 Yuxin Dong
Yuxin Dong Yulei Gao
Yulei Gao Yanfen Chai
Yanfen Chai- Department of Emergency Medicine, Tianjin Medical University General Hospital, Tianjin, China
A body temperature >38.3°C that lasts ≥3 weeks and lacks a clear diagnosis after 1 week of standard hospital examination and treatment is called “fever of unknown origin” (FUO). The main causes of FUO are infections, hematological diseases, autoimmune diseases, and other non-infectious inflammatory diseases. In recent years, quantitative metagenomics next-generation sequencing (Q-mNGS) has been used widely to detect pathogenic microorganisms, especially in the contribution of rare or new (e.g., severe acute respiratory syndrome-coronavirus-2) pathogens. This review addresses the undetermined cause of fever and its evaluation by Q-mNGS.
Introduction
Fever of unknown origin (FUO) can be caused by various diseases. More than 200 causes of FUO have been reported (Horowitz, 2013). In 1961, Petersdorf and Beeson were the first to define FUO as a state of febrile illness for more than 3 weeks, with a body temperature greater than 38.3°C (101°F) on several occasions and an uncertain diagnosis after 1 week of standard hospital examination and treatment (Petersdorf and Beeson, 1961). In 1991, Durak and Street re-defined FUO into four groups: “classic,” “nosocomial,” “neutropenic,” and “human immunodeficiency virus (HIV)-associated.” They proposed three outpatient visits and related investigations as an alternative to “1 week of hospitalization” (Durack and Street, 1991). In 1997, Arnow and Flaherty updated the FUO definition and considered the “minimum diagnostic evaluation to qualify as FUO” to be: comprehensive history-taking; repeated physical examination; complete blood count (including differential and platelet counts); routine blood chemistry (including lactate dehydrogenase, bilirubin, and liver enzymes); urinalysis (including microscopic examination); chest radiograph; erythrocyte sedimentation rate (ESR); antinuclear antibodies; rheumatoid factor; angiotensin-converting enzyme; routine blood cultures (×3) while not receiving antibiotics; cytomegalovirus immunoglobulin-M antibodies or virus detection in blood; heterophile antibody test in children and young adults; tuberculin skin test; computed tomography (CT) of the abdomen or radionuclide scan; HIV antibodies or virus-detection assay; further evaluation of abnormalities detected by the tests stated above (Arnow and Flaherty, 1997). Because of the complicated clinical characteristics and lack of laboratory indicators of a disease, the diagnosis is difficult and contributes to a high cost of hospitalization.
Infections, neoplasms, non-infectious inflammatory diseases, and other conditions are the four primary etiological groups for FUO (Mourad et al., 2003). Obtaining a detailed medical history and undertaking examinations to evaluate the cause of fever are crucial. The standard diagnosis and treatment process of FUO have not yet been proposed, but they should be performed in a specific order when carrying out the examination and diagnosis (Figure 1). Identification of pathogenic bacteria is crucial for targeted anti-infective medication (Haidar and Singh, 2022; Messacar et al., 2017). In patients with prolonged fever, empiric therapy is not recommended because it can mask symptoms, delay the diagnosis, and obstruct decision-making regarding optimal treatment (Unger et al., 2016). Only a few exceptions exist if treatment must be initiated based solely on diagnostic suspicion: antibiotics for culture-negative endocarditis, tuberculostatic agents for active tuberculosis, and glucocorticoids for temporal arteritis with a risk of vision loss (Bryan and Ahuja, 2007). Culture and testing of body fluids are common for a microbial diagnosis, but such cultures and tests are positive in only ∼40% of cases. Also, implementation and interpretation of blood cultures require time, which delays the information obtained by clinicians (Tromp et al., 2012). The sensitivity and specificity of PCR-based detection is based on the genomic sequence of known pathogenic bacteria, provides limited information, and is suboptimal for detection of mixed infections (Reuwer et al., 2019). Medication mistakes or treatment delays may arise due to the limits of clinical testing.
