 Davide Mangioni1,2†
Davide Mangioni1,2† Valeria Fox3†
Valeria Fox3† Paola Saltini1,2*
Paola Saltini1,2* Andrea Lombardi1,2
Andrea Lombardi1,2 Linda Bussini4,5Francesco Carella4,5Lisa Cariani6Agnese Comelli1
Linda Bussini4,5Francesco Carella4,5Lisa Cariani6Agnese Comelli1 Caterina Matinato6Antonio Muscatello1Antonio Teri6Leonardo Terranova7
Caterina Matinato6Antonio Muscatello1Antonio Teri6Leonardo Terranova7 Valeria Cento5,8
Valeria Cento5,8 Sara Carloni5,9
Sara Carloni5,9 Michele Bartoletti4,5
Michele Bartoletti4,5 Claudia Alteri3
Claudia Alteri3 Alessandra Bandera1,2
Alessandra Bandera1,2- 1Infectious Diseases Unit, Foundation Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Department of Pathophysiology and Transplantation, University of Milano, Milan, Italy
- 3Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy
- 4Infectious Disease Unit, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Humanitas Research Hospital, Milan, Italy
- 5Department of Biomedical Sciences, Humanitas University, Milan, Italy
- 6Microbiology Laboratory, Clinical Pathology, Foundation Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
- 7Respiratory Unit and Adult Cystic Fibrosis Center, Department of Internal Medicine, Foundation Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Cà Granda Ospedale Maggiore Policlinico, Milan, Italy
- 8Microbiology and Virology, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Humanitas Research Hospital, Milan, Italy
- 9Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Humanitas Research Hospital, Rozzano, Italy
Background: Group A Streptococcus (GAS) causes multiple clinical manifestations, including invasive (iGAS) or even life-threatening (severe-iGAS) infections. After the drop in cases during COVID-19 pandemic, in 2022 a sharp increase of GAS was reported globally.
Methods: GAS strains collected in 09/2022–03/2023 in two university hospitals in Milan, Italy were retrospectively analyzed. Clinical/epidemiological data were combined with whole-genome sequencing to: (i) define resistome/virulome, (ii) identify putative transmission chains, (iii) explore associations between emm-types and clinical severity.
Results: Twenty-eight isolates were available, 19/28 (67.9%) from adults and 9/28 (32.1%) from pediatric population. The criteria for iGAS were met by 19/28 cases (67.9%), of which 11/19 (39.3%) met the further criteria for severe-iGAS. Pediatric cases were mainly non-invasive infections (8/9, 88.9%), adult cases were iGAS and severe-iGAS in 18/19 (94.7%) and 10/19 (52.6%), respectively. Thirteen emm-types were detected, the most prevalent being emm1 and emm12 (6/28 strains each, 21.4%). Single nucleotide polymorphism (SNP) analysis of emm1.0 and emm12.0 strains revealed pairwise SNP distance always >10, inconsistent with unique transmission chains. Emm12.0-type, found to almost exclusively carry virulence factors speH and speI, was mainly detected in children and in no-iGAS infections (55.6 vs. 5.3%, p = 0.007 and 66.7 vs. 0.0%, p < 0.001, respectively), while emm1.0-type was mainly detected in severe-iGAS (0.0 vs. 45.5%, p = 0.045).
Conclusions: This study showed that multiple emm-types contributed to a 2022/2023 GAS infection increase in two hospitals in Milan, with no evidence of direct transmission chains. Specific emm-types could be associated with disease severity or invasiveness. Overall, these results support the integration of classical epidemiological studies with genomic investigation to appropriately manage severe infections and improve surveillance.
Introduction
Streptococcus pyogenes (Group A Streptococcus, GAS) is a human-specific Gram-positive coccus responsible for a broad spectrum of diseases differing in clinical presentation and severity (Brouwer et al., 2023). Primary infection sites are usually upper airways and skin, where non-invasive GAS infections occur and from where the pathogen can be transmitted to a new host or can disseminate causing invasive disease (iGAS) such as empyema (infection of the pleural cavity), streptococcal toxic shock syndrome (STSS) or necrotising fasciitis (Efstratiou and Lamagni, 2022; Stevens and Bryant, 2022a,b; GOV.UK, 2023). iGAS infections can rapidly progress and often require prompt surgical treatment to obtain rapid and adequate source control (Stevens et al., 2014; Johnson and LaRock, 2021). They are also characterized by high morbidity and mortality rates, with ~8–23% of deaths within 7 days from infection (Walker et al., 2014). Incidence of GAS diseases varies with season and geographical location. Yet, while outbreaks of non-invasive infections are fairly common in occurence, iGAS are fortunately rare conditions (Efstratiou and Lamagni, 2022; Brouwer et al., 2023).
The mechanisms of GAS infection are complex and multifactorial, related to a large number of virulence determinants. A major virulence factor is represented by the M-protein, a surface protein which forms fibrils on the surface of the bacteria (Ghosh, 2011), encoded by the emm gene and basis of the most widely used method of epidemiological genotyping (Beall et al., 1996; Facklam et al., 1999). Certain emm types are linked to a higher risk of developing worse clinical manifestations, such as emm1, which has been related to iGAS cases including necrotizing fasciitis and STSS (Friães et al., 2012). Additional important virulence factors include T-cell superantigens, which are able to cross-link the major histocompatibility complex (MHC) class II with the T-cell receptor (TCR) on antigen presenting cells inducing extensive T-cell activation and proliferation contributing to pathologies such as streptococcal toxic shock syndrome (Proft and Fraser, 2022). GAS carry various superantigens, some found on the chromosome, and some associated with prophages (McShan and Nguyen, 2022).
In late 2022, several countries of the European Region and the United Kingdom reported a marked increase of scarlet fever and iGAS infections, mostly affecting children under 10 years of age (ECDC, 2022; GOV.UK, 2022; WHO, 2022). This was presumed to be caused by an early start of the GAS infection season together with lowered immunity following 2 years of strict social distancing and reduced GAS infections during COVID-19 pandemic (de Gier et al., 2019, 2023; GOV.UK, 2022; Lassoued et al., 2023). This surge in infections where investigated was found to be caused by GAS of multiple emm-types, and in general was not dominated by the emergence and expansion of a single clone. This sharp increase of GAS infections seems not determined by specific new emm-type or obvious expansion of a single clone (de Gier et al., 2023; Guy et al., 2023).
Between the last quarter of 2022 and the 1st months of 2023, an increased incidence of iGAS infections was observed in our hospitals in the Milan area (Italy), especially in adult patients. To provide exploratory information regarding epidemiological, clinical, and microbial characteristics of GAS infections during this timeframe, we conducted this retrospective, observational study on cases with bacterial strain available for genomic characterization. To provide background information on incidence of GAS infections in the two centers, we also calculated the temporal trend of laboratory-identified cases during the preceding 7 years.
Methods
Study setting and population
All the available bacterial strains of Streptococcus pyogenes at Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico of Milan and IRCCS Humanitas Research Hospital collected from September 2022 to March 2023 were included. The two centers are large university hospitals located in Milan, Northern Italy. For the period investigated, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico of Milan archived as for clinical practice bacteria strains from both invasive cases (blood cultures, intraoperative cultures) and non-invasive infections (pharyngeal swabs, nasopharyngeal aspirates), and for IRCCS Humanitas Research Hospital only blood culture isolates were obtained.
Clinical and epidemiological data were retrospectively collected from hospital records and databases.
Patients below 18 years of age were considered pediatric cases. Non-invasive GAS (non-iGAS) infections and iGAS infections were defined based on microbiological and clinical criteria according to literature (Stevens and Bryant, 2022a,b; GOV.UK, 2023). Non-iGAS infections included upper airway respiratory infections, scarlet fever, and superficial acute bacterial skin and skin structure infections (ABSSSIs; Stevens and Bryant, 2022a). iGAS infections included cases with isolation of Streptococcus pyogenes from normally sterile body sites as well as severe clinical presentations caused by GAS even if not isolated from a normally sterile material (Stevens and Bryant, 2022b; GOV.UK, 2023). Within iGAS group, we defined as severe iGAS the life-threatening conditions associated with the highest mortality such as sepsis/septic shock, necrotizing fasciitis and STSS (Stevens and Bryant, 2022b). Temporal trends of culture-confirmed non-iGAS and iGAS cases were calculated from 2015 to the first quarter of 2023.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing after MALDI-ToF identification (VITEK-MS, bioMérieux or Bruker Diagnostics) was performed by automated systems (VITEK2 compact automated system, bioMérieux or Phoenix automated microbiology system, Becton Dickinson Diagnostic Systems). Susceptible, intermediate and resistant categories were assigned according to the EUCAST breakpoint table (version 13.0, available at https://www.eucast.org/clinical_breakpoints).
