 Daria Shafranskaya
Daria Shafranskaya Varsha Kale
Varsha Kale Rob Finn2
Rob Finn2 Alla L. Lapidus
Alla L. Lapidus Anton Korobeynikov
Anton Korobeynikov Andrey D. Prjibelski
Andrey D. Prjibelski- 1Center for Algorithmic Biotechnology, Saint Petersburg State University, Saint Petersburg, Russia
- 2European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Cambridge, United Kingdom
- 3Department of Computer Science, University of Helsinki, Helsinki, Finland
While metagenome sequencing may provide insights on the genome sequences and composition of microbial communities, metatranscriptome analysis can be useful for studying the functional activity of a microbiome. RNA-Seq data provides the possibility to determine active genes in the community and how their expression levels depend on external conditions. Although the field of metatranscriptomics is relatively young, the number of projects related to metatranscriptome analysis increases every year and the scope of its applications expands. However, there are several problems that complicate metatranscriptome analysis: complexity of microbial communities, wide dynamic range of transcriptome expression and importantly, the lack of high-quality computational methods for assembling meta-RNA sequencing data. These factors deteriorate the contiguity and completeness of metatranscriptome assemblies, therefore affecting further downstream analysis.
Here we present MetaGT, a pipeline for de novo assembly of metatranscriptomes, which is based on the idea of combining both metatranscriptomic and metagenomic data sequenced from the same sample. MetaGT assembles metatranscriptomic contigs and fills in missing regions based on their alignments to metagenome assembly. This approach allows to overcome described complexities and obtain complete RNA sequences, and additionally estimate their abundances. Using various publicly available real and simulated datasets, we demonstrate that MetaGT yields significant improvement in coverage and completeness of metatranscriptome assemblies compared to existing methods that do not exploit metagenomic data. The pipeline is implemented in NextFlow and is freely available from https://github.com/ablab/metaGT.
Introduction
Metagenome sequencing gained noticeable popularity in the past decade, as multiple projects shed light on microbial communities in various ecosystems (Poretsky et al., 2005; Nowinski et al., 2019) and eukaryotic microbiomes (Turnbaugh et al., 2007; Arumugam et al., 2011; Lloyd-Price et al., 2019). However, these studies required the development of novel software tools, as the previously designed methods for conventional sequencing data analysis appeared to be underperforming on large and complex metagenomic datasets. Thus, multiple tools, such as de novo assemblers (Li et al., 2015; Nurk et al., 2017), sequence binners (Uritskiy et al., 2018; Kang et al., 2019; Nissen et al., 2021), annotation pipelines (Seemann, 2014, Keegan et al., 2016) and various pipelines for metagenomic downstream analysis (Caporaso et al., 2010; Mitchell et al., 2020) were developed in the past years.
Although metagenomic sequencing may provide insights on species abundances and gene content, it does not show which members of the community and which genes are active, and how this activity depends on external conditions. To analyze gene expression in the microbial community researchers perform RNA-Seq experiments, which may include sequencing of samples under different conditions, time series, as well as complementary metagenomic and metatranscriptomic sequencing.
As complete genomes of the organisms in the community of interest are often unknown, both metagenomics and metatranscriptomics studies heavily rely on de novo sequence assembly. While assembly of metagenomes is typically performed with community-established tools, such as MEGA-HIT (Li et al., 2015) and metaSPAdes (Nurk et al., 2017), metatranscriptome assembly software remains at an early stage and no pipeline is currently regarded as a golden standard (Shakya et al., 2019). Among available tools one can name IDBA-MT (Leung et al., 2013) and its derivative version IDBA-MTP (Leung et al., 2015), which utilizes a database of known proteins to reconstruct complete transcript sequences. Another tool, TAG (Ye and Tang, 2016), exploits the fact that metatranscriptomes are often sequenced along with the metagenomic data from the same sample. TAG maps RNA-Seq reads onto a metagenome assembly graph using and further restores paths corresponding to transcripts. Unfortunately, all listed tools appear to be unmaintained for several years and challenging to run under modern environments. Thus, some of the current studies exploit conventional RNA-Seq assemblers, such as Trinity (Grabherr et al., 2011) and rnaSPAdes (Bushmanova et al., 2019), performance of which remains under-examined on metatranscriptomic data.
In this work we present MetaGT, a user-friendly pipeline for de novo assembly of metatranscriptomes, which follows the concept of TAG assembler by simultaneous usage of both metagenomic and metatranscriptomic sequencing data obtained from the same sample. We demonstrate that using metagenomic data greatly improves completeness of assembled transcripts compared to sequences assembled solely from metatranscriptomic data.
