 Daniel Santana de Carvalho1,2†
Daniel Santana de Carvalho1,2† Ana Paula Trovatti Uetanabaro1,3†
Ana Paula Trovatti Uetanabaro1,3† Rodrigo Bentes Kato1,2
Rodrigo Bentes Kato1,2 Flávia Figueira Aburjaile2
Flávia Figueira Aburjaile2 Arun Kumar Jaiswal2
Arun Kumar Jaiswal2 Rodrigo Profeta2
Rodrigo Profeta2 Rodrigo Dias De Oliveira Carvalho2
Rodrigo Dias De Oliveira Carvalho2 Sandeep Tiwar2
Sandeep Tiwar2 Anne Cybelle Pinto Gomide2
Anne Cybelle Pinto Gomide2 Eduardo Almeida Costa4
Eduardo Almeida Costa4 Olga Kukharenko5
Olga Kukharenko5 Iryna Orlovska5
Iryna Orlovska5 Olga Podolich5
Olga Podolich5 Oleg Reva6
Oleg Reva6 Pablo Ivan P. Ramos7
Pablo Ivan P. Ramos7 Vasco Ariston De Carvalho Azevedo2
Vasco Ariston De Carvalho Azevedo2 Bertram Brenig8
Bertram Brenig8 Bruno Silva Andrade9
Bruno Silva Andrade9 Jean-Pierre P. de Vera10
Jean-Pierre P. de Vera10 Natalia O. Kozyrovska5
Natalia O. Kozyrovska5 Debmalya Barh2,11*
Debmalya Barh2,11* Aristóteles Góes-Neto1*
Aristóteles Góes-Neto1*
- 1Laboratory of Molecular and Computational Biology of Fungi, Department of Microbiology, Department of Genetics, Ecology and Evolution, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
- 2Laboratory of Cellular and Molecular Genetics, Department of Genetics, Ecology and Evolution, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 3Postgraduate Program in Biology and Biotechnology of Microorganisms, Department of Biological Sciences, State University of Santa Cruz, Ilhéus, Brazil
- 4Computational Biology and Biotechnological Information Management Center (NBCGIB), State University of Santa Cruz, Ilhéus, Brazil
- 5Institute of Molecular Biology and Genetics of NASU, Kyiv, Ukraine
- 6Department of Biochemistry, Genetics and Microbiology, Centre for Bioinformatics and Computational Biology, University of Pretoria, Pretoria, South Africa
- 7Center for Data and Knowledge Integration for Health (CIDACS), Institute Gonçalo Moniz, Oswaldo Cruz Foundation (FIOCRUZ-Bahia), Salvador, Brazil
- 8Institute of Veterinary Medicine, Burckhardtweg, University of Göttingen, Göttingen, Germany
- 9Laboratory of Bioinformatics and Computational Chemistry, Department of Biological Sciences, State University of Southwest Bahia (UESB), Jequié, Brazil
- 10German Aerospace Center (DLR) Berlin, Institute of Planetary Research, Planetary Laboratories, Astrobiological Laboratories, Berlin, Germany
- 11Centre for Genomics and Applied Gene Technology, Institute of Integrative Omics and Applied Biotechnology, Purba Medinipur, India
Komagataeibacter is the dominant taxon and cellulose-producing bacteria in the Kombucha Microbial Community (KMC). This is the first study to isolate the K. oboediens genome from a reactivated space-exposed KMC sample and comprehensively characterize it. The space-exposed genome was compared with the Earth-based reference genome to understand the genome stability of K. oboediens under extraterrestrial conditions during a long time. Our results suggest that the genomes of K. oboediens IMBG180 (ground sample) and K. oboediens IMBG185 (space-exposed) are remarkably similar in topology, genomic islands, transposases, prion-like proteins, and number of plasmids and CRISPR-Cas cassettes. Nonetheless, there was a difference in the length of plasmids and the location of cas genes. A small difference was observed in the number of protein coding genes. Despite these differences, they do not affect any genetic metabolic profile of the cellulose synthesis, nitrogen-fixation, hopanoid lipids biosynthesis, and stress-related pathways. Minor changes are only observed in central carbohydrate and energy metabolism pathways gene numbers or sequence completeness. Altogether, these findings suggest that K. oboediens maintains its genome stability and functionality in KMC exposed to the space environment most probably due to the protective role of the KMC biofilm. Furthermore, due to its unaffected metabolic pathways, this bacterial species may also retain some promising potential for space applications.
Importance
Nowadays, there is an increased effort to study the possibility of life on Mars and the ability of organisms to survive the Mars environment to assess the possibility of colonizing the planet. Recent studies have shown that bacterial cellulose is a strong material that has a variety of uses and applications, such as medicine, tissue regeneration, fabrics, electronics, and biotechnology in general. In this study we were able to expose a cellulose producing bacteria (Komagataeibacter oboediens) to the space/Mars-like environment, and our results show that K. oboediens was able to survive being exposed to the space environment for 18 months and that its genome remains mainly conserved after spaceflight. This suggests that K. oboediens is a strong candidate for cellulose production in Mars colonies’ industry.
Introduction
One of the most intriguing questions is the capability of an organism to survive and resist the extremely harsh extraterrestrial outer space conditions, such as cosmic radiation, microgravity, temperature extremes, substrates, atmospheric pressure, and vacuum (Horneck et al., 2010; de Vera et al., 2014; Schulze-Makuch et al., 2015). In order to find an answer, the European Space Agency (ESA) conducted a series of multi-user space biology exposure experiments, using the EXPOSE-R2 platform installed outside the International Space Station (ISS) that provides an environment for sets of long-term experiments (1.5 years). One of the biological samples elected to be sent to the ISS was dehydrated kombucha biofilm in the project BIOMEX (BIOlogy and Mars EXperiment) (de Vera et al., 2019; de Vera, 2020).
Kombucha multimicrobial community (KMC) is a natural assemblage of bacteria and yeasts living in metabolic cooperation and streaky in a cellulose-based pellicle film (Pothakos et al., 2016; May et al., 2019; Barbosa et al., 2021). In astrobiology, KMC is used as a model for researching a natural probiotic drink for astronauts during space travel (Kozyrovska et al., 2012, 2021). Recent BIOMEX results revealed that key KMC community members were capable of surviving under space and Mars-like conditions (Podolich et al., 2019) and that the bacterial cellulose (BC) produced by KMC retained its robustness under the impact of Mars-like stressors (Orlovska et al., 2021). Our post-flight metagenomic studies proved that the genus Komagataeibacter was the dominant bacterial taxon both in the ground-based reference KMC ecotype and reactivated cultured KMC samples exposed to Mars-like conditions outside the ISS (Góes-Neto et al., 2021; Lee et al., 2021). Moreover, we observed that the key KMC strain K. oboediens, which survives under such extraterrestrial stresses, retains its cellulose synthesizing bcs operon genes intact (Góes-Neto et al., 2021). This observation led us to analyze the entire K. oboediens genome from space-exposed KMC samples to understand its genome stability under extra-terrestrial conditions. Little is known about the K. oboediens genome, and for this reason, herein, we isolated, sequenced, and characterized the K. oboediens IMBG180 (from ground based KMC culture) and K. oboediens IMBG185 (from space exposed KMC samples) and compared their genomes to understand their genomic components. In this study, we addressed the question if there are significant differences between K. oboediens strains under post-flight space/Mars-like and standard terrestrial conditions by: (i) identifying and analyzing all the shared and exclusive genes, (ii) evaluating Single Nucleotide Variations (SNVs) in protein-coding genes, and (iii) performing global metabolic reconstruction of the two strains.
Materials and Methods
Exposure Conditions, Reactivation of Space-Returned Kombucha Microbial Community Samples, and Bacterial Strains
All the methodological workflow (from the original KMC consortium until the genome sequencing of isolated strains) is graphically depicted as Figure 1. The original kombucha microbial culture was obtained from the collection of microorganisms of the Institute of Molecular Biology and Genetics (Kyiv, Ukraine). It was maintained in filter-sterilized black tea (Lipton, 1.2%, w/v) with white sugar (3%, w/v) (BTS) at 28°C, prior to preparation as dry organo-mineral KMC pellicle fragments samples previously described in Podolich et al. (2019). The KMC samples were placed in a three-level tray hosting four samples (independent sampling units or biological replicates) on either level (top, middle, and bottom) and mounted on an EXPOSE-2 platform outside the ISS as described in de Vera et al. (2019). These samples were maintained for 18 months (1.5 years) within the tray under a simulated Mars atmosphere (95.55% CO2, 2.70% N2, 1.60% Ar, 0.15% O2, ∼370 ppm H2O), under pressure of 980 Pa. No UV filters were used in the kombucha experiments, and space varied between 4.5 × 106 and 8.4 × 106 kJ/m2 (de Vera et al., 2019). A set of analogous samples were kept in the laboratory at room temperature in darkness. Returned and ground reference KMC samples were cultivated for reactivation in filter-sterilized sugared (7%) black tea infusions at 28°C until a cellulose-based pellicle appeared, meaning that bacteria from the genus Komagataeibacter were active. In reference samples, pellicles appeared in 7 days, and returned samples formed pellicles after 60 days of a culturing. After KMC sample reactivation, K. oboediens IMBG185 was isolated from the exposed top-level KMC samples and opened to UV irradiation, and K. oboediens IMBG180 from initial reference KMC (iKMC) was kept in the lab during the spaceflight experiment, desiccated and stored at room temperature. Both samples were sequenced together to avoid variation due to different sequencing runs. Both strains were cultured in the HS medium (Hestrin and Schramm, 1954) at 28°C for 3 days before extraction of genomic DNA.
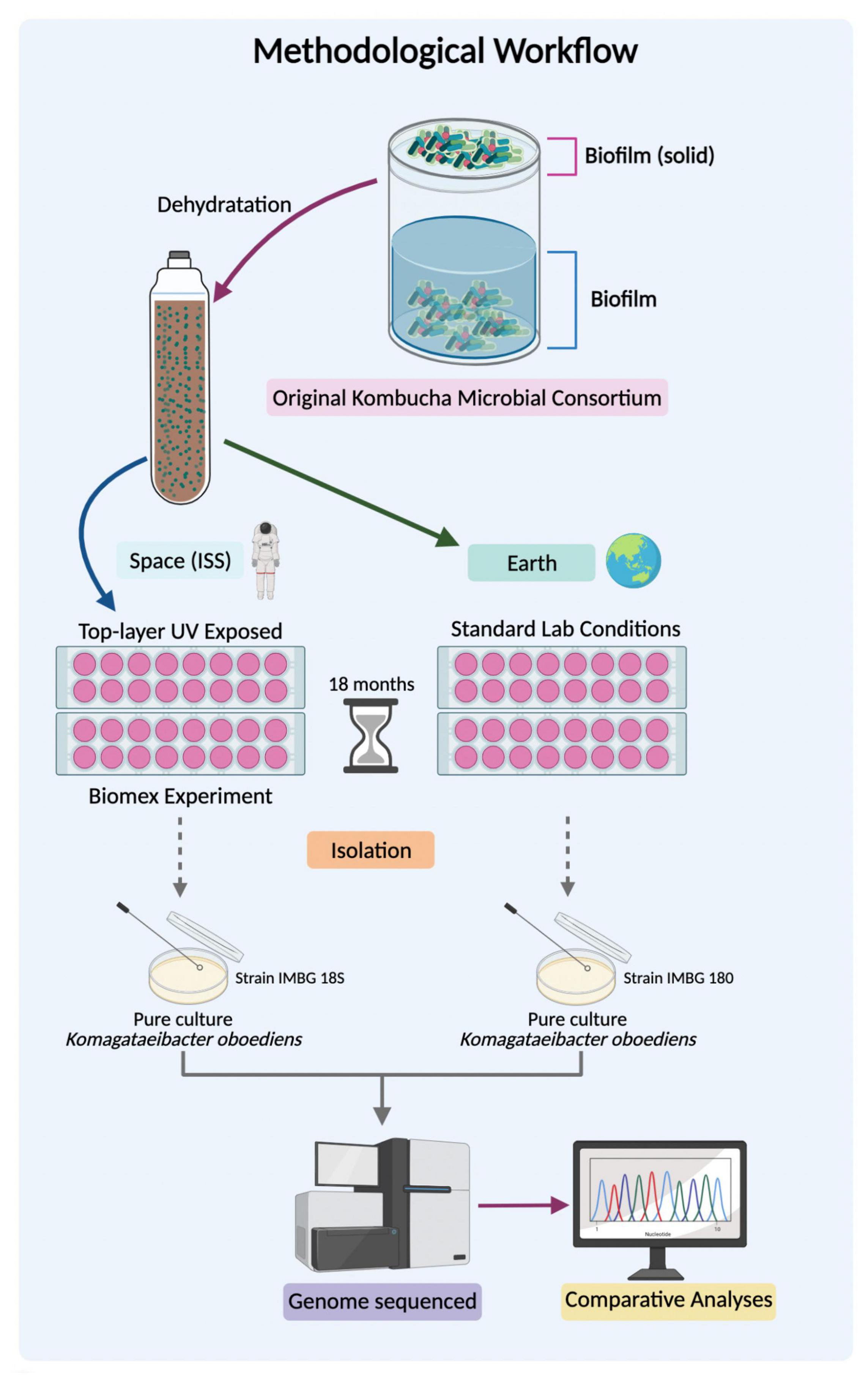
Figure 1. Methodological workflow.
Genomic DNA Extraction, Library Construction, and Whole-Genome Sequencing
Total DNA from the planktonic cells of KMC was isolated by using the innuSPEED Bacteria/Fungi DNA isolation kit (Analytik Jena AG, Germany) during the stage of biofilm formation (7th day). The nucleic acids were quantified and qualified by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States). A 450 bp library was prepared from genomic DNA with the NEBNext Fast DNA Fragmentation and Library Preparation Kit (New England Biolabs, Ipswich, MA, United States) following the manufacturer’s instructions. A library of 450 bp was prepared with the NEBNext Fast DNA Fragmentation and Library Preparation Kit (New England Biolabs). Library quality was checked with Agilent 2100 Bioanalyzer. The genomes were sequenced using Illumina HiSeq 2500 (Illumina, San Diego, CA, United States) to obtain paired-end reads. The obtained final genome coverage of IMBG185 (715.0x) and IMBG180 (1964.0x).