Whole genome sequencing analysis and bacterial typing
Genomic DNA was extracted from pure isolates using ZymoBIOMICS DNA Miniprep Kit (Zymo Reseach) in accordance with manufacturer's instructions. Libraries for whole genome sequencing were generated using Illumina DNA Library Prep kit (Illumina, Inc., San Diego, CA, USA) and sequenced on Illumina MiSeq sequencing platform (Illumina, San Diego, CA, USA) using MiSeq Reagent Kit v2 to obtain 150-bp paired-end reads. Raw reads were trimmed for adapters and filtered for quality (average phred score>20) with Fastp (v0.20.1) and quality checked after trimming with FastQC (v0.11.9; Andrews, 2010; Chen et al., 2018). Kraken (v1.1.1) was used to screen for potential contaminations (Wood and Salzberg, 2014). Whole genome sequencing reads were assembled de novo using SPAdes (v3.14.1), the quality of the assemblies was evaluated using Quast (v5.1) and was annotated using Prokka (v1.14.6; Bankevich et al., 2012; Gurevich et al., 2013; Seemann, 2014). In silico emm typing and Multilocus Sequence Typing (MLST) were performed with emm-typer (v.0.2.0) and mlst (v2.11), respectively (Jolley and Maiden, 2010; Microbiological Diagnostic Unit Public Health Laboratory, 2021; Seemann, 2022). The investigation of antibiotic resistance (AMR) genes and virulence factors was carried out with ABRicate (v0.4) by using the Comprehensive Antibiotic Resistance Database (CARD) with 90% coverage (–mincov) and 90% identity (–minid) parameters (Jia et al., 2017; Lacey et al., 2020; Seemann, 2020; Alcolea-Medina et al., 2023). Virulence factors were investigated with a combination of ABRicate (v0.4) by using the Virulence Factor Database (VFDB; Chen et al., 2016) and a BLAST search for the main virulence factors described for S. pyogenes in literature (namely, fbaa, fbp54, fctA, fctB, grab, hasA, hasB, hasC, hylA, ideS, lepA, lmb, prtf2, sagA, scpA/scpB, sda1, sda2, sdn, sfb1, sfbX, sic, ska, sla, slo, smeZ, sof, speA, speB, speC, speG, speH, speI, speJ, speK, speL, speM, speQ, speR, spd, spd1, spd3, srtC1, and ssa), with 70% coverage and 70% identity parameters (Lacey et al., 2020). The presence of intact bacteriophages was investigated using geNomad (Camargo et al., 2023) and manually curated by searching the identified sequences with the Microbial Nucleotide BLAST (available at https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=MicrobialGenomes), using the bacteriophages database, while mobile genetic elements (MGEs) identification and plasmid identification were carried out using the MOB-suite tool (v3.1.4; Robertson and Nash, 2018; Robertson et al., 2020).
Phylogenetic analyses
Single nucleotide polymorphism (SNP) calling was performed with Snippy (v4.6.0; Seemann, 2015), using the S. pyogenes MGAS5005 genome (GenBank accession number NC_007297) as reference, after masking phage regions from the alignment and removing regions predicted as possible recombinogenic regions by Gubbins (v3.2.1; Croucher et al., 2015). Reference strains for the various emm types available from the PATRIC database (Wattam et al., 2017) and from the NCBI database (https://www.ncbi.nlm.nih.gov/) were also incorporated. The core SNPs identified relative to MGAS5005 were used to infer phylogenetic relationships among the whole genome sequences by Maximum-Likelihood (ML) using IQTREE (v2.0.6) with 1,000 bootstrap replicates under the best nucleotide substitution model (TVM+F+ASC+R2) determined by ModelFinder (Kalyaanamoorthy et al., 2017; Nguyen et al., 2017). The ML tree was visualized and annotated using iTOL (v6.5.2; Letunic and Bork, 2021).
To more accurately evaluate genetic differences among the most prevalent emm types (emm1 and emm12), SNPs were identified by mapping trimmed reads against the MGAS5005 and MGAS2096 (GenBank accession number CP000261) reference strains, respectively, using Snippy (v4.6.0; Seemann, 2015). Pairwise SNP distances were calculated using snp-dists tool (https://github.com/tseemann/snp-dists) and genetic relationships among the isolates were inferred by Minimum Spanning Trees using Grapetree (v1.5.0; Zhou et al., 2018). Potential transmission chains were defined by the presence of at least three strains clustering together in the ML tree with a bootstrap value of 100% and displaying an SNP distance of <10, consistently with other already published articles on S. pyogenes (Metcalf et al., 2022; Xie et al., 2023).
Statistical analysis
Statistical associations between emm types and virulence factors were evaluated with respect to iGAS, severe iGAS and severe outcome by Fisher exact test or Mann-Whitney test, as appropriate. To adjust significance for multiple comparisons and confirm potential associations, a Benjamini-Hochberg correction was applied after a chi square test (SPSS v.28.0.1.1).
Ethical and regulatory aspects
The study was registered by the Milan Area 2 Ethical Committee (#332_2023) and was conducted in accordance with standards of the Helsinki Declaration. At the two hospitals, informed consent for pseudonymized data processing for future research purposes was provided by all patients at the time of hospital admission, as routine procedure. Specific written informed consent was waived because of the retrospective nature of the analysis.
Results
7-year epidemiological surveillance of laboratory-identified GAS infections in the two centers
Cases of GAS between January 2015 and March 2023 by quarters are shown in Figure 1, overall (panel A) and stratified according to invasiveness (panels B and C) and age group (panels D and E).
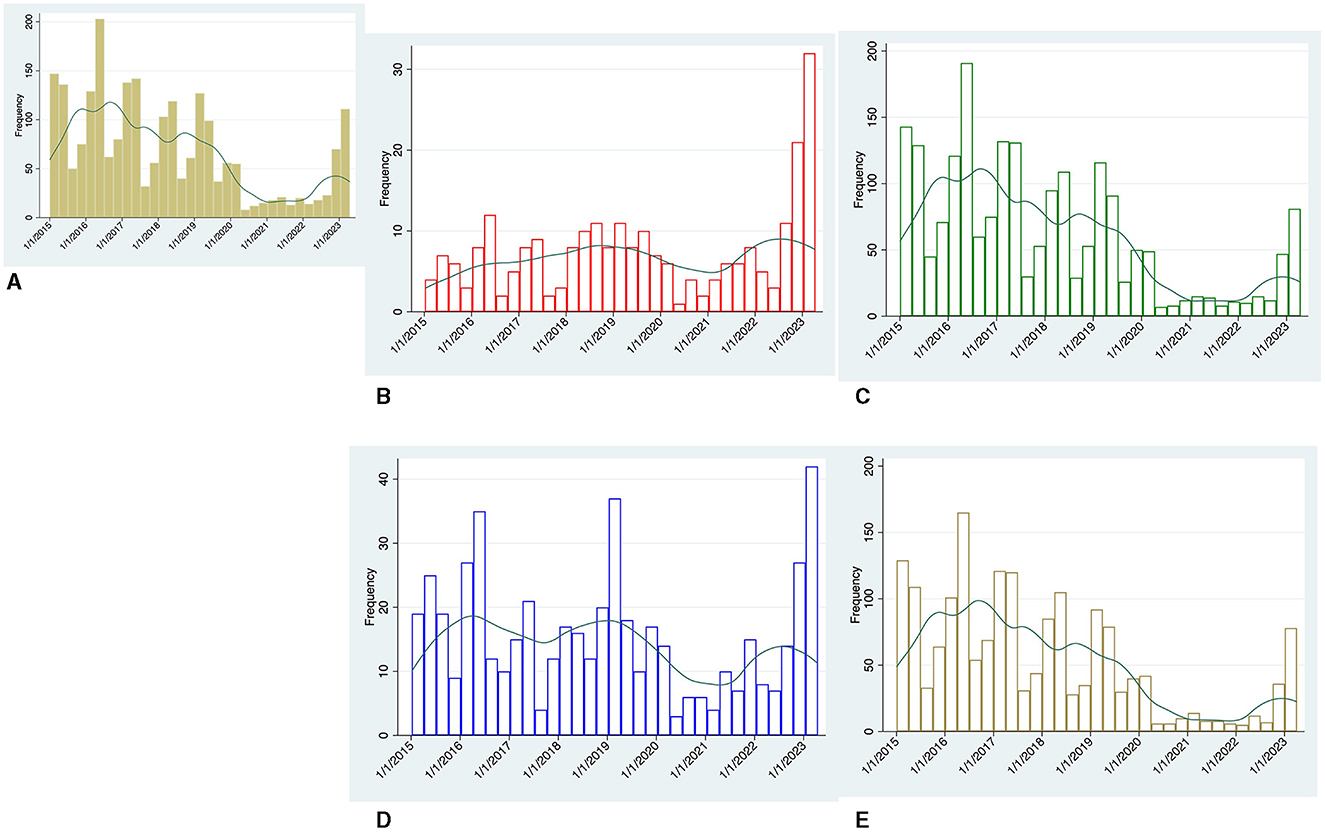
Figure 1. Temporal trend by quarters of microbiologically-confirmed GAS cases between January 2015 and March 2023, Green line represents the kernel density estimation generated for each group. (A) Overall cases, (B) invasive (iGAS) cases, (C) non-iGAS cases, (D) adult cases, and (E) pediatric cases.
