 Yongxiong Zhang1†
Yongxiong Zhang1† Haiying Gu1,2*†Zhouhong Shi3Weiqin Chen3Airu Li2Weiwei Ye3Cheng Zhang3Huikun Yuan2Mingming Zhao1
Haiying Gu1,2*†Zhouhong Shi3Weiqin Chen3Airu Li2Weiwei Ye3Cheng Zhang3Huikun Yuan2Mingming Zhao1- 1Health Science Center, Ningbo University, Ningbo, Zhejiang, China
- 2Laboratory of Gastroenterology, The Affiliated Hospital of Medical School, Ningbo University, Ningbo, Zhejiang, China
- 3Department of Gastrology, Ninghai First Hospital, Ningbo, Zhejiang, China
This study used multilocus sequence typing (MLST) to investigate the prevalence of Helicobacter pylori (H. pylori) mixed infections and H. pylori mixed infections involving unrelated strains; and determined the phylogeographic groups of H. pylori recovered from patients in Ningbo, China. A total of 156 H. pylori isolates were obtained from a convenience sample of 33 patients with culture-positive H. pylori infection. MLST was used to classify 150 H. pylori clinical isolates and 12 methodological control strains (6 clinical isolates and 6 strains of American Type Culture Collection H. pylori) into 43 and 12 sequence types (STs), respectively. In this study, 246 new alleles and 53 new STs were identified by MLST. The prevalence of mixed infections was 41% (11/27). The prevalence of H. pylori mixed infections involving unrelated strains was 46% (5/11) and the prevalence of H. pylori mixed infections involving completely unrelated strains (strains with all 7 housekeeping genes different) was 36% (4/11). A phylogenetic tree was created to determine the evolutionary relationships between different strains. The STs in this study were clustered within the hspEAsia subgroup (98%) and hpEurope group (2%). H. pylori mixed infections were common in Ningbo, China. The H. pylori isolates belonging to the hpEurope group were recovered from three different biopsy samples in a native Chinese patient. Most of H. pylori strains colonizing the antrum, corpus, and duodenum bulb were homologous.
Introduction
Helicobacter pylori (H. pylori) is a human pathogen that colonizes the gastrointestinal mucosa and causes chronic active gastritis, peptic ulcers, lymphoid tissue lymphoma, and gastric adenocarcinoma (Blaser and Atherton, 2004; Burkitt et al., 2017; Camilo et al., 2017). It is a gram-negative microaerophilic bacterium and has a high prevalence worldwide.
The gastrointestinal mucosa can be colonized by single or multiple H. pylori strains (Blaser and Berg, 2001; Kusters et al., 2006; Talebi Bezmin Abadi and Perez-Perez, 2016). H. pylori mixed infections and heteroresistance may lead to the failure of eradication treatment, based on different resistance of isolates from the antrum and corpus to antimicrobial agents (Farzi et al., 2019). H. pylori mixed infections are a cause for concern. The prevalence of H. pylori mixed infections varies from 0 to 100% in different geographic regions (Ben Mansour et al., 2016). The evolutionary relationships between different H. pylori isolates recovered from individual hosts are unclear. Previous studies have described four patterns of mixed infection: (i) most H. pylori isolates are related and exhibit slightly different patterns, and only few isolates are unrelated (Kim et al., 2003; Carroll et al., 2004; Kao et al., 2014; Farzi et al., 2015; Mendoza-Elizalde et al., 2019); (ii) all isolates are related (Patra et al., 2012; Ren et al., 2012; Kibria et al., 2015); (iii) isolates are predominantly unrelated (70%), with a few (30%) related isolates exhibiting slightly different patterns (Seo et al., 2019); (iv) all isolates are unrelated or show two independent populations of H. pylori (Carroll et al., 2004; Matteo et al., 2007; Salama et al., 2007). The prevalence of mixed infections varies according to the geographic region and the participant selection criteria (Kersulyte et al., 2000; Wong et al., 2001; Ben Mansour et al., 2016). H. pylori isolates with slightly different DNA fingerprinting patterns from a single host have revealed the mechanisms of microevolution (Marshall et al., 1998; Kim et al., 2003; Carroll et al., 2004; Mendoza-Elizalde et al., 2019). Due to its limitations, the DNA fingerprinting method cannot determine the evolutionary relationships of isolates from a single host (Carroll et al., 2004; Matteo et al., 2007).
Multilocus sequence typing (MLST) is based on comparison of the sequences of seven housekeeping genes (atpA, efp, mutY, ppa, trpC, ureI, and yphC) (Osaki et al., 2015; Raaf et al., 2017; Mendoza-Elizalde et al., 2019) and has the advantages of high repeatability, high resolution, and normalization. Moreover, this method can be used to determine the evolutionary relationships of isolates recovered from a single patient with H. pylori mixed infections (Mendoza-Elizalde et al., 2019). The prevalence of H. pylori mixed infections involving strains that are unrelated according to MLST is unclear. Importantly, the geographical location of the human host can be inferred from the MLST results (Mégraud et al., 2016; Gutiérrez-Escobar et al., 2017; Vazirzadeh et al., 2022). However, there is a lack of data on the geographic grouping of H. pylori isolates from patients in southern urban China.
Therefore, the purpose of this study was to characterize H. pylori mixed infections by MLST. More specifically, we investigated the prevalence of H. pylori mixed infections involving unrelated strains and phylogeographic groups of H. pylori isolates recovered from single patients in Ningbo, China.
Materials and methods
Ethics statement
Ethical approval for this study was obtained from the Medical Research Committee of the Ningbo University School of Medicine (approval number: NBU-2021-129).
Patients and specimens
A convenience sample of 33 patients from the Ninghai First Hospital and the Affiliated Hospital of Ningbo University School of Medicine who were culture positive for H. pylori participated in the study. Patients were excluded if they had taken antibiotics, proton pump inhibitors, H2-receptor antagonists, non-steroidal anti-inflammatory drugs, or bismuth-containing compounds within the 15 days prior to endoscopy. All patients provided written informed consent to participate in the study. Six patients were assigned to the control group, in which the prevalence of H. pylori mixed infections was not compared. Two or three mucosal biopsy specimens were collected from the antrum, corpus, and/or duodenal bulb of each patient for bacterial culture. The specimens were immediately placed in a transport medium (Gu’s Kit for Preservation of Helicobacter pylori, TianKuo, Ningbo Xunjian Biotechnology Co., Ltd., Ningbo, China) and transported to the laboratory at 2–8°C. Two other biopsy specimens were collected from each patient for histological examination.
Isolation and identification of H. pylori
The biopsy specimens were homogenized and inoculated on Gu’s plates (Gu’s Medium for Rapid Isolation of Helicobacter pylori, TianKuo, Ningbo Xunjian Biotechnology Co., Ltd., Ningbo, China; PCT WO2022178982A1). The plates were incubated at 37°C for 2–5 days in a microaerophilic environment (3–5% O2, 5–10% CO2, 5–10% H2, 75–87% N2) with 100% humidity (Anoxomat; MART Microbiology BV, Drachten, Netherlands). A pool of colonies was selected from the primary culture plates for subculturing. Single colonies were obtained from biopsy specimens of different sites of each patient using the colony suspension dilution method, and 1–10 single colonies were selected and passaged separately to obtain single colony isolates.
One of the three isolation schemes a, b, and c was selected to analyze H. pylori mixed infections in each patient, as follows: (a) One H. pylori colony was isolated from each of multiple biopsy specimens of a single patient; (b) Multiple (3–11) single colonies were obtained from one biopsy specimen of a single patient; (c) Multiple (2–10) single colonies were isolated from each of several biopsy specimens of a single patient.