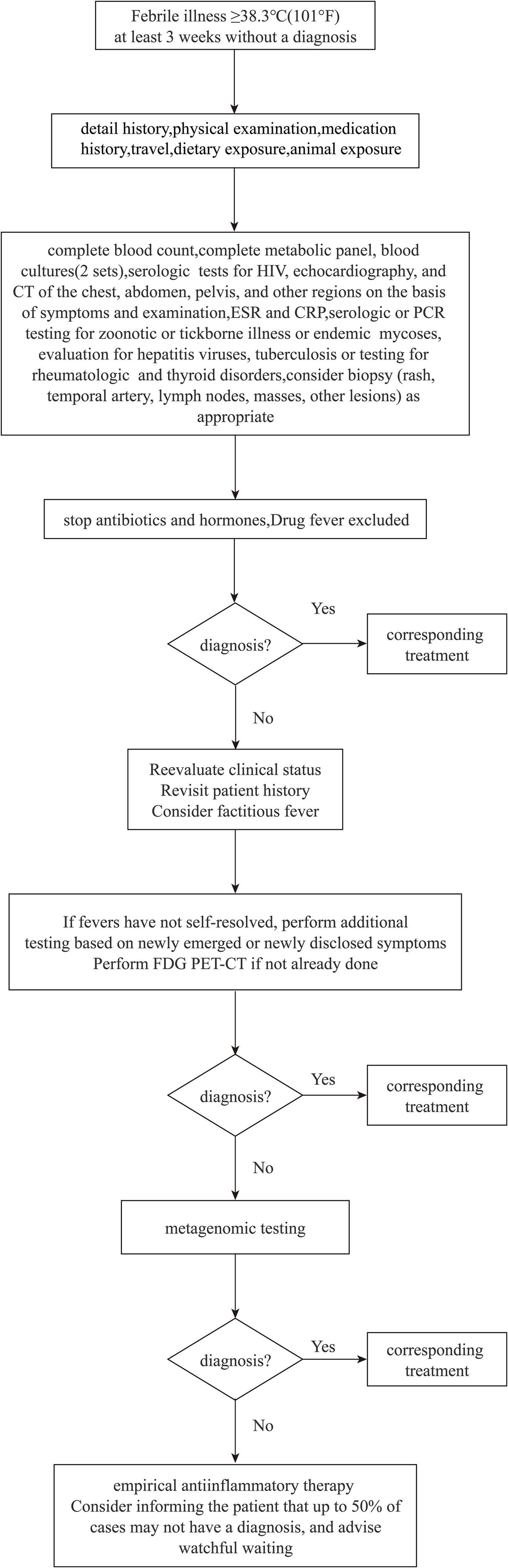
Figure 1. FUO diagnosis and treatment flow chart.
According to two systematic reviews conducted from 1995 to 2004 (Gaeta et al., 2006) and 2005 to 2015 (Fusco et al., 2019), infections are the leading cause of FUO. Screening and diagnostic processes must be developed to detect the pathogens that cause infection-related FUO. Quantitative metagenomics next-generation sequencing (Q-mNGS) is a current method to detect infection-related FUO pathogens. Quantitative metagenomics next-generation sequencing, also known as “high-throughput sequencing” or “massive parallel sequencing,” is a type of technology that allows for the simultaneous and independent sequencing of hundreds to billions of DNA fragments (Morganti et al., 2019). Q-mNGS has many uses in clinical microbiological testing, and provides an unbiased method for pathogen detection. Recent studies have shown that Q-mNGS could be used to diagnose various infectious diseases, including coronavirus disease 2019 (COVID-19) (Ren et al., 2020), pneumonia due to Chlamydia psittaci infection (Chen et al., 2020), Ebola virus (EBOV) infection (Li et al., 2019), and talaromycosis (Shi et al., 2021).
Revolution in DNA-Sequencing: From Sanger Sequencing to Quantitative Metagenomics Next-Generation Sequencing
The “first generation” of gene-sequencing technology was born with the advent of the chain-termination method described by Sanger and Coulson (1975) and the chain-degradation method described by Maxam and Gilbert (1977). Gilbert and Sanger built the first sequencer in 1977 and used it to sequence the first full-length genome, phage X174, with 5,375 bases (Maxam and Gilbert, 1977; Aoyama et al., 1981). First-generation sequencing can produce a sequence of 700–1,000 bases at a time, so it cannot keep up with the pressing need for biological gene sequences.
