 Francisco Salvà-Serra1,2,3,4,5
Francisco Salvà-Serra1,2,3,4,5 Priyank Nimje6
Priyank Nimje6 Beatriz Piñeiro-Iglesias2,3Leonarda Achá Alarcón1,3,4Sofia Cardew2,3Elisabeth Inganäs2,3Susanne Jensie-Markopoulos2,3Maria Ohlén2,3Hanna-Sophia Sailer2,3Christel Unosson2,3
Beatriz Piñeiro-Iglesias2,3Leonarda Achá Alarcón1,3,4Sofia Cardew2,3Elisabeth Inganäs2,3Susanne Jensie-Markopoulos2,3Maria Ohlén2,3Hanna-Sophia Sailer2,3Christel Unosson2,3 Víctor Fernández-Juárez7
Víctor Fernández-Juárez7 Cesar O. Pacherres8
Cesar O. Pacherres8 Michael Kühl8
Michael Kühl8 Edward R. B. Moore1,2,3,4
Edward R. B. Moore1,2,3,4 Nachiket P. Marathe6*
Nachiket P. Marathe6*- 1Department of Infectious Diseases, Institute for Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 2Department of Clinical Microbiology, Sahlgrenska University Hospital, Gothenburg, Sweden
- 3Culture Collection University of Gothenburg (CCUG), Department of Clinical Microbiology, Sahlgrenska University Hospital and Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 4Centre for Antibiotic Resistance Research (CARe), University of Gothenburg, Gothenburg, Sweden
- 5Methodology Textiles and Medical Technology, Division Materials and Production, RISE Research Institutes of Sweden, Gothenburg, Sweden
- 6Institute of Marine Research (IMR), Bergen, Norway
- 7Department of Biology and Nordic Center for Earth Evolution (NordCEE), University of Southern Denmark, Odense, Denmark
- 8Marine Biological Section, Department of Biology, University of Copenhagen, Copenhagen, Denmark
Three β-lactam resistant bacterial strains isolated from gut samples of wild Atlantic mackerel (Scomber scombrus) collected from the northern North Sea were characterized by polyphasic analyses. The strains were determined to belong to the genus Pseudomonas but could not be assigned to a known species. The nearly-complete 16S rRNA gene sequence showed the highest similarity (99.9%) to four different species, although partial rpoD sequence exhibited relatively low similarities to Pseudomonas proteolytica (93.4%) and other Pseudomonas spp. Genome sequencing and subsequent digital DNA–DNA hybridization (dDDH), average nucleotide identity (ANI) analysis and core genome analysis confirmed that these strains represent a novel species within the genus Pseudomonas. The three strains demonstrated ANIb values >99.5% with each other, confirming that all three strains (CCUG 74779T = CECT 30571T, CCUG 74780 and CCUG 74781) belong to the same genomospecies. Phylogenomic analysis confirmed that the strains form a distinct genomic clade, representing a novel taxonomic species, for which the name Pseudomonas imrae sp. nov., is proposed, with strain CCUG 74779T (=CECT 30571T) designated as the type strain. We report the complete genome sequence of the type strain of P. imrae sp. nov. and show that it carries a gene encoding a novel variant of a chromosomally-encoded class C β-lactamase, which has been designated as PFL-7.
Introduction
The species of the genus Pseudomonas are aerobic, oxidase-positive, Gram-negative bacilli. They are present in a wide variety of environments, including soil, water, plants, humans, and other animals, and have a broad metabolic diversity (Palleroni, 2015). Many Pseudomonas species exhibit beneficial functions in the environment, including biodegradation of complex compounds and enhanced plant growth (Dong et al., 2023; Pieterse et al., 2021; Peix et al., 2018), while some species are opportunistic pathogens, with P. aeruginosa being the most virulent and clinically-relevant human pathogen of the genus (Qin et al., 2022; Ikuta et al., 2022). The genus Pseudomonas is highly diverse and taxonomically complex, comprising more than 340 validly published species names,1 which can be further subdivided into several groups and subgroups (Lalucat et al., 2020; Lalucat et al., 2022). Some members of the genus Pseudomonas have been recently reclassified into several novel genera, such as Atopomonas, Halopseudomonas, Stutzerimonas and Trinickia, by whole genome phylogenetic analyses (Rudra and Gupta, 2024). However, multiple groups remain within the genus, such as the P. fluorescens group, which can further be divided into several subgroups, such as the P. gessardii subgroup (Lalucat et al., 2020; Lalucat et al., 2022). Species of the P. gessardii subgroup have been mostly described based on strains isolated from water samples, soil or plants, and strains isolated from fish (Duman et al., 2021). Although reports on infections are scattered, they are related to the P. fluorescens subgroup, which encompasses species that apart from multiple environments, have been isolated from clinical samples (Scales et al., 2014).
Pseudomonas aeruginosa and other clinically important Pseudomonas spp. are known to harbor natural and acquired resistance genes against different antibiotics, including β-lactams (Lodge et al., 1990; Girlich et al., 2004; Fajardo et al., 2014; Zhao and Hu, 2010). They exhibit resistance to several clinically important antimicrobials such as β-lactam antibiotics, including penicillin and cephalosporins, owing to the presence of AmpC β-lactamases and drug efflux pumps. Thus, infections caused by pathogenic species of the genus are often difficult to treat.
In this study, we used a polyphasic approach, including phylogenomic analyses, to confirm that three bacterial strains, previously isolated from gut samples of wild Atlantic mackerel (Scomber scombrus) from the northern North Sea (Marathe et al., 2022; Nimje and Marathe, 2023), represent a novel species within the P. gessardii subgroup of the genus Pseudomonas. The name Pseudomonas imrae sp. nov. is proposed. The strains exhibited high minimum inhibitory concentrations for several antibiotics and carry a gene encoding a novel variant of class C β-lactamase, blaPFL-7.
Materials and methods
Strain isolation and identification
Strains 16FHM2T (=CCUG 74779T=CECT 30571T), 15FMM2 (=CCUG 74780) and 15FMM3 (=CCUG 74781) were isolated from gut contents of two specimens of wild Atlantic mackerel (Scomber scombrus) collected in the northern North Sea (International Council for the Exploration of the Sea, ICES region 4.a, in November 2018) (Marathe et al., 2022). The three strains were isolated on Mueller-Hinton (MH) agar containing meropenem (0.125 μg mL−1), incubated at 30°C, for 36 h.
Initial identification was performed, using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), using Bruker MALDI Biotyper® (Bruker Daltonics, Bremen, Germany), as previously described (Grevskott et al., 2024); the strains were identified as belonging to the genus Pseudomonas but could not be assigned to any described species. Total genomic DNA was extracted from strain 16FHM2T, using the previously described “heat-shock” protocol (Welinder-Olsson et al., 2000). The nearly complete 16S rRNA gene was amplified by PCR, using modified versions of primers (16F28, 5’-AGAGTTTGATCKTGGCTCAG-3′ and 16R1494, 5’-TACGGYTA CCTTGTTACGAC-3′) (Lane, 1991; Hauben et al., 1997), as described previously (Carvalheira et al., 2020). The PCR products were purified and sequenced, as described previously (Jaén-Luchoro et al., 2020), using modified versions sequencing primers (16F530, 5’-TTCGTGCCAGCAG CCGCGG-3′ 16R806, 5’-GGACTACCAGGGTATCTAAT-3′; 16F1103, 5’-TGTTGGGTTAAGTCCCGCAAC-3′, and 16R1494 5’-TACGGYTA CCTTGTTACGAC-3′) (Lane, 1991; Hauben et al., 1997; Buchholz-Cleven et al., 1997) and an Applied Biosystems SeqStudio 8 Flex Genetic Analyzer system (Thermo Fisher Scientific, Waltham, MA, United States). The partial rpoD sequence was determined, using previously-described primers and protocols for amplification and sequencing (Mulet et al., 2009). The 16S rRNA and rpoD gene sequences were analyzed using EzBiocloud (Yoon et al., 2017), and NCBI BLAST (Altschul et al., 1990) against sequences of type material of the Nucleotide collection (nr/nt) of GenBank (Sayers et al., 2024), respectively.
Growth assays and biochemical tests
The strains were grown overnight on MH Agar medium and were characterized by biochemical profiling, using the CCUG NFX worksheet,2 including API ZYM and API 20NE commercial panels (bioMérieux, Marcy-l’Étoile, France). Bacterial growth was evaluated by streaking the strains on MH Agar plates and incubating at different temperatures ranging from 4 to 42°C for as long as 96 h.
Microscopy analyses
Cell sizes and morphology were examined after 1 day incubation on Blood Agar medium, at 30°C, using a digital holotomographic microscope (DHM; HT-2, Tomocube Inc., Daejeon, South Korea), following previously described procedures (Fernández-Juárez et al., 2023). This enables non-invasive, label-free 3D imaging of bacterial cell morphology, based on obtaining tomographic refractive index (RI) data, which can subsequently be segmented to highlight particular cell structures based on their distinct RI signatures. For holotomographic imaging, bacterial colonies were recovered from the agar medium and re-suspended in phosphate-buffered saline (PBS). Then, 30 μL of the bacterial suspension was placed in a Tomodish (Tomocube Inc., Daejeon, South Korea) and covered with a coverslip in preparation for imaging. The resulting tomographic imaging data were visualized and analyzed, using TomoStudio and TomoAnalysis software (Tomocube Inc., Daejeon, South Korea). An average of 20 cells were imaged and analyzed, and the resulting images were exported to ImageJ software for additional analysis and figure preparation.
