 Irina Bessarab1
Irina Bessarab1 Abdul Majid Maszenan2,3
Abdul Majid Maszenan2,3 Mindia A. S. Haryono1Krithika Arumugam4
Mindia A. S. Haryono1Krithika Arumugam4 Nay Min Min Thaw Saw4
Nay Min Min Thaw Saw4 Robert J. Seviour5
Robert J. Seviour5 Rohan B. H. Williams1*
Rohan B. H. Williams1*
- 1Singapore Centre for Environmental Life Sciences Engineering, National University of Singapore, Singapore, Singapore
- 2Nanyang Environment & Water Research Institute (NEWRI), Nanyang Technological University, Singapore, Singapore
- 3NUS Environmental Research Institute, National University of Singapore, Singapore, Singapore
- 4Singapore Centre for Environmental Life Sciences Engineering, Nanyang Technological University, Singapore, Singapore
- 5School of Life Sciences, La Trobe University, Melbourne, VIC, Australia
Members of the genus Defluviicoccus occur often at high abundances in activated sludge wastewater treatment plants designed to remove phosphorus, where biomass is subjected to alternating anaerobic feed/aerobic famine conditions, believed to favor the proliferation of organisms like Ca. Accumulibacter and other phosphate-accumulating organisms (PAO), and Defluviicoccus. All have a capacity to assimilate readily metabolizable substrates and store them intracellularly during the anaerobic feed stage so that under the subsequent famine aerobic stage, these can be used to synthesize polyphosphate reserves by the PAO and glycogen by Defluviicoccus. Consequently, Defluviicoccus is described as a glycogen-accumulating organism or GAO. Because they share a similar anaerobic phenotype, it has been proposed that at high Defluviicoccus abundance, the PAO are out-competed for assimilable metabolites anaerobically, and hence aerobic P removal capacity is reduced. Several Defluviicoccus whole genome sequences have been published (Ca. Defluviicoccus tetraformis, Defluviicoccus GAO-HK, and Ca. Defluviicoccus seviourii). The available genomic data of these suggest marked metabolic differences between them, some of which have ecophysiological implications. Here, we describe the whole genome sequence of the type strain Defluviicoccus vanusT, the only cultured member of this genus, and a detailed comparative re-examination of all extant Defluviicoccus genomes. Each, with one exception, which appears not to be a member of this genus, contains the genes expected of GAO members, in possessing multiple copies of those for glycogen biosynthesis and catabolism, and anaerobic polyhydroxyalkanoate (PHA) synthesis. Both 16S rRNA and genome sequence data suggest that the current recognition of four clades is insufficient to embrace their phylogenetic biodiversity, but do not support the view that they should be re-classified into families other than their existing location in the Rhodospirillaceae. As expected, considerable variations were seen in the presence and numbers of genes encoding properties associated with key substrate assimilation and metabolic pathways. Two genomes also carried the pit gene for synthesis of the low-affinity phosphate transport protein, pit, considered by many to distinguish all PAO from GAO. The data re-emphasize the risks associated with extrapolating the data generated from a single Defluviicoccus population to embrace all members of that genus.
Introduction
All continuous flow enhanced biological phosphorus removal wastewater treatment plants (EBPR) operate by passing the biomass continuously between anaerobic and aerobic reactors, a plant configuration considered essential for removing phosphorus (P) microbiologically by advantaging selectively the populations involved in its removal (Seviour and McIlroy, 2008; Stokholm-Bjerregaard et al., 2017; Nielsen et al., 2019; Roy et al., 2021). One popular but not universally accepted explanation as to why EBPR plants often perform badly is that the polyphosphate-accumulating organisms (PAO) are out-competed in the EBPR anaerobic “feed” stage by other bacteria known collectively as the glycogen-accumulating organisms (GAO) (Cech and Hartman, 1993; Liu et al., 1996; Law et al., 2016; Nielsen et al., 2019). These populations share with the PAO a similar anaerobic phenotype in being able to assimilate readily metabolizable substrates present in the plant influent, which are used to synthesize intracellular storage compounds of a diverse chemical composition. Intracellular polyphosphate (polyP) reserves, an anaerobic TCA cycle, and glycogen are used by the PAO as anaerobic sources of energy and reducing power for both substrate assimilation and subsequent intracellular biosynthesis of storage products (McMahon et al., 2010; Zhou et al., 2010), and orthophosphate is released into the bulk liquid. Storage compounds varying in their chemical composition (see below) are then reutilized in the subsequent aerobic “famine” stage, thus providing energy to support PAO growth, the assimilation of phosphate from the bulk liquid and subsequent synthesis of polyP granules as energy stores needed to support their subsequent anaerobic metabolism (Oehmen et al., 2007; Seviour and McIlroy, 2008). Stored P is then removed from the system by sludge wasting.
The GAO also assimilate metabolites under anaerobic conditions, but now, in the absence of polyphosphate reserves, use the stored glycogen as their main source of energy and reducing power from the activity of the Embden Meyerhof Parnas (EMP) pathway. As with the PAO, this storage material is then metabolized to support GAO growth under subsequent aerobic conditions, and to synthesize and store glycogen for use during the subsequent anaerobic phase. Hence, as no intracellular polyP is synthesized and stored intracellularly, then at high abundances, the GAO are thought to reduce EBPR capacity (Oehmen et al., 2007, 2010; Mielczarek et al., 2013; Nielsen et al., 2019). However, emerging data from full-scale systems suggest that any PAO–GAO competition occurs primarily in ecological niches generated by PAO enrichment protocols (Law et al., 2016; Nielsen et al., 2019), highlighting the need to maintain a broader view of the ecophysiological impact of GAO populations on EBPR capacity.
The GAO phenotype is found in phylogenetically diverse bacterial populations. These include the gammaproteobacterial Ca. Competibacter (Kong et al., 2006) and Ca. Contendobacter (McIlroy et al., 2014, 2015), the betaproteobacterial Ca. Propionivibrio aalborgensis (Albertsen et al., 2016) and Spb280 (Kong et al., 2007), the actinobacterial Micropruina glycogenica (Shintani et al., 2000; McIlroy et al., 2018), and the alphaproteobacterial Defluviicoccus vanus (Maszenan et al., 2005). Of these, only M. glycogenica and D. vanus have been cultured. As with the known PAO, considerable metabolic diversity exists between individual GAO populations. Thus, while M. glycogenica utilize and store sugars and amino acids generated by fermentation, Ca. Propionivibrio aalborgensis (Albertsen et al., 2016), an unusually aerobic member of this genus and closely related phylogenetically to the PAO Ca. Accumulibacter phosphatis (Stokholm-Bjerregaard et al., 2017), possesses the same anaerobic phenotype as this PAO, in assimilating short-chain fatty acids and storing them as poly-ß-hydroxy alkanoates (PHA). This same anaerobic phenotype is seen in Ca Competibacter and Ca. Contendobacter (McIlroy et al., 2014) and all members of the genus Defluviicoccus examined to date (Nittami et al., 2009; Nobu et al., 2014; Wang et al., 2014; Stokholm-Bjerregaard et al., 2017; Onetto et al., 2019).
Based on 16S rRNA sequence data, Defluviicoccus is placed currently in the family Rhodospirillaceae in the order Rhodospirillales (Maszenan et al., 2005), where members of this genus fall at present into four clades (Wong et al., 2004; Meyer et al., 2006; Burow et al., 2007; McIlroy and Seviour, 2009; Nittami et al., 2009). However, this phylogenetic diversity has not always been recognized in many Defluviicoccus publications based on partial 16S rRNA amplicon sequencing, and in the absence of any accompanying FISH analyses. D. vanusT, the type species, which was isolated from an EBPR wastewater treatment plant (WWTP) treating mainly brewery wastes in Pilsen, Czechia showing poor P removal capacity (Maszenan et al., 2005), is a member of clade 1, as is Ca. Defluviicoccus tetraformis (Nobu et al., 2014), while Defluviicoccus strain GAO-HK (Wang et al., 2014) belongs to clade II. These and clade IV (McIlroy and Seviour, 2009) members of this genus, and visualized in situ with clade targeted FISH probes, have the characteristic morphology of cocci arranged in tetrads (Figure 1) (McIlroy and Seviour, 2009; McIlroy et al., 2011). However, Ca. Defluviicoccus seviourii, a member of clade III, is filamentous (Nittami et al., 2009), sharing the distinctive Nostocoida limicola II morphotype described by Liu et al. (2000) and Seviour et al. (2002).
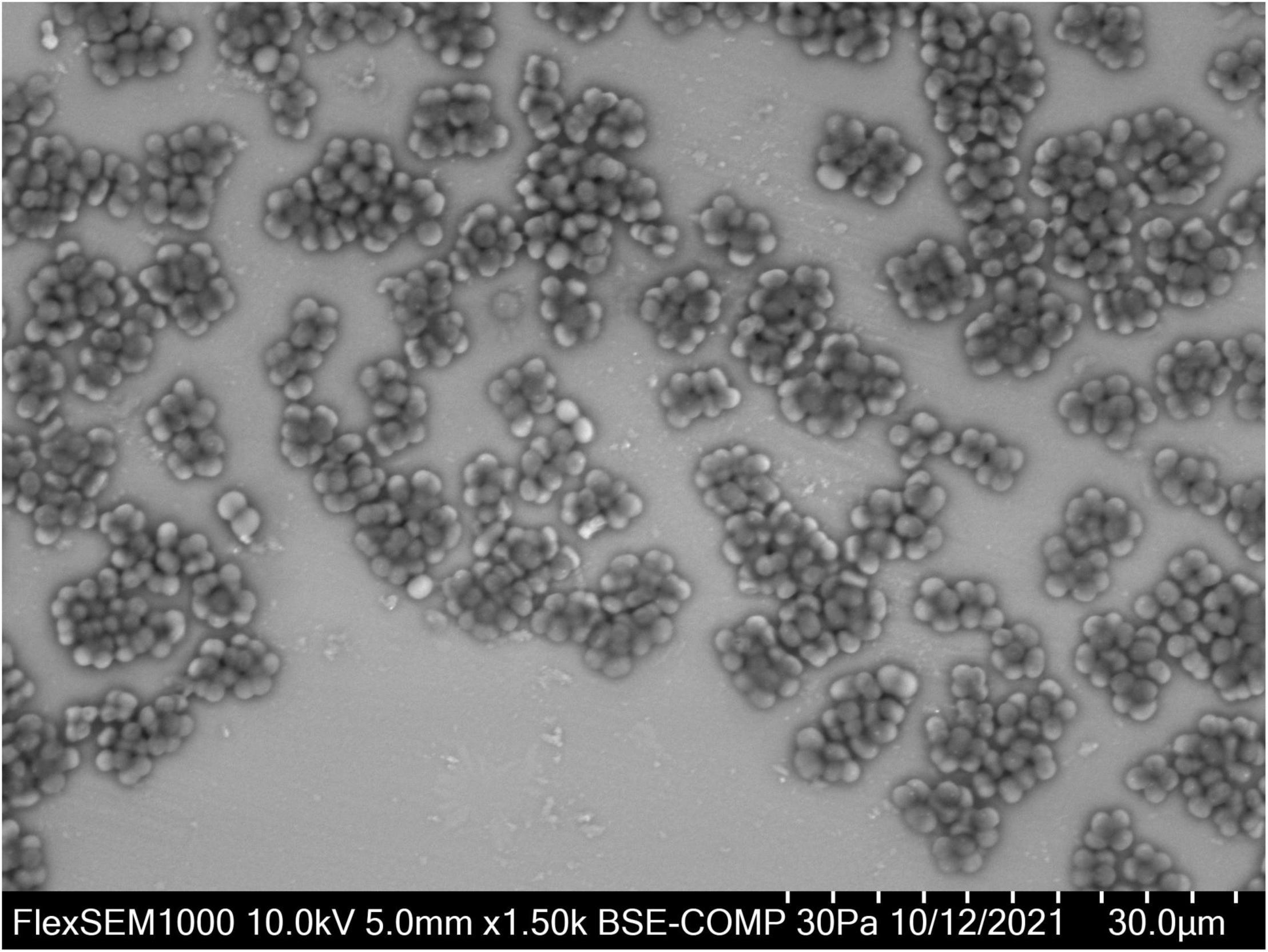
Figure 1. Defluviicoccus vanus showing its distinctive cocci arranged in tetrads, and clusters of tetrads. For VP-SEM imaging, the D. vanus cells were grown on GS agar for 3 weeks. Individual colonies were selected and transferred onto a glass slide cut to the size of the specimen stub. Samples were visualized uncoated in a variable-pressure scanning electron microscope VP-SEM (Hitachi FlexSEM 1000 II) at 30 Pa and accelerating voltage of 10 kV, using a BSE detector.
Draft genomes of Defluviicoccus strain GAO-HK, Ca. D. tetraformis, Ca. D. seviourii, and SSA4 have been generated from metagenome assemblies (Nobu et al., 2014; Wang et al., 2014; Arumugam et al., 2019, 2021; Onetto et al., 2019), and in general support the earlier metabolic models of Lopez-Vazquez et al. (2009) and Oehmen et al. (2010). They also confirm the presence of many of the metabolic processes detailed in earlier physiological and metabolic inhibitor studies of members of this genus (Saunders et al., 2007; Burow et al., 2008a,b, 2009). Furthermore, each genome harbors the genes encoding the same pathways expected of GAO associated with carbon recycling, involving glycogen and PHA synthesis and their degradation. These early genomic sequencing data also revealed several ecologically important variations among them, the most notable being a lack of the glyoxylate cycle pathway in Ca. D. seviourii, which instead possessed the anaplerotic ethylmalonyl CoA pathway (Onetto et al., 2019). It alone of the three was thought to have the gene encoding a Na/glutamate symporter, while a gene (pit) encoding a low-affinity phosphate transporter pit was seen only in the genome of strain GAO-HK (clade II) (Nobu et al., 2014; Wang et al., 2014; Onetto et al., 2019).
Consequently, we undertook to sequence the genome of the type strain D. vanusT, and to compare it to all available genomes of other uncultured Defluviicoccus populations. The aim was to understand better their ecophysiology and expose the genomic differences existing among them, especially those of potential ecological importance.
Materials and Methods
Culture Revival, DNA Extraction, and Identification Confirmation
Defluviicoccus vanus (strain Ben 114T) was obtained from the NCIMB (National Collection of Industrial, Food and Marine Bacteria, Aberdeen, United Kingdom) culture deposited there in 2003. The freeze-dried culture was revived in modified glucose sulfite (GS) agar (Williams and Unz, 1985; Maszenan, 2001) according to the culture revival procedure recommended by NCIMB. After ca. 3 weeks of incubation at 25°C, cells with the distinctive tetrad morphology expected of strain Ben 114T were seen (Figure 1). The strain was stored on GS agar at 4°C and maintained at 25°C. DNA was extracted from D. vanus cells grown on solid media. For DNA extraction, the biomass was collected from the GS agar plates with sterile GS medium (Williams and Unz, 1985), pelleted by centrifugation and washed 1–2× with sterile PBS. Washed biomass was pre-treated either with Proteinase K (Sigma, >10 mg/ml) at 55°C for 30 min followed by 80°C for 10 min, or with MetaPolyzyme (Sigma) at 37°C for 4–6 h. Total DNA was extracted from pre-treated biomass using two methods: FastDNA™ SPIN kit for soil (MP Biomedicals), using Lysing Matrix E and 2 × bead beating with a FastPrep homogenizer (MP Biomedicals), or with DNeasy PowerBiofilm kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. To confirm that the revived culture was D. vanus strain Ben 114T, a nearly full-length 16S rRNA gene sequence was generated by PCR amplification of extracted genomic DNA with primers 27F (5′-AGAGTTTCMTGGCTCAG-3′) and 1429R (5′-TACGGYTACCTTGTTACGACTT-3′) and KapaHiFi Hot Start Ready Mix (Roche Sequencing, Pleasanton, CA, United States). The nucleotide sequence of the PCR product was determined by Sanger sequencing (ABI PRISM 3500 Genetic Analyzer, Applied Biosystems, Waltham, MA, United States). The paired-end ABI-Sanger PCR products were trimmed and merged using SeqMan NGen (version 17.0.22 with default parameters), and Sanger Sequence Assembly Software from DNASTAR (14-day trial version). The merged reads were annotated against SILVA database (SSURef_NR99_132_SILVA_13_12_17_opt.arb) (Quast et al., 2013) using sina-1.6.0-linux running default settings except -t -v –log-file –meta-fmt csv, and with –lca-fields set for all five databases, namely, tax_slv, tax_embl, tax_ltp, tax_gg, and tax_rdp.