Genome Assembly
De novo genome assemblies of K. oboediens strains were carried out with a customized pipeline (Figure 2). Using this pipeline, the read quality was checked using FastQC v0.11.81, followed by seven different assembly strategies, executed with distinct software and parameters. The first assembly was carried out by Edena v3.131028 (Hernandez et al., 2008), in which the overlap-layout-consensus approach was employed to assemble the raw paired short reads solely exploiting the overlaps between them. The second assembly was executed using SPAdes v3.13 (Bankevich et al., 2012), which breaks the raw paired short reads into k-mers (substrings of size k) and applies De Bruijn graphs, which were generated to perform genome assemblies. The k-mers used for this second assembly were the best three predicted by KmerStream version 1.1 (Melsted and Halldórsson, 2014). The third assembly is a hybrid between Edena and SPAdes, where the contigs produced in the first assembly were employed as trusted contigs in a new assembly performed by SPAdes, again using the raw paired short reads and the three best k-mers detected by KmerStream. In the fourth assembly, SPAdes was executed again after quality control of the short reads performed by AdapterRemoval v2 (Schubert et al., 2016) and k-mer prediction using KmerStream. AdapterRemoval was employed to collapse paired short reads that overlap by at least 11 base pairs, remove adapters, and trim base pairs from the 3′ end with quality lower than Phred 20 and, ultimately, exclude short reads that were left with less than 31 nucleotides after trimming. Trimmed paired reads were passed as parameters to SPAdes in conjunction with the singleton and collapsed short reads. The singleton and collapsed short reads were also used in the fifth assembly, but this time, the raw paired reads were used as parameters to SPAdes besides the contigs produced by Edena, which were used as trusted contigs. The k-mers used were the top 3 predicted by KmerStream. In the sixth assembly, which was also performed by SPAdes, we employed the singleton, collapsed, and paired short reads enhanced by AdapterRemoval along with the contigs assembled by Edena, which were used as trusted contigs. The top 3 k-mers predicted by KmerStream were also used for the last assembly. The seventh assembly was performed by Unicycler v.0.4.5 and Spades v.3.9.1. Afterward, we ran QUAST (Gurevich et al., 2013) to evaluate these seven distinct assemblies for each genome, considering the N50 score, lesser numbers of contigs, and the expected genome size. Finally, the genome obtained from Unicycler v. 0.4.5 was considered as the best assembled for each of the two genomes (IMBG180 and IMBG185).
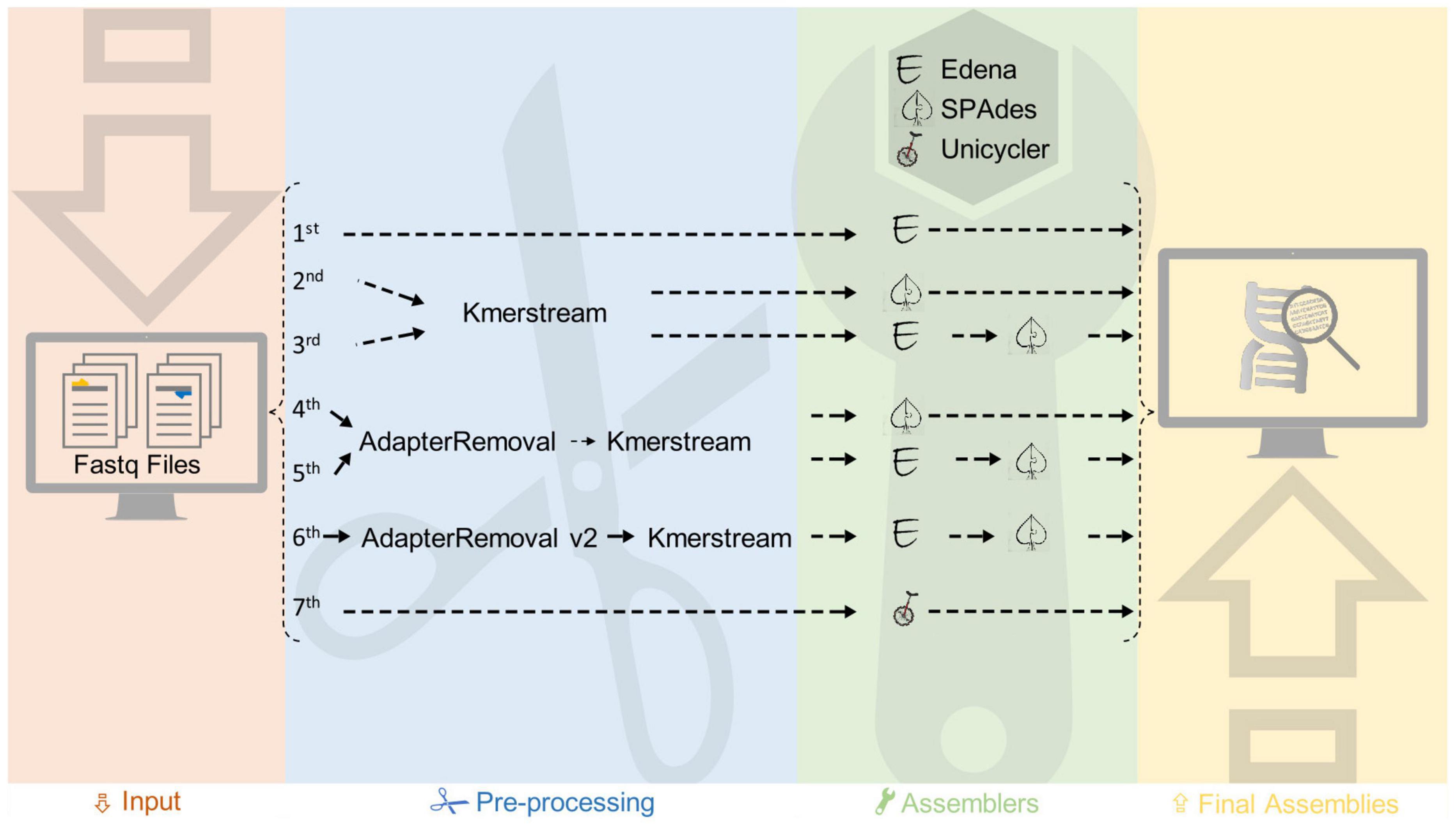
Figure 2. Steps of the customized pipeline used for assembly of ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
Genome Annotation
The genome sequences were annotated using Prokka v. 1.14.5 prokaryotic genome annotation (Seemann, 2014), as well as using BUSCO v. 4.0.1 to predict the completeness of single-copy orthologs (Seppey et al., 2019), and MAUVE v. 2.4.0 (Darling et al., 2004) to verify the synteny of the genes between the two genomes. The predicted contigs were also analyzed with GO FEAT (Araujo et al., 2018), an on-line platform for functional annotation and enrichment of genomic data integrated with UNIPROT2, INTERPRO3, PFAM4, NCBI5, and KEGG6 databases.
The draft genomes of K. oboediens strains, both the space/Mars-like conditions (IMBG185) and the reference ground (IMBG180), were deposited in NCBI with the following accession numbers: K. oboediens IMBG185 (BioProject: PRJNA632016, BioSample: SAMN14942470) and K. oboediens IMBG180 (BioProject: PRJNA633374, BioSample: SAMN14942824, Accession: JABLUV000000000), respectively.
Taxonomic Identification, Phylogenetic (Small-Scale), and Phylogenomic (Large-Scale) Analyses
For species identification, a multilocus sequence analysis (MLSA), a small-scale phylogenetic analysis, was performed based on concatenated partial sequences of the housekeeping genes: 16S rRNA, dnaK, rpoB, and groEL, as previously described (Liu et al., 2018). These gene sequences were extracted from the strains IMBG185 and IMBG180 of deposited genomes and other 16 Komagataeibacter species (Table 1) from NCBI and aligned with MAFFT v7.310 (Katoh and Standley, 2013). Acetobacter aceti was used as the outgroup. The phylogeny was reconstructed using the maximum likelihood method implemented in IQ-TREE v1.6.12 (Nguyen et al., 2015), and the robustness of the branches was evaluated by bootstrap analysis based on 1000 iterations. For large-scale phylogenomic analysis, MUMmer (NUCmer) was used to align the genome sequences, and Pyani tool version 0.2.x was used to calculate the percentage nucleotide identity for the analysis of average nucleotide identity (ANI) and to perform the biplot dendrogram associated to a color matrix.
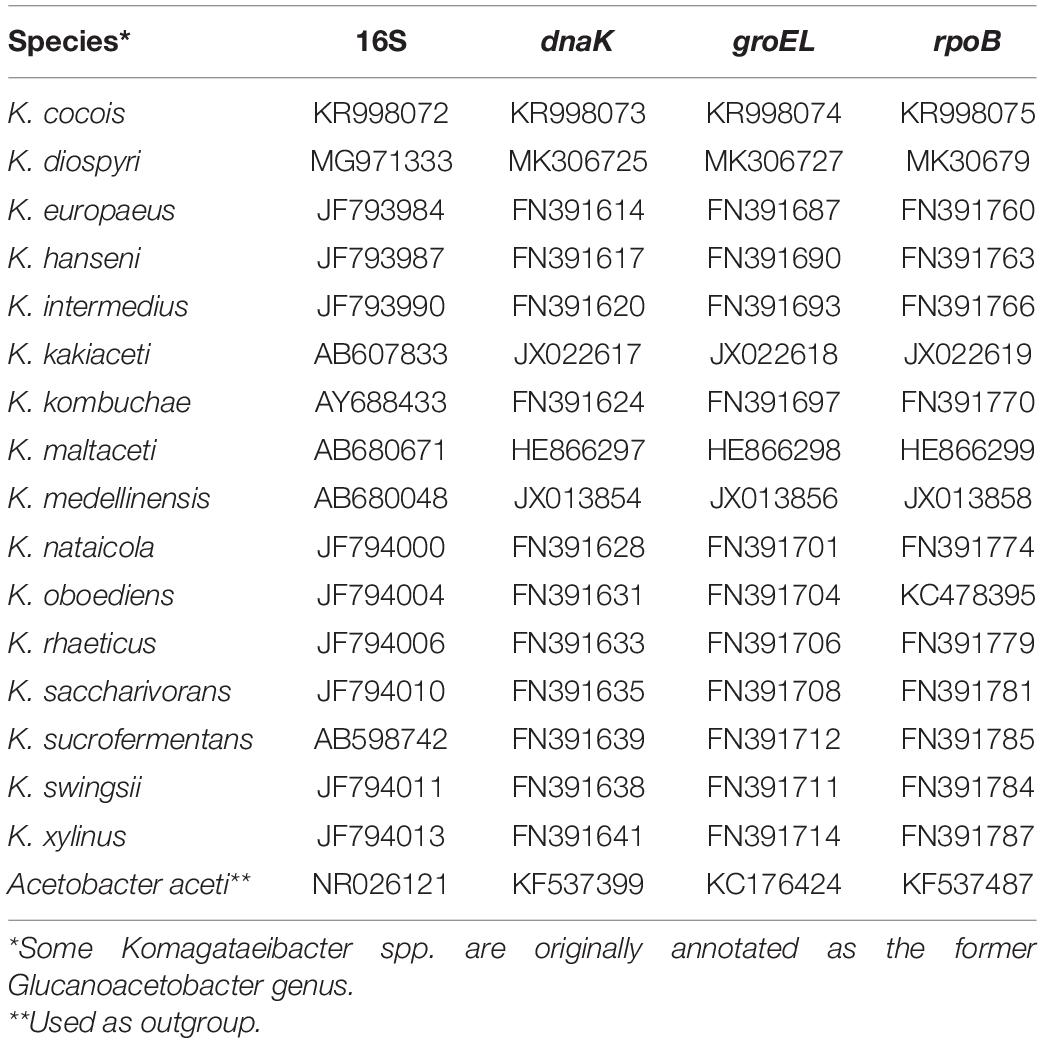
Table 1. Komagataeibacter spp. gene sequences from GenBank (NCBI) used for phylogeny reconstruction of ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
Prediction of Genomic Islands, Transposases, and Prion Aggregating Regions
The prediction of genomic islands and transposases in the genomes were performed using the Genomic Islands Prediction Software GIPSy (Soares et al., 2016), using K. saccharivorans strain CV1 as a reference genome. This tool provides information about the genes present in these islands (symbiosis, pathogenicity and virulence, antibiotic resistance, and metabolic) and their respective location in the genomes. Only genomic islands with normal and strong classifications were considered. In addition, GIPSy also detects transposase genes based on HMMER3 (Eddy, 2011) with E-value ≤ 1 × 10–4. The online tool PAPA (Prion aggregation prediction algorithm) (Toombs et al., 2012) was used to predict candidate prion aggregating regions in the sequences annotated as “Transcription termination factor Rho” in both genomes.
Prophage and Plasmid Characterization
Prophage Hunter tool (Song et al., 2019) was used for prediction and classification of prophages. Circular contigs with high depth coverage, predicted by Unicycler (Wick et al., 2017), were considered plasmids. Non-circular contigs were assessed using a combination of three different tools: (i) Mob-recon (reference-based approach, version 2.1.0) (Robertson and Nash, 2018), (ii) HyAsP (hybrid approach, version 1.0.0) (Müller and Chauve, 2019), and (iii) Gplas (version 0.5.0) (Arredondo-Alonso et al., 2020). Non-circular contigs were considered plasmids only when predicted by the majority of these three tools.
Prediction of CRISPR-Cas
The prediction of Cas proteins and classification of CRISPR-Cas system subtype in both genomes were performed via CRISPRCas Identifier (Padilha et al., 2020).
Evaluation and Integration of the Metabolic Repertoire of Komagataeibacter oboediens
The KAAS web server (Moriya et al., 2007) was used to predict KEGG Orthology (KO) identifiers for both the sequenced strains. The representative set of organisms was selected as the default for prokaryotes, in addition to the closely related species Gluconacetobacter diazotrophicus (three-letter code: gdi), Acetobacter pasteurianus (apt), and Granulibacter bethesdensis (gbe). The bidirectional best-hit approach was used as an assignment method. In order to complement KEGG analysis, the MetaCyc (Caspi et al., 2020) ontology was also employed to perform metabolic prediction. For this analysis, the replicons and associated annotation for both genomes were input to Pathway Tools (Karp et al., 2002), which creates a Pathway/Genome Database (PGDB) containing the predicted metabolic pathways for the organisms. Metabolic reconstruction included determining gene-protein-reaction associations, which are primarily based on the corresponding enzyme commission (EC) numbers. The pathway score yielded by Pathway Tools was used as a proxy for the presence or absence of a given pathway.