A total of 2,900 cases were identified. Pediatric cases accounted for most of the non-iGAS infections [total cases 2,039: adults 310 (15.2%), children 1,729 (84.8%)], while adult cases accounted for most of the iGAS infections [total cases 251: adults 208 (82.9%), children 43 (17.1%)].
Prior to COVID-19 pandemic, GAS infections peaked in the first two quarters of each year. Unlike non-iGAS infections, the number of iGAS cases was less affected by temporal fluctuations and settled at values permanently below 13 cases every quarter.
During the pandemic years (March 2020 to September 2022), overall GAS cases fell well below the nadir seen in previous years. This trend applies especially to non-iGAS infections, and to a lesser extent to iGAS cases.
Over the last quarter of 2022 and first quarter of 2023 our centers experienced a steep increase in GAS cases. Of note, the largest increase was observed in iGAS infections, which reached 34 cases in the first quarter of 2023 over 3-fold higher than the average case numbers of the same seasonal period in the pre-pandemic years (Figure 1B).
Description of GAS cases detected during the study period
Between September 2022 and March 2023, 179 laboratory-confirmed GAS infections were identified in the two study hospitals (54 iGAS and 125 non-iGAS). Among them, bacterial strains were available for sequencing in 28 cases. Demographic and clinical features of the study population are detailed in Table 1, Supplementary Table 1.
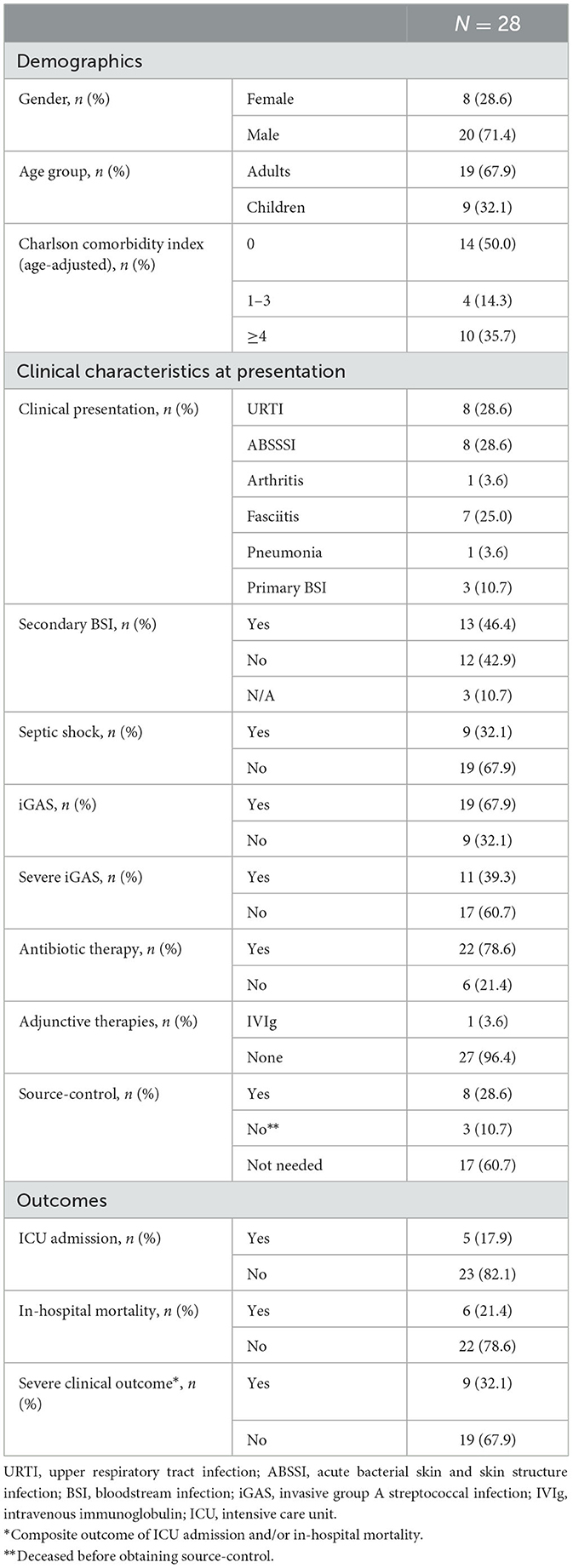
Table 1. Demographic and clinical characteristics and outcomes of the study population.
Patients were mainly male (20/28, 71.4%) and adults over 18 years of age (19/28, 67.9%). Median age in adults was 58.5 (Q1–Q3: 44–82) years, in children 5 (Q1–Q3: 3–6) years. Frailty was low on average, with age-adjusted Charlson Comorbidity Index <4 in almost two thirds of cases (18/28, 64.3%). GAS clinical presentation was represented mainly by mild to moderate upper respiratory tract infections (URTI; 8/28, 28.6%), ABSSSI (8/28, 28.6%) and necrotizing fasciitis (7/28, 25.0%). Arthritis was detected in one patient, as was pneumonia (3.6%), while primary bloodstream infections (BSI) without known origin occurred in 3/28 (10.7%) cases. Secondary BSI was diagnosed in almost half of the cohort (13/28, 46.4%), septic shock at presentation in 9/28 (32.1%) cases. Diagnostic criteria of iGAS and severe iGAS infections were met in 19/28 (67.9%) and 11/28 (39.3%) cases, respectively. Five patients (17.9%) required intensive care unit (ICU) admission and 6/28 (21.4%) died during hospitalization. Severe clinical outcome involving ICU admission and/or in-hospital mortality was met in 9/28 (32.1%) cases (Table 1).
Characteristics of patients according to age group are reported in Supplementary Table 1. Pediatric cases were mainly URTI (8/9, 88.9%), while in adults clinical characteristics varied and also presented higher severity, with iGAS and severe iGAS infections diagnosed in 18/19 (94.7%) and 10/19 (52.6%) cases, respectively.
Bacterial typing and genome characteristics
Whole-genome assemblies of the isolates displayed a median number of contigs of 61 (Q1–Q3: 53–69), with a median N50 of 164,198 bp (Q1–Q3: 130,587–203,753 bp), while the isolates' total genome sizes ranged from 1.75 to 1.93 Mb.
Among the 28 strains, a total of 13 emm types were detected, demonstrating the co-circulation of genetically divergent GAS strains in the 8 months analyzed. The most prevalent were emm1.0 and emm12.0, with 6 strains each (21.4%), followed by emm28.0 and emm164.2 with 3 strains each (10.7%). Other emm types were detected in one strain each (3.6%), and included emm types 4.0 (with sub-type 4.19 also observed), 11.0, 22.0, 58.0, 60.1, 82.0, 87.0, 89.0, and 92.0.
By MLST, each ST was found to be unique for each emm type, except for ST53, which was found both in strains of emm types 60.1 and 164.2 (Figure 2).