Following a revolution in traditional sequencing technology, second-generation sequencing technology, known as “next-generation sequencing” (NGS), can be employed to obtain the sequences of hundreds of thousands to millions of nucleic-acid molecules in a single run. With NGS introduction, the transcriptome and genome of a species can be investigated in great detail. Jonathan Rothberg developed the biotechnology company 454 Life Sciences (Branford, CT, United States) in 2005 (Margulies et al., 2005). Other technologies, such as sequencing by oligonucleotide ligation and detection (SOLiD) (Applied Biosystems, Foster City, CA, United States) and Solexa (Illumina, San Diego, CA, United States), emerged subsequently. A total of 454 Life Sciences was acquired by Roche (Basel, Switzerland) in 2007.
The basic principles of this technology are that the DNA fragment does not need to be fluorescently labeled, there is no need for electrophoresis, and the sequence is changed by synthesis. A pyrophosphate group is removed when a base is added to the sequence, so pyrosequencing is also known as the detection of pyrophosphate bases (Nyrén et al., 1993; Ronaghi et al., 1998). Sequencing by SOLiD technology is based on ligase sequencing (Pérez-Enciso and Ferretti, 2010). Solexa technology (which is also used for sequencing-by-synthesis) was developed first by Illumina and is now used by the second-generation sequencer developed by Illumina (Pérez-Enciso and Ferretti, 2010; Strub et al., 2011).
Metagenomics (also known as “microbial environmental genomics”) creates a metagenomic “library” by extracting the DNA or RNA of all microorganisms from environmental samples directly and studying them using genomics research strategies. Metagenomics based on NGS has become the focus of clinical research since the development of gene-sequencing technology.
Q-mNGS is a method for analyzing the genetic material of microbes and hosts from patient samples to diagnose infectious diseases. Q-mNGS has become the focus of clinical research due to the rapid advancement of gene-sequencing technology.
Third-generation sequencing technology includes the Pacific Bioscience (Levene et al., 2003) and Oxford Nanopore (Eisenstein, 2012) platforms, which are single-molecule technologies. Single-molecule sequencing (which does not require PCR amplification and which can, theoretically, determine nucleic-acid sequences of any length) is most notable when compared with first-generation and second-generation sequencing technologies (Figure 2).
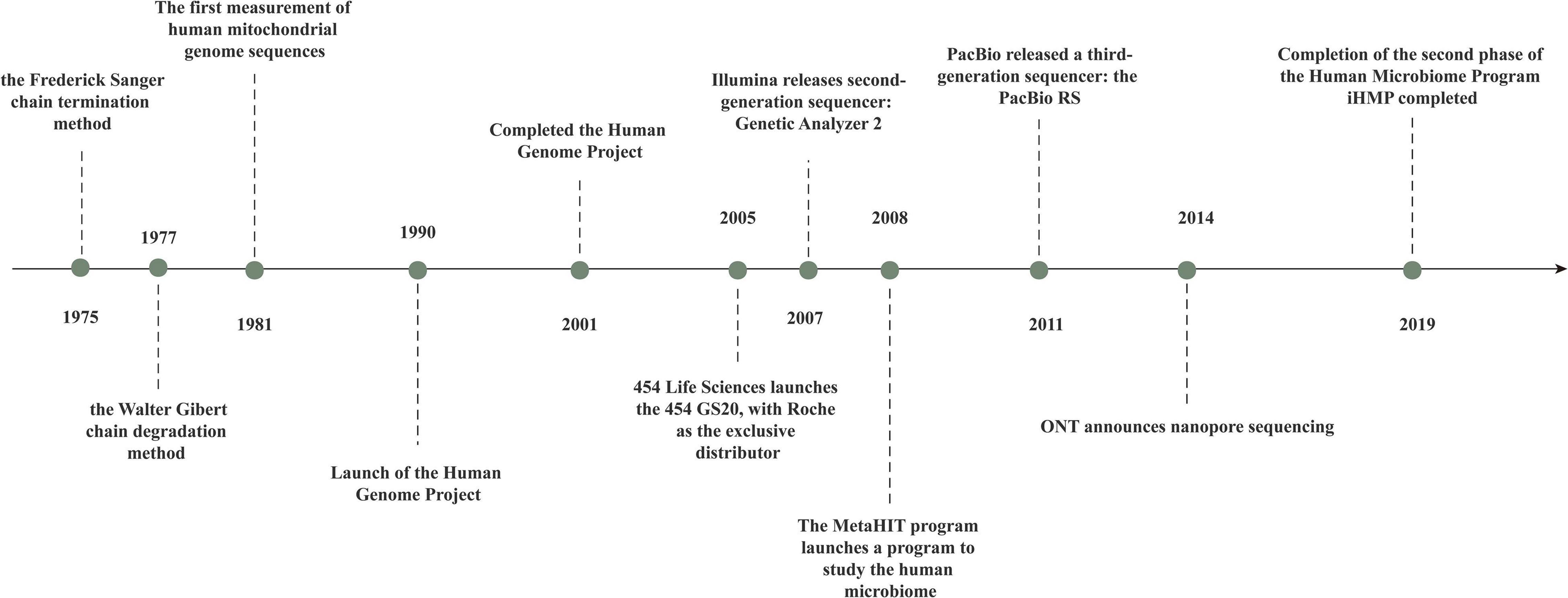
Figure 2. The history of gene sequencingtechnology.