Pathway Tools was also employed to thoroughly study the key cellulose synthesis bcs operon, which was mined in K. oboediens using the bcs operon identified in K. xylinus as template (PMID: 31983038; PMID: 30761107). BLAST analyses against the UniProt database (PMID: 15215342) were also performed to search for orthologs related to this metabolic function.
Stress-related genes and pathways were identified by manual literature searches as well as by the retrieval of curated proteins deposited in UniProtKB/Swiss-Prot, which were associated to the “Stress response” keyword (KW-0346) and belonged to either Archaea or Bacteria domains. In order to reduce the redundancy of this protein set (originally composed of 7,724 sequences), the proteins were clustered at 90% sequence identity using UniRef90 clusters, yielding a final set of 2,687 stress-related sequences. Subsequently, for retrieving orthologs in K. oboediens, BLASTp was performed using each predicted K. oboediens proteome set as a query against the reference stress-related proteins, followed by manual curation of the alignments.
A principal component analysis (PCA) was also conducted to assess the metabolic profiles of K. oboediens and related bacteria. In order to perform this task, the enzymatic content (as predicted by KEGG) was used as input to the prcomp function in R7, which employs the singular value decomposition method to extract principal components. The compared bacteria (and corresponding labels, when applicable) were Acetobacter aceti (Aac), A. cerevisae (Ace), A. malorum (Ama), A. pasteurianus (Apa), A. pomorum (Apo), Clostridium kluyveri, Escherichia coli, E. albertii, E. fergusonii, Komagataeibacter cocoi (Kco), K. diospyri (Kdi), K. europaeus (Keu), K. hansenii (Kha), K. intermedius (Kin), K. kakiaceti (Kka), K. maltaceti (Kma), K. medellinensis (Kme), K. nataicola (Kna), K. oboediens LMG 18849 (Kob 18849), K. rhaeticus (Krh), K. saccharivorans (Ksa), K. sucrofermentais (Ksu), K. swingsii (Ksw), K. kylinus (Kxy), Salmonella bongori, S. enterica, Shigella boydii, Sh. flexneri, and Sh. sonnei.
Single Nucleotide Variation Analysis
A comparison between coding sequences (CDS), as well as corresponding plasmid CDS, of K. oboediens in space and terrestrial conditions was performed to search for SNV between their genomes and their plasmids. An in-house Python script (see Availability of data and materials) was used to find the corresponding genes in both genomes based on the following criteria: (i) both genes had to present exactly the same 60 first nucleotides and (ii) have the same gene length. These criteria were applied assuming that the space stress would not have caused severe mutations that would make these criteria to fail the analysis, as such stress would possibly be lethal to the bacteria.
Furthermore, a list of corresponding pairs of genes present in both genomes was generated, as well as a list of genes without a match in the other genome. The list of genome-specific, putative exclusive genes was then further manually curated. The lists generated are available in Supplementary Material 1.
Protein Modeling and Molecular Dynamics
In order to evaluate the K. oboediens pyruvate phosphate dikinase (ppdk) single mutation amino acid, we performed ppdk homology modeling and molecular dynamics for discovering the influence of the amino acid SER287 on the ATP/ADP site. Firstly, the K. oboediens PPDK amino acid FASTA sequence was submitted to The Swiss Model Workspace (Arnold et al., 2006) for automated modeling using the PDB template 5LU48, because it is complexed with the ADP molecule, as well as Mg++ cofactor. Additionally, we have considered the template resolution below 3.0 Å, identity, and sequence coverage above 50%. After QMEAN validation, we submitted both the ppdk model and its template to a structural alignment using the UCSF Chimera (Pettersen et al., 2004) program to identify the catalytic and other important binding site positions, as well as to verify if the ppdk mutation could affect the enzyme function. Considering the ATP/ADP binding site’s high identity between both the ppdk model and its 5LU4 template, and, since ppdk SER287 was aligned with 5LU4 GLY285, we decided to generate a single mutation in 5LU4 (GLY285 to SER285) in order to use this crystallographic structure complexed with ADP and Mg++ ligands and performed a molecular dynamics simulation for describing how this mutation would affect the ATP/ADP binding site, which would be important for enzyme activation and its transition state (Minges et al., 2017b). In this case, using a homologous crystallographic structure avoids a possible bias, which could happen only if a modeled protein docked with its respective ATP/ADP ligand was used.
For the Molecular Dynamic experiments, we used the GROMACS 2020 program. The mutated complex 5LU4, bound to ADP and Mg++, was subjected to a stability MD in water solvent for 50 ns. Subsequently, we followed the GROMACS tutorial for protein–ligand complexes (Lemkul, 2019) with adaptations, as well as with the program manual (Berendsen et al., 1995) for all the simulation steps and analyses. The MD graphs were generated using the Grace 5.1.25 program9, and the output data were used for creating a video of the simulation using the VMD program (Humphrey et al., 1996).
Results
IMBG180 and IMBG185 Belong to Komagataeibacter oboediens
A total of two distinct and highly supported clades (bootstrap values: 99–100%) were retrieved in multigene phylogenetic analysis of the genus Komagataeibacter: one of them (clade 1), including the well-studied and representative species, K. hansenii, and the other one (clade 2) comprising another much studied and representative species, K. xylinus. Clade 2, in turn, encompassed two differently and highly supported clades: clade 3, with 76% bootstrap, including, besides K. xylinus and other related species, K. medellinensis and K. saccharivorans, and clade 4, encompassing K. oboediens, K. intermedius and related species (Figure 3A). Moreover, the analysis clearly revealed that both strains (IMBG180 and IMBG185) are very closely related to K. oboediens (Figure 3A), thus confirming that they belong to the same species K. oboediens. Furthermore, this finding of the small-scale phylogenetic analysis (MLSA) was fully corroborated with large-scale phylogenomic analysis based on global genomic similarity (ANI), showing that both strains (IMBG180 and IMBG185) and all the K. oboediens available genomes are the most identical (the large red cluster) amongst all the other genomes of Komagataeibacter species (Figure 3B). Moreover, ANI (average nucleotide identity) index between K. oboediens IMBG180 and IMBG185 were 99.9%, suggesting that they are almost identical.
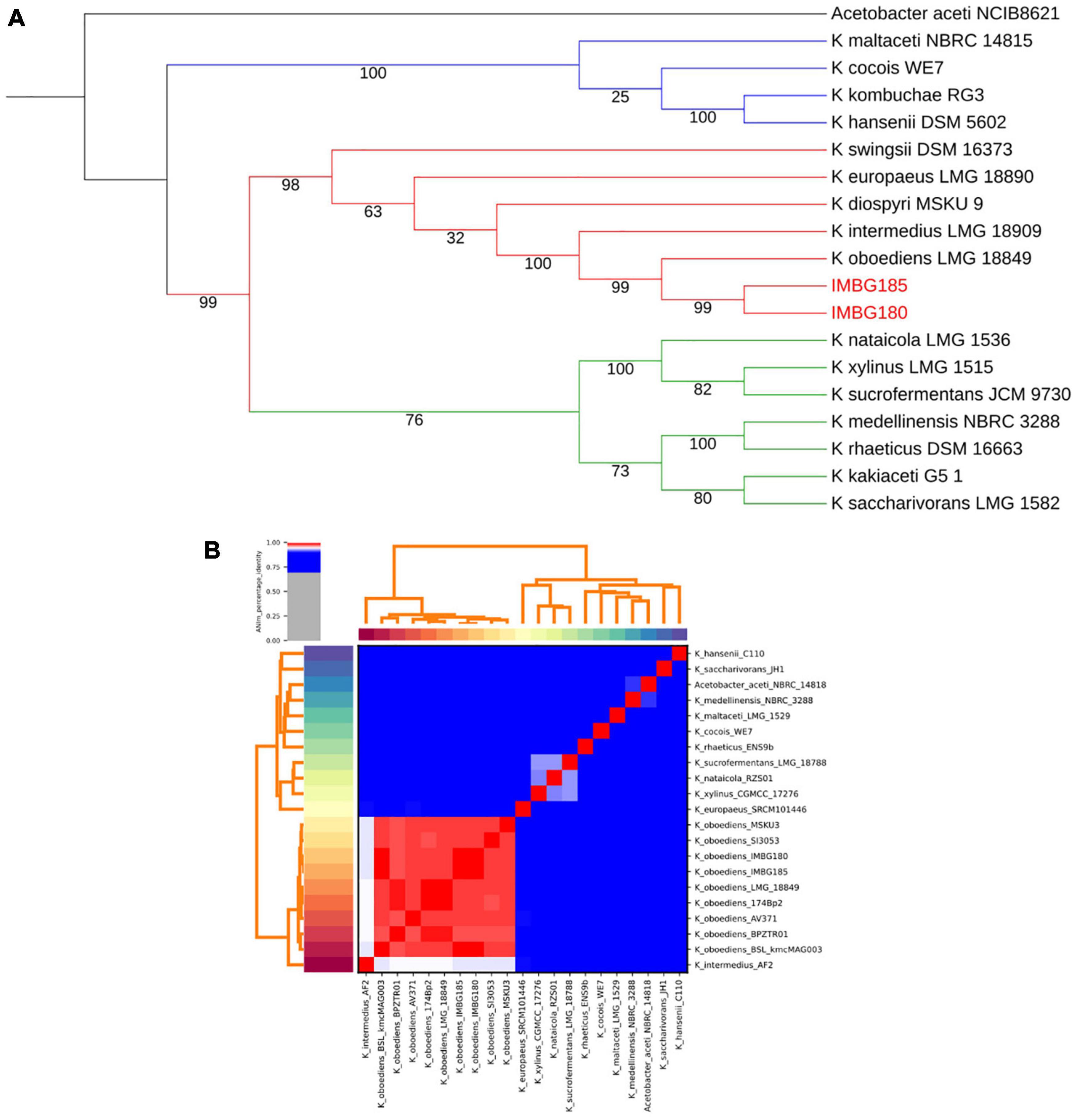
Figure 3. (A) Maximum likelihood phylogenetic tree of IMBG185 and IMBG180 strains and Komagataeibacter species from GenBank (NCBI). The tree represents concatenated partial sequences of the housekeeping genes, 16S rRNA, dnaK, rpoB, and groEL. Bootstrap support values (1,000 replicates) are indicated below the branches. Clade 1 (blue); Clade 2 (red + green); Clade 3 (green); Clade 4 (red). (B) Large-scale (phylogenomics) global similarity analysis (ANI) of IMBG185 and IMBG180 strains and available Komagataeibacter species from GenBank (NCBI). The large red cluster contains all the genomes of the most globally similar Komagataeibacter species, which includes IMBG185 and IMBG180 strains and all the available Komagataeibacter oboediens.
Notable Similarities Between Komagataeibacter oboediens IMBG180 and IMBG185 Genomes
A remarkable similarity between IMBG185 (space/Mars-like conditions) and IMBG180 (ground reference) strains of sequenced genomes (Figure 4 and Supplementary Material 2) was noticed. Very small differences in size (3,560,605 and 3,562,447 bp), total gene prediction (3,282 and 3,275), total number of protein-coding genes (3,230 and 3,222), and other features between samples IMBG185 and IMBG180, respectively, may be because they are not of the same strain or because of minor changes in IMBG185 due to the stressful conditions it was exposed to. Besides, both strains displayed exactly the same G + C content (61.85%) and the same number of rRNA genes (3) (Table 2).
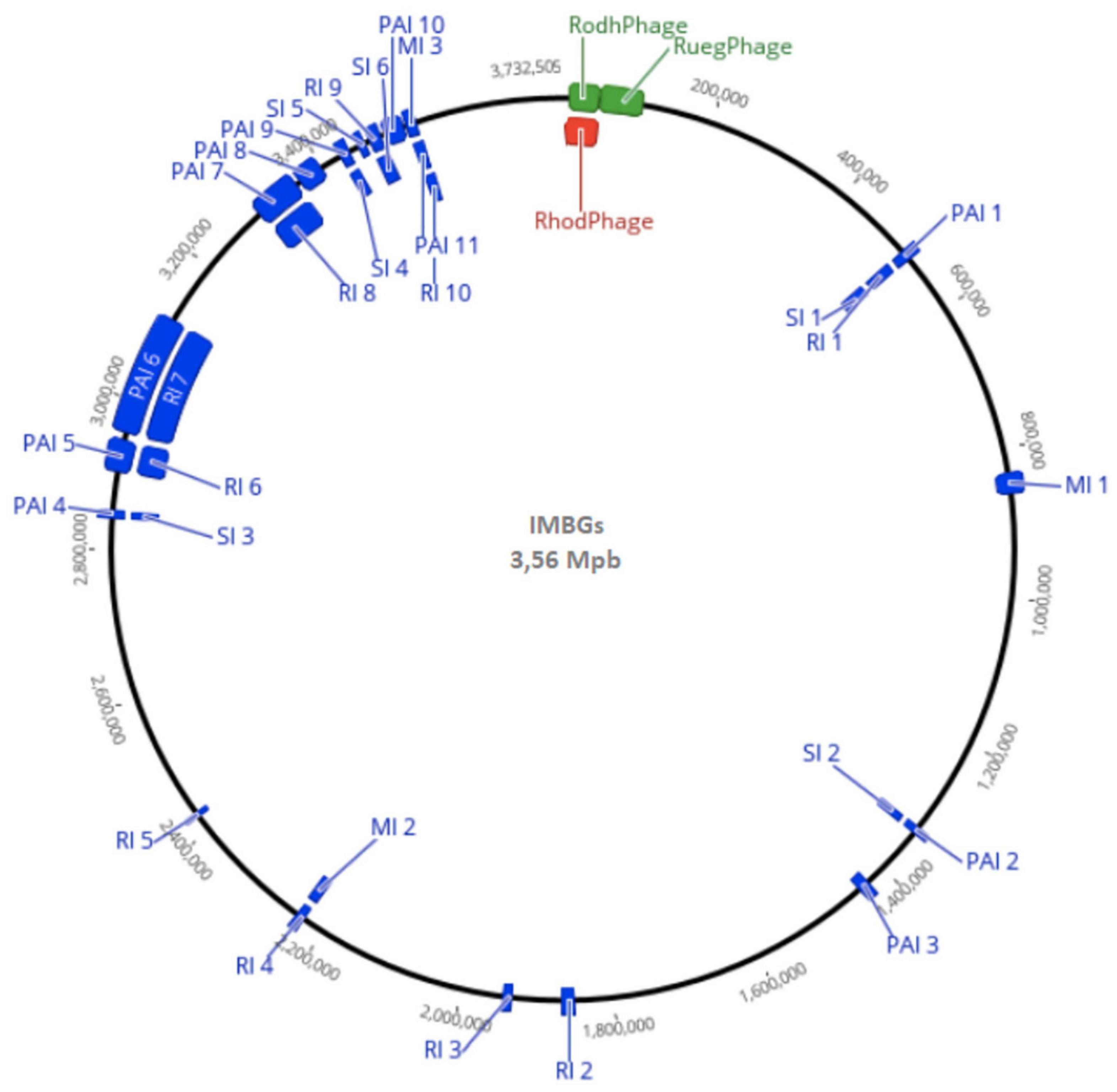
Figure 4. General whole genome representation of IMBGs (IMBG180 – ground reference, and IMBG185 – space/Mars-like conditions), with Genomic Islands highlighted in blue (MI: Metabolic, PI: Pathogenic Islands, RI: Resistance Islands, and SI: Symbiotic Islands) and active prophages highlighted in green (IMBG180) and red (IMBG185). RhodPhage: Rhodovulum phage RS1; RuegPhage: Ruegeria phage DSS3-P1.