Long Read Genome Sequencing and Data Analyses
To obtain the complete genome sequence of D. vanusT, nanopore sequencing was performed on a MinION Mk1B instrument (Oxford Nanopore Technologies) using a SpotON FLO MIN106 FAK45997 flow cell with R9.4 chemistry. A sequencing library was constructed from 400 ng of D. vanus genomic DNA using Rapid Barcoding Sequencing Kits SQK-RBK004 supplied by Oxford Nanopore Technologies, with two barcodes to differentiate gDNA obtained with the different extraction protocols. The sequencing run was operated for approximately 48 h. Data acquisition was performed using MinKNOW software (release 19.06.7) without live base calling on an HP ProDesk 600G2 computer (64-bit, 16 GB RAM, 2 Tb SSD HD). Long reads were base called using guppy (CPU version 3.2.1) and the base called reads were trimmed for adaptors using Porechop (version 0.2.2) with default settings except -v 3 -t 20. Long reads were assembled using Unicycler (version 0.4.7) (Wick et al., 2017) with default settings except -t 40 –keep 3. We refer to assembled sequences here as long read assembled contigs (LRAC). DIAMOND (version 0.9.24) (Buchfink et al., 2015, 2021) was used to perform alignment of LRAC sequences (with default settings except -f 100 -p 40 -v –log –long-reads -c1 -b12) against the NBCI-NR database (February, 2019). From the MEGAN Community Edition suite (version 6.17.0), Daa-meganizer was used to format the .daa output file for use in the MEGAN GUI (version 6.17.0) (Huson et al., 2018). Within MEGAN, LRAC sequences were exported with the “Export Frame-Shift Corrected Reads” option to obtain frameshift corrected sequences. The frameshift corrected LRAC sequences were annotated using Prokka version 1.13 (Seemann, 2014) with default settings except –debug –addgenes –rfam. Genome quality statistics were obtained using CheckM (version 1.0.11; Parks et al., 2015).
Coverage profiles were generated from long read data against the LRAC sequences using minimap2 (version 2.17) with the following flags -ax map-ont. Sorted .bam files were processed subsequently using bedtools genomeCoverageBed (version 2.26.0) (Quinlan and Hall, 2010) with the following flags -d. Alignments to the genome sequences were then examined using the Integrated Genome Viewer (IGV version 2.4.14) (Robinson et al., 2011) to evaluate genome integrity and to identify the presence of any misassembled regions.
16S-SSU rRNA genes were identified using the Prokka workflow and annotated against SILVA database (SURef_NR99_132_SILVA_13_12_17_opt.arb) (Quast et al., 2013) using sina-1.6.0-linux running default settings except -t -v –log-file –meta-fmt csv and with –lca-fields set for all five databases, namely, tax_slv, tax_embl, tax_ltp, tax_gg, and tax_rdp.
Short Read Sequencing and Data Analyses
Genomic DNA Library preparation was performed using a modified version of the Illumina TruSeq DNA Sample Preparation protocol. A MiSeq sequencing run was then performed with a read length of 301 bp (paired-end). The raw FASTQ files were processed with cutadapt (version 2.5) and the following arguments: –overlap 10 -m 30 -q 20,20 –quality-base 33. Reads were assembled using SPAdes (version 3.13.0, executed with default parameters except -k 21,33,55,77,99,127 –meta -t 44) (Bankevich et al., 2012). The SPAdes contig fasta file was processed using the R package RKXM1 and the chromosomal genome was manually binned in the GC-coverage plane. Genome quality statistics were obtained again using CheckM (version 1.0.11) (Parks et al., 2015). The concordance statistic was computed between contigs in short read assembly and the long read assembled chromosome using the R package srac2lrac (Arumugam et al., 2019, 2021). Coverage profiles of short read data against the long read assembly was achieved using the same methods described in the immediately preceding section, with minimap2 settings -ax sr -a -t 20.
Genomes of Other Members of the Genus Defluviicoccus Used in the Study
The following Defluviicoccus genomes were examined in this study: (1) the draft genome of clade III Ca. Defluviicoccus seviourii from Onetto et al. (2019); (2) the draft genome of Defluviicoccus clade II GAO-HK obtained by Wang et al. (2014); (3) the draft genome of clade I Ca. D. tetraformis strain TFO71 of Nobu et al. (2014); (4) the draft genome attributed to Defluviicoccus obtained by Slaby et al. (2017), and of uncertain classification (see later), and denoted as bin 129 in that paper; (5) a Defluviicoccus genome obtained previously by us from long read metagenome data from a PAO enrichment reactor (Arumugam et al., 2019, 2021), denoted as Defluviicoccus sp. SSA4; and (6) three long read metagenome-assembled genomes from members of the genus Defluviicoccus recovered by Singleton et al. (2021), and denoted here as Defluviicoccus FRED MAXAC 307, Defluviicoccus FRED MAXAC 378 and Defluviicoccus KALU MAXAC 148. The PRM01 genome of Onetto et al. (2019) was excluded from this study because of its high levels of sequence contamination (Onetto et al., 2019). We computed average nucleotide identity (ANI) value comparisons between these genomes using FastANI (Jain et al., 2018).
Genome Annotation
All genome assemblies were annotated using Prokka version 1.13 default settings except –debug –addgenes –rfam (Seemann, 2014). To document the presence or absence of canonical metabolic pathways, translated gene sequences from the Prokka (.faa files) were put into the KEGG Mapper Reconstruction workflow of the BlastKOALA webserver (Kanehisa et al., 2016) selecting KEGG Modules with the “including any incomplete” option, in order to fully document the degree of completeness of each selected pathway. The following nomenclature was used to describe the presence and completeness of pathways; (1) pathways labeled with “C” are considered complete, and classified as such by BlastKOALA, or determined to be complete from additional manual annotation (see below); (2) pathways largely intact (no more than 2 missing blocks as defined by BlastKOALA) are labeled with “+”; (3) pathways classified by BlastKOALA as incomplete or missing more than 2 blocks are labeled with “?”; and (4) pathways for which no gene products could be identified in the genome are labeled with “–”.
In the case of key pathways that were classified as largely intact (category 2 above), the possibility that partial incompleteness had resulted from false-negative annotations of individual gene products was examined by manually cross-referencing them against annotations available from Prokka. Any ambiguities of gene-product annotation were tested using the NCBI blastp webserver searching against the BLAST nr database. All annotation data are provided as Supplementary Data Files as tab-delimited text files combining gene-product level annotations from Prokka and BlastKOALA (Supplementary Data File 1).
Comparative Analysis of 16S-SSU rRNA Genes
All 16S-SSU rRNA sequences annotated to members of genus Defluviicoccus (n = 83) from the SILVA database (version 132) (Quast et al., 2013) were downloaded and combined with the 1116S-SSU rRNA sequences harbored on contigs from each of the draft genomes listed earlier.
For testing hypotheses about the taxonomic placement of these Defluviicoccus populations, full-length 16S SSU-rRNA sequences were downloaded from the ‘The All-Species Living Tree’ Project (LTP) (file: LTP_04_2021_compressed.fasta from the download page2). From this database, all sequences annotated to members of Rhodospirillales (n = 344), which include the existing sequence from D. vanusT and two sequences from members of the genus Tistrella (Tistrella bauzanesis and Tistrella mobilis) were augmented with the set of 11 16S sequences from the nine Defluviicoccus genomes listed above and a set of 19 16S sequences annotated to the order Tistrellales derived from cultured isolates.
All subsequent phylogenetic analyses were conducted with the SILVA ACT webserver3 (Pruesse et al., 2012) using RAxML with “Model to use” set to GTR and “Rate model for likelihoods” set to “Gamma” (settings as available within the “Compute tree” option, with “Denovo with user sequences only” selected). The output .tree file was visualized using the plot .phylo function in the R library ape (Paradis and Schliep, 2019). The output.in_fasta files were imported into R using the read .alignment function in the R package seqinr, and converted to character matrices using the R/seqinr function as .matrix.alignment. The percent (sequence) identity (PID) was computed for each pairwise combination of sequences using a custom R script that implemented PID as the number of position-wide identical nucleotides divided by the total number of aligned nucleotides, not counting the occurrence of shared indels.
Comparative Analyses of Defluviicoccus Genomes
Phylogenetic analyses based on whole genome sequences were conducted with GTDB-Tk v0.3.2 (Parks et al., 2018, 2020; Chaumeil et al., 2019) using the de novo workflow and 120 concatenated-marker genes selective for bacteria. In addition to the nine Defluviicoccus draft genomes (Table 1), these analyses included other members of the Rhodospirillales (using the following arguments –taxa_filter o__Rhodospirillales, o__Rhodospirillales_A, o__Rhodospirillales_B), and following Onetto et al. (2019), the genomes from members of the genus Gemmatimonas were assigned as the outgroup taxa (using the argument –outgroup_taxon g__Gemmatimonas).
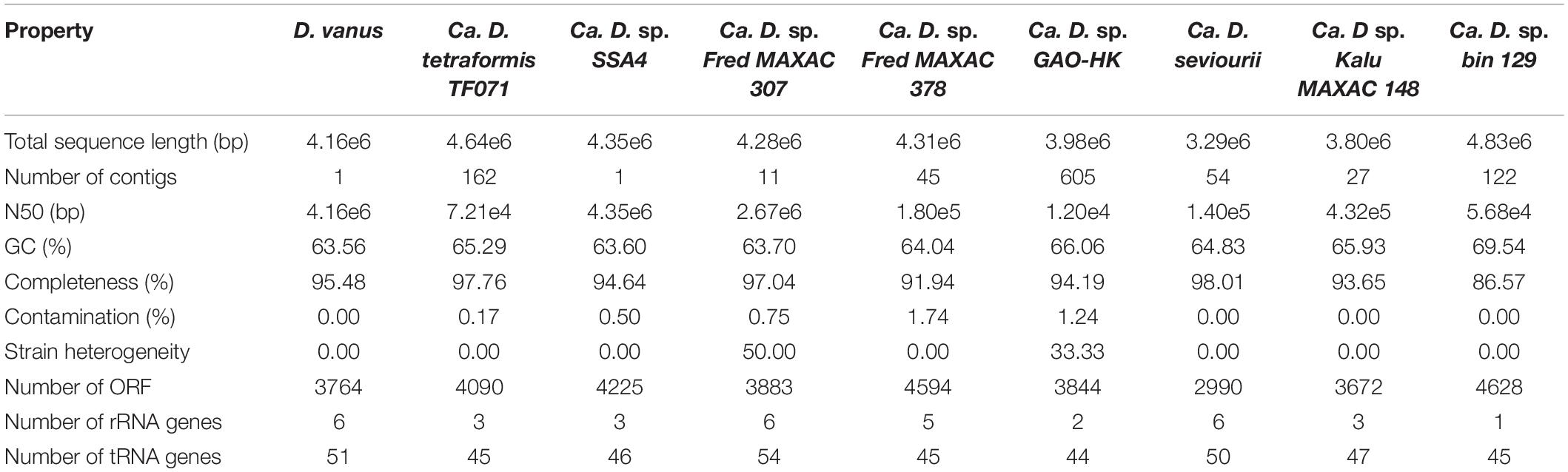
Table 1. Properties of Defluviicoccus genomes examined in this study.
Data Availability
Raw sequence data from both long and short read sequencing are available from NCBI Short Read Archive via BioProject accession PRJNA635277. The D. vanusT chromosomal and plasmid genomes are available via GenBank accessions CP053923.1 and CP053924.1, respectively.
Results and Discussion
Genome Assembly, Annotation, and Overview of Gene Level Annotation for D. vanusT
Assembly of the long read genome sequence data from D. vanusT yielded two circular contigs. The longer contig was 4.1 Mbp in length (coverage 100× from long read data, and 800× from short read). The other was 70 kbp in length (coverage 30× from long read and 150× from short read data) and is discussed below. The longer contig met the MIMAG criteria (Bowers et al., 2017) of a high-quality genome, as supported by CheckM-derived completeness and contamination values, observed as 95.48 and 0.0%, respectively. A total of 3,764 protein-encoding genes, including two complete ribosomal RNA gene operons and 51 tRNA encoding genes, 1 tmRNA gene, and 86 miscellaneous RNA genes were identified, together with four repeat regions associated with CRISPR repeat sequences (Table 1 and Figure 2A). The structure of the cumulative GC plot (Grigoriev, 1998) was consistent with the presence of a single replication origin (Figure 2B).
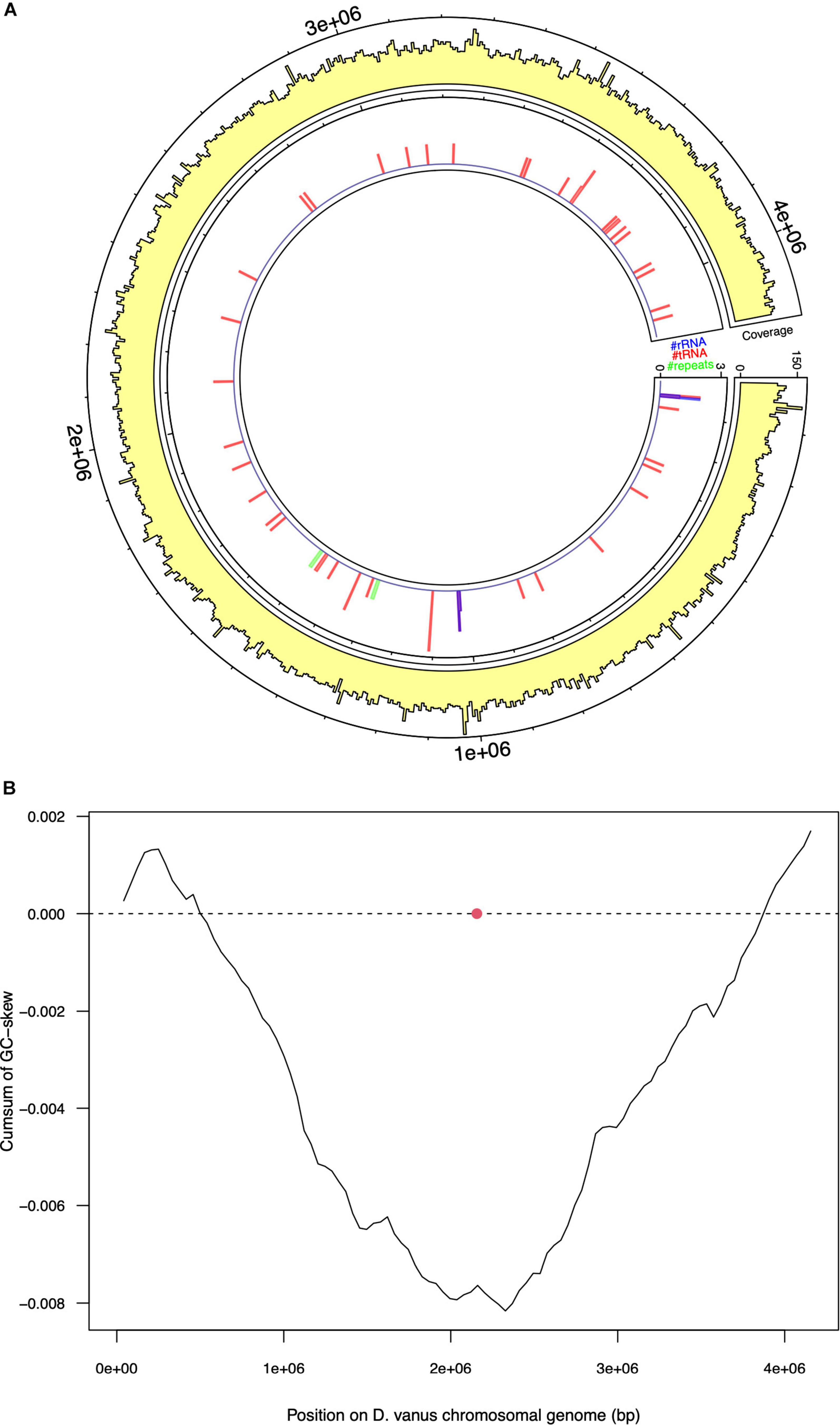
Figure 2. Selected features of the structure and composition of the D. vanus chromosomal genome. (A) Circular visualization of the D. vanus chromosomal genome. The outmost tracks show the mean coverage estimated from mapping long reads to the genome, with per-base coverage binned into 5-kbp non-overlapping windows; the innermost track shows positions of rRNA genes (blue), tRNA genes (blue), and repeat regions (green) with counts obtained within the same 5-kbp windows as above. (B) Cumulative GC-plots showing the origin and terminus of replication, with the red closed circle showing the genomic position of the chromosomal replication initiator protein (dnaA) gene (CDGBEKEE_02056; Supplementary Data File 1).