Antimicrobial susceptibility testing
The strains were grown overnight on MH Agar medium with ampicillin (100 μg mL−1) and analyzed for antimicrobial sensitivity, using broth dilution method on Sensititre™ plates (Thermo Fisher Scientific, Waltham, MA, United States), following the manufacturer’s instructions, as described previously (Grevskott et al., 2024). Briefly, suspensions were prepared from freshly grown cultures to an optical density of 0.5 McFarland. Ten microliters of suspensions were added to 10 mL of MH Broth and added to sensititre plates. The plates were incubated at 30°C (optimal temperature for the strains) for 24 h and read manually. Minimum inhibitory concentrations (MICs) were determined as the lowest tested antimicrobial concentrations with no observed growth, as recommended by the EUCAST reading guide for broth microdilution (Version 5.0, January 2024).
Cell fatty acid-fatty acid methyl ester analysis
The strains were cultivated on Trypticase Soy Agar medium at 28°C for 24 h for cellular fatty acid-fatty acid methyl ester (CFA-FAME) analysis. The CFA-FAME profiles were determined, using a gas chromatograph (HP 5890; Hewlett-Packard, Palo Alto, CA, United States), following a standardized protocol similar to that of the MIDI Sherlock MIS system (Sasser, 2001), as described previously (Zamora et al., 2012).
Whole genome sequencing and genome assembly
Strains were cultivated on MH Agar medium with ampicillin 100 μg mL−1 at 30°C for 24 h, and genomic DNA was prepared, using a DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany), for Illumina short-read sequencing, and a modified version (Salvà-Serra et al., 2018) of a previously described protocol (Marmur, 1961) for Oxford Nanopore long-read sequencing. For Illumina sequencing, a paired-end library was prepared, using a Nextera DNA Flex library prep kit, and sequenced on an Illumina MiSeq platform (Illumina, Inc., San Diego, CA, United States) at the Norwegian Sequencing Centre in Oslo. For Oxford Nanopore sequencing, a library was prepared, using a Rapid Barcoding Sequencing kit (SQK-RBK004), and sequenced for 72 h on a MinION device (Oxford Nanopore Technologies, Ltd., Oxford, United Kingdom), at the Research Lab of the Culture Collection University of Gothenburg (CCUG), using a FLO-MIN106 (version R9.4.1) flow cell and analyzed, using the software MinKNOW version 3.6.5 (Oxford Nanopore Technologies), with default parameters. The raw Oxford Nanopore reads were base-called, using Guppy version 3.4.5 and evaluated, using NanoPlot version 1.26.3 (De Coster et al., 2018). The Oxford Nanopore sequence reads were assembled de novo following a previously described protocol (Wick et al., 2023). Briefly, four subsets of the Oxford Nanopore reads were created, using Trycycler v0.5.5 (Wick et al., 2021). Each subset was assembled de novo, using Flye v2.9.5 (Kolmogorov et al., 2019), Raven v1.8.3 (Vaser and Šikić, 2021) and Canu v2.2 (Koren et al., 2017). A consensus assembly was generated, using Trycycler v0.5.5, and polished using three different methods: Medaka v1.11.3 (i.e., using the Oxford Nanopore reads)3, with the mode r941_min_high_g344; Polypolish v0.6.0 (i.e., using the Illumina reads) (Wick and Holt, 2022); and POLCA, using Masurca v4.1.0 (Zimin and Salzberg, 2020; Zimin et al., 2013). Manual observations of read mappings were performed, using UGENE v48.1 (Okonechnikov et al., 2012). The Illumina-only assemblies were performed, using SPAdes version v3.13 (Bankevich et al., 2012), and evaluated, using QUAST version 5.2 (Gurevich et al., 2013). The genome assemblies were annotated, using PGAP v6.3 and v6.9 (Tatusova et al., 2016).
β-lactamase sequence analysis
The amino acid sequences of possible β-lactamases were analyzed, using TBLASTN against the entire Core nucleotide database (core_nt) of NCBI GenBank (Sayers et al., 2024). A second search was conducted, against the Nucleotide collection (nr/nt), restricting the search space to the genus Pseudomonas (Taxonomy ID: 286). Sequences were also analyzed using BLASTP against the Beta-Lactamase DataBase (Naas et al., 2017). The genetic context was analyzed, using Unipro UGENE v48.1 (Okonechnikov et al., 2012).
Overall genome relatedness indices
Digital DNA–DNA hybridization (dDDH) (Auch et al., 2010) values were determined, using the Genome-to-Genome Distance Calculator (GGDC) v3.0 (Meier-Kolthoff et al., 2013). Average nucleotide identity values, based on BLAST (ANIb) (Goris et al., 2007; Altschul et al., 1990), were calculated, using the webserver JSpeciesWS (Richter et al., 2016). For each comparison, ANIb values were determined bi-directionally and the average calculated.
Average nucleotide identity (ANI)-based dendrogram
The matrix containing the average ANIb values was used to generate a dendrogram, using the software PermutMatrix v1.9.3 (Caraux and Pinloche, 2005). The dendrogram was constructed, using Pearson’s distance correlation and hierarchical clustering with an average linkage method (UPGMA). The dendrogram was displayed, using the Interactive Tree of Life (iTOL) v7.1 (Letunic and Bork, 2024).
Core genome-based phylogenomic analysis
A core genome-based phylogenomic tree was constructed, including type strains of species of the P. fluorescens and P. gessardii subgroups (Supplementary Table 1). Briefly, the genome sequences were annotated, using Prokka v1.14.6 (Seemann, 2014), and the annotated proteins sequences were compared, using the Software GET_HOMOLOGUES v17112020 (Contreras-Moreira and Vinuesa, 2013), with BLASTP (all vs. all) (Altschul et al., 1990). The sequences were clustered by applying a 70/70 criterion (i.e., 70% of identity over 70% of the length), using three clustering algorithms: BDBH; COGtriangles (Kristensen et al., 2010); and OrthoMCL (Li et al., 2003). The intersection of the three algorithms was used to determine a consensus core genome formed by single-copy orthologous sequences. The sequences were aligned, using Clustal Omega v1.2.0 (Sievers et al., 2011), and the alignment was trimmed, using Gblocks v0.91b (Castresana, 2000). Subsequently, the alignments were concatenated and a phylogenomic tree was constructed, using PhyML v20120412 (Guindon et al., 2010) and a Shimodaira-Hasegawa-like approximate likelihood-ratio test (SH-aLRT) for branching statistical support (Anisimova and Gascuel, 2006). The phylogenomic tree was visualized, using iTOL v7.1 (Letunic and Bork, 2024).
Ecological distribution
Additional related strain genome sequences were searched by analyzing the partial rpoD sequence, using BLASTN (Altschul et al., 1990), against the Nucleotide collection (nt) of NCBI. Metagenome-assembled genomes (MAGs) of the proposed novel species were searched using Protologger v2 (Hitch et al., 2021). The Branchwater Metagenome Query platform (Irber et al., 2022) was used to search in more than one million metagenomic datasets from the Sequence Read Archive (SRA) (Leinonen et al., 2011).
Results
Strain isolation and characterization
During a screening for β-lactam-resistant bacteria in gut samples of wild Atlantic mackerel (Scomber scombrus) from the northern North Sea, three Pseudomonas spp. strains were isolated. The rod-shaped cells were Gram-stain-negative, forming smooth, translucent (2–3 mm wide) colonies when grown for 36 h on MH agar medium with ampicillin.
MALDI-TOF MS typing analysis could not identify the strains to the species level. The 16S rRNA gene sequence showed relatedness to P. libanensis (99.9%), as well as to P. synxantha, P. gessardii and P. shahriarae (99.9%) while partial rpoD sequence indicated distant relationships to P. proteolytica (93.4%) and P. mucidolens (92.7%) and, thus, could not be assigned to any existing species of the genus Pseudomonas (Girard et al., 2020). The discrepancies observed in the identifications by 16S rRNA gene and rpoD sequence analyses indicated a high probability that the strains represented a novel species.
The strains are Gram-negative and motile. The strains grow between 10 and 35°C but not at 4 or 42°C on MH Agar medium. Only one of the strains was able to grow when cultivated on blood agar at 37°C, suggesting that persistence in humans might be possible. The strains demonstrated good growth at salinities up to 3% (w/v) NaCl, while variable growth was observed at 4.5 and 5% NaCl. The strains are catalase- and oxidase-positive and exhibit gelatine hydrolysis, nitrate reduction and esterase activities. Additional phenotypic traits, including the results of multiple growth assays and biochemical analyses, are presented in Supplementary Table 2. The cell morphology was studied by digital holotomographic microscopy and the cell size was determined to be 1.6 ± 0.3 μm by 0.8 ± 0.05 μm (Figure 1).