Helicobacter pylori was identified based on colony morphology and Gram staining as a gullwing-shaped bacterium; positive reactions for catalase, oxidase, and urease (Gu’s Kit for Rapid Identification of Helicobacter pylori, TianKuo, Ningbo Xunjian Biotechnology Co., Ltd., Ningbo, China); and H. pylori antigen testing (H. pylori Antigen Rapid Test, Abon Biopharm, Hangzhou, China). Six American Type Culture Collection (ATCC) H. pylori reference strains (ATCC 43629, ATCC 700392, ATCC 51932, ATCC 700824, ATCC 43579, and ATCC 49503) were used as controls.
DNA extraction
Genomic DNA was extracted using a HiPure Bacterial DNA Kit (Guangzhou Magen Biotechnology Co., Ltd., Guangzhou, China) according to the manufacturer’s instructions. The extracted genomic DNA was stored at −20°C until being amplified using polymerase chain reaction (PCR).
Multilocus sequence typing
The extracted DNA was used as a template for PCR amplification of seven housekeeping genes (atpA, efp, mutY, ppa, trpC, ureI, and yphC). The primers used for the MLST housekeeping genes are shown in Table 1. The PCR amplification conditions used were as described by Achtman et al. (1999). The PCR products were detected, purified, and sequenced using Sanger sequencing (QingKe Biotechnology Co., Ltd., Hangzhou, China). The sequence peaks were interpreted using Chromas software to remove miscellaneous peaks, and the resulting sequences were imported into DNA Star software and assembled using the SeqMan function. The trimmed sequences of the seven housekeeping genes were imported into the H. pylori PubMLST database1 to identify allelic matches. The STs were identified based on the combination of MLST alleles. Nucleotide sequences that did not match existing PubMLST sequences were submitted to the database for new number assignment and sequence typing.
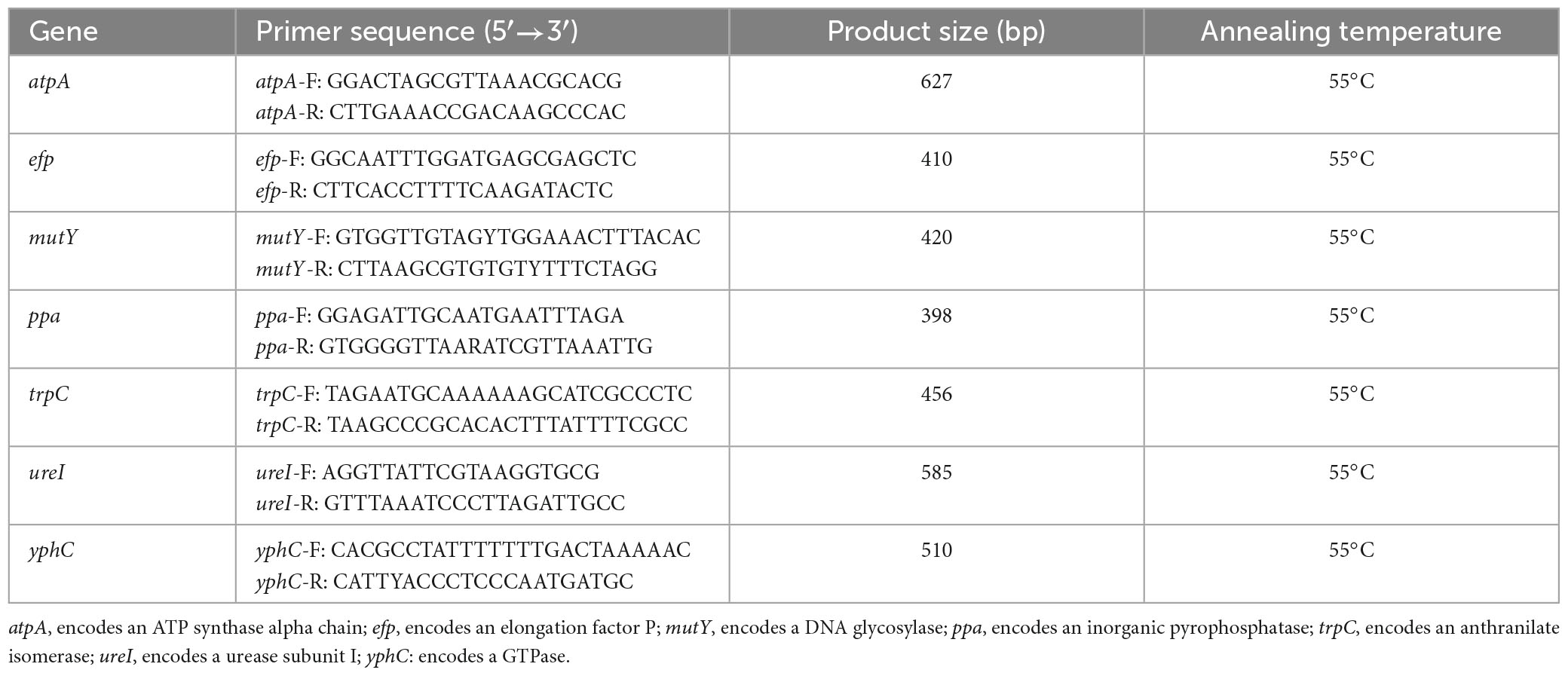
Table 1. Primers for the multilocus sequence typing (MLST) housekeeping gene.
Phylogeny, genealogic analysis, and genetic diversity analysis
The genealogic relationships among H. pylori isolates from the patients with mixed infections were investigated using PHYLOViZ (Mendoza-Elizalde et al., 2019). The goeBURST algorithm was used to demonstrate the relationship between clonal complexes per patient at the triple locus variants (TLV) level. The neighbor-joining algorithm with the Hamming distance was utilized to calculate the phylogenetic distance among strains using the Saitou and Nei (1987) criterion.
A phylogenetic tree was constructed using the MEGA 11 software for evolutionary analysis (Tamura et al., 2021). The concatenated nucleotide sequences of seven housekeeping genes in the studied H. pylori clinical isolates and six reference strains (ATCC 43629, ATCC 700392, ATCC 51932, ATCC 700824, ATCC 43579, and ATCC 49503) were aligned in ClustalW. Phylogenetic distances were computed using the neighbor-joining method (Saitou and Nei, 1987) with the Kimura 2-parameter model of nucleotide substitution (Kimura, 1980). The reliability of clustering was assessed using a bootstrap test (1,000 bootstrap replications). Polymorphisms in the housekeeping genes of H. pylori isolates obtained from patients with mixed infections were analyzed using DnaSP v6, which included polymorphic sites (S), nucleotide diversity (Pi), number of haplotypes (h), and haplotype diversity (Hd).
In addition, we downloaded 284 concatenated nucleotide sequences of the seven housekeeping genes of H. pylori strains from the PubMLST database2 as representatives of different geographical groups to determine the geographical type of H. pylori strains obtained in this study. After aligning the concatenated nucleotide sequences of the seven housekeeping genes in the studied strains (55 STs) and reference sequences (284 STs) by ClustalW, a phylogenetic tree was constructed in MEGA 11. The reference sequences for the geographical groups were as follows: hpEurope, 75 sequences; hpAsia2, 18 sequences; hspMaori, 35 sequences; hspEAsia, 50 sequences; hspAmerind, 9 sequences; hpAfrica2, 3 sequences; hspSAfrica, 25 sequences; hspWAfrica, 29 sequences; hpNEAfrica, 20 sequences; and hpSahul, 20 sequences.