Materials and methods
Pipeline overview
MetaGT is a pipeline for de novo assembly of metatranscriptomes, which is based on the idea of combining both metatranscriptomic and metagenomic data sequenced from the same sample. First, MetaGT pipeline assembles metatranscriptomic and metagenomic reads individually with rnaSPAdes (Bushmanova et al., 2019) and metaSPAdes (Nurk et al., 2017) respectively. Metagenomic contigs are then annotated with Prokka pipeline (Seemann, 2014). Alternatively, a user may provide assemblies and the annotation obtained with software of their choice. Further, MetaGT aligns transcriptomic contigs to the genomic fragments with minimap2 (Li, 2018). These alignments are used to extend, merge and correct assembled transcriptomic contigs into full-length transcripts. Optionally, unaligned contigs are annotated with Transdecoder1 and clustered with previously obtained full-length transcripts with MMseqs2 (Steinegger and Söding, 2017) in order to avoid duplications. Finally, the resulting set of transcripts is quantified using Kallisto (Bray et al., 2016). The scheme of the pipeline is presented in Figure 1.
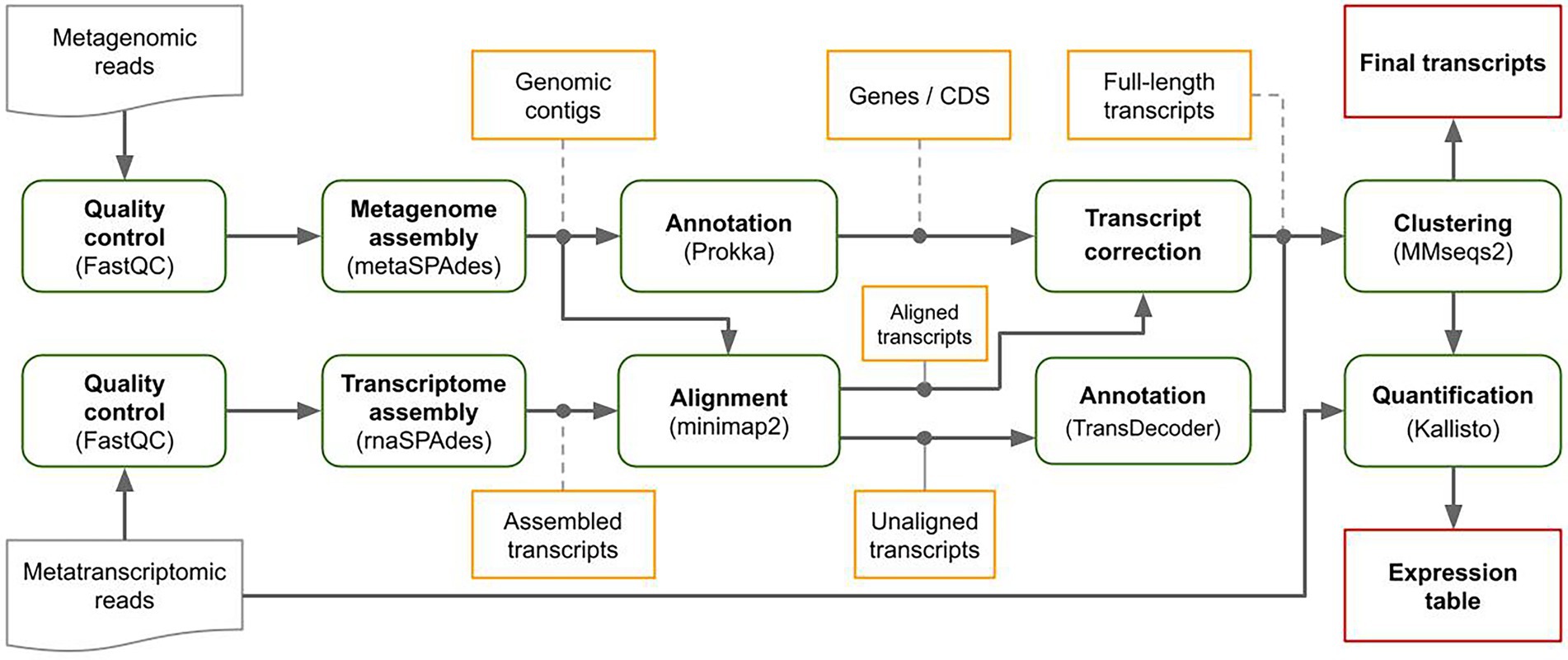
Figure 1. MetaGT pipeline.
Transcript correction
Due to extremely uneven coverage depth and presence of homologous genes in diverse microbial communities, assemblers tend to generate incomplete and fragmented transcript sequences. For example, during the analysis of various metatranscriptomic samples we detected such assembly artifacts as: (i) fragmented or overlapping transcripts (Figure 2A), (ii) incompletely assembled transcripts (Figures 2B,C) and (iii) extended transcripts containing intergenic sequences (Figure 2D). To improve a metatranscriptome assembly MetaGT implements a procedure that corrects assembled transcripts based on their alignments to the annotated metagenomic contigs.