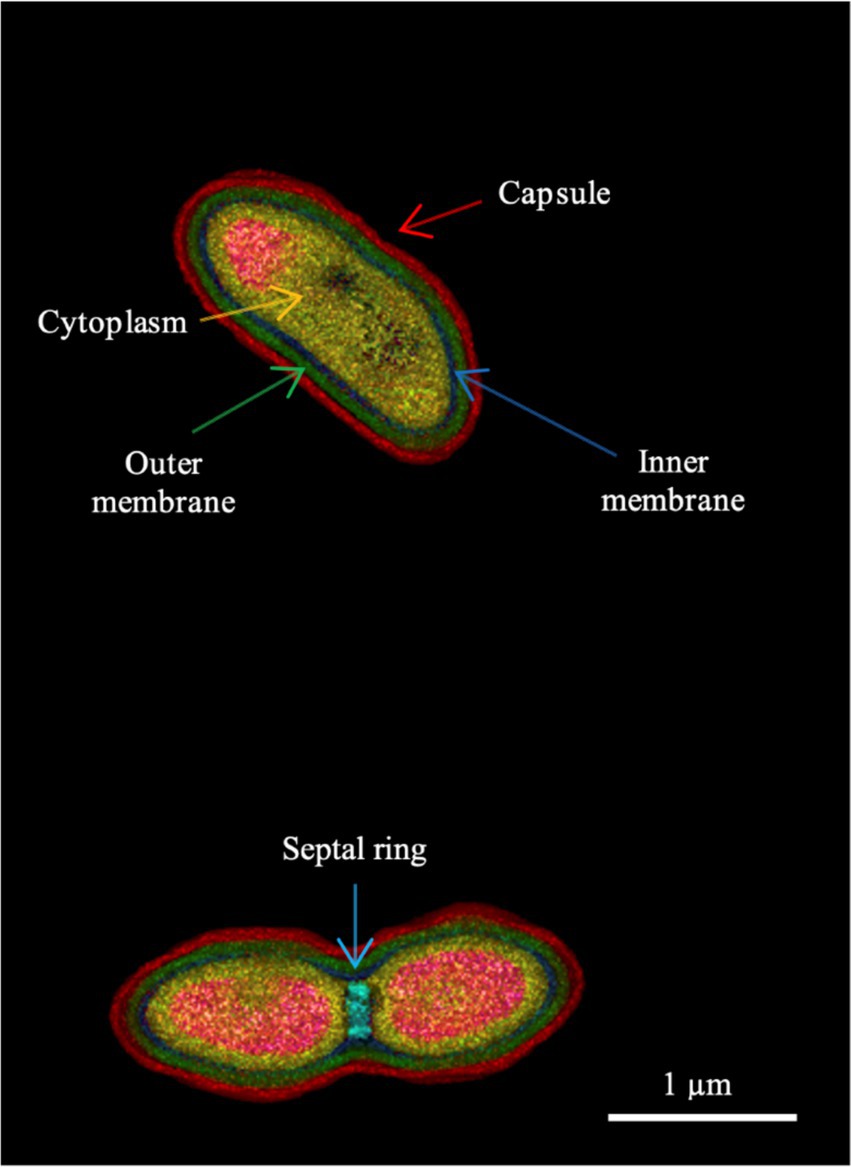
Figure 1. Holotomographic microscopy image of strain 16FHM2T showing different morphological features of the cells, indicated by different color arrows. The lower part of the figure shows a dividing cell. The colors represent different refractive index ranges (see Supplementary Figure 1 for the refractive index scale). Strain 16FHM2T was cultivated on Blood Agar medium, for 1 day, at 30°C.
Antimicrobial susceptibility
The strains exhibited high MIC values for ampicillin (>64 μg mL−1), cephalosporins, such as cefazolin, cefuroxime and cefoxitin (>32 μg mL−1), and carbapenems, such as ertapenem (8 μg mL−1), as well as against azithromycin (8 μg mL−1) and trimethoprim (>16 μg mL−1), while the strains exhibited low MIC values for ciprofloxacin, gentamycin and tigecycline. The MICs for different antimicrobials tested are presented in Supplementary Table 3.
Cell fatty acids
Strains 16FHM2T, 15FMM2 and 15FMM3 presented CFA-FAME profiles characteristic of species of the genus Pseudomonas: palmitic acid (C16:0), C10:0 3-OH, C12:0 2-OH and C12:0 3-OH. The major CFAs of the strains were C16:1 ω7c (36.0–37.2%), C16:0 (21.6–23.4%), followed by the summed feature formed by C18:1ω7c, 12 t and/or 9 t (11.7–12.9%) and C17:0 cyclo (10.9–11.8%) (Table 1). Compared with P. mucidolens CCUG 1424T, the three strains present similar CFA-FAME patterns overall, although they displayed higher proportions of C17:0 cyclo and lower proportions of C18:1ω7c/12t/9t.
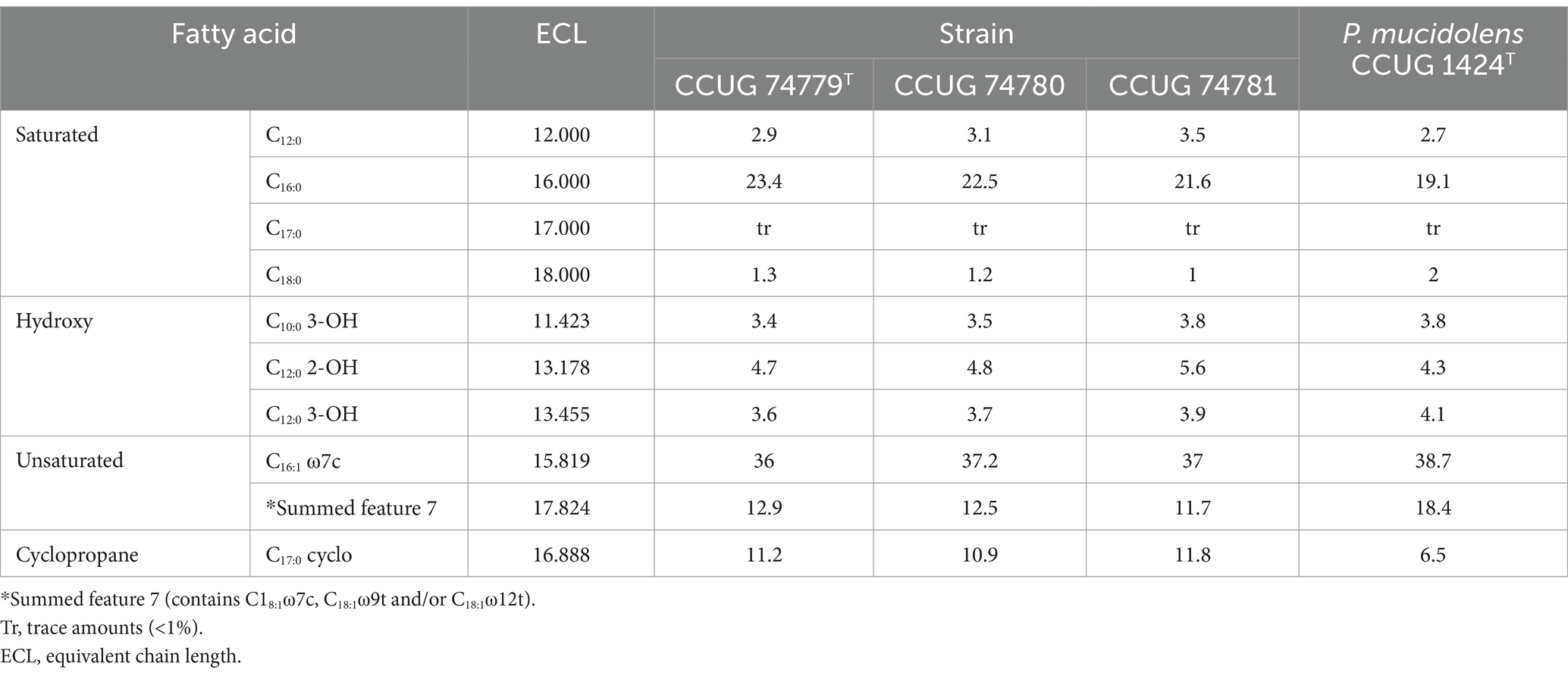
Table 1. Cellular fatty acid compositions (%) of the three strains of the proposed novel species, P. imrae sp. nov. and the type strain of P. mucidolens, its most closely-related species.
Whole-genome sequencing and overall genome relatedness indices
The genomes of the three strains were sequenced, using an Illumina MiSeq platform. Additionally, strain 16FHM2T was sequenced, using also an Oxford Nanopore MinION device. The assembly of strain 16FHM2T resulted in a single complete sequence of 5,444,440 bp (Table 2). The assemblies of strains 15FMM2 and 15FMM3 resulted in two draft genome sequences of 5,406,388 and 5,405,281 bp, respectively. The number of coding sequences per genome ranged from 4,904 to 4,906 and the GC contents of the genomes were determined to be 58.9%.