Results
H. pylori isolation
A total of 156 clinical isolates of H. pylori were obtained from 33 patients with dyspepsia (18 males and 15 females) at two hospitals: Ninghai First Hospital and the Affiliated Hospital of Ningbo University School of Medicine. Clinical data showed that among the 33 patients with gastric diseases investigated by gastric endoscopy, 6 (18%) had gastric ulcer, 4 (12%) had duodenal ulcer, and 2 (6%) had compound ulcer, whereas the remaining 13 (39%), 7 (21%), and 1 (3%) were diagnosed with chronic active gastritis, chronic superficial gastritis, and chronic atrophic gastritis by pathology, respectively. The clinical data of the 33 patients and information on the sampling of H. pylori isolates and the number of isolates are summarized in Supplementary Table 1.
Multilocus sequence typing results
Multilocus sequence typing (MLST) was used to classify 162 H. pylori strains into 55 sequence types (STs). The STs of the six H. pylori isolates in the clinical control group were ST3689, ST3744, ST4096, ST4097, ST3715, and ST3696. The STs of the six ATCC reference strains were ATCC 43629 for ST4089, ATCC 700392 for ST181, ATCC 51932 for ST4091, ATCC 700824 for ST3496, ATCC 43579 for ST4092, and ATCC 49503 for ST4095. MLST separated the six clinical control isolates and the six ATCC reference strains into twelve STs. This indicated that isolates from different patients possessed different STs and that MLST genotyping in this study was accurate. The STs of 156 H. pylori isolates from the 33 patients as well as 6 ATCC strains are shown in Table 2. The analysis of seven housekeeping genes in these strains revealed 246 new alleles (atpA, 39; efp, 37; mutY, 37; ppa, 23; trpC, 38; ureI, 37; yphC, 35) and 53 new STs.
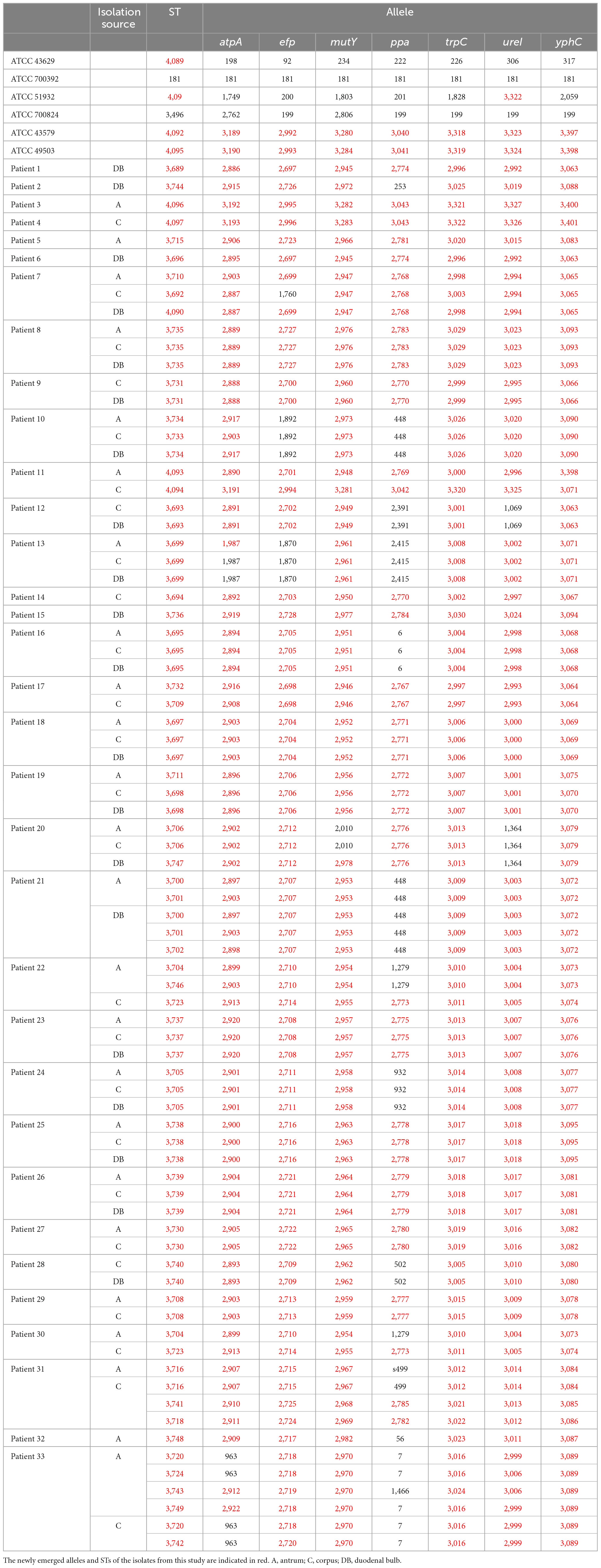
Table 2. Dataset of alleles and STs of Helicobacter pylori in this study.
Evolutionary relationships between isolates in patients with mixed infections
The different STs identified in each patient with mixed infections were used as genotyping data for the PHYLOViZ platform (Mendoza-Elizalde et al., 2019). The evolutionary relationships between isolates in the 11 patients with mixed infections are shown in Figure 1. The goeBURST algorithm was used at the TLV level to define clonal relationships. In the case of Patient 7, ST4090 was the major clonal complex with two linked STs (ST3710 and ST3692). The neighbor-joining algorithm was used to define the evolutionary distance; the longer the line, the greater the genetic distance. The number on the line represents how many housekeeping genes differ between these STs. For example, Patient 7 had one different housekeeping gene between ST3710 and ST4090, two differences between ST4090 and ST3692, and three differences between ST3710 and ST3692.
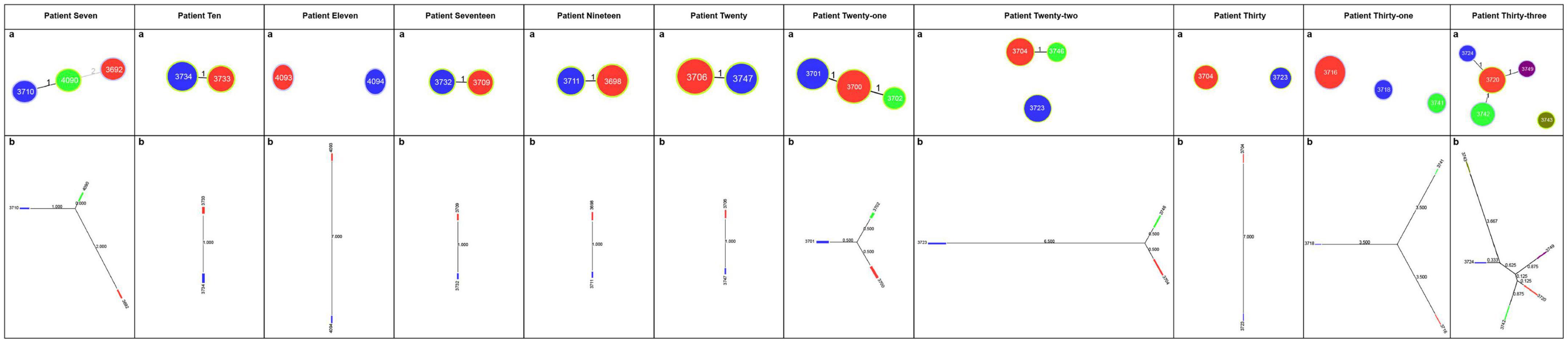
Figure 1. Evolutionary relationships among STs of H. pylori in patients with mixed infections. (A) Image showing the clonal relationships between the STs of H. pylori. Each circle represents an ST, and the size of the circle indicates the number of clinical isolates in the ST. The number on each line indicates the number of alleles with mutations. The unlinked STs imply that there are more than three different housekeeping genes between them. PHYLOViZ (goeBURST algorithm) was used to define the clonal relationships. (B) Image showing the evolutionary distance between the STs of H. pylori. Each square represents an ST; the larger the square, the more clinical isolates are included. Moreover, the longer the line and the larger the value on the line, the greater the genetic distance. PHYLOViZ (neighbor-joining algorithm) was used to define the evolutionary distance.