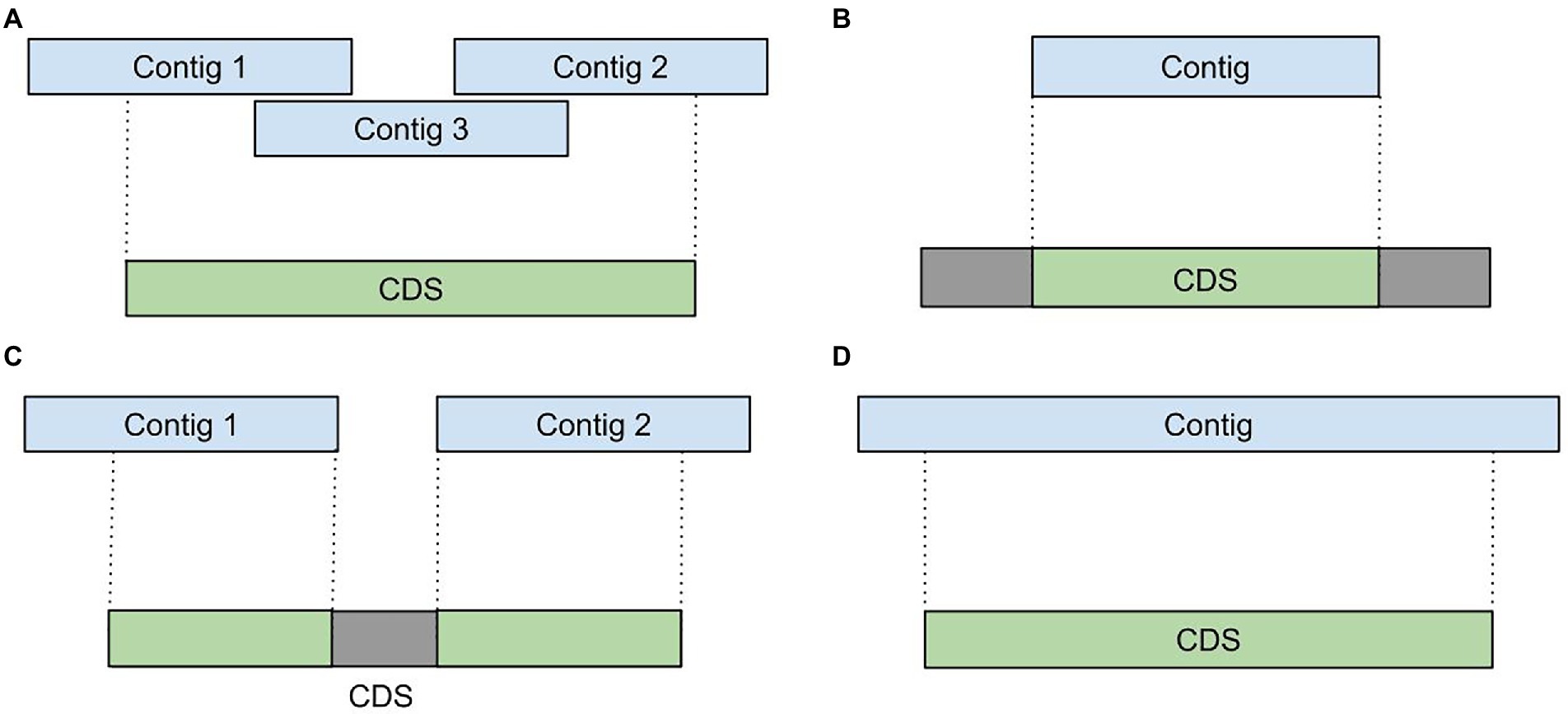
Figure 2. Assembly artifacts typical for metatranscriptome de novo assembly. (A) Fragmented transcripts. (B) Partially assembled transcripts. (C) Fragmented and partially assembled transcripts. (D) Transcripts containing intergenic sequences.
First, coordinates of the aligned transcripts are compared against positions of the сoding regions predicted by Prokka in the metagenomic contigs. This allows to identify incompletely assembled and fragmented transcripts. For further processing MetaGT selects only coding regions that have at least 50% of their bases covered by the assembled transcripts. This threshold is merely a default parameter and can be modified by the user.
Further, MetaGT merges selected coding regions with the respective assembled sequences by concatenating fragmented transcripts and filling in missing sequences. Moreover, MetaGT removes excessive fragments that map outside of coding regions. Thus, transcriptomic contigs representing operons with several coding regions are split into individual sequences. During the consensus step MetaGT prioritizes assembled sequences in order to preserve any variants and modifications detected in RNA-Seq data. The output of this procedure is a set of transcripts, in which each of them includes a single complete coding region.
