Maclean, R. C., and San Millan, A. (2021). Beyond horizontal gene transfer: the role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 19, 347–359. doi: 10.1038/s41579-020-00497-1
Roeschlin, R. A., Uviedo, F., Garcia, L., Molina, M. C., Favaro, M. A., Chiesa, M. A., et al. (2019). PthA4(AT), a 7.5-repeats transcription activator-like (TAL) effector from Xanthomonas citri ssp. citri, triggers citrus canker resistance. Mol. Plant Pathol. 20, 1394–1407. doi: 10.1111/mpp.12844
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Ruh, M., Briand, M., Bonneau, S., Jacques, M. A., and Chen, N. W. G. (2017). Xanthomonas adaptation to common bean is associated with horizontal transfers of genes encoding TAL effectors. BMC Genomics 18:670. doi: 10.1186/s12864-017-4087-6
Teper, D., and Wang, N. (2021). Consequences of adaptation of TAL effectors on host susceptibility to Xanthomonas. PLoS Genet. 17:e1009310. doi: 10.1371/journal.pgen.1009310
Timilsina, S., Pereira-Martin, J. A., Minsavage, G. V., Iruegas-Bocardo, F., Abrahamian, P., Potnis, N., et al. (2019). Multiple recombination events drive the current genetic structure of Xanthomonas perforans in Florida. Front. Microbiol. 10:448. doi: 10.3389/fmicb.2019.00448
Timilsina, S., Potnis, N., Newberry, E. A., Liyanapathiranage, P., Iruegas-Bocardo, F., White, F. F., et al. (2020). Xanthomonas diversity, virulence and plant-pathogen interactions. Nat. Rev. Microbiol. 18, 415–427. doi: 10.1038/s41579-020-0361-8
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Triplett, L. R., Hamilton, J. P., Buell, C. R., Tisserat, N. A., Verdier, V., Zink, F., et al. (2011). Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl. Environ. Microbiol. 77, 3930–3937. doi: 10.1128/AEM.00028-11
Turrini, P., Artuso, I., Tescari, M., Lugli, G. A., Frangipani, E., Ventura, M., et al. (2021). Draft genome sequence and secondary metabolite biosynthetic potential of the Lysobacter niastensis type strain DSM 18481. Microbiol. Resour. Announc. 10:e01296-20. doi: 10.1128/MRA.01296-20
Vinatzer, B. A., Monteil, C. L., and Clarke, C. R. (2014). Harnessing population genomics to understand how bacterial pathogens emerge, adapt to crop hosts, and disseminate. Annu. Rev. Phytopathol. 52, 19–43. doi: 10.1146/annurev-phyto-102313-045907
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, J. R., Holt, J., Mcmillan, L., and Jones, C. D. (2018). FMLRC: hybrid long read error correction using an FM-index. BMC bioinformatics 19:50. doi: 10.1186/s12859-018-2051-3
Wang, P., Meng, F., Moore, B. M., and Shiu, S. H. (2021). Impact of short-read sequencing on the misassembly of a plant genome. BMC Genomics 22:99. doi: 10.1186/s12864-021-07397-5
Wu, Y. F., Hsu, S. T., and Tzeng, K. C. (1995). Association of plasmid with copper resistance in strains of Xanthomonas campestris pv. vesicatoria from Taiwan. Plant Prot. Bull. 37, 209–218.
Yen, M. R., Lin, N. T., Hung, C. H., Choy, K. T., Weng, S. F., and Tseng, Y. H. (2002). oriC region and replication termination site, dif, of the Xanthomonas campestris pv. campestris 17 chromosome. Appl. Environ. Microbiol. 68, 2924–2933. doi: 10.1128/AEM.68.6.2924-2933.2002
Zhang, Y., Jalan, N., Zhou, X., Goss, E., Jones, J. B., Setubal, J. C., et al. (2015). Positive selection is the main driving force for evolution of citrus canker-causing Xanthomonas. ISME J. 9, 2128–2138. doi: 10.1038/ismej.2015.15
Keywords: Xanthomonas citri subsp. citri, copper resistance, whole-genome sequence, TALE, plasmid, plasticity
Citation: Huang C-J, Wu T-L, Zheng P-X, Ou J-Y, Ni H-F and Lin Y-C (2021) Comparative Genomic Analysis Uncovered Evolution of Pathogenicity Factors, Horizontal Gene Transfer Events, and Heavy Metal Resistance Traits in Citrus Canker Bacterium Xanthomonas citri subsp. citri. Front. Microbiol. 12:731711. doi: 10.3389/fmicb.2021.731711
Received: 28 June 2021; Accepted: 18 August 2021;
Published: 07 September 2021.
Edited by:
Jean-Yves Matroule, University of Namur, BelgiumReviewed by:
Yong-Qiang He, Guangxi University, ChinaDawei Xin, Northeast Agricultural University, China
Copyright © 2021 Huang, Wu, Zheng, Ou, Ni and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chien-Jui Huang, Y2hpZW5qdWkuaHVhbmdAbWFpbC5uY3l1LmVkdS50dw==; Yao-Cheng Lin, eWFsaW5Ac2luaWNhLmVkdS50dw==