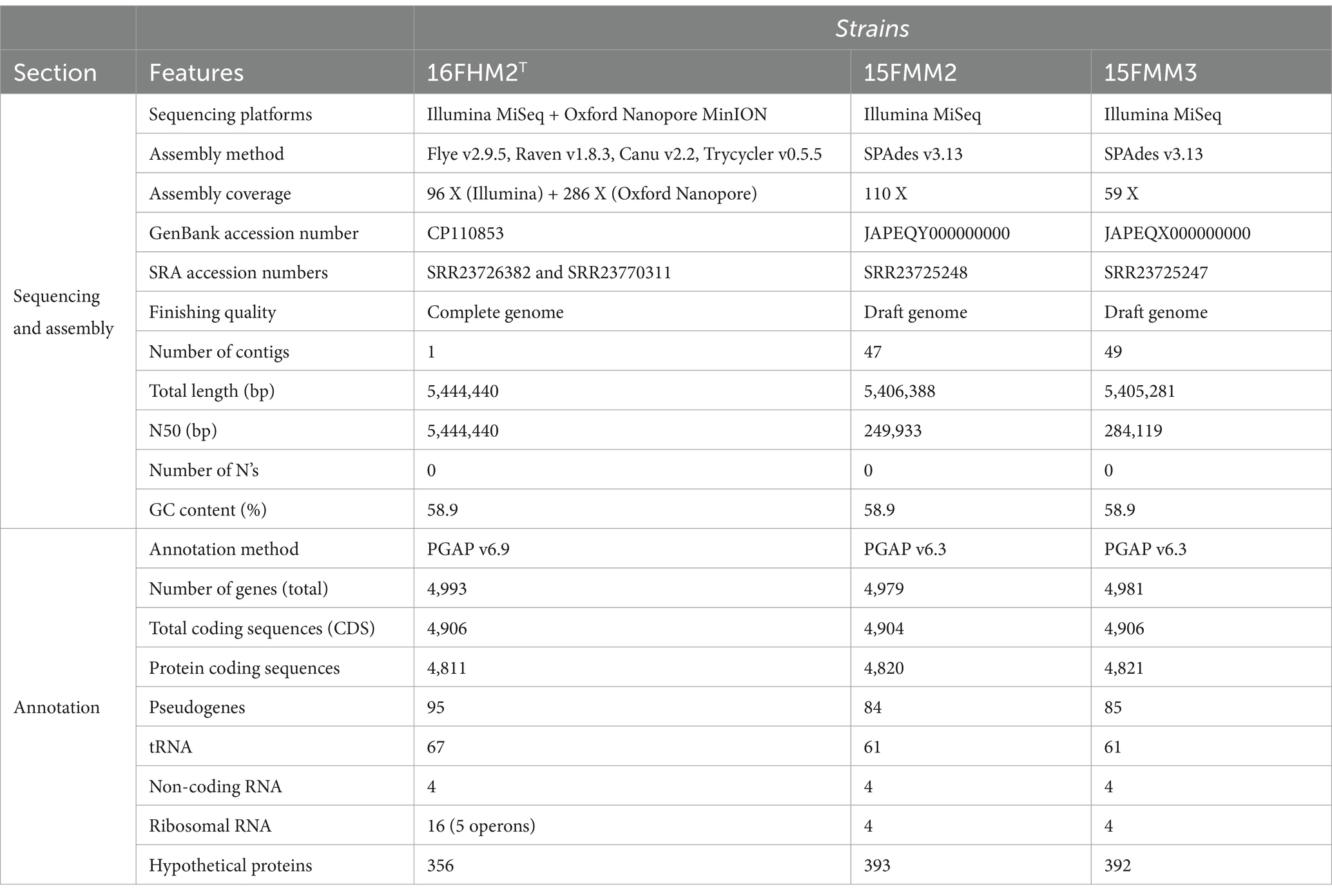
Table 2. Genome features of the three strains of the proposed novel species, P. imrae sp. nov.
ANIb and dDDH were calculated between the three strains of the proposed novel species, and between strain 16FHM2T and the type strains of 55 closely-related Pseudomonas species belonging to the P. gessardii and P. fluorescens subgroups of the P. fluorescens group. The ANIb and the dDDH values between the three strains of the proposed novel species were 99.99 and 100%, respectively, confirming that the three strains are very similar and closely related to each other. The ANIb values between the genome sequence of strain 16FHM2T and those of the type strains of species of the P. fluorescens and P. gessardii subgroups with validly published names ranged from 87.4 to 82.2%, while the dDDH values ranged from 35.3 to 26.8% (Table 3). The analyses confirmed that the most closely related species is P. mucidolens. These data indicate that strain 16FHM2T represents a novel species within the P. gessardii subgroup of the genus Pseudomonas.
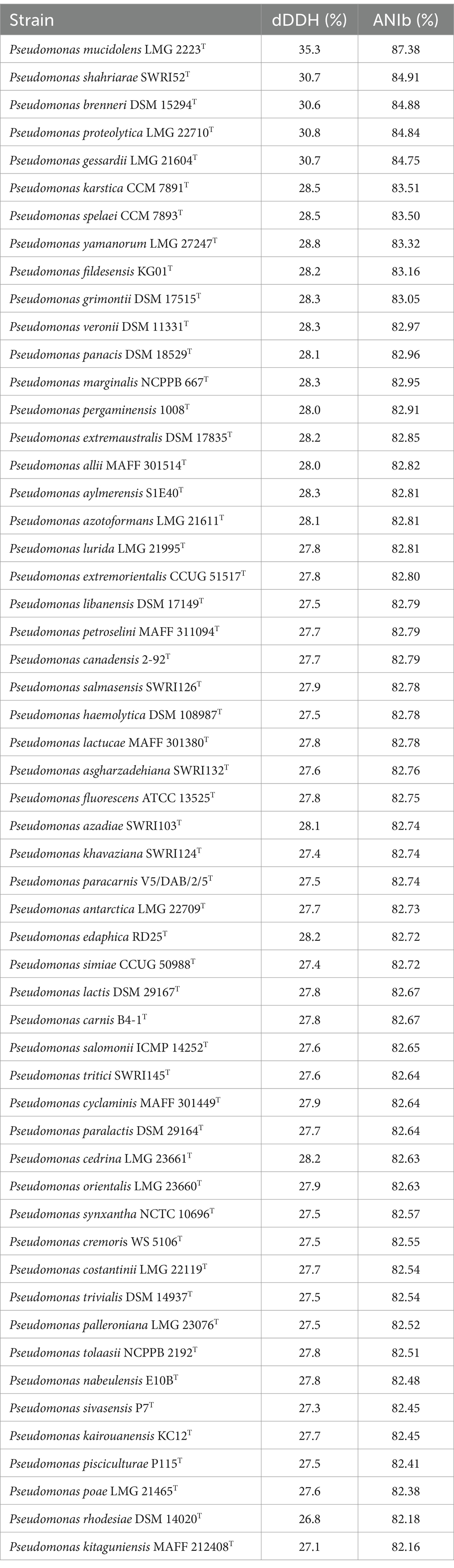
Table 3. ANIb and dDDH values determined between the genome sequence of strain 16FHM2T and the genome sequences of the type strains of species of the P. gessardii and P. fluorescens subgroups.
Whole-genome sequence ANIb dendrogram
The ANIb values were determined between (all vs. all) genome sequences of type strains of the species of the P. gessardii and P. fluorescens subgroups. The values ranged from 82.02% (between P. mucidolens and P. kitaguniensis) to 96.44% (between P. panacis and P. marginalis). The ANIb values of strain 16FHM2T compared with the type strains of other species ranged from 87.34% (P. mucidolens LMG 2223T) and 82.16% (P. kitaguniensis MAFF 212408T), which suggests that P. mucidolens is the most closely related species. Indeed, the dendrogram also shows the relationship of P. mucidolens to the proposed novel species, within the cluster formed by species of the P. gessardii subgroup (Figure 2).