Analysis of unaligned transcripts
Metatranscriptome sequencing may also capture mRNAs that are not produced by the microbial community itself, for example, products of food from gut microbiome samples. Since assembled sequences corresponding to such mRNAs are unlikely to map to the metagenomic contigs, they are processed separately. For all unaligned sequences MetaGT uses TransDecoder2 to identify reliable transcript candidates. To avoid duplicated transcripts in its output, MetaGT further uses MMSeqs2 (Steinegger and Söding, 2017) to cluster transcripts reported by TransDecoder with full-length transcripts obtained via correction step. If a user wishes to exclude unaligned sequences from the analysis, this step can be turned off via command line options. The resulting full-length transcript sequences are saved to a FASTA file, in which transcripts are marked as aligned or unaligned.
Datasets used for testing
To test MetaGT we simulated metagenomic and metatranscriptomic data based on 3 sets of genomes with various complexity: (i) Simple7: 7 bacterial genomes, (ii) Medium20: 20 bacterial genomes, and (iii) Complex32: 32 bacterial genomes, some of which belong to closely related strains (Table 1). The composition of the simulated data was selected in order to test the applicability of the developed software to bacterial communities with different properties. Indeed, these obtained sets of genomes are synthetic and do not reflect any actual bacterial communities known to date.

Table 1. Datasets used in this work.
During the simulation each bacteria was assigned a random relative abundance value, such that the abundance distribution within the simulated community resembles a distribution of the real one. These values were used to simulate metagenomic Illumina reads with InSilicoSeq software (Gourlé et al., 2019). Further, each gene was assigned an arbitrary expression value in a similar manner: artificial gene expression patterns should be similar to ones observed in real-life data. To obtain the resulting gene abundances for simulation, generated expression levels were multiplied by the respective species abundance. Metatranscriptomic reads were then simulated using the RSEM simulator (Li and Dewey, 2011).
We also exploited the Mock16 dataset containing real sequencing data obtained from a synthetic mix of 16 different bacteria (Ternus et al., 2021; Table 1). Finally, to test MetaGT in a real-life environment we used sequencing data from the human and snail gut microbiomes: HumanGut (Lloyd-Price et al., 2019) and SnailGut datasets (Yang et al., 2022).
Quality evaluation
To evaluate assembled sequences we aligned them against the true set of transcripts with minimap2 (Li, 2018). These alignments are processed to estimate the number of unmapped contigs and percentage of captured transcripts. A reference transcript is considered to be captured if 95% of its bases are covered by a single alignment. For simulated data the ground truth set contained only transcripts with expression level TPM ≥ 1. For Mock16 we used the entire reference set as the true expression is unknown.
Further, to access assembly correctness we estimated the number of misassemblies using rnaQUAST (Bushmanova et al., 2016) and simply by mapping assembled transcripts to the reference genomes with minimap2. A contig is considered as misassembled if its parts (i) either align to different genomes, or (ii) map to the same genome at least 1 kbp apart (Gurevich et al., 2013).
For real sequencing data we also estimate assembly completeness using predicted coding regions in genomic contigs. We similarly map resulting transcripts to the predicted CDS using minimap2, and for each CDS we compute its fraction captured by a single transcript alignment.
Results
Table 2 demonstrates that on simulated data simultaneous use of metagenomic and metatranscriptomic data allows to reduce the total number of assembled sequences by 12% and the amount of unaligned contigs by 15% on average compared to assemblies obtained solely from RNA-Seq data. Importantly, MetaGT shows a 16% average increase in the number of captured reference transcripts. Moreover, the difference appears to be more significant for complex communities, where homologous genes are frequent and de novo assembly becomes challenging: for Complex32 simulated dataset MetaGT restored 25% more complete RNA sequences compared to rnaSPAdes. With respect to assembly correctness, both rnaSPAdes and MetaGT show a rather low number of misassembled contigs according to rnaQUAST. However, MetaGT is being somewhat less accurate on Medium20 and Complex35 datasets according to the genomic approach to misassembly detection (see “Materials and methods”).
