 Emanuel J. Muruaga1,2
Emanuel J. Muruaga1,2 Paula J. Uriza1,2
Paula J. Uriza1,2 Gonzalo A. K. Eckert1,2
Gonzalo A. K. Eckert1,2 María V. Pepe1,2Cecilia M. Duarte1,2
María V. Pepe1,2Cecilia M. Duarte1,2 Mara S. Roset1,2*
Mara S. Roset1,2* Gabriel Briones1,2*
Gabriel Briones1,2*- 1Instituto de Investigaciones Biotecnológicas, Universidad Nacional de San Martín (UNSAM)- Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
- 2Escuela de Bio y Nanotecnologías (EByN), Universidad Nacional de San Martín, Buenos Aires, Argentina
Introduction: The S-layer proteins are a class of self-assembling proteins that form bi-dimensional lattices named S-Layer on the cell surface of bacteria and archaea. The protein SlpA, which is the major constituent of the Lactobacillus acidophilus S-layer, contains in its C-terminus region (SlpA284 − 444), a protein domain (named here as SLAPTAG) responsible for the association of SlpA to the bacterial surface. SLAPTAG was adapted for the development of a novel affinity chromatography method: the SLAPTAG-based affinity chromatography (SAC).
Methods: Proteins with different molecular weights or biochemical functions were fused in-frame to the SLAPTAG and efficiently purified by a Bacillus subtilis-derived affinity matrix (named Bio-Matrix or BM). Different binding and elution conditions were evaluated to establish an optimized protocol.
Results: The binding equilibrium between SLAPTAG and BM was reached after a few minutes of incubation at 4°C, with an apparent dissociation constant (KD) of 4.3μM. A reporter protein (H6-GFP-SLAPTAG) was used to compare SAC protein purification efficiency against commercial immobilized metal affinity chromatography. No differences in protein purification performance were observed between the two methods. The stability and reusability of the BM were evaluated, and it was found that the matrix remained stable for more than a year. BM could be reused up to five times without a significant loss in performance. Additionally, the recovery of bound SLAP-tagged proteins was explored using proteolysis with a SLAP-tagged version of the HRV-3c protease (SLAPASE). This released the untagged GFP while the cut SLAPTAG and the SLAPASE were retained in the BM. As an alternative, iron nanoparticles were linked to the BM, resulting in BMmag. The BMmag was successfully adapted for a magnetic SAC, a technique with potential applications in high-throughput protein production and purification.
Discussion: The SAC protocol can be adapted as a universal tool for the purification of recombinant proteins. Furthermore, the SAC protocol utilizes simple and low-cost reagents, making it suitable for in-house protein purification systems in laboratories worldwide. This enables the production of pure recombinant proteins for research, diagnosis, and the food industry.
Introduction
Proteins are biopolymers formed by a particular amino acid sequence that determines a given atomic spatial distribution, also known as protein conformation. Far from being a static structure, proteins behave as “nano-machines” performing precise molecular activities due to internal movements of the protein parts or protein domains (Wood, 2004). In addition, proteins can interact intramolecularly or intermolecularly with different macromolecules such as proteins, DNA, RNA, polysaccharides, or small compounds (Buxbaum, 2015).
Proteins perform vital functions in life. For instance, some proteins break down food into nutrients during the process of digestion in the human body, while other proteins transport critical compounds for supporting life (like hemoglobin that transports oxygen to the cells), shape the cellular structure (like actin, tubulin, or keratin), function as hormones (like insulin), or defend the organism against pathogens (like the antibodies) (Buxbaum, 2015).
In addition to the multiplicity of existent polypeptides in nature, molecular biology and biotechnology have generated a myriad of chimeric proteins by combining different protein domains (Baldo, 2015). These artificial proteins can be used as therapeutic tools to treat cancer, autoimmunity, or different medical conditions (Wen et al., 2009).
However, for various applications such as biochemical, industrial, or medical purposes, proteins need to be purified, stabilized, and concentrated to a high degree to be useful. This condition is reached by a series of steps oriented to isolate and purify a desired protein from the other proteins in the mixture, free of contaminants, and preserving its biological activity. The purification process exploits differences in size, charge, hydrophobicity, ligand-binding affinity, or specific sequences (Schales, 1942; Freitag and Horváth, 1996; Wood, 2014). Several fractionations of chromatographic steps can be combined to efficiently enrich or purify a particular protein.
Affinity chromatography is a special type of liquid chromatography that exploits the existence of natural (Kuntz et al., 1999) or artificial (Mouratou et al., 2015) affinities between two moieties: the molecular target (or tag) and its ligand (the affinity ligand) which is usually immobilized onto a chromatographic stationary phase to generate a chromatography matrix. Thus, proteins of interest can be fused in-frame to different molecular tags (such as His-6X, GST, and MBP) expressed and purified ideally in a single step from a complex mixture of proteins. Additionally, there is a type of affinity chromatography that allows the purification of antibodies without the need for a molecular tag. This method employs proteins such as protein A, protein G, or different synthetic proteins that exhibit a high affinity for the Fc region of IgG antibodies (Mouratou et al., 2015).
Since it was developed by Starkestein in 1910 (Hais, 1986), affinity chromatography was gaining popularity and centrality for many industrial processes such as the purification of proteins for diagnosis, research, and therapeutic purposes (Rodriguez et al., 2020). Regulatory requirements for the purity and quality of the proteins vary greatly depending on the area of application. For instance, bio-products can be used with little purification for industrial use. In addition, the recombinant protein produced for research, diagnosis, or non-clinical purposes has a less stringent regulatory approval process than proteins designed as biopharmaceuticals.
The most common strategy for affinity chromatography is the ON/OFF format. In the “ON” phase, a biological sample containing the protein of interest (that can be fused in-frame to a molecular tag) is formulated in a specific application buffer that will favor the binding process to a ligand. Then, the sample is placed in contact with a chromatography material (or chromatography matrix) associated with the affinity ligand that will consequently recognize and retain the tagged protein. Finally, the chromatography matrix is washed several times to remove all the unbound protein. In the “OFF” phase (or elution phase), an elution buffer is passed or incubated to release the tagged protein by changing the pH or the ionic strength modifying the affinity of the interaction (non-specific elution) or by the addition of the free ligand that will out-compete the retained tagged protein (bio-specific elution) (Rodriguez et al., 2020). The simplicity and specificity of affinity chromatography have made this technique central for the purification of biomolecules and biopharmaceuticals.
Here, we explored the adaptation of a protein domain present in the carboxy terminus of the Lactobacillus acidophilus protein SlpA as a molecular tag. In silico analysis of this region (SlpA284 − 444) allowed us to identify a tandem of two copies of the 60-aminoacid protein domain named SLP-A (pfam03217) which is necessary for the association (Supplementary Figure 1). Here, this dual SLP-A domain was named SLAPTAG, and the adaptation as a molecular tag was evaluated. Recently, we have characterized the binding properties of the SLAPTAG and characterized its association with the cell wall of live Lactobacillus for vaccine purposes (Uriza et al., 2020). Thus, the SLAPTAG was fused in-frame to a chimeric antigen derived from the Shiga toxin-producing Escherichia coli (STEC) formed by the peptides EspA36 − 192, Intimin653 − 953, and Tir240 − 378 (or EIT). The resulting chimeric antigen (EIT-SLAPTAG) recombinantly expressed and purified was able to associate with the bacterial cell wall of L. acidophilus, a process that we named decoration. Thus, EIT-decorated L. acidophilus after oral administration was able to deliver the EIT antigen to the intestinal mucosa eliciting a protective immune response that controls an experimental STEC infection in mice (Uriza et al., 2020). Remarkably, the decoration process does not modify genetically the Lactobacillus genome preserving its GRAS (Generally Recognized as Safe) status, a trait that is important for vaccine purposes.
Here, we explore the adaptation of the SLAPTAG for the development of novel affinity batch chromatography, the SLAP affinity chromatography (SAC), and a comprehensive protocol is presented.
Materials and methods
Strains and plasmids
All the bacterial strains and plasmids used here are summarized in Table 1. Bacteria Escherichia coli and Bacillus subtilis strains were grown in Luria Bertani (LB) medium (Sigma, St. Louis, MO, the United States) at 37°C and 180 rpm. Bacterial plasmid vectors were transformed into E. coli DH5α or E. coli BL21 (DE3) for protein expression. Pichia pastoris was grown in Yeast Extract–Peptone–Dextrose (YPD) medium at 28°C and 180 rpm. HEK293F cells were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco modified Eagle medium (DMEM) supplemented with 5% fetal bovine serum and streptomycin (50 μg/ml)–penicillin (50 U/ml). The SARS-CoV-2 Spike ectodomain Hexa-pro construction (Table 1) was a gift from Jason McLellan (Addgene # 154754; http://n2t.net/addgene:154754; RRID: addgene_154754) (Hsieh et al., 2020).