Annotation of the entire contig sequence with GTDB-Tk and individual 16S rRNA encoding genes (see further analyses below), using SILVA, confirmed its placement to the genus Defluviicoccus. Based on these data, we conclude that the longer contig is the chromosomal genome sequence of D. vanusT, which showed a high degree of similarity to the draft genome recovered from the short read sequence obtained from the same DNA aliquot (Supplementary Figure 1), with the concordance (κ) statistic holding a value of 0.98 (Arumugam et al., 2019, 2021) (Supplementary Figure 2).
Of the 3,764 protein encoding genes on the chromosomal genome, 1,989 (52.8%) were assigned a functional annotation by Prokka, with the remaining 1,775 (47.2%) classified as hypothetical proteins. From a separate analysis against KEGG, 2,476/3,764 genes (65.8%) were assigned a KEGG Orthology (KO) annotation (representing 1,642 unique KO identifiers) and 386 were annotated to 299 unique KO identifiers that were members of 139 KEGG Modules (Supplementary Data File 1).
The shorter contig was hypothesized to be a non-chromosomal replicon, because of its short length and circularity. A total of 67 protein-coding genes were detected on this shorter contig, of which 10 were classified as hypothetical proteins. All 67 were annotated to a KEGG Orthology identifier but with no KEGG Modules being implicated (Supplementary Data File 2). The contig contained gene modules known to be associated with replication (rep, par) and propagation (Type IV secretion system), and consequently is most probably a conjugative plasmid (Norman et al., 2009). Adaptation modules encoding various metal resistance systems, including a complete czc operon were observed. BLAST nucleotide analysis of this plasmid genome showed limited sequence similarity to any sequences in NCBI nt (5.8% query cover on the top ranked subject sequence; see Supplementary Data File 3), suggesting that it had not been described previously.
Phylogenetic Analyses of D. vanusT and Other Defluviicoccus Genomes Using 16S rRNA and Whole Genome Sequences
The topology of the 16S rRNA tree constructed from sequences from these nine Defluviicoccus showed the expected pattern groupings of previously identified clades I–IV members (Figure 3), with the 16S rRNA gene from the recovered D. vanusT genome being closest to those recovered earlier from this isolate (Maszenan et al., 2005). D. vanusT fell into the same clade, Clade 1, as did Ca. D. tetraformis TF071, while clade 2 contained Defluviicoccus GAO-HK, as expected, together with Defluviicoccus SSA4, and two recovered genomes from the treatment plant at Fredericia Jutland in Denmark (FRED MAXAC 307 and 378). The sequence from Ca. D seviourii located within the Clade III, as expected (McIlroy et al., 2010), while the bin129 genome sequence obtained from the marine habitat, and attributed to Defluviicoccus (Slaby et al., 2017) and the sequence of the other Danish bacteria population from the Kalundborg and Sjaelland (KALU MAXAC 148) wastewater treatment plants appeared in separate clades, and quite distinct from pre-existing clades members I–IV (Figure 3). Thus, we propose here the formation of a new clade (V) to accommodate the KALU MAXAC 148 population. The systematics of the source organism of the bin129 genome will be addressed later.

Figure 3. Ribosomal SSU-rRNA gene phylogram showing clusters containing members of the genus Defluviicoccus. Note the recapitulation of previously defined Cluster groups (I–IV) as well as evidence of a new cluster (V) associated typified by 16S rRNA sequences from the sample from Kalundborg WWTP (for further details, see Section “Materials and Methods: Comparative Analysis of 16S-SSU rRNA Genes”).
Average nucleotide identity (ANI) comparisons between all pairs of the nine Defluviicoccus genomes (Table 1) were within the range: 76–80%, consistent with each being, at minimum, a distinct species, according to the interpretation of Jain et al. (2018).
A whole genome phylogenetic analysis (Figure 4 and Supplementary Figure 3), while having fewer and more sparsely populated clusters, reflects closely the relationships revealed by the 16S rRNA gene sequence analyses. Thus, the D. vanusT genome sequence appears in the same clade as that of Ca. D. tetraformis TF701, and those from the populations in Hong Kong (GAO-HK), Singapore (SSA4), and the Fredericia plant in Denmark (FRED MAXAC 307 and 378), all clustered in the same clade. The Ca. D. seviourii genome was adjacent to these six genomes, and that from the Kalundborg WWTP, Sjaelland Danish plant (KALU MAXAC 148) was adjacent to the other seven genomes above. The bin 129 genome sequence reported by Slaby et al. (2017) was distinct from all these Defluviicoccus sequences, assuming a distant position to other members in the tree (Supplementary Figure 3).

Figure 4. Whole genome phylogenetic structure of members of the genus Defluviicoccus, based on genomes listed in Table 1 and closely related draft and reference genomes. The Defluviicoccus specific cluster is shown here and the full tree is provided as Supplementary Figure 3. Note that the Slaby bin 129 genomes assumed a distal position with respect to this region and is not shown (see Section “Methods: Comparative Analysis of Defluviicoccus Genomes”).
Are Members of Defluviicoccus Misplaced in the Family Rhodospirillaceae or Should They Be Relocated in the Geminococceae as Proposed by Hördt et al. (2020)?
Based on Genome Taxonomy Database (GTDB) systematics of the Ca. D. seviourii extant genome, Onetto et al. (2019) suggested that Defluviicoccus should be removed from the family Rhodospirillaceae and instead reclassified into a candidate family 2-12-FULL-67-15. Furthermore, in a recent reorganization of the Alphaproteobacteria, based on 16S rRNA gene sequences, it was proposed that members of the genus Defluviicoccus should be placed within the family Geminicoccaceae, with the caveat that Defluviicoccus may be assigned to its own family if sufficient evidence became available from additional whole genome data (Hördt et al., 2020).
The 16S rRNA sequence analysis was expanded here to examine more closely the proposals by Hördt et al. (2020), where Defluviicoccus and Tistrella were considered for placement within a single family, the Geminicoccaceae (order Rhodospirillales), rather than their existing location within the family Rhodospirillaceae (Figure 5). Direct comparison of per-group proportion nucleotide identity (PNI) statistics (Figure 5) showed that the median differences in PNI between both Defluviicoccus and Tistrella and Geminicoccaceae were larger than those observed between both Defluviicoccus and Tistrella and the Rhodospirillaceae, with the former distances being larger than the median of the inter-family distances observed within members of the Rhodospirillales. However, the majority are still above the conservative lower threshold for inter-family variation as defined by Yarza et al. (2014). The differences between Defluviicoccus and Tistrella are consistent with these two genera being related at either intra-family or intra-order level. Therefore, while it seems clear that sequences from members of the genus Defluviicoccus and those from members of the genus Tistrella show a magnitude of difference inconsistent with them both belonging to the family Geminicoccaceae, as proposed by Hördt et al. (2020), these findings support the view that members of the genus Defluviicoccus are best considered as a separate family within the order Rhodospirillales.
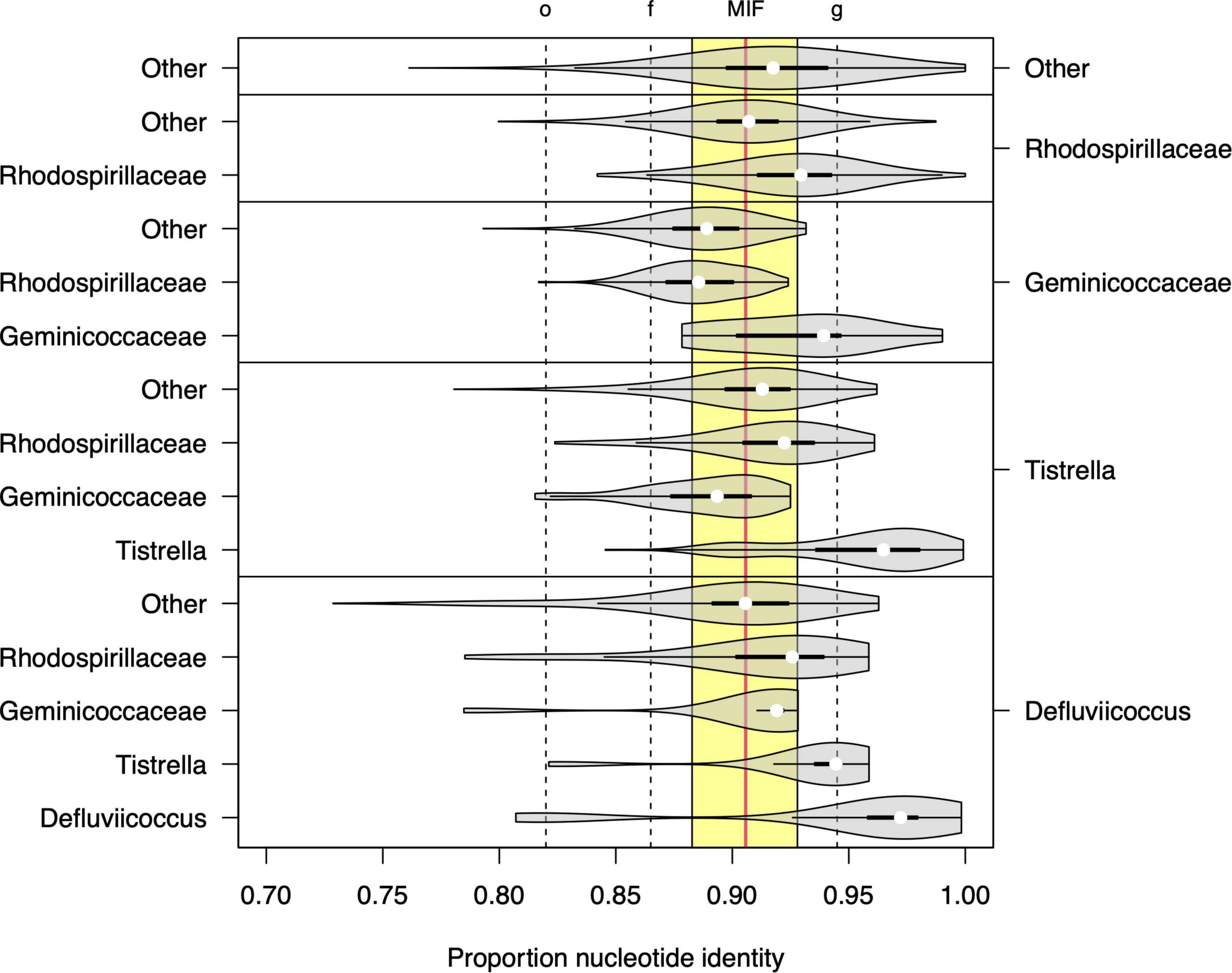
Figure 5. Sequence identity statistics of full length 16S SSU-rRNA sequences among members of genera Defluviicoccus and Tistrella, the families Geminicoccaceae and Rhodospirillaceae, and other members of the order Rhodospirillales. Each violin plot shows the distribution of the proportion of nucleotide identity (PNI) between members of two microbial groups (listed on the right and left axes, respectively). The vertical dashed lines show the values of the minimal inclusion thresholds for different taxonomic levels (g, genus; f, family; o, order) defined by Yarza et al. (2014) and the yellow region delineates the range of mean PNI values from intra-family groups within order Rhodospirillales, with the overall mean shown as vertical solid red line (denoted as MIF: median inter-family).
This situation highlights that much is still to be learned about Defluviicoccus systematics, shown by a recent global survey of activated sludge plants of many different configurations (Dueholm et al., 2021) and based on 16S rRNA sequence data to consist of at least 18 putative new species. What is not yet clear is whether they all belong to the currently existing five clades, or additional clades are required to accommodate them.
General Features of the Defluviicoccus Genomic Sequence Data
The genomic data reveal potential aspects of behavior of these organisms not exposed by experimental data, suggesting that some of these genes may not be expressed in wastewater treatment plants, at least. Thus, all the Defluviicoccus genomes, with one exception (genome bin 129), but including the filamentous Ca. D. seviourii, contained genes encoding the production of components of bacterial flagellar synthesis (Supplementary Data File 1). This is despite Defluviicoccus never having been seen to show motility or to possess such organelles. They also possess several genes encoding proteins dedicated to chemotaxis (Supplementary Data File 1), although not in the genomes of D. vanusT, Defluviicoccus clone SS4, Ca. D. seviourii clone, and genome bin129.
Equally interesting is that genomes of all except Defluviicoccus bin 129 possess the kaiC gene, and, except in the case of the KALU MAXAC 148 genome, the kaiB gene, which encode circadian clock-related proteins (Dvornyk et al., 2003). Furthermore, some have the genes associated with conjugation. Thus, D. vanusT, Ca. D. tetraformis, Defluviicoccus GAO-HK, and FRED MAXAC 307 and 378 have the conjugal transfer protein encoding gene traG, while Defluviicoccus SSA4, Ca. D. seviourii, genome bin 129, and MAXAC 148 possess the genes encoding the pilus assembly protein cpfA and the conjugal transfer protein cpaF. The bin 129 genome alone has the gene for the conjugal mating pair stabilization protein traN (Supplementary Data File 1). With the exception of D. vanusT, Defluviicoccus GAO-HK, and Defluviicoccus strains MAXAC 307 and 378, they also possess the genes for complete or partial synthesis of pili.
Genes Involved in Substrate Uptake in Defluviicoccus
The D. vanusT genome contains the genes acsA (acetyl-CoA synthetase, EC6.2.1.1), ackA (acetate kinase, EC 2.7.2.1), and pta (phosphate acetyl/butyryltransferase, EC2.3.1.8), allowing assimilation and activation of acetate and involving the cation/acetate symporter actP (Table 2). These were also present in all but the bin129 genome, which lacked genes ackA and pta, although their copy numbers varied markedly in the remaining members (Supplementary Data File 1). Genes encoding acetyl CoA synthetase were also present in all Defluviicoccus genomes in multiple copies (Table 2 and Supplementary Data File 1). Additionally, all Defluviicoccus genomes, except KALU MAXAC 148 and that of bin129 genome, contained a gene encoding the succinate-acetate/proton symporter satP (Sun et al., 2018).