Quantitative Metagenomics Next-Generation Sequencing in Fever of Unknown Origin or Infectious Diseases
The diagnostic value of NGS has been investigated in retrospective studies for patients suffering from fever (Table 1). The effectiveness of detection of NGS is higher than that of traditional methods. Fu et al. (2021) undertook a retrospective study on 175 patients with FUO to compare Q-mNGS with culture and traditional methods, including smears, serological tests, and amplification of nucleic acids (traditional PCR, Xpert MTB/RIF, and Xpert MTB/RIF Ultra). In comparison with culture and conventional methods, the authors concluded that Q-mNGS of blood might increase the overall rate of detection of novel organisms by 22.9 and 19.79%, and enhance the diagnostic rate by 38.0 and 32.0%, respectively. Zou et al. (2022) evaluated 12 patients with tuberculosis following renal transplantation, and Q-mNGS was helpful in 67% of cases.
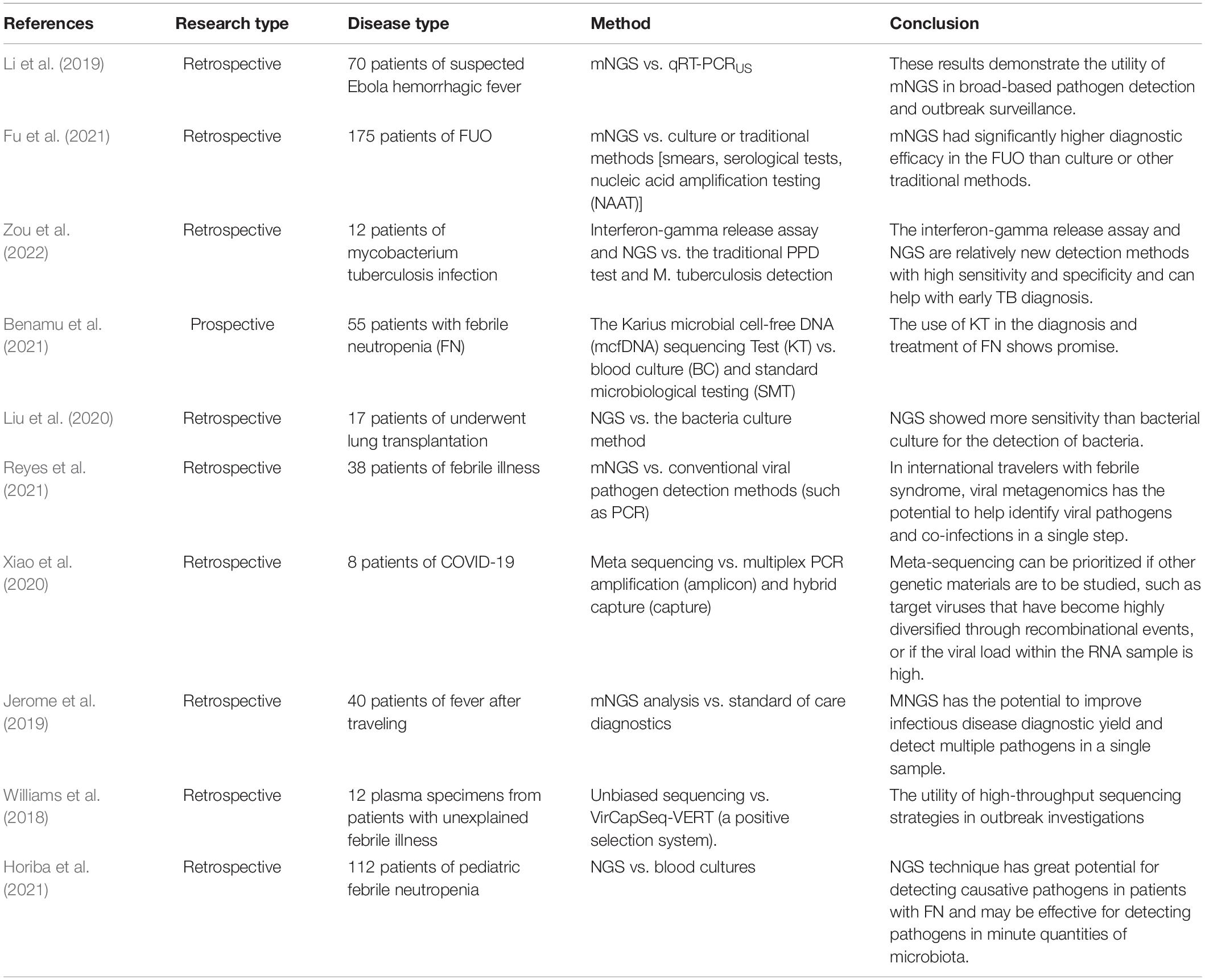
Table 1. List of sequencing associated with fever or infectious disease’ study.
Benamu et al. (2021) evaluated 55 patients with febrile neutropenia to compare the results of blood culture and standard microbiological testing within 24 h of fever onset and every 2–3 days. The Karius microbial cell-free DNA sequencing test (KT) sensitivity and specificity were 85% (41/48) and 100% (14/14), respectively. The calculated time-to-the-diagnosis was, in general, shorter with KT (87%). Adjudicators determined real-time KT results have allowed early optimization of antimicrobial agents in 47% of patients. Liu et al. (2020) retrospectively evaluated 17 patients who received a lung transplant. The proportion of bacteria detected in the lungs of donors was 52.9 and 35.3% by NGS and bacterial culture, respectively. NGS was more sensitive for bacterial detection than the classic bacterial culture. Reyes and colleagues Xiao undertook a retrospective study on 38 patients. In eight of the 38 patients (21%), all viral pathogens detected by 42 conventional assays were also detected by Q-mNGS, and Q-mNGS resulted in additional pathogenic findings in two patients (5%).