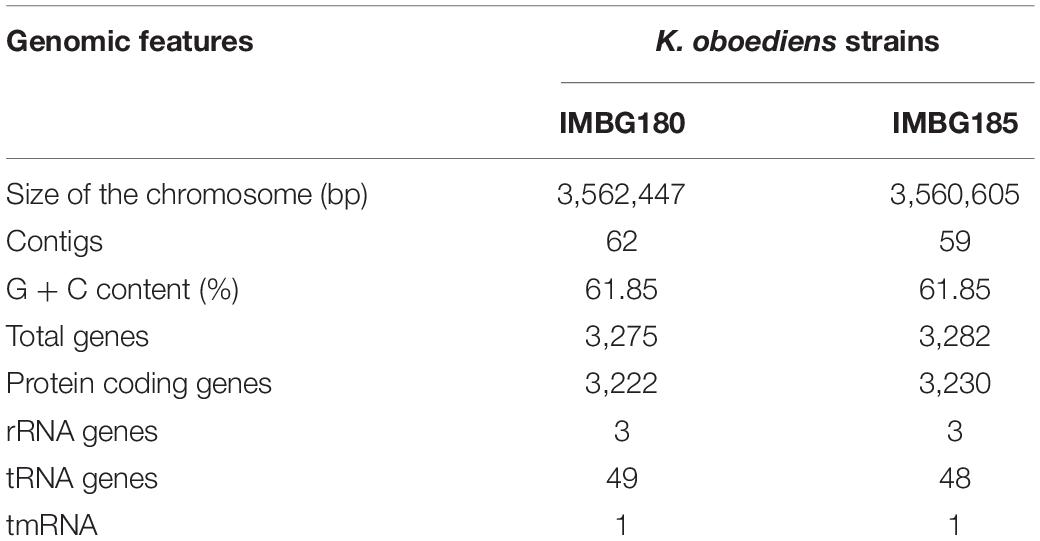
Table 2. General features of K. oboediens strains of ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
Both Genomes Have 427 Orthologous Genes
In the genome completeness analysis, 432 orthologous genes were found in the two analyzed genomes (IMBG185 and IMBG180), according to the following annotation databases: tBLASTn 2.2+, Augustus 3.2, Prodigal, and HMMER3.1. The genomes also showed 427 complete orthologous genes divided by categories: singles-copy (425), duplicated (2), fragmented (1), and missing (1). These results revealed the very high similarity between IMBG185 and IMBG180 sequenced genomes (Supplementary Material 2).
Identical Genomic Islands, Transposases, and Prion-Like Proteins
The number of symbiosis (4), metabolic (4), and virulence + pathogenicity islands (7), and antibiotic resistance (6) islands found in both IMBGs were identical. Furthermore, considering only the identifications with identity ≥ 30% and E-values ≤ 1 × 10–7, the number of protein-coding genes inside all the genomic islands were also similar (Figure 4 and Supplementary Material 3). Moreover, the prediction of transposases retrieved 39 potential protein sequences for both IMBGs. For Prion-like proteins, the transcription terminator Rho gene had been detected in both IMBGs, but the Cb-Rho cPrD (prion-forming) domain was not found in the IMBG180 and IMBG185 genomes. This may mean that Cb-Rho does not behave as a prion in IMBGs.
IMBG180 and IMBG185 Genomes Show Very Little Difference in Number of Prophages
A total of 96 candidates for prophage regions in the IMBGs were predicted, and these regions have 92 inactive, three active, and one ambiguous prophage (Figure 3 and Supplementary Material 4). Regarding the regions containing active prophages, 62 genes found in IMBGs displayed high similarity to Rhodovulum phage RS1 (score = 0.87), and 52 genes from postflight (IMBG185) and 60 genes from reference (IMBG180) genomes showed high similarity to Ruegeria phage DSS3-P1 (score = 0.97). Moreover, 18 genes from the genome IMBG185 contained similar sequences to Streptomyces phage (score = 0.92), whereas in the IMBG180 genome, 19 prophage genes in unrelated regions with other bacterial species were also characterized.
Both Genomes Have Two Plasmids but Differ in Sequence Length
Plasmid prediction of the assembled contigs of K. oboediens IMBGs genomes resulted in two plasmids (1 and 2) on each strain, with a variation of only 20 nucleotides in one of the contigs of plasmid 2. Although only plasmid 1 was confirmed as circular, plasmid 2, which contains two contigs, was characterized as a mob conjugative plasmid (Table 3).
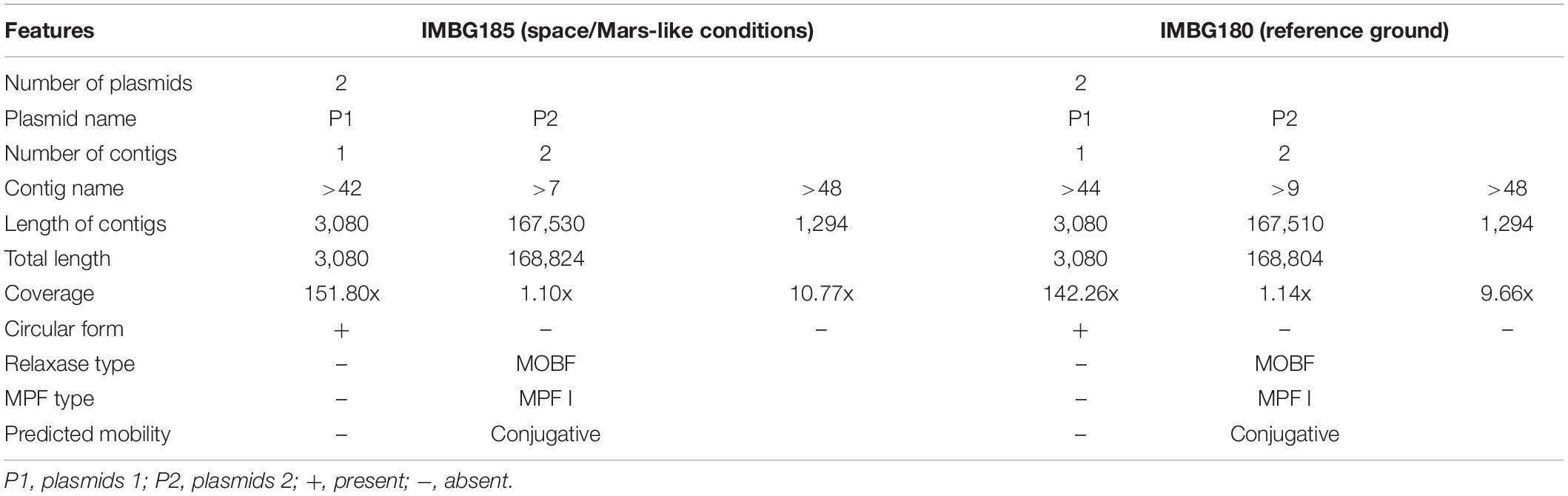
Table 3. General features of K. oboediens plasmids prediction in samples of space/Mars-like (IMBG185) conditions and reference ground (IMBG180) samples.
IMBG180 and IMBG185 Show Two CRISPR-Cas Cassettes, but the Locations of the cas Genes Differ
Komagataeibacter oboediens IMBGs strains genome exhibited two CRISPR-Cas cassettes, which were classified as CAS-VI-B and one as CAS-III-D (or Type I, according to Makarova et al., 2020) subtype. The only difference between the isolates was the position of some cas genes along the genome sequence (Supplementary Material 4).
Moderate Difference in Genome-Specific Genes and Single Nucleotide Variations
The comparison of IMBG genomes resulted in a list of 3,234 corresponding genes present in both samples. This represents 98.5% (3,234/3,282) of IMBG185 (space/Mars-like) and 98.7% (3,234/3,275) of IMBG180 (ground reference) coding sequences. The IMBG185 genome exhibited 48 putative genome-specific genes and the IMBG180 genome displayed 41 putative specific genes. Furthermore, among the corresponding genes, only two of these genes presented SNVs: pyruvate phosphate dikinase (ppdk) (1 non-synonymous SNV), and sugar phosphatase (yidA) (3 synonymous SNVs) genes (Table 4). The plasmid CDS SNV analysis did not find any variation between the strains, indicating that the CDS plasmid sequences of both strains were identical. The non-synonymous substitution SER285 (transition) that was identified in the ppdk gene of IMBG185 was further analyzed with protein modeling and molecular dynamics in order to evaluate if this SNV could affect the predicted binding capacity of the ADP molecule and pyruvate metabolism.
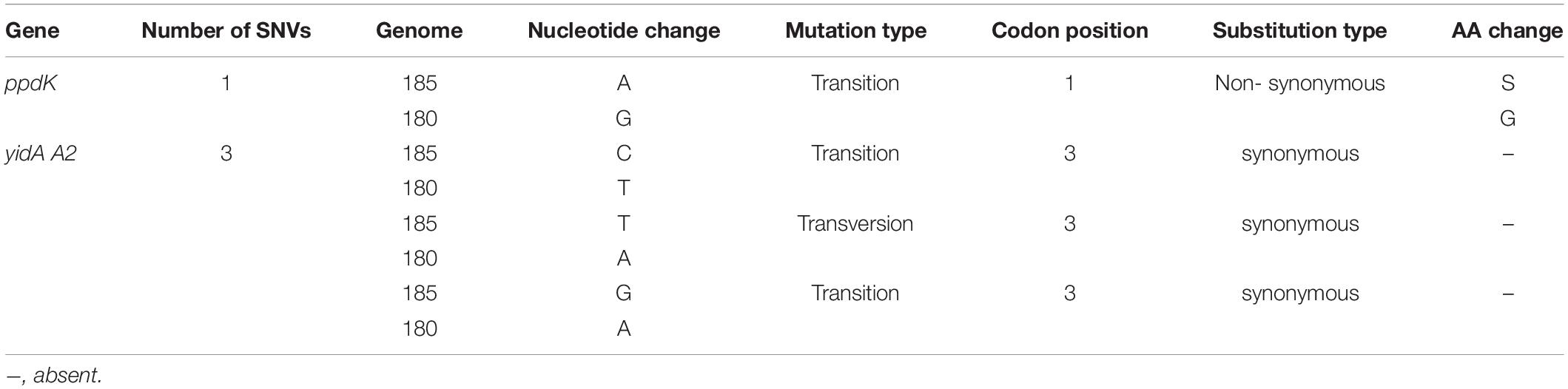
Table 4. Four single nucleotide variations detected in IMBG185 strain as compared to IMBG180.
SER285 in Pyruvate Phosphate Dikinase May Not Affect ADP Binding and Pyruvate Metabolism
The K. oboediens PPDK 3D model was constructed and structurally aligned with its respective template (5LU4). The region where the mutation occurred is well-aligned with its template corresponding amino acids (5LU4 ALA 255 to HIS 390), which are involved with binding to ADP and Mg++ ligands (Minges et al., 2017a,b; Figure 5). RMSD between K. oboediens PPDK chain B and 5LU4 chain A was 0.93 Å with overall identity of 57%. Figure 6 clearly shows the structural alignment between K. oboediens PPDK (magenta) and 5LU4 (dark gray) with the amino acids SER287 and GLY285 (respectively on nearby positions) as they appear surrounding the ADP/MG++ binding site. Additionally, the amino acids responsible for binding to these ligands in the ADP/MG++ binding site are in the same positions between the two proteins.
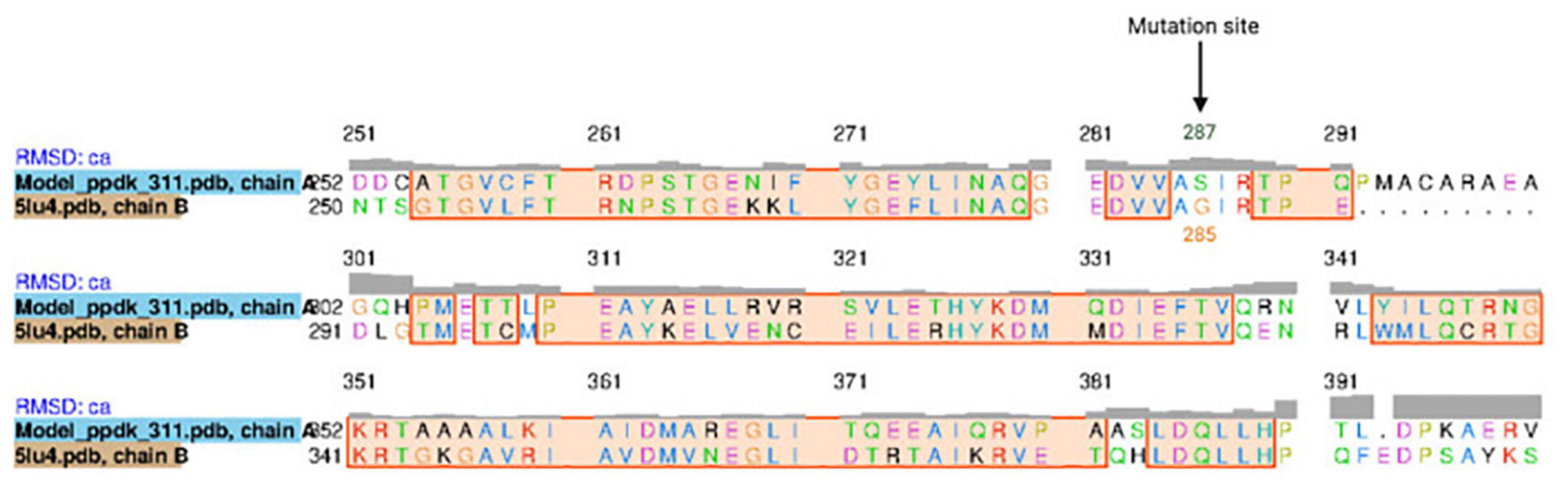
Figure 5. Sequence alignment between K. oboediens IMBG185 PPDK and 5LU4 template, emphasizing the ADP/Mg++ binding site. The black arrow indicates the mutation site Glycine/Serine.