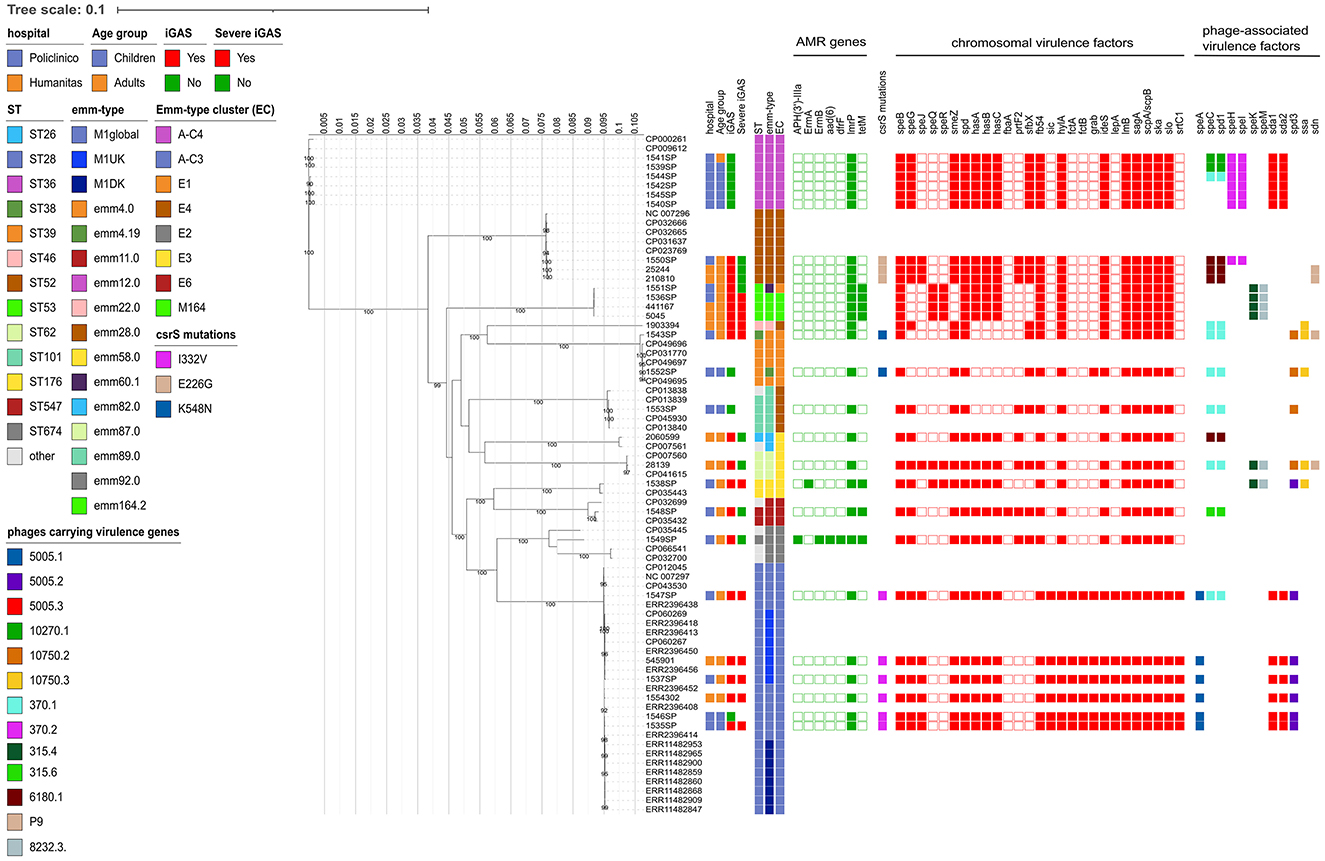
Figure 2. Estimated maximum likelihood phylogenetic analysis of Streptococcus pyogenes isolates (n = 28) and reference genomes (n = 45). The phylogeny was estimated on a coreSNP of 12,596 bp with IqTree using the best-fit model of nucleotide substitution TVM + F + ASC + R2 with 1,000 replicates fast bootstrapping. Leaves number represent the sample IDs, bootstraps values higher than 90 are shown on branches. Information regarding the samples were reported: hospital, age group, presence of invasive GAS infection (iGAS), presence of severe iGAS, Sequence Type (ST), emm-type, emm-type cluster (EC), the presence (filled squared) or absence of antimicrobial resistance genes, csrS mutations identified, presence (filled square) or absence of virulence factors, divided in chromosomal (red) or phage-associated (color based on phage on which they are present).
When looking at the emm-type cluster (EC) classification, which is based on related M proteins sharing similar binding and structural properties, strains mainly belonged to A-C3 (6/28, 21.4%) and A-C4 (6/28, 21.4%), followed by E4 (5/28, 17.9%), E1 (3/27, 10.7%), E3 (3/28, 10.7%), and M164 (3/28, 10.7%).
Antimicrobial resistance, virulence, and mobile genetic elements
Antimicrobial resistance gene lmrP was present in all the GAS strains. In addition, tet(M), conferring phenotypic resistance to tetracyclines, was found in 7/28 strains (25.0%, four of them belonging to the 3 emm164.2 and emm60.1 types), followed by erm(A), conferring the Macrolide-Lincosamide-Streptogramin B (MLSB) resistance phenotype, found in the emm58.0 strain. The erm(B) gene, conferring the MLSB resistance phenotype, the genes aad(6) and APH(3′)-III, conferring resistance to aminoglycosides, and dfrF gene, causing resistance to diaminopyrimidine antibiotics, were found together in the emm type 92.0 strain (ID 1549SP; Supplementary Table 2). All these data are in line with the phenotypic antimicrobial susceptibility profile of the strains (Supplementary Table 3).
Although the phenotypic test did not detect any large variation in the penicillin resistance, as a full susceptibility was observed for all strains for which the test was available (Supplementary Table 3), mutations at the level of penicillin-binding proteins (PBPs) 1A, 1B, 2A, and 2X were identified (Supplementary Table 4). In particular, the PBP2X mutations included M593T, found in 2/6 emm12 strains, mutations I502V, P676S, and K708E, found in the emm22 strain, S562T found in the emm89 strain and mutations V241I, K213N, and T246A, found in the emm58 strain. The M593T mutation was previously found to result in a 2-fold increase in penicillin G and ampicillin MIC when present in combination with the I502V, P676S, and K708E mutations (Yu et al., 2023). However, in our strains this mutation was always present as single PBP2X mutation, probably explaining why no change in the penicillin susceptibility could be observed.
Virulence factors identified were 40 in total, of which 10 present in all strains (Figure 2, Supplementary Table 2). Of the remaining 30, 20 were localized on the chromosome and 10 were phage-associated, and included genes coding for surface proteins, DNases, pilus machinery, capsule synthesis and superantigens (Figure 2, Supplementary Tables 2, 5).
Regarding superantigens, 12 different types could be identified, of which five chromosomally encoded and seven carried by prophages. Among the chromosomal superantigens, smeZ was found in all strains (24/28 strains, 85.7%) except for the four strains belonging to emm60.1 and 164.2, speG was found in 22 strains (78.6%, absent in strains belonging to emm types 60.1, 164.2, 4 and sub-type 4.19), speJ was found in 10 strains (35.7%, belonging to emm type 28, 87 and 1), and, speQand speR were always found together in 6 strains (21.4%, belonging to emm types 60.1, 164.2, 87 and 58).
The seven prophage-associated superantigens included speA, found in all six strains belonging to emm1 (21.4%), speC, found in 14/28 strains always together with DNase spd1 (50.0%, of which 1/6 emm1, 3/6 emm12, and 3/3 emm28 strains, as well as emm types 4, sub-type 4.19, 11, 22, 82, 87, and 92), speH and speI, always found together in 7/28 strains (25.0%, including 6/6 emm12 strains and only 1/3 emm28 strain), speK and speM, present together in six strains (21.4%, belonging to emm types 60.1, 164.2, 87 and 58), and ssa, present in 5/28 strains (17.9%, belonging to emm types 4 and sub-type 4.19, 22, 58 and 87; Figure 2, Supplementary Table 2). While most of phage-associated superantigens were carried by different phages and emm types, in our cohort, speA was always found on the Streptococcus phage 5,005.1 (GenBank acc. n. NC_007297.2, positions 983,974–1,022,694) and only in strains belonging to emm1.0 (Supplementary Table 5). Similarly, six out of the 7 speH-I (85.7%) were carried by Streptococcus phage 370.2 (GenBank acc. n. AE004092.2, positions 778,520–821,004) in the six emm12.0 strains (Supplementary Table 5).