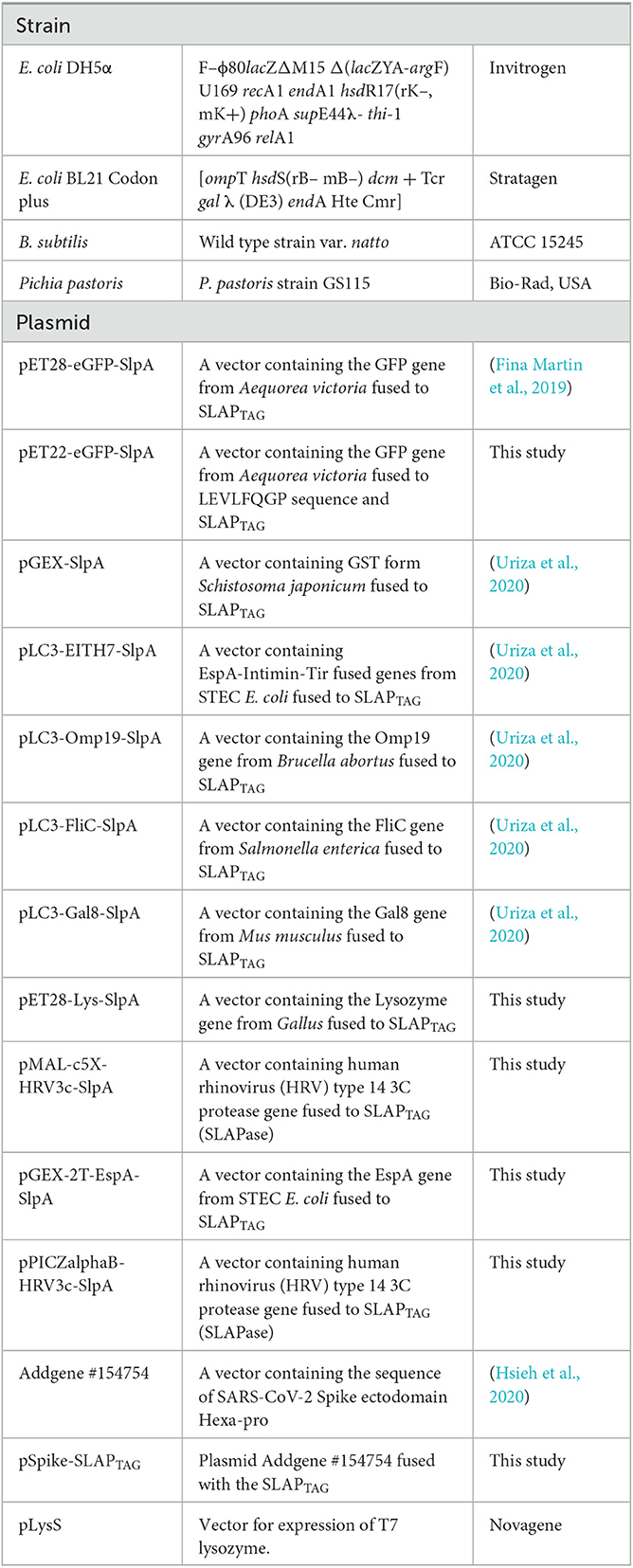
Table 1. Bacterial strains and plasmids used in this study.
Cloning
Lysozyme gene was amplified from the pLysS plasmid (Millipore Sigma, Novagen) with the oligonucleotide primers Fw-lys-NdeI (CCCATATGG CTCGTGTACAGTTTAAACAACGTG) and Rv-lys-SalI (CGGTCGACTCCACGGTCAGAAGTGACCAGTTCG). The PCR product was digested with the restriction enzymes NdeI and SalI and then cloned into the pLC3-EITH7-SlpA vector in the same restriction sites, and the recombinant plasmid was transformed into E. coli DH5α. In addition, the lysozyme gene was subcloned into pET28-e GFP-SlpA at the NdeI and NotI restriction sites and transformed into E. coli BL21 by electroporation.
EspA gene was amplified with the following primers: pRvEspAXbaI (GCTCTAGATTTACCAAGGGATATTGCTG) and pFwEITSalI (ACGCGTCGACGATATGAATGAGGCATCTAAA). After digestion with XbaI and SalI, the gene was cloned into pGEX-SlpA (Table 1), which was previously digested with the same enzymes. The plasmid was introduced by electroporation into E. coli BL21.
The gene encoding the human rhinovirus (HRV) type 14 3C protease fused to SLAPTAG was synthesized and cloned in pMAL-c5X and pPICZ alpha B for protein expression in E. coli and P. pastoris, respectively (Gene Universal, Delaware—USA).
For P. pastoris GS115 transformation, 5 μg of pPICZ alpha B—hrv-3c plasmid was linearized with SacI restriction enzyme and transformed into cells through electroporation using 2 mm gap cuvettes (1,500 V, 125 X, 50 lF). Transformed cells were selected by plating on a YPD medium containing Zeocin 100 ug/ml for resistance selection. Isolated colonies were further grown in test tubes containing YPD broth, and after 24 h, protein expression was induced by adding 1% (v/v) pure methanol every 24 h. The supernatants were sampled after 72 h of induction, and the best producer clones were chosen for further experiments.
The pET28-eGFP-SLAP was digested with BamHI and XhoI restriction enzymes, and the SLAPTAG-containing band was subcloned into SARS-CoV-2 S HexaPro plasmid (Addgene#154754) with the same restriction sites.
Protein expression
E. coli BL21 were grown at 37°C and 180 rpm in liquid LB supplemented with antibiotics until they reached OD600 = 0.6. Then, 0.1 mM IPTG was added, and bacteria were grown for another 20 h at 18°C and 180 rpm. Finally, bacteria were harvested and lysed by sonication. The bacterial lysates were clarified by centrifugation.
For Pichia, pastoris protein expression cells were induced with methanol. In brief, P. pastoris cultures were grown in 50 ml of YPD for 48 h until the dextrose was consumed. Then, methanol pulses (200 ul) were supplied every 24 h for 5 days. Finally, P. pastoris were harvested, and the supernatants were collected.
HEK293 cells were transfected using polyethyleneimine (PEI) for protein expression. In brief, 30,000 cells per well were seeded in 24 well plates and incubated at 37°C, 5% CO2 for 24 h. For transfection, 4 μl of PEI was diluted in 40 μl of DMEM. 500 ng of DNA was added, and the transfection mix was incubated for 20 min at room temperature. Then, the transfection mix was transferred to the cells and incubated for 48 h. Finally, the supernatant was collected to check protein expression.
6xHis-tag purification method
Purification of 6xHis-tagged proteins was carried out according to the manufacturer's protocol. In brief, benchtop columns were equilibrated with the binding buffer (200 mM NaCl, 50 mM Tris-HCl pH 7.5). Columns were loaded with the bacterial lysate. After being washed, columns were eluted in steps with 10-, 50-, 100-, 300-, and 500-mM imidazole in the equilibration buffer. When compared with the SAC, the IMAC batch format was adopted. Thus, Ni-NTA superflow resin (Qiagen) was equilibrated with buffer (200 mM NaCl, 50 mM Tris-HCl pH 7.5) in 1.5 ml tubes. Cleared bacterial lysates were loaded and mixed in an orbital shaker for 1 h at 4°C. The samples were centrifugated, and, after being washed, the resin was eluted with 500-mM imidazole in an equilibration buffer.
Bio-Matrix preparation
B. subtilis natto was grown in 200 mL of Luria Bertani broth at 37°C and 180 rpm for 48 h. Then, the culture was centrifugated, and the bacteria were washed twice with PBS. The culture was resuspended in PBS with 2% glutaraldehyde and incubated overnight with soft agitation. Next, fixed bacteria were washed twice with PBS and stored in 20 % ethanol. 1 mL of BM correspond to DO600 = 30 of B. subtilis.
BM was weighted in a drying scale (KERN MLS-D), and a calibration curve for dry weight vs. optical density was performed.
SLAPTAG purification method
To establish an optimized SAC protocol, BM was equilibrated in an initial binding buffer (50 mM Tris-HCl, pH 7.5). Then, the samples containing SLAP-tagged proteins were incubated with the BM at different times and temperatures. After incubation, the samples were washed three times by centrifugation (8,000 rpm) using the binding buffer. Finally, as shown in the results section, elution was evaluated by incubating at different times and temperatures with different elution buffers. Based on our experimental optimization studies, we have determined that the optimal binding conditions are 200 mM NaCl and 50 mM Tris-HCl at pH 7.5 and a temperature of 4°C for 5 min. For the elution step, we found that using a carbonate-bicarbonate buffer (0.1M) with a pH of 10 and a concentration of 200 mM NaCl for 5 min was optimal.
Once the protocol was set, proteins expressed in Escherichia coli, HEK293, and Pichia pastoris were purified from bacterial lysate or cellular supernatant using the final protocol described here.
Bio-Matrix time stability and reusability
To analyze stability in time, aliquots of BM were frozen at −20°C. At different times, the samples were unfrozen and used for purifying GFP-SLAP following the protocol developed in this study. To analyze reusage, an aliquot of BM was used for purifying GFP-SLAP following the protocol developed in this study. After elution, the BM was washed with two volumes of the elution Buffer (0.1M carbonate-bicarbonate buffer) with a pH of 10 and a concentration of 200 mM NaCl and then two volumes of binding buffer (200 mM NaCl, 50 mM Tris-HCl pH 7.5). The cleaning process was repeated each time after elution.
Protein analysis
Protein samples were dissolved in a cracking buffer and incubated for 5 min at 100°C. Protein electrophoresis was performed at 120 V on 12% SDS-PAGE gel. Gels were stained in a Coomassie Blue solution (20% methanol and 10% acetic acid).
For Western blot analysis, proteins were transferred to a nitrocellulose membrane for 55 min at 15 V using a semi-dry electroblotting transfer unit (Bio-Rad, Hercules, CA, the United States). Membranes were incubated for 1 h with blocking buffer (1% dry skim milk and 0.1% Tween in PBS). Then, membranes were incubated for 1 h with primary antibody diluted in blocking buffer (1/500). After washing with PBS-0.1% Tween, membranes were incubated for 1 h with IRDye fluorophore-labeled secondary antibodies (LI-COR, Lincoln, NE, the United States) diluted in a blocking buffer (1/20,000). Finally, membranes were scanned using the Odyssey Imaging System (LI-COR).
Fluorescence measurements
GFP fluorescence measurements were performed at 485/535 nm excitation–emission wavelength, respectively, using FilterMax F5 Microplate Reader in Black 96 Well Plates (Thermo).
Protein modeling
(H6)-GFP-SLAPTAG structure was predicted by AlphaFold2 (Jumper et al., 2021). ColabFold web interface was employed using standard settings (five models and no templates).
Adsorption isotherm
Adsorption isotherms for (H6)-GFP-SLAPTAG on BM were performed using batch experiments. BM was equilibrated with binding buffer (50 mM Tris-HCl, 200 mM NaCl, and pH 7.6). Purified (H6)-GFP-SLAPTAG protein at 3 mg/ml in binding buffer was used as a stock solution. Different concentrations of protein were incubated with 10 μl of BM. After reaching equilibrium, the samples were centrifugated and GFP fluorescence from the supernatant was measured. Unbound protein in equilibrium with the BM was calculated using GFP fluorescence. Bound protein was estimated by the difference between input and unbound protein. The adsorption isotherm data were then fitted to the Langmuir isotherm equation to calculate the parameters Qmax and KD.