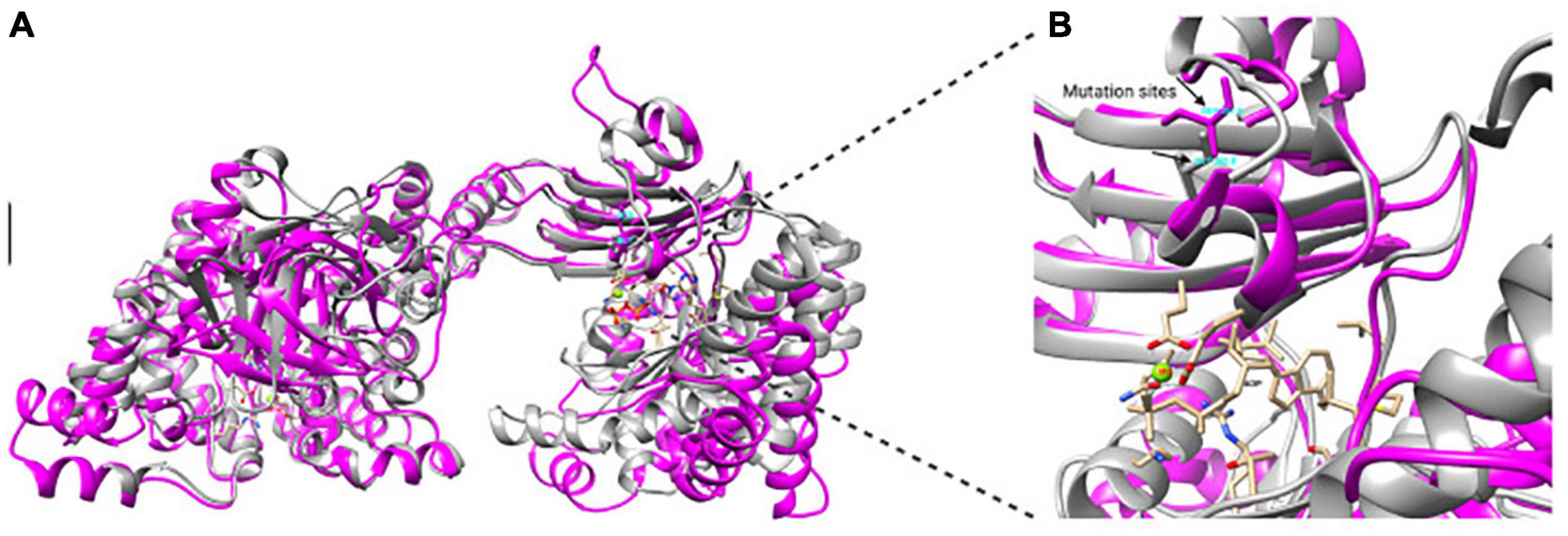
Figure 6. Structural alignment between K. oboediens IMBG185 PPDK (magenta) and 5LU4 (dark gray). (A) In the overall alignment, both chains displayed similar conformation. (B) Detailed ADP/Mg++ binding site showing both SER287 (magenta) and GLY285 (dark gray) amino acids in similar positions between the two enzymes, as well as the cavity conformation.
After 50 ns of solvent-accessible molecular dynamics of the complex 5LU4 mutated G285S, the behavior of the ADP ligand and the influence of serine on the nucleotide-binding positioning were evaluated. The root mean square fluctuations (RMSF) of all the chain B amino acids were analyzed (Figure 7A), displaying that the amino acid S285 had an RMSF of 0.3 nm, which indicated that its sidechain possibly had freely rotational movements. This observation was further confirmed by the MD video (see Supplementary Material 510), showing the rotational movements of this amino acid. Additionally, the binding stability of the ADP molecule to the protein backbone (Figure 7B) was also evaluated, revealing that the root mean square deviation of this ligand was below 0.4 nm, which indicates that the ligand is stable inside its binding site. Furthermore, the MD video also confirmed the ADP positioning inside its 5LU4 binding site for all the 50 ns time of the simulation. The Coulombic interaction energy between ADP and 5LU4 (Figure 7C) showed that they had a high affinity with each other, with an average of −191.586 KJ/mol. Therefore, it is unlikely that they have been affected by the mutated SER285 amino acid. Furthermore, ADP formed a high number of hydrogen bonds (Figure 7D) with the enzyme active amino acids, varying from 1 to 8, during all the simulation. Altogether, it is unlikely that the mutated SER285 can affect the binding capacity of the ADP molecule or the enzyme metabolism.
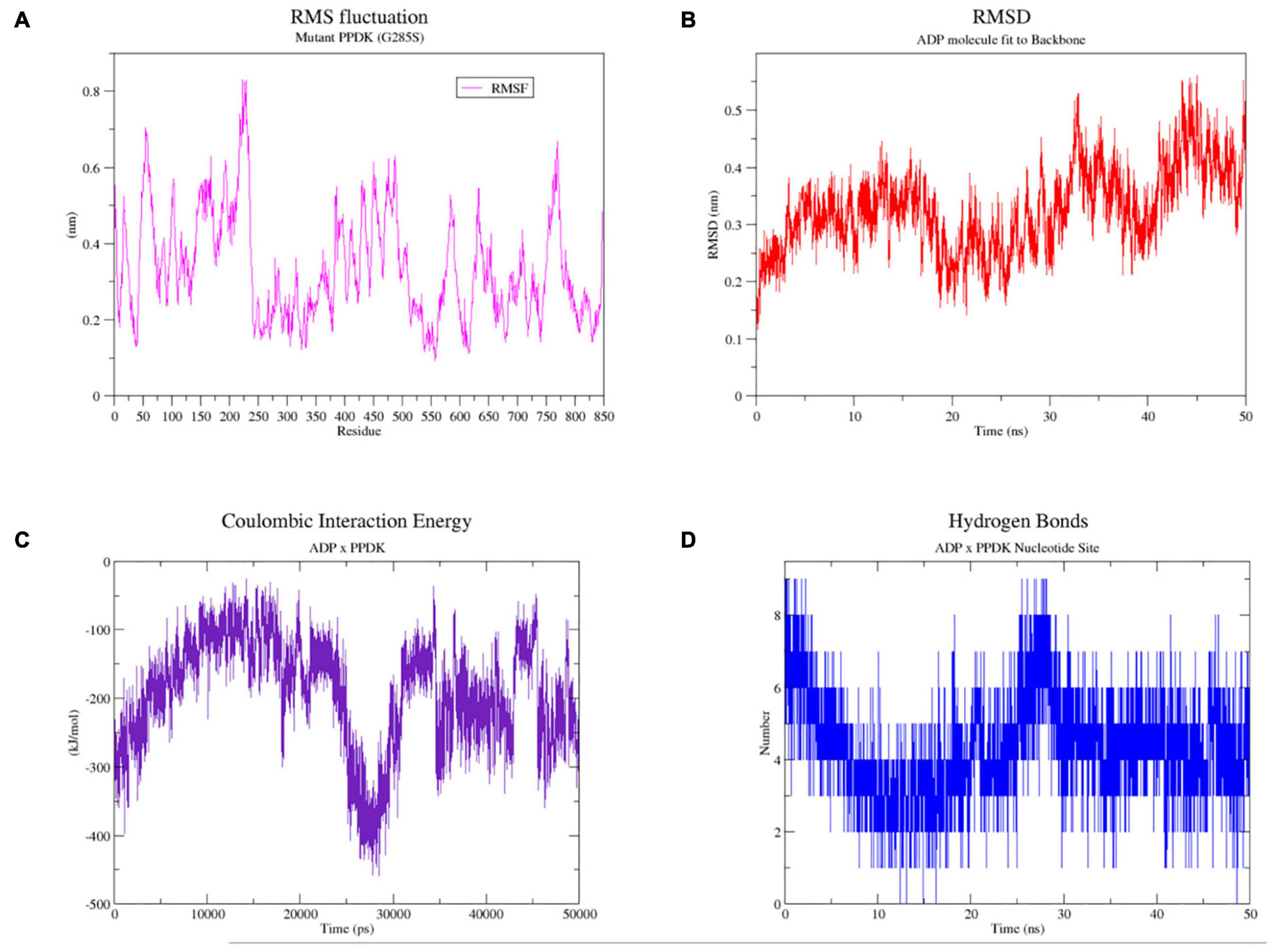
Figure 7. Molecular dynamics results for 50 ns of simulation of the 5LU4 mutated (G285S) in complex with ADP and Mg++. (A) RMS amino acid fluctuation graph for the 5LU4 chain B during the 50 ns simulation. (B) RMSD graph of the ADP ligand bound to the 5LU4 backbone during all the MD simulation. (C) Short-range coulombic energy graph of interaction between the ADP ligand and 5LU4 enzyme. (D) Hydrogen bond graph for the interaction between the ADP molecule and the 5LU4 amino acid side chains.
Difference in Total Gene Content Does Not Affect Metabolic Profiles of Komagataeibacter oboediens IMBG180 and IMBG185
The genome-scale metabolic reconstructions of K. oboediens species comprised 1,894 enzymatic reactions associated with 1,111 genes and 1,418 metabolites (small-molecules in the MetaCyc ontology), covering 33.9% of the annotated open reading frames (Supplementary Material 6). Furthermore, we evaluated differences of the metabolic repertoire between the two isolates of K. oboediens IMBG180 and IMBG185 using KEGG and MetaCyc ontologies for metabolic reconstructions.
The genome-based analysis revealed the absence of gene content differences that could impact the metabolic capacities of post-flight K. oboediens IMBG185, since the predicted content of metabolic genes of the ground reference (IMBG180) and strains exposed to space/Mars-like conditions (IMBG185) was quite similar, independent of the metabolic ontology applied (KEGG or MetaCyc) (Supplementary Material 6). Subsequently, we compared the metabolic functions within the Komagataeibacter genus, as well as how it compares to other bacteria. As expected, there is extensive conservation, in terms of metabolic capacities, within the genus (Figure 8). Furthermore, bacteria with similar metabolic profiles are also closely related to Komagataeibacter, particularly the acetic acid bacteria within the Acetobacter genus (Figure 8). We also included in these analyses representatives from Enterobacteriaceae, which clustered together in a single group but have strong metabolic dissimilarities when compared to the acetic acid bacteria (Figure 7), probably reflecting different lifestyles of these organisms. It is worth noting that K. kakiaceti was the only Komagataeibacter representative that did not group with the remaining bacteria of this genus (Figure 8). Next, we evaluated pathways of interest coded in the genomes of K. oboediens IMBGs that could be important to the overall functioning of the KMC.
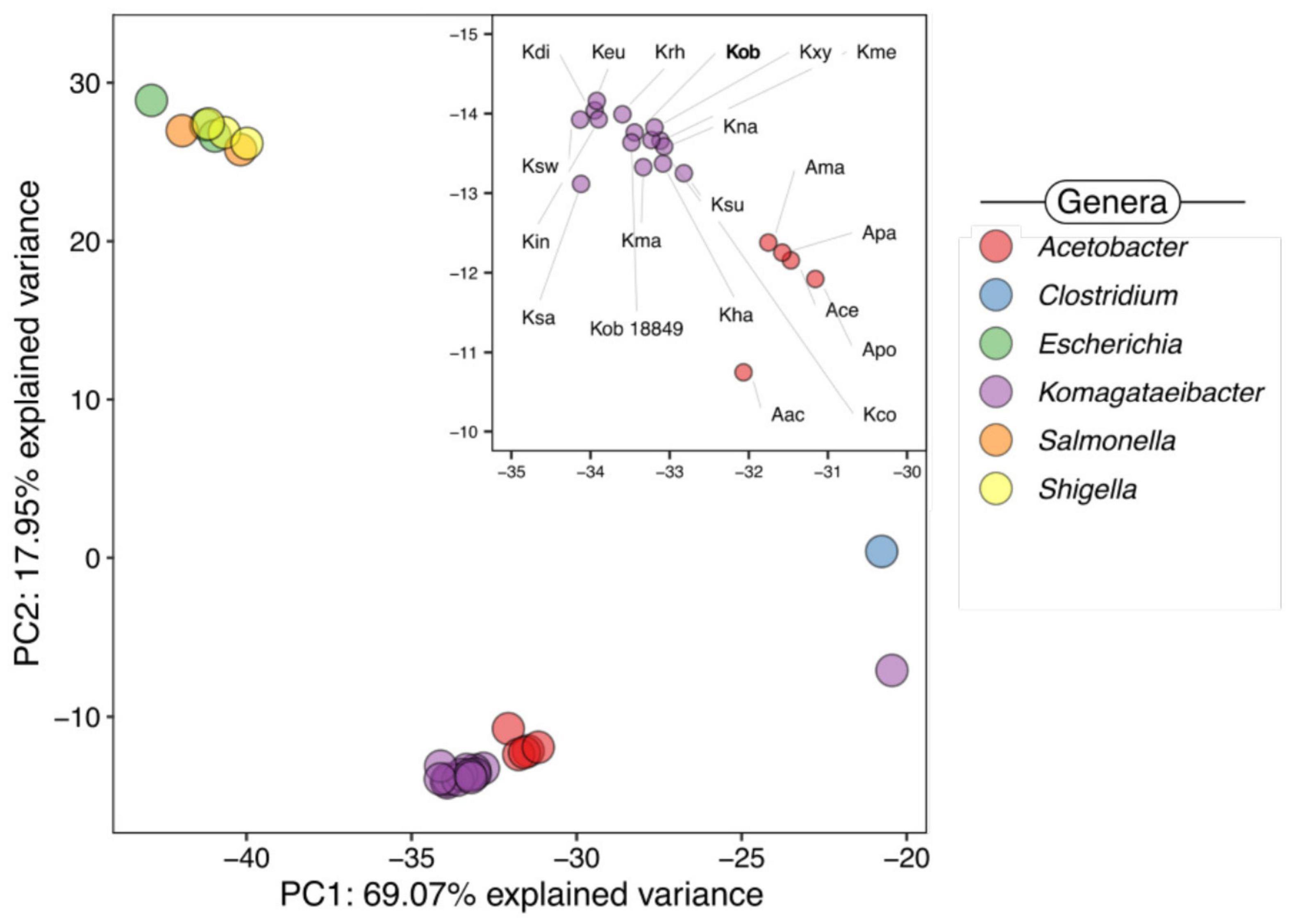
Figure 8. Principal component analysis based on KEGG enzymatic predictions of Komagataeibacter genus relating acetic acid bacteria and Enterobacteriaceae representatives. Acetobacter aceti (Aac), A. cerevisae (Ace), A. malorum (Ama), A. pasteurianus (Apa), A. pomorum (Apo), Clostridium kluyveri, Escherichia coli, E. albertii, E. fergusonii, K. cocoi (Kco), K. diospyri (Kdi), K. europaeus (Keu), K. hansenii (Kha), K. intermedius (Kin), K. kakiaceti (Kka), K. maltaceti (Kma), K. medellinensis (Kme), K. nataicola (Kna), K. oboediens LMG 18849 (Kob 18849), K. rhaeticus (Krh), K. saccharivorans (Ksa), K. sucrofermentais (Ksu), K. swingsii (Ksw), K. kylinus (Kxy), Salmonella bongori, S. enterica, Shigella boydii, Sh. flexneri, and Sh. sonnei.