NGS provides more information than conventional diagnostic tests. Xiao et al. (2020) were the first to systematically investigate inter- and intra-individual variations in severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) using amplicon- and capture-based whole-genome sequencing; it was also the first comparative study using multiple approaches. The study illustrated that ultra-high-throughput metatranscriptomic (meta) sequencing uncovered rich genetic information in clinical samples besides SARS-CoV-2, and provided references for clinical diagnostics and therapeutics. In June 2020, the US Food and Drug Administration granted Emergency Use Authorization for a Q-mNGS test for COVID-19 manufactured by Illumina, the first such authorization for a NGS diagnostic News in Brief (2020). Li et al. (2019) demonstrated that Q-mNGS of field-collected samples could be used to recover nine genomes from the EBOV outbreak in Boende (Democratic Republic of Congo) in 2014 (>50% coverage), detect the EBOV with a high sequencing depth of 17.3 ± 4.7 SD million reads with comparable sensitivity to PCR, and identify co-infections from well-recognized (Plasmodium falciparum) and novel/uncommon (e.g., Orungo virus) pathogens. Jerome et al. (2019) prospectively included 40 returning travelers presenting with fever (≥38°C) whose plasma samples were sequenced: 11 of 40 patients were diagnosed with a viral infection. Five viral infections were detected by Q-mNGS that were also revealed in standard-of-care diagnostics, but two patients infected with the Chikungunya virus and one patient with the mumps virus were also diagnosed by Q-mNGS only. Williams et al. (2018) investigated the plasma virome from cases of unexplained febrile illness in Tanzania from 2013 to 2014 by sequencing methods. The latter could aid detection of viral coinfections, such as the nearly complete genomes of dengue virus-2 and human pestivirus. Horiba et al. (2021) evaluated 87 patients with febrile neutropenia. Putative pathogens were detected by Q-mNGS in 17.2% of patients, but all had negative blood cultures. Pathogenic detection methods (e.g., PCR) require clinicians to first suspect a specific bacterial infection before carrying out the corresponding detection. However, NGS technology can be employed to detect pathogenic bacteria in patient samples with high sensitivity, thereby providing recommendations for clinical treatment.