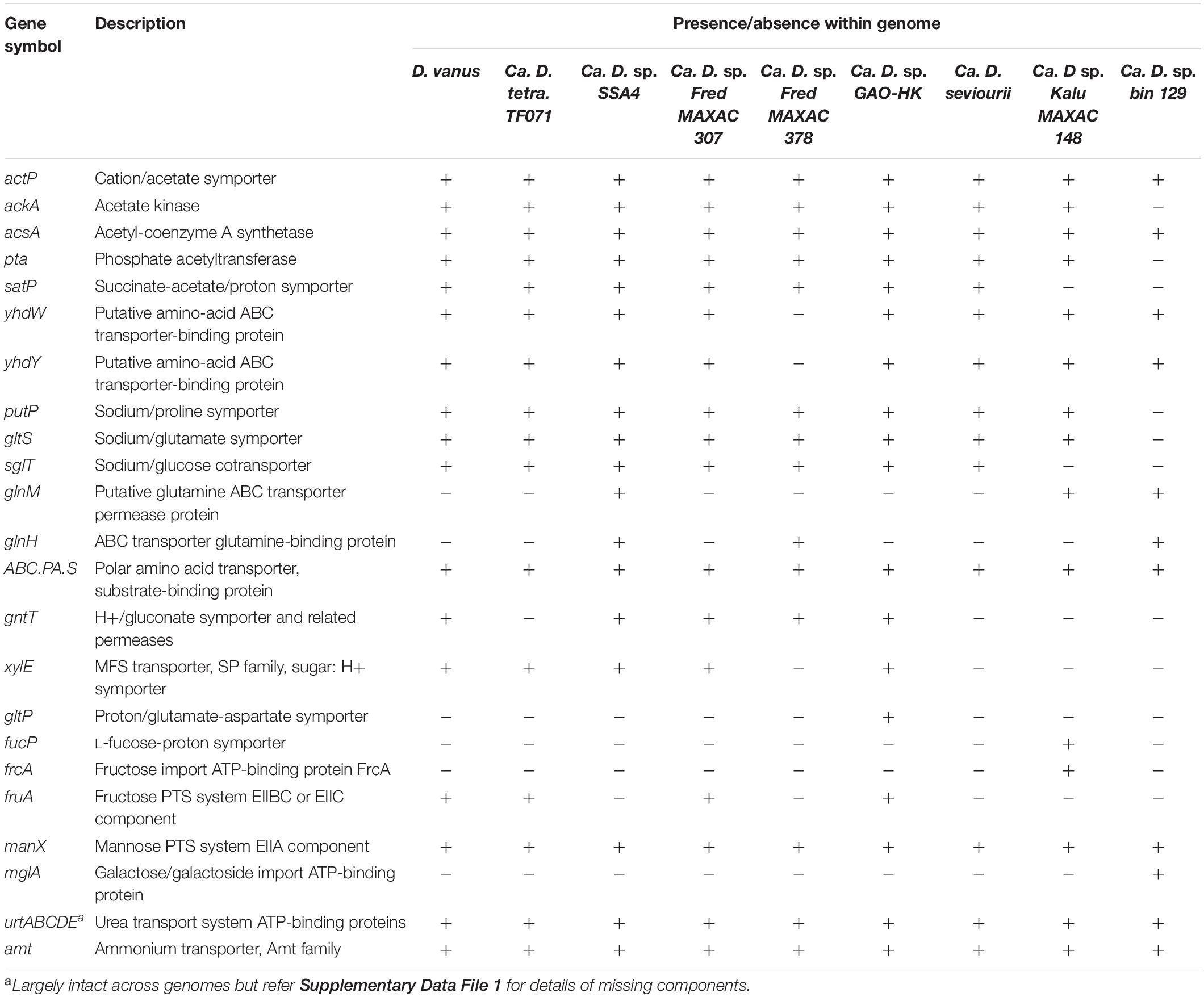
Table 2. Selected genes related to substrate and amino acid uptake present in genomes from members of the genus Defluviicoccus.
All Defluviicoccus genomes except bin129 contain the genes encoding a propionyl CoA synthetase, responsible for activation of propionate and a propionyl CoA carboxylase, which converts propionyl CoA to methyl malonyl CoA. However, no corresponding genes encoding the cation/propionate symporter (pctP) were identified, and while the succinate-acetate/proton symporter, satP was observed in some genomes, there is no direct evidence that it can transport propionate (Sun et al., 2018). Thus, all genomes have dedicated genes for uptake and subsequent metabolism of acetate but not for propionate (Supplementary Table 1), which seems to suggest, as Burow et al. (2008a) did, that acetate and propionate share the same transporter/s in Defluviicoccus. Its members appear to assimilate propionate at a higher rate than acetate (Oehmen et al., 2005a; Dai et al., 2007), and is the preferred substrate when both are available. However, although Ca. Competibacter has a negligible rate for assimilation of propionate, its assimilation by Ca. Accumulibacter is faster than Defluviicoccus (Oehmen et al., 2005a,b), suggesting different Km values for the two substrates. Furthermore, unlike Defluviicoccus, there was no delay in their assimilation rates when supplied alternatively in the feed (Lu et al., 2006), which has led to the suggestion (Oehmen et al., 2005a,2006) that regularly alternating the feed substrate would provide an elegant strategy for controlling the abundances of both Ca. Competibacter and Defluviicoccus GAO. However, its feasibility for use in full-scale EBPR plants has received little interest.
Whether Defluviicoccus can assimilate butyrate under anaerobic conditions is still the subject of controversy, and the scant literature available suggests that not all members have this ability (Burow et al., 2008a; He and McMahon, 2011; Begum and Batista, 2014; Izadi et al., 2020). This character may be one of many that are not present universally in all Defluviicoccus populations. Certainly, the enzymes potentially involved in butyrate metabolism were not distributed equally among these genomes (Supplementary Data File 1). Thus, genome bin 129 alone encoded genes (bcd; EC1.3.8.1) for butyryl CoA dehydrogenase. Only Ca. D. tetraformis lacked that encoding 3-hydroxybutyryl CoA dehydrogenase (EC 1.1.1.157; K00074 synonyms paaH, hbd, fadB, and mmgB) enabling the remainder to convert 3-hydroxybutyrate to butyryl-CoA. Genes encoding isobutyryl-CoA mutase (EC 5.4.99.13; K11942, icmF) were found in all genomes except Ca. Defluviicoccus HK-GAO, Ca. D. tetraformis, and genome bin 129, while 4-hydroxybutyryl dehydratase (EC 4.2.1.120; K14534, abfD) was encoded in all genomes except Defluviicoccus MAXAC 307 and 378, and the genome bin 129. No genes encoding butyryl-CoA synthetase (EC 6.2.1.2) were found in any of these genomes, suggesting that growth on butyrate is unlikely for the strains examined here, although whether the same system/s used to transport and activate acetate and propionate are used for butyrate remains unresolved by this analysis. In Escherichia coli, the satP transporter discussed above has been shown to have co-selectivity for butyrate (Sun et al., 2018), although no related data yet exist for D. vanusT. Wang et al. (2021a) showed that at high temperatures, where Defluviicoccus was thought to have a competitive advantage over Accumulibacter PAO (Lopez-Vazquez et al., 2009), butyrate impacted negatively on Defluviicoccus abundance. More recently, however, Wang et al. (2021b) have shown that at these high temperatures, highly enriched cultures of D. vanusT assimilated both butyrate and isobutyrate, albeit at much slower rates of assimilation than either acetate and propionate, and uncommon mid-chain (6–14 carbons) poly-β-hydroxyalkanoates, polyhydroxy-4-methylvalerate (PH4MV), polyhydroxyhexanoate (PHHX), and polyhydroxy-2-methyl hexanoate (H2MHX) were synthesized by them anaerobically.
The genome sequence data presented here suggest that D. vanusT and all the other Defluviicoccus genomes contain genes encoding diverse ABC amino acid transporters (Supplementary Data File 1), and with the exception of Defluviicoccus FRED MAXAC 378, the aap systems for transporting L-amino acids, namely, yhdW and yhdY (Table 2), permitting them to assimilate a wide range of amino acids, despite not always being implicated in earlier FISH/MAR data (Burow et al., 2008a). Not all the encoding genes for these transport systems were present universally in these genomes, and so while all possessed the genes (liv family) for transport of branched amino acids (Supplementary Data File 1), those encoding the Na+-linked symporters, namely, Na+/proline (putP; K03307), Na+/glucose (sglT; K03307), and Na+/glutamate (gltS; K03312) symporters, were present only in D. vanusT, Defluviicoccus GAO-HK, Ca. D. seviourii (consistent with the analysis of Onetto et al., 2019), and Ca. D. tetraformis (Table 2). Equally, individual copy numbers of most of these genes varied with the strain (Supplementary Data File 1). Polar amino acid dedicated transporter genes (glnM, COG0765, and K09970) were found in Defluviicoccus SS4, genome bin129, and KALU MAXAC 148, while glnH (COG0834) was found in Defluviicoccus SS4, genome bin 129, and FRED MAXAC 378. Substrate binding polar amino acids (ABC.PA.S; COG0834, K02030) were present in all Defluviicoccus genomes (Table 2).
Considerable disagreement exists in the literature as to whether all Defluviicoccus can grow anaerobically on glucose or other sugars as sole carbon sources, although FISH/MAR has shown that Ca. Competibacter denitrificans can assimilate glucose under both aerobic and anaerobic conditions (McIlroy et al., 2014). Maszenan et al. (2005) managed to culture D. vanusT with glucose aerobically as sole carbon source, but it grew very slowly. Wong and Liu (2007) have shown with FISH/MAR that Defluviicoccus-related tetrads in a laboratory reactor could utilize acetate, lactate, propionate, and pyruvate, but not aspartic acid and glucose into PHA under anaerobic condition, yet when D. vanusT was cultured in continuous anaerobic–aerobic conditions, it took up glucose with concurrent glycogen consumption and PHA production, the assumption being that it was the same strain.
Genomic analyses revealed the presence of a gene encoding a fructose import ATP binding protein in Defluviicoccus KALU MAXAC 148 (frcA; K10554), a fructose PTS system (ELLBC/fruA, EC:2.7.1.202) in D. vanusT, Ca. D tetraformis, Defluviicoccus GAO-HK, and Defluviicoccus FRED MAXAC 307, while the mannose PTS system (ELLA component, manX, EC:2.7.1.191) was present in all genomes. A galactose/galactoside import encoding ATP protein (mglA) was seen only in the genome bin129.
Genomes of D. vanusT, Ca. D. tetraformis, Defluviicoccus sp. SSA4, and Defluviicoccus FRED MAXAC 378 each possessed a single gene copy encoding a glucose/mannose co-transporter (glcP, Table 2). It was not identified in the genomes of Ca. Defluviicoccus GAO-HK, Ca. D. seviourii, or Defluviicoccus KALU MAXAC 148. Given that both Defluviicoccus GAO-HK and Defluviicoccus KALU MAXAC 148 possessed one or more copies of a sodium/glucose symporter gene (sglT), it seems probable that potentially these strains may assimilate glucose, while those with neither gene, i.e., Defluviicoccus FRED MAXAC 307, nor the genome bin29 may be unable to.
All possessed transporter encoding often several copies of the genes for urea (urt family) and ammonium (amt family) transport, although a sulfate transporter gene was found only in the genomes of D. vanusT, Ca. D tetraformis, and Defluviicoccus KALU MAXAC 148 and FRED MAXAC 378 (Supplementary Data File 1).
Mechanisms of Anaerobic and Aerobic Substrate Uptake in Defluviicoccus Strains
Active transport processes are required in Defluviicoccus for anaerobic acetate and propionate membrane transport, where acetate and propionate probably share the same transporter. Burow et al. (2008a,b) used metabolic inhibitors in attempts to resolve details of the transport mechanisms with a highly enriched culture of clade 1 Defluviicoccus. While inhibitor selectivity is always a concern with such approaches, their data were consistent with anaerobic acetate assimilation depending on both a proton motive force (pmf) and a Na+ potential, with the former being the main contributor. They suggested that ATPases and P efflux through the membrane (involving the pit transporter) played no role in pmf establishment and thus was quite different to the situation in Ca. Accumulibacter PAO and Ca. Competibacter (Saunders et al., 2007; Seviour and McIlroy, 2008; Oehmen et al., 2010). Instead, the pmf was more likely established by H+ efflux coupled to electron transport-linked reduction of fumarate in the reductive branch of the TCA cycle (see below). Genes encoding fumarate reductases were present in all the Defluviicoccus genomic sequences examined here, including D. vanusT (Supplementary Data File 1).
Burow et al. (2008a,b) also suggested that the anaerobic Na+ motive force (smf) could be generated by extrusion of a methylmalonyl-CoA decarboxylation-linked efflux of Na+ across the cell membrane. Methylmalonyl-CoA is an intermediate in the succinate–propionate pathway, with the encoding genes for methylmalonyl-CoA decarboxylase present in all except the bin 129 genome, and leading to the formation of 3-hydroxy valeryl-CoA (Seviour and McIlroy, 2008). The key enzyme in this pathway, methylmalonyl-CoA mutase, converts methylmalonyl-CoA to propionyl-CoA, and genes encoding it were detected in all the Defluviicoccus genomes, with the exception of genome bin 129. Again, neither process is found in Ca. Accumulibacter, although a fumarate reductase generated membrane pmf potential was reported in Ca. Competibacter (Saunders et al., 2007; McMahon et al., 2010). The same system probably exists in those Defluviicoccus strains possessing the genes encoding for methylmalonyl-CoA decarboxylase. Although this enzyme is absent from both genome bin 129 and Defluviicoccus KALU MAXAC 148 genomes (Supplementary Data File 1), a fumarate reductase-generated pmf may well be used instead.
Under aerobic conditions, Ca. Accumulibacter can assimilate P by a process driven by a pmf established by P efflux of Pi across the membrane (Saunders et al., 2007; McIlroy et al., 2014) involving the low-affinity pit, and operating when Pi is plentiful. A high-affinity pst system operates at lower Pi levels, or possibly both are used simultaneously (Burow et al., 2008a). It has long been thought (Seviour and McIlroy, 2008; McMahon et al., 2010) that possession of a pit gene distinguished all PAO from the GAO, in that pit appeared to be absent from Ca. Competibacter (McIlroy et al., 2014) and some earlier published Defluviicoccus genomes (Nobu et al., 2014; Onetto et al., 2019), Furthermore, while the pst gene occurred in all genomes examined here, often in multiple copies (Supplementary Data File 1), the pit gene was also detected, but only in Defluviicoccus FRED MAXAC 307, the genome bin 129 genomes, and in Defluviicoccus GAO-HK, as reported previously by Wang et al. (2014). No GAO can store polyphosphate under the aerobic conditions tested to date. Whether these pit genes are homologous to the pit in the PAO, or whether they are expressed are not known. What seems clear is that much remains to be clarified about regulation of P metabolism in Defluviicoccus and the other GAO.
Source of Reducing Power and Energy for Polyhydroxyalkanoate Synthesis by Defluviicoccus Under Anaerobic Conditions
This aspect of GAO molecular physiology has been controversial, as it has been for Ca. Accumulibacter PAO (Oehmen et al., 2007; Burow et al., 2009; Zhou et al., 2010). It is likely to differ fundamentally from the situation in PAO since Defluviicoccus possess glycogen and not polyphosphate as their major stored energy source. Equally controversial is how they balance their intracellular redox under anaerobic conditions. Experimental and genomic data suggest that Ca. Accumulibacter can operate the TCA cycle anaerobically to generate energy in addition to that arising from polyphosphate hydrolysis, and reducing power, since it possesses an unusual cytochrome b/b6, which allows succinate dehydrogenase to function in the forward direction to produce fumarate (García Martín et al., 2006; Skennerton et al., 2015). This transformation step is not possible in Defluviicoccus. Debate has focused on whether these GAO obtain all or only some of their energy and reducing power for PHA synthesis from anaerobic glycogen catabolism via the Embden Meyerhoff Parnas pathway (Burow et al., 2009; Zhou et al., 2010). What now seems more likely is that Defluviicoccus can use the reductive branch of the TCA cycle to balance its intracellular redox. Key features involve fumarate reductase, which converts fumarate to succinate and the associated succinate–propionate pathway leading via methylmalonyl-CoA decarboxylase and methylmalonyl-CoA to PHA storage products. While PHA production from an acetate feed in Ca. Accumulibacter is 3-polyhydroxybutyrate (PHB) only, Defluviicoccus produces both 3-polyhydroxyvalerate (PHV, 25%) and 3-polyhydroxybutyrate (73%) (Zhou et al., 2008). With propionate as substrate, an increased production of propionyl CoA (Oehmen et al., 2007) and the subsequent operation of the succinate–propionate pathway supplying precursors for the methylmalonyl-CoA pathway lead to the synthesis of 3-polyhydroxyvalerate and 3-hydroxy 2 methylvalerate. As Ca. Accumulibacter lacks the methylmalonyl-CoA pathway (Oehmen et al., 2007, 2010), no 3-hydroxy 2 methylvalerate is produced (Seviour and McIlroy, 2008). The genes encoding fumarate reductase, propionyl-CoA-carboxylase, and both methylmalonyl-CoA-decarboxylase, converting methylmalonyl-CoA to propionyl-CoA, and its mutase, converting succinyl-CoA to methylmalonyl-CoA, are found in most of the Defluviicoccus genomes examined here, including D. vanusT (Supplementary Data File 1).