Small Difference Found in Central Carbohydrate and Energy Pathways
The main pathways found in IMBGs as glycolysis, pyruvate oxidation, Krebs cycle, pentose phosphate pathway (oxidative and non-oxidative phases), phosphoribosyl diphosphate (PRPP – an important intermediate in cellular metabolism), and Entner-Doudoroff pathway had their KOs completely matched (100%) with the reference pathway of KEGG. Six of seven of the gluconeogenesis potential proteins were detected (86%), and only one protein was not complete (86%) considering the pentose phosphate pathway (7 proteins). Other carbohydrate pathways found were the De Ley-Doudoroff pathway (D-galactonate degradation), nucleotide sugar biosynthesis, and UDP-N-acetyl-D-glucosamine biosynthesis (Supplementary Material 7).
Fermentative Pathways Common to IMBG180 and IMBG185
For both IMBGs, several fermentative complete pathways were identified through the genome potential enzymes. Key enzymes of the fermenting processes were found, such as phosphate acetyltransferase (pta) and acetate kinase (ackA) for acetate production, pyruvate decarboxylase (pdc) and alcohol dehydrogenase (adh) for ethanol production, acetolactate synthase (ilv) for 2-acetolactate production, α-acetolactate decarboxylase (budA) and diacetyl reductase (budC) for acetoin production, and finally the enzyme diacetyl reductase (budC) to complete the acid homeostasis in cells (Supplementary Material 7).
No Difference Is Observed in Medium-Chain Carboxylate Pathways
The putative presence of genes related to the fatty acid biosynthesis (FAB) route for MCCA synthesis such as acetyl-CoA carboxylase (accABCD) (all the subunits) and ortholog coding for malonyltransferase (fabD) (at least one), ketoacyl-ACP synthase (fabF; fabH), ketoacyl-ACP reductase (fabG), hydroxyacyl-ACP dehydratase (fadZ), and enoyl-ACP reductase (fabI; fabK) enzymes was detected (Supplementary Material 8).
Stress-Related Pathways Are Intact in Both Genomes
In order to disclose general stress-related genes coded in IMBGs genomes, we firstly conducted a large-scale analysis by leveraging information on all the stress-associated, manually reviewed proteins available in UniProtKB/Swiss-Prot and compared both sequenced strains. This resulted in the identification of highly conserved genes in IMBGs genomes (Supplementary Material 9), and no differences in gene content between both IMBGs were observed in this analysis. Genes with chaperone activities, such as groL, groS, dnaJ, dnaK, and hfq were detected, as well as the products of recA and uvrABC systems involved against DNA damage and other resistance properties. Protease activities coded by hslVU, lon, and clp were detected in the genomes, and also hrcA and lexA, both of which regulate the expression of part of these genes. Trehalose biosynthesis pathway (otsA/otsB) genes associated with ROS scavenging (katB, oxyR) were also identified in both genomes (Supplementary Material 9).
No Topological Change in Cellulose Biosynthesis Operons
The cellulose synthesis key bcs operons following the K. xylinus (a close member of K. oboediens) as described by Liu et al. (2018), Gullo et al. (2019) were constructed and can be accessed on Gullo et al. (2019), La China et al. (2020). There was no topological change in any of the bcs operon genes between postflight and reference genomes, and all the genes of the four operons had 100% identity at both the DNA and amino acid levels, as recently characterized by Lee et al. (2021).
Equal Number of Nitrogen-Fixing Genes Found in Both IMBG180 and IMBG185
A total of five protein-coding genes associated with biological nitrogen fixation was retrieved in both genomes of IMBG185 (space/Mars-like conditions) and IMBG180 (ground reference) strains (Table 5). One gene (nifU) product is involved in the assembly and incorporation of iron into the metalloclusters of a nitrogenase, whereas the other four genes either encode transcription factors that regulate (positively and negatively) nitrogenase (such as fixK and vnfA) or code for proteins that regulate the transcription factors of nitrogenase (such as the PII proteins glnB and glnK).
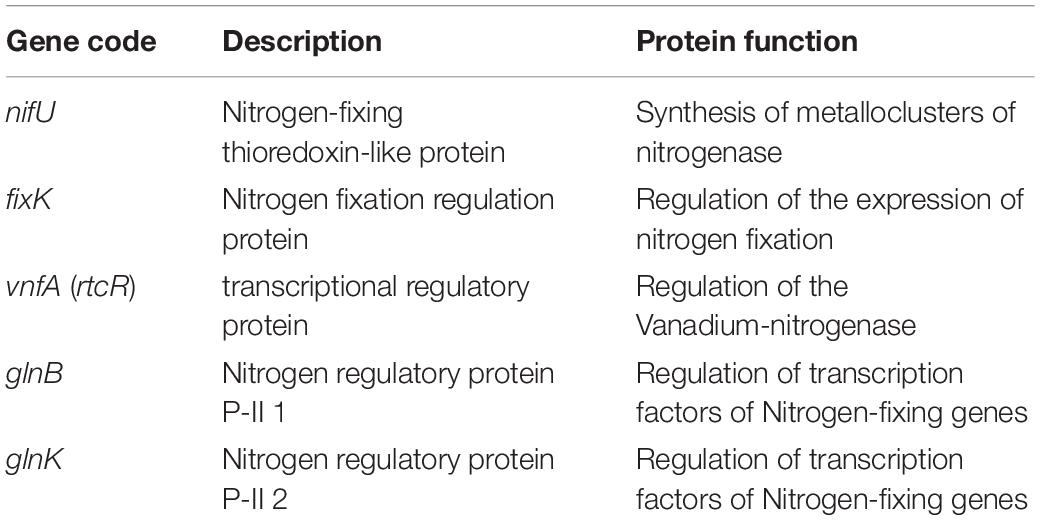
Table 5. Genes associated with biological nitrogen fixation in ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
No Change Is Observed in Hopanoid Lipid Biosynthesis Gene Number or Gene Sequence
There are five enzyme-coding genes directly associated with the biosynthesis of hopanoid lipids in both genomes (Table 6). One of the genes (sqc) codes for the enzyme responsible for the synthesis of hopanoid lipids, whereas another gene (hpnR) encodes an enzyme that modifies these hopanoids. Moreover, the other three genes (hpnC, hpnD, and hpnE) code for enzymes that jointly participate in the conversion of all trans-farnesyl diphosphate to squalene.
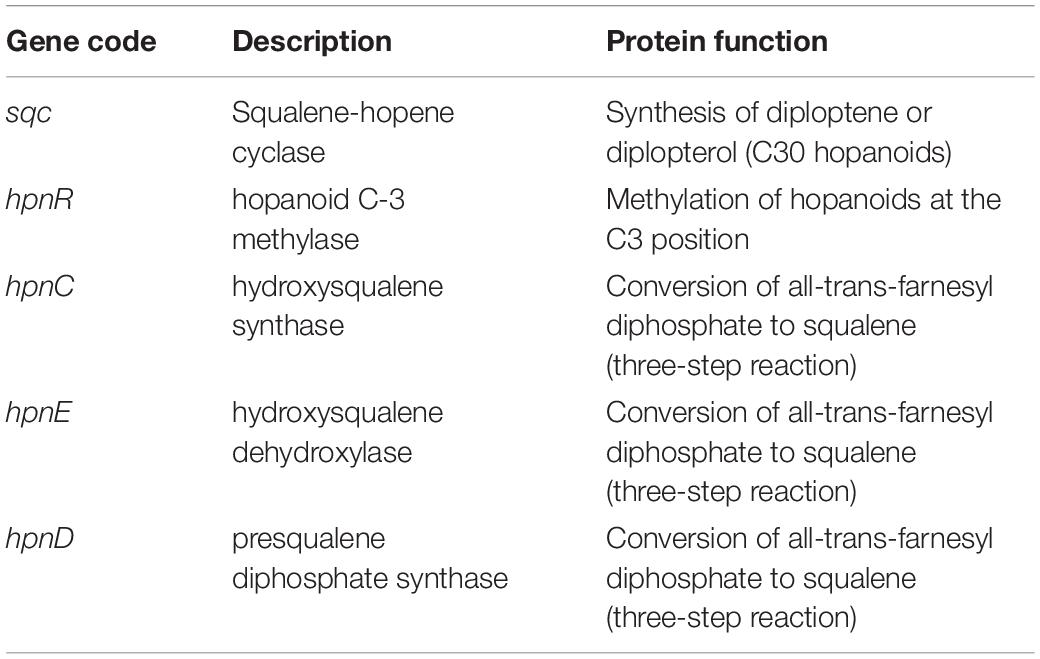
Table 6. Genes associated with biosynthesis of hopanoid lipids ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
Discussion
A growing body of evidence indicates that exposure of bacteria in culture to the spaceflight environmental stressors results in stress-induced mutations (Li et al., 2014; Guo et al., 2015; Fajardo-Cavazos et al., 2018; Zhao et al., 2019). Direct damage of DNA by cosmic radiation and indirect consequences due to reactive oxygen species generation affect the genetic apparatus of exposed microorganisms. Genomics methods revealed a number of mutant microbial strains after spaceflights. For instance, in Gram-positive bacteria Bacillus subtilis (Fajardo-Cavazos et al., 2018), Staphylococcus aureus (Guo et al., 2015), and Gram-negative Klebsiella pneumoniae (Li et al., 2014), many differences were found between spaceflight and ground control samples. Conversely, comparative genomic analysis of Acinetobacter baumannii flight (33 days on the Shenzhou 11 spacecraft) strain yielded only a few mutations compared with the reference strain (Zhao et al., 2019). Only one SNV was identified in the flight strain, suggesting that these strains underwent the genomic changes in short-term spaceflight.
Simulated Martian conditions on the ISS also induced mutagenesis in dehydrated bacterial material. After a 1.5-year exposure to Mars-like conditions, the rates of induced mutations to rifampicin resistance and sporulation deficiency in B. subtilis spore DNA increased (Moeller et al., 2012). In the BIOMEX project, KMC samples were exposed to space/Mars-like conditions, and genomes of re-isolated K. oboediens from KMC (ground reference strain IMBG180 and space exposed strain IMBG185) were remarkably similar (Table 2 and Supplementary Material 2), indicating that K. oboediens tolerated extra-terrestrial conditions.
Genomes of K. oboediens strains show some differences in genome size and rRNA + tRNA (K. oboediens 174Bp2 and: ∼3.56 Mpb and 96, K. oboediens LMG 18849: ∼4.18 Mpb and 51, K. oboediens BPZTR0: 2,64 Mbp and 54) (Andrés-Barrao et al., 2011; Ryngajłło et al., 2019; Pinar et al., 2020), and most of these differences in the same species of K. oboediens are probably due to transferable mobile elements DNA (Darmon and Leach, 2014). In both samples of IMBGs, we also observed a similar trend and not much difference at genomic level (genome size ∼3.56 Mpb and the rRNA + tRNA is 51–52) (Tables 2, 3).
Using the genomic islands classification proposed by Soares et al. (2016), we have predicted four symbiotic, four metabolic, seven virulence + pathogenicity, and six antibiotic resistance islands in both genomes. We identified symbiosis island genes in both the strains encoding nitrogen fixation proteins such as nitrogen fixation regulation protein (FixK), molybdenum permease, and Fe3+ siderophore transport system. Gluconobacter kombuchae (now K. hansenii) isolated from Kombucha tea has been shown to have nitrogen-fixing capabilities (Dutta and Gachhui, 2007). Nevertheless, putative nitrogen fixation/regulation genes such as nifH in K. hansenii JR-02 (Li et al., 2019) and nifU, ntrB, ntrC, ntrX, and ntrY in BC producer K. rhaeticus ENS9a are reported, which are not homologous to nif HDK of Klebsiella oxytoca or other nitrogen-fixing bacteria. In K. oboediens IMBG180, no structural genes of nitrogenase were found, and, to date, no experimental evidence of N-fixation by these species was reported. Free-living bacteria carry the nif-genes originated from symbiotic ancestors (carrying both nif- and nod-genes) (Tao et al., 2021), and this transition is characterized by losses of symbiosis-related genes, most of which were in the symbiosis island. This loss probably occurred in K. oboediens, which now harbors only remnants of nif- and nod-genes clusters.
Antimicrobial activity through bacteriocins has been detected in lactic acid bacteria isolated from kombucha (Matei et al., 2018; Pei et al., 2020). Thus, the acquisition and maintenance of antibiotic resistance genes probably through phages by KMC may be necessary (Góes-Neto et al., 2021), which could be supported by the presence of antibiotic resistance islands found here.
Genomic islands often harbor transposases that affect the activity of nearby genes (Soares et al., 2016). A thermo-adapted strain of K. oboediens MSKU 3 (39°C) isolated from vinegar displayed, among other mutations, a transposon insertion into cellulose synthase catalytic subunit homolog (MSKU 3_1013) (Taweecheep et al., 2019), showing the stimulation of transposase in higher temperature. In both of our strains, the number of transposase genes was 39 (Supplementary Material 4), while 47 were found in K. oboediens 174Bp2 (Andrés-Barrao et al., 2011). The reason behind the smaller number of transposons in our samples is unknown.