Often, NGS has been undertaken without using a structured diagnostic protocol, and at different stages of FUO. Zhu et al. (2021) found that use of Q-mNGS for blood as the first-line investigation could increase the diagnosis rate of FUO by 10.9% compared with that using culture, and that using Q-mNGS as the second-line investigation could improve the diagnosis rate of concurrent infection by 66.7 and 12.5% for non-bloodstream infection.
Ultimately, the cost of FUO assessment can be reduced by Q-mNGS application because the diagnosis will be achieved early because unnecessary and costly diagnostic tests will not be carried out. Chai et al. (2018) investigated the cost–benefit relationship of Q-mNGS in FUO in which a cause could not be found despite appropriate investigations. A decision tree was created to describe systematically the costs and benefits associated with NGS introduction. Each diagnostic pathway was made until a first- or second-line investigation was positive. NGS was introduced into the pathway as a supplement to first- or second-line investigations. Chai and colleagues reported NGS use as the first-line investigation assuming a probability of detecting the cause of cost-effectiveness in all cases of ≥60% using unit costs of diagnostic tests and procedures in Singapore dollars in 2016. In that analysis, using a rational set of rates for a second-line investigation, the total expected cost of using NGS as a second-line investigation was greater than that using it as a first-line investigation. Although that analysis excluded the costs associated with hospitalization duration, the faster and more definitive answers provided by NGS may enable additional cost savings.
Conclusion
Q-mNGS is a sensitive diagnostic method for FUO evaluation. It could become a routine procedure in the diagnostic workup of FUO. Q-mNGS appears to be cost-effective in FUO because it avoids unnecessary investigations and reduces the duration of hospitalization.
Author Contributions
Y-LG and S-TS conceived this study. Y-XD and Y-LG collected clinical data and were responsible for patient care, and drafted the manuscript. Y-LG, S-TS, and Y-FC revised the manuscript critically. All authors have revised the final version of the manuscript and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81871593 and 81701931). This work was also funded by the Tianjin Key Medical Discipline (Specialty) Construction Project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Shuzhang Cui (Emergency Department of Tianjin Medical University General Hospital) for help in study design.
References
Aoyama, A., Hamatake, R. K., and Hayashi, M. (1981). Morphogenesis of Phi X174: in vitro synthesis of infectious phage from purified viral components. Proc. Natl. Acad. Sci. U.S.A. 78, 7285–7289.
Benamu, E., Gajurel, K., Anderson, J. N., Lieb, T., Gomez, C. A., Seng, H., et al. (2021). Plasma microbial cell-free DNA next generation sequencing in the diagnosis and management of febrile neutropenia. Clin. Infect. Dis. 74, 1659–1668. doi: 10.1093/cid/ciab324
Bryan, C. S., and Ahuja, D. (2007). Fever of unknown origin: is there a role for empiric therapy? Infect. Dis. Clin. North Am. 21, 1213–1220. doi: 10.1016/j.idc.2007.08.007
Chai, J. H., Lee, C. K., Lee, H. K., Wong, N., Teo, K., Tan, C. S., et al. (2018). Cost-benefit analysis of introducing next-generation sequencing (metagenomic) pathogen testing in the setting of pyrexia of unknown origin. PLoS One 13:e0194648. doi: 10.1371/journal.pone.0194648
Chen, X., Cao, K., Wei, Y., Qian, Y., Liang, J., Dong, D., et al. (2020). Metagenomic next-generation sequencing in the diagnosis of severe pneumonias caused by Chlamydia psittaci. Infection 48, 535–542. doi: 10.1007/s15010-020-01429-0
Durack, D. T., and Street, A. C. (1991). Fever of unknown origin – reexamined and redefined. Curr. Clin. Top. Infect. Dis. 11, 35–51.