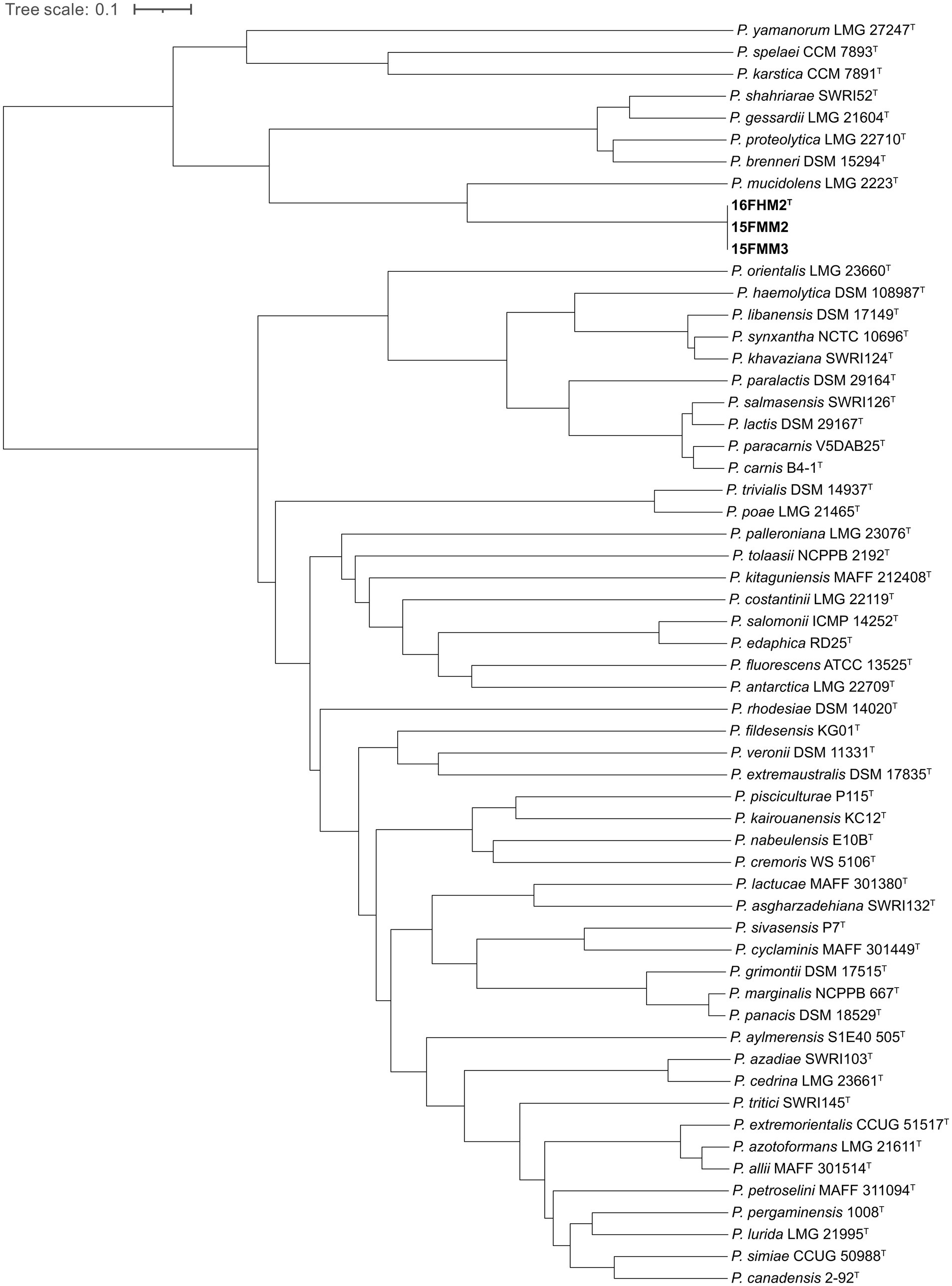
Figure 2. Dendrogram generated from whole-genome sequence ANIb determinations, demonstrating the estimated relationships of the three strains of the proposed novel species and type strains of species of the P. gessardii and P. fluorescens subgroups.
Core genome-based phylogenomic analysis
A total of 381,851 amino acid positions, encoded by 1,361 single-copy shared genes, were used to construct the core genome-based phylogenomic tree, including the genome sequences of the type strains of species of the P. gessardii and P. fluorescens subgroups. The core genome confirms that the species are divided in two well-defined clusters, corresponding to the two subgroups included in the analysis, and that the proposed novel species is a member of the P. gessardii subgroup. Additionally, the analysis confirms that P. mucidolens is the most closely related species (Figure 3).
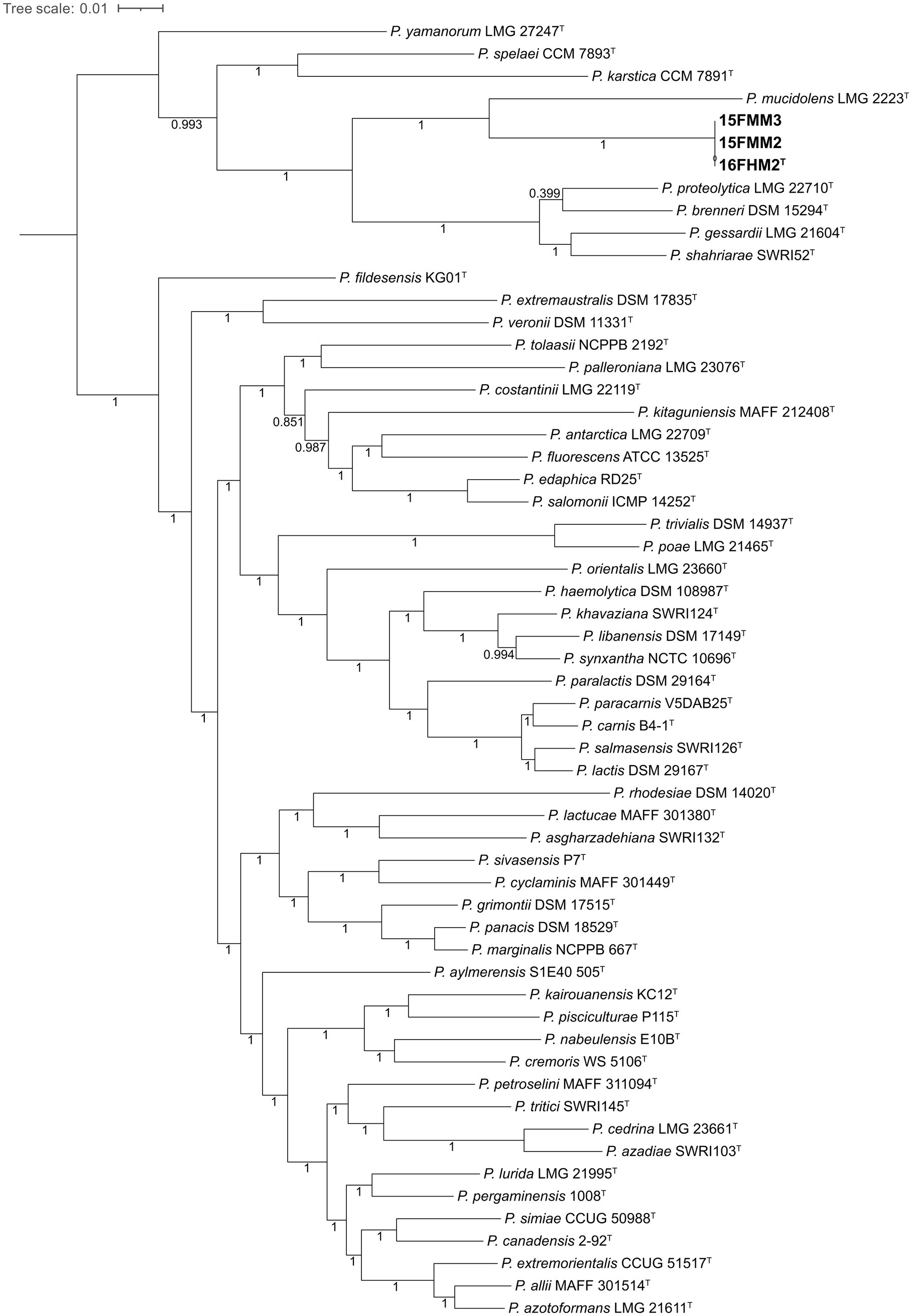
Figure 3. Core genome-based phylogenomic tree of the three strains of the proposed novel species and type strains of species of the P. gessardii and P. fluorescens subgroups. The tree was constructed, using Maximum Likelihood and the Shimodaira-Hasegawa-like approximate likelihood-ratio test (SH-aLRT). The numbers at the nodes indicate the SH-aLRT support values.
Class C β-lactamase
We detected a new variant of class C β-lactamase in our strains (NCBI Reference Sequence accession number: WP_410017811.1). This variant showed 85.3% amino acid identity (query coverage 100%) to a Class C β-lactamase from P. mucidolens. The search against the Beta-Lactamase DataBase revealed that it belongs to the family PFL and therefore it was designated PFL-7. The most closely related listed variant was PFL-5 (WP_017475175.1), from Pseudomonas sp. PAMC 26793, with 76% of amino acid identity. PFL-7 has a LysR family regulator (WP_410017810.1) encoded upstream of its gene, which shows 92.7% amino acid identity to the regulator from P. mucidolens. Thus, PFL-7 forms an AMP-C-AMP-R system detected in many bacteria (Balasubramanian et al., 2012).
Ecological distribution
The rpoD sequence-based search of the Nucleotide collection (nt) of NCBI did not detect any additional strain of the proposed novel species. No MAGs of the proposed novel species were found when screening thousands of MAGs, using Protologger. The Branchwater Metagenome Query search did not yield any match with a containment ANI (cANI) score larger than 0.97, which often represents a species-level match, but yielded 41 matches with a cANI score between 0.95 (33 matches) and 0.96 (8 matches) (Supplementary Table 4). These matches probably do not represent members of the same species but might represent related strains of related taxa within the analyzed metagenomic datasets. The samples originated from fresh water (n = 13), wastewater (n = 21) and food samples (leafy greens, chicken and a meat factory) (n = 7).