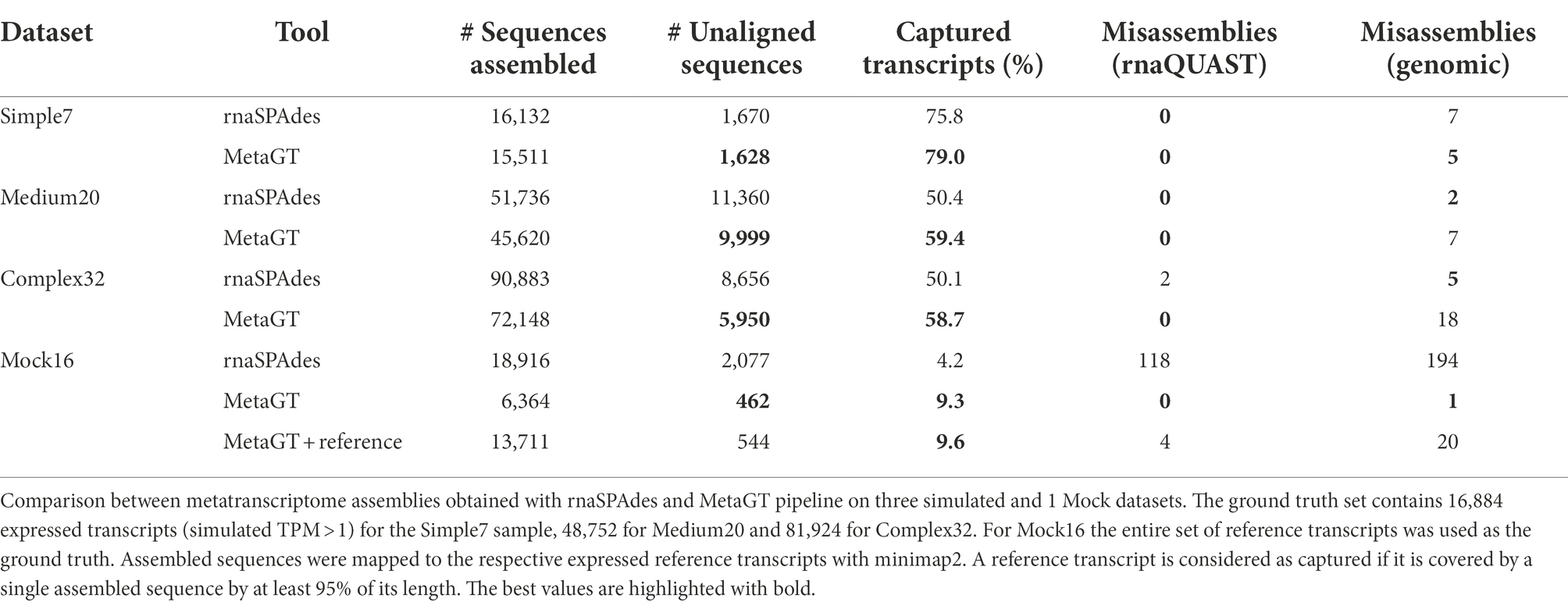
Table 2. Assembly results on simulated data and synthetic community.
Importantly, on the synthetic Mock16 dataset MetaGT yields a more significant improvement: a 4-fold drop in unaligned contigs and almost a double increase in captured reference transcripts. Moreover, Table 2 shows that MetaGT eliminated almost all misassembled contigs from the initial rnaSPAdes assembly (0 vs. 118 according to rnaQUAST).
Since in real-life metagenomic projects researchers obtain metagenome-assembled genomes (MAGs), we also tested MetaGT on Mock16 data by providing reference genomes instead of raw genomic reads. Interestingly, using the genome assembly of a significantly better quality results only in a marginal improvement in transcriptome assembly. As per Table 2, this approach allowed to capture only 121 additional transcripts (0.3%) and resulted in a few more misassemblies compared to the original read-based approach. However, we predict that for more complex bacterial communities exploiting MAGs instead of draft genome assembly could potentially lead to a more noticeable improvement.
For real sequencing data (Mock16 and HumanGut) we estimated completeness of assembled transcripts using predicted genes as described in the Quality evaluation section. Expectedly, combined usage of metatranscriptomic and metagenomic data yields a significantly higher percentage of fully captured transcripts on both datasets (Figures 3A,B respectively). On Mock16 MetaGT reconstructs 6,425 full-length transcripts (95% of bases captured), which is 2.5-fold more than assembled by rnaSPAdes (2,596). Similarly, for real HumanGut dataset MetaGT reports 2-fold more complete transcripts compared to rnaSPAdes (7,465 vs. 3,649), thus proving that the developed pipeline is efficient on real data and allows to significantly reduce fragmentation of the assembly. It is important to point out that this striking improvement also demonstrates that real sequencing data is noticeably more challenging for de novo transcriptome assembly compared to simulated data.