According to the evolutionary relationships and the polymorphisms of housekeeping genes of these isolates (Table 3), four of the eleven patients with mixed infections had isolates with seven different housekeeping genes, among which three patients (Patients 11, 22, and 30) showed different STs colonized at different sites (antrum, corpus), and one patient (Patient 31) exhibited three different STs at the same site (corpus). Moreover, two patients (Patients 7 and 33) had isolates with up to three and five different housekeeping genes, respectively. Eight of the eleven patients had isolates with only one housekeeping gene change, with atpA accounting for 75% (6/8), mutY for 12.5% (1/8) and yphC for 12.5% (1/8). Based on clonal relationships at the TLV level, related strains had the same STs or STs that differed by only very few alleles, whereas unrelated strains had STs that differed by more than three alleles. Therefore, the isolates from five patients (Patients 11, 22, 30, 31 and 33) were considered unrelated strains, and even isolates from four of these patients were considered completely unrelated strains (strains with all seven housekeeping genes different).
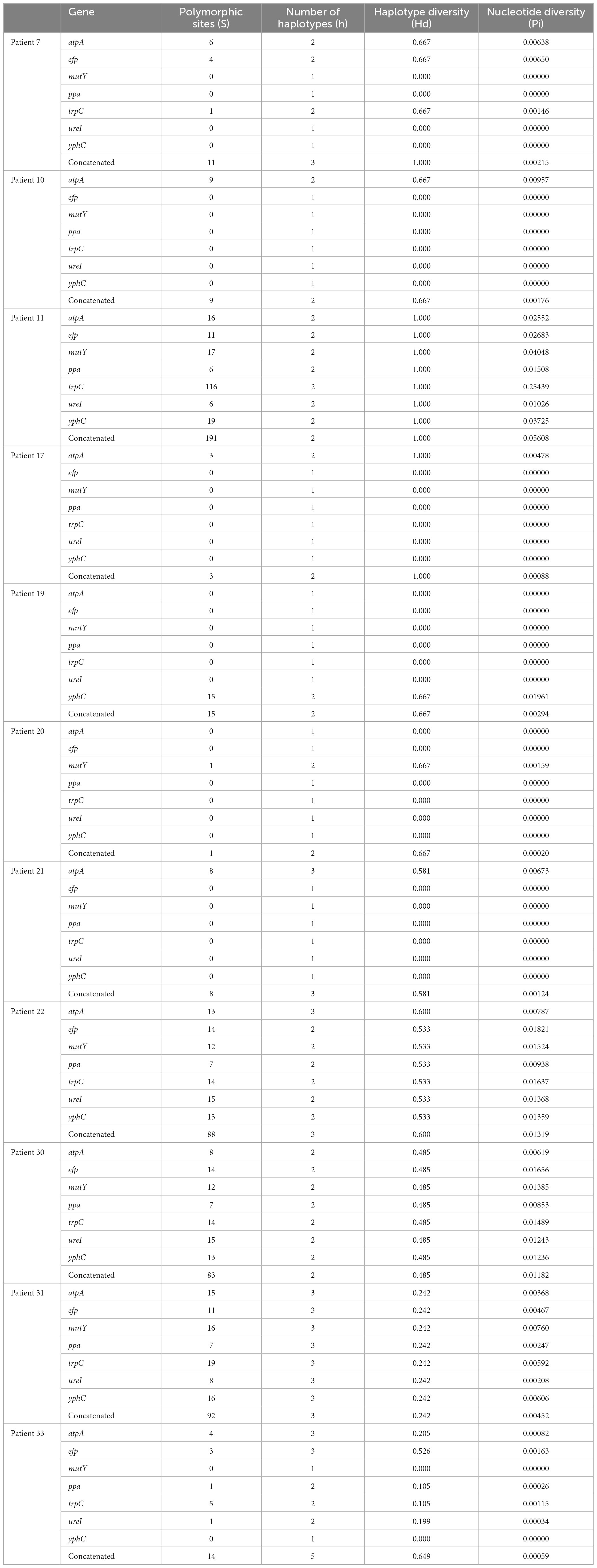
Table 3. Polymorphisms of housekeeping genes of Helicobacter pylori isolates obtained from 11 patients with mixed infections.
Phylogenetic analysis (Figure 2) showed that among the patients with mixed infections, the strains from four patients (Patients 11, 22, 30, and 31) were genetically distant and did not cluster together, indicating no evolutionary relatedness. The seven housekeeping genes of the isolates from these four patients were all different.