Discussion
Three β-lactam-resistant isolates were obtained from gut samples of wild Atlantic mackerel (Scomber scombrus) from the northern North Sea and could not be assigned to any previously described species. Using a polyphasic approach, including phenotypic, chemotaxonomic, phylogenetic and phylogenomic analyses, we have demonstrated that these three isolates represent a novel species of Pseudomonas within the P. gessardii subgroup of the P. fluorescens group, for which the name Pseudomonas imrae sp. nov. is proposed. The species belonging to the genus Pseudomonas and, more particularly, species of the P. gessardii subgroup, are widespread in aquatic environments, including marine habitats. P. proteolytica was isolated from water bodies in Antarctica (Reddy et al., 2004), P. gessardii and P. brenneri from natural mineral waters (Baida et al., 2001), P. yamanorum from soil on the coast of Observatorio island in south Patagonia (subantarctic environment), P. karstica and P. spelaei from caves (Arnau et al., 2015; Švec et al., 2020), and additional strains of species of the subgroup were isolated from fish (Duman et al., 2021). These observations are in accordance with the results of the Branchwater Metagenome Query, in which 33 of the 41 metagenomic samples containing related strains originated from fresh water or wastewater. This suggests that subgroup-specific adaptations for aquatic environments might be observed in the P. gessardii subgroup, although some species were found that were associated with plants and animals, such as P. mucidolens (isolated from egg) (Levine and Anderson, 1932) or P. shahriarae, isolated from rhizosphere of wheat (Girard et al., 2021).
Pseudomonas has a complex taxonomy with several species showing high similarities in biochemical analyses, as well as in studies using molecular markers, such as the 16S rRNA gene sequence. To overcome difficulties in classifying and differentiating Pseudomonas species, using such methods, alternative protocols, such as multilocus sequence analysis (MLSA) and whole genome sequence analysis have been proposed (Gomila et al., 2015). MLSA, using gene sequences, such as gyrB, rpoB and rpoD, and whole genome sequence analysis, using parameters, such as ANIb and core genome-based phylogeny, have made it possible to resolve the taxonomic spectrum of the genus Pseudomonas. Using these tools, several species have been recently reclassified (Lalucat et al., 2022; Rudra and Gupta, 2024). Our study adds to the recognition of the diversity of Pseudomonas species and highlights the use of for whole genome sequence analysis for the definitive resolution of the taxonomy of genus Pseudomonas. We further determined the complete genome sequence of the type strain of the proposed novel species. Using the described polyphasic approach, we have demonstrated that the three strains isolated and characterized in our study represent a novel species in the P. gessardii subgroup of Pseudomonas, for which the name Pseudomonas imrae sp. nov. is proposed.
Pseudomonas spp. are known to be intrinsically resistant to several antimicrobials including commonly used antibiotics and disinfectants (Poole, 2011). PFL-7 forms a typical AmpC-AmpR system detected in many bacteria (Balasubramanian et al., 2012). Amp-R is a regulator that modulates ampC expression. High expression of ampC, due to higher expression of ampR, has been shown in previous studies to be associated with resistance against a variety of β-lactam antibiotics, including penicillins, cephalosporins and, sometimes, carbapenems (Tariq et al., 2023). Along with activation of AmpC, AmpR also regulates genes responsible for recycling of cell wall/peptidoglycan on stimuli of cell wall damage, thus, emphasizing the presence of this system in a variety of bacteria including the genus Pseudomonas (Gyger et al., 2024; Balasubramanian et al., 2015). The presence of AmpC-AmpR system in our strain, may thus, explain the high MIC observed for P. imrae against different β-lactam antibiotics.
Description of Pseudomonas imrae sp. nov.
Pseudomonas imrae (im’rae. N.L. gen. n. imrae, formed from IMR, acronym for Institute of Marine Research, Norway, where the first strains were isolated and studied).
Cells are Gram-negative, rod shaped, non-spore forming, and strictly aerobic, positive for catalase and oxidase. Optimum temperature for growth is 25–30°C, with no growth observed at 4°C and 42°C. The strains are positive for catalase, oxidase, acid phosphatase, esterase, gelatin hydrolysis, L-Arginine dihydrolase activity, and nitrate reduction, while they are negative for urease, α-fucosidase activity, α-mannosidase activity, N-acetyl-β-glucosaminidase activity, β-glucosidase activity, α-glucosidase activity, β-glucuronidase activity, β-galactosidase, α-galactosidase, acetamide utilization, DNAse activity, indole production and esculin hydrolysis. The strains can grow in the presence of 3% NaCl and show variable growth at 4.5 and 5% NaCl with no growth observed above 6% NaCl. The predominant cell fatty acids are C16:1 ω7c and C16:0, followed by C18:1ω7c/12 t/9 t and C17:0 cyclo, which are present in lower levels.
The 16S rRNA gene sequence is highly similar to P. libanensis, P. synxantha, P. gessardii and P. shahriarae (99.9%), while the partial rpoD sequence shows highest similarity to P. proteolytica (93.4%). Members belong to class Gammaproteobacteria, order Pseudomonadales, family Pseudomonadaceae, genus Pseudomonas, P. fluorescens group, P. gessardii subgroup. The type strain of the species is Pseudomonas imrae strain 16FHM2T (=CCUG 74779T = CECT 30571T); strains15FMM2 (=CCUG 74780) and 15FMM3 (= CCUG 74781) are other representatives. The strains were isolated from gut contents of two specimens of wild Atlantic mackerel (Scomber scombrus) collected in the northern North Sea, ICES region 4.a, in November 2018.
Conclusion
Using a combination of phenotyping methods, genomics and phylogenomics, Pseudomonas imrae sp. nov. is described as a novel species of the genus Pseudomonas, belonging to the P. gessardii subgroup of the P. fluorescens group, isolated from the gut contents of Atlantic mackerel in Norway. The three characterized strains of P. imrae carry a novel class C β-lactamase gene variant. Our study highlights the importance of whole genome sequencing in bacterial taxonomy.
Data availability statement
The strains are deposited and available at the Culture Collection University of Gothenburg (CCUG) under the accession numbers CCUG 74779T (=16FHM2T), CCUG 74780 (=15FMM2) and CCUG 74781 (=15FMM3). The type strain is also deposited and available at the Spanish Type Culture Collection (CECT, Valencia, Spain) under the accession number CECT 30571T. The genome sequences of the strains CCUG 74779T (=16FHM2T), CCUG 74780 (=15FMM2) and CCUG 74781 (=15FMM3) have been deposited in DDBJ/ENA/GenBank under the accession numbers CP110853, JAPEQY000000000 and JAPEQX000000000, respectively. The Illumina and the Oxford Nanopore sequence reads are deposited and publicly available at the Sequence Read Archive (SRA) under the accession numbers SRR23726382, SRR23770311, SRR23725248 and SRR23725247. The nearly-complete 16S rRNA gene sequence and the partial rpoD gene sequence for strain 16FHM2T, determined by Sanger sequencing, are deposited in DDBJ/ENA/GenBank under the accession numbers PQ479520 and PQ505025, respectively.
Author contributions
FS-S: Data curation, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Formal analysis, Writing – review & editing. PN: Formal analysis, Investigation, Methodology, Writing – review & editing. BP-I: Investigation, Writing – review & editing. LA: Investigation, Writing – review & editing, Methodology. SC: Investigation, Writing – review & editing. EI: Investigation, Writing – review & editing, Methodology. SJ-M: Investigation, Writing – review & editing. MO: Investigation, Writing – review & editing. H-SS: Investigation, Writing – review & editing. CU: Investigation, Writing – review & editing. VF-J: Investigation, Writing – review & editing. CP: Investigation, Writing – review & editing. MK: Investigation, Methodology, Writing – review & editing. EM: Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. NM: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Formal analysis, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Institute of Marine Research under the Ocean health initiative (project number 15495) and marine microbiota project (project number 15930). This study was supported by the Culture Collection University of Gothenburg (CCUG; www.ccug.se), Project: Genomics and Proteomics Research on Bacterial Diversity. The CCUG is supported by the Department of Clinical Microbiology, Sahlgrenska University Hospital, Gothenburg, Region Västra Götaland, Sweden. Additional support for the tomographic imaging was from the Carlsberg Foundation (project number CF21-0599; MK), and the European Union (Marie Skłodowska-Curie grant agreements no. 101073507 to MK and no.101108420 to CP).
Acknowledgments
We acknowledge the Culture Collection University of Gothenburg (CCUG, Gothenburg, Sweden) and the Spanish Type Culture Collection (CECT, Valencia, Spain) for maintaining the strains. The computations were performed, using resources provided by the Swedish National Infrastructure for Computing (SNIC), through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX), under project SNIC 2019/8-176. We thank Arne Levsen for providing mackerel from research cruise on surveillance of fish parasites. We thank Valeria Ruffo (University of Copenhagen), for strain cultivation in preparation for holotomography analysis. We acknowledge Aharon Oren at the Hebrew University of Jerusalem for assistance with the nomenclature of the novel taxon.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1530878/full#supplementary-material
SUPPLEMENTARY FIGURE 1 | Reconstruction of Pseudomonas imrae strain 16FHM2T using its refractive index, obtained with holotomographic imaging.