Confocal fluorescence microscopy
The samples were incubated for 30 min in plates treated with poly-L-Lysine (Sigma, St. Louis, MO, the United State). After treatment with PFA (4% in PBS), the samples were washed twice with PBS. Finally, the samples were observed with a confocal laser-scanning microscope Olympus FV1000 using a PlanApo N (60 × 1.42 NA) oil objective.
Cleavage of SLAPTAG with SLAPASE protease
Cleavage buffer recommended for commercial HRV3c protease was prepared: 50 mM Tris-HCl, pH 7.0, 150 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol. GFP-LEVLFQGP-SLAPTAG protein was bound to the BM. After binding, BM was washed with the same buffer thrice at 4°C. SLAPASE was added and incubated at 4°C for 60 min. Percolate containing GFP protein without SLAPTAG was recovered. As a control, the protease and the cut SLAPTAG were eluted with Bio-Matrix elution buffer after cleavage using 2M LiCl.
Synthesis of iron nanoparticles
Iron nanoparticles were synthesized by reverse co-precipitation as described by Nadi et al. (2019). In summary, the precursor was prepared by dissolving 0.89 g of FeCl2.4H2O in 90 mL of water. The sample was incubated in stirring for 15 min and sonicated for 10 min to ensure complete dissolution of the salt. The solution was then poured over a solution of ammonium hydroxide diluted 1:2 with water and stirred for an hour. Finally, the sample was washed repeatedly with deionized water.
Magnetic Bio-Matrix generation
In brief, 5 ml of Bio-Matrix were resuspended in PBS and iron nanoparticles were added at a final concentration of 40 g/L. The sample was mixed with gentle stirring for 30 min. Then, it was washed repeatedly with PBS 1X and preserved in 20% ethanol.
Antibody generation
BALB/c mice were immunized intraperitoneally with 10 μg of the different purified recombinant proteins (GST-SLAPTAG or GFP) using aluminum hydroxide as an adjuvant. Boosters with 5 μg of protein were further performed at 2 and 4 weeks. One week after the last immunization, the mice were bled, and the serum was stored at −20°C for later use.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 software. Statistical significance was analyzed by one-way ANOVA with Bonferroni.
Results
SLAPTAG binds rapidly and efficiently to the Bio-Matrix (BM)
As described in the Methods section, the SLAPTAG (Supplementary Figure 1) was fused in-frame to the carboxy terminus of the green fluorescent protein (GFP) to generate the chimeric protein (H6)-GFP-SLAPTAG adapted here as a SLAPTAG reporter protein. As shown in Figure 1A, the structure of the chimeric protein (H6)-GFP-SLAPTAG was predicted based on the AlphaFold2 method (Jumper et al., 2021). The beta-barrel corresponding to GFP and the two globular domains corresponding to SLAPTAG were predicted with high performance, and as expected, linkers and 6xHis-tag, which are flexible regions, showed low prediction values.
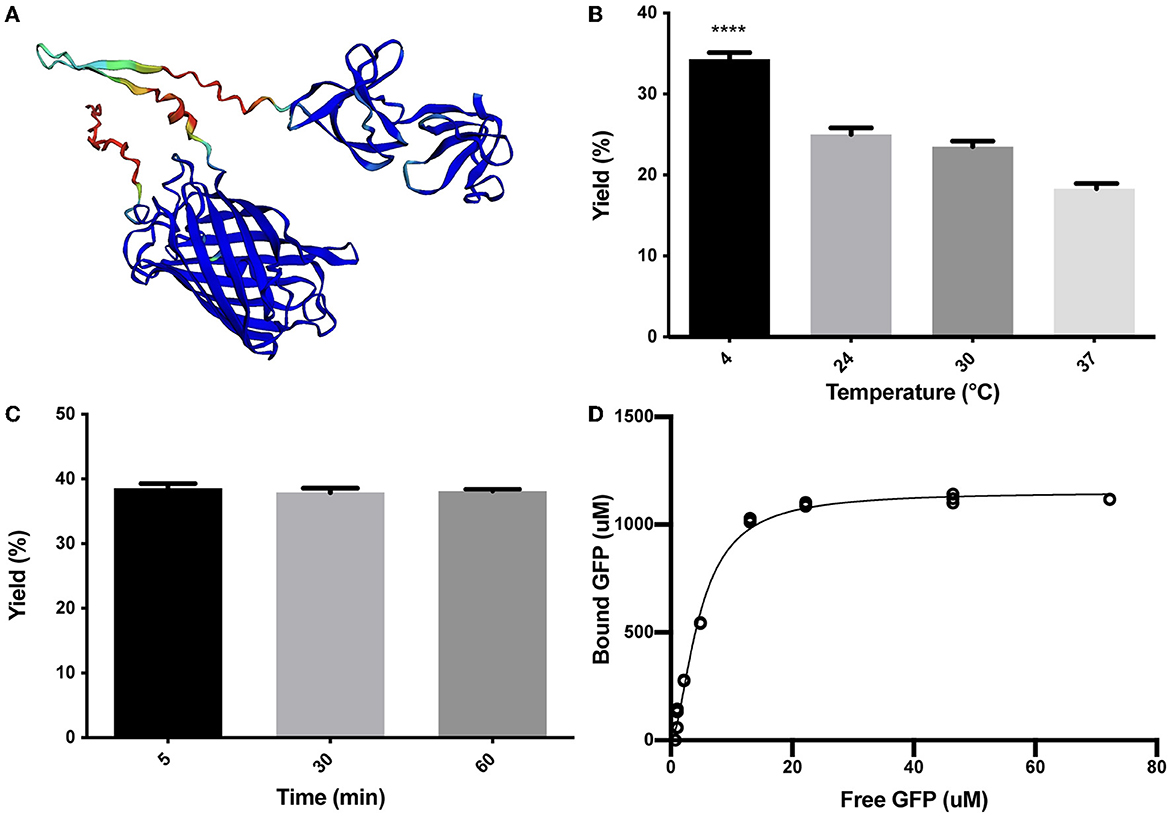
Figure 1. Characterization of (H6)-GFP-SLAPTAG binding to Bio-Matrix. (A) The AlphaFold2 model of (H6)-GFP-SLAPTAG fusion protein. AlphaFold2 predicted structure was automatically colored by the pLDDT confidence measure. High accuracy is colored in blue, while low accuracy is in red. Although SLAPTAG has not been crystallized, structure prediction showed a good performance. (B) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography for different temperatures of the binding process. (C) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography for different times of binding incubation. (D) Adsorption isotherm of (H6)-GFP-SLAPTAG onto Bio-Matrix. Asterisk (****) denotes significant differences using the ANOVA method, Bonferroni test (p < 0.0001).
As a working chromatographic matrix, a culture of Bacillus subtilis natto was processed as described in the Materials and Methods section to generate a bacterial-derived affinity chromatography matrix, named here Bio-Matrix (BM). Interestingly, although B. subtilis has no S-layer, we have demonstrated previously that this bacterium can be externally covered with SLAP-tagged proteins to generate a recombinant S-layer on its bacterial cell wall, a process that we called decoration (Uriza et al., 2020). Decoration of B. subtilis with SLAP-tagged proteins is possible because teichoic acid and lipoteichoic acid (which are the molecules responsible for the SLAPTAG association) have the same chemical composition as Lactobacillus acidophilus. Interestingly, in B. subtilis and L. acidophilus, teichoic acid and lipoteichoic acid are distributed homogeneously on their cell wall.
To characterize the binding properties of the SLAPTAG, the reporter protein (H6)-GFP-SLAPTAG was recombinantly expressed in Escherichia coli, purified by affinity chromatography mediated by its Hisx6 tag (H6), and further incubated with BM under a variety of conditions. Initially, as shown in Figure 1B, the optimal binding temperature of (H6)-GFP-SLAPTAG to the BM was evaluated, finding that the best binding efficiency was reached when the incubation was performed at 4°C. To get insights into the SLAPTAG association dynamics, (H6)-GFP-SLAPTAG was incubated for 5, 30, and 60 min with BM, and as shown in Figure 1C, the maximal binding of (H6)-GFP-SLAPTAG to BM was reached very rapidly (5 min), showing no significant increments in its association at longer time points. These results indicated that the SLAPTAG binds very rapidly and with an apparent high affinity to the BM. To quantify the affinity of the interaction between the SLAPTAG with the BM, an adsorption isotherm was performed to determine the apparent equilibrium dissociation constant as described in the Materials and Methods section. As shown in Figure 1D, the apparent KD was estimated as 4.7 μM. Interestingly, this dissociation constant value was in the same order as other KD described for different microbial S-layer proteins and their respective bacterial cell walls (Garduno et al., 1995; Mader et al., 2004; Li et al., 2009). In addition, the maximum adsorption capacity (Bmax) of BM was estimated as 1.152 mmol of (H6)-GFP-SLAPTAG or 53.9 mg of protein per milliliter of BM or 0.0815 g of SLAPTAG protein/g of wet BM.