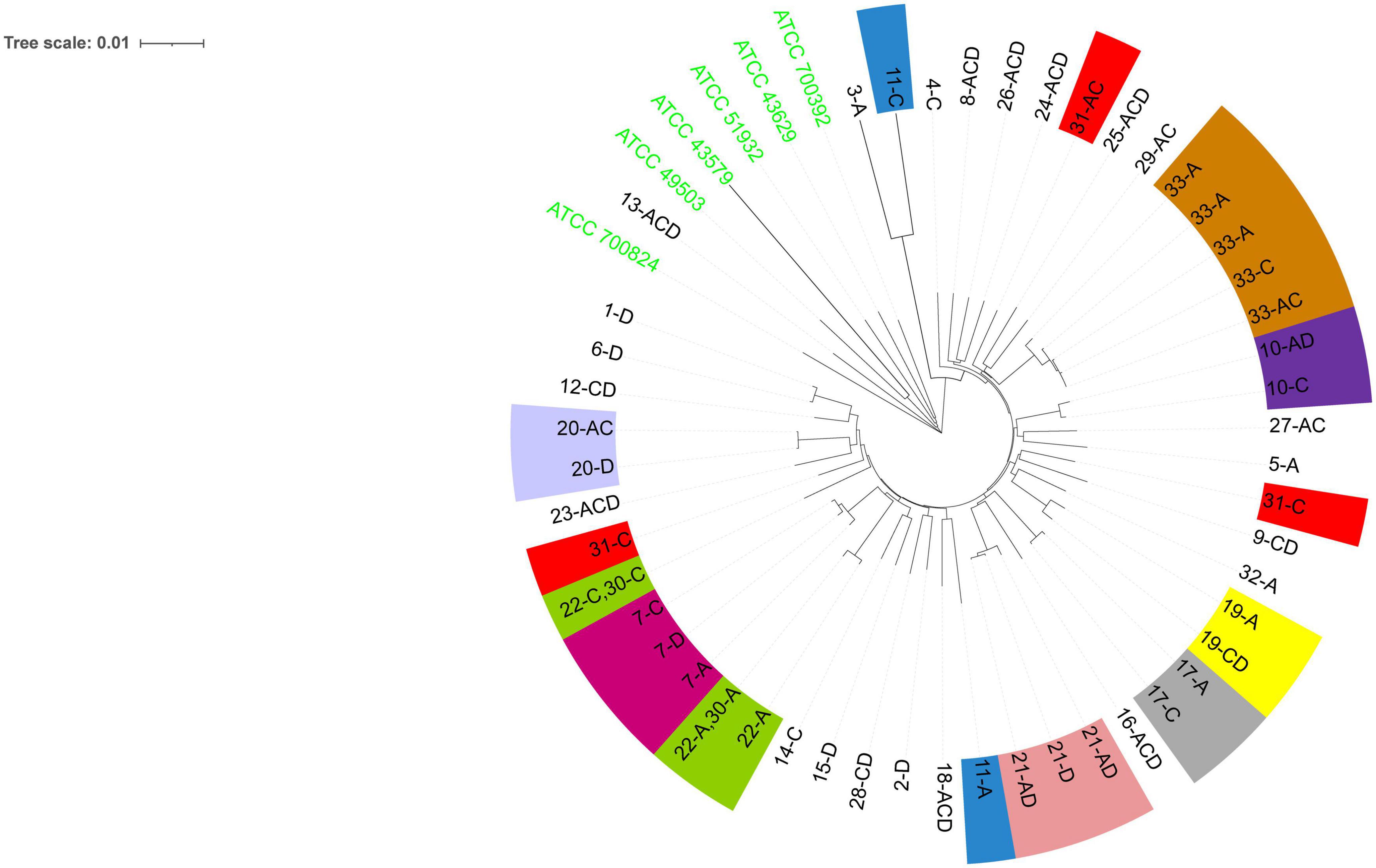
Figure 2. The evolutionary history was inferred using the neighbor-joining method (Saitou and Nei, 1987). The optimal tree is shown. The evolutionary distances were computed using the Kimura (1980) 2-parameter method and were in the units of the number of base substitutions per site. This analysis involved 55 nucleotide sequences. All positions containing gaps and missing data were eliminated (complete deletion option). A total of 3,406 positions were included in the final dataset. Evolutionary analyses were conducted using MEGA11 (Tamura et al., 2021). The six ATCC strains shown are the reference strains. Numbers 1–33 are patient numbers. Strains from the same patient are indicated by the same color. A, antrum; C, corpus; D, duodenum bulb.
Phylogeography
The phylogenetic tree revealed that the majority (153) of the 156 H. pylori clinical isolates belonged to the hspEAsia subgroup, whereas the three isolates (ST3699) from Patient 13 belonged to the hpEurope group (Figure 3). Among the reference strains, ATCC 700824 belonged to hspWAfrica, and the rest belonged to hpEurope.
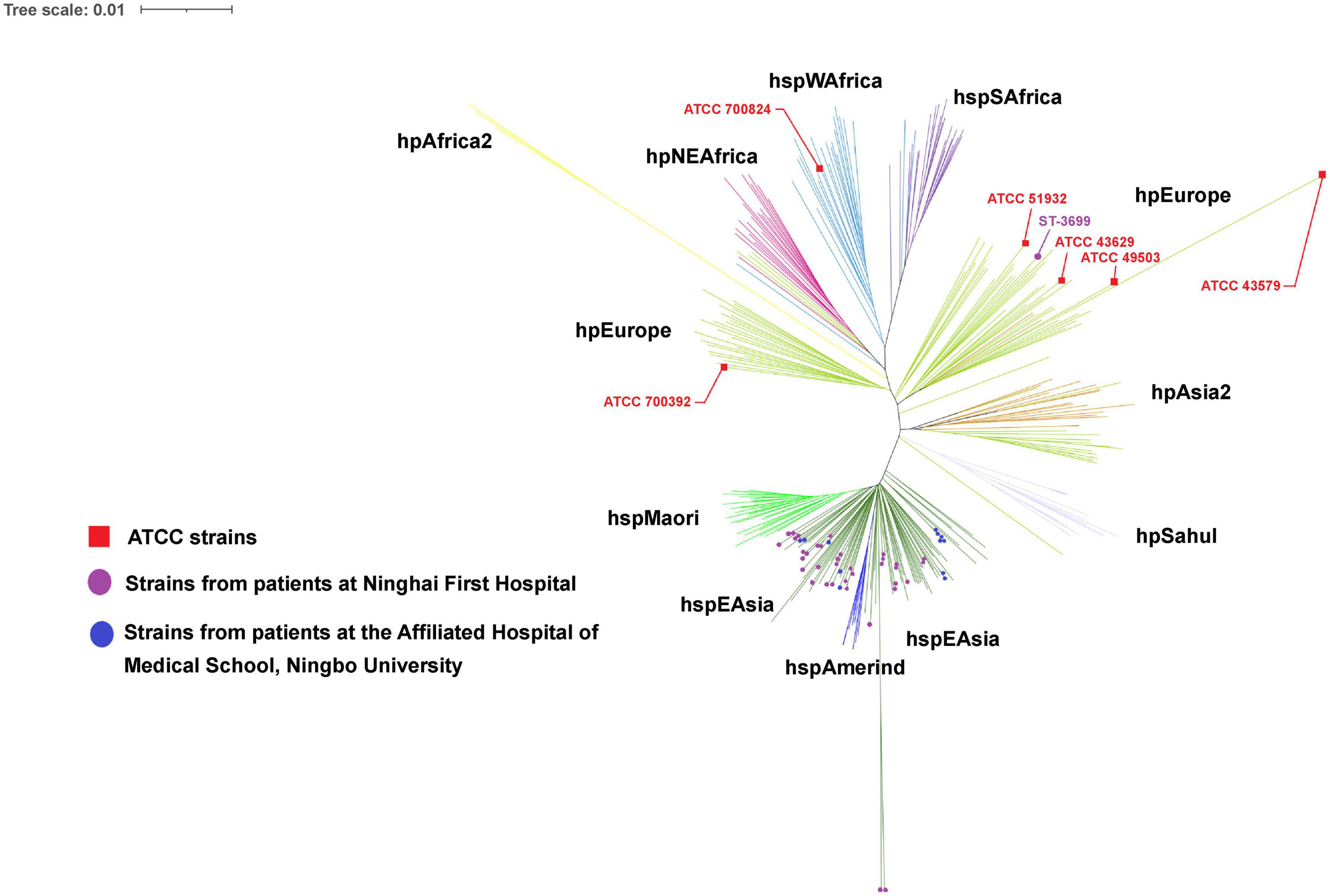
Figure 3. Phylogeography of the analyzed strains. The phylogeography was inferred using the neighbor-joining method (Saitou and Nei, 1987). The optimal tree is shown. The evolutionary distances were computed using the Kimura (1980) 2-parameter method and were in the units of the number of base substitutions per site. This analysis involved 55 (in this study) + 284 [from the Helicobacter pylori PubMLST database (http://pubmlst.org/organisms/helicobacter-pylori)] nucleotide sequences. All positions containing gaps and missing data were eliminated (complete deletion option). A total of 3,406 positions were included in the final dataset. Phylogeographical analyses were conducted using MEGA11 (Tamura et al., 2021).