SUPPLEMENTARY TABLE 1 | List of genome sequences of type strains of species of the Pseudomonas gessardii and Pseudomonas fluorescens subgroups included in the study.
SUPPLEMENTARY TABLE 2 | Phenotypic characteristics of the three strains of the proposed novel species and the type strain of Pseudomonas mucidolens.
SUPPLEMENTARY TABLE 3 | Minimum Inhibitory Concentrations (MIC) of different antimicrobials determined using SensititreTM Standard AST Plates (Thermo Scientific, United States) for the three strains of the proposed novel species.
SUPPLEMENTARY TABLE 4 | Top matches from the search of strain 16FHM2T using the Branchwater Metagenome Query platform.
Abbreviations
ANIb, average nucleotide identity based on BLAST; cANI, containment average nucleotide identity; CFA-FAME, cellular fatty acid – fatty acid methyl ester; dDDH, digital DNA–DNA hybridization; ICES, International Council for the Exploration of the Sea; MALDI-TOF MS, Matrix-Assisted Laser Desorption/Ionization-Time Of Flight Mass Spectrometry; MAG, metagenome-assembled genome; MH, Mueller-Hinton; MIC, minimal inhibitory concentration; MLSA, multilocus sequence analysis.
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Arnau, V. G., Sánchez, L. A., and Delgado, O. D. (2015). Pseudomonas yamanorum sp. nov., a psychrotolerant bacterium isolated from a subantarctic environment. Int. J. Syst. Evol. Microbiol. 65, 424–431. doi: 10.1099/ijs.0.065201-0
Auch, A. F., Von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital Dna-Dna hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Baida, N., Yazourh, A., Singer, E., and Izard, D. (2001). Pseudomonas brenneri sp. nov., a new species isolated from natural mineral waters. Res. Microbiol. 152, 493–502. doi: 10.1016/S0923-2508(01)01223-2
Balasubramanian, D., Kumari, H., and Mathee, K. (2015). Pseudomonas aeruginosa AmpR: an acute–chronic switch regulator. Pathogens Dis. 73, 1–14. doi: 10.1111/2049-632X.12208
Balasubramanian, D., Schneper, L., Merighi, M., Smith, R., Narasimhan, G., Lory, S., et al. (2012). The regulatory repertoire of Pseudomonas aeruginosa AmpC ß-lactamase regulator AmpR includes virulence genes. PLoS One 7:e34067. doi: 10.1371/journal.pone.0034067
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Buchholz-Cleven, B. E. E., Rattunde, B., and Straub, K. L. (1997). Screening for genetic diversity of isolates of anaerobic Fe(ii)-oxidizing Bacteria using Dgge and whole-cell hybridization. Syst. Appl. Microbiol. 20, 301–309. doi: 10.1016/S0723-2020(97)80077-X
Caraux, G., and Pinloche, S. (2005). PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21, 1280–1281. doi: 10.1093/bioinformatics/bti141
Carvalheira, A., Gonzales-Siles, L., Salvà-Serra, F., Lindgren, Å., Svensson-Stadler, L., Thorell, K., et al. (2020). Acinetobacter portensis sp. nov. and Acinetobacter guerrae sp. nov., isolated from raw meat. Int. J. Syst. Evol. Microbiol. 70, 4544–4554. doi: 10.1099/ijsem.0.004311
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
De Coster, W., Dhert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dong, X., Rao, Z., Wu, S., Peng, F., Xie, Z., and Long, Y. (2023). Pseudomonas benzopyrenica sp. nov., isolated from soil, exhibiting high-efficiency degradation of benzo(a)pyrene. Int. J. Syst. Evol. Microbiol. 73:6034. doi: 10.1099/ijsem.0.006034
Duman, M., Mulet, M., Altun, S., Saticioglu, I. B., Ozdemir, B., Ajmi, N., et al. (2021). The diversity of Pseudomonas species isolated from fish farms in Turkey. Aquaculture 535:736369. doi: 10.1016/j.aquaculture.2021.736369
Fajardo, A., Hernando-Amado, S., Oliver, A., Ball, G., Filloux, A., and Martinez, J. L. (2014). Characterization of a novel Zn2+−dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 69, 2972–2978. doi: 10.1093/jac/dku267
Fernández-Juárez, V., Hallstrøm, S., Pacherres, C. O., Wang, J., Coll-Garcia, G., Kühl, M., et al. (2023). Biofilm formation and cell plasticity drive diazotrophy in an anoxygenic phototrophic bacterium. Appl. Environ. Microbiol. 89, e01027–e01023. doi: 10.1128/aem.01027-23
Girard, L., Lood, C., Höfte, M., Vandamme, P., Rokni-Zadeh, H., Van Noort, V., et al. (2021). The ever-expanding Pseudomonas genus: description of 43 new species and partition of the Pseudomonas putida group. Microorganisms 9:1766. doi: 10.3390/microorganisms9081766
Girard, L., Lood, C., Rokni-Zadeh, H., Van Noort, V., Lavigne, R., and De Mot, R. (2020). Reliable identification of environmental Pseudomonas isolates using the rpoD gene. Microorganisms 8:1166. doi: 10.3390/microorganisms8081166
Girlich, D., Naas, T., and Nordmann, P. (2004). Biochemical characterization of the naturally occurring Oxacillinase Oxa-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48, 2043–2048. doi: 10.1128/AAC.48.6.2043-2048.2004
Gomila, M., Peña, A., Mulet, M., Lalucat, J., and García-Valdés, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). Dna-Dna hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grevskott, D. H., Radisic, V., Salvà-Serra, F., Moore, E. R. B., Akervold, K. S., Victor, M. P., et al. (2024). Emergence and dissemination of epidemic-causing Oxa-244 carbapenemase-producing Escherichia coli St38 through hospital sewage in Norway, 2020–2022. J. Hosp. Infect. 145, 165–173. doi: 10.1016/j.jhin.2023.12.020
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of Phyml 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). Quast: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Gyger, J., Torrens, G., Cava, F., Bernhardt, T. G., and Fumeaux, C. (2024). A potential space-making role in cell wall biogenesis for SltB1and DacB revealed by a beta-lactamase induction phenotype in Pseudomonas aeruginosa. MBio 15, e01419–e01424. doi: 10.1128/mbio.01419-24
Hauben, L., Vauterin, L., Swings, J., and Moore, E. R. (1997). Comparison of 16S ribosomal Dna sequences of all Xanthomonas species. Int. J. Syst. Bacteriol. 47, 328–335. doi: 10.1099/00207713-47-2-328
Hitch, T. C. A., Riedel, T., Oren, A., Overmann, J., Lawley, T. D., and Clavel, T. (2021). Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. Isme Communications 1:16. doi: 10.1038/s43705-021-00017-z
Ikuta, K. S., Swetschinski, L. R., Robles Aguilar, G., Sharara, F., Mestrovic, T., Gray, A. P., et al. (2022). Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the global burden of disease study 2019. Lancet 400, 2221–2248. doi: 10.1016/S0140-6736(22)02185-7
Irber, L., Pierce-Ward, N. T., and Brown, C. T. (2022). Sourmash Branchwater enables lightweight petabyte-scale sequence search. [Epubh ahead of preprint]. doi: 10.1101/2022.11.02.514947
Jaén-Luchoro, D., Gonzales-Siles, L., Karlsson, R., Svensson-Stadler, L., Molin, K., Cardew, S., et al. (2020). Corynebacterium sanguinis sp. nov., a clinical and environmental associated corynebacterium. Syst. Appl. Microbiol. 43:126039. doi: 10.1016/j.syapm.2019.126039
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kristensen, D. M., Kannan, L., Coleman, M. K., Wolf, Y. I., Sorokin, A., Koonin, E. V., et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487. doi: 10.1093/bioinformatics/btq229
Lalucat, J., Gomila, M., Mulet, M., Zaruma, A., and García-Valdés, E. (2022). Past, present and future of the boundaries of the Pseudomonas genus: proposal of Stutzerimonas gen. Nov. Syst. Appl. Microbiol. 45:126289. doi: 10.1016/j.syapm.2021.126289
Lalucat, J., Mulet, M., Gomila, M., and García-Valdés, E. (2020). Genomics in bacterial taxonomy: impact on the genus Pseudomonas. Genes 11:139. doi: 10.3390/genes11020139
Lane, D. J. (1991). “16S/23S rrna sequencing” in Nucleic acid techniques in bacterial systematics. eds. E. G. Stackebrandt and Michael (New York: Wiley).
Leinonen, R., Sugawara, H., and Shumway, M.International Nucleotide Sequence Database Collaboration (2011). The sequence read archive. Nucleic Acids Res. 39, D19–D21. doi: 10.1093/nar/gkq1019
Letunic, I., and Bork, P. (2024). Interactive tree of life (itol) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/nar/gkae268
Levine, M., and Anderson, D. Q. (1932). Two new species of Bacteria causing mustiness in eggs. J. Bacteriol. 23, 337–347. doi: 10.1128/jb.23.4.337-347.1932
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). Orthomcl: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lodge, J. M., Minchin, S. D., Piddock, L. J. V., and Busby, S. J. W. (1990). Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC β-lactamase. Biochem. J. 272, 627–631
Marathe, N. P., Salvà-Serra, F., Nimje, P. S., and Moore, E. R. B. (2022). Novel plasmid carrying mobile colistin resistance gene mcr-4.3 and mercury resistance genes in Shewanella baltica: insights into mobilization of mcr-4.3 in Shewanella species. Microbiol. Spectr. 10, e02037–e02022. doi: 10.1128/spectrum.02037-22
Marmur, J. (1961). A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3:208-In1. doi: 10.1016/S0022-2836(61)80047-8
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformat. 14:60. doi: 10.1186/1471-2105-14-60
Mulet, M., Bennasar, A., Lalucat, J., and Garcia-Valdes, E. (2009). An rpoD-based Pcr procedure for the identification of Pseudomonas species and for their detection in environmental samples. Mol. Cell. Probes 23, 140–147. doi: 10.1016/j.mcp.2009.02.001
Naas, T., Oueslati, S., Bonnin, R. A., Dabos, M. L., Zavala, A., Dortet, L., et al. (2017). Beta-lactamase database (Bldb) – structure and function. J. Enzyme Inhib. Med. Chem. 32, 917–919. doi: 10.1080/14756366.2017.1344235
Nimje, P. S., and Marathe, N. P. (2023). Genome sequence of Vibrio anguillarum isolates carrying a novel class a β-lactamase Van-1: do migratory fish transport novel resistance factors? J. Glob. Antimicrob. Resist. 32, 152–154. doi: 10.1016/j.jgar.2022.10.017
Okonechnikov, K., Golosova, O., and Fursov, M.Team, U (2012). Unipro Ugene: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Palleroni, N. J. (2015). “Pseudomonas” in Bergey's manual of systematics of Archaea and Bacteria. eds. M. E. Trujillo, S. Dedysh, P. Devos, B. Hedlund, P. Kämpfer, and F. A. Rainey, et al. doi: 10.1002/9781118960608.gbm01210
Peix, A., Ramírez-Bahena, M. H., and Velázquez, E. (2018). The current status on the taxonomy of Pseudomonas revisited: an update. Infect. Genet. Evol. 57, 106–116. doi: 10.1016/j.meegid.2017.10.026
Pieterse, C. M. J., Berendsen, R. L., De Jonge, R., Stringlis, I. A., Van Dijken, A. J. H., Van Pelt, J. A., et al. (2021). Pseudomonas simiae Wcs417: star track of a model beneficial rhizobacterium. Plant Soil 461, 245–263. doi: 10.1007/s11104-020-04786-9
Poole, K. (2011). Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. doi: 10.3389/fmicb.2011.00065
Qin, S., Xiao, W., Zhou, C., Pu, Q., Deng, X., Lan, L., et al. (2022). Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 7:199. doi: 10.1038/s41392-022-01056-1
Reddy, G. S. N., Matsumoto, G. I., Schumann, P., Stackebrandt, E., and Shivaji, S. (2004). Psychrophilic pseudomonads from Antarctica: Pseudomonas antarctica sp. nov., Pseudomonas meridiana sp. nov. and Pseudomonas proteolytica sp. nov. Int. J. Syst. Evol. Microbiol. 54, 713–719. doi: 10.1099/ijs.0.02827-0
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rudra, B., and Gupta, R. S. (2024). Phylogenomics studies and molecular markers reliably demarcate genus Pseudomonas sensu stricto and twelve other Pseudomonadaceae species clades representing novel and emended genera. Front. Microbiol. 14:3665. doi: 10.3389/fmicb.2023.1273665
Salvà-Serra, F., Svensson-Stadler, L., Busquets, A., Jaén-Luchoro, D., Karlsson, R., Moore, R. B. E., et al. (2018). A protocol for extraction and purification of high-quality and quantity bacterial Dna applicable for genome sequencing: a modified version of the Marmur procedure. Protocol Exchange. doi: 10.1038/protex.2018.084
Sasser, M. (2001). “Identification of Bacteria by gas chromatography of cellular fatty acids” in Technical note #101. ed. M. Sasser (Newark, DE, USA: MIDI, Inc.).
Sayers, E. W., Beck, J., Bolton, E. E., Brister, J. R., Chan, J., Comeau, D. C., et al. (2024). Database resources of the National Center for biotechnology information. Nucleic Acids Res. 52, D33–D43. doi: 10.1093/nar/gkad1044
Scales, B. S., Dickson, R. P., Lipuma, J. J., and Huffnagle, G. B. (2014). Microbiology, genomics, and clinical significance of the Pseudomonas fluorescens species complex, an unappreciated colonizer of humans. Clin. Microbiol. Rev. 27, 927–948. doi: 10.1128/CMR.00044-14
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7:539.doi: 10.1038/msb.2011.75
Švec, P., Kosina, M., Zeman, M., Holochová, P., Králová, S., Němcová, E., et al. (2020). Pseudomonas karstica sp. nov. and Pseudomonas spelaei sp. nov., isolated from calcite moonmilk deposits from caves. Int. J. Syst. Evol. Microbiol. 70, 5131–5140. doi: 10.1099/ijsem.0.004393
Tariq, F. N., Shafiq, M., Khawar, N., Habib, G., Gul, H., Hayat, A., et al. (2023). The functional repertoire of AmpR in the AmpC β-lactamase high expression and decreasing β-lactam and aminoglycosides resistance in Esbl Citrobacter freundii. Heliyon 9:e19486. doi: 10.1016/j.heliyon.2023.e19486
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). Ncbi prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vaser, R., and Šikić, M. (2021). Time- and memory-efficient genome assembly with raven. Nat. Comput. Sci. 1, 332–336. doi: 10.1038/s43588-021-00073-4
Welinder-Olsson, C., Kjellin, E., Badenfors, M., and Kaijser, B. (2000). Improved microbiological techniques using the polymerase chain reaction and pulsed-field gel electrophoresis for diagnosis and follow-up of Enterohaemorrhagic Escherichia coli infection. Eur. J. Clin. Microbiol. Infect. Dis. 19, 843–851. doi: 10.1007/s100960000380
Wick, R. R., and Holt, K. E. (2022). Polypolish: short-read polishing of long-read bacterial genome assemblies. PLoS Comput. Biol. 18:e1009802. doi: 10.1371/journal.pcbi.1009802
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Meric, G., Vezina, B., et al. (2021). Trycycler: Consensus long-read assemblies for bacterial genomes. [Epubh ahead of preprint]. doi: 10.1101/2021.07.04.451066
Wick, R. R., Judd, L. M., and Holt, K. E. (2023). Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput. Biol. 19:e1010905. doi: 10.1371/journal.pcbi.1010905
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rrna gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Zamora, L., Fernández-Garayzábal, J. F., Svensson-Stadler, L. A., Palacios, M. A., Domínguez, L., Moore, E. R., et al. (2012). Flavobacterium oncorhynchi sp. nov., a new species isolated from rainbow trout (Oncorhynchus mykiss). Syst. Appl. Microbiol. 35, 86–91. doi: 10.1016/j.syapm.2011.11.007
Zhao, W.-H., and Hu, Z.-Q. (2010). β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 36, 245–258. doi: 10.3109/1040841X.2010.481763
Zimin, A. V., Marçais, G., Puiu, D., Roberts, M., Salzberg, S. L., and Yorke, J. A. (2013). The MaSurca genome assembler. Bioinformatics 29, 2669–2677. doi: 10.1093/bioinformatics/btt476
Keywords: Pseudomonas fluorescens group, Pseudomonas gessardii subgroup, β-lactamase novel variant, novel species, polyphasic taxonomy, phylogenomics, whole-genome sequencing
Citation: Salvà-Serra F, Nimje P, Piñeiro-Iglesias B, Alarcón LA, Cardew S, Inganäs E, Jensie-Markopoulos S, Ohlén M, Sailer H-S, Unosson C, Fernández-Juárez V, Pacherres CO, Kühl M, Moore ERB and Marathe NP (2025) Description of Pseudomonas imrae sp. nov., carrying a novel class C β-lactamase gene variant, isolated from gut samples of Atlantic mackerel (Scomber scombrus). Front. Microbiol. 16:1530878. doi: 10.3389/fmicb.2025.1530878
Edited by:
Jin Zhou, Tsinghua University, ChinaReviewed by:
Muhammed Duman, Bursa Uludağ University, TürkiyeLeonid Valentovich, The National Academy of Sciences of Belarus, Belarus
Copyright © 2025 Salvà-Serra, Nimje, Piñeiro-Iglesias, Alarcón, Cardew, Inganäs, Jensie-Markopoulos, Ohlén, Sailer, Unosson, Fernández-Juárez, Pacherres, Kühl, Moore and Marathe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nachiket P. Marathe, bmFjaGlrZXQubWFyYXRoZUBoaS5ubw==