Prophages can provide both resistance mechanisms as well as some metabolic advantages, allowing the bacteria more chances to survive (Fortier and Sekulovic, 2013). Furthermore, phenotypic diversity among bacterial strains of the same species is typically connected with the mobile genome elements of DNA (Darmon and Leach, 2014). Three intact prophages were found in both IMBGs genomes, which is in accordance with the range retrieved by Ryngajłło et al. (2019) who reported 0–4 intact prophages in three Komagataeibacter species from distinct clades of K. oboediens. Some differences between IMBG185 and IMBG180 strains for the prophage profile were also found, and interestingly, 18 genes similar to Streptomyces phage were uniquely found in IMBG185 (Figure 3 and Supplementary Material 10).
Plasmids appear to have a high mobility within the Komagataeibacter genus. In our study, two plasmids (P1 and P2) were predicted in both strains (Table 3) which is similar in number reported in K. xylinus DSM 2325 (Jang et al., 2019). Nonetheless, Komagataeibacter species show a variable number of plasmids that ranges from 3 to 14 (Ogino et al., 2011; Kubiak et al., 2014; Zhang et al., 2017; Lu et al., 2020; Ryngajłło et al., 2020). Plasmid P1 in our samples is circular; however, relaxase and mpf sequences were not found. Therefore, this plasmid is probably non-mobilizable in nature. On the other hand, plasmid P2 exhibits a linear form and has a set of mobility (mob) and mating pair formation (mpf1) genes that are essential for conjugation and self-transmission (Smillie et al., 2010).
Conversely, (Ryngajłło et al., 2019) solely detected CRISPR/Cas system of the Type I–E (Cas3) in only K. hansenii and K. medellinensis NBRC; both our K. oboediens strains display Cas proteins cassettes of two distinct subtypes: CAS-VI-B and CAS-III-D subtypes (although Makarova et al., 2020 classified the latter as Type I) and located in different positions in the genomes (Supplementary Material 10). To date and to our knowledge, it is the first time the subtype CAS-VI-B has been described for this genus. The CRISPR-Cas system maintains the genome stability in acetic acid bacteria, such as Acetobacter pasteurianus (Wang et al., 2015). Therefore, the presence of CRISPR-Cas system in IMBGs suggests that mobile genetic elements are, at least, under control of this specific protective molecular system. Nonetheless, additional studies are required to investigate whether the predicted cassettes are rare subtypes of CRISPR/Cas systems and whether they are functional.
Prions can serve as heritable bet-hedging devices for diversifying microbial phenotypes (Newby and Lindquist, 2013). The bacterial transcription terminator Rho of Clostridium botulinum (Cb-Rho) could form a prion and expresses amyloidogenicity (Yuan and Hochschild, 2017). Despite protein aggregates being detected in IMBGs cultures (data not shown), and the transcription terminator Rho gene being detected, the Cb-Rho cPrD domain was not found in the transcription terminator Rho gene of IMBG180 and IMBG185 genomes.
Another interesting finding is the presence of protein-coding genes associated with hopanoids biosynthesis in both the genomes. Hopanoid lipids might have a crucial importance in tolerance of space/Mars-like stress conditions, since KMC UV-irradiated samples displayed the highest read relative abundance of the protein coding gene squalene-hopene cyclase (shc) (Góes-Neto et al., 2021). In fact, both K. oboediens IMBGs (derived from KMC) not only contain shc but also other genes involved in the synthesis and modification of hopanoids. Moreover, as hopanoids can intercalate into lipid bilayers of membranes because of their both planar and hydrophobic structure, they may also decrease cellular permeability to oxygen (Poger and Mark, 2013), which could cause a decrease of the intracellular oxygen concentration.
Hopanoid-enriched membranes may facilitate aerobic nitrogen fixation by providing a physical barrier that restricts oxygen diffusion and may permit the maintenance of low intracellular O2 concentrations (Cornejo-Castillo and Zehr, 2019). Furthermore, the typical KMC cellulose biofilm mainly formed by cellulose-producing bacteria (such as K. oboediens) may also act not only as a protection of the liquid phase from colonization by competitors but also as a physical barrier to oxygen diffusion to that phase (May et al., 2019; Góes-Neto et al., 2021), creating a microaerophilic environment that would be compatible with nitrogen fixation. Taken together, KMC may be a good candidate system for future Mars colonization.
Finally, in our study, K. oboediens did not exhibit considerable genomic changes after exposure to a space/Mars-like environment. One possible explanation could be that the K. oboediens genome was not affected by the Mars-like conditions experienced outside the ISS due to biofilm and mineral shielding of latent bacterial cells in KMC. Cellulose-based biofilm is considered a protective shield against environmental cues, including UV (Williams and Cannon, 1989). Despite the stability of genomic apparatus under the exposure experiment, K. oboediens exhibited altered fitness after spaceflight that expressed a slower growth rate and cellulose synthesis (Orlovska et al., 2021), a changed rate of enzyme activities (dehydrogenases, nucleases), cellular membrane lipidome structure, and a mode of outer-membrane vesicle production (Podolich et al., 2020). Since genomic content does not show extensive changes in Mars-like conditions, we may assume that the set of changes observed in K. oboediens are associated with cytoplasmically inheritable damage but still have functional biomolecules and protein aggregates, which are associated with the impact of environmental stressors (Govers et al., 2018; Wang et al., 2020). Additionally, the lack of mutations in K. oboediens from KMC biofilm exposed to space or Mars-like conditions also correlates with conserved core microbiome of KMC exposed to the same conditions as observed in our recent study (Góes-Neto et al., 2021). Although the mutations found do not seem to affect the genomes, as well as the maintenance and conservation of the metabolic pathways, we suggest that the genome is stable under space conditions. Nonetheless, future studies are needed to support this claim.
Conclusion
Our study suggests that the genome of K. oboediens either is stable in KMC exposed to space or that space-exposed KMC recovers most of its functions after upon successive cultures in the lab after return from space. Comprehensive comparative genomics of two strains: K. oboediens IMBG180 (ground-based reference) from the laboratory-kept KMC sample and K. oboediens IMBG185 (space-exposed) from post-flight KMC revealed that their genomes are highly similar both structurally and functionally, without any striking changes. Both the K. oboediens IMBG genomes harbor hopanoid lipids as well as nitrogen-fixing genes, which may make a microaerobic environment compatible with biological nitrogen fixation. The observed genome integrity of K. oboediens in the space-exposed KMC samples may be also due to the protecting biofilm of the KMC. Notably, if the biofilm is responsible for protecting the K. oboediens genome, this KMC biofilm needs attention to explore its potential for space-related technologies.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
NK, DB, and AG-N designed the study. DS, RK, FA, AJ, RP, RD, ST, AC, EA, OK, IO, OP, OR, PR, VD, BB, BA, ATU, DB, and J-PV performed the analyses. DB, DS, ATU, NK, and AG-N wrote, reviewed, and edited the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was partially supported by the National Academy of Sciences of Ukraine (grant 49/2019). Scientific productivity scholarships were supported by the CNPq (Brazil) awarded to AG-N (310764/2016-5), VD (312045/2020-4), and ATU (104327/2019-7). DS was funded by a CAPES/PrInt (Brazil) grant (88887.364931/2019-00 and 88887.511110/2020-00).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Graduate Programs of Microbiology (https://www.microbiologia.icb.ufmg.br/pos/) and Bioinformatics (https://www.pgbioinfo.icb.ufmg.br), and the Pro-Rectory of Research of Universidade Federal de Minas Gerais (UFMG). Authors dedicate this study to all the innocent victims of COVID-19 pandemic and war and appeal to the global community for peace and prosperity of our mother Earth.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.782175/full#supplementary-material
Supplementary Material 1 | List of shared and specific genes in samples IMBG180 and IMBG185.
Supplementary Material 2 | Genome completeness of K. oboediens IMBG180 and IMBG185 compared with K. oboediens BPZTR01 using a BUSCO analysis.
Supplementary Material 3 | Spreadsheet of genomic islands found in both genomes IMBG180 and IMBG185.
Supplementary Material 4 | Spreadsheet of prophages with gene annotation.
Supplementary Material 5 | Molecular dynamics video of PPDK.
Supplementary Material 6 | Annotation, using KEGG ontology, of the KO identifiers for both genomes of Komagataeibacter oboediens, IMBG180 and IMBG185.
Supplementary Material 7 | Pathway conservation and completeness, evaluated using the KEGG ontology, of strains Komagataeibacter oboediens IMBG185 and K. oboediens IMBG180. Both genomes share the same set of KO orthologs; for this reason, the key represents conservation across both genomes. Complete pathways are highlighted in bold.
Supplementary Material 8 | Orthologs, and respective loci IDs, of key genes related to the synthesis of medium-chain carboxylates (MCAA) through the fatty acid biosynthesis pathway detected in Komagataeibacter oboediens genomes IMBG185 and IMBG180.
Supplementary Material 9 | Orthologs, and respective loci IDs, of key genes related to the stress response detected in Komagataeibacter oboediens genomes IMBG185 and IMBG180.
Supplementary Material 10 | Prediction of Cas proteins and classification of CRISPR-Cas system subtypes of ground reference (IMBG180) and space/Mars-like conditions (IMBG185) genomes.
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://www.uniprot.org/
- ^ https://www.ebi.ac.uk/interpro/
- ^ https://pfam.xfam.org/
- ^ https://www.ncbi.nlm.nih.gov/
- ^ https://www.genome.jp/kegg/
- ^ https://R-project.org
- ^ https://www.rcsb.org/structure/5LU4
- ^ https://plasma-gate.weizmann.ac.il/Grace/
- ^ https://drive.google.com/file/d/1ME25mtaLUbhPYKtFJRKlXGNI_vsAhZN5/view?usp=sharing
References
Andrés-Barrao, C., Falquet, L., Calderon-Copete, S. P., Descombes, P., Pérez, R. O., and Barja, F. (2011). Genome sequences of the high-acetic acid-resistant bacteria Gluconacetobacter europaeus LMG 18890T and G. europaeus LMG 18494 (reference strains), G. europaeus 5P3, and Gluconacetobacter oboediens 174Bp2 (isolated from vinegar). J. Bacteriol. 193, 2670–2671. doi: 10.1128/JB.00229-11
Araujo, F. A., Barh, D., Silva, A., Guimarães, L., and Ramos, R. T. J. (2018). GO FEAT: a rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 8:1794. doi: 10.1038/s41598-018-20211-9
Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. doi: 10.1093/bioinformatics/bti770
Arredondo-Alonso, S., Bootsma, M., Hein, Y., Rogers, M. R., Corander, J., Willems, R. J., et al. (2020). gplas: a comprehensive tool for plasmid analysis using short-read graphs. Bioinformatics 36, 3874–3876. doi: 10.1093/bioinformatics/btaa233
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barbosa, C. D., Uetanabaro, A. P. T., Santos, W. C. R., Caetano, R. G., Albano, H., Kato, R., et al. (2021). Microbial-physicochemical integrated analysis of kombucha fermentation. LWT 148:111788.
Berendsen, H. J., van der Spoel, D., and van Drunen, R. (1995). GROMACS: a message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91, 43–56. doi: 10.1016/0010-4655(95)00042-e
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., et al. (2020). The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 48, D445–D453. doi: 10.1093/nar/gkz862
Cornejo-Castillo, F. M., and Zehr, J. P. (2019). Hopanoid lipids may facilitate aerobic nitrogen fixation in the ocean. Proc. Natl. Acad. Sci. U S A. 116, 18269–18271. doi: 10.1073/pnas.1908165116
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darmon, E., and Leach, D. R. (2014). Bacterial genome instability. Microbiol. Mol. Biol. Rev. 78, 1–39. doi: 10.1128/mmbr.00035-13
de Vera, J. P. (2020). Astrobiology on the International Space Station. Berlin: Springer International Publishing.
de Vera, J. P., Alawi, M., Backhaus, T., Baqué, M., Billi, D., Böttger, U., et al. (2019). Limits of life and the habitability of Mars: the ESA space experiment BIOMEX on the ISS. Astrobiology 19, 145–157. doi: 10.1089/ast.2018.1897
de Vera, J. P., Schulze-Makuch, D., Khan, A., Lorek, A., Koncz, A., Möhlmann, D., et al. (2014). Adaptation of an Antarctic lichen to Martian niche conditions can occur within 34 days. Plan. Space Sci. 98, 182–190. doi: 10.1016/j.pss.2013.07.014
Dutta, D., and Gachhui, R. (2007). Nitrogen-fixing and cellulose-producing Gluconacetobacter kombuchae sp. nov., isolated from Kombucha tea. Int. J. Systematic Evol. Microbiol. 57, 353–357. doi: 10.1099/ijs.0.64638-0
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Fajardo-Cavazos, P., Leehan, J. D., and Nicholson, W. L. (2018). Alterations in the spectrum of spontaneous rifampicin-resistance mutations in the Bacillus subtilis rpoB gene after cultivation in the human spaceflight environment. Front. Microbiol. 9:192. doi: 10.3389/fmicb.2018.00192
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Góes-Neto, A., Kukharenko, O., Orlovska, I., Podolich, O., Imchen, M., Kumavath, R., et al. (2021). Shotgun metagenomic analysis of kombucha mutualistic community exposed to mars-like environment outside the international space station. Environ. Microbiol. 23, 3727–3742. doi: 10.1111/1462-2920.15405
Govers, S. K., Mortier, J., Adam, A., and Aertsen, A. (2018). Protein aggregates encode epigenetic memory of stressful encounters in individual Escherichia coli cells. PLoS Biol. 16:e2003853. doi: 10.1371/journal.pbio.2003853
Gullo, M., La China, S., Petroni, G., Di Gregorio, S., and Giudici, P. (2019). Exploring K2G30 genome: a high bacterial cellulose producing strain in glucose and mannitol based media. Front. Microbiol. 10:58. doi: 10.3389/fmicb.2019.00058
Guo, J., Han, N., Zhang, Y., Wang, H., Zhang, X., Su, L., et al. (2015). Use of genome sequencing to assess nucleotide structure variation of Staphylococcus aureus strains cultured in spaceflight on Shenzhou-X, under simulated microgravity and on the ground. Microbiol. Res. 170, 61–68. doi: 10.1016/j.micres.2014.09.001
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hernandez, D., François, P., Farinelli, L., Østerås, M., and Schrenzel, J. (2008). De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 18, 802–809. doi: 10.1101/gr.072033.107
Hestrin, S., and Schramm, M. J. B. J. (1954). Synthesis of cellulose by Acetobacter xylinum. 2. preparation of freeze-dried cells capable of polymerizing glucose to cellulose. Biochem. J. 58:345. doi: 10.1042/bj0580345
Horneck, G., Klaus, D. M., and Mancinelli, R. L. (2010). Space microbiology. Microbiol. Mol. Biol. Rev. 74, 121–156.