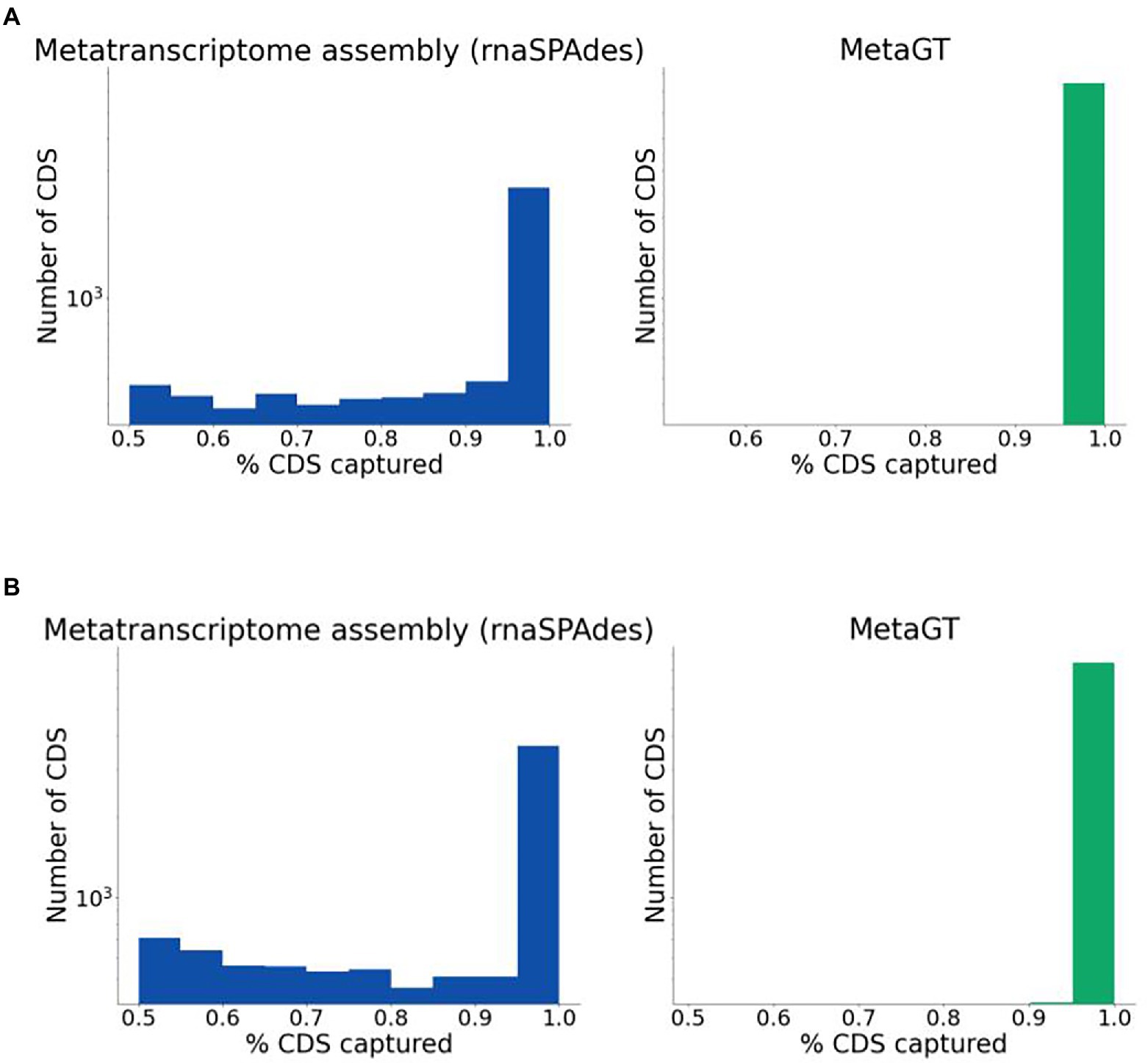
Figure 3. Completeness of sequences assembled from real data. (A) Histogram for percentage of coding regions captured by rnaSPAdes (left) and MetaGT (right) assemblies on the Mock16 dataset. The histogram is shown in logarithmic scale. (B) Same as (A), but for the HumanGut dataset.
To assess MetaGT ability to estimate gene expression levels, we reproduced quantification results from a recent metatranscriptomic study of a deep-sea snail gut microbiome (Yang et al., 2022). Abundances generated with the MetaGT pipeline were compared against the approach exploited in the original work, in which Salmon (Patro et al., 2017) was used to quantify genes predicted by Prodigal (Hyatt et al., 2010). In addition, we also computed expression levels for the same set of genes with an alignment-based approach by using minimap2 (Li, 2018) and featureCounts from the Subread package (Liao et al., 2014). We then estimated similarity between counts provided by MetaGT and two other approaches by computing Spearman’s rank correlation coefficient (Figures 4A,B respectively). In both cases the results appear to be highly similar with Spearman’s Rho > 0.98 (p-values < 2.2 × 10−16), which suggests that MetaGT provides meaningful quantification results as well.