Elution of (H6)-GFP-SLAPTAG protein can be performed with different buffers
As it was mentioned, the SLAPTAG contains the protein region responsible for the association of SlpA to the L. acidophilus cell wall. As reported, L. acidophilus SlpA has the natural ability to self-assemble on the bacterial surface to generate a proteinaceous layer named S-layer (Lortal et al., 1992), a highly ordered wall structure that functions as a protective barrier against bacteriophages, resistance to low pH and proteases, and bacterial adhesion. It was characterized that removal of the S-layer can be performed efficiently by the addition of chaotropic agents like LiCl, a compound that disrupts hydrogen bonds leading to a partial denaturation of proteins and the consequent detachment of the S-layer (Lortal et al., 1992). To study (H6)-GFP-SLAPTAG elution from BM, LiCl solution was selected as the positive control. As shown in Figure 2A while PBS has no effect on protein elution, the detachment of BM-associated (H6)-GFP-SLAPTAG was achieved with high efficiency with a 2M LiCl solution at room temperature, in a short lapse of 5 min (Figure 2B). As it was mentioned above, lithium solutions have certain deleterious effects on proteins, which is an undesired effect for protein purification, especially for enzyme purification. Therefore, to elute SLAPTAG-tagged proteins preserving protein structure and therefore their function, different milder alternatives of buffers were explored. As shown in Figure 2C, a series of sugars (monosaccharide and disaccharides) described previously (Fina Martin et al., 2019) were tested for the elution of the SLAP reporter observing only a partial elution efficiency when compared with the LiCl elution control (Figure 2C). Considering that the cationic nature of the SLAPTAG at neutral pH is critical for the interaction of SLAPTAG with its membrane-bound ligand, the teichoic acid, a set of cationic compounds were tested for the elution. As shown in Figure 2C, 0.3M of CTAB was able to elute (H6)-GFP-SLAPTAG with similar efficiency as the LiCl solutions. Although CTAB was very efficient in this step, we found that this compound was difficult to remove downstream from the elution fraction. Then, taking into consideration that SLAPTAG has a theoretical isoelectric point (pI) value of 9.92, consequently, we explored whether the modification of pH can be adapted as an elution method. As shown in Figure 2C, when the pH of the carbonate-bicarbonate buffer was close to the theoretical SLAPTAG isoelectric point (pI), (H6)-GFP-SLAPTAG was efficiently eluted from BM. In addition, after establishing the carbonate-bicarbonate buffer at pH 10 as an elution buffer, we explore whether the addition of NaCl can improve the elution process. As shown in Figure 2C, the addition of 200 mM of NaCl was able to maximize the yield of recovery of (H6)-GFP-SLAPTAG in the eluate. Interestingly, at pH 7.6, the same concentration of NaCl (present in the binding/washing buffer) has no effect on detaching SLAP-tagged proteins from BM.
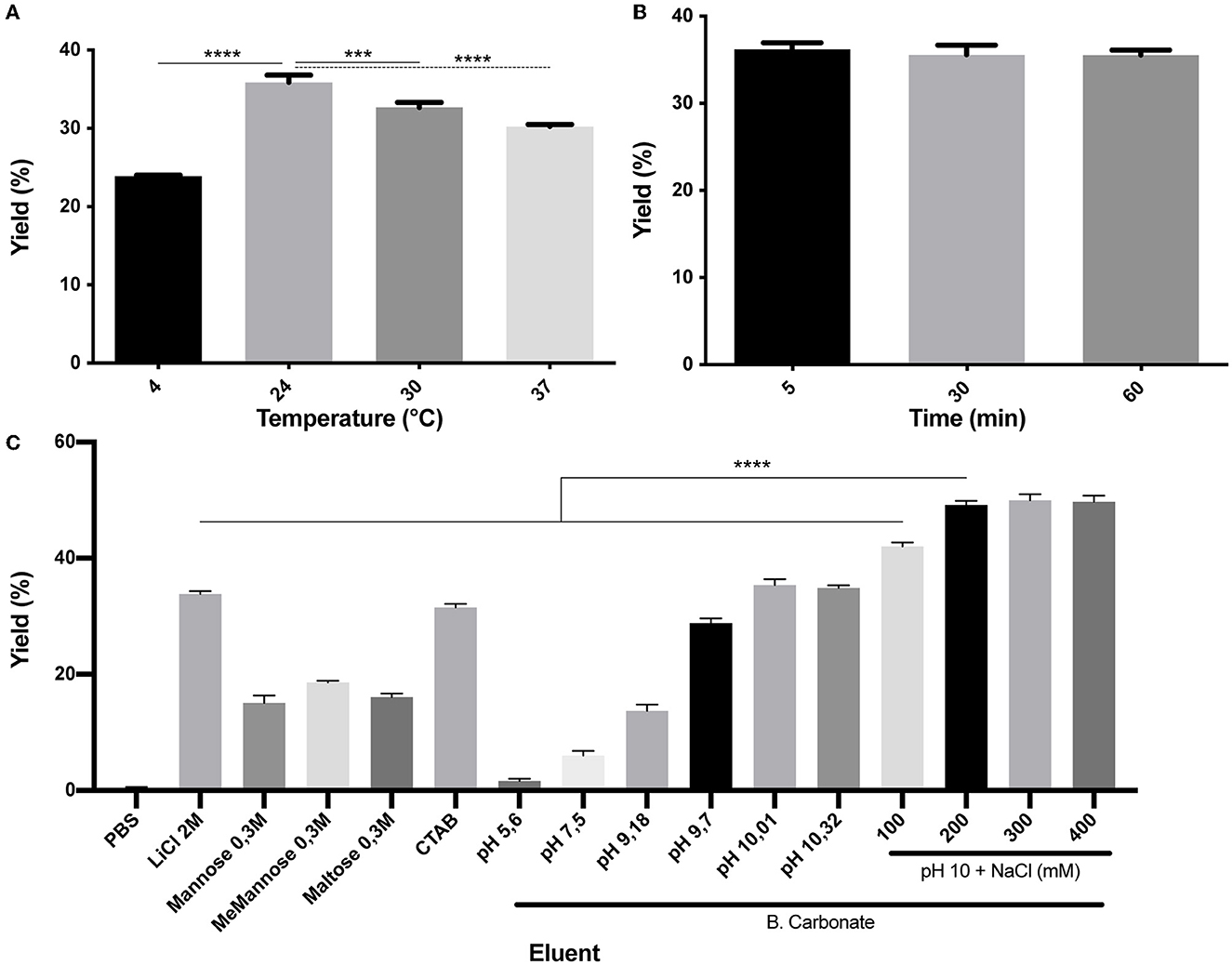
Figure 2. Characterization of (H6)-GFP-SLAPTAG elution. (A) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography for different temperatures of elution. (B) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography for different times of elution incubation. (C) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography system for different eluents. The asterisk denotes significant differences using the ANOVA method, the Bonferroni test (***p = 0,0003; ****p < 0.0001).
The optimized SAC protocol allowed the efficient purification of proteins with similar efficiency to the high-performance Ni2+-charged agarose matrix (IMAC)
With all the experimental information obtained, an optimized SAC protocol for the purification of SLAP-tagged proteins was established as shown in Figure 3A. Remarkably, the entire process of protein purification can be performed in 15 min. To have a direct observation of the purification process of (H6)-GFP-SLAPTAG, the BM was fixed and immobilized on coverslips at different steps of the process of protein purification to be observed by confocal microscopy. As shown in Figure 3B, before the incubation of BM with (H6)-GFP-SLAPTAG, only a red native autofluorescence was observed from fixed Bacillus subtilis natto cells present in BM. Remarkably, after the incubation of BM with (H6)-GFP-SLAPTAG, it was possible to detect the adhesion of the reporter protein to the BM establishing a recombinant fluorescent S-layer that covers completely the bacterial surface (Figure 3C). As expected, after the addition of the elution buffer, the (H6)-GFP-SLAPTAG was completely removed from the bacterial surface (Figure 3D). In addition, the whole process of purification can also be monitored by direct observation under UV light (Figure 3E).
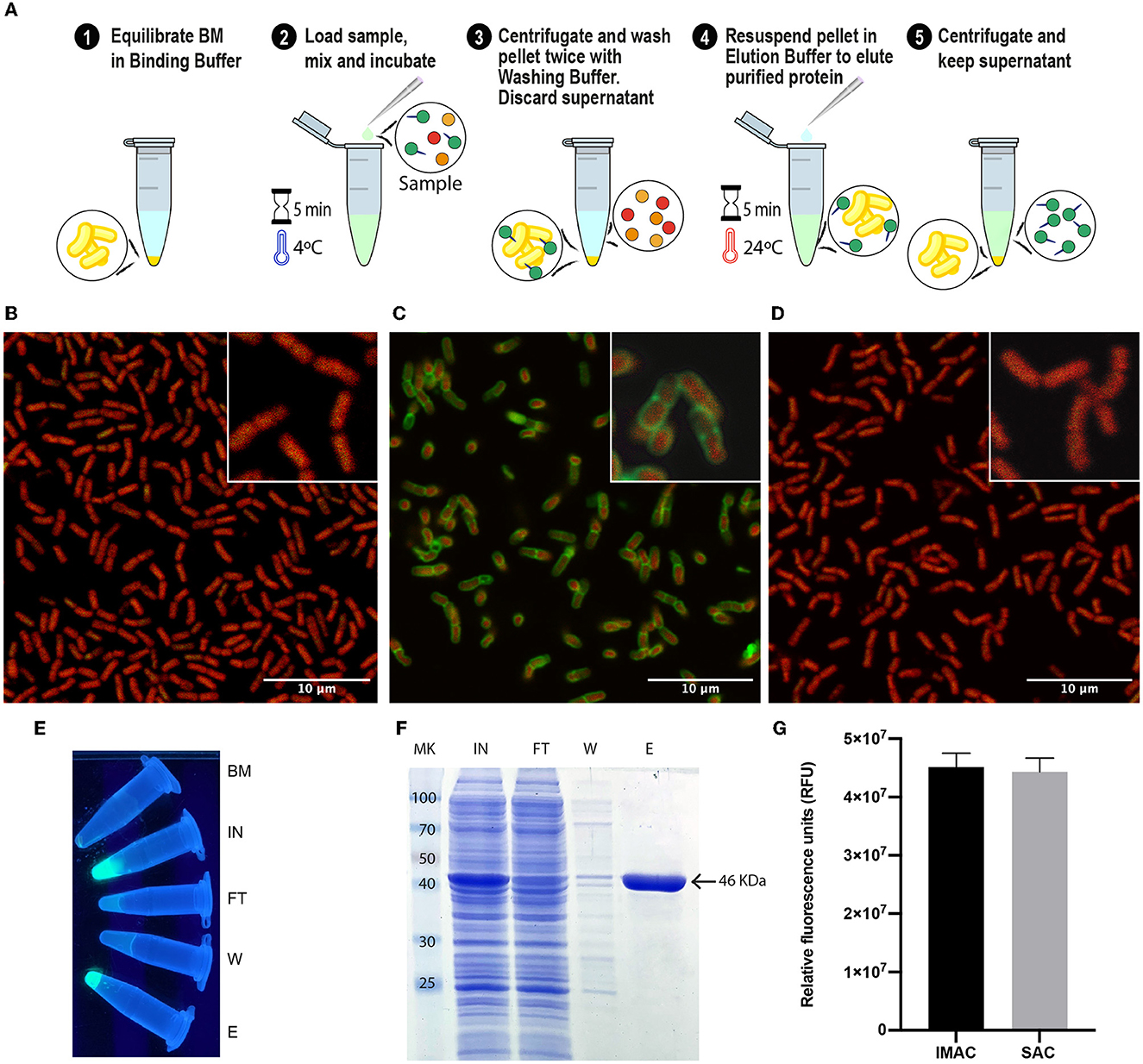
Figure 3. Optimized protocol for (H6)-GFP-SLAPTAG purification using the Bio-Matrix and comparison with the high-performance Ni2+-charged agarose matrix. (A) Graphical description of the SLAPTAG-based affinity chromatography protocol. (B) Confocal microscopy of the Bio-Matrix. Bacillus subtilis red native autofluorescence is observed. (C) Confocal microscopy of (H6)-GFP-SLAPTAG bound to the Bio-Matrix. GFP is visualized on the Bacillus surface. (D) Confocal microscopy of the Bio-Matrix after elution of (H6)-GFP-SLAPTAG. (E) Tubes containing purification fractions seen under UV light. GFP input fluorescence is recovered in the eluate. (F) Coomassie Blue staining analysis of (H6)-GFP-SLAPTAG scaled purification. BM, Bio-Matrix; MK, protein marker (kDa); IN, input; FT, flow-through; W, wash; E, elution. (G) Immobilized metal affinity chromatography (IMAC) and SLAPTAG-based affinity chromatography systems are compared in their capacity to purify (H6)-GFP-SLAPTAG. Relative fluorescence of total (H6)-GFP-SLAPTAG recovered in elution fraction is shown.