Eisenstein, M. (2012). Oxford Nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 30, 295–296. doi: 10.1038/nbt0412-295
Fu, Z.-F., Zhang, H.-C., Zhang, Y., Cui, P., Zhou, Y., Wang, H.-Y., et al. (2021). Evaluations of clinical utilization of metagenomic next-generation sequencing in adults with fever of unknown origin. Front. Cell. Infect. Microbiol. 11:745156. doi: 10.3389/fcimb.2021.745156
Fusco, F. M., Pisapia, R., Nardiello, S., Cicala, S. D., Gaeta, G. B., and Brancaccio, G. (2019). Fever of unknown origin (FUO): which are the factors influencing the final diagnosis? A 2005–2015 systematic review. BMC Infect. Dis. 19:653. doi: 10.1186/s12879-019-4285-8
Gaeta, G. B., Fusco, F. M., and Nardiello, S. (2006). Fever of unknown origin: a systematic review of the literature for 1995-2004. Nucl. Med. Commun. 27, 205–211. doi: 10.1097/00006231-200603000-00002
Haidar, G., and Singh, N. (2022). Fever of unknown origin. N. Engl. J. Med. 386, 463–477. doi: 10.1056/NEJMra2111003
Horiba, K., Torii, Y., Okumura, T., Takeuchi, S., Suzuki, T., Kawada, J.-I., et al. (2021). Next-generation sequencing to detect pathogens in pediatric febrile neutropenia: a single-center retrospective study of 112 cases. Open Forum Infect. Dis. 8:ofab223. doi: 10.1093/ofid/ofab223
Horowitz, H. W. (2013). Fever of unknown origin or fever of too many origins? N. Engl. J. Med. 368, 197–199. doi: 10.1056/NEJMp1212725
Jerome, H., Taylor, C., Sreenu, V. B., Klymenko, T., Filipe, A. D. S., Jackson, C., et al. (2019). Metagenomic next-generation sequencing aids the diagnosis of viral infections in febrile returning travellers. J. Infect. 79, 383–388. doi: 10.1016/j.jinf.2019.08.003
Levene, M. J., Korlach, J., Turner, S. W., Foquet, M., Craighead, H. G., and Webb, W. W. (2003). Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299, 682–686.
Li, T., Mbala-Kingebeni, P., Naccache, S. N., Thézé, J., Bouquet, J., Federman, S., et al. (2019). Metagenomic next-generation sequencing of the 2014 Ebola virus disease outbreak in the democratic republic of the Congo. J. Clin. Microbiol. 57:e00827-19. doi: 10.1128/JCM.00827-19
Liu, D., Zhang, J., Wu, B., Liu, F., Ye, S., Wang, H., et al. (2020). Impact of donor lung colonized bacteria detected by next-generation sequencing on early post-transplant outcomes in lung transplant recipients. BMC Infect. Dis. 20:689. doi: 10.1186/s12879-020-05393-w
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Maxam, A. M., and Gilbert, W. (1977). A new method for sequencing DNA. Proc. Natl. Acad. Sci. U.S.A. 74, 560–564.
Messacar, K., Parker, S. K., Todd, J. K., and Dominguez, S. R. (2017). Implementation of rapid molecular infectious disease diagnostics: the role of diagnostic and antimicrobial stewardship. J. Clin. Microbiol. 55, 715–723. doi: 10.1128/JCM.02264-16
Morganti, S., Tarantino, P., Ferraro, E., D’Amico, P., Viale, G., Trapani, D., et al. (2019). Complexity of genome sequencing and reporting: next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit. Rev. Oncol. Hematol. 133, 171–182. doi: 10.1016/j.critrevonc.2018.11.008
Mourad, O., Palda, V., and Detsky, A. S. (2003). A comprehensive evidence-based approach to fever of unknown origin. Arch. Intern. Med. 163, 545–551.
News in Brief (2020). First Ngs-based COVID-19 diagnostic. Nat. Biotechnol. 38:777. doi: 10.1038/s41587-020-0608-y
Nyrén, P., Pettersson, B., and Uhlén, M. (1993). Solid phase DNA minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Anal. Biochem. 208, 171–175. doi: 10.1006/abio.1993.1024
Pérez-Enciso, M., and Ferretti, L. (2010). Massive parallel sequencing in animal genetics: wherefroms and wheretos. Anim. Genet. 41, 561–569. doi: 10.1111/j.1365-2052.2010.02057.x
Petersdorf, R. G., and Beeson, P. B. (1961). Fever of unexplained origin: report on 100 cases. Medicine 40, 1–30.