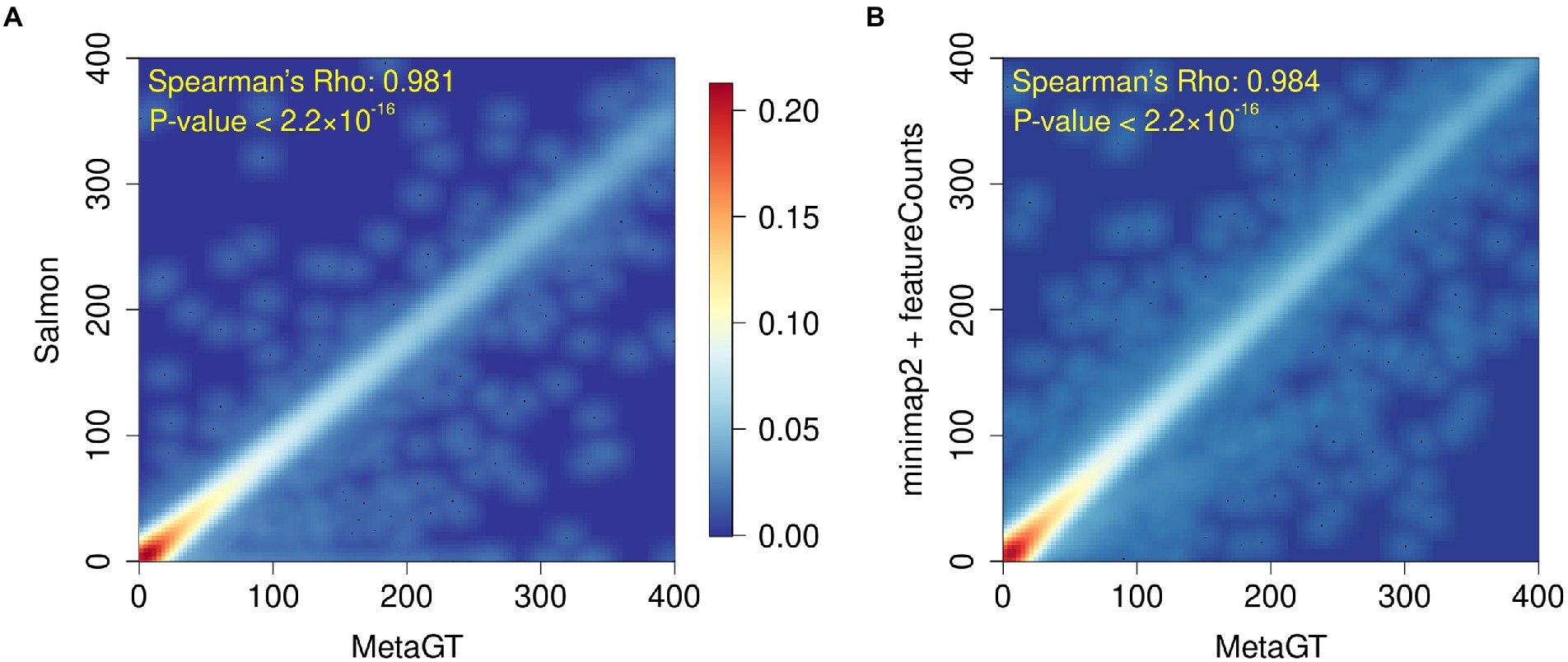
Figure 4. Heatmaps showing comparison between gene counts obtained with MetaGT and (A) approach used in the original study (Prodigal and Salmon), and (B) alignment based approach that involves Prodigal, mimimap2 and featureCounts.
Discussion
While combining metatranscriptomic data with metagenome assemblies obtained from the same sample seems to be intuitive, to the best of our knowledge no modern bioinformatics software implements such an idea, with a solo exception of TAG assembler, support of which has been unfortunately discontinued a long time ago. As described previously, TAG maps RNA-Seq reads using Bowtie2 (Langmead and Salzberg, 2012) and k-mer matching to the metagenomic de Bruijn graph, and further derives transcript sequences from the corresponding alignment paths in the graph. In comparison to MetaGT, the approach implemented in TAG can be useful for reconstructing transcripts with extremely low coverage, i.e., when the number of reads is insufficient for de novo transcriptome assembly. At the same time, TAG may output incomplete and fragmented transcripts when RNA-Seq reads do not cover the entire coding region. In contrast, MetaGT exploits predicted genes to fill in the gaps and restore complete transcripts sequences.
In this work we present a pipeline that performs de novo assembly of metagenome and metatranscriptome sequencing data using existing software and combines the results in order to reconstruct and further quantify full-length transcripts. Providing complete coding sequences as the result of the assembly pipeline may significantly improve quality of the downstream analysis, such as functional annotation, gene ontology and differential expression analysis. In the view of growing popularity of metatranscriptomic sequencing we believe that MetaGT will be a useful instrument in the field and will allow researchers to perform high-quality studies without spending time developing custom in-house pipelines.
Data availability statement
The datasets analyzed in this study can be found in the NCBI short read archive (SRA) under accession numbers SRR5947833, SRR5947907, SRR10175815, SRR10175826, SRR8397925, SRR8416101. Simulated data is published on Zenodo with DOI: 10.5281/zenodo.7152149. The pipeline is implemented in NextFlow and is freely available at https://github.com/ablab/metaGT.
Author contributions
DS designed and developed MetaGT software. DS and VK tested the software. DS, AK, and AP wrote the manuscript. RF and VK provided the data for testing. AK and AL acquired funding. RF, AK, and AP managed the project and supervised the research. All authors contributed to the article and approved the submitted version.
Funding
The research was carried out in part by computational resources provided by the Resource Center “Computer Center of SPbU.” The reported study was funded by the Russian Scientific Foundation, project number 19-14-00172.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8, 1–13. doi: 10.1093/gigascience/giz100
Bushmanova, E., Antipov, D., Lapidus, A., Suvorov, V., and Prjibelski, A. D. (2016). rnaQUAST: a quality assessment tool for de novo transcriptome assemblies. Bioinformatics 32, 2210–2212. doi: 10.1093/bioinformatics/btw218
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Gourlé, H., Karlsson-Lindsjö, O., Hayer, J., and Bongcam-Rudloff, E. (2019). Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics 35, 521–522. doi: 10.1093/bioinformatics/bty630
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1038/NBT.1883
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 1–11. doi: 10.1186/1471-2105-11-119
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019:e7359. doi: 10.7717/PEERJ.7359/SUPP-3
Keegan, K. P., Glass, E. M., and Meyer, F. (2016). MG-RAST, a metagenomics Service for Analysis of microbial community structure and function. Methods Mol. Biol. 1399, 207–233. doi: 10.1007/978-1-4939-3369-3_13
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leung, H. C., Yiu, S. M., and Chin, F. Y. (2015). IDBA-MTP: a hybrid metatranscriptomic assembler based on protein information. J. Comput. Biol. 22, 367–376.
Leung, H. C. M., Yiu, S.-M., Parkinson, J., and Chin, F. Y. L. (2013). IDBA-MT: de novo assembler for metatranscriptomic data generated from next-generation sequencing technology. J. Comput. Biol. 20, 540–550. doi: 10.1089/cmb.2013.0042
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 1–16. doi: 10.1186/1471-2105-12-323/tables/6
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lloyd-Price, J., Arze, C., Ananthakrishnan, A. N., Schirmer, M., Avila-Pacheco, J., Poon, T. W., et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. doi: 10.1038/s41586-019-1237-9
Mitchell, A. L., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., et al. (2020). MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578. doi: 10.1093/NAR/GKZ1035
Nissen, J. N., Johansen, J., Allesøe, R. L., Sønderby, C. K., Armenteros, J. J. A., Grønbech, C. H., et al. (2021). Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560. doi: 10.1038/s41587-020-00777-4
Nowinski, B., Smith, C. B., Thomas, C. M., Esson, K., Marin, R., Preston, C. M., et al. (2019). Microbial metagenomes and metatranscriptomes during a coastal phytoplankton bloom. Sci. Data 6:129. doi: 10.1038/S41597-019-0132-4
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Poretsky, R. S., Bano, N., Buchan, A., LeCleir, G., Kleikemper, J., Pickering, M., et al. (2005). Analysis of microbial gene transcripts in environmental samples. Appl. Environ. Microbiol. 71, 4121–4126. doi: 10.1128/AEM.71.7.4121-4126.2005
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shakya, M., Lo, C. C., and Chain, P. S. G. (2019). Advances and challenges in metatranscriptomic analysis. Front. Genet. 10:904. doi: 10.3389/fgene.2019.00904/bibtex
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Ternus, K. L., Keplinger, N. C., Kappell, A. D., Godbold, G. D., Palsikar, V., Acevedo, C. A., et al. (2021). Detection of ESKAPE pathogens and Clostridioides difficile in simulated skin transmission events with metagenomic and Metatranscriptomic sequencing. bioRxiv [Epub ahead of preprint], doi: 10.1101/2021.03.04.433847
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Yang, Y., Sun, J., Chen, C., Zhou, Y., Van Dover, C. L., Wang, C., et al. (2022). Metagenomic and metatranscriptomic analyses reveal minor-yet-crucial roles of gut microbiome in deep-sea hydrothermal vent snail. Animal Microbiome 4, 1–17. doi: 10.1186/s42523-021-00150-z
Keywords: metatranscriptomics, metagenomics, RNA-Seq, de novo assembly, computational pipeline
Citation: Shafranskaya D, Kale V, Finn R, Lapidus AL, Korobeynikov A and Prjibelski AD (2022) MetaGT: A pipeline for de novo assembly of metatranscriptomes with the aid of metagenomic data. Front. Microbiol. 13:981458. doi: 10.3389/fmicb.2022.981458
Edited by:
Richard Allen White III, University of North Carolina at Charlotte, United StatesReviewed by:
Yu-Wei Wu, Taipei Medical University, TaiwanMircea Podar, Oak Ridge National Laboratory (DOE), United States
Copyright © 2022 Shafranskaya, Kale, Finn, Lapidus, Korobeynikov and Prjibelski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrey D. Prjibelski, YW5kcmV3cHJ6aEBnbWFpbC5jb20=