Since SAC was efficient in protein purification, we compared this new technique against an established affinity purification protocol. For this, we took advantage of (H6)-GFP-SLAPTAG which also has a His-tag that can be purified by a metal affinity chromatography or IMAC. As described in Materials and Methods, a bacterial lysate (H6)-GFP-SLAPTAG was divided into two fractions, and both protocols were performed accordingly in parallel, confirming that SAC optimized protocol was able to purify (H6)-GFP-SLAPTAG with high efficiency (Supplementary Table 1; Figure 3F) and with a similar yield to the one obtained with the commercial IMAC (Figure 3G).
Proteins of different biological sources, molecular weights, biochemical functions, or expressed by prokaryotic or eukaryotic expression systems can be efficiently purified by the SAC
To evaluate whether SLAPTAG and SAC can be adapted as a universal protein purification system, a set of the selected proteins were fused to the SLAPTAG (Figure 4).
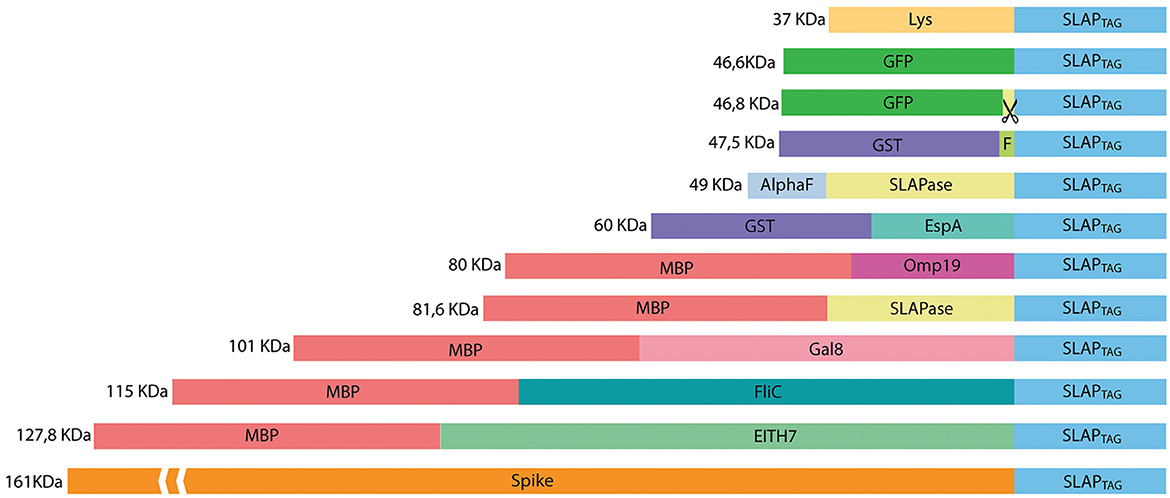
Figure 4. Representation of SLAP-tagged recombinant protein structure. Lys, bacteriophage T4 lysozyme; GFP, Aequorea victoria green fluorescent protein; GST, glutathione-s-transferase; HRV-3c, human Rhinovirus 3C Protease; EspA, E. coli EspA protein; Omp19, B. abortus Omp19 protein; MBP, maltose-binding protein; Gal8, mouse Galectin-8; EITH7, EspA, Intimin and Tir fusion protein from the Shiga toxin-producing E. coli; Spike, SARS-CoV-2 spike protein.
Thus, bacterial proteins such as Salmonella flagellin (Figure 5A), the STEC proteins, EspA (Figure 5B), the chimeric EIT (Figure 5C), the B. abortus Omp19 (Figure 5D), the bacteriophage protein T7 lysozyme (Figure 5E), the human viral proteins Rhinovirus 3C Protease (Figure 5E), the mouse Galectin-8 (Figure 5G), and the commercial molecular tag GST (Figure 5H) were fused in-frame with the SLAPTAG.
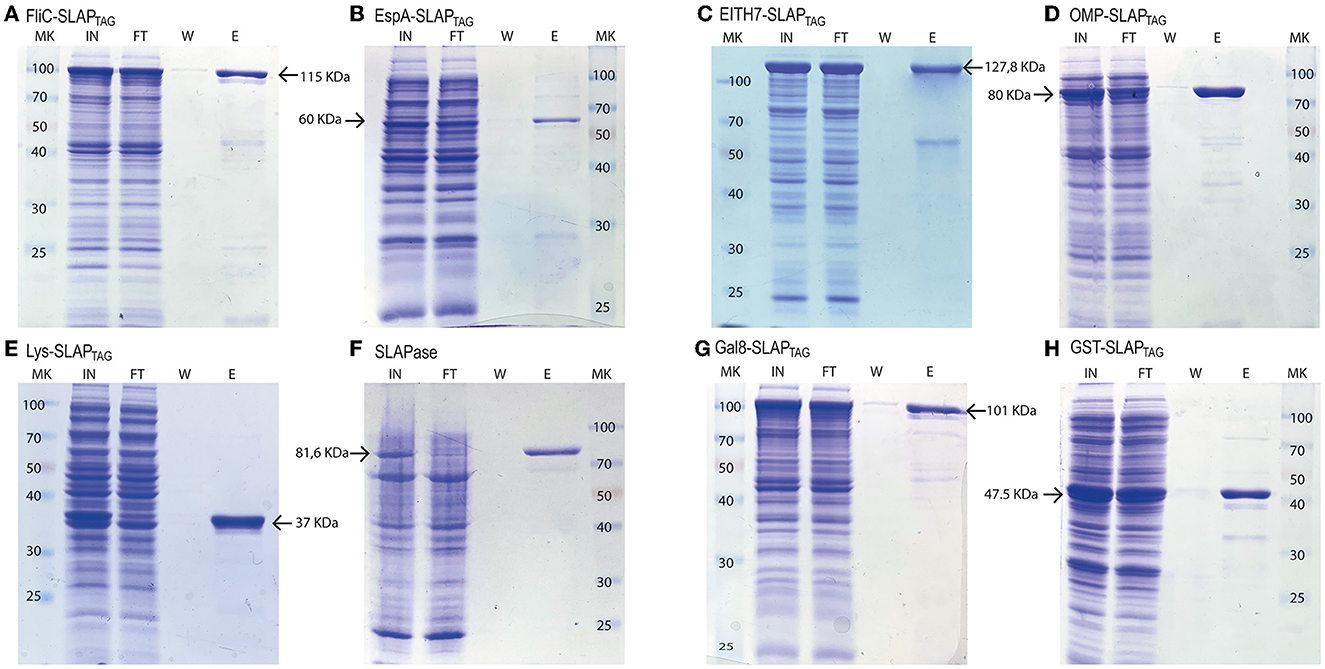
Figure 5. Analysis of different SLAP-tagged protein purifications expressed in E. coli. Coomassie Blue stained SDS-PAGE of purification fractions of SLAPTAG-based affinity chromatography for (A) FliC-SLAPTAG, (B) EspA-SLAPTAG, (C) EITH7-SLAPTAG, (D) Omp19-SLAPTAG, (E) Lys-SLAPTAG, (F) SLAPASE (human rhinovirus 3c fused to SLAPTAG), (G) Gal8-SLAPTAG, and (H) GST-SLAPTAG. MK, protein marker (kDa); IN, input; FT, flow-through; W, wash; E, elution.
As shown in Figure 5, all the selected proteins were purified efficiently with SAC. In addition to the bacterial expression system, different protein expressions systems (yeast and mammalian cells) were also evaluated (Figure 6). As shown in Figure 6A, the viral HRV-3C protease fused to the SLAPTAG was expressed and purified from Pichia pastoris supernatant. In addition, the SARS-CoV-2 SPIKE fused to the SLAPTAG was purified from the supernatant of transfected HEK293 (Figure 6B). These results confirmed that the SLAPTAG can be widely adopted for affinity chromatography purification. It is interesting to note that the protein SPIKE-SLAPTAG was efficiently purified from a sample with a high protein content, such as a culture medium containing FBS, using SAC (Supplementary Figure 2). These findings indicate that SAC can be potentially employed as a first step to enrich a protein of interest directly from an unclarified feedstock derived from a host cell culture medium in downstream protein production processes.