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Jang, W. D., Kim, T. Y., Kim, H. U., Shim, W. Y., Ryu, J. Y., Park, J. H., et al. (2019). Genomic and metabolic analysis of Komagataeibacter xylinus DSM 2325 producing bacterial cellulose nanofiber. Biotechnol. Bioeng. 116, 3372–3381. doi: 10.1002/bit.27150
Karp, P. D., Paley, S., and Romero, P. (2002). The pathway tools software. Bioinformatics 18(Suppl._1), S225–S232.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kozyrovska, N. O., Reva, O. M., Goginyan, V. B., and De Vera, J. P. (2012). Kombucha microbiome as a probiotic: a view from the perspective of post-genomics and synthetic ecology. Âiopolym Cell 28, 103–113.
Kozyrovska, N., Reva, O., Podolich, O., Kukharenko, O., et al. (2021). To other planets with upgraded millennial kombucha in rhythms of sustainability and health support. Front. Astron. Space Sci. 8:701158. doi: 10.3389/fspas.2021.701158
Kubiak, K., Kurzawa, M., Jędrzejczak-Krzepkowska, M., Ludwicka, K., Krawczyk, M., Migdalski, A., et al. (2014). Complete genome sequence of Gluconacetobacter xylinus E25 strain valuable and effective producer of bacterial nanocellulose. J. Biotechnol. 176, 18–19. doi: 10.1016/j.jbiotec.2014.02.006
La China, S., Bezzecchi, A., Moya, F., Petroni, G., Di Gregorio, S., and Gullo, M. (2020). Genome sequencing and phylogenetic analysis of K1G4: a new Komagataeibacter strain producing bacterial cellulose from different carbon sources. Biotechnol. Lett. 42, 807–818. doi: 10.1007/s10529-020-02811-6
Lee, I., Barh, D., Podolich, O., Brenig, B., Tiwari, S., Azevedo, V., et al. (2021). Metagenome-Assembled genome sequences obtained from a reactivated kombucha microbial community exposed to a mars-like environment outside the international space station. Microbiol Resource Announcements 10:e0054921. doi: 10.1128/MRA.00549-521
Lemkul, J. (2019). From proteins to perturbed Hamiltonians: a suite of tutorials for the GROMACS-2018 molecular simulation package [article v1. 0]. Living J. Computational Mol. Sci. 1:5068.
Li, J., Chen, G., Zhang, R., Wu, H., Zeng, W., and Liang, Z. (2019). Production of high crystallinity type-I cellulose from Komagataeibacter hansenii JR-02 isolated from Kombucha tea. Biotechnol. Appl. Biochem. 66, 108–118. doi: 10.1002/bab.1703
Li, J., Liu, F., Wang, Q., Ge, P., Woo, P. C., Yan, J., et al. (2014). Genomic and transcriptomic analysis of NDM-1 Klebsiella pneumoniae in spaceflight reveal mechanisms underlying environmental adaptability. Sci. Rep. 4:6216. doi: 10.1038/srep06216
Liu, L. X., Liu, S. X., Wang, Y. M., Bi, J. C., Chen, H. M., Deng, J., et al. (2018). Komagataeibacter cocois sp. nov., a novel cellulose-producing strain isolated from coconut milk. Int. J. Systematic Evol. Microbiol. 68, 3125–3131. doi: 10.1099/ijsem.0.002947
Lu, T., Gao, H., Liao, B., Wu, J., Zhang, W., Huang, J., et al. (2020). Characterization and optimization of production of bacterial cellulose from strain CGMCC 17276 based on whole-genome analysis. Carbohydrate Polymers 232:115788. doi: 10.1016/j.carbpol.2019.115788
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J., et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Matei, B., Salzat, J., Diguta, C. F., Cornea, C. P., Luta, G., Utoiu, E. R., et al. (2018). Lactic acid bacteria strains isolated from kombucha with potential probiotic effect. Rom Biotechnol. Lett. 23, 1–15. doi: 10.3390/foods9121780
May, A., Narayanan, S., Alcock, J., Varsani, A., Maley, C., and Aktipis, A. (2019). Kombucha: a novel model system for cooperation and conflict in a complex multi-species microbial ecosystem. PeerJ 7:e7565. doi: 10.7717/peerj.7565
Melsted, P., and Halldórsson, B. V. (2014). KmerStream: streaming algorithms for k-mer abundance estimation. Bioinformatics 30, 3541–3547. doi: 10.1093/bioinformatics/btu713
Minges, A., Ciupka, D., Winkler, C., Höppner, A., Gohlke, H., and Groth, G. (2017a). Structural intermediates and directionality of the swiveling motion of Pyruvate Phosphate Dikinase. Sci. Rep. 7:45389. doi: 10.1038/srep45389
Minges, A., Höppner, A., and Groth, G. (2017b). Trapped intermediate state of plant pyruvate phosphate dikinase indicates substeps in catalytic swiveling domain mechanism. Protein Sci. 26, 1667–1673. doi: 10.1002/pro.3184
Moeller, R., Schuerger, A. C., Reitz, G., and Nicholson, W. L. (2012). Protective role of spore structural components in determining Bacillus subtilis spore resistance to simulated mars surface conditions. Appl. Environ. Microbiol. 78, 8849–8853. doi: 10.1128/AEM.02527-12
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35(Suppl._2), W182–W185. doi: 10.1093/nar/gkm321
Müller, R., and Chauve, C. (2019). HyAsP, a greedy tool for plasmids identification. Bioinformatics 35, 4436–4439. doi: 10.1093/bioinformatics/btz413
Newby, G. A., and Lindquist, S. (2013). Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 23, 251–259. doi: 10.1016/j.tcb.2013.01.007
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ogino, H., Azuma, Y., Hosoyama, A., Nakazawa, H., Matsutani, M., Hasegawa, A., et al. (2011). Complete genome sequence of NBRC 3288, a unique cellulose-nonproducing strain of Gluconacetobacter xylinus isolated from vinegar. J. Bacteriol. 193, 6997–6998. doi: 10.1128/JB.06158-11
Orlovska, I., Podolich, O., Kukharenko, O., Zaets, I., Reva, O., Khirunenko, L., et al. (2021). Bacterial cellulose retains robustness but its synthesis declines after exposure to the Mars-like environment simulated outside the International Space Station. Astrobiology 21, 706–717. doi: 10.1089/ast.2020.2332
Padilha, V. A., Alkhnbashi, O. S., Shah, S. A., De Carvalho, A. C., and Backofen, R. (2020). CRISPRCasIdentifier: machine learning for accurate identification and classification of CRISPR-Cas systems. GigaScience 9:giaa062. doi: 10.1093/gigascience/giaa062
Pei, J., Jin, W., Abd El-Aty, A. M., Baranenko, D. A., Gou, X., Zhang, H., et al. (2020). Isolation, purification, and structural identification of a new bacteriocin made by Lactobacillus plantarum found in conventional kombucha. Food Control 110:106923.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pinar, O., Bozdaǧ, G., Yemişer, Ý, Büyük, E., and Kazan, D. (2020). Complete genome sequence of a cellulose-producing bacteria Komagataeibacter oboediens BPZTR01. Genetics Appl. 4, 38–40.
Podolich, O., Kukharenko, O., Haidak, A., Zaets, I., Zaika, L., Storozhuk, O., et al. (2019). Multimicrobial kombucha culture tolerates Mars-like conditions simulated on low Earth orbit. Astrobiology 19, 183–196. doi: 10.1089/ast.2017.1746
Podolich, O., Kukharenko, O., Zaets, I., Orlovska, I., Palchykovska, L., Zaika, L., et al. (2020). Fitness of outer membrane vesicles from Komagataeibacter intermedius is altered under the impact of simulated Mars-like stressors outside the International Space Station. Front. Microbiol. 11:1268. doi: 10.3389/fmicb.2020.01268
Poger, D., and Mark, A. E. (2013). The relative effect of sterols and hopanoids on lipid bilayers: when comparable is not identical. J. Phys. Chem. B 117, 16129–16140. doi: 10.1021/jp409748d
Pothakos, V., Illeghems, K., Laureys, D., Spitaels, F., Vandamme, P., and De Vuyst, L. (2016). “Acetic acid bacteria in fermented food and beverage ecosystems,” in Acetic Acid Bacteria, eds K. Matsushita, H. Toyama, N. Tonouchi, and A. Okamoto-Kainuma (Tokyo: Springer), 73–99. doi: 10.1007/978-4-431-55933-7_3
Robertson, J., and Nash, J. H. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microbial Genom. 4:e000206. doi: 10.1099/mgen.0.000206
Ryngajłło, M., Jędrzejczak-Krzepkowska, M., Kubiak, K., Ludwicka, K., and Bielecki, S. (2020). Towards control of cellulose biosynthesis by Komagataeibacter using systems-level and strain engineering strategies: current progress and perspectives. Appl. Microbiol. Biotechnol. 104, 6565–6585. doi: 10.1007/s00253-020-10671-3
Ryngajłło, M., Kubiak, K., Jędrzejczak-Krzepkowska, M., Jacek, P., and Bielecki, S. (2019). Comparative genomics of the Komagataeibacter strains—efficient bionanocellulose producers. Microbiologyopen 8:e00731. doi: 10.1002/mbo3.731
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Schulze-Makuch, D., Schulze-Makuch, A., and Houtkooper, J. M. (2015). The physical, chemical and physiological limits of life. Life 5, 1472–1486. doi: 10.3390/life5031472
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245.
Smillie, C., Garcillán-Barcia, M. P., Francia, M. V., Rocha, E. P., and de la Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452.
Soares, S. C., Geyik, H., Ramos, R. T., de Sá, P. H., Barbosa, E. G., Baumbach, J., et al. (2016). GIPSy: genomic island prediction software. J. Biotechnol. 232, 2–11. doi: 10.1016/j.jbiotec.2015.09.008
Song, W., Sun, H. X., Zhang, C., Cheng, L., Peng, Y., Deng, Z., et al. (2019). Prophage Hunter: an integrative hunting tool for active prophages. Nucleic Acids Res. 47, W74–W80. doi: 10.1093/nar/gkz380
Tao, J., Wang, S., and Liao, T. (2021). Evolutionary origin, and ecological implication of a unique nif island in free-living Bradyrhizobium lineages. ISME J. 15, 3195–3206. doi: 10.1038/s41396-021-01002-z
Taweecheep, P., Naloka, K., Matsutani, M., Yakushi, T., Matsushita, K., and Theeragool, G. (2019). In vitro thermal and ethanol adaptations to improve vinegar fermentation at high temperature of Komagataeibacter oboediens MSKU 3. Appl. Biochem. Biotechnol. 189, 144–159. doi: 10.1007/s12010-019-03003-3
Toombs, J. A., Petri, M., Paul, K. R., Kan, G. Y., Ben-Hur, A., and Ross, E. D. (2012). De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. U S A. 109, 6519–6524. doi: 10.1073/pnas.1119366109
Wang, B., Shao, Y., Chen, T., Chen, W., and Chen, F. (2015). Global insights into acetic acid resistance mechanisms and genetic stability of Acetobacter pasteurianus strains by comparative genomics. Sci. Rep. 5:18330. doi: 10.1038/srep18330
Wang, X., Cole, C. G., DuPai, C. D., and Davies, B. W. (2020). Protein aggregation is associated with Acinetobacter baumannii desiccation tolerance. Microorganisms 8:343. doi: 10.3390/microorganisms8030343
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Williams, W. S., and Cannon, R. E. (1989). Alternative environmental roles for cellulose produced by Acetobacter xylinum. Appl. Environ. Microbiol. 55, 2448–2452. doi: 10.1128/aem.55.10.2448-2452.1989
Yuan, A. H., and Hochschild, A. (2017). A bacterial global regulator forms a prion. Science 355, 198–201.
Zhang, H., Ye, C., Xu, N., Chen, C., Chen, X., Yuan, F., et al. (2017). Reconstruction of a genome-scale metabolic network of Komagataeibacter nataicola RZS01 for cellulose production. Sci. Rep. 7:7911. doi: 10.1038/s41598-017-06918-1
Keywords: comparative genomics, Acetobacteraceae, metabolic reconstruction, single nucleotide variation, nitrogen fixation, hopanoids, cellulose biosynthesis
Citation: Santana de Carvalho D, Trovatti Uetanabaro AP, Kato RB, Aburjaile FF, Jaiswal AK, Profeta R, De Oliveira Carvalho RD, Tiwar S, Cybelle Pinto Gomide A, Almeida Costa E, Kukharenko O, Orlovska I, Podolich O, Reva O, Ramos PIP, De Carvalho Azevedo VA, Brenig B, Andrade BS, de Vera J-PP, Kozyrovska NO, Barh D and Góes-Neto A (2022) The Space-Exposed Kombucha Microbial Community Member Komagataeibacter oboediens Showed Only Minor Changes in Its Genome After Reactivation on Earth. Front. Microbiol. 13:782175. doi: 10.3389/fmicb.2022.782175
Received: 23 September 2021; Accepted: 01 February 2022;
Published: 11 March 2022.
Edited by:
Elena Perrin, University of Florence, ItalyReviewed by:
Stefano Amalfitano, Water Research Institute, National Research Council, ItalyNiamh O’Hara, SUNY Downstate Medical Center, United States
Copyright © 2022 Santana de Carvalho, Trovatti Uetanabaro, Kato, Aburjaile, Jaiswal, Profeta, De Oliveira Carvalho, Tiwar, Cybelle Pinto Gomide, Almeida Costa, Kukharenko, Orlovska, Podolich, Reva, Ramos, De Carvalho Azevedo, Brenig, Andrade, de Vera, Kozyrovska, Barh and Góes-Neto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Debmalya Barh, ZHIuYmFyaEBnbWFpbC5jb20=; Aristóteles Góes-Neto, YXJpZ29lc25ldG9AaWNiLnVmbWcuYnI=
†These authors have contributed equally to this work