Central Carbon Metabolism in the D vanusT Genome; How Do They Compare to Other Members of the Genus Defluviicoccus?
The D. vanusT genome encodes a complete Emden–Meyerhoff pathway (EMP), tricarboxylic cycle (TCA), gluconeogenesis pathway, and a complete module for pyruvate oxidation to acetyl-CoA (Table 3). These pathways are largely recapitulated across other genomes in members of the genus Defluviicoccus, albeit with missing components, notably in Defluviicoccus FRED MAXAC 378, Defluviicoccus GAO_HK, and the genome bin 129. Such gaps most probably are consequences of fractionated genome assemblies in the case of those constructed from short read data, and instances of reduced gene sequence quality with those constructed with long read data. In contrast to these pathways, the pentose phosphate pathway, the Enter–Doudoroff pathway, and the photorespiration pathway all had substantive numbers of missing enzymes and appear to be non-functional (Table 3).
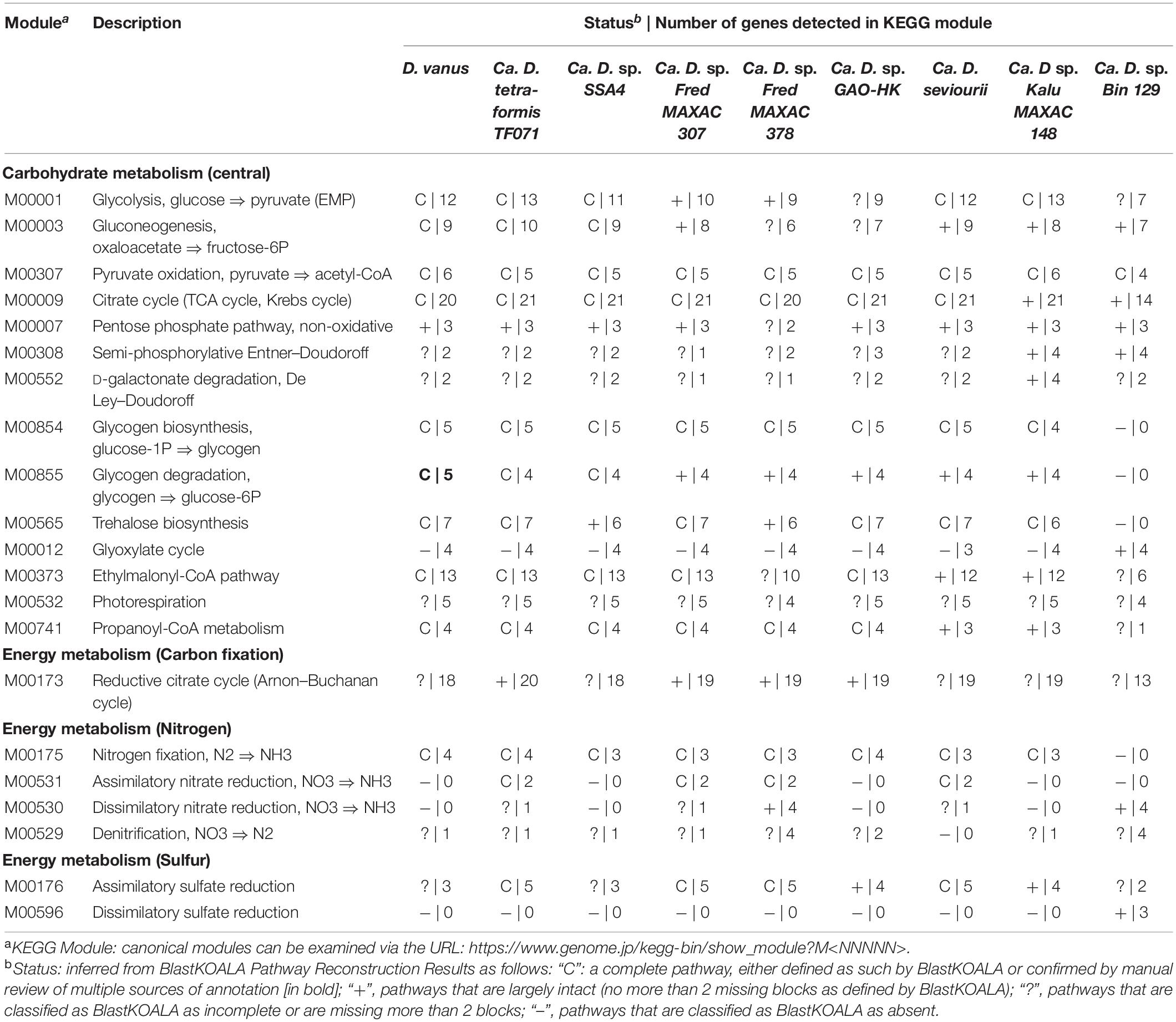
Table 3. Summary of KEGG modules detected in genomes of members of genus Defluviicoccus.
The glyoxylate shunt pathway (KEGG M00012) represents an important mechanism for the conversion of acetyl-CoA into both gluconeogenic and anaplerotic precursors (Ensign, 2006; Renilla et al., 2012), and was classified by BlastKOALA here as only partially complete in all Defluviicoccus genomes, including D. vanusT. With the exception of the genome bin 129, this arose from the absence of the gene encoding isocitrate lyase (aceA; KEGG orthology K01637, EC: 4.1.3.1; also not annotated by Prokka), which is a diagnostic enzyme for this pathway (Supplementary Data File 1). Furthermore, Ca. D. seviourii lacked the gene encoding malate synthase (KEGG Orthology K01638 and EC: 2.3.3.9), as noted previously (Onetto et al., 2019; Supplementary Data File 1). Because all other gene-encoded enzymes of the pathway are also members of the TCA cycle, it was therefore considered to be absent in all genomes, except genome bin 129, where it was also classified as incomplete in missing the mdh gene (malate dehydrogenase; K00024 and EC:1.1.1.37) (Table 3 and Supplementary Data File 1). The presence of this pathway has also been revealed for Ca. Accumulibacter (Burow et al., 2009) based on targeted enzyme inhibitor studies and genome sequence data for Ca. C. denitrificans (McIlroy et al., 2014).
In the absence of the glyoxylate shunt pathway, the ethylmalonyl-CoA pathway (KEGG M00373) is thought to provide an alternative mechanism for conversion of C2 compounds, notably acetyl-CoA (Alber, 2011; Anthony, 2011; Schada von Borzyskowski et al., 2020) to the intermediates malate and succinyl-CoA in the TCA cycle. As annotated by BlastKOALA, the gene encoding (3S)-malyl-CoA thioesterase (mcl2; K14451, EC:3.1.2.30), which converts (3S)-malyl-CoA to malate, was a false negative and its presence was confirmed subsequently in all Defluviicoccus genomes by examining annotations from Prokka (in D. vanusT see gene CDGBEKEE_03341; Supplementary Data File 1). Hence, in the case of D. vanusT, Ca. D. tetraformis TF071, Defluviicoccus SSA4, Defluviicoccus FRED MAXAC 307, and Defluviicoccus GAO-HK, we conclude that the ethylmalonyl-CoA pathway is complete (Table 3 and Supplementary Data File 1). This pathway showed various degrees of incompleteness for the remaining genomes. Hence, the Defluviicoccus FRED MAXAC 378 genome was missing the key genes encoding crotonyl-CoA reductase (ccr; K14446), (2R), and ethylmalonyl-CoA mutase, the diagnostic gene for this pathway (ecm; K14447), and (2S) methyl succinyl-CoA dehydrogenase (mcd K14448), while both Defluviicoccus KALU MAXAC 148 and Ca. D. seviourii genomes lacked the key gene encoding methylmalonyl-CoA/ethylmalonyl-CoA epimerase (epi K05606, EC:5.1.99.1). Thus, either these genes are redundant, the ethylmalonyl-Co-A pathway cannot function in these strains, or they are false negatives as a result of draft genome incompleteness.
Comparative Genomics of Glycogen and Trehalose Metabolism Within Genus Defluviicoccus
Glycogen recycling is a key feature of the GAO phenotype, expressed in the cyclical anaerobic–aerobic conditions operative in EBPR bioprocesses (Lv et al., 2014). Glycogen degradation is thought to be the primary source of reducing equivalents in Defluviicoccus for formation of intracellular PHA, synthesized from either carbon substrates assimilated in the anaerobic feed phase and/or from organic acids derived from catabolism of glycogen (Liu et al., 1997). As mentioned earlier, aerobic degradation of PHA in the aerobic phase provides substrates for glycogen synthesis through gluconeogenesis for which the complete pathway is present in D. vanusT (see above). As annotated by BlastKOALA (Table 3 and Figure 6), and as expected, all other Defluviicoccus genomes with the exception of genome bin 129 contain a complete canonical pathway for glycogen synthesis (KEGG M00854). In the case of glycogen degradation, BlastKOALA appears to have misannotated the glycogen debranching enzyme (glgX) as a false negative. Its presence in all Defluviicoccus genomes was confirmed subsequently from examining the Prokka annotation (CDGBEKEE_01736 in D. vanusT; Supplementary Data File 1).
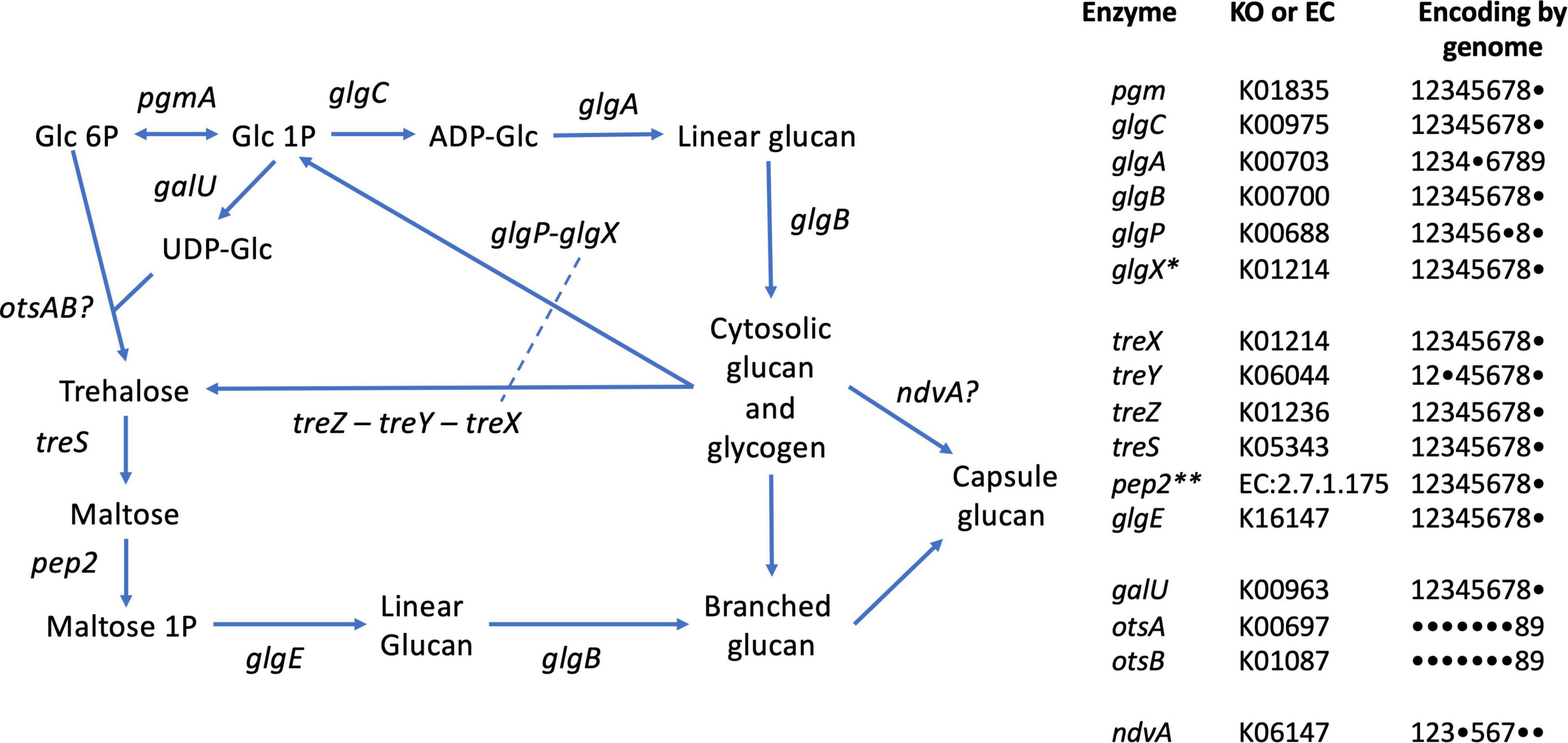
Figure 6. Glycogen and trehalose biosynthetic pathways conserved in members of the genus Defluviicoccus, based on Figure 1 of Chandra et al. (2011). The tabulation on the right lists the enzymes and presence of encoding genes in the 9 genomes analyzed in this study, using the following numeric key: 1: Defluviicoccus vanus; 2: Ca. Defluviicoccus tetraformis TF071; 3: Ca. Defluviicoccus. sp. SSA4; 4: Ca. Defluviicoccus Fred MAXAC 307; 5: Ca. Defluviicoccus Fred MAXAC 378; 6: Defluviicoccus GAO-HK; 7: Ca. D. seviourii; 8: Ca. Defluviicoccus Kalu MAXAC 148; 9: Defluviicoccus bin129. A closed circle • denotes genes not identified in the corresponding genome. Full enzyme names as follows: pgm, phosphoglucomutase; glgC, glucose-1-phosphate adenylyltransferase; glgA, glycogen synthase; glgB, 1,4-alpha-glucan branching enzyme; glgP, glycogen phosphorylase; glgX, glycogen debranching enzyme; treX, maltooligosyltrehalose synthase; treY, maltooligosyl trehalose synthase; treZ, malto-oligosyltrehalose trehalohydrolase; treS, trehalose synthase/amylase; pep2, maltokinase; glgE, alpha-1,4-glucan:maltose-1- phosphate maltosyltransferase; galU, UTP-glucose-1-phosphate uridylyltransferase; otsA, trehalose-6-phosphate synthase; otsB, trehalose-phosphate phosphatase; ndvA, beta-(1– > 2)glucan export ATP-binding/permease protein. *This appears to be a single type of glycogen debranching enzyme in the genus Defluviicoccus; dashed line in pathway schematic. **As described by Chandra et al. (2011), in all genomes except D. vanus, pep2 (maltokinase) formed a single continuous sequence with treS and identity was confirmed by sequence alignments; see Supplementary Data File 4.
The presence of the trehalose biosynthesis pathway in Ca. D. seviourii was noted by Onetto et al. (2019) and is thought to provide flexibility in energy storage and protection against stress (Onetto et al., 2019). Evidence for trehalose acting as an osmolyte in Ca. Accumulibacter has been reported (de Graaff et al., 2021), providing tolerance for saline environments in this group. A similar function for trehalose may be served in Defluviicoccus. D vanusT also contains the genes for the complete trehalose biosynthetic pathway as defined by KEGG (M00565; Table 3 and Figure 6), where the treX (glgX)-treY-treZ genes encode the conversion of glycogen to trehalose (de Graaff et al., 2021). This pathway is present in genomes of the other genus members (Table 3 and Figure 6).