Surface proteins identified included fibronectin binding proteins fbp54, present in all strains (28/28, 100%), sfbX, present in 16/28 strains (57.1%, belonging to emm types 4, 11, 12, 22, 28, 87, 89, 92), prtF2, present in 8/28 strains (28.6%, belonging to emm types 11, 28, 89, 82, 87, and 92), fbaA, present only in the emm11 strain, and the laminin-binding protein lmB, present in all strains (28/28, 100%). Several DNases were also detected, namely spd, present in all strains (28/28, 100%), spd1 in 14 strains (50.0%), spd3 in 11 strains (39.3%, belonging to emm types 1, 4, sub-type 4.19, 58, 89, and 87), sda1 and sda2, always found together in 12/28 strains (42.9%, of which 6/6 emm1 and 6/6 emm12 strains), and sdn in four strains (14.3%, including emm type 4, 87 and 2/3 emm28 strains). Capsule synthesis genes, hasA-C, were absent in four strains, belonging to emm types 4, sub-type 4.19, 22, and 89, while grab gene was present only in seven strains, comprising 6/6 emm1 strains and the emm4.19 sub-type (Figure 2, Supplementary Table 2).
Since the control of several streptococcal virulence factors is mediated by a 2-component regulatory system known as CsrR/CsrS (capsule synthesis regulation), the presence of mutations in the csrR and csrS genes was evaluated. No csrR mutations were identified in any of the 28 strains, whereas three different mutations could be detected in the csrS gene. In particular, the I332V mutation was found in all the 6 emm1 strains, the E226G mutation in all the 3 emm28 strains, and the K548N mutation in the 2 emm4 and sub-type 4.19 strains (Figure 2). No N498K mutations, previously found to be associated with invasive infections (Lin et al., 2014), were identified.
Plasmid analysis revealed the presence of 2 non-mobilizable plasmids, both carrying a putative bacteriocin: the pA996 (GenBank acc.n. KC895877) in strain 1549SP (emm92.0) and pA852 (acc n. KC895878) in strain 1548SP (emm11.0).
Phylogenetic analysis and genomic characterization of the most common emm types
The ML phylogenetic tree, inferred from a coreSNP alignment of 12,596 bp and constructed including reference genomes available for the emm types identified, confirmed the distribution of the strains based on the emm type (Figure 2).
Of the 6 strains belonging to emm1.0, 4 were from Policlinico Hospital and 2 from Humanitas Hospital. They were characterized by the presence of virulence genes speA, sic (Streptococcal inhibitor of complement), fctA-fctB (Major pilins), lepA (Signal peptidase I), and srtC1 (sortase), all absent in the other study strains. The ID 1547SP also carried the spd1 and speC virulence factors, carried by the same phage (Streptococcus phage 370.1, GenBank acc. n. AE004092.2, positions 529,587 to 570,504). As showed by the Minimum Spanning Tree, the median SNP distance characterizing the six strains was 69 (Q1–Q3: 61–83), with no strains displaying a pairwise distance of <10 SNPs, suggesting that they did not belong to a unique transmission chain (Supplementary Figure 1A). By further investigating the genetic diversity, four strains (66.7%) were found to belong to the M1global clone, whereas two strains (namely ID 1537SP, from Policlinico Hospital, and 545,901 from Humanitas Hospital) were found to belong to the M1UK clone, as they carried 27/27 of the clone-defining SNPs. The two M1UK strains differed from each other for 41 SNPs and from the nearest M1UK reference (ERR2396456) by 35 and 20 SNPs, respectively, of which 8 present in both strains (Supplementary material). Among these eight common SNPs, one fell in intergenic regions, three caused missense mutations, and four caused synonymous mutations. The missense mutations were: C423006T, falling in an ABC-2 family transporter; C931062A, falling in an ABC transporter ATP-binding protein; and G1296882A, falling in a Cof-type HAD-IIB family hydrolase (Supplementary material).
The 6 strains belonging to emm12.0 exclusively came from Policlinico Hospital and shared the same virulence factors, except for three strains that also carried the phage-associated spd1 and speC virulence factors (Figure 2). Despite sharing the same hospital origin, the Minimum Spanning Tree strains revealed high diversity among the 6 emm12.0 strains, which were characterized by a high number of SNPs [median 185 (Q1–Q3: 97–192)], and with the pairwise SNP distance always higher than 10 SNPs, suggesting also in this case that they did not belong to a unique transmission chain (Supplementary Figure 1B, Supplementary material).
Regarding the less prevalent emm types, the three strains belonging to emm28.0 shared the same virulence factors, with only one strain also carrying the phage associated spd1-speC genes. These strains were characterized by high number of SNPs [median 90 (min-max: 11–95)], suggesting once more that they did not belong to a single transmission chain.
The four strains belonging to emm164.2 and emm60.1 (localized in the same clade in the phylogenetic tree) were characterized by the presence of tetM, the same set of chromosomal virulence factors, and the absence of any phage-associated virulence factors. Among these strains, the three belonging to emm164.2 were characterized by a low genetic divergence, with a median SNP distance of 6 (min-max: 5–9). On account of their low numbers, it is rather challenging to speculate on potential transmission chains.
Associations between emm types and virulence factors with age and clinical severity
Table 2 reports the associations between emm types and virulence factors with age and clinical severity.
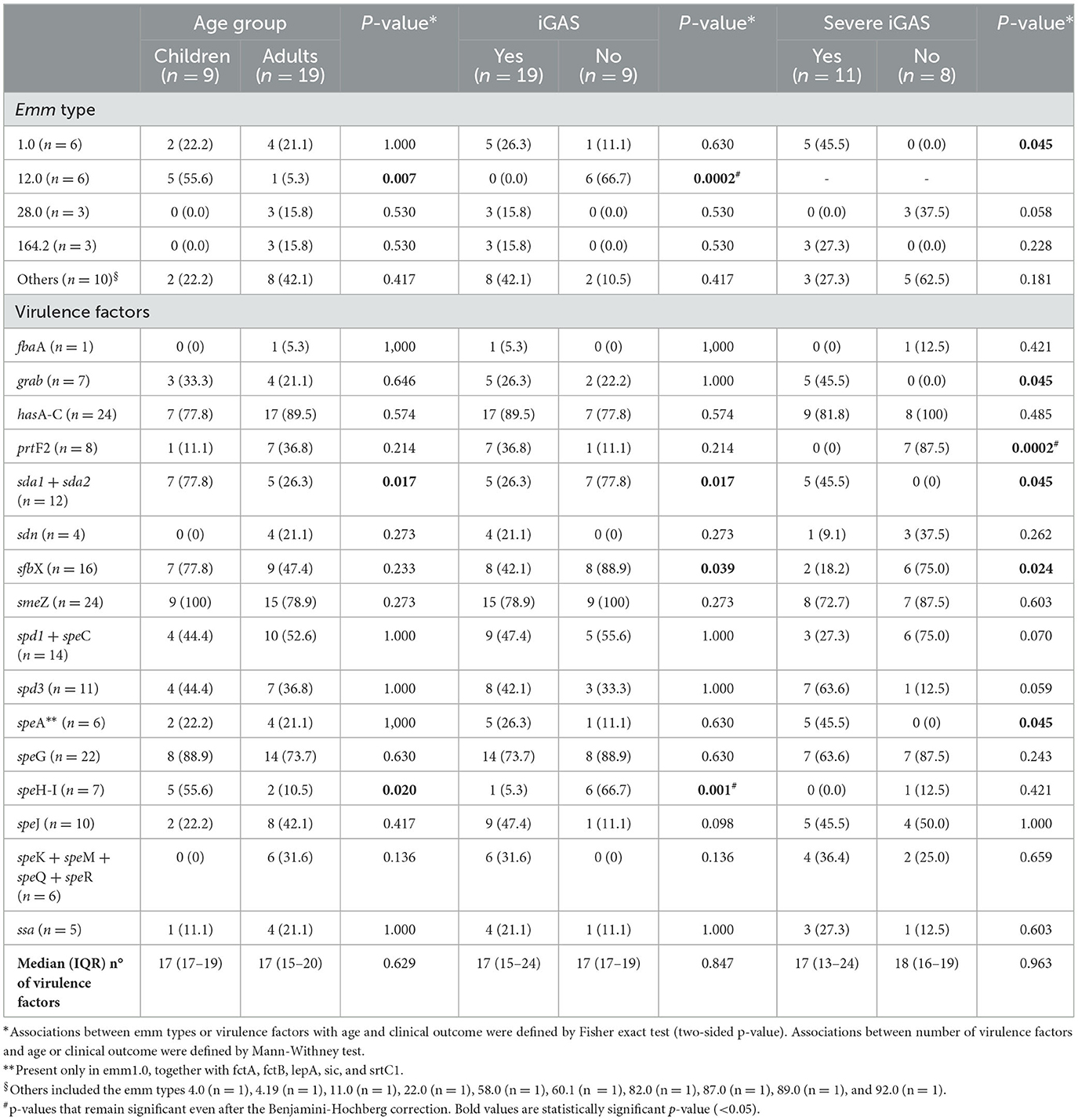
Table 2. Associations between emm types and virulence factors with age and clinical parameters.
No significant associations between emm type and age group were observed, with the exception of a higher prevalence of emm12 infections among pediatric cases [adults 1/19 (5.3%), children 5/9 (55.6%), P = 0.007, although not confirmed after the Benjamini-Hochberg correction for multiple comparisons was applied]. iGAS presentation was never found in emm12.0 strains, which were found to almost exclusively carry the phage-associated speH-I, virulence factors found to be negatively associated with iGAS (p = 0.0002). Of note, this association was confirmed also after the Benjamini-Hochberg correction.
A higher number of severe iGAS cases were detected from strains belonging to emm1.0 strains (P = 0.045, although not confirmed after the Benjamini-Hochberg correction).
Only one virulence factor, namely prtF2, was found to be negatively associated with severe iGAS (P = 0.0002, confirmed also after the Benjamini-Hochberg correction), but not to iGAS (P = 0.214).
All the associations found between emm types and virulence factors with severe iGAS infections were confirmed with severe clinical outcome (data not shown), defined as ICU admission and/or in-hospital mortality.
No significant associations were found between the median number of virulence factors and clinical parameters.
Discussion
This study describes the increased number of GAS infections in two university hospitals in Milan, Italy, in the last quarter of 2022 and first quarter of 2023, during the surge of invasive cases reported across the European Region (de Gier et al., 2019, 2023; ECDC, 2022; GOV.UK, 2022; WHO, 2022; Guy et al., 2023; Lassoued et al., 2023). We performed genomic characterization of the available GAS strains and investigated relatedness and transmission chains, and the potential association of bacterial genomic characteristics with clinical severity.
Over the last few months, several studies have reported an increase of GAS and iGAS infections starting from early 2022 and mainly affecting the pediatric population. This surge in infections may be related to decreased exposure to GAS and common respiratory viral infection during the COVID-19 pandemic due to physical distancing, which may have led to lower levels of immunity (GOV.UK, 2022). After 2 years of low incidence, an early start of the GAS infection season coupled with high circulating rates of respiratory viruses, which may increase the risk of iGAS diseases (de Gier et al., 2019), is likely to have amplified the resurgence of GAS infections (Lynskey et al., 2019; ECDC, 2022; WHO, 2022; de Gier et al., 2023; Guy et al., 2023; Lassoued et al., 2023).
In our centers, we also observed a sharp increase of GAS and iGAS cases starting from the last quarter of 2022. In contrast to other cohorts, severe infections occurred mostly in adults (almost 95% of iGAS cases) with necrotizing fasciitis and deep ABSSSI with bacteremia as major manifestations. In more than half of cases these represented life-threatening infections (severe iGAS), and mortality occurred in over a quarter of iGAS cases during hospital stay.
Clusters of iGAS infections in adults have been described, mostly found to be caused by outbreaks of specific emm-types in hospitals or long-term care facilities (Chalker et al., 2016; Coelho et al., 2019; Trell et al., 2020) or among at-risk population such as homeless people or intravenous drug users (Cornick et al., 2017; Bubba et al., 2019). Our cohort differed for epidemiological characteristics and was not predominated by the clonal expansion of a single emm-type, as confirmed by the fact that the 28 strains belonged to 13 different emm types and, in the majority of cases, were characterized by a distance higher than 10 SNPs.
Among the detected emm types, the most prevalent were emm1.0 and emm12.0, with 6 strains each. The same emm types were found to be the most prevalent in a recently described outbreak in London (Alcolea-Medina et al., 2023), where emm1.0 strains were found to cause a higher proportion of invasive cases and predominantly to affect children. Nevertheless, our results showed some differences. First, even though emm1.0 strains caused over a quarter of iGAS infections (26.3%) and almost half of all severe iGAS infections (45.5%), they were mostly found in adults. Second, in the London outbreak, 7/9 emm1.0 strains were found to belong to the M1UK clone, an emm1.0 sublineage characterized by increased SpeA toxin production, while in our samples we found only 2/6 M1UK clone, suggesting a higher differentiation of M1 lineage in our samples. In line with these findings, although M1UK has been described as the predominant clone in emm1.0 strains (Demczuk et al., 2019; Li et al., 2020a; Davies et al., 2023), in the UK as well as in other European countries (Rümke et al., 2020; van der Putten et al., 2023; Zhi et al., 2023), recent evidence from Denmark suggested additional differentiation of the M1 lineage with the rising of new M1 clones (Johannesen et al., 2023). In line with that observed in Denmark (Johannesen et al., 2023), the emm12.0, the second most prevalent emm type identified in our study, mostly detected in children, was found to be negatively associated to invasive infections.
When looking at potential transmission chains in the two most prevalent emm types, we found that for both the emm1 and emm12 strains investigated, strains differed within an emm type by more than 10 core SNPs, indicating that they were not linked by recent direct transmission events, and were not likely part of a common transmission chain. When looking at the pairwise SNP distance also in the emm28 and emm164.2 types, found in three individuals each, only emm164.2 showed a limited genetic divergence. Due to the low number of emm164.2 strains detected, we cannot speculate about the existence of a real transmission chain, even if all the three strains belonged to males, aged from 57 to 101, affected by severe iGAS, and hospitalized for comorbidities in Humanitas (n = 2) and Policlinico (n = 1) hospitals. Thus, consistent with studies conducted after COVID-19 pandemic in the pediatric population (de Gier et al., 2023; Guy et al., 2023), the increased incidence of iGAS cases, which we observed predominantly in adult patients, mainly reflects a high circulating rate of several GAS strains in the community rather than direct transmission chains among patients. Understanding and differentiating between these two scenarios might reflect also in the different management of patients, supporting the role of rapid and accurate identification and handling of suspected cases during seasonal epidemics, before enhancing the infection control measures proposed in previous iGAS clusters, such as the screening and treatment of healthcare workers and household contacts (Chalker et al., 2016; Coelho et al., 2019).
As for GAS virulence factors, our findings were in line with what has already been observed in literature, since we found a correlation between emm type and the presence of specific superantigen genes. In particular, speA was identified only in the 6 emm1.0 strains (Lynskey et al., 2019; Davies et al., 2023), and speH and speI almost exclusively in the 6 emm12.0 strains (Li et al., 2020b). As already reported, emm12.0 was predominantly found in the pediatric population (Imöhl et al., 2017), while emm1.0, and the virulence factors identified in these strains, were found to be associated to severe iGAS infections (Lynskey et al., 2019).
The major limitation of our study is that it included only a subgroup (28/179) of GAS isolated in the two hospitals within the study period. This is due to the retrospective nature of the study and the absence of an active biobanking of all GAS strains. Nevertheless, the majority of missing cases belongs to non-iGAS infections, especially in children. This is due to the characteristics of the two centers (Humanitas does not have a pediatric department) and to the fact that these infections are often diagnosed with rapid antigenic tests without the obtainment of the bacterial isolate (e.g., upper airway respiratory infections, scarlet fever). Moreover, genotyped strains corresponded predominantly to invasive infections (19/28). For this reason, although the results might consistently describe the strains responsible for clinically severe cases in the two hospitals, the data obtained may not be very representative of the circulation of S. pyogenes strains in the community at large. Therefore, this could have definitely impacted the ability to detect transmission chains. As further limitation, the modest sample size of our study and the predominance of iGAS infections do not permit us to comprehensively discuss the significant correlations between emm-types and clinical manifestations, or to perform other comparisons between patient subsets. Extending the study period and increasing the sample availability are therefore necessary in order to draw definitive conclusions. Nevertheless, we were able to confirm previous evidence on emm1.0 and speA correlation with severe iGAS cases (Lynskey et al., 2019). This opens interesting perspectives on the employment of bacterial genome sequencing in clinical practice for clinical risk stratification and patients' management, besides its use for outbreak characterization.
In conclusion, through the integration of genome analysis with clinical and epidemiological patient data, our retrospective study provides a preliminary characterization of iGAS infections that we observed during the surge of invasive cases reported globally. Our results describe the predominant involvement of adult patients in infections requiring hospitalization and give insights into bacterial typing and genomic diversity of the isolates. Integrated genomic studies such as the present emphasize the crucial need to apply genomic surveillance in clinical practice, in order to provide valuable insights on the genomic and evolution of pathogens, enable identification and tracking of emerging strains, and implement targeted infection control measures. In fact, integrating classical epidemiological studies with genomic investigation allowed us to exclude transmission chains during GAS outbreak, helping in ensuring the appropriate infection control strategies and in properly managing severe infections.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB63359.
Ethics statement
The studies involving humans were approved by Milan Area 2 Ethical Committee (#332_2023). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
DM: Conceptualization, Methodology, Writing – original draft. VF: Conceptualization, Formal analysis, Methodology, Writing – original draft. PS: Data curation, Writing – original draft. AL: Conceptualization, Formal analysis, Methodology, Writing – review & editing. LB: Writing – review & editing, Validation. FC: Data curation, Formal analysis, Writing – review & editing. LC: Conceptualization, Data curation, Writing – review & editing. AC: Conceptualization, Methodology, Writing – review & editing. CM: Writing – review & editing. AM: Writing – review & editing. AT: Conceptualization, Data curation, Writing – review & editing. LT: Data curation, Writing – review & editing. VC: Data curation, Writing – review & editing. SC: Writing – review & editing. MB: Writing – review & editing. CA: Conceptualization, Methodology, Supervision, Writing – review & editing. AB: Conceptualization, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partially funded by the Italian Ministry of Health—Current research IRCCS, by project STOP COVID financed by COVID-19 emergency donations and by EU funding within the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT).
Acknowledgments
We would like to thank all the physicians and nurses of IRCCS Ca' Granda Ospedale Maggiore Policlinico of Milano and IRCCS Humanitas Research Hospital that were involved in the management of GAS infections. We are also grateful to the whole staff of the Microbiology Laboratories of the Hospitals involved in the study for outstanding technical support in samples collection, processing, and data management. We also thank F. McDonald for English discourse revision and editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1287522/full#supplementary-material
References
Alcolea-Medina, A., Snell, L. B., Alder, C., Charalampous, T., Williams, T. G. S., Laboratory Group, S. M., et al. (2023). The ongoing Streptococcus pyogenes (Group A Streptococcus) outbreak in London, United Kingdom in December 2022, a molecular epidemiology study. Clin. Microbiol. Infect. 2023, S1198-743X(23)00115-5. doi: 10.1016/j.cmi.2023.03.001
Andrews, S. (2010). FastQC: a Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed May 24, 2023).
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beall, B., Facklam, R., and Thompson, T. (1996). Sequencing emm-specific PCR products for routine and accurate typing of group A streptococci. J. Clin. Microbiol. 34, 953–958.
Brouwer, S., Rivera-Hernandez, T., Curren, B. F., Harbison-Price, N., De Oliveira, D. M. P., Jespersen, M. G., et al. (2023). Pathogenesis, epidemiology and control of Group A Streptococcus infection. Nat. Rev. Microbiol. 6, 1–17. doi: 10.1038/s41579-023-00939-6
Bubba, L., Bundle, N., Kapatai, G., Daniel, R., Balasegaram, S., Anderson, C., et al. (2019). Genomic sequencing of a national emm66 group A streptococci (GAS) outbreak among people who inject drugs and the homeless community in England and Wales, January 2016-May 2017. J. Infect. 79, 435–443. doi: 10.1016/j.jinf.2019.08.009
Camargo, A. P., Roux, S., Schulz, F., Babinski, M., Xu, Y., Hu, B., et al. (2023). Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 23, 1953. doi: 10.1038/s41587-023-01953-y
Chalker, V. J., Smith, A., Al-Shahib, A., Botchway, S., Macdonald, E., Daniel, R., et al. (2016). Integration of genomic and other epidemiologic data to investigate and control a cross-institutional outbreak of Streptococcus pyogenes. Emerg. Infect. Dis. 22, 973–980. doi: 10.3201/eid2204.142050
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016, hierarchical and refined dataset for big data analysis−10 years on. Nucl. Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Coelho, J. M., Kapatai, G., Jironkin, A., Al-Shahib, A., Daniel, R., Dhami, C., et al. (2019). Genomic sequence investigation Streptococcus pyogenes clusters in England (2010–2015). Clin. Microbiol. Infect. 25, 96–101. doi: 10.1016/j.cmi.2018.04.011
Cornick, J. E., Kiran, A. M., Vivancos, R., Van Aartsen, J., Clarke, J., Bevan, E., et al. (2017). Epidemiological and molecular characterization of an invasive group A Streptococcus emm32.2 outbreak. J. Clin. Microbiol. 55, 1837–1846. doi: 10.1128/JCM.00191-17
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucl. Acids Res. 43, e15. doi: 10.1093/nar/gku1196
Davies, M. R., Keller, N., Brouwer, S., Jespersen, M. G., Cork, A. J., Hayes, A. J., et al. (2023). Detection of Streptococcus pyogenes M1UK in Australia and characterization of the mutation driving enhanced expression of superantigen SpeA. Nat. Commun. 14, 1051. doi: 10.1038/s41467-023-36717-4
de Gier, B., Marchal, N., de Beer-Schuurman, I., Te Wierik, M., Hooiveld, M., de Melker, H. E., et al. (2023). Increase in invasive group A streptococcal (Streptococcus pyogenes) infections (iGAS) in young children in the Netherlands, 2022. Euro Surveill. 28, 2200941. doi: 10.2807/1560-7917.ES.2023.28.1.2200941
de Gier, B., Vlaminckx, B. J. M., Woudt, S. H. S., van Sorge, N. M., and van Asten, L. (2019). Associations between common respiratory viruses and invasive group A streptococcal infection: a time-series analysis. Influenza Other Respir Viruses 13, 453–458. doi: 10.1111/irv.12658
Demczuk, W., Martin, I., Domingo, F. R., MacDonald, D., and Mulvey, M. R. (2019). Identification of Streptococcus pyogenes M1UK clone in Canada. Lancet Infect Dis. 19, 1284–1285. doi: 10.1016/S1473-3099(19)30622-X
ECDC (2022). Increase in Invasive Group A Streptococcal Infections Among Children in Europe, Including Fatalities. Available online at: https://www.ecdc.europa.eu/en/news-events/increase-invasive-group-streptococcal-infections-among-children-europe-including (accessed August 30, 2023).
Efstratiou, A., and Lamagni, T. (2022). “Chapter 19. Epidemiology of Streptococcus pyogenes,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd Edn, eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City, OK: University of Oklahoma Health Sciences Center), 19.
Facklam, R., Beall, B., Efstratiou, A., Fischetti, V., Johnson, D., Kaplan, E., et al. (1999). Emm typing and validation of provisional M types for group A streptococci. Emerg. Infect. Dis. 5, 247–253.
Friães, A., Pinto, F. R., Silva-Costa, C., Ramirez, M., and Melo-Cristino, J. (2012). Portuguese group for the study of streptococcal infections. Group A streptococci clones associated with invasive infections and pharyngitis in Portugal present differences in emm types, superantigen gene content and antimicrobial resistance. BMC Microbiol. 12, 280. doi: 10.1186/1471-2180-12-280
Ghosh, P. (2011). The nonideal coiled coil of M protein and its multifarious functions in pathogenesis. Adv. Exp. Med. Biol. 715, 197–211. doi: 10.1007/978-94-007-0940-9_12
GOV.UK (2022). Group A Streptococcal Infections: Report on Seasonal Activity in England, 2022 to 2023. Available online at: https://www.gov.uk/government/publications/group-a-streptococcal-infections-activity-during-the-2022-to-2023-season/group-a-streptococcal-infections-report-on-seasonal-activity-in-england-2022-to-2023 (accessed August 30, 2023).
GOV.UK (2023). UK Guidelines for the Management of Contacts of Invasive Group A Streptococcus (iGAS) Infection in Community Settings V2.0. Available online at: https://www.gov.uk/government/publications/invasive-group-a-streptococcal-disease-managing-community-contacts (accessed August 30, 2023).
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Guy, R., Henderson, K. L., Coelho, J., Hughes, H., Mason, E. L., Gerver, S. M., et al. (2023). Increase in invasive group A streptococcal infection notifications, England. Euro Surveill. 28, 2200942. doi: 10.2807/1560-7917.ES.2023.28.1.2200942
Imöhl, M., Fitzner, C., Perniciaro, S., and van der Linden, M. (2017). Epidemiology and distribution of 10 superantigens among invasive Streptococcus pyogenes disease in Germany from 2009 to 2014. PLoS ONE 12, e0180757. doi: 10.1371/journal.pone.0180757
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017, expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucl. Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Johannesen, T. B., Munkstrup, C., Edslev, S. M., Baig, S., Nielsen, S., Funk, T., et al. (2023). Increase in invasive group A streptococcal infections and emergence of novel, rapidly expanding sub-lineage of the virulent Streptococcus pyogenes M1 clone, Denmark, 2023. Euro Surveill. 28, 291. doi: 10.2807/1560-7917.ES.2023.28.26.2300291
Johnson, A. F., and LaRock, C. N. (2021). Antibiotic treatment, mechanisms for failure, and adjunctive therapies for infections by group A streptococcus. Front. Microbiol. 12, 760255. doi: 10.3389/fmicb.2021.760255
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 11, 595. doi: 10.1186/1471-2105-11-595
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Lacey, J. A., James, T. B., Tong, S. Y. C., and Davies, M. R. (2020). “Whole genome sequence analysis and population genomics of group A streptococci,” in Group A Streptococcus. Methods in Molecular Biology, eds T. Proft and J. Loh (New York, NY: Humana), 2136.
Lassoued, Y., Assad, Z., Ouldali, N., Caseris, M., Mariani, P., Birgy, A., et al. (2023). Unexpected increase in invasive group A streptococcal infections in children after respiratory viruses outbreak in France: a 15-year time-series analysis. Open For. Infect. Dis. 10, ofad188. doi: 10.1093/ofid/ofad188
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5, an online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, H., Zhou, L., Zhao, Y., Ma, L., Liu, X., and Hu, J. (2020b). Molecular epidemiology and antimicrobial resistance of group A streptococcus recovered from patients in Beijing, China. BMC Infect. Dis. 20, 507. doi: 10.1186/s12879-020-05241-x
Li, Y., Nanduri, S. A., Van Beneden, C. A., and Beall, B. W. (2020a). M1UK lineage in invasive group A streptococcus isolates from the USA. Lancet Infect. Dis. 20, 538–539. doi: 10.1016/S1473-3099(20)30279-6
Lin, J. N., Chang, L. L., Lai, C. H., Lin, H. H., and Chen, Y. H. (2014). Association between polymorphisms in the csrRS two-component regulatory system and invasive group A streptococcal infection. Eur. J. Clin. Microbiol. Infect. Dis. 33, 735–743. doi: 10.1007/s10096-013-2005-7
Lynskey, N. N., Jauneikaite, E., Li, H. K., et al. (2019). Emergence of dominant toxigenic M1T1 Streptococcus pyogenes clone during increased scarlet fever activity in England: a population-based molecular epidemiological study. Lancet Infect. Dis. 19, 1209–1218. doi: 10.1016/S1473-309930446-3
McShan, W. M., and Nguyen, S. V. (2022). “Chapte 9. The bacteriophages of Streptococcus pyogenes,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd Edn, eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City, OK: University of Oklahoma Health Sciences Center), 9.
Metcalf, B., Nanduri, S., Chochua, S., Li, Y., Fleming-Dutra, K., McGee, L., et al. (2022). Cluster transmission drives invasive group A streptococcus disease within the united states and is focused on communities experiencing disadvantage. J. Infect. Dis. 226, 546–553. doi: 10.1093/infdis/jiac162
Microbiological Diagnostic Unit Public Health Laboratory (2021). Emmtyper—Emm Automatic Isolate Labeller (v0.2.0). Available online at: https://github.com/MDU-PHL/emmtyper (accessed May 24, 2023).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2017). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Proft, T., and Fraser, J. D. (2022). “Chapter 14. Streptococcus pyogenes superantigens: biological properties and potential role in disease,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd Edn, eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City, OK: University of Oklahoma Health Sciences Center), 14.
Robertson, J., Bessonov, K., Schonfeld, J., and Nash, J. H. E. (2020). Universal whole-sequence-based plasmid typing and its utility to prediction of host range and epidemiological surveillance. Microb. Genom. 6, mgen000435. doi: 10.1099/mgen.0.000435
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4, e000206. doi: 10.1099/mgen.0.000206
Rümke, L. W., de Gier, B., Vestjens, S. M. T., van der Ende, A., van Sorge, N. M., Vlaminckx, B. J. N., et al. (2020). Dominance of M1UK clade among Dutch M1 Streptococcus pyogenes. Lancet Infect. Dis. 20, 539–540. doi: 10.1016/S1473-309930278-4
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2015). Snippy: Fast Bacterial Variant Calling From NGS Reads. Available online at: https://github.com/tseemann/snippy (accessed May 24, 2023).
Seemann, T. (2020). Abricate. Available online at: https://github.com/tseemann/abricate (accessed May 24, 2023).
Seemann, T. (2022). mlst Tool. Available online at: https://github.com/tseemann/mlst (accessed May 24, 2023).
Stevens, D. L., Bisno, A. L., Chambers, H. F., Dellinger, E. P., Goldstein, E. J. C., Gorbach, S. L., et al. (2014). Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the infectious diseases society of America. Clin. Infect. Dis. 59, e10–e52. doi: 10.1093/cid/ciu444
Stevens, D. L., and Bryant, A. E. (2022a). “Chapter 23. Streptococcus pyogenes impetigo, erysipelas, and cellulitis,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd Edn, eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City, OK: University of Oklahoma Health Sciences Center), 23.
Stevens, D. L., and Bryant, A. E. (2022b). “Chapter 24. Severe Streptococcus pyogenes infections,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd Edn, eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City, OK: University of Oklahoma Health Sciences Center), 24.
Trell, K., Jörgensen, J., Rasmussen, M., and Senneby, E. (2020). Management of an outbreak of postpartum Streptococcus pyogenes emm75 infections. J. Hosp. Infect. 105, 752–756. doi: 10.1016/j.jhin.2020.05.040
van der Putten, B. C. L., Vlaminckx, B. J. M., de Gier, B., Freudenburg-de Graaf, W., and van Sorge, N. M. (2023). Group A streptococcal meningitis with the M1UK variant in the Netherlands. J. Am. Med. Assoc. 329, 1791–1792. doi: 10.1001/jama.2023.5927
Walker, M. J., Barnett, T. C., McArthur, J. D., Cole, J. N., Gillen, C. M., Henningham, A., et al. (2014). Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin. Microbiol. Rev. 27, 264–301. doi: 10.1128/CMR.00101-13
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucl. Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
WHO (2022). Increased Incidence of Scarlet Fever and Invasive Group A Streptococcus Infection—Multi-country. Available online at: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON429 (accessed August 30, 2023).
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46. doi: 10.1186/gb-2014-15-3-r46
Xie, O., Zachreson, C., Tonkin-Hill, G., Price, D. J., Lacey, J. A., Morris, J. M., et al. (2023). Overlapping transmission of group A and C/G Streptococcus facilitates inter-species mobile genetic element exchange. MedRxiv 2023, 23294027. doi: 10.1101/2023.08.17.23294027
Yu, D., Guo, D., Zheng, Y., and Yang, Y. (2023). A review of penicillin binding protein and group A Streptococcus with reduced-β-lactam susceptibility. Front. Cell. Infect. Microbiol. 13, 1117160. doi: 10.3389/fcimb.2023.1117160
Zhi, X., Li, H. K., Li, H., Loboda, Z., Charles, S., Vieira, A., et al. (2023). Emerging invasive group A streptococcus M1UK lineage detected by allele-specific PCR, England, 20201. Emerg. Infect. Dis. 29, 1007–1010. doi: 10.3201/eid2905.221887
Keywords: Streptococcus pyogenes, GAS, necrotizing fasciitis, virulence factors, WGS, genomic surveillance
Citation: Mangioni D, Fox V, Saltini P, Lombardi A, Bussini L, Carella F, Cariani L, Comelli A, Matinato C, Muscatello A, Teri A, Terranova L, Cento V, Carloni S, Bartoletti M, Alteri C and Bandera A (2024) Increase in invasive group A streptococcal infections in Milan, Italy: a genomic and clinical characterization. Front. Microbiol. 14:1287522. doi: 10.3389/fmicb.2023.1287522
Received: 01 September 2023; Accepted: 04 December 2023;
Published: 11 January 2024.
Edited by:
Thomas Proft, The University of Auckland, New ZealandReviewed by:
Luke Blagdon Snell, King's College London, United KingdomMark Davies, The University of Melbourne, Australia
Stephen B. Beres, Houston Methodist Research Institute, United States
Copyright © 2024 Mangioni, Fox, Saltini, Lombardi, Bussini, Carella, Cariani, Comelli, Matinato, Muscatello, Teri, Terranova, Cento, Carloni, Bartoletti, Alteri and Bandera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Saltini, cGFvbGEuc2FsdGluaUB1bmltaS5pdA==
†These authors have contributed equally to this work and share first authorship