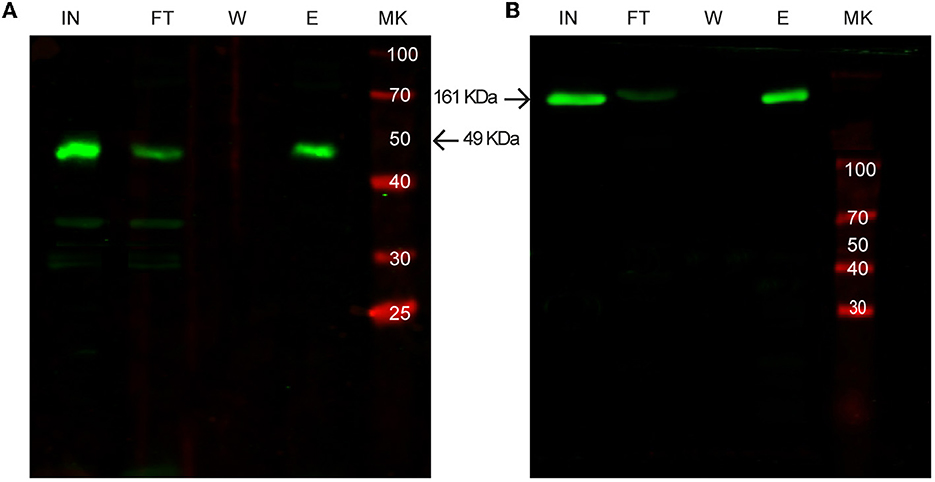
Figure 6. Western blot analysis of purification of SLAP-tagged proteins using different expression systems. (A) Purification of SLAPASE (human rhinovirus 3c fused to SLAPTAG) protease expressed in Pichia pastoris. (B) Purification of SARS-CoV-2 Spike-SLAPTAG protein expressed in HEK293 cells. MK, protein marker (kDa); IN, input; FT, flow-through; W, wash; E, elution.
BM is a reusable chromatography matrix with long-term stability
As described in the Materials and Methods section, a batch of BM was produced, and several aliquots were frozen at −20°C to study time stability. As shown in Figure 7A, at different times, a few aliquots were unfrozen and tested for protein purification of the reporter protein (H6)-GFP-SLAPTAG being the BM stable for more than a year that was tested (14 months). In addition, BM reusability capacity was evaluated determining that the matrix can be reused five times with no modification of the protein purification yield (Figure 7B).
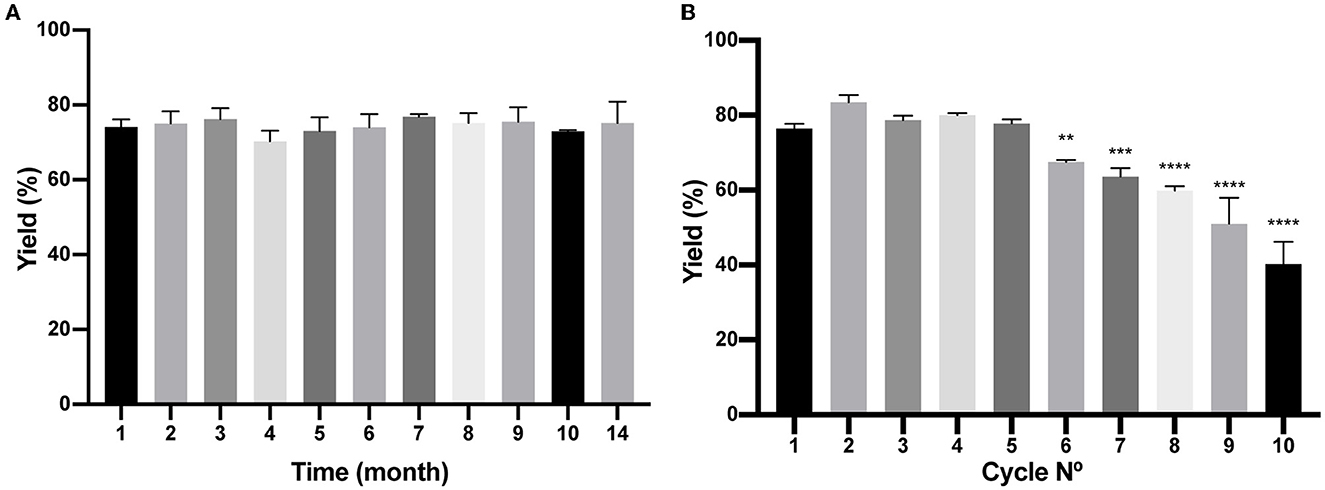
Figure 7. Analysis of Bio-Matrix stability in time and reuse. (A) The figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography at different times when it is conserved at−20°C. (B) Figure compares (H6)-GFP-SLAPTAG purification yield of SLAPTAG-based affinity chromatography for different cycles of reuse. The asterisk denotes a significant difference using the ANOVA method, Bonferroni test (**p = 0,0059; ***p = 0,0002; ****p < 0.0001)
Use of the Human Rhinovirus 3C protease fused to the SLAPTAG (SLAPASE) to release the SLAP-tagged proteins from BM
As shown in Figures 4, 5, the gene sequence of the viral protease HRV-3C was fused to the SLAPTAG (named SLAPASE), recombinantly expressed, and purified (Figures 8B, D, lane SLAPase) to evaluate its activity. A reporter protein for SLAPASE activity was generated (GFP-LEVLFQGP-SLAPTAG) (Table 1), and a protocol for tag removal by SLAPase was set as described in the Methods section. As shown in Figure 8A, the GFP-LEVLFQGP-SLAPTAG was expressed (Figures 8B, C, lane Input or IN) and mixed with the BM for 5 min allowing the binding process. After binding, a purified SLAPASE was added to the mix and incubated for 60 min at 4°C, releasing a tag-less GFP (Figures 8B, C, E, lanes flow-through or FT) by proteolysis. Cut SLAPTAG is not observed in the flow-through indicating that was retained by the BM along with the SLAPase. To confirm these steps, post-proteolysis BM was incubated with LiCl solution to recover any residual bound SLAP-tagged protein. As shown in Figures 8B, D (lane Elution or E), the LiCl solution released the SLAPase and the cut SLAPTAG.
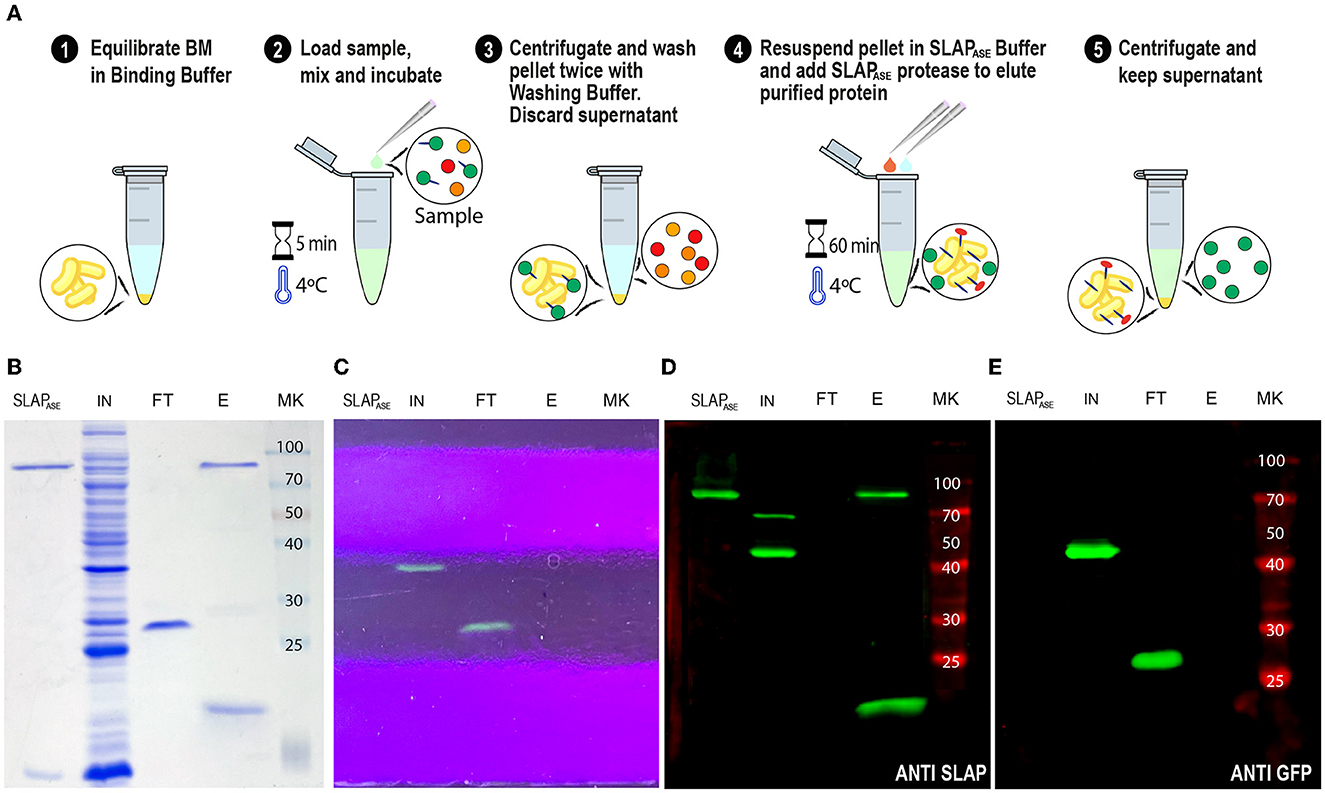
Figure 8. SLAPTAG removal. (A) Graphical description of the protocol to remove the SLAPTAG using SLAPTAG-based affinity chromatography. SDS-PAGE of purification fractions of SLAPTAG removal protocol stained with Coomassie Blue solution (B) or under UV light (C). Western Blot of purification fractions of SLAPTAG removal protocol revealed with anti-SLAPTAG (D) or anti-GFP (E) antibodies. SLAPASE, purified protease; IN, input; FT, flow-through; E, elution; MK, protein marker (KDa).
Adaptation of SAC to magnetic affinity chromatography
Since we confirmed the efficiency and universality of SAC for the purification of recombinant proteins, we explored a magnetic affinity chromatography alternative for SAC (Figure 9A). As shown in Figures 9B, C, and Supplementary Video 1, the (H6)-GFP-SLAPTAG was purified in a few and easy steps. As shown in the SDS-PAGE (Figure 9B), the adapted SAC protocol for magnetic chromatography (BMmag) was able to purify the reporter protein (H6)-GFP-SLAPTAG very efficiently (Figure 9B, lane Elution or E). As shown in Supplementary Table 2, BMmag is identical to BM in terms of purification performance. These results indicate that the binding capacity of BM is not modified by its association with magnetic particles.
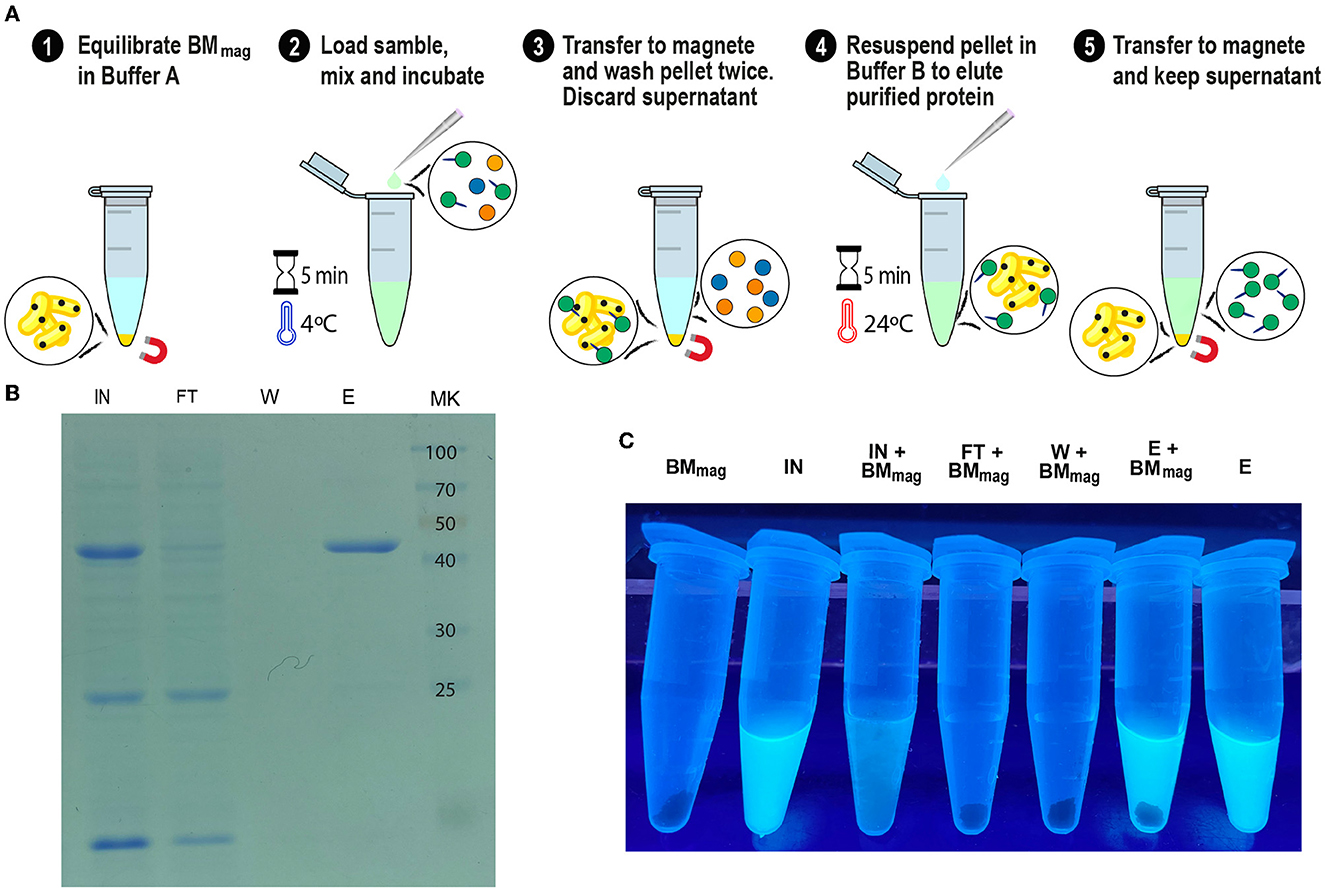
Figure 9. GFP-SLAPTAG purification using Magnetic Bio-Matrix. (A) Graphical description of the purification protocol for magnetic Bio-Matrix. (B) Photograph of 1.5 ml tubes containing fractions of GFP-SLAPTAG magnetic purification under UV light. BMmag, magnetic Bio-Matrix; IN, input; FT, flow-through; W, wash; E, elution. (C) Tubes containing magnetic purification fractions are seen under UV light. GFP input fluorescence is recovered in the eluate.
Interestingly, this protocol can be adapted to commercial devices that use magnetic force for protein or DNA purification (like King Fisher Flex from Thermo Fisher). In Figure 9C, the entire process of purification was monitored by direct observation under UV light (Supplementary Video 1). Noteworthy, the binding of the BMmag to GFP quenched the fluorescence of this protein, an effect that was described for transition metal binding to GFP or binding of iron cations to fluorescent proteins (Figure 9C, tube IN+BMmag) (Richmond et al., 2000; Kim et al., 2021).
Discussion
The B. subtilis-derived matrix, named here as Bio-Matrix (BM), showed a high performance in its binding and elution capacities, combined with good purification parameters for our reporter protein (H6)-GFP-SLAPTAG. The results presented here show that the SAC protocol can be potentially adapted as a universal tool for recombinant protein purification. Thus, proteins from different origins, with different molecular weights, or produced by different recombinant expression systems (bacteria, yeast, or cells) can be efficiently purified by SAC. In addition to its universal application, we demonstrate here that SAC was able to achieve a protein yield similar to those obtained by commercial affinity chromatography systems such as the immobilized metal affinity chromatography or IMAC. As reported, in protein production bioprocess, chromatography is the most expensive step (Wong et al., 2018). Of interest, the SAC protocol only uses simple and low-cost reagents, consequently presenting an economic advantage in protein purification over current commercial systems. In addition, since the Bio-Matrix is “grown” instead of being chemically synthesized, the production of larger quantities of chromatography matrix can be achieved easily by simply scaling up the volume of the bioreactor. In addition, small research laboratories can easily produce an in-house BM version by the protocol provided here.
One critical step in the generation of the affinity matrix is the immobilization of the affinity ligand to the chromatography matrix. Initially, at the beginning of the affinity chromatography development, the immobilization of affinity ligands was performed by covalent modification using diazo coupling (Rodriguez et al., 2020). This procedure allows to immobilize different haptens or certain proteins to isolate antibodies. A second breakthrough in affinity chromatography was the development of the cyanogen bromide (CNBr) immobilization method which allows the easy cross-link of proteins or peptides to the activated agarose matrix (Rodriguez et al., 2020). These two major advances were combined in 1969 by Cuatreacasas et al. where the term affinity chromatography was used for the first time (Cuatrecasas et al., 1968).
In contrast, in our approach, the affinity ligands (LTA and teichoic acid) are naturally integrated into the surface of the BM (Lortal et al., 1992), and consequently, no chemical reactions to cross-link the affinity ligands are required. Remarkably, no toxic chemicals or solvents are required for BM production.
The protein purification procedures described in this study were conducted in a batch format, utilizing either centrifugation or magnetic separation for the various steps of the process. Although SAC in a batch format proved to be a very efficient and straightforward method, the adaptation of SAC to the column format was more cumbersome because BM tended to clog the column, resulting in slow flow rates. To address this limitation, we explored the immobilization of BM on various supports that allow high-flow chromatography, such as polyurethane sponges, polystyrene beds, glass beads, and cellulose, but with limited success (data not shown). Additionally, we investigated different potential chromatography materials that can directly bind SLAPTAG-tagged proteins, which could potentially be useful for SAC in the column format. Our preliminary findings indicate that chitosan is a promising candidate (Supplementary Figure 3).
One outstanding aspect of SAC was its ability to efficiently capture SLAP-tagged proteins from a high-concentration protein solution, such as an unclarified crude sample (e.g., HEK295 cell supernatant) (Supplementary Figure 2). Importantly, this characteristic allows us to propose adapting SAC for expanded-bed adsorption (EBA) chromatography (Chase, 1994; Hjorth et al., 1998). EBA has been demonstrated to be a useful method, particularly for protein capture in a continuous protein purification process from unclarified feedstocks in the recovery of enzymes and therapeutic proteins from a variety of expression hosts, without the need for extensive clarification steps (Schneider et al., 2022). Interestingly, a magnetic version of EBA (EBA/MSFBs) has been explored (Tong and Sun, 2003). Magnetically susceptible chromatography supports are forced to low back-mixing by applying a weak, external magnetic field that oriented the magnetic particles axially or transversely relative to the flow (Schneider et al., 2022).
As shown here, BM was able to efficiently capture SLAP-tagged proteins directly from different feeds such as bacterial lysates, Pichia pastoris supernatant, or HEK293 cell supernatant in a single step (Figures 3, 5, 6; Supplementary Figure 2). In addition, a magnetically susceptible Bio-Matrix (BMmag) was generated that showed the same purification properties than BM (Supplementary Tables 1, 2). Both results made SAC an interesting technique for a potential EBA or EBA/MSFBs adaptation.
In the last decade, the introduction of single-use technologies has enlightened the potential for reduced regulatory and operational costs associated with chromatography. The researchers point to its potential for simpler operation, shorter processing times, and decreased buffer consumption, leading to better economics (Hester et al., 2021). In addition, the lack of need for cleaning over repeat-use cycles significantly reduces costs. SAC could be potentially adapted as a single-use alternative chromatography for some industries, with the benefit of being more eco-friendly than those available on the market, as it is a biologically based and biodegradable matrix.
SAC proved to successfully adapt to protease tag removal. Although new technologies are being developed for tag removal (e.g., inteins), the enzymatic cleavage of the tag is still preferred as it is the most controlled process, with no premature cleaving and the best yields are obtained (Pina et al., 2014). Moreover, new technologies might be compatible with SAC.
Therefore, we propose SLAPTAG affinity chromatography for protein purification in industries with permissive regulations. SAC can be adapted as an in-house protein purification system, available for any laboratory around the globe, to produce pure recombinant proteins for research, diagnosis, and the food industry. Although so far regulatory issues might preclude the use of the SAC for proteins used as biotherapeutics, new efforts have been performed to develop a new version of matrix chromatography suitable for more stringent industrial or clinical purposes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The experimental procedure of this study (Permit Number CICUAE UNSAM 15/2018) was approved by the Committee on the Ethics of Animal Experiments of the Universidad Nacional de San Martín (UNSAM), under the recommendations for animal experimentation (Helsinki Declaration and its amendments, Amsterdam Protocol of Welfare and Animal Protection and National Institutes of Health, USA NIH, Guidelines: Guide for the Care and Use of Laboratory Animals).
Author contributions
EM performed most of experimental work and also generate figures and contributed to discussion of manuscript. PU performed molecular cloning of multiples proteins used. GE perform experiments regarding mammalian cells proteins expression. MP perform microscopy results. CD perform Pichia pastoris experiments. MR contribute in the manuscript writing and results discussion of the manuscript. GB is the P.I. and director of the project, contributing to experiment design, result discussion, and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica, Buenos Aires, Argentina, PICT-2018-0778, PICT-2021-CAT-I-00049, and CONICET (PUE-0086). EM, PU, and CD are doctoral fellows from CONICET. MP is a postdoctoral fellow from the Agencia Nacional de Promoción Científica y Tecnológica. MR and GB are members of the Research Career of CONICET.
Acknowledgments
We would like to thank Dr. Juan E. Ugalde for his careful and critical reading of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1210898/full#supplementary-material
References
Baldo, B. A. (2015). Chimeric fusion proteins used for therapy: indications, mechanisms, and safety. Drug Saf. 38, 455–479. doi: 10.1007/s40264-015-0285-9
Buxbaum, E. (2015). “Enzymes are biocatalysts,” in Fundamentals of Protein Structure and Function (Cham: Springer). doi: 10.1007/978-3-319-19920-7_4
Chase, H. A. (1994). Purification of proteins by adsorption chromatography in expanded beds. Trends Biotechnol. 12, 296–303. doi: 10.1016/0167-7799(94)90046-9
Cuatrecasas, P., Wilchek, M., and Anfinsen, C. B. (1968). Selective enzyme purification by affinity chromatography. Proc. Natl. Acad. Sci. U. S. A. 61, 636–643. doi: 10.1073/pnas.61.2.636
Fina Martin, J., Palomino, M. M., Cutine, A. M., Modenutti, C. P., Fernández Do Porto, D. A., Allievi, M. C., et al. (2019). Exploring lectin-like activity of the S-layer protein of Lactobacillus acidophilus ATCC 4356. Appl. Microbiol. Biotechnol. 103, 4839–4857. doi: 10.1007/s00253-019-09795-y
Freitag, R., and Horváth, C. (1996). Chromatography in the downstream processing of biotechnological products. Adv. Biochem. Eng. Biotechnol. 53, 17–59. doi: 10.1007/BFb0102324
Garduno, R. A., Phipps, B. M., and Kay, W. W. (1995). Physical and functional S-layer reconstitution in Aeromonas salmonicida. J. Bacteriol. 177, 2684–2694. doi: 10.1128/jb.177.10.2684-2694.1995
Hais, I. M. (1986). Biospecific sorption, Prague, 1910: Emil Starkenstein (1884-1942). J. Chromatogr. B Biomed. Sci. Appl. 376, 5–9. doi: 10.1016/S0378-4347(00)80820-8
Hester, J. F., Lu, X., Calhoun, J. D., Hochstein, R. A., and Olson, E. J. (2021). Orthogonal pre-use and post-use efficiency testing for single-use anion exchange chromatography. J. Chromatogr. A 1654, 462445. doi: 10.1016/j.chroma.2021.462445
Hjorth, R., Leijon, P., Frej, A. -K. B., and Jägersten, C. (1998). “Expanded bed adsorption chromatography,” in Bioseparation and Bioprocessing, ed G. Subramanian. doi: 10.1002/9783527619641.ch9
Hsieh, C.-L., Goldsmith, J. A., Schaub, J. M., DiVenere, A. M., Kuo, H.-C., Javanmardi, K., et al. (2020). Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505. doi: 10.1126/science.abd0826
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583. doi: 10.1038/s41586-021-03819-2
Kim, I. J., Xu, Y., and Nam, K. H. (2021). Spectroscopic analysis of Fe Ion-induced fluorescence quenching of the green fluorescent protein ZsGreen. J. Fluoresc. 31, 307–314. doi: 10.1007/s10895-020-02656-2
Kuntz, I. D., Chen, K., Sharp, K. A., and Kollman, P. A. (1999). The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 96, 9997–10002. doi: 10.1073/pnas.96.18.9997
Li, J., Hu, X., Yan, J., and Yuan, Z. (2009). Species-specific cell wall binding affinity of the S-layer proteins of mosquitocidal bacterium Bacillus sphaericus C3-41. Appl. Environ. Microbiol. 75, 3891–3895. doi: 10.1128/AEM.00356-09
Lortal, S., Van Heijenoort, J., Gruber, K., Sleytr, U. B., Lortal,' Jean Van Heijenoort, S., Gruber3, K., et al (1992). S-layer of Lactobacillus Helveticus atcc 12046: Isolation, Chemical Characterization and Re-formation After Extraction With Lithium Chloride. Available online at: https://hal.inrae.fr/hal-02713428 (accessed April 2, 2023).
Mader, C., Huber, C., Moll, D., Sleytr, U. B., and Sára, M. (2004). Interaction of the crystalline bacterial cell surface layer protein SbsB and the secondary cell wall polymer of geobacillus stearothermophilus PV72 assessed by real-time surface plasmon resonance biosensor technology. J. Bacteriol. 186, 1758–1768. doi: 10.1128/JB.186.6.1758-1768.2004
Mouratou, B., Béhar, G., and Pecorari, F. (2015). Artificial affinity proteins as ligands of immunoglobulins. Biomolecules 5, 60–75. doi: 10.3390/biom5010060
Nadi, A., Boyer, D., Charbonnel, N., Boukhriss, A., Forestier, C., and Gmouh, S. (2019). Immobilisation of bacteria onto magnetic nanoparticles for the decolorisation and degradation of azo dyes. IET Nanobiotechnol. 13, 144–149. doi: 10.1049/iet-nbt.2018.5026
Pina, A. S., Batalha, Í. L., and Roque, A. C. A. (2014). Affinity tags in protein purification and peptide enrichment: an overview. Methods Mol. Biol. 1129, 147–168. doi: 10.1007/978-1-62703-977-2_14
Richmond, T. A., Takahashi, T. T., Shimkhada, R., and Bernsdorf, J. (2000). Engineered metal binding sites on green fluorescence protein. Biochem. Biophys. Res. Commun. 268, 462–465. doi: 10.1006/bbrc.1999.1244
Rodriguez, E. L., Poddar, S., Iftekhar, S., Suh, K., Woolfork, A. G., Ovbude, S., et al. (2020). Affinity chromatography: a review of trends and developments over the past 50 years. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1157. doi: 10.1016/j.jchromb.2020.122332
Schales, O. (1942). Preparation and properties of renin. J. Am. Chem. Soc. 64, 561–564. doi: 10.1021/ja01255a028
Schneider, A., Herlevi, L. M., Guo, Y., and Fernandez Lahore, H. M. (2022). Perspectives on adsorption technology as an effective strategy for continuous downstream bioprocessing. J. Chem. Technol. Biotechnol. 97, 2305–2316. doi: 10.1002/jctb.6923
Tong, X. D., and Sun, Y. (2003). Application of magnetic agarose support in liquid magnetically stabilized fluidized bed for protein adsorption. Biotechnol. Prog. 19, 1721–1727. doi: 10.1021/bp030028p
Uriza, P. J., Trautman, C., Palomino, M. M., Fina Martin, J., Ruzal, S. M., Roset, M. S., et al. (2020). Development of an antigen delivery platform using lactobacillus acidophilus decorated with heterologous proteins: a sheep in wolf's clothing story. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.509380
Wen, F., Rubin-Pitel, S. B., and Zhao, H. (2009). “Engineering of therapeutic proteins,” in Protein Engineering and Design, eds S. J. Park and J. R. Cochran (Boca Raton: CRC Press), 153–177.
Wong, F. W. F., Ariff, A. B., and Stuckey, D. C. (2018). Downstream protein separation by surfactant precipitation: a review. Crit. Rev. Biotechnol. 38, 31–46. doi: 10.1080/07388551.2017.1312266
Wood, D. W. (2014). New trends and affinity tag designs for recombinant protein purification. Curr. Opin. Struct. Biol. 26, 54–61. doi: 10.1016/j.sbi.2014.04.006
Keywords: affinity chromatography, S-layer proteins, Bacillus subtilis natto, SLAPTAG, applied microbiology and biotechnology
Citation: Muruaga EJ, Uriza PJ, Eckert GAK, Pepe MV, Duarte CM, Roset MS and Briones G (2023) Adaptation of the binding domain of Lactobacillus acidophilus S-layer protein as a molecular tag for affinity chromatography development. Front. Microbiol. 14:1210898. doi: 10.3389/fmicb.2023.1210898
Received: 23 April 2023; Accepted: 19 May 2023;
Published: 13 June 2023.
Edited by:
Randhir Makkar, Guild BioSciences, United StatesReviewed by:
David W. Wood, The Ohio State University, United StatesHieu Tran-Van, Vietnam National University, Ho Chi Minh City, Vietnam
Copyright © 2023 Muruaga, Uriza, Eckert, Pepe, Duarte, Roset and Briones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mara S. Roset, bXJvc2V0QGlpYi51bnNhbS5lZHUuYXI=; Gabriel Briones, Z2JyaW9uZXNAaWliLnVuc2FtLmVkdS5hcg==