An alternative pathway facilitating reconversion of trehalose to a branched glucan via the production of maltose (Chandra et al., 2011) and mediated by proteins encoded by treS-pep2-glgE/glgE1-glgB is also present in both D. vanusT and all genomes studied here (Table 3 and Figure 6). This latter pathway may be involved in formation of its exocellular capsular material (Chandra et al., 2011). We observed the ndvA glucan export transporter in 6 of the 9 genomes (Figure 6), giving at least some insight into the possible mechanisms underpinning capsule formation.
In contrast, the genes encoding the classical galU-OtsA-OtsB trehalose biosynthetic pathway was only observed in the case of the Kalu MAXAC 148 genome (Figure 6). Furthermore, the Rv3032/Rv3031 non-classical pathway, present in Mycobacterium tuberculosis and associated with the production of specialized glucans (reviewed in Chandra et al., 2011), was not observed in any Defluviicoccus genome.
Pathways Related to Polyhydroxyalkanoate Metabolism in Genus Defluviicoccus
As discussed above, PHA cycling, and its complementary relationships to glycogen cycling in EBPR plants also represent key components of the GAO phenotype. As shown by earlier genomic analyses of other members of the genus Defluviicoccus (Nobu et al., 2014; Wang et al., 2014; Onetto et al., 2019), the D. vanusT genome contains the genes encoding synthesis of the monomers PHB and PHV from acyl-CoA compounds derived from acetate and propionate metabolism, respectively. These are namely phaA and phaB from acetyl-CoA, and fabF and fabG from propionyl-CoA, and most likely butyryl-Co-A (Brune and Schink, 1992), although as discussed above, the fate of butyrate in some members of this genus remains unclear.
D. vanusT also possesses the genes for pathways for alternate PHA-monomer synthesis, related to fatty acid metabolism. These are fabG and phaJ (see genes CDGBEKEE_02222 and CDGBEKEE_02420; Table 4 and Supplementary Data File 1) (Wang et al., 2014). Two PHA synthases, phaC and phaE, respectively, and co-located in the genome, were identified, together with the phaZ PHA depolymerase (KO05973) (Table 4). D. vanusT also harbors genes encoding three PaaU-like proteins, capable of reducing acetoacetyl-CoA to (S)-3-HB-CoA, with the R-3-HB-CoA isomer synthesized by phaB (Sagong et al., 2018). Other PHA proteins identified in the D. vanusT genome include a PhaR-like repressor protein, a putative esterase-type PHB depolymerase and three short phasin proteins that possibly play a role in regulation and formation of the PHA granules (Grage et al., 2009). As discussed previously, catabolic degradation of PHA most probably proceeds through conversion of 3-HB-CoA to acetoacetyl-CoA via crotonyl-CoA (Grage et al., 2009), and the acetyl-CoA can then enter central metabolism with glycogen formation occurring via the gluconeogenesis pathway, as discussed above.
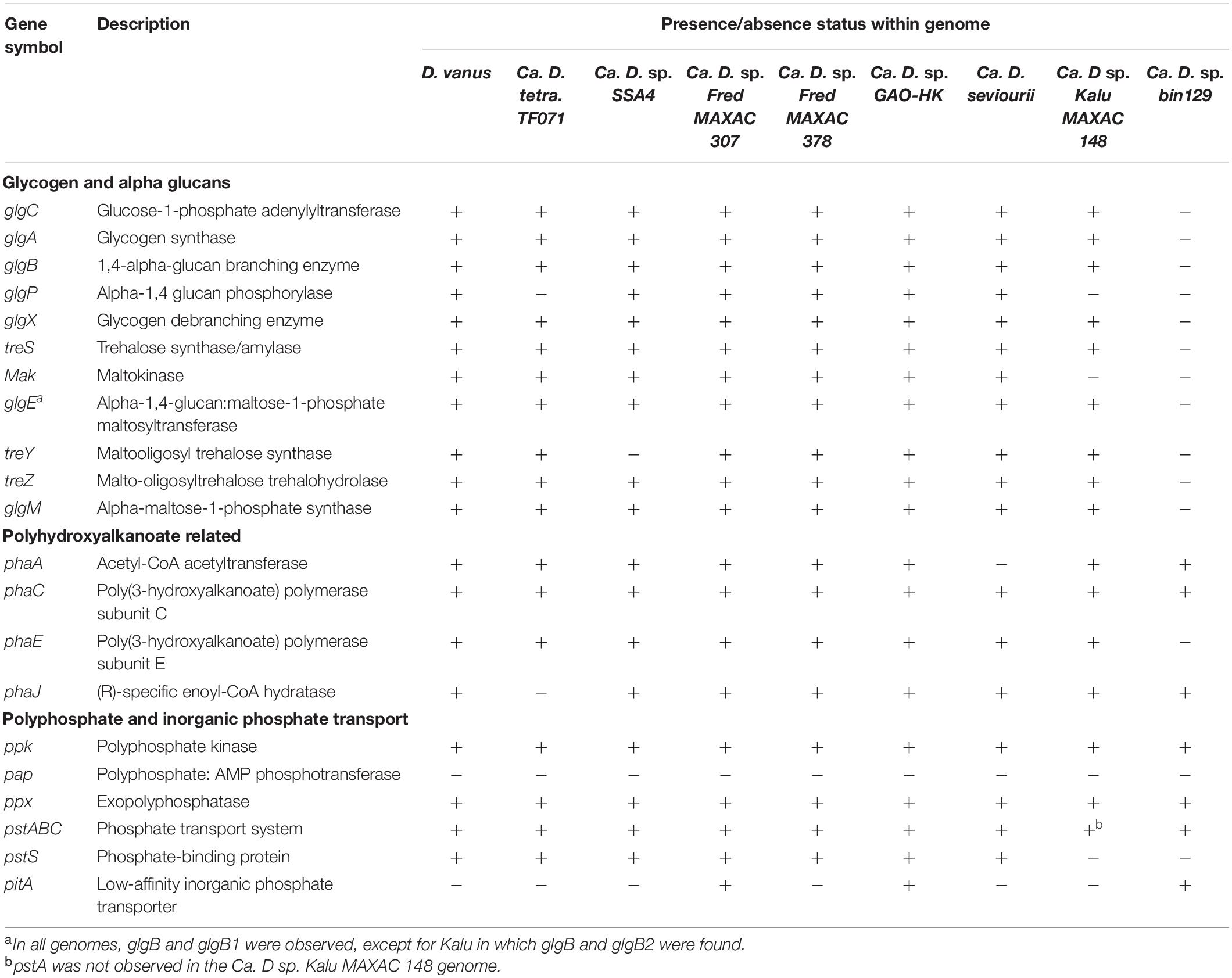
Table 4. Selected genes related to polymeric metabolism in genomes from members of the genus Defluviicoccus.
Pathways Involving Utilization of Phosphate, Nitrogen, and Sulfur
The D. vanusT genome also contains genes essential for polyphosphate synthesis and degradation, as do all Defluviicoccus genomes described here (Table 4).
Although the genes encoding the metabolic pathways discussed above appear mainly to be conserved across the genomes studied here, some variations become evident when examining the metabolism of nitrogen and sulfur compounds. While nitrogen fixation (KEGG M00175) appeared universally present, with the nifHDK operon being present in all Defluviicoccus genomes (Supplementary Data File 1), in D. vanusT, Ca. D. tetraformis, and Defluviicoccus GAO_HK, a complete vnfDGK operon encoding an alternative nitrogenase (Supplementary Data File 1) was present. Of the genes in the canonical denitrification pathway (KEGG Module M00529), only those for the nitrate reductase (nar), nitrite reductase (nir), and nitric oxide reductase (nor) gene families were observed, either singly or in various combinations (Supplementary Figure 4), with the sole exception of Ca. D. seviourii, which contained no genes from this canonical pathway. No other genome encoded a full denitrification pathway (Table 3 and Supplementary Data File 1), in agreement with all earlier physiological data (e.g., Wang et al., 2014). In the bin 129 genome, a partial denitrification pathway was observed, composed of narG, narI, nary, and nirK. Multiple genes in the nar family, namely, narG, narI, and narY, along with norB, were present in the Defluviicoccus FRED MAXAC 378 genome. Across all nine genomes, the most commonly observed gene was norB, which was present in all genomes except those of bin 129 and Defluviicoccus KALU MAXAC 148, suggesting that NO detoxification may be a common capability in members of this genus. In addition to norB, the Defluviicoccus GAO_HK genome also contained the norC gene. In the case of Defluviicoccus KALU MAXAC 148, the only denitrification-related gene found was nirK.
Marked differences were seen with the genes encoding assimilatory nitrate reduction, where those for that pathway were present in Ca D. tetraformis, Defluviicoccus FRED MAXAC 307, and FRED MAXAC 378 as well as Ca. D. seviourii (Table 3).
No genome studied here encoded a dissimilatory sulfate reduction, unlike Ca. C. denitrificans (McIlroy et al., 2014) (Table 3), although variations in genes encoding the assimilatory sulfate reduction pathway were observed in these genomes (Table 3). Thus, only those of Ca. D tetraformis, Ca. D. seviourii, and Defluviicoccus FRED MAXAC 307 and FRED MAXAC 378 possessed the full gene complement. Collectively, these observations highlight the potential for varied niche flexibility among members of genus Defluviicoccus.
Conclusion
This paper reports the recovery of the complete, closed chromosomal genome of the GAO Defluviicoccus vanusT taking advantage of new long read DNA sequencing technology. This approach has also facilitated recovery of a complete, closed genome of a conjugative plasmid, which can be challenging otherwise to reconstruct from short read DNA sequencing data alone (Arredondo-Alonso et al., 2017). We have provided here a complete set of gene annotations for all the currently available Defluviicoccus genomes in a form that can be used for systematic comparative analysis and genomic mining (Supplementary Data File 1). Both the raw sequencing data and the assembled genome sequences are publicly available from NCBI.
Phylogenomic functional comparisons in combination with recent whole genome sequences from other members of genus Defluviicoccus (Nobu et al., 2014; Wang et al., 2014; Onetto et al., 2019) were then conducted. The 16S rRNA gene and whole genome sequence comparisons give outcomes consistent with the previously proposed clade structures of members of this genus, as well as also suggesting the presence of at least one previously unrecognized clade/cluster, which we denote here as clade V (Figure 4). The proposed new clade is most evident in the 16S analysis, and includes Defluviicoccus Kalu MAXAC 148, but not the Bin 129 genome, and is consistent with phylogenetic analyses conducted using the more sparsely sampled whole genome datasets. As discussed above, we anticipate further delineation of the taxonomic structure of genus Defluviicoccus, as a consequence of the increasing numbers of both complete genome sequences, and full-length ribosomal SSU-rRNA sequences, that are becoming available (Dueholm et al., 2020; Arumugam et al., 2021; Singleton et al., 2021). The data presented here have confirmed much, but not all, of the earlier data generated for the then recognized clades by experimental methods (Burow et al., 2007, 2008a,b; Wong and Liu, 2007). Comparative analysis against 16S rRNA sequences from all members of the order Rhodospirillales does not support the view that Defluviicoccus should be moved into the family Geminicoccaceae, but is consistent with the notion that Defluviicoccus forms a distinct family within the Rhodospirillales.
Our interpretations are reliant on the quality of genome sequences included in this analysis. Of the genomes analyzed, only two are complete, closed (single chromosome) sequence, namely, those of D. vanusT and Defluviicoccus SSA4, with the remainder being fractionated with contig numbers varying between 11 and 605 (Table 1). With the exception of the bin 129 genomes, all other genomes analyzed here are classified as high-quality draft genomes (as defined by the accepted MIMAG criteria). Notwithstanding that in the case of metagenome-assembled genomes (MAG), such fractionated genomes may harbor contaminant sequences (false positives) from non-cognate genomes and/or remain incomplete from the failure to incorporate all cognate sequences in the draft genome (false negatives), and both may occur in some of the genome sequence data described here.
Interestingly, the bin 129 genome derives from a marine, not wastewater source, and this might contribute to its substantive differences with the other eight, resulting from the imposition of quite different selection pressures. Alternatively, the distant position of sequence bin 129 in both the 16S rRNA and genome phylograms suggests that this genome arises from another group within the Rhodospirillaceae (Figure 4). Notably, the bin 129 genome lacks the genes considered to define the GAO phenotype, with none detected encoding the pathways for glycogen biosynthesis or degradation. At an estimated 86% completeness and <1% contamination, the bin 129 genome is close to being assessed as “high quality” and so the absence of genes for those pathways is probably real and does not result from false negatives associated with draft genome incompleteness.
Although these phylogenomic data support the presence of the extant metabolic features expected of populations possessing the GAO phenotype, one striking feature of their genomes is the functional diversity among them, especially in terms of their substrate preferences and possession of the Pit gene. This is not surprising given the similar clade diversity known to occur in both Ca. Accumulibacter (Flowers et al., 2013; Kolakovic et al., 2021) and Ca. Competibacter (McIlroy et al., 2014). Our analysis of glucan metabolism (glycogen and trehalose) shows that both classical (glgC-glgA) and non-classical pathways (treS-pep2-glgE) are present (Chandra et al., 2011); it remains open as to whether some members possess additional alternative pathways (i.e., the Kalu MAXAC 148 genome contains the galU-otsA-otsB trehalose biosynthesis system, not observed in the other genomes), as seen, for example, in members of the genus Mycobacterium (Koliwer-Brandl et al., 2016). A related area of exploration should involve possible interrelationships between intracellular and extracellular forms of glucan polymers, which may shed more light on their mechanisms of ecophysiological adaptation.
These functional variations are seen even among members of the same clade and serve to emphasize again how dangerous it is to make generalized predictions of their behavior in wastewater treatment plants. Consequently, any studies involving them should identify unequivocally which Defluviicoccus clade member is under examination, which is now possible from full-length 16S rRNA amplicon sequencing (Dueholm et al., 2020). The data here and those of Dueholm et al. (2020) should permit new, highly targeted FISH probes to be designed to cover the increasing diversity of Defluviicoccus now known to exist.
The analysis of the gene content of the D. vanusT plasmid sequence clearly suggests that it is classifiable as a conjugative plasmid, based on the presence of gene clusters for its replication and propagation (Norman et al., 2009). The plasmid also contained at least one metal resistance operon (czc), which may convey tolerance to mercury. Recently Ma et al. (2019) have documented the presence of a Defluviicoccus population in mercury-contaminated soil, although its precise identity remains unknown.
Whether the sequence data can provide clues on how each of these organisms might be better grown in axenic condition remains to be seen: the advantage of having pure cultures of these and other strains is of paramount value. For example, there would be no need to rely on highly enriched populations in attempts to understand in situ physiology under different conditions, and the generation of genome sequence data would be less equivocal than in the case of metagenome-assembled genomes. With pure cultures, it would be easier to elucidate the factors affecting regulation of enzyme synthesis and activity of key enzymes, using transcriptome and proteomic approaches, and interpretation of complementary functional assays, such as FISH/MAR (Carr et al., 2006), nanoSIMS (Musat et al., 2012), or Raman-FISH spectroscopy (Huang et al., 2007; Fernando et al., 2019). For example, the difficulty experienced growing D. vanusT on GS medium, with glucose, chosen somewhat arbitrarily as its carbon source (Maszenan et al., 2005), may be overcome by replacing it with either acetate and or propionate under alternating anaerobic/aerobic conditions and fine-tuning medium composition based on its gene complement and known phenotype.
Data Availability Statement
The D. vanus chromosomal and plasmid genomes are available via GenBank accessions CP053923.1 and CP053924.1, respectively. The corresponding raw sequence data is being released via NCBI BioProject accession PRJNA635277.
Author Contributions
RS proposed the study. IB, AM, and RW designed the experiments. IB and NS performed experimental work. IB performed long read sequencing. IB, AM, RS, and RW designed the analyses. IB, MH, KA, and RW performed data analysis. All authors were involved in data interpretation. RS and RW primarily wrote the manuscript, with inputs from other authors in specific areas.
Funding
This work was supported by the Singapore National Research Foundation and Ministry of Education under the Research Centre of Excellence Programme.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Stuart Lorimer (NCIMB) for provision of freeze-dried D. vanusT culture; Kathryn Eales, Tong Zhang, Zhiping Wang, Wen-Tso Liu, Caitlin Singleton, Per Halkjaer Nielsen, and Mads Albertsen for providing Defluviicoccus whole genome sequence data; Daniela Drautz-Moses and members of the SCELSE sequencing laboratory for their assistance in obtaining short read Illumina data; and Art Matysik and Foo Yong Hwee for assistance with imaging work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.834906/full#supplementary-material
Supplementary Figure 1 | GC-coverage plot of the short read assembly from Defluviicoccus vanusT. The rectangular region delineated with dashed lines contains the contigs considered to arise from the chromosomal genome, which is denoted as bin 0.2 in the analysis presented in Supplementary Figure 2.
Supplementary Figure 2 | Summary of the concordance-statistic analysis between the D. vanus genome recovered from long read sequence data (denoted as LR-chr 1) and the D. vanus genome recovered short read data (denoted as bin 0.2; see Supplementary Figure 1). Both sets of sequence were obtained from the same DNA aliquot. The concordance statistic (κ, kappa) was computed using the srac2lrac from BLASTN alignments (see Section “Methods: Short Read Sequencing and Data Analyses and Arumugam et al. (2021) for Further Details]. (A) Distribution of κ-statistics from comparison of LR-chr 1 against genome bins recovered from the corresponding short read assembly; the highest scoring genome bin (bin 0.2; see Supplementary Figure 1) is indicated, indicating the high degree of recapitulation between each draft genome. Other statistics shown arise from alternative bins shown in Supplementary Figure 1. (B) Coverage-GC plot for the short read assembly, with bin 2 highlighted (closed black circles and dark gray convex hull; other bins are highlighted by light gray convex hulls). (C) Short read (SR: black crosses) and long read (LR: gray crosses) coverage profiles across LR-chr 1, showing expected uniform coverage across LR-chr 1. (D–F) BLASTN statistics for alignments of short read contigs from bin 0.2 against LR-chr 1. Horizontal segments show alignment position on the long read sequence (horizontal axis) and the height of the segment is value of corresponding statistic (vertical–axis) namely percent identity (PID) in (D), the ratio of alignment length to query length (al2ql) in (E) and log10 bitscore in (F). Note the presence of full length, high quality alignments of contig sequence from bin 0.2 to LR-chr 1. (G) GC content as a function of position on LR-chr 1 (gray closed circles, computed in adjacent windows of length 46,700 bp) and for aligned short read contigs (black closed circles), showing highly correlated patterning of GC content between the two sets of sequences. (H–K) Distribution of four component statistics of κ (refer Arumugam et al., 2021), with the position of the top scoring short read bin (bin 0.2) highlighted. (H) Proportion of short read contigs in bin aligned to LR-chr 1 (psrac). (I) Mean percent identity (pid) and (J) mean ratio of alignment length to query length (al2ql) and (K) proportion of the long read contig (LR-chr 1) that is covered by an alignment from bin 0.2 (paln). Approximately 10% of the LR-chr 1 sequence is not covered by aligned short read contigs, most likely due to incompleteness of the latter genome bin.
Supplementary Figure 3 | Whole genome sequence phylogram from genomes from genus Defluviicoccus (n = 9; Table 1) and genomes from members of order Rhodospirillales (n = 85) available in the GTDB database. Whole genome distance statistics were computed within GTDB-Tk. Following Onetto et al. (2019), genomes from members of the genus Gemmatimonas (n = 5) were assigned as the outgroup taxa. See Section “Materials and Methods: Comparative Analyses of Defluviicoccus genomes” for details.
Supplementary Figure 4 | Network representation of the occurrence of genes in the KEGG among the genomes from genus Defluviicoccus. Genes are shown as red nodes and genomes are shown as blue nodes. Edges are weighted by the copy number of the gene in the cognate genome, in this case ranging from one copy to three copies. Full node names as follows; 307: Ca. Defluviicoccus Fred MAXAC 307; 378, Ca. Defluviicoccus Fred MAXAC 378; HK, Defluviicoccus GAO-HK; 148, Ca. Defluviicoccus Kalu MAXAC 148; SSA4, Ca. Defluviicoccus. sp. SSA4; 129, Defluviicoccus bin 129; TF071, Ca. Defluviicoccus tetraformis TF071; Vanus, Defluviicoccus vanusT; narG, nitrate reductase, alpha subunit; narI, respiratory nitrate reductase 1 gamma chain; narY, respiratory nitrate reductase 2 beta chain; nirK, copper-containing nitrite reductase; nirS, nitrite reductase; norB, nitric oxide reductase, subunit B; norC, nitric oxide reductase, subunit B.
Supplementary Data File 1 | Combined functional annotations for all protein-coding genes in the nine Defluviicoccus genomes listed in Table 1. Source data from Prokka and BlastKOALA as specified in the column names.
Supplementary Data File 2 | Combined functional annotations for all protein-encoding genes in the plasmid sequence recovered from Defluviicoccus vanusT. Source data from Prokka and BlastKOALA as specified in the column names.
Supplementary Data File 3 | BLASTN analysis of the plasmid sequence covered from D. vanus against the NCBI nt database.
Supplementary Data File 4 | Sequence alignment results relating to the identification of pep2-treS fused protein sequence; see legend for Figure 6. Protein sequence annotated to pep2 were sourced from the .faa files generated by Prokka in each of the 9 genomes listed in Table 1. In all cases except D. vanus, where a single adjacent pair of treS (D. vanus gene: CDGBEKEE_01737; length 1,764 bp) and pep2/mak (CDGBEKEE_01738; length 1,491 bp) were detected, in the remaining genomes pep2/mak was absent and treS was approximately 3,300 bp in length (the sole exception being the Kalu 148 genome, in which a second copy of treS was present, being 1,617 bp in length, but without a flanking pep2/mak gene). Herein these are referred to “long” treS sequences. Given the consistency of these observations with the common occurrence of “fused” treS-pep2 sequence documented by Chandra et al. (2011), we tested the hypothesis that the long versions of treS contained the pep2/mak using BLASTP with the D. vanus pep2/mak protein sequence as a query and the complete set of 10 treS sequences as the subjects. The resulting alignment are provided in the this file; clearing showing that in the case of the long treS sequences, the D. vanus pep2/mak is highly aligned with distal 40% of the treS sequence, consistent with analysis of Chandra et al. (2011).
Footnotes
- ^ https://github.com/rbhwilliams/RKXM
- ^ imedea.uib-csic.es/mmg/ltp/
- ^ https://www.arb-silva.de/aligner/
References
Alber, B. E. (2011). Biotechnological potential of the ethylmalonyl-CoA pathway. Appl. Microbiol. Biotechnol. 89, 17–25. doi: 10.1007/s00253-010-2873-z
Albertsen, M., McIlroy, S. J., Stokholm-Bjerregaard, M., Karst, S. M., and Nielsen, P. H. (2016). “Candidatus Propionivibrio aalborgensis”: a novel glycogen accumulating organism abundant in full-scale enhanced biological phosphorus removal plants. Front. Microbiol. 7:1033. doi: 10.3389/fmicb.2016.01033
Anthony, C. (2011). How half a century of research was required to understand bacterial growth on C1 and C2 compounds; the story of the serine cycle and the ethylmalonyl-CoA pathway. Sci. Prog. 94, 109–137. doi: 10.3184/003685011X13044430633960
Arredondo-Alonso, S., Willems, R. J., van Schaik, W., and Schürch, A. C. (2017). On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genom. 3:e000128. doi: 10.1099/mgen.0.000128
Arumugam, K., Bağci, C., Bessarab, I., Beier, S., Buchfink, B., Górska, A., et al. (2019). Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data. Microbiome 7:61. doi: 10.1186/s40168-019-0665-y
Arumugam, K., Bessarab, I., Haryono, M. A. S., Liu, X., Zuniga-Montanez, R. E., Roy, S., et al. (2021). Recovery of complete genomes and non-chromosomal replicons from activated sludge enrichment microbial communities with long read metagenome sequencing. NPJ Biofilms Microb. 7:23. doi: 10.1038/s41522-021-00196-6
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Begum, S. A., and Batista, J. R. (2014). Impact of butyrate on microbial selection in enhanced biological phosphorus removal systems. Environ. Technol. 35, 2961–2972. doi: 10.1080/09593330.2014.927531
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731.
Brune, A., and Schink, B. (1992). Anaerobic degradation of hydroaromatic compounds by newly isolated fermenting bacteria. Arch. Microbiol. 158, 320–327. doi: 10.1007/BF00245360
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Meth. 18, 366–368. doi: 10.1038/s41592-021-01101-x
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Meth. 12, 59–60. doi: 10.1038/nmeth.3176
Burow, L. C., Kong, Y., Nielsen, J. L., Blackall, L. L., and Nielsen, P. H. (2007). Abundance and ecophysiology of Defluviicoccus spp., glycogen-accumulating organisms in full-scale wastewater treatment processes. Microbiology 153, 178–185. doi: 10.1099/mic.0.2006/001032-0
Burow, L. C., Mabbett, A. N., Borrás, L., and Blackall, L. L. (2009). Anaerobic central metabolic pathways active during polyhydroxyalkanoate production in uncultured cluster 1 Defluviicoccus enriched in activated sludge communities. FEMS Microbiol. Lett. 298, 79–84. doi: 10.1111/j.1574-6968.2009.01695.x
Burow, L. C., Mabbett, A. N., McEwan, A. G., Bond, P. L., and Blackall, L. L. (2008a). Bioenergetic models for acetate and phosphate transport in bacteria important in enhanced biological phosphorus removal. Environ. Microbiol. 10, 87–98. doi: 10.1111/j.1462-2920.2007.01432.x
Burow, L. C., Mabbett, A. N., and Blackall, L. L. (2008b). Anaerobic glyoxylate cycle activity during simultaneous utilization of glycogen and acetate in uncultured Accumulibacter enriched in enhanced biological phosphorus removal communities. ISME J. 2, 1040–1051. doi: 10.1038/ismej.2008.45
Carr, E. L., Eales, K. L., and Seviour, R. J. (2006). Substrate uptake by Gordonia amarae in activated sludge foams by FISH-MAR. Water Sci. Technol. 54, 39–45. doi: 10.2166/wst.2006.369
Cech, J. S., and Hartman, P. (1993). Competition between polyphosphate and polysaccharide accumulating bacteria in enhanced biological phosphorus removal systems. Water Res. 27, 1219–1225. doi: 10.1016/0043-1354(93)90014-9
Chandra, G., Chater, K. F., and Bornemann, S. (2011). Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology 175, 1565–1572. doi: 10.1099/mic.0.044263-0
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Dai, Y., Yuan, Z., Wang, X., Oehmen, A., and Keller, J. (2007). Anaerobic metabolism of Defluviicoccus vanus related glycogen accumulating organisms (GAOs) with acetate and propionate as carbon sources. Water Res. 41, 1885–1896. doi: 10.1016/j.watres.2007.01.045
de Graaff, D. R., van Loosdrecht, M. C. M., and Pronk, M. (2021). Trehalose as an osmolyte in Candidatus Accumulibacter phosphatis. Appl. Microbiol. Biotechnol. 105, 379–388. doi: 10.1007/s00253-020-10947-8
Dueholm, M. S., Andersen, K. S., McIlroy, S. J., Kristensen, J. M., Yashiro, E., Karst, S. M., et al. (2020). Generation of comprehensive ecosystem-specific reference databases with species-level resolution by high-throughput full-length 16S rRNA gene sequencing and automated taxonomy assignment (AutoTax). mBio 11:e01557-20. doi: 10.1128/mBio.01557-20
Dueholm, M. S., Nierychlo, M., Andersen, K. S., Rudkjøbing, V., Knutsson, S., The MiDAS Global Consortium, et al. (2021). MiDAS 4: A global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. bioRxiv. [preprint]. doi: 10.1101/2021.07.06.451231
Dvornyk, V., Vinogradova, O., and Nevo, E. (2003). Origin and evolution of circadian clock genes in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 100, 2495–2500. doi: 10.1073/pnas.0130099100
Ensign, S. A. (2006). Revisiting the glyoxylate cycle: alternative pathways for microbial acetate assimiliation. Mol. Microbiol. 61, 274–276. doi: 10.1111/j.1365-2958.2006.05247.x
Fernando, E. Y., McIlroy, S. J., Nierychlo, M., Herbst, F. A., Petriglieri, F., Schmid, M., et al. (2019). Resolving the individual contribution of key microbial populations to enhanced biological phosphorus removal with Raman–FISH. ISME J. 13, 1933–1946. doi: 10.1038/s41396-019-0399-7
Flowers, J. J., He, S., Malfatti, S., del Rio, T. G., Tringe, S. G., Hugenholtz, P., et al. (2013). Comparative genomics of two ’Candidatus Accumulibacter’ clades performing biological phosphorus removal. ISME J. 7, 2301–2314. doi: 10.1038/ismej.2013.117
García Martín, H., Ivanova, N., Kunin, V., Warnecke, F., Barry, K. W., McHardy, A. C., et al. (2006). Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat. Biotechnol. 24, 1263–1269. doi: 10.1038/nbt1247
Grage, K., Jahns, A. C., Parlane, N., Palanisamy, R., Rasiah, I. R., Atwood, J. A., et al. (2009). Bacterial polyhydroxyalkanoate granules: biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 10, 660–669. doi: 10.1021/bm801394s
Grigoriev, A. (1998). Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 26, 2286–2290.
He, S., and McMahon, K. D. (2011). Microbiology of ‘Candidatus Accumulibacter’ in activated sludge. Microb. Biotechnol. 4, 603–619. doi: 10.1111/j.1751-7915.2011.00248.x
Hördt, A., López, M. G., Meier-Kolthoff, J. P., Schleuning, M., Weinhold, L. M., Tindall, B. J., et al. (2020). Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 11:468. doi: 10.3389/fmicb.2020.00468
Huang, W. E., Stoecker, K., Griffiths, R., Newbold, L., Daims, H., Whiteley, A. S., et al. (2007). Raman-FISH: combining stable-isotope Raman spectroscopy and fluorescence in situ hybridization for the single cell analysis of identity and function. Environ. Microbiol. 9, 1878–1889. doi: 10.1111/j.1462-2920.2007.01352.x
Huson, D., Albrecht, B., Bagci, C., Bessarab, I., Gorska, A., Jolic, D., et al. (2018). MEGAN-LR: New algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biol. Direct 13:6. doi: 10.1186/s13062-018-0208-7
Izadi, P., Izadi, P., and Eldyasti, A. (2020). Design, operation and technology configurations for enhanced biological phosphorus removal (EBPR) process: a review. Rev. Environ. Sci. BioTechnol. 19, 561–593. doi: 10.1007/s11157-020-09538-w
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731.
Kolakovic, S., Freitas, E. B., Reis, M. A. M., Carvalho, G., and Oehmen, A. (2021). Accumulibacter diversity at the sub-clade level impacts enhanced biological phosphorus removal performance. Water Res. 199:117210. doi: 10.1016/j.watres.2021.117210
Koliwer-Brandl, H., Syson, K., van de Weerd, R., Chandra, G., Appelmelk, B., Alber, M., et al. (2016). Metabolic network for the biosynthesis of intra- and extracellular α-glucans required for virulence of Mycobacterium tuberculosis. PLoS Pathog. 12:e1005768. doi: 10.1371/journal.ppat.1005768
Kong, Y., Xia, Y., Nielsen, J. L., and Nielsen, P. H. (2006). Ecophysiology of a group of uncultured Gammaproteobacterial glycogen-accumulating organisms in full-scale enhanced biological phosphorus removal wastewater treatment plants. Environ. Microbiol. 8, 479–489. doi: 10.1111/j.1462-2920.2005.00914.x
Kong, Y., Xia, Y., Nielsen, J. L., and Nielsen, P. H. (2007). Structure and function of the microbial community in a full-scale enhanced biological phosphorus removal plant. Microbiology 153, 4061–4073. doi: 10.1099/mic.0.2007/007245-0
Law, Y. Y., Kirkegaard, R. H., Cokro, A. A., Liu, X., Arumugam, K., Xie, C., et al. (2016). Integrative microbial community analysis reveals full-scale enhanced biological phosphorus removal under tropical conditions. Sci. Rep. 6:25719. doi: 10.1038/srep25719
Liu, J. R., Burrell, P., Seviour, E. M., Soddell, J. A., Blackall, L. L., and Seviour, R. J. (2000). The filamentous bacterial morphotype ‘Nostocoida limicola’ I contains at least two previously described genera in the low G+C Gram positive bacteria. Syst. Appl. Microbiol. 23, 528–534. doi: 10.1016/S0723-2020(00)80027-2
Liu, W. T., Mino, T., Nakamura, K., and Matsuo, T. (1996). Glycogen accumulating population and its anaerobic substrate uptake in anaerobic-aerobic activated sludge without biological phosphorus removal. Water Res. 30, 75–82.
Liu, W. T., Nakamura, K., Matsuo, T., and Mino, T. (1997). Internal energy-based competition between polyphosphate-and glycogen-accumulating bacteria in biological phosphorus removal reactors—effect of PC feeding ratio. Water Res. 31, 1430–1438.
Lopez-Vazquez, C. M., Oehmen, A., Hooijmans, C. M., Brdjanovic, D., Gijzen, H. J., Yuan, Z., et al. (2009). Modeling the PAO-GAO competition: effects of carbon source, pH and temperature. Water Res. 43, 450–462. doi: 10.1016/j.watres.2008.10.032
Lu, H., Oehmen, A., Virdis, B., Keller, J., and Yuan, Z. (2006). Obtaining highly enriched cultures of Candidatus Accumulibacter phosphates through alternating carbon sources. Water Res. 40, 3838–3848. doi: 10.1016/j.watres.2006.09.004
Lv, J. H., Yuan, L. J., Chen, X., Liu, L., and Luo, D. C. (2014). Phosphorus metabolism and population dynamics in a biological phosphate-removal system with simultaneous anaerobic phosphate stripping. Chemosphere 117, 715–721. doi: 10.1016/j.chemosphere.2014.10.018
Ma, M., Du, H., Sun, T., An, S., Yang, G., and Wang, D. (2019). Characteristics of archaea and bacteria in rice rhizosphere along a mercury gradient. Sci. Total Environ. 650, 1640–1651. doi: 10.1016/j.scitotenv.2018.07.175
Maszenan, A. M. (2001). The Occurrence, Characterization and Biodiversity of “G-Bacteria” in Activated Sludge Systems. [PhD thesis]. Melbourne: La Trobe University Australia.
Maszenan, A. M., Seviour, R. J., Patel, B. K. C., Janssen, P. H., and Wanner, J. (2005). Defluvicoccus vanus gen. nov., sp. nov., a novel Gram-negative coccus/coccobacillus in the ’Alphaproteobacteria’ from activated sludge.”. Int. J. Syst. Evol. Microbiol. 55, 2105–2111.
McIlroy, S., Nittami, T., Seviour, E. M., and Seviour, R. J. (2010). Filamentous members of cluster III Defluviicoccus have the in situ phenotype expected of a glycogen-accumulating organism in activated sludge. FEMS Microbiol. Ecol. 74, 248–256. doi: 10.1111/j.1574-6941.2010.00934.x
McIlroy, S., and Seviour, R. J. (2009). Elucidating further phylogenetic diversity among the Defluviicoccus-related glycogen-accumulating organisms in activated sludge. Environ. Microbiol. Rep. 1, 563–568. doi: 10.1111/j.1758-2229.2009.00082.x
McIlroy, S., Speirs, L. B. M., Tucci, J., and Seviour, R. J. (2011). In situ profiling of microbial communities in full-scale aerobic sequencing batch reactors treating winery waste in Australia. Environ. Sci. Technol. 45, 8794–8803.
McIlroy, S. J., Albertsen, M., Andresen, E. K., Saunders, A. M., Kristiansen, R., Stokholm-Bjerregaard, M., et al. (2014). Candidatus Competibacter lineage genomes retrieved from metagenomes reveal functional metabolic diversity. ISME J. 8, 613–624. doi: 10.1038/ismej.2013.162
McIlroy, S. J., Nittami, T., Kanai, E., Fukuda, J., Saunders, A. M., and Nielsen, P. H. (2015). Reappraisal of the phylogeny and fluorescence in situ hybridization probes for the analysis of the Competibacteraceae in wastewater treatment systems. Environ. Microbiol. Rep. 7, 166–174. doi: 10.1111/1758-2229.12215
McIlroy, S. J., Onetto, C. A., McIlroy, B., Herbst, F. A., Dueholm, M. S., Kirkegaard, R. H., et al. (2018). Genomic and in situ analyses reveal the Micropruina spp. as abundant fermentative glycogen accumulating organisms in enhanced biological phosphorus removal systems. Front. Microbiol. 9:1004. doi: 10.3389/fmicb.2018.01004
McMahon, K. D., He, S., and Oehmen, A. (2010). “The microbiology of phosphorus removal,” in Microbial Ecology of Activated Sludge, eds R. J. Seviour and P. H. Nielsen (London: IWA Publishing), 281–319.
Meyer, R. L., Saunders, A. M., and Blackall, L. L. (2006). Putative glycogen-accumulating organisms belonging to the Alphaproteobacteria identified through rRNA-based stable isotope probing. Microbiology 152(Pt 2), 419–429. doi: 10.1099/mic.0.28445-0
Mielczarek, A. T., Nguyen, H. T. T., Nielsen, J. L., and Nielsen, P. H. (2013). Population dynamics of bacteria involved in enhanced biological phosphorus removal in Danish wastewater treatment plants. Water Res. 47, 1529–1544. doi: 10.1016/j.watres.2012.12.003
Musat, N., Foster, R., Vagner, T., Adam, B., and Kuypers, M. M. (2012). Detecting metabolic activities in single cells, with emphasis on nanoSIMS. FEMS Microbiol. Rev. 36, 486–511. doi: 10.1111/j.1574-6976.2011.00303.x
Nielsen, P. H., McIIroy, S. J., Albertsen, M., and Nierychlo, M. (2019). Re-evaluating the microbiology of the enhanced biological phosphorus removal process. Curr. Opinion Biotechnol. 57, 111–118. doi: 10.1016/j.copbio.2019.03.008
Nittami, T., McIlroy, S., Seviour, E. M., Schroeder, S., and Seviour, R. J. (2009). Candidatus Monilibacter spp., common bulking filaments in activated sludge, are members of Cluster III Defluviicoccus. Syst. Appl. Microbiol. 32, 480–489. doi: 10.1016/j.syapm.2009.07.003
Nobu, M. K., Tamaki, H., Kubota, K., and Liu, W. T. (2014). Metagenomic characterization of ‘Candidatus Defluviicoccus tetraformis strain TFO71’, a tetrad-forming organism, predominant in an anaerobic– aerobic membrane bioreactor with deteriorated biological phosphorus removal. Environ. Microbiol. 16, 2739–2751.
Norman, A., Hansen, L. H., and Sørensen, S. J. (2009). Conjugative plasmids: vessels of the communal gene pool. Phil. Trans. R. Soc. B 364, 2275–2289.
Oehmen, A., Carvalho, G., Lopez-Vazquez, C. M., van Loosdrecht, M. C. M., and Reis, M. A. M. (2010). Incorporating microbial ecology into the metabolic modelling of polyphosphate accumulating organisms and glycogen accumulating organisms. Water Res. 44, 4992–5004. doi: 10.1016/j.watres.2010.06.071
Oehmen, A., Lemos, P. C., Carvalho, G., Yuan, Z., Keller, J., Blackall, L. L., et al. (2007). Advances in enhanced biological phosphorus removal: from micro to macro scale. Water Res. 41, 2271–2300. doi: 10.1016/j.watres.2007.02.030
Oehmen, A., Saunders, A. M., Vives, M. T., Yuan, Z., and Keller, J. (2006). Competition between polyphosphate and glycogen accumulating organisms in enhanced biological phosphorus removal systems with acetate and propionate as carbon sources. J. Biotechnol. 123, 22–32. doi: 10.1016/j.jbiotec.2005.10.009
Oehmen, A., Zeng, R. J., Yuan, Z., and Keller, J. (2005a). Anaerobic metabolism of propionate by polyphosphate-accumulating organisms in enhanced biological phosphorus removal systems. Biotechnol. Bioeng. 91, 43–53. doi: 10.1002/bit.20480
Oehmen, A., Yuan, Z., Blackall, L. L., and Keller, J. (2005b). Comparison of acetate and propionate uptake by polyphosphate accumulating organisms and glycogen accumulating organisms. Biotechnol. Bioeng. 91, 162—-168. doi: 10.1002/bit.20500
Onetto, C. A., Grbin, P. R., McIlroy, S. J., and Eales, K. L. (2019). Genomic insights into the metabolism of ‘Candidatus Defluviicoccus seviourii’, as a member of Defluviicoccus cluster III abundant in industrial activated sludge. FEMS Microbiol. Ecol. 95:fiy231. doi: 10.1093/femsec/fiy231
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528.
Parks, D., Chuvochina, M., Waite, D., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Chuvochina, M., Chaumeil, P. A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genom. Res. 25, 1043–1055.
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596.
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842.
Renilla, S., Bernal, V., Fuhrer, T., Castaño-Cerezo, S., Pastor, J. M., Iborra, J. L., et al. (2012). Acetate scavenging activity in Escherichia coli: interplay of acetyl–CoA synthetase and the PEP–glyoxylate cycle in chemostat cultures. Appl. Microbiol. Biotechnol. 93, 2109–2124. doi: 10.1007/s00253-011-3536-4
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26.
Roy, S., Qiu, G., Zuniga-Montanez, R., Williams, R. B. H., and Wuertz, S. (2021). Recent advances in understanding the ecophysiology of enhanced biological phosphorus removal. Curr. Opinion Biotechnol. 67, 166–174. doi: 10.1016/j.copbio.2021.01.011
Sagong, H. Y., Son, H. F., Choi, S. Y., Lee, S. Y., and Kim, K. J. (2018). Structural insights into polyhydroxyalkanoates biosynthesis. Trends Biochem. Sci. 43, 790–805. doi: 10.1016/j.tibs.2018.08.005
Saunders, A. M., Mabbett, A. N., McEwan, A. G., and Blackall, L. L. (2007). Proton motive force generation from stored polymers for the uptake of acetate under anaerobic conditions. FEMS Microbiol. Lett. 274, 245–251. doi: 10.1111/j.1574-6968.2007.00839.x
Schada von Borzyskowski, L., Bernhardsgrütter, I., and Erb, T. J. (2020). Biochemical unity revisited: microbial central carbon metabolism holds new discoveries, multi-tasking pathways, and redundancies with a reason. Biol. Chem. 401, 1429–1441. doi: 10.1515/hsz-2020-0214
Seviour, R. J., Liu, J. R., Seviour, E. M., McKenzie, C. A., Blackall, L. L., and Saint, C. P. (2002). The “Nostocoida limicola” story: resolving the phylogeny of this morphotype responsible for bulking in activated sludge. Water Sci. Technol. 46, 105–110.
Seviour, R. J., and McIlroy, S. (2008). The microbiology of phosphorus removal in activated sludge processes - the current state of play. J. Microbiol. 46, 115–124. doi: 10.1007/s12275-008-0051-0
Shintani, T., Liu, W. T., Hanada, S., Kamagata, Y., Miyaoka, S., Suzuki, T., et al. (2000). Micropruina glycogenica gen. nov., sp. nov., a new Gram-positive glycogen-accumulating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 50(Pt 1), 201–207. doi: 10.1099/00207713-50-1-201
Singleton, C. M., Petriglieri, F., Kristensen, J. M., Kirkegaard, R. H., Michaelsen, T. Y., Andersen, M. H., et al. (2021). Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 12:2009. doi: 10.1038/s41467-021-22203-2
Skennerton, C. T., Barr, J. J., Slater, F. R., Bond, P. L., and Tyson, G. W. (2015). Expanding our view of genomic diversity in Candidatus Accumulibacter clades. Environ. Microbiol. 17, 1574–1585. doi: 10.1111/1462-2920.12582
Slaby, B. M., Hackl, T., Horn, H., Bayer, K., and Hentschel, U. (2017). Metagenomic binning of a marine sponge microbiome reveal unity in defense but metabolic specialization. ISME J. 11, 2465–2478. doi: 10.1038/ismej.2017.101
Stokholm-Bjerregaard, M., McIlroy, S. J., Nierychlo, M., Karst, S. M., Albertsen, M., and Nielsen, P. H. (2017). A critical assessment of the microorganisms proposed to be important to enhanced biological phosphorus removal in full-scale wastewater treatment systems. Front. Microbiol. 8:718. doi: 10.3389/fmicb.2017.00718
Sun, P., Li, J., Zhang, X., Guan, Z., Xiao, Q., Zhao, C., et al. (2018). Crystal structure of the bacterial acetate transporter SatP reveals that it forms a hexameric channel. J. Biol. Chem. 293, 19492–19500. doi: 10.1074/jbc.RA118.003876
Wang, L., Liu, J., Oehmen, A., Le, C., Geng, Y., and Zhou, Y. (2021a). Butyrate can support PAOs but not GAOs in tropical climates. Water Res. 193:116884. doi: 10.1016/j.watres.2021.116884
Wang, L., Oehmen, A., Le, C., Liu, J., and Zhou, Y. (2021b). Defluviicoccus vanus glycogen-accumulating organisms (DvGAOs) are less competitive than polyphosphate-accumulating organisms (PAO) at high temperature. ACS EST Water 1, 319–327.
Wang, Z., Guo, F., Mao, Y., Xia, Y., and Zhang, T. (2014). Metabolic characteristics of a glycogen-accumulating organism in Defluviicoccus cluster II revealed by comparative genomics. Microb. Ecol. 68, 716–728.
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Williams, T. M., and Unz, R. F. (1985). Isolation and characterization of filamentous bacteria present in bulking activated sludge. Appl. Microbiol. Biotechnol. 22, 273–282. doi: 10.1007/BF00252030
Wong, M. T., and Liu, W. T. (2007). Ecophysiology of Defluviicoccus-related tetrad-forming organisms in an anaerobic-aerobic activated sludge process. Environ. Microbiol. 9, 1485–1496. doi: 10.1111/j.1462-2920.2007.01267.x
Wong, M. T., Tan, F. M., Ng, W. J., and Liu, W. T. (2004). Identification and occurrence of tetrad-forming Alphaproteobacteria in anaerobic-aerobic activated sludge processes. Microbiology 150, 3741–3748. doi: 10.1099/mic.0.27291-0
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Zhou, Y., Pijuan, M., Oehmen, A., and Yuan, Z. (2010). The source of reducing power in the anaerobic metabolism of polyphosphate accumulating organisms (PAOs) - a mini-review. Water Sci. Technol. 61, 1653–1662. doi: 10.2166/wst.2010.983
Keywords: activated sludge, glycogen accumulating organisms, Defluviicoccus, Defluviicoccus vanus, enhanced biological phosphorus removal (EBPR)
Citation: Bessarab I, Maszenan AM, Haryono MAS, Arumugam K, Saw NMMT, Seviour RJ and Williams RBH (2022) Comparative Genomics of Members of the Genus Defluviicoccus With Insights Into Their Ecophysiological Importance. Front. Microbiol. 13:834906. doi: 10.3389/fmicb.2022.834906
Received: 14 December 2021; Accepted: 02 February 2022;
Published: 12 April 2022.
Edited by:
Nikolai Ravin, Institute of Bioengineering, Research Center of Biotechnology of the Russian Academy of Sciences, RussiaReviewed by:
Juan Antonio Baeza, Universitat Autònoma de Barcelona, SpainYu Xia, Huazhong University of Science and Technology, China
Copyright © 2022 Bessarab, Maszenan, Haryono, Arumugam, Saw, Seviour and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohan B. H. Williams, bHNpcmJod0BudXMuZWR1LnNn