Discussion
Helicobacter pylori is the most common infectious pathogen causing gastrointestinal diseases in humans, and can cause chronic gastritis, peptic ulcer, and gastric cancer (Blaser and Atherton, 2004; Burkitt et al., 2017; Camilo et al., 2017). Individuals can be infected by one or more types of H. pylori, which is known as multiple infections (Didelot et al., 2013; Palau et al., 2020), co-infection (Didelot et al., 2013; Seo et al., 2019; Mi et al., 2021), or mixed infections (Kao et al., 2014; Lai et al., 2016). Ben Mansour et al. (2016) distinguished the concepts of multiple infections and mixed infections, and defined multiple infections as an individual with two or more genetically different H. pylori isolates, whereas they defined mixed infections as an individual with two or more H. pylori isolates with different antibiotic sensitivity characteristics. It would contradict much of the literature to define mixed infections as simply different strains involving differences in antibiotic susceptibility characteristics. In essence, mixed infections are the colonization of the upper gastrointestinal mucosa of an individual by strains with heterogeneous biological characteristics, which can be reflected in the phenotypic characteristics of the strains, such as virulence factors, heteroresistance (Lai et al., 2016; Mi et al., 2021), and as well as strains with heterogeneous DNA.
The diagnosis of H. pylori mixed infection is mainly through genotyping techniques, including detection of virulence genes (cagA, vacA, iceA), random amplified polymorphic DNA (RAPD), MLST, and whole-genome sequencing (WGS). WGS is the gold standard for research, but its application is limited due to its high cost and complexity of analysis (Wang and Gu, 2018). MLST is the standard method of molecular typing of bacteria because of its high resolution and repeatability (Raaf et al., 2017). In addition, it can be used to determine the genotype of ancestral strains in individual patients (Mendoza-Elizalde et al., 2019). Although RAPD is low cost and rapid, the PCR-based RAPD fingerprinting pattern has low reproducibility across different experiments (Yokota et al., 2015). Moreover, RAPD has limitations in determining H. pylori mixed infections: researchers can only subjectively determine whether there is relatedness between different strains by DNA fingerprinting and cannot determine the evolutionary relationships of these isolates from a single host. It is unclear whether strain diversity is a result of multiple-strain infections or microevolution occurring in a single ancestral strain. In this study, we used MLST to explore the occurrence of strain diversity. We concatenated seven housekeeping genes of each H. pylori strain and used the PHYLOViZ and neighbor-joining algorithm to show the evolutionary relationships among the strains. We found that most of the colonized strains of patients with mixed infections differed by only one housekeeping gene, suggesting that different genotypes may originate from a single ancestral strain. The evolutionary relatedness of strains can be identified by comparing STs (Mendoza-Elizalde et al., 2019), and in this study, five patients had more than three different housekeeping genes between their respective STs, which indicated that they were infected with unrelated strains. Moreover, four of these patients had isolates with seven different housekeeping genes, indicating infection with completely unrelated strains. The prevalence of H. pylori mixed infections involving completely unrelated strains was non-negligible. Notably, we found that unrelated H. pylori strains could colonize the same gastric site (corpus) or different sites (antrum and corpus), indicating that both the gastric antrum and corpus can be colonized by mixed unrelated strains, and that the same site can be infected with multiple different types of H. pylori. Previous studies have revealed the existence of a microevolutionary mechanism through slightly different DNA fingerprinting patterns among H. pylori isolates from a single host (Wong et al., 2001; Kim et al., 2003; Carroll et al., 2004; Farzi et al., 2019), and our study further revealed a mechanism of mixed infections based on MLST technology.
The prevalence of H. pylori mixed infections varies widely according to region. For example, the prevalence of mixed infections in developed countries/regions is generally lower than that in developing countries/regions (Ben Mansour et al., 2016). In this study, we used MLST and determined that the prevalence of H. pylori mixed infections among patients in Ningbo, China, was 41%, which is higher than that reported in other regions of China, such as Hong Kong (24% by RAPD) (Wong et al., 2001) and Taiwan (28.6% by detection of virulence genes) (Lai et al., 2016), but lower than that in Guiyang (76.9% by RAPD) (Mi et al., 2021). It remains to be investigated whether the differences in prevalence are related to the measurement method, other than the geographic region. This study confirmed that MLST is more reliable than RAPD for determining the prevalence of mixed infections. In this study, 12 different STs were obtained by MLST from H. pylori isolates isolated from 6 unrelated patient control groups and 6 ATCC controls, whereas only two different profiles from the clinical isolates were obtained by RAPD (data not shown), indicating that four isolates were not distinguished as different based on the RAPD type. However, almost all studies on mixed infections have not included controls and reference strains for H. pylori isolates from patients and ATCC strains (Wong et al., 2001; Kim et al., 2003; Carroll et al., 2004; Kao et al., 2014; Ben Mansour et al., 2016; Lai et al., 2016; Seo et al., 2019; Palau et al., 2020; Mi et al., 2021).
It is unclear whether the prevalence of H. pylori mixed infections may varies according to the presence of disease, and sex. The prevalence of mixed infections in duodenal ulcer patients was higher than other diseases (Lai et al., 2016), but other studies did not support this conclusion (Kim et al., 2004; Ben Mansour et al., 2016). One report suggested that the prevalence of H. pylori mixed infections was significantly higher in women than in men (Kibria et al., 2015), but other reports confirmed that the prevalence was unrelated to sex (Ben Mansour et al., 2016). The data from the Supplementary Table 1 showed that, in this study, the prevalence of H. pylori mixed infection was independent of disease status or sex. Whether these inconsistent findings are caused by different assay methods needs to be confirmed.
Reasons for the variation in the prevalence of mixed infections may also include the effects of the sampling strategies, such as biopsy specimens from one vs. several gastrointestinal sites and one vs. multiple colonies from each biopsy. The assay using H. pylori only from the antrum may not be representative of H. pylori populations in the entire stomach, and Wong et al. (2001) suggested that several different sites need to be assayed. In this study, we used H. pylori isolates from the gastric antrum, corpus, and/or duodenum bulb for the assays.
In this study, we identified 246 new alleles and 53 new STs associated with H. pylori infection. H. pylori is a highly genetically diverse species (Suerbaum et al., 1998). Analysis of the polymorphisms of housekeeping genes of H. pylori obtained from patients with mixed infections showed higher Hd values, which indicated that haplotypes forming the populations in each patient were very divergent, thus confirming the high diversity of H. pylori. The number of polymorphic sites revealed that H. pylori can mutate, making H. pylori more adaptable to specific ecological niches in individual hosts (Kang and Blaser, 2006; Naito et al., 2006).
Phylogenetic analyses of the seven concatenated housekeeping genes revealed different geographical groups of H. pylori. In this study, H. pylori was most prominently represented by the hspEAsia subgroup, and for the first time, three isolates (ST3699) recovered from a local patient with no history of living abroad were found to belong to the hpEurope group. MLST is a useful tool for tracking human migration (Yamaoka, 2009), enabling us to infer human migration routes or colonization history based on the geographic grouping of H. pylori. H. pylori strains that colonize in patients from Ningbo belong predominantly to the hspEAsia subgroup, which is consistent with the geographical location of Ningbo (eastern Asia).
In this study, we used a self-developed kit, Gu’s kit, for rapid isolation of H. pylori, which is a new technique for obtaining isolates rapidly and efficiently. Studies have reported that some genotypes were lost during culture passages (Ren et al., 2012) and eventually failed to obtain isolates (Arévalo-Jaimes et al., 2019), which is detrimental to the study of mixed infections. This new and rapid culture technique can effectively shorten culture time and reduce the loss of genetic representation during subculture passages, which can truly reflect the patient’s H. pylori mixed infections.
The main limitation of this study is the small sample size, which did not enable us to analyze the effect of the specific isolation scheme on the prevalence of mixed infections detected. Therefore, these results need to be confirmed by larger studies.
In conclusion, the prevalence of H. pylori mixed infections in patients from Ningbo, China, is high. Moreover, the prevalence of H. pylori mixed infections involving unrelated strains was 46%, and the prevalence of H. pylori mixed infections involving completely unrelated strains was 36%, which may be related to the use of new H. pylori isolation and culture techniques. In this study, 246 new alleles and 53 new STs were discovered for the first time. Most H. pylori strains in Ningbo belong to the hspEAsia subgroup, whereas very few strains belong to the hpEurope group.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in this article/Supplementary material.
Ethics statement
The studies involving humans were approved by the Medical Research Committee of the Ningbo University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HG conceived the study, collected gastrointestinal biopsy samples, and reviewed the manuscript. YZ performed the experiment, generated the sequence data, performed bioinformatics analysis, and prepared the manuscript. ZS, WC, WY, and CZ collected gastrointestinal biopsy samples. AL participated in the analysis of the results. MZ generated the sequence data. HY performed some of the experiments. All authors read and approved the final manuscript.
Funding
This study was supported by the Zhejiang Provincial Natural Science Foundation (LZ14H200001). The funder had no role in the design of the study or collection, analysis, and interpretation of data or in writing the manuscript.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1207878/full#supplementary-material
Abbreviations
ATCC, American Type Culture Collection; atpA, gene encoding an ATP synthase alpha chain; efp, gene encoding an elongation factor P; h, number of haplotypes; Hd, haplotype diversity; MLST, multilocus sequence typing; mutY, gene encoding a DNA glycosylase; PCR, polymerase chain reaction; Pi, nucleotide diversity; ppa, gene encoding an inorganic pyrophosphatase; RAPD, random amplified polymorphism DNA; S, number of polymorphic sites; ST, sequence type; TLV, triple locus variants; trpC, gene encoding an anthranilate isomerase; ureI, gene encoding a urease subunit I; yphC, gene encoding a GTPase.
Footnotes
References
Achtman, M., Azuma, T., Berg, D. E., Ito, Y., Morelli, G., Pan, Z. J., et al. (1999). Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 32, 459–470. doi: 10.1046/j.1365-2958.1999.01382.x
Arévalo-Jaimes, B. V., Rojas-Rengifo, D. F., Jaramillo, C. A., de Molano, B. M., Vera-Chamorro, J. F., and Del Pilar Delgado, M. (2019). Genotypic determination of resistance and heteroresistance to clarithromycin in Helicobacter pylori isolates from antrum and corpus of Colombian symptomatic patients. BMC Infect. Dis. 19:546. doi: 10.1186/s12879-019-4178-x
Ben Mansour, K., Fendri, C., Battikh, H., Garnier, M., Zribi, M., Jlizi, A., et al. (2016). Multiple and mixed Helicobacter pylori infections: Comparison of two epidemiological situations in Tunisia and France. Infect. Genet. Evol. 37, 43–48. doi: 10.1016/j.meegid.2015.10.028
Blaser, M. J., and Atherton, J. C. (2004). Helicobacter pylori persistence: Biology and disease. J. Clin. Invest. 113, 321–333. doi: 10.1172/JCI20925
Blaser, M. J., and Berg, D. E. (2001). Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Invest. 107, 767–773. doi: 10.1172/JCI12672
Burkitt, M. D., Duckworth, C. A., Williams, J. M., and Pritchard, D. M. (2017). Helicobacter pylori-induced gastric pathology: Insights from in vivo and ex vivo models. Dis. Model. Mech. 10, 89–104. doi: 10.1242/dmm.027649
Camilo, V., Sugiyama, T., and Touati, E. (2017). Pathogenesis of Helicobacter pylori infection. Helicobacter 22, 24–31. doi: 10.1111/hel.12405
Carroll, I. M., Ahmed, N., Beesley, S. M., Khan, A. A., Ghousunnissa, S., Moráin, C. A. Ó, et al. (2004). Microevolution between paired antral and paired antrum and corpus Helicobacter pylori isolates recovered from individual patients. J. Med. Microbiol. 53, 669–677. doi: 10.1099/jmm.0.05440-0
Didelot, X., Nell, S., Yang, I., Woltemate, S., van der Merwe, S., and Suerbaum, S. (2013). Genomic evolution and transmission of Helicobacter pylori in two South African families. Proc. Natl. Acad. Sci. U. S. A. 110, 13880–13885. doi: 10.1073/pnas.1304681110
Farzi, N., Behzad, C., Hasani, Z., Alebouyeh, M., Zojaji, H., and Zali, M. R. (2019). Characterization of clarithromycin heteroresistance among Helicobacter pylori strains isolated from the antrum and corpus of the stomach. Folia Microbiol. 64, 143–151. doi: 10.1007/s12223-018-0637-9
Farzi, N., Malekian, T., Alebouyeh, M., Vaziri, F., and Zali, M. R. (2015). Genotype diversity and quasispecies development of Helicobacter pylori in a single host. Jpn. J. Infect. Dis. 68, 176–180. doi: 10.7883/yoken.JJID.2014.165
Gutiérrez-Escobar, A. J., Trujillo, E., Acevedo, O., and Bravo, M. M. (2017). Phylogenomics of Colombian Helicobacter pylori isolates. Gut Pathog. 9:52. doi: 10.1186/s13099-017-0201-1
Kang, J., and Blaser, M. J. (2006). Bacterial populations as perfect gases: Genomic integrity and diversification tensions in Helicobacter pylori. Nat. Rev. Microbiol. 4, 826–836. doi: 10.1038/nrmicro1528
Kao, C. Y., Lee, A. Y., Huang, A. H., Song, P. Y., Yang, Y. J., Sheu, S. M., et al. (2014). Heteroresistance of Helicobacter pylori from the same patient prior to antibiotic treatment. Infect. Genet. Evol. 23, 196–202. doi: 10.1016/j.meegid.2014.02.009
Kersulyte, D., Mukhopadhyay, A. K., Velapatiño, B., Su, W., Pan, Z., Garcia, C., et al. (2000). Differences in genotypes of Helicobacter pylori from different human populations. J. Bacteriol. 182, 3210–3218. doi: 10.1128/JB.182.11.3210-3218.2000
Kibria, K. M., Hossain, M. E., Sultana, J., Sarker, S. A., Bardhan, P. K., Rahman, M., et al. (2015). The prevalence of mixed Helicobacter pylori infections in symptomatic and asymptomatic subjects in Dhaka, Bangladesh. Helicobacter 20, 397–404. doi: 10.1111/hel.12213
Kim, J. J., Kim, J. G., and Kwon, D. H. (2003). Mixed-infection of antibiotic susceptible and resistant Helicobacter pylori isolates in a single patient and underestimation of antimicrobial susceptibility testing. Helicobacter 8, 202–206. doi: 10.1046/j.1523-5378.2003.00145.x
Kim, J. W., Kim, J. G., Chae, S. L., Cha, Y. J., and Park, S. M. (2004). High prevalence of multiple strain colonization of Helicobacter pylori in Korean patients: DNA diversity among clinical isolates from the gastric corpus, antrum and duodenum. Korean. J. Intern. Med. 19, 1–9. doi: 10.3904/kjim.2004.19.1.1
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kusters, J. G., van Vliet, A. H., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490. doi: 10.1128/CMR.00054-05
Lai, C. H., Huang, J. C., Chiang-Ni, C., Li, J. P., Wu, L. T., Wu, H. S., et al. (2016). Mixed infections of Helicobacter pylori isolated from patients with gastrointestinal diseases in Taiwan. Gastroenterol. Res. Pract. 2016:7521913. doi: 10.1155/2016/7521913
Marshall, D. G., Dundon, W. G., Beesley, S. M., and Smyth, C. J. (1998). Helicobacter pylori – A conundrum of genetic diversity. Microbiology 144, 2925–2939. doi: 10.1099/00221287-144-11-2925
Matteo, M. J., Granados, G., Pérez, C. V., Olmos, M., Sanchez, C., and Catalano, M. (2007). Helicobacter pylori cag pathogenicity island genotype diversity within the gastric niche of a single host. J. Med. Microbiol. 56, 664–669. doi: 10.1099/jmm.0.46885-0
Mégraud, F., Lehours, P., and Vale, F. F. (2016). The History of Helicobacter pylori: From phylogeography to paleomicrobiology. Clin. Microbiol. Infect. 22, 922–927. doi: 10.1016/j.cmi.2016.07.013
Mendoza-Elizalde, S., Cortés-Márquez, A. C., Zuñiga, G., Cerritos, R., Valencia-Mayoral, P., Sánchez, A. C., et al. (2019). Inference from the analysis of genetic structure of Helicobacter pylori strains isolates from two paediatric patients with recurrent infection. BMC Microbiol. 19:184. doi: 10.1186/s12866-019-1554-z
Mi, M., Wu, F., Zhu, J., Liu, F., Cui, G., Wen, X., et al. (2021). Heterogeneity of Helicobacter pylori strains isolated from patients with gastric disorders in Guiyang, China. Infect. Drug Resist. 14, 535–545. doi: 10.2147/IDR.S287631
Naito, M., Yamazaki, T., Tsutsumi, R., Higashi, H., Onoe, K., Yamazaki, S., et al. (2006). Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology 130, 1181–1190. doi: 10.1053/j.gastro.2005.12.038
Osaki, T., Konno, M., Yonezawa, H., Hojo, F., Zaman, C., Takahashi, M., et al. (2015). Analysis of intra-familial transmission of Helicobacter pylori in Japanese families. J. Med. Microbiol. 64, 67–73. doi: 10.1099/jmm.0.080507-0
Palau, M., Piqué, N., Comeau, A. M., Douglas, G. M., Ramírez-Lázaro, M. J., Lario, S., et al. (2020). Detection of Helicobacter pylori microevolution and multiple infection from gastric biopsies by housekeeping gene amplicon sequencing. Pathogens 9:97. doi: 10.3390/pathogens9020097
Patra, R., Chattopadhyay, S., De, R., Ghosh, P., Ganguly, M., Chowdhury, A., et al. (2012). Multiple infection and microdiversity among Helicobacter pylori isolates in a single host in India. PLoS One 7:e43370. doi: 10.1371/journal.pone.0043370
Raaf, N., Amhis, W., Saoula, H., Abid, A., Nakmouche, M., Balamane, A., et al. (2017). Prevalence, antibiotic resistance, and MLST typing of Helicobacter pylori in Algiers, Algeria. Helicobacter 22, e12446. doi: 10.1111/hel.12446
Ren, L., Liao, Y. L., Song, Y., Guo, Y., Mao, X. H., Xie, Q. H., et al. (2012). High frequency variations of Helicobacter pylori isolates in individual hosts in a Chinese population. Int. J. Infect. Dis. 16, e358–e363. doi: 10.1016/j.ijid.2012.01.006
Saitou, N., and Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Salama, N. R., Gonzalez-Valencia, G., Deatherage, B., Aviles-Jimenez, F., Atherton, J. C., Graham, D. Y., et al. (2007). Genetic analysis of Helicobacter pylori strain populations colonizing the stomach at different times postinfection. J. Bacteriol. 189, 3834–3845. doi: 10.1128/JB.01696-06
Seo, J. W., Park, J. Y., Shin, T. S., and Kim, J. G. (2019). The analysis of virulence factors and antibiotic resistance between Helicobacter pylori strains isolated from gastric antrum and body. BMC Gastroenterol. 19:140. doi: 10.1186/s12876-019-1062-5
Suerbaum, S., Smith, J. M., Bapumia, K., Morelli, G., Smith, N. H., Kunstmann, E., et al. (1998). Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 95, 12619–12624. doi: 10.1073/pnas.95.21.12619
Talebi Bezmin Abadi, A., and Perez-Perez, G. (2016). Role of dupA in Virulence of Helicobacter pylori. World J. Gastroenterol. 22, 10118–10123. doi: 10.3748/wjg.v22.i46.10118
Tamura, K., Stecher, G., and Mega, K. S. (2021). MEGA11: Molecular evolutionary genetics analysis, version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Vazirzadeh, J., Karbasizadeh, V., Falahi, J., Moghim, S., Narimani, T., and Rafiei, R. (2022). Genetic diversity of Helicobacter pylori isolates from patients with gastric diseases in Isfahan. Adv. Biomed. Res. 11:4. doi: 10.4103/abr.abr_25_21
Wang, J., and Gu, H. (2018). Research progress on genotyping of Helicobacter pylori. J. Zhejiang Univ. Med. Sci. 47, 97–103. doi: 10.3785/j.issn.1008-9292.2018.02.14
Wong, B. C., Wang, W. H., Berg, D. E., Fung, F. M., Wong, K. W., Wong, W. M., et al. (2001). High prevalence of mixed infections by Helicobacter pylori in Hong Kong: Metronidazole sensitivity and overall genotype. Aliment. Pharmacol. Ther. 15, 493–503. doi: 10.1046/j.1365-2036.2001.00949.x
Yamaoka, Y. (2009). Helicobacter pylori typing as a tool for tracking human migration. Clin. Microbiol. Infect. 15, 829–834. doi: 10.1111/j.1469-0691.2009.02967.x
Yokota, S., Konno, M., Fujiwara, S., Toita, N., Takahashi, M., Yamamoto, S., et al. (2015). Intrafamilial, preferentially mother-to-child and intraspousal, Helicobacter pylori infection in Japan determined by mutilocus sequence typing and random amplified polymorphic DNA fingerprinting. Helicobacter 20, 334–342. doi: 10.1111/hel.12217
Keywords: Helicobacter pylori, mixed infections, unrelated strains, evolutionary relationships, homologous, multilocus sequence typing, phylogeographic groups
Citation: Zhang Y, Gu H, Shi Z, Chen W, Li A, Ye W, Zhang C, Yuan H and Zhao M (2023) High prevalence of Helicobacter pylori mixed infections identified by multilocus sequence typing in Ningbo, China. Front. Microbiol. 14:1207878. doi: 10.3389/fmicb.2023.1207878
Received: 18 April 2023; Accepted: 25 July 2023;
Published: 08 August 2023.
Edited by:
Paulo Jorge Dias, University of Lisbon, PortugalReviewed by:
Abbas Yadegar, Shahid Beheshti University of Medical Sciences, IranAmin Talebi Bezmin Abadi, Tarbiat Modares University, Iran
Sílvia A. Sousa, Institute for Bioengineering and Biosciences, Portugal
Copyright © 2023 Zhang, Gu, Shi, Chen, Li, Ye, Zhang, Yuan and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiying Gu, Z3VoYWl5aW5nQG5idS5lZHUuY24=
†These authors have contributed equally to this work