Ren, L.-L., Wang, Y.-M., Wu, Z.-Q., Xiang, Z.-C., Guo, L., Xu, T., et al. (2020). Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin. Med. J. 133, 1015–1024. doi: 10.1097/CM9.0000000000000722
Reuwer, A. Q., van den Bijllaardt, W., Murk, J. L., Buiting, A. G. M., and Verweij, J. J. (2019). Added diagnostic value of broad-range 16s PCR on periprosthetic tissue and clinical specimens from other normally sterile body sites. J. Appl. Microbiol. 126, 661–666. doi: 10.1111/jam.14156
Reyes, A., Carbo, E. C., Harinxma Thoe Slooten, J. S. V., Kraakman, M. E. M., Sidorov, I. A., Claas, E. C. J., et al. (2021). Viral metagenomic sequencing in a cohort of international travellers returning with febrile illness. J. Clin. Virol. 143:104940. doi: 10.1016/j.jcv.2021.104940
Ronaghi, M., Uhlén, M., and Nyrén, P. (1998). A sequencing method based on real-time pyrophosphate. Science 281, 363–365.
Sanger, F., and Coulson, A. R. (1975). A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94, 441–448.
Shi, J., Yang, N., and Qian, G. (2021). Case report: metagenomic next-generation sequencing in diagnosis of talaromycosis of an immunocompetent patient. Front. Med. 8:656194. doi: 10.3389/fmed.2021.656194
Strub, T., Giuliano, S., Ye, T., Bonet, C., Keime, C., Kobi, D., et al. (2011). Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene 30, 2319–2332. doi: 10.1038/onc.2010.612
Tromp, M., Lansdorp, B., Bleeker-Rovers, C. P., Gunnewiek, J. M. K., Kullberg, B. J., and Pickkers, P. (2012). Serial and panel analyses of biomarkers do not improve the prediction of bacteremia compared to one procalcitonin measurement. J. Infect. 65, 292–301. doi: 10.1016/j.jinf.2012.06.004
Unger, M., Karanikas, G., Kerschbaumer, A., Winkler, S., and Aletaha, D. (2016). Fever of unknown origin (FUO) revised. Wien Klin. Wochenschr. 128, 796–801.
Williams, S. H., Cordey, S., Bhuva, N., Laubscher, F., Hartley, M.-A., Boillat-Blanco, N., et al. (2018). Investigation of the plasma virome from cases of unexplained febrile illness in Tanzania from 2013 to 2014: a comparative analysis between unbiased and vircapseq-vert high-throughput sequencing approaches. mSphere 3:e00311-18. doi: 10.1128/mSphere.00311-18
Xiao, M., Liu, X., Ji, J., Li, M., Li, J., Yang, L., et al. (2020). Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 12:57. doi: 10.1186/s13073-020-00751-4
Zhu, H., Zhu, M., Lei, J.-H., Xiao, Y.-L., and Zhao, L.-M. (2021). Metagenomic next-generation sequencing can clinch diagnosis of non-tuberculous mycobacterial infections: a case report. Front. Med. 8:679755. doi: 10.3389/fmed.2021.679755
Keywords: quantitative metagenomics next-generation sequencing, fever of unknown origin, infections, pathogen, rare disease
Citation: Dong Y-x, Gao Y-l, Chai Y-f and Shou S-t (2022) Use of Quantitative Metagenomics Next-Generation Sequencing to Confirm Fever of Unknown Origin and Infectious Disease. Front. Microbiol. 13:931058. doi: 10.3389/fmicb.2022.931058
Received: 28 April 2022; Accepted: 15 June 2022;
Published: 04 July 2022.
Edited by:
Hezhao Ji, Public Health Agency of Canada (PHAC), CanadaReviewed by:
Eswarappa Pradeep Bulagonda, Sri Sathya Sai Institute of Higher Learning (SSSIHL), IndiaBinhua Liang, Public Health Agency of Canada (PHAC), Canada
Copyright © 2022 Dong, Gao, Chai and Shou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songtao Shou, c3RzaG91QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship