 Zhongchi Cui
Zhongchi Cui Wenyue Wu
Wenyue Wu Fan Fan
Fan Fan Fei Wang
Fei Wang Daqun Liu
Daqun Liu Dianping Di
Dianping Di Haiyan Wang
Haiyan Wang- 1College of Plant Protection, Technological Innovation Center for Biological Control of Crop Diseases and Insect Pests of Hebei Province, Hebei Agricultural University, Baoding, Hebei, China
- 2Plant Protection Institute, Hebei Academy of Agricultural and Forestry Sciences, Baoding, Hebei, China
Introduction: Wheat leaf rust caused by Puccinia triticina (Pt) remains one of the most destructive diseases of common wheat worldwide. Understanding the pathogenicity mechanisms of Pt is important to control wheat leaf rust.
Methods: The urediniospores of Pt race PHNT (wheat leaf rust resistance gene Lr19-avirulent isolate) were mutagenized with ethyl methanesulfonate (EMS), and two Lr19-virulent mutants named M1 and M2 were isolated. RNA sequencing was performed on samples collected from wheat cultivars Chinese Spring and TcLr19 infected with wild-type (WT) PHNT, M1, and M2 isolates at 14 days post-inoculation (dpi), respectively. Screening AvrLr19 candidates by quantitative reverse transcription PCR (qPCR) and Agrobacterium-mediated transient assays in Nicotiana benthamiana.
Results: 560 genes with single nucleotide polymorphisms (SNPs) and insertions or deletions (Indels) from non-differentially expressed genes were identified. Among them, 10 secreted proteins were screened based on their fragments per kilobase of exon model per million mapped reads (FPKM) values in the database. qPCR results showed that the expression profiles of 7 secreted proteins including PTTG_27471, PTTG_12441, PTTG_28324, PTTG_26499, PTTG_06910, PTTG_26516, and PTTG_03570 among 10 secreted proteins in mutants were significantly different with that in wild-type isolate after infection wheat TcLr19 and might be related to the recognition between Lr19 and AvrLr19. In addition, a total of 216 differentially expressed genes (DEGs) were obtained from three different sample comparisons including M1-vs-WT, M2-vs-WT, and M1-vs-M2. Among 216 DEGs, 15 were predicted to be secreted proteins. One secreted protein named PTTG_04779 could inhibit programmed progress of cell death (PCD) induced by apoptosis-controlling genes B-cell lymphoma-2 associated X protein (BAX) on Nicotiana benthamiana, indicating that it might play a virulence function in plant. Taken together, total 8 secreted proteins, PTTG_04779, PTTG_27471, PTTG_12441, PTTG_28324, PTTG_26499, PTTG_06910, PTTG_26516, PTTG_03570 are identified as AvrLr19 candidates.
Discussion: Our results showed that a large number of genes participate in the interaction between Pt and TcLr19, which will provide valuable resources for the identification of AvrLr19 candidates and pathogenesis-related genes.
1. Introduction
Wheat leaf rust caused by Puccinia triticina (Pt) is one of the most common and severe diseases in the wheat-growing regions worldwide. The yield losses of wheat infected with Pt ranges from 5–15% and the yield of wheat infected with leaf rust at the seedling stage can be reduced by 50% or even more (Bolton et al., 2008; Prasad et al., 2020). Considering the impacts of global climate change, temperature and humidity conditions may be more suitable for the proliferation and epidemic of Pt in the future, which will further deleteriously impact wheat yields. Control of leaf rust mainly relies on the fungicide application and deployment of cultivars carrying resistance genes. Genetic resistance is the most effective, environmentally safe, and economically feasible approach to reduce the damage caused by Pt. However, monoculture of select resistant varieties leads to the host selection pressures that drive Pt evolution and promote the continuous emergence of new virulent races, which often leads to the decline of wheat resistant varieties after several years of planting.
To date, over 100 genes conferring leaf rust resistance derived from wheat (Triticum) cultivars have been identified and designated as Lr1 to Lr79 (Qureshi et al., 2018). Among them, Lr9, Lr19, Lr24, Lr34, Lr37, Lr38, Lr46, Lr47, Lr51, Lr53, Lr68 and Lr42 are known to confer effective resistance to leaf rust according to the field survey in Sichuan province, China (Gao et al., 2019). Lr19 was derived from the grass Agropyron elongatum and was transferred to the long arm of wheat chromosome 7D (Pourkhorshid et al., 2022). Although resistance of Lr19 has been overcome by some virulent Pt races in Mexico (Huerta and Singh, 1994), Russia (Lind and Gultyaeva, 2007), Ukraine (Elyasi-Gomari and Panteleev, 2006) and India (Bhardwaj et al., 2005), it still showed high resistance to all the collected Pt races in China (Zhang et al., 2020). It has been reported that Lr19 holds the potential to be deployed in combination with other Lr genes in the field to confer durable resistance against leaf rust (Mccallum et al., 2021). It has great potential in wheat breeding program in China. Although Lr19 has not been cloned, yet many molecular markers closely linked to the Lr19 gene have been developed, such as SCS265, SCS253, GB, Xwmc221, XustSSR2001-7DL, Xgwm37 and Xgwm44, and two of them (GB and Xwmc221) proved to be specific to Lr19 (Kiel et al., 2020). To prevent Lr19 from being out-competed by newly emergent virulent races and enable the long-term deployment of Lr19 in leaf rust-resistant wheat varieties, it is key to clone avirulent gene AvrLr19 that can be recognized by wheat leaf rust resistance gene Lr19, determine the molecular mechanism of AvrLr19 and Lr19 interactions, and monitor the natural Pt population changes in response to Lr19 resistance.
Genetic mutation is the most important avenue for creating new rust races and genotypes. Ethyl methanesulfonate (EMS) is an alkylating chemical mutagen that generates single nucleotide polymorphisms (SNPs) and insertions and deletions (Indels), resulting in amino acid sequence variation, and finally leads to phenotype changes (Anderson et al., 2016). Mutagenesis integrated with genomic sequencing is an efficient way to study the relationships between phenotypic traits and associated genotypes, leading to the identification of fungal effectors or Avr genes. For example, Salcedo et al. (2017) obtained stripe rust mutants through EMS-induced mutation and performed genome sequencing to obtain AvrSr35 candidate genes, and then verified the candidate genes through co-expression of AvrSr35 and its corresponding resistance gene in tobacco and wheat to trigger cell death. Li et al. (2020) screened 30 mutant variants from the least virulent isolate generated by EMS mutagenesis and candidates Avr genes were determined by sequencing. Transcriptomics has proven to be an instrumental molecular tool to help identify virulence effectors and Avr genes (Elmore et al., 2020; Wei et al., 2020; Xu et al., 2020). In the process of the host and pathogen interaction, the underlying transcriptional regulation of gene expression in the plant and pathogen provides clues to their defense and virulence mechanisms, respectively (Rutter et al., 2017).
The gene-for-gene hypothesis proposed by Flor (2003) indicates that only the host with an “R gene” is resistant to the corresponding Avr gene in pathogens. Jones and Dangl (2006) proposed the famous “ZigZag” model in 2006 to analyze the molecular mechanism of interaction between plants and their pathogens. In order to inhibit plant defense responses, pathogens secrete a series of effector proteins through the haustorium, the structure with which the fungus attaches to and extracts nutrients from its host plant to interfere with or ablate the plant defense response, so as to meet their own growth needs. In recent years, with the continuous improvement of sequencing technology and the reduction of sequencing costs, and the development and application of prediction software such as SignalP (Almagro Armenteros et al., 2019), TargetP (Emanuelsson et al., 2000), TMHMM (Krogh et al., 2001), EffectorP (Sperschneider et al., 2016) and Pfam (Finn et al., 2014) have improved the screening efficiency of candidate Avr genes of rust. To date, a handful of Avr genes have been identified in rust pathogens, including AvrL567, AvrP123, AvrP4, AvrM, AvrL2 and AvrM14 from the flax rust pathogen (Anderson et al., 2016; Prasad et al., 2019), as well as RTP1 in bean rust (Kemen et al., 2005), and PGTAUSPE10–1 from wheat stem rust (Petre et al., 2014). Three articles reported the successful cloning of the Avr genes AvrSr50 (Chen et al., 2017) and AvrSr27 (Upadhyaya et al., 2021) of wheat stem rust, and preliminarily revealed their interaction with corresponding resistance genes (Segovia et al., 2016; Bakkeren and Szabo, 2020). Some effector candidates for Lr26, Lr9, Lr24 and Lr20 were obtained from Pt (Wu et al., 2017; Figueroa et al., 2020), but their biological functions have not been determined, so, no known Avr genes have been identified in Pt so far.
In this study, we aim to obtain Lr19-virulent mutants using EMS mutagenization, then identify differentially expressed genes (DEGs) and the genes with SNPs and Indel associated with the Pt infection. Our results will provide resources for identification of AvrLr19 candidates and pathogenicity-related genes, and lay a foundation for revealing the pathogenic mechanism of Pt.
2. Materials and methods
2.1. Plant materials and Pt isolates
Wheat seedlings of near-isogenic lines were requested from the Cereal Disease Lab of the USDA located at the University of Minnesota and were preserved in the laboratory of Leaf rust, Hebei Agricultural University. Pt race 07–10–426-1 (PHNT race), collected from China and preserved in our laboratory, was used to inoculate wheat according to Roelfs (1984) standards, it was incompatible with the resistant variety TcLr19 (origin of grain resource: Tc*6/RL6040, containing wheat leaf rust resistance gene Lr19, Lr19+), but compatible with the susceptible variety Chinese Spring (Lr19-). Seedlings (14 days old) from wheat lines Chinese Spring (Lr19-) inoculated by spraying with a suspension of Pt urediniospores were used as controls.
2.2. Staining and microscopic observation
To identify fungal tissues and the presence of dead cells in the leaf mesophyll of infected plants, the samples were stained via the Rohringer (1997) fluorescent staining method. Stained tissues were observed under optical microscopy, and at least 10 fungal infection sites from three biological replicates were analyzed for each stained leaf sample.
2.3. EMS mutagenesis of Pt urediniospores of the PHNT race
The Lr19-avirulent Pt race PHNT was used to create Lr19-virulent mutants by treatment with EMS as previously described (Salcedo et al., 2017). Four different concentrations of EMS were used for mutagenesis: 0.1 M, 0.05 M, 0.01 M and 0.005 M. After 2 hours (h) of treatment with EMS, then the urediniospores was washed 1 L of ddH2O and dried for 12–14 h at room temperature (22–25°C). The germination rate of spores was observed using microscopy and untreated Pt spores were used as control. Spores with at least 50% germination rate were applied with a brush to the surface of the primary leaves of differential wheat near-isogenic lines that carry different Lr genes to validate that only Lr19-virulence was affected by EMS mutagenesis. After inoculation, the plants were kept at 100% relative humidity in the dark for 24 h at 20°C, and then were incubated in a growth chamber at 22°C with a 16 h/8 h (day/night) photoperiod. In addition, the molecular marker of Lr19 was used to detect the validity of TcLr19 (Gupta et al., 2006).
2.4. Transcriptome sequencing and identification of the AvrLr19 candidates
The Pt pathotypes PHNT mutants and PHNT were purified, and then inoculated onto the TcLr19 and Chinese Spring, respectively. We sampled the inoculated leaf at 14 days post-inoculation (dpi) showing severe disease phenotypes for extraction of total RNA of Pt. Transcriptome sequencing and analyses were conducted by OE Biotech Co., Ltd. (Shanghai, China). The RNA was isolated using an RNA isolation kit (RNeasy Plant MiniKit, Tiangen, Beijing) according to the manufacturer’s instructions. Sequencing was carried out on the Illumina HiSeqTM 2500 platform. Then, the clean reads were mapped to reference genome “Pt 1–1 BBBD Race 1” (Kiran et al., 2016) using Hisat2 (Trapnell et al., 2010) to obtain the location information on the reference genome or gene, as well as the specific sequence feature information of the sequenced sample. The read counts of each gene were obtained by HTSeq-count (Anders et al., 2015). DEGs were identified using the DESeq R package (Wang et al., 2009). “FDR-adjusted value of p <0.05” and “|fold Change| >2” were set as the thresholds for significantly differential expression. Hierarchical clustering analysis of DEGs was performed to explore gene expression patterns. GO enrichment and KEGG (Kanehisa et al., 2008) pathway enrichment analysis of DEGs were, respectively, performed using R based on the hypergeometric distribution. We detected the presence of an N-terminal signal peptide through SignalP 5.0. The expression of genes with SNPs and Indels in interaction between Pt and wheat were determined. The expression of genes was profiled based on their fragments per kilobase of exon model per million mapped reads (FPKM) values in the transcriptome database (PRJNA605036). Conserved domains analysis was performed using InterProScan1 and HMMER2. Effector P 3.03 was used to predict the effector candidate.
2.5. qPCR analysis
Total RNA was extracted from leaves infected by Pt at 0, 12, 48 and 96 hours post inoculation (hpi). First-strand cDNA was synthesized from an equal amount of RNA using a PrimeScript Reverse Transcriptase kit (TaKaRa, Beijing, China). The β-actin gene (GenBank accession ADAS02000088.1: 291774-292,960) encoding β-actin protein was used as an internal reference gene, and 3 independent biological replicates were used in the experiments. The quantitative reverse transcription PCR (qPCR) reactions were conducted using a TransStart R Top Green qPCR SuperMix (TransGen, Beijing, China) with a Roche-LightCycler 96 qPCR instrument (Roche, Basel, Switzerland) using different primers designed according to different candidates. The transcriptional abundance of genes was quantitated relative to that of the β-actin following the 2−ΔCT method (Ridout et al., 2006).
2.6. Agrobacterium-mediated transient assays in Nicotiana benthamiana (N. benthamiana)
To generate constructs for transient expression in N. benthamiana, 4 secreted proteins genes were amplified using primers listed in Supplementary Table S6. The resulting PCR products were cloned into the in-house ligation-independent cloning vector pCamA carries a CaMV 35S promoter The resulting pCamA::secreted proteins constructs were used for Agrobacterium transformation, as described previously (Ma et al., 2012).
2.7. Yeast secretion trap assay
The predicted coding sequences of the signal peptides of selected effector candidates were amplified from cDNA using the primers listed in Supplementary Table S6 and cloned into the yeast secretion trap vector pSUC2 using EcoR I and Xho I restriction sites. Ps87 were used as a positive control, Mg87 and YTK12 as negative control. The resulting constructs were transformed into the yeast strain YTK12 to examine secretion following the method described previously (Haddadi et al., 2016). And the reduction of colorless TTC to red-colored insoluble TFP, as described by Yin et al. (2018).
3. Results
3.1. Lr19 Triggers a resistance response at the early stages of infection source identification initiative
Leaf rust resistance conferred by Lr19 is best expressed in all stages of the wheat plant and culminates in a hypersensitive response (HR) at the infection site which is also known as race-specific resistance. In order to confirm the recognition between Lr19 and its respective Avr gene in the Lr19-avirulent Pt race PHNT, the phenotype and histopathology examination of Pt-infected leaf tissues from resistant TcLr19 (Lr19+) and susceptible Chinese Spring (Lr19–) wheat lines were analyzed. Compared to Chinese Spring (Lr19–) wheat lines, the development of fungal infection hyphae (stained blue) stopped before the formation of a haustorium, the structure with which the fungus extracts nutrients from its host plant, in the resistant wheat line TcLr19 (Lr19+). Imaging at 48 hpi revealed fungal growth in susceptible Chines Spring but no further fungal growth in TcLr19, the dead host cells in TcLr19 were stained green, and no dead cells were revealed in Chinese Spring (Figures 1A,B). This early immune response is consistent with pronounced HR symptoms and suggested early expression of a fungal gene recognized by Lr19, which demonstrated that Lr19 triggers a resistance response at the early stages of infection.
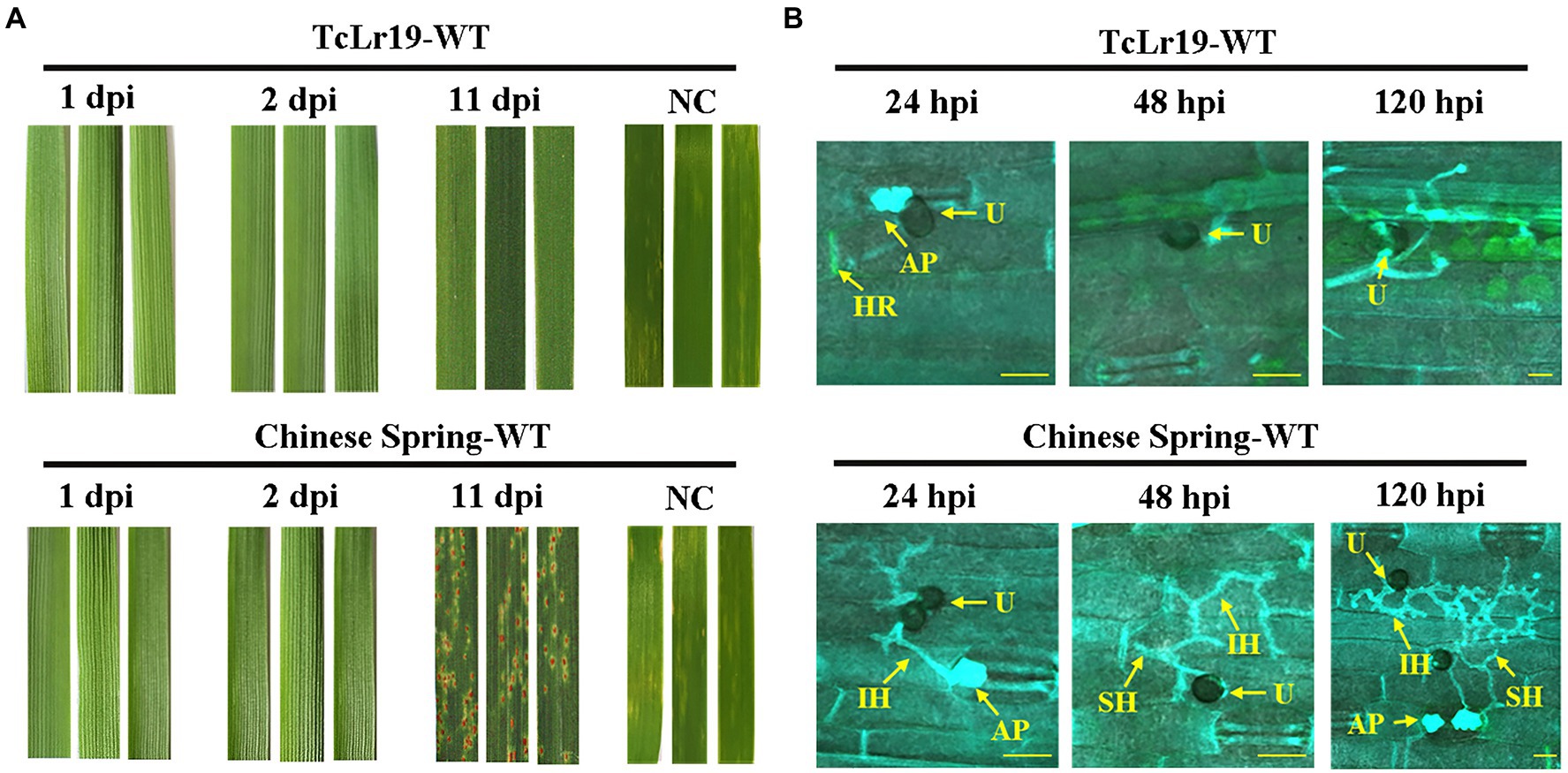
Figure 1. Lr19 provides prehaustoria resistance against Pt. (A) Infected leaves of susceptible cultivar Chinese Spring (Lr19-) and resistant line TcLr19 (Lr19+) were observed; NC: negative control, non-inoculation. (B) Fungal infection hyphae (IH) (stained blue) entered the leaf mesophyll tissue (stained blue) through the plant stoma (S) in both wheat lines at 24 hpi. Fungal haustoria (H) developed only in susceptible Chinese Spring. Imaging at 48 hpi revealed fungal growth in susceptible Chinese Spring, but no further fungal growth in TcLr19. At 48 hpi revealed presumably dead host cells (stained green) in TcLr19; no dead cells were revealed in Chinese Spring. U, spores; AP, appressorium. Scale bars, 100 μm.
3.2. Lr19-virulent mutants were obtained and confirmed
The Lr19-avirulent Pt race PHNT was used to create Lr19-virulent mutants via EMS mutagenesis. The germination effect of PHNT urediniospores under different EMS concentrations was observed, and control check (CK) were carried out using 0 M EMS. The results showed that the germination rate of urediniospores was 50% at an EMS concentration of 0.005 M (Figure 2A). Two Pt mutants virulent to the Lr19 allele were isolated, named M1 and M2, suggesting that they contained mutations in the Avr factor (Figure 2B). Confocal microscopy of wheat leaves from the compatible Chinese Spring cultivar infected with the wild-type (WT) and mutant Pt strains. The length of the hyphae in Chinese Spring inoculated with M1 and M2 was the same as Chinese Spring inoculated with WT. The results showed that the AvrLr19 gene mutation did not change the virulence of the pathogen (Supplementary Figure S1). In order to ensure the reliability of experimental materials of TcLr19, molecular markers of Lr19 were used to detect the existence of Lr19 (Figure 2C). To validate that only Lr19-virulence was affected by the EMS mutagenesis, M1 and M2 were analyzed by infecting a standard full set of near-isogenic lines that carry different Lr genes, with the WT PHNT race inoculated as controls (Figure 2D; Supplementary Table S1). Only TcLr19 inoculated with M1 and M2 demonstrated a change from resistant (‘0’) to susceptible (‘3 +’), while the susceptibility of other near-isogenic lines remained unchanged, indicating that AvrLr19 was altered in M1 and M2 alleles and that Lr19-virulent mutant strains were successfully obtained.
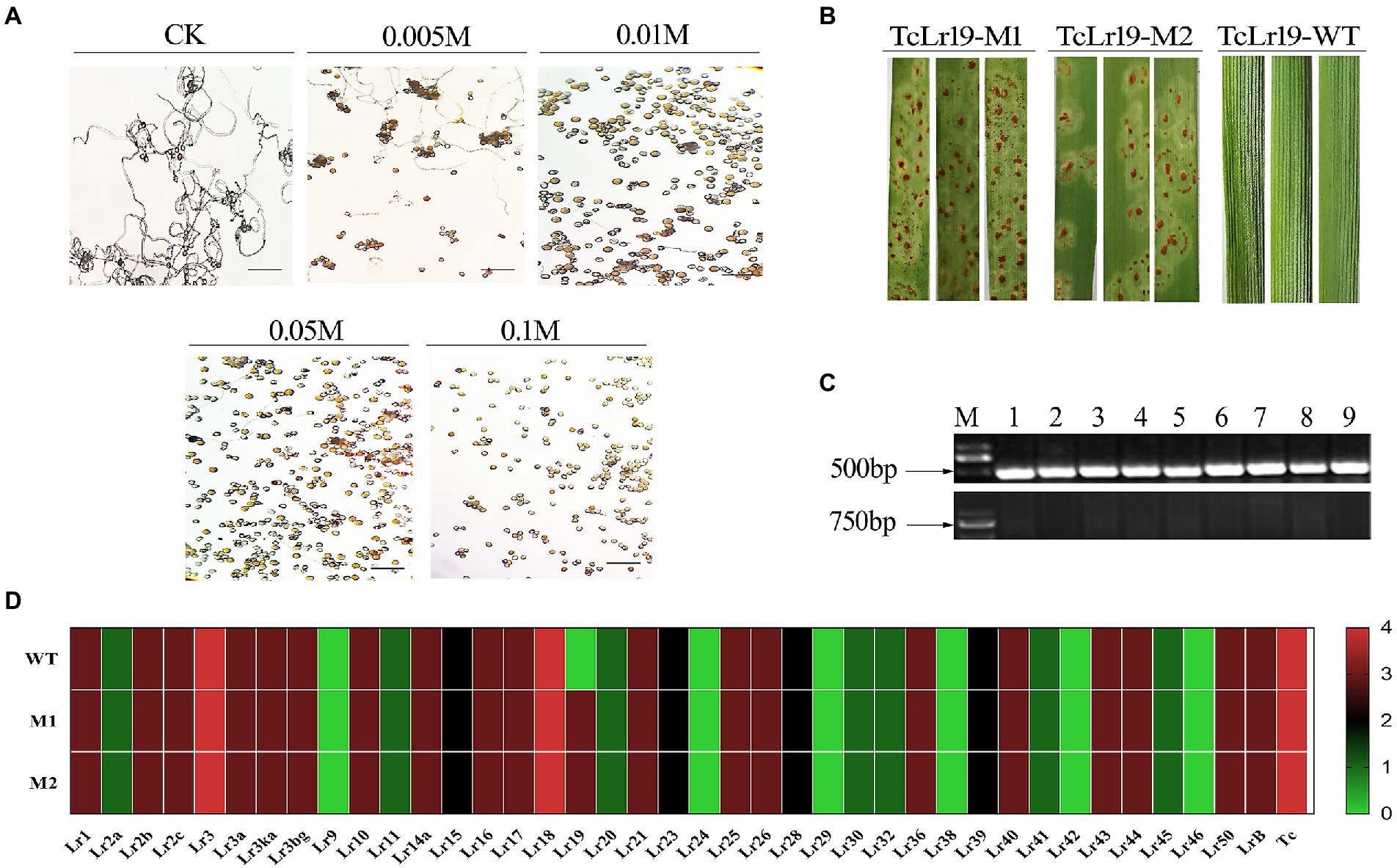
Figure 2. Lr19-virulent Mutants were obtained and confirmed. (A) The germination effect of Pt spores of PHNT race under different EMS concentration; CK, control check. (B) The phenotypes of Lr19-virulent Mutants M1 and M2 were compared with wild-type (WT). (C) Molecular markers detection of Lr19. There was a specific band at 500 bp for forward primer detection and there was no specific band at 750 bp for reverse primer detection, M is the abbreviation of marker, 1–9 represents 9 test samples. (D) Heatmap of WT and its mutants (M1 and M2) of Pt based on infection types, the virulence characterization of all isolates was conducted on the 36 wheat Lr single-gene differentials. Its 0 to 4 were transformed to the color key ranging from green to red, which indicate avirulent (resistant) to virulent (susceptible) reactions.
3.3. RNA sequencing (RNA-Seq) analyses display the information of M1, M2, and WT
As there is no virulent strain of Lr19 in the field, Pt-Chinese Spring which was compatible interaction was used as wildtype isolates to compare with Pt-TcLr19 mutants. To characterize the genes containing SNPs and Indels between Pt-Chinese Spring and Pt-TcLr19 mutants during infection, RNA-seq of TcLr19 or Chinese Spring inoculated with M1, M2, and WT were performed. Following inoculation and incubation, leaves displaying infection symptoms were harvested, then the total RNA was extracted from M1, M2, and WT successfully (Supplementary Figure S2A) and was used for RNA-seq cDNA library construction. Quality statistics of sequencing data and the percentages of reads for each sample were mapped to the Pt BBBD race reference genome (Supplementary Tables S2, S3). Based on the density plot (Supplementary Figure S2B), box-whisker plot (Supplementary Figure S3A), and the expected number of FPKM expression distribution plot (Supplementary Figure S3B) of gene expression levels. The results suggested that the differences in the distributions were low between the three repeat libraries of each sample, it is clear that the expression level and number of protein-coding genes in each sample is highly similar. Variability among the samples was determined by preparing a principal component analysis (PCA). The WT-sample was located in the third quadrant. The M1-sample was located in the second and fourth quadrants. The M2-sample is located fourth quadrant and the edge of the third quadrant (Supplementary Figure S3C). MT-sample is separated from WT-sample as shown by PCA analysis. PCA displayed a clear distinction between the transcriptomes of mutant and WT strains.
3.4. Characterization of genes containing SNPs and indels derived from whole transcriptome
EMS mutagenesis generates SNPs and Indels, thereby inducing base substitutions in the genome, and eventually leading to phenotype changes based on mutations. SNPs were screened by selecting only CG to TA mutations since mutations caused by EMS had the strong CG to TA transition bias. Firstly, considering that AvrLr19 might be non-functional in two mutants and be strongly repressed in expression, alignment and mutational analysis was done in whole transcriptome level except the DEGs. After comparison of sequences and keeping only the EMS-induced SNPs and Indels, a total of 396 genes containing SNPs and 164 genes containing Indels were identified from non-differentially expressed genes (Figures 3A,B), of which 45 genes encoding secreted proteins were predicted to be candidate genes (Table 1). Missense_variants were the predominant (96.67%) among all the deleterious mutations. Similarly, the effects of Indels were mostly in 5’-UTRs (26.67%), 3’-UTRs (26.67%), upstream (20.00%) and frameshift_variant (20.00%) (Supplementary Tables S4, S5). Through the analysis of KEGG enrichment, we explored the main biochemical metabolic pathways and signal transduction pathways involved in secreted proteins. The results show that 4 genes are distributed among “protein processing in endoplasmic reticulum,” “starch and sucrose metabolism,” “lysosome” and “protein processing in endoplasmic reticulum” pathways (Figure 3C). GO analysis was performed to classify the 45 genes into 34 classes categories, 12% genes are enriched in terms of “integral component of membrane” (Figure 3D).
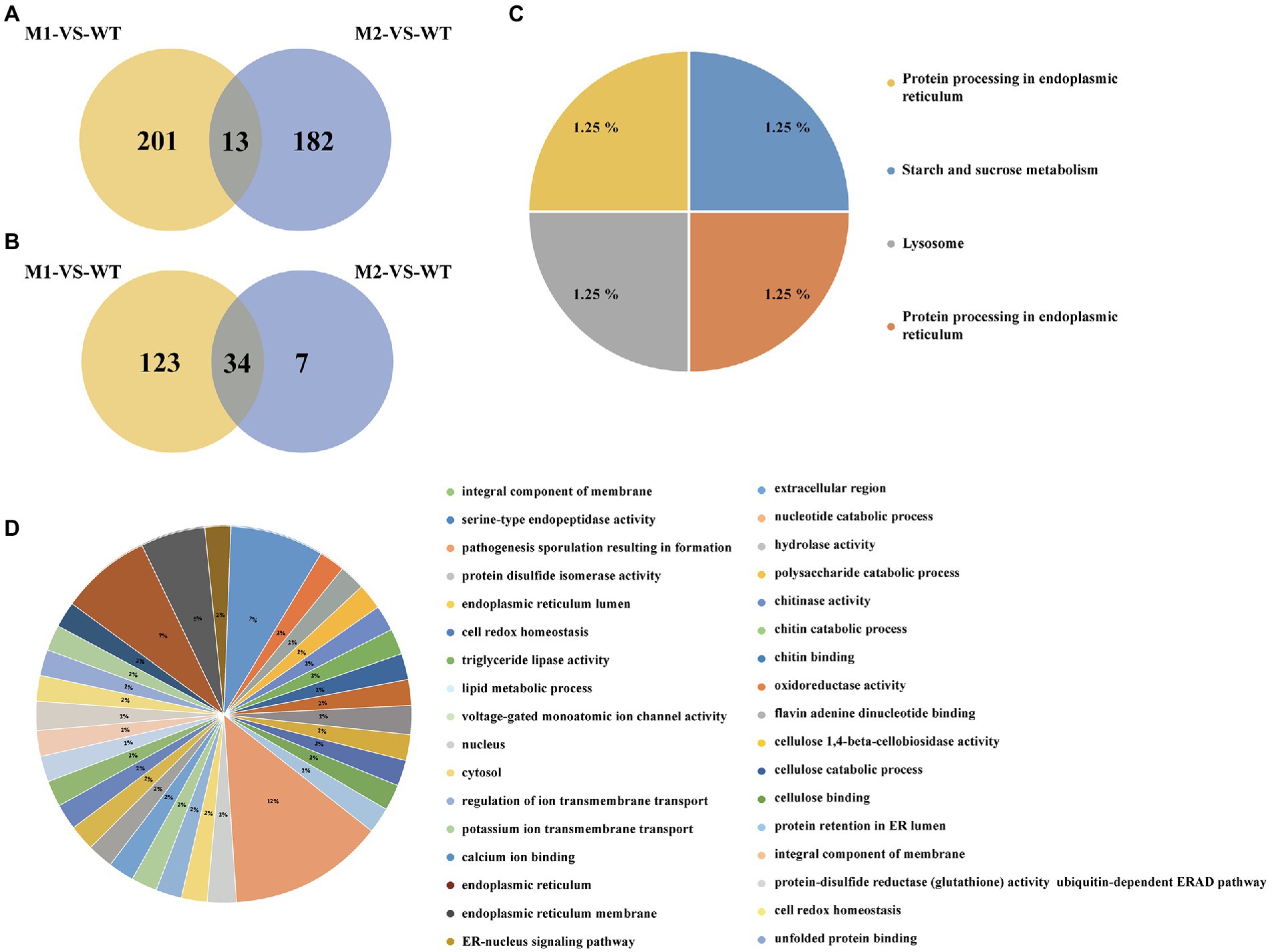
Figure 3. The types and numbers of SNPs in M1, M2, and WT among not differentially expressed genes. (A) Numbers of SNPs from the M1-VS-WT and M2-VS-WT. (B) Numbers of Indels from the M1-VS-WT and M2-VS-WT. (C) Results of KEGG analysis of secreted proteins. (D) GO annotation for secreted proteins.
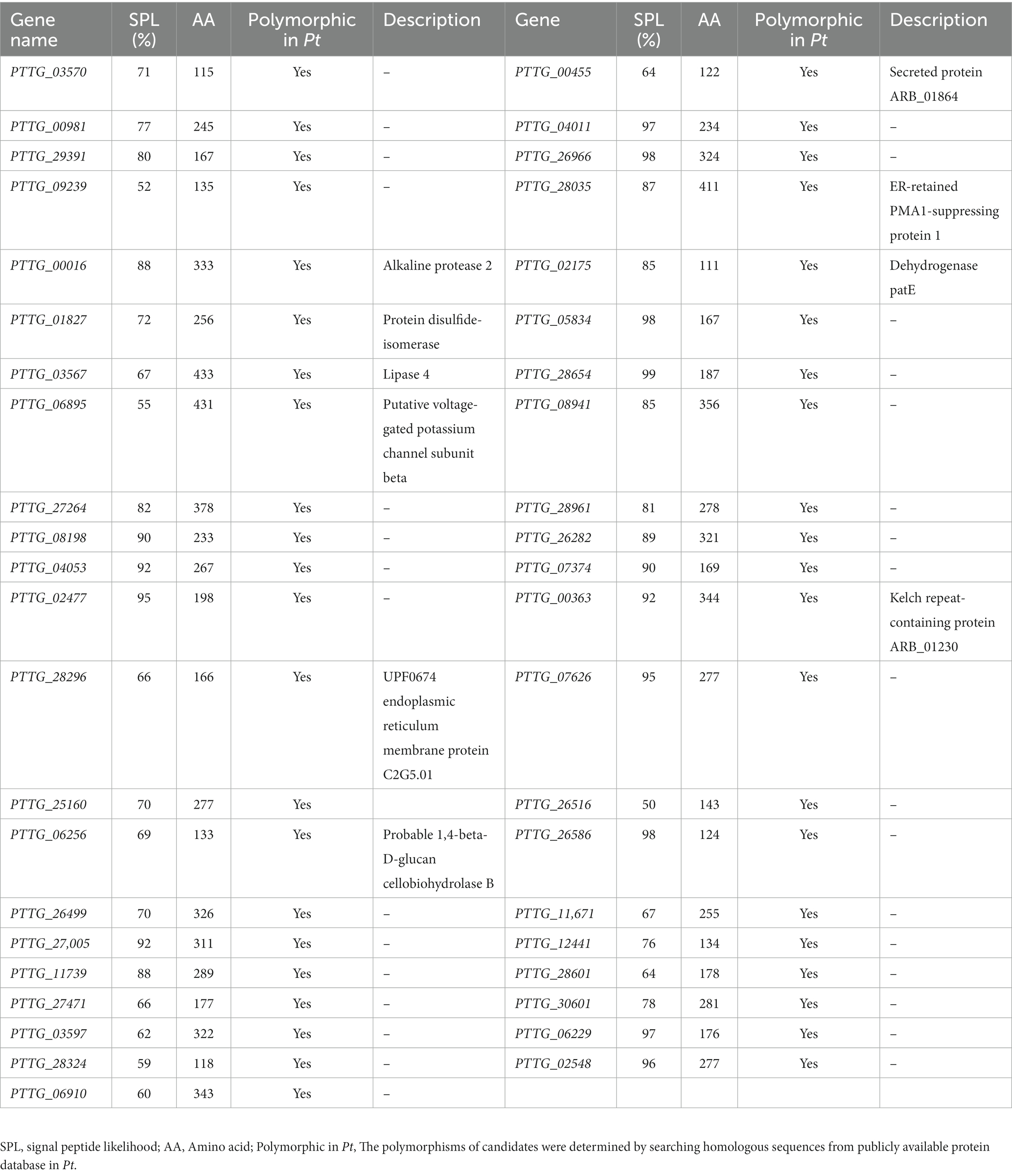
Table 1. 45 secreted protein genes contain SNPs and Indels among non-differentially expressed genes.
3.5. Expression profiles of genes containing SNPs and indels in MT-VS-WT
In order to confirm whether 45 secreted proteins containing SNPs and Indels are related to the infection of Pt, their expression in the interaction between Pt race PHNT and wheat was identified based on their FPKM values in a transcriptome database (PRJNA605036). Among them, 9 genes including PTTG_27471, PTTG_27,005, PTTG_28324, PTTG_06910, PTTG_26499, PTTG_11671, PTTG_26282, PTTG_12441, and PTTG_26516 with SNPs were significantly upregulated during the early stage (4–6 dpi) of Pt infection, and one gene PTTG_03570 containing Indels were significantly upregulated during 4–6 dpi of Pt infection (Figure 4A). All the 10 genes highly expressed at 4–6 dpi deserve more attention than those highly expressed at GT stage.
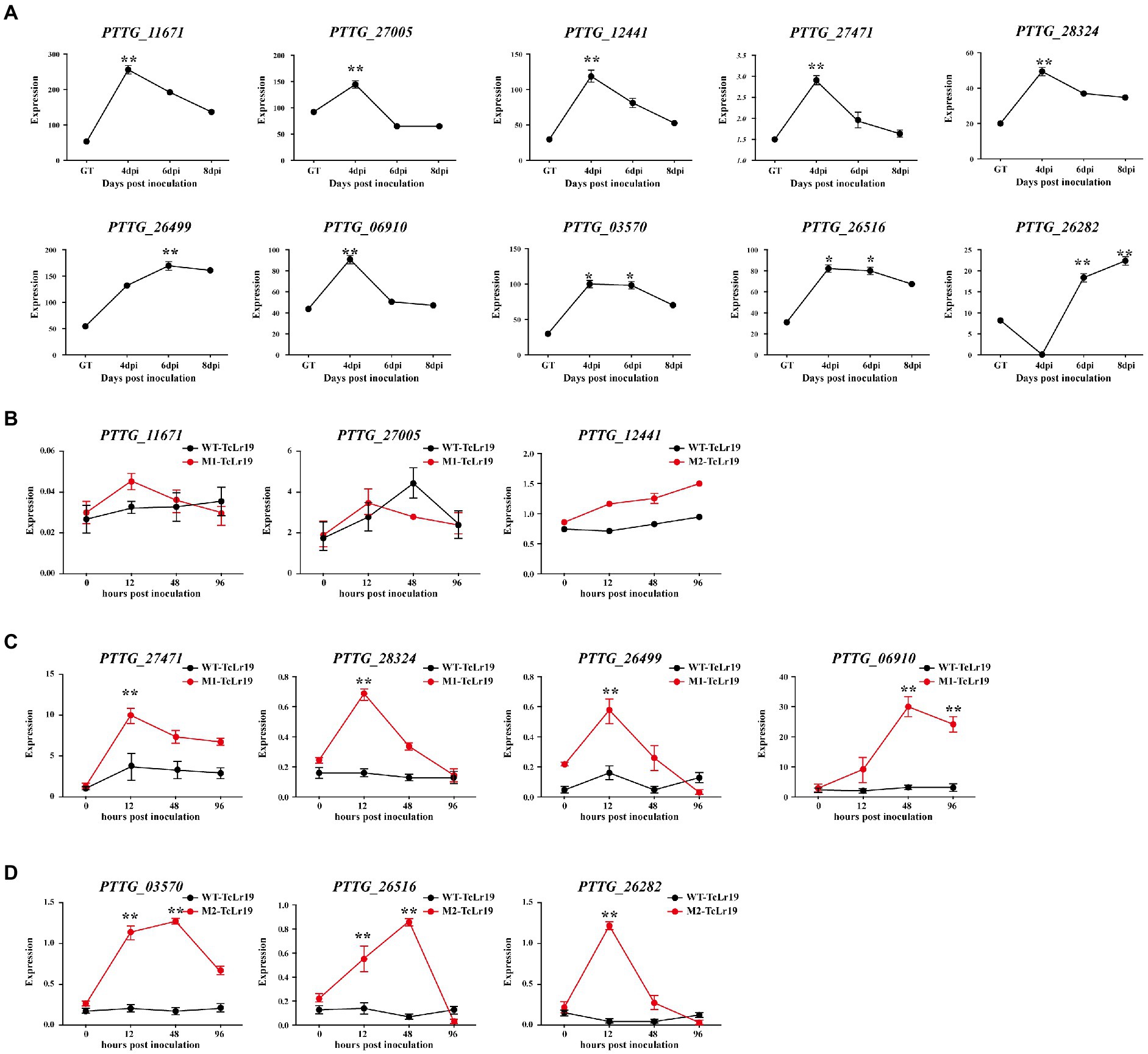
Figure 4. The genes containing SNPs and Indels with different expression patterns were selected. (A) Their expression patterns were generated based on corresponding FPKM values from the transcriptome database. The transcript levels of the genes containing SNPs and Indels selected during Pt infection at 4, 6, and 8 days post-inoculation (dpi). (B–D) Transcriptional profile of genes during the Pt pathotypes infection measured by qPCR. The transcript levels of the 14 secreted proteins during Pt infection at 0, 12, 48, and 96 hpi (Hour post inoculation) were determined by qPCR assay. The transcript levels for all genes were expressed as linearized fold-Pt actin levels, which were calculated according to the formula 2−△CT. Data were expressed as mean values ± SE from four biological replicates. An asterisk (*p < 0.05,**p < 0.01) indicates.
qPCR was conducted in order to test whether the expression of these 10 genes were affected by EMS mutation. The results displayed that the expression of PTTG-11671 and PTTG-27005 in M1-TcLr19 displayed similar profiling to that in WT-TcLr19, and the expression of PTTG_12441 in M2-TcLr19 also displayed similar profiling to that in WT-TcLr19 (Figure 4B), which indicated that PTTG-11671, PTTG-27005 and PTTG_12441 expression was not affected by EMS mutation. However, the expression profiling’s of the other 7 genes including PTTG_27471, PTTG_03570, PTTG_26282, PTTG_28324, PTTG_06910, PTTG_26516, and PTTG_26499 in MT-TcLr19 were significantly different with that in WT-TcLr19 at 12–48 hpi. The expression level of PTTG_27471, PTTG_28324 and PTTG_26499 in M1 was about 4-fold higher than that in WT at 12 hpi, and PTTG_06910 expression peaked at 48 hpi in M1 when the expression level was 30-fold higher than that in WT (Figure 4C). In M2, PTTG_03570 and PTTG_26516 expression peaked at 48 hpi, and the expression level of PTTG_03570 and PTTG_26516 in M2 was about 5-fold higher than that in WT. The transcript quantity of PTTG_26282 in M2 peaked at 12 hpi which was almost 15-fold higher compared with that in WT (Figure 4D). Taken together, EMS mutation affected the expression of these 7 genes in Lr19-virulent mutants, these 7 genes should be further study as AvrLr19 candidate genes.
3.6. Characterization of Pt secreted proteins in DEGs
The function of Avr genes might be affected by changes in expression rather than sequence changes (Upadhyaya et al., 2021), so the secreted proteins in DEGs were analyzed. In order to understand the overall distribution of DEGs, volcano maps of DEGs were constructed (Supplementary Figure S4A). The cluster diagram of sample to sample distance matrix was prepared for DEGs to calculate the direct correlation of samples (Supplementary Figure S4B). From the comparison made between M1 and WT (M1-vs-WT), a total number of 168 DEGs were obtained, 74 of which were up-regulated and 94 of which were down-regulated. From the comparison made between M2 and WT (M2-vs-WT), 76 DEGs were obtained, 45 of which were up-regulated and 31 of which were down-regulated. From the comparison made between M1 and M2 (M1-vs-M2), 4 DEGs were obtained, 2 of which were up-regulated and 2 of which were down-regulated (Figure 5A). Similar DEGs found among the comparisons were shown in a Venn diagram (Figure 5B). Finally, a total of 216 genes were identified as differentially expressed. To understand the roles of various biological processes and pathways throughout the infection process, we analyzed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of the 216 DEGs. GO analysis was performed to classify the 216 DEGs into 3 major biological categories. The 216 DEGs belonged to 18 classes in the biological process category, 11 classes in the cellular component category, and 7 classes in the molecular function category (Figure 5C). In total, 15 secreted proteins were identified among the 216 proteins encoded by the DEGs using SignalP 5.0 (Table 2). 4 proteins among 15 proteins were predicated to be secreted proteins, PTTG_25982, PTTG_25534, PTTG_04712 and PTTG_04779 were identified as cysteine-rich proteins with the percentage of cysteine greater than or equal to 3% (Table 3).
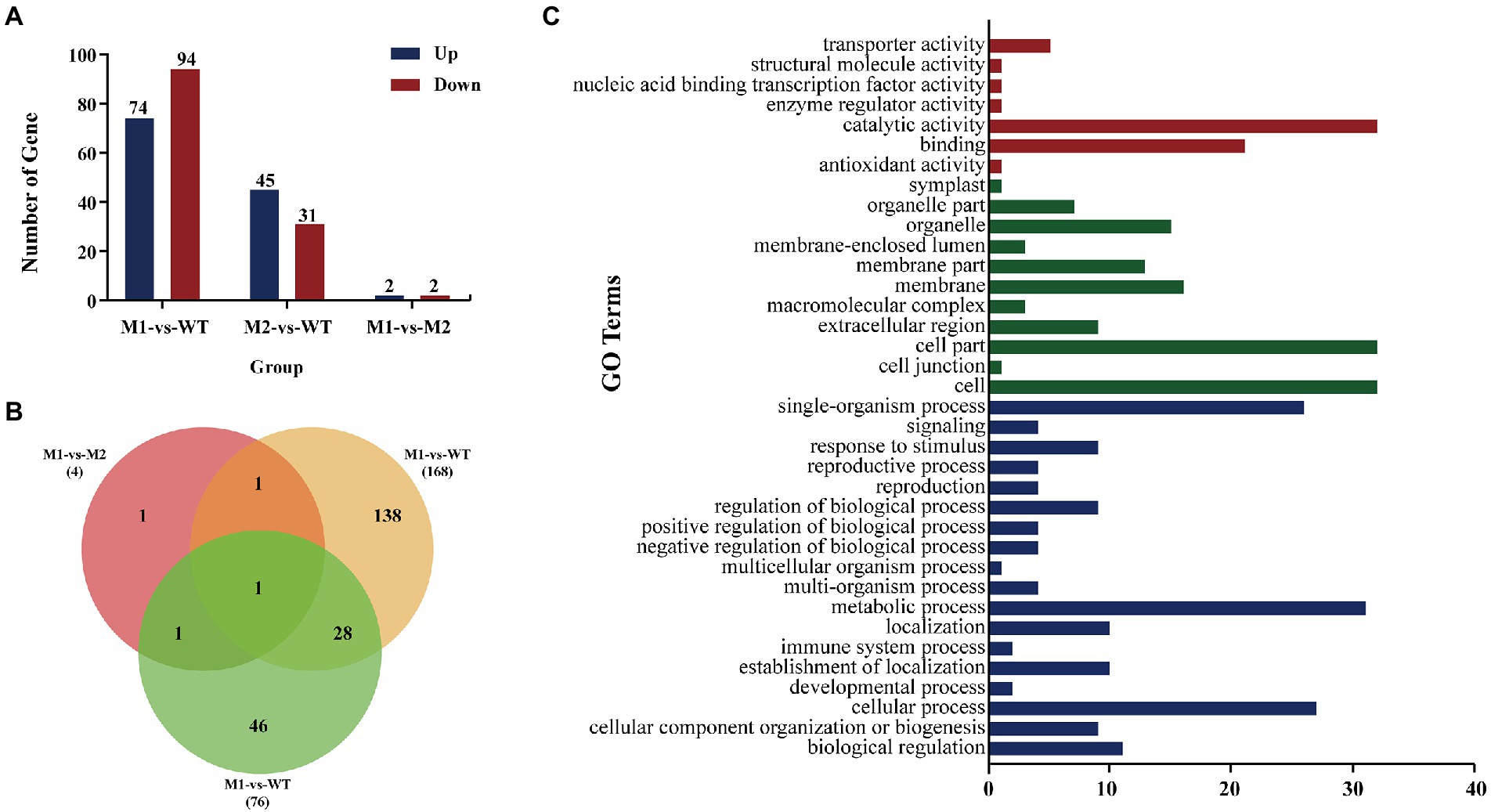
Figure 5. The differentially expressed genes (DEGs). (A) Number of up-regulated and down regulated DEGs indifferent samples. (B) DEGs that are unique or shared among various samples comparisons in mutants (M1, M2) and WT. The numbers of DEGs are noted in each section of the Venn diagrams. (C) Bar graph showing the number of 216 DEGs identified utilizing gene ontology (GO) enrichment analysis for involvement in specific molecular function, cellular component and biological processes. In the figure, red color represents molecular function, green color represents molecular function, blue color represents molecular function, the Y coordinate is the name of GO term, and the X coordinate is number of gene.
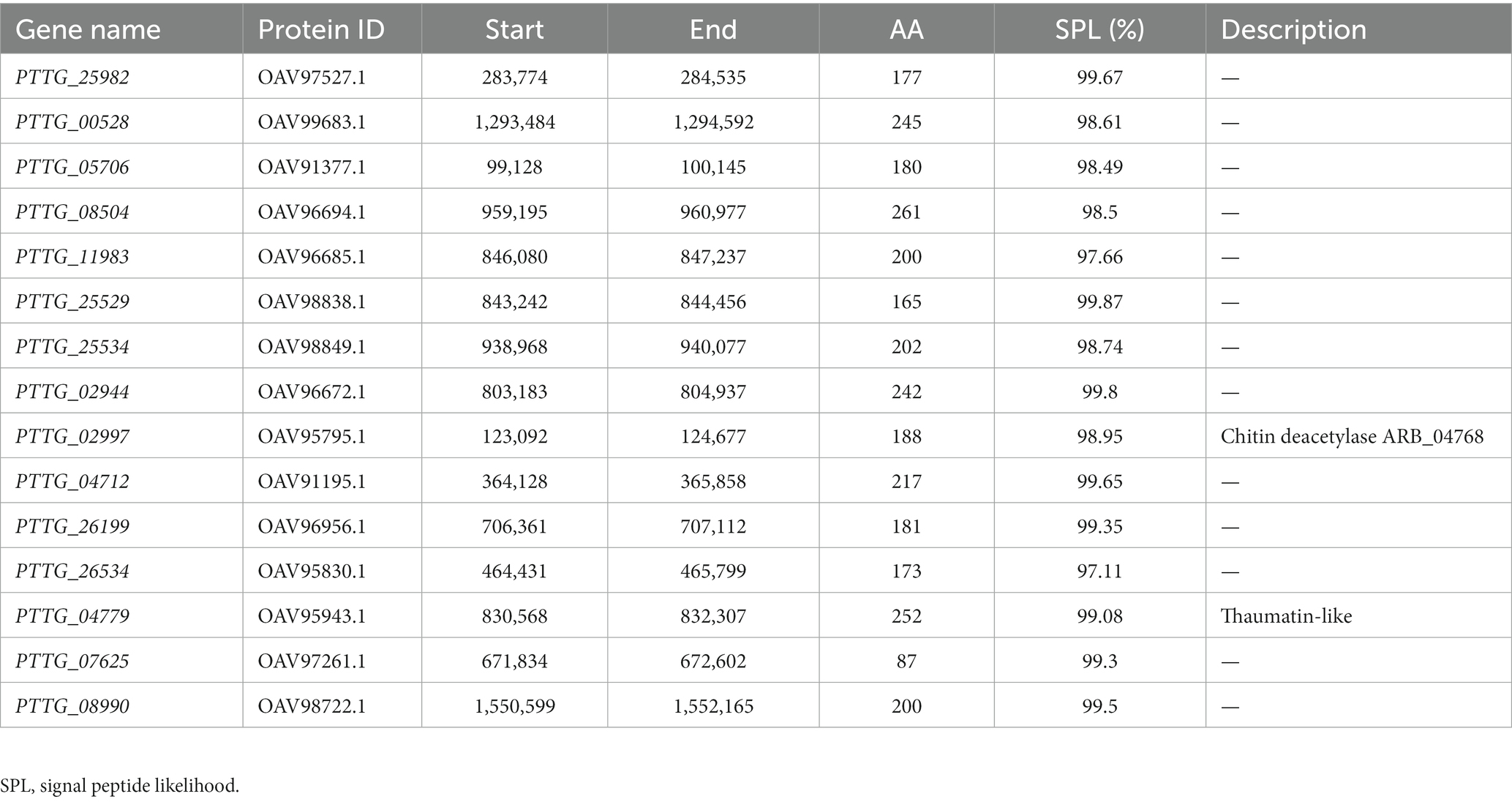
Table 2. Characterization of 15 secreted proteins of Pt.
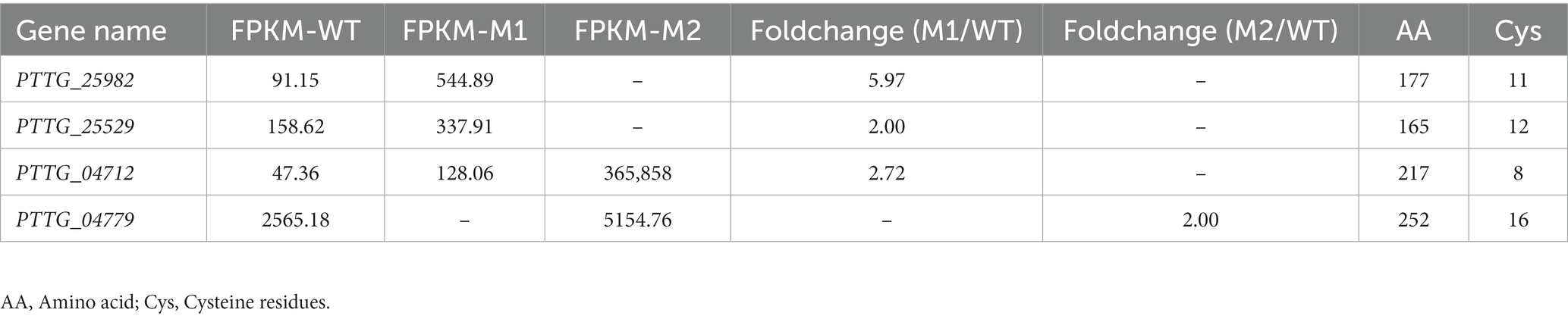
Table 3. Characterization of 4 secreted proteins, which identified as cysteine-rich proteins.
3.7. PTTG_04779 inhibition the progress of cell death (PCD) induced by B-cell lymphoma-2 associated X protein (BAX) in N. benthamiana
Numerous avirulence proteins such as AvrM and AvrL567 from Melampsora lini (Ml) have been confirmed to be related to virulence of the pathogen (Ellis et al., 2007). To examine the function of secreted proteins, we test whether the secreted proteins could inhibit PCD. After transiently expressed the coding regions of 4 secreted proteins in N. benthamiana using Agrobacterium infiltration, the results showed that leaf regions co-expressing pCamA as a negative control and BAX developed PCD (Figure 6A), PTTG_25982, PTTG_25534 and PTTG_04712 could not suppressed PCD triggered by the expression of BAX gene (GenBank accession NP_031553) in N. benthamiana leaves (Supplementary Figure S5). However, there was no necrosis at the sites where BAX and PTTG_04779 were infiltrated together (Figure 6A), indicating that PTTG_04779 inhibited BAX function, preventing PCD from being induced, with potential toxic function.
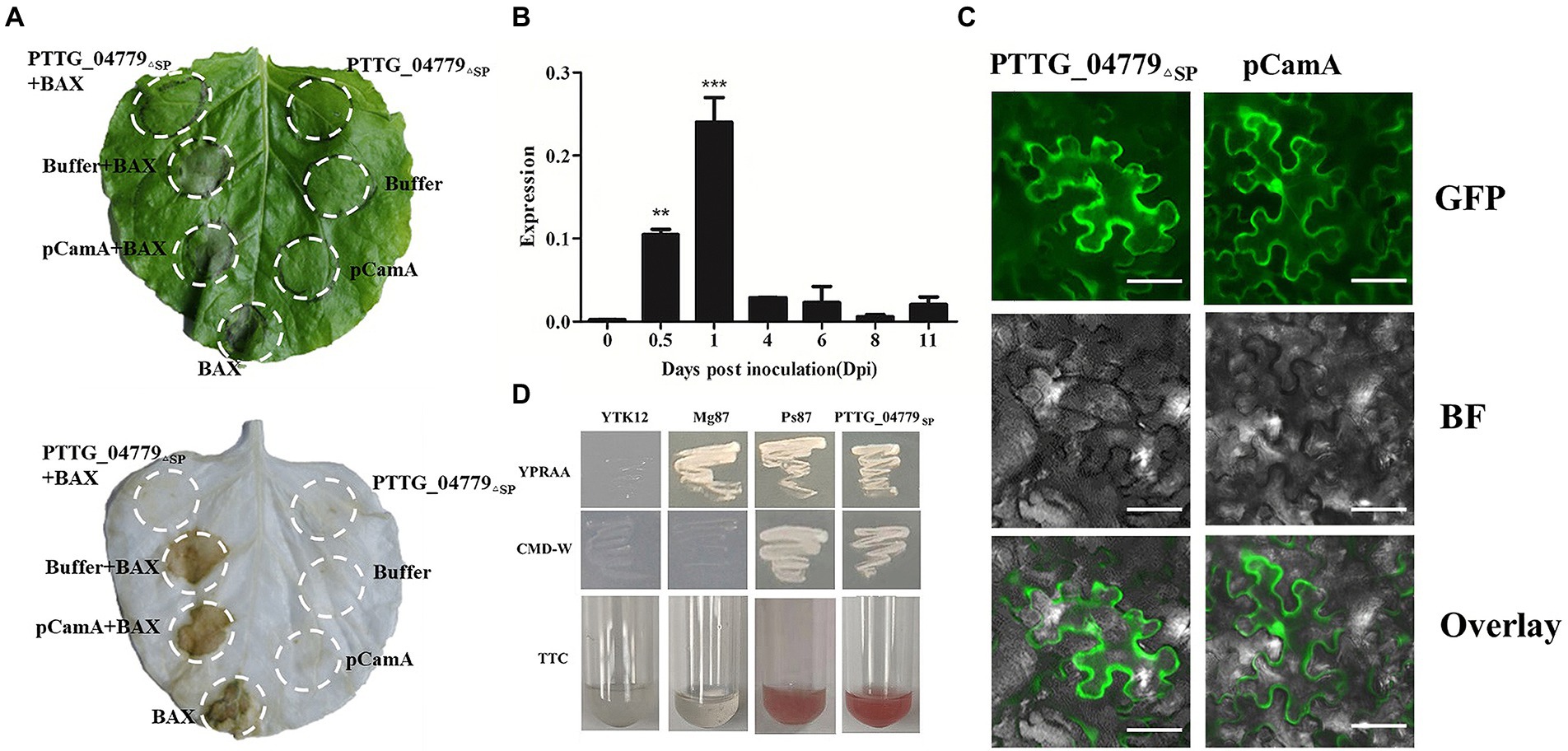
Figure 6. Characterization of secreted protein PTTG_04779. (A) PTTG_04779 can suppress BAX-induced cell death. PTTG_04779 was infiltrated into N. benthamiana leaves, followed 24 h later by infiltration with A. tumefaciens carrying the Bax gene. pCamA and buffer served as a negative. (B) Transcriptional profile of genes during the Pt pathotypes infection measured by qPCR. (C) Expressed fluorescently tagged PTTG_04779ΔSP location in the N. benthamiana leaves epidermal cells. The pCamA-PTTG_04779ΔSP (with GFP tag) in N. benthamiana cells and GFP field show green. Scale bar, 50 μm. (D) Yeast secretion trap assay of the secreted protein PTTG_04779. The predicted signal peptide coding sequences of PTTG_04779 (PTTG_04779SP) were cloned into the yeast secretion trap vector pSUC2. The Ps87 was used as a positive control, empty vector and Mg87 was used as a negative control.
qPCR was used to detect the expression levels of PTTG_04779 at different time after Pt infection, and the results displayed that PTTG_04779 expression peaked at 1 dpi which was 118-fold higher than that at 0 dpi (Figure 6B). Subcellular localization study showed that PTTG_04779△SP (without signal peptide) located in the cytoplasm and nuclear of N. benthamiana cell (Figure 6C). To examine the function of the predicted signal peptide of PTTG_04779, a yeast signal sequence trap system was employed. Consistent with the positive control Ps87, YTK12 strains carrying the predicted signal-peptide coding sequences of PTTG_04779 (PTTG_04779SP) fused to pSUC2 were able to grow on YPRAA plates (Figure 6D). As expected, they were also able to catalyze the conversion of colorless 2,3,5-triphenyltetrazolium chloride (TTC) to red-colored triphenylformazan (TFP) (Figure 6D). Conversely, the negative control (YTK12 with empty vector) and Mg87 was not able to grow on a YPRAA plate and did not induce a TTC to TFP color change (Figure 6D). These results demonstrated that the predicted signal peptides of PTTG_04779 are functional. All these results suggest that PTTG_04779 should be further studied as AvrLr19 candidate gene. Taken together with 7 genes in Lr19-virulent mutants whose expression were affected by EMS mutation, there are total 8 genes as AvrLr19 candidate (Table 4).
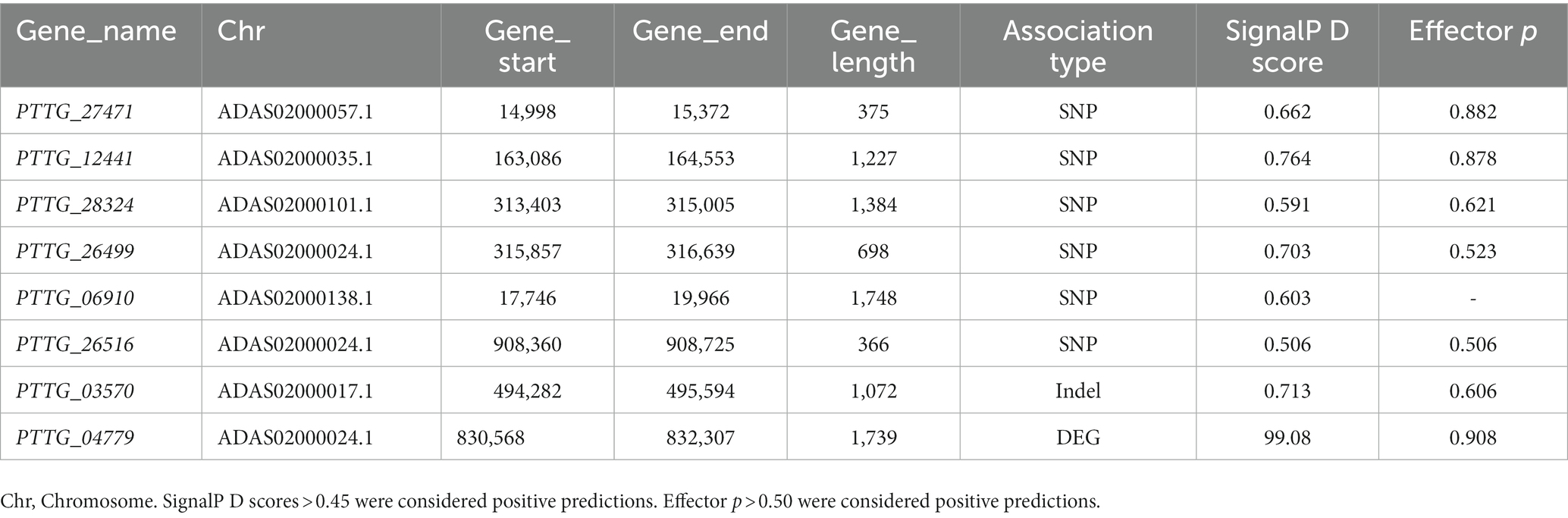
Table 4. A list of candidate genes for being AvrLr19.
4. Discussion
Pt is the causative agent of leaf rust, the most widespread and devastating disease of wheat, which resulted in yield losses of more than 50% in a single crop season. The obligate biotrophy and the lack of a transformation system for Pt complicates validation of the function of Avr gene from Pt and their host targets. Thus, different approaches to address the questions of virulence and avirulence in Pt had to be used. EMS mutagenesis have proved to be a useful tool to provide these answers. Li et al. (2019) developed a Puccinia striiformis f. sp. tritici (Pst) mutant population through EMS mutagenesis and characterized the population with virulence and molecular markers. Salcedo et al. (2017) obtained EMS induced urediniospore mutants from the wheat stem rust pathogen Puccinia graminis f. sp. tritici (Pgt) Ug99, which led to the cloning of Avr gene AvrSr35. Mutagenesis integrated with genomic sequencing is an efficient way to study the relationships between phenotypic traits and associated genes, leading to the identification of fungal effectors or avirulence genes. The innovation of the present study is that we developed two Lr19-virulent mutants via EMS mutagenesis, M1 and M2, and confirmed that the mutation site only altered the virulence of Pt race PHNT to Lr19, but not other Lr genes, which benefits the screening of AvrLr19 candidates.
With the rapid development of sequencing technologies, the genome sequences of pathogen are available, which makes it possible to further understand the pathogenesis of the obligate biotrophic fungal parasite (Steuernagel et al., 2016). McDonald and Linde (2002) identified five Pst candidate effector genes from 2,999 predicted secreted protein genes. Xia et al. (2017) predicted a set of 25 Pst Avr candidate genes from 2,146 predicted secreted protein by combining comparative genomics with association analyses. Similar approaches were also used in detecting Avr candidate genes in Pt (Wu et al., 2017). Mutations in the AvrLr19 coding region may lead to amino acid changes and loss of protein interaction with Lr19. To determine and characterize the potential Avr genes in Pt, whole-genome sequences of EMS-induced mutants were generated and analyzed. The reads of each isolate were mapped directly against the Pt Race 1 reference genome for SNP detection in a similar way to several previous studies (Cantu et al., 2013). An average of 240, 945 SNPs and 22, 361 Indels per isolate was found. This rate of mutation site matches that reported for the Pt race 1 genome (Persoon et al., 2014). This feature has been consistently noted in the genomes of other rust fungi, such as M. larici-populina, Pgt and Pst (Upadhyaya et al., 2021).
It was reported that a frequency of 99% G/C to-A/T transitions observed in Arabidopsis (Persoon et al., 2014). Thus, C/G-to-T/A mutations from the variants were selected, the other types of mutations were filtered out in this study, which is the same strategy used on the mutation screening work on tomato (Zheng et al., 2013), and fungal pathogen Pgt (Salcedo et al., 2017). 560 common genes containing SNPs and Indels mutation with SignalP D scores higher than 0.50 were predicted as secreted proteins by SignalP 5.0 (Mori et al., 2015; Nielsen et al., 2019). Salcedo et al. (2017) identified a fungal secreted protein named AvrSr35 that is required for Sr35 avirulence by whole-genome sequencing of EMS mutagenized and natural Pgt isolates. AvrSr35 had a nonsynonymous mutation producing valine to isoleucine (V128I) substitution in mutation. In this study, 7 genes, PTTG_27471, PTTG_03570, PTTG_26282, PTTG_28324, PTTG_06910, PTTG_26516 and PTTG_26499, from 560 common genes occurred SNPs and Indels mutation resulting amino acids, and the expression levels of these genes were significantly higher in Lr19-virulent mutants than that in WT, which indicated that they might be involved in the infection of TcLr19 and should be further study as AvrLr19 candidate genes. Domain analysis was performed using InterProScan (Supplementary Figures S6A–G) and HMMER (Supplementary Figures S7A–G), no conserved domains were found in these 7 genes. Lots of fungi protein without known domain can play a pathogenic or avirulent role (Salcedo et al., 2017; Qi et al., 2019) and affect the process of fungi infection. In this study, the function of the candidate genes without known conserved domain will be further verified in the follow-up study.
It is attainable that the use of expression profiling to prioritize candidate genes for avirulence function in rust fungi. 216 DEGs obtained in this study may play a certain role in infection of M1 and M2 on TcLr19, and 15 proteins were predicated to be secreted proteins from them. It has been reported that cysteine is important for the function of proteins (Huang et al., 2019; Zhang et al., 2021). Therefore, PTTG_25982, PTTG_25534, PTTG_04712 and PTTG_04779 which identified as cysteine-rich proteins were paid much attention in this study. Among them, PTTG_04779 can suppress PCD, and its expression level is peaked at the stage of leaf rust infection, indicating that it might play a virulence function in plant. Domain analysis showed that there is a conserved domain of thaumatin in PTTG_04779 (Supplementary Figures S6H, S7H). It has been reported that the thaumatin-like family can regulate reactive oxygen species (ROS) production in plants and affect the disease resistance response of plants (Cui et al., 2021). Whether PTTG_04779 may play a thaumatin-like function in leaf rust similar to plants need to be test in the future. Moreover, to identify whether total 8 candidate genes conform to the characteristics of the effector, Effector P 3.0 was used. The results showed that 7 candidates were identified as effectors except PTTG_06910 (Table 4). However, some atypical secretory proteins do not conform to the characteristics of effector, but can affect the immune response of plants as pathogenic factors (Liu et al., 2014), so the function of PTTG_06910 need to be test.
As we all know the genes with signal peptide have a higher priority functional verification in the transcriptome sequencing data, but the candidates without signal peptide should not been discarded, because we found that not all the identified avirulence genes encoding secreted proteins. For example, Magnaporthe oryzae (M. oryzae) Avr gene ACE1 is predicted to be non-secreted and localizes in the cytoplasm of appressoria (Greene et al., 2003). Therefore, non-SP genes obtained from variants with high associations to avirulence will be evaluated in the future. By analyzing the transcriptomes of all samples, we found that the ratio of M1_sample 1, M1-sample 3, and M2-sample 6 to the reference genome were lower than other samples. It may be due to insufficient spores in the sample. It was reported that 15 Sr35-virulent mutants generated by EMS were used to clone AvrSr35 (Salcedo et al., 2017). More Lr19-virulent mutants need to be obtained and sequenced in order to improve the power of this study. Moreover, tools such as the effector-to-host analyser Pseudomonas fluorescens (EtHAn) strain-mediated effector delivery system (a bacterial type III secretion system) provide useful approaches for functional characterization of rust fungal effectors (Upadhyaya et al., 2014). The function of AvrLr19 candidates and pathogenesis-related genes of Pt will be identified by using the bacteria type III secretion system in the future. By defining AvrLr19 and its key functions, AvrLr19 can be used to develop gene-specific molecular markers for directly monitoring Pt population changes in the field, which can be used for real-time monitoring of leaf rust races combined with marker-assisted selection (MAS) of wheat leaf rust resistance genes to predict the resistance of varieties and inform the correct use of resistant varieties. Our results will provide valuable resources for identifying and characterizing AvrLr19 candidates and pathogenic genes, and lay a foundation for further elucidating the pathogenic mechanism of Pt and analyzing the disease-resistance mechanism of Lr19.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
DD, DL, and HW conceived and coordinated the study, designed the experiments, provided materials and resources, interpreted data, and revising the manuscript. WW and ZC participated in developing and increasing urediniospores of the mutant isolates, data analyses, draft the manuscript, and bioinformatic analysis of RNAseq data. FW and ZC participated in selecting isolate, producing spores, inoculating plants, and virulence data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32172384, 31501623), the Natural Science Foundation of Hebei (C2020204028). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Acknowledgments
We say thanks to OE Biotech Co., Ltd. (Shanghai, China) for performance RNA-seq and analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1062548/full#supplementary-material
Footnotes
References
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq--a python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Anderson, C., Khan, M. A., Catanzariti, A. M., Jack, C. A., Nemri, A., Lawrence, G. J., et al. (2016). Genome analysis and avirulence gene cloning using a high-density RADseq linkage map of the flax rust fungus, Melampsora lini. BMC Genom. 17, 667–687. doi: 10.1186/s12864-016-3011-9
Bakkeren, G., and Szabo, L. J. (2020). Progress on molecular genetics and manipulation of rust fungi. Phytopathology 110, 532–543. doi: 10.1094/PHYTO-07-19-0228-IA
Bhardwaj, S. C., Prashar, M., Kumar, S., Jain, S. K., and Datta, D. (2005). Lr19 resistance in wheat becomes susceptible to Puccinia triticina in India. Plant Dis. 89:1360. doi: 10.1094/PD-89-1360A
Bolton, M. D., Kolmer, J. A., and Garvin, D. F. (2008). Wheat leaf rust caused by Puccinia triticina. Mol. Plant Pathol. 9, 563–575. doi: 10.1111/j.1364-3703.2008.00487.x
Cantu, D., Segovia, V., MacLean, D., Bayles, R., Chen, X., Kamoun, S., et al. (2013). Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genom. 14, 270–288. doi: 10.1186/1471-2164-14-270
Chen, J., Upadhyaya, N. M., Ortiz, D., Sperschneider, J., Li, F., Bouton, C., et al. (2017). Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 358, 1607–1610. doi: 10.1126/science.aao4810
Cui, Z., Liang, F., Zhang, J., Wang, F., and Wang, H. (2021). Transgenic expression of TaTLP1, a thaumatin-like protein gene, reduces susceptibility to common root rot and leaf rust in wheat. Crop J. 9, 1214–1218. doi: 10.1016/j.cj.2021.03.021
Ellis, J. G., Dodds, P. N., and Lawrence, G. J. (2007). Flax rust resistance gene specificity is based on direct resistance-avirulence protein interactions. Annu. Rev. Phytopathol. 45, 289–306. doi: 10.1146/annurev.phyto.45.062806.094331
Elmore, M. G., Banerjee, S., Pedley, K. F., Ruck, A., and Whitham, S. A. (2020). De novo transcriptome of phakopsora pachyrhizi uncovers putative effector repertoire during infection. Physiol. Mol. Plant Pathol. 110, 101464–101473. doi: 10.1016/j.pmpp.2020.101464
Elyasi-Gomari, S., and Panteleev, V. K. (2006). Virulence polymorphism of Puccinia recondita f. sp. tritici and effectiveness of Lr genes for leaf rust resistance of wheat in Ukraine. Plant Dis. 90, 853–857. doi: 10.1094/PD-90-0853
Emanuelsson, O., Nielsen, H., Brunak, S., and Heijne, G. V. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016. doi: 10.1006/jmbi.2000.3903
Figueroa, M., Dodds, P. N., and Henningsen, E. C. (2020). Evolution of virulence in rust fungi—multiple solutions to one problem. Curr. Opin. Plant Biol. 56, 20–27. doi: 10.1016/j.pbi.2020.02.007
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Flor, H. H. (2003). Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. doi: 10.1146/annurev.py.09.090171.001423
Gao, P., Zhou, Y., Gebrewahid, T., Zhang, P., Yan, X., Li, X., et al. (2019). Identification of known leaf rust resistance genes in common wheat cultivars from Sichuan province in China. Crop Prot. 115, 122–129. doi: 10.1016/j.cropro.2018.09.012
Greene, E. A., Codomo, C. A., Taylor, N. E., Henikoff, J. G., Till, B. J., Reynolds, S. H., et al. (2003). Spectrum of chemically induced mutations from a large-scale reverse genetic screen in Arabidopsis. Genetics 164, 731–740. doi: 10.1093/genetics/164.2.731
Gupta, S. K., Charpe, A., Prabhu, K. V., and Haque, M. R. (2006). Identification and validation of molecular markers linked to the leaf rust resistance gene Lr19 in wheat. Theor. Appl. Genet. 113, 1027–1036. doi: 10.1007/s00122-006-0362-7
Haddadi, P., Ma, L., Wang, H., and Borhan, M. H. (2016). Genome-wide transcriptomic analyses provide insights into the lifestyle transition and effector repertoire of Leptosphaeria maculans during the colonization of Brassica napus seedlings. Mol. Plant Pathol. 17, 1196–1210. doi: 10.1111/mpp.1235645
Huang, S., Zhang, Z., Liu, W., Tao, H., Zhang, Y., Shi, N., et al. (2019). Expression of the small cysteine-rich protein SCR96 from Phytophthora cactorum in mammalian cells: phytotoxicity and exploitation of its polyclonal antibody. Biotechnol. Lett. 42, 125–133. doi: 10.1007/s10529-019-02754-7
Huerta, E. J., and Singh, R. P. (1994). First report of virulence to wheat with leaf rust resistance gene Lr19 in Mexico. Plant Dis. 78:640C. doi: 10.1094/PD-78-0640C
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kemen, E., Kemen, A. C., Rafiqi, M., Hempel, U., Mendgen, K., Hahn, M., et al. (2005). Identification of a protein from rust fungi transferred from haustoria into infected plant cells. Mol. Plant-Microb Interact. 18, 1130–1139. doi: 10.1094/MPMI-18-1130
Kiel, A., Weigt, D., Kurasiak-Popowska, D., Niemann, J., and Karpińska, M. (2020). An analysis of the functionality of molecular markers related to the Lr19 gene conditioning resistance to wheat leaf rust. Zemdirbyste 107, 63–70. doi: 10.13080/z-a.2020.107.009
Kiran, K., Rawal, H. C., Dubey, H., Jaswal, R., Devanna, B. N., Gupta, D. K., et al. (2016). Draft genome of the wheat rust pathogen (Puccinia triticina) unravels genome-wide structural variations during evolution. Genome Biol. Evol. 8, 2702–2721. doi: 10.1093/gbe/evw197
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Li, Y., Wang, M., See, D. R., and Chen, X. (2019). Ethyl-methanesulfonate mutagenesis generated diverse isolates of Puccinia striiformis f. sp. tritici, the wheat stripe rust pathogen. World J. Microbiol. Biotechnol. 35:28. doi: 10.1007/s11274-019-2600-6
Li, Y., Xia, C., Wang, M., Yin, C., and Chen, X. (2020). Whole-genome sequencing of Puccinia striiformis f. sp. tritici mutant isolates identifies avirulence gene candidates. BMC Genom. 21, 247–269. doi: 10.1186/s12864-020-6677-y
Lind, V., and Gultyaeva, E. (2007). Virulence frequencies of Puccinia triticina in Germany and the European regions of the Russian federation. J. Phytopathol. 155, 13–21. doi: 10.1111/j.1439-0434.2006.01182.x
Liu, T., Song, T., Zhang, X., Yuan, H., Su, L., Li, W., et al. (2014). Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis. Nat. Commun. 5:4686. doi: 10.1038/ncomms5686
Ma, L., Lukasik, E., Gawehns, F., and Takken, F. L. W. (2012). The use of agroinfiltration for transient expression of plant resistance and fungal effector proteins in Nicotiana benthamiana leaves. Methods Mol. Biol. 835, 61–74. doi: 10.1007/978-1-61779-501-5_4
Mccallum, B. D., Reimer, E., Mcnabb, W., Foster, A., and Xue, A. (2021). Physiologic specialization of Puccinia triticina, the causal agent of wheat leaf rust, in Canada in 2015-2019. Can. J. Plant Pathol. 43, S333–S346. doi: 10.1080/07060661.2021.1888156
McDonald, B. A., and Linde, C. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379. doi: 10.1146/annurev.phyto.40.120501.101443
Mori, A., Hara, S., Sugahara, T., Kojima, T., Iwasaki, Y., Kawarasaki, Y., et al. (2015). Signal peptide optimization tool for the secretion of recombinant protein from Saccharomyces cerevisiae. J. Biosci. Bioeng. 120, 518–525. doi: 10.1016/j.jbiosc.2015.03.003
Nielsen, H., Konstantinos, D., Sorenvon, B., and von Heijne, G. (2019). A brief history of protein sorting prediction. Protein J. 38, 200–216. doi: 10.1007/s10930-019-09838-3
Persoon, A., Morin, E., Delaruelle, C., Thibaut, P., Fabien, H., Pascal, F., et al. (2014). Patterns of genomic variation in the poplar rust fungus Melampsora larici-Populina identify pathogenesis-related factors. Front. Plant Sci. 5, 450–466. doi: 10.3389/fpls.2014.00450
Petre, B., Joly, D. L., and Duplessis, S. (2014). Effector proteins of rust fungi. Front. Plant Sci. 5, 416–422. doi: 10.3389/fpls.2014.00416
Pourkhorshid, Z., Dadkhodaie, A., and Shamloo-Dashtpagerdi, R. (2022). Molecular analyses in wheat and Aegilops tauschii reveals a new orthologue of the leaf rust resistance gene Lr19 on 7DL chromosome of ae. Tauschii. J. Phytopathol. 170, 255–263. doi: 10.21203/rs.3.rs-1080624/v1
Prasad, P., Savadi, S., Bhardwaj, S. C., Gangwar, O. P., and Kumar, S. (2019). Rust pathogen effectors: perspectives in resistance breeding. Planta 250, 1–22. doi: 10.1007/s00425-019-03167-6
Prasad, P., Savadi, S., Bhardwaj, S. C., and Gupta, P. K. (2020). The progress of leaf rust research in wheat. Fungal Biol. 124, 537–550. doi: 10.1016/j.funbio.2020.02.013
Qi, T., Guo, J., Liu, P., He, F.-X., Wan, C.-P., Islam, M. A., et al. (2019). Stripe rust effector PstGSRE1 disrupts nuclear localization of ROS-promoting transcription factor TaLOL2 to defeat ROS-induced defense in wheat. Mol. Plant 12, 1624–1638. doi: 10.1016/j.molp.2019.09.01
Qureshi, N., Bariana, H., Kumran, V. V., Muruga, S., Forrest, K. L., Hayden, M. J., et al. (2018). A new leaf rust resistance gene Lr79 mapped in chromosome 3BL from durum wheat landrace Aus26582. Theor. Appl. Genet. 131, 1091–1098. doi: 10.1007/s00122-018-3060-3
Ridout, C. J., Skamnioti, P., Porritt, O., Sacristan, S., Jones, J. D., and Brown, J. K. (2006). Multiple avirulence paralogues in cereal powdery mildew fungi may contribute to parasite fitness and defeat of plant resistance. Plant Cell 18, 2402–2414. doi: 10.1105/tpc.106.043307
Roelfs, A. P. (1984). Race specificity and methods of study. Cereal Rusts 2, 131–164. doi: 10.1016/B978-0-12-148401-9.50011-X
Rohringer, R. (1997). Calcofluor: an optical brightener for fluorescence microscopy of fungal plant parasites in leaves. Phytopathology 77, 808–810. doi: 10.1094/Phyto-67-808
Rutter, W. B., Salcedo, A., Akhunova, A., He, F., Wang, S., and Liang, H. (2017). Divergent and convergent modes of interaction between wheat and Puccinia graminis f. sp. tritici isolates revealed by the comparative gene co-expression network and genome analyses. BMC Genomics 18, 291–311. doi: 10.1186/s12864-017-3678-6
Salcedo, A., Rutter, W., Wang, S., Akhunova, A., Bolus, S., Chao, S., et al. (2017). Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 358, 1604–1606. doi: 10.1126/science.aao7294
Segovia, V., Bruce, M., Rupp, J. L., Huang, L., Bakkeren, G., Trick, H. N., et al. (2016). Two small secreted proteins from induce reduction of ß-glucoronidase transient expression in wheat isolines containing and Lr9, Lr24 and Lr26. Can. J. Plant Pathol. 38, 91–102. doi: 10.1080/07060661.2016.1150884
Sperschneider, J., Gardiner, D. M., Dodds, P. N., Tini, F., Covarelli, L., Singh, K. B., et al. (2016). EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 210, 743–761. doi: 10.1111/nph.13794
Steuernagel, B., Periyannan, S. K., Hernandez-Pinzon, I., Witek, K., Rouse, M. N., Yu, G., et al. (2016). Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 34, 652–655. doi: 10.1038/nbt.3543
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNASeq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Upadhyaya, N. M., Mago, R., Panwar, V., Hewitt, T., and Dodds, P. N. (2021). Genomics accelerated isolation of a new stem rust avirulence gene–wheat resistance gene pair. Nat. Plants. 7, 1220–1228. doi: 10.1038/s41477-021-00971-5
Upadhyaya, N. M., Mago, R., Staskawicz, B. J., Ayliffe, M. A., Ellis, J. G., and Dodds, P. N. (2014). A bacterial type III secretion assay for delivery of fungal effector proteins into wheat. Mol. Plant Microbe Interact. 27, 255–264. doi: 10.1094/MPMI-07-13-0187-FI
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2009). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wei, J., Cui, L., Zhang, N., Du, D., Meng, Q., and Yan, H. (2020). Puccinia triticina pathotypes THTT and THTS display complex transcript profiles on wheat cultivar Thatcher. BMC Genet. 21, 48–59. doi: 10.21203/rs.2.12168/v3
Wu, J. -Q., Sakthikumar, S., Dong, C. -M., Zhang, P., Cuomo, C. A., and Park, R. F. (2017). Comparative genomics integrated with association analysis identifies candidate effector genes corresponding to Lr20 in phenotype-paired Puccinia triticina isolates from Australia. Front. Plant Sci. 8, 148–166. doi: 10.3389/fpls.2017.00148
Xia, C. -J., Wang, M. -N., Cornejo, O. E., Jiwan, D. A., See, D. R., and Chen, X. -M. (2017). Secretome characterization and correlation analysis reveal putative pathogenicity mechanisms and identify candidate avirulence genes in the wheat stripe rust fungus Puccinia striiformis sp. tritici. Front Microbiol. 8:2394. doi: 10.3389/fmicb.2017.02394
Xu, Q., Tang, C., Wang, L., Zhao, C., Kang, Z., and Wang, X. (2020). Haustoria–arsenals during the interaction between wheat and Puccinia striiformis f. sp. tritici. Mol. Plant Pathol. 21, 83–94. doi: 10.1111/mpp.12882
Yin, W., Wang, Y., Chen, T., Lin, Y., and Luo, C. (2018). Functional evaluation of the signal peptides of secreted proteins. BIO Protoc. 8:e2839. doi: 10.21769/BioProtoc.2839
Zhang, Z., Jin, J., Sheng, G., Xing, Y., Liu, W., Zhou, X., et al. (2021). A small cysteine-rich phytotoxic protein of phytophthora capsica functions as both plant defense elicitor and virulence factor. Mol. Plant Microbe Interact. 34, 891–903. doi: 10.1094/MPMI-01-21-0025-R
Zhang, L., Shi, C., Li, L., Li, M., Meng, Q., and Yan, H. (2020). Race and virulence analysis of Puccinia triticina in China in 2014 and 2015. Plant Dis. 104, 455–464. doi: 10.1094/PDIS-05-19-1051-RE
Keywords: wheat, Puccinia triticina, single nucleotide polymorphism, sequence insertion or deletion, AvrLr19 candidates
Citation: Cui Z, Wu W, Fan F, Wang F, Liu D, Di D and Wang H (2023) Transcriptome analysis of Lr19-virulent mutants provides clues for the AvrLr19 of Puccinia triticina. Front. Microbiol. 14:1062548. doi: 10.3389/fmicb.2023.1062548
Edited by:
Ashish Srivastava, Amity University, IndiaReviewed by:
Vishal Srivastava, Cleveland Clinic, United StatesPramod Prasad, ICAR-Indian Institute of Wheat and Barley Research, Regional Station, India
Sonali Mishra, Washington University in St. Louis, United States
Copyright © 2023 Cui, Wu, Fan, Wang, Liu, Di and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianping Di, Y2htcmR2QDE2My5jb20=; Haiyan Wang, bmR3YW5naGFpeWFuQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship