 Qikai Zhang1,2†
Qikai Zhang1,2† Chunxi Cheng1†
Chunxi Cheng1† Baoqian Lyu1,2*
Baoqian Lyu1,2* Yulin Gao3Lilin Chen4Liang Peng1
Yulin Gao3Lilin Chen4Liang Peng1 Zhengfu Yue1
Zhengfu Yue1 Hui Lu1Jihong Tang1,2Bin Jiao1,2
Hui Lu1Jihong Tang1,2Bin Jiao1,2 Shen Liu1,2
Shen Liu1,2- 1Key Laboratory of Integrated Pest Management of Tropical Crops, Ministry of Agriculture and Rural Affairs, Environment and Plant Protection Institute, Chinese Academy of Tropical Agricultural Sciences, Haikou, China
- 2Hainan Key Laboratory for Biosafety Monitoring and Molecular Breeding in the Nanfan Area, Sanya Research Institute, Chinese Academy of Tropical Agricultural Sciences, Sanya, China
- 3State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 4State Key Laboratory of Ecological Pest Control for Fujian and Taiwan Crops, College of Plant Protection, Fujian Agriculture and Forestry University, Fuzhou, China
Introduction: Thrips are key vectors for plant viruses, representing a significant challenge to the cultivation of cucurbits and other vegetables in tropical agriculture. This study investigates the diversity of viromes carried by thrips and their ecological roles in viral epidemics affecting specific crops.
Methods: We identify thrips populations in tropical regions and perform a comprehensive virome analysis through high-throughput sequencing.
Results: Our findings reveal that the predominant thrips species associated with these crops are Frankliniella intonsa, Thrips palmi, and Megalurothrips usitatus. The sequencing efforts identified 19 viruses within these thrips, including previously undocumented viruses, such as a double-stranded RNA virus and several positive- and negative-sense single-stranded RNA viruses. Notably, detection rates of specific plant viruses—Melon yellow spot virus (MYSV), Watermelon silver mottle virus (WSMoV), and Telosma mosaic virus (TeMV)—exhibit significant correlations with thrips population density in cucurbits and other vegetables.
Discussion: This study lays the groundwork for future research into the ecological relationships between thrips and plant viruses, offering valuable insights for developing targeted disease management strategies in tropical agricultural systems.
1 Introduction
Outbreaks of plant viruses present a serious threat to the production of cucurbits and other vegetables (He and Creasey Krainer, 2020). Viral infections in plants can lead to various symptoms, such as dwarfing, chlorosis, necrosis, yellowing, and mosaic patterns. Infected fruits often exhibit reduced size and surface deformities or mottling, significantly lowering their market value (Ertunç, 2020). Climate change, shifts in planting structures, and evolving farming systems have expanded the cultivation of cucurbits and vegetables, amplifying the impact of viral diseases, which have now become the primary disease challenge for these crops (Jones, 2021). These viruses frequently co-occur in crop fields, resulting in mixed infections that pose a significant risk to production, especially within the Cucurbitaceae, Solanaceae, Fabaceae, Brassicaceae, and Malvaceae families (Sánchez-Sánchez et al., 2024).
Thrips (Thysanoptera) are small, cryptic, piercing-sucking insects known for rapid reproduction and high insecticide resistance, making them prominent agricultural pests worldwide (Mound and Morris, 2007). Importantly, thrips act as vectors for plant viruses, especially in cucurbits and other vegetables, where viral infections they transmit often cause far greater harm than direct feeding damage (Whitfield et al., 2005). Although only a subset of thrips species are virus vectors, they carry an exceptionally diverse range of viruses (Rotenberg et al., 2015). In cucurbits and vegetables, thrips are known to transmit over 15 harmful viruses [including Tobacco streak virus (TSV), Tobacco ringspot virus (TRSV), and Sowbane mosaic virus (SoMV)]. The most impactful of these belong to the Tospovirus genus (now Orthotospovirus, containing 26 recognized species), along with viruses from four additional genera (Nichol et al., 2005). This virus group alone contributes to substantial global economic losses annually (Wu et al., 2018; He et al., 2020). The extensive spread of these viruses underscores thrips’ critical role in viral disease epidemics affecting cucurbits and other vegetables.
Theoretically, vector population size directly impacts transmission efficiency, as larger populations can amplify both the scope and rate of virus spread, assuming the proportion of viruliferous individuals remains relatively stable (Cunniffe et al., 2021). The relationship between thrips density and viral disease prevalence is well-documented (He et al., 2020). Field studies have shown that epidemics of viruses such as Tomato spotted wilt virus (TSWV), Impatiens necrotic spot virus (INSV), and Tomato chlorotic spot virus (TCSV) are closely associated with outbreaks of Frankliniella intonsa (Karthikeyan et al., 2021). Similarly, viral disease prevalence in sweet peppers is highly correlated with adult densities of Thrips palmi (Zhang et al., 2016). In tropical agriculture, the warm and humid conditions favor rapid disease spread, making viral infections particularly challenging (Sastry and Zitter, 2014). While previous studies highlight the role of thrips in virus transmission, systematic analyses of the viromes they carry and their ecological relationships with viral epidemics in cucurbits and other vegetables are still lacking.
In this study, we investigated tropical vegetable fields on Hainan Island, China’s only tropical island. We first identified and classified thrips populations across these fields in Hainan Island. We then conducted a comprehensive virome analysis to detect and classify viruses within these populations, exploring their phylogenetic relationships. Finally, we analyzed correlations between virus presence in thrips viromes and thrips population densities in cucurbit and vegetable crops. Through these efforts, this study aims to provide foundational insights into the diversity of viral spectra within thrips populations and to elucidate their ecological roles in viral epidemics affecting cucurbits and vegetables.
2 Materials and methods
2.1 Sampling thrips in cucurbits and other vegetables in Hainan Province, China
Random sampling was conducted in cucurbit and other vegetable fields across Hainan Province, China, a region with a tropical monsoon climate that remains consistently warm and humid year-round. On the island, cucurbits and vegetables hold significant economic importance. Sampling involved selecting five points within each field and examining three plants at each point. Thrips were collected by gently tapping the flowers, leaves, and shoot tips of plants over a white enamel pan, ensuring comprehensive sampling of all accessible plant structures. Dislodged thrips were transferred as a group into a 0.5 L glass jar with a breathable lid using a fine brush. Simultaneously, plant parts hosting thrips were also collected, with records kept of the collection location, host plant species, and relevant field details (Supplementary Table 1). In the laboratory, thrips were examined microscopically for morphological identification (Supplementary Table 2).
For specimens not identifiable by morphology, DNA was extracted using an animal DNA extraction kit (Tiangen Biotech, Beijing, China) for molecular identification. PCR amplification of the COI gene sequence was conducted using universal primers, LepF1 and LepR1, as outlined by Hebert et al. (2004), producing a ~700 bp fragment. Post-amplification, 2 μL of the product was analyzed by gel electrophoresis for verification. The amplified sequences were then compared with those in the NCBI nucleotide database. Thrips were identified to the species level if sequence similarity to reference sequences exceeded 95%. Morphological identification was additionally performed as a complementary approach to molecular methods, further confirming species identity and enhancing the overall accuracy of the results.
2.2 Analysis of viromes in thrips
2.2.1 RNA extraction, library construction, and sequencing
Thrips samples were collected from 16 distinct sites on Hainan Island, encompassing various crops, including cucurbits, with sampling conducted between July and December. Total RNA was extracted separately from each thrips population at these sites, with each population containing a minimum of 50 individuals. When multiple thrips species were present at a site, individuals were pooled by species for RNA extraction and analysis. RNA extraction was carried out using the Trizol method, and RNA quantity was assessed with a NanoDrop 2000 spectrophotometer.
To ensure data representativeness and scientific rigor, RNA from the 16 thrips populations was pooled into six composite RNA samples based on thrips species and host plant. This was due to limited RNA, high costs, and our broad research aim. These pooled samples were prepared for metatranscriptomic sequencing. Total RNA samples underwent ribosomal RNA removal using the Ribo-Zero Gold kit (specific for human/mouse/rat rRNA), and paired-end 150 bp sequencing was performed on the Illumina NovaSeq™ 6000 platform. Library preparation and sequencing were completed by Lianchuan Biotechnology Co., Ltd. (Hangzhou, China), yielding 81 GB of raw sequencing data. This sequencing depth provides robust data for the identification and comprehensive analysis of thrips virome diversity.
2.2.2 Sequence assembly and discovery of new viruses
After assessing the quality of raw sequencing data from each library, data trimming was performed using Trimmomatic v0.32 (Bolger et al., 2014) to remove low-quality reads and adapter sequences, resulting in a set of clean reads. To efficiently detect viral fragments, these clean reads were initially aligned to the host genome (F. intonsa reference genome) using Bowtie2 v2.4.4 (Langmead and Salzberg, 2012), allowing for the removal of host-derived sequences. The remaining, non-aligned reads were then assembled de novo using Trinity v2.8.5, which generated preliminary viral contigs.
The assembled contigs were annotated using the blastX algorithm in DIAMOND v3.12 with an E-value threshold of 1 × 10−5, balancing sensitivity and specificity to ensure reliable viral identification while minimizing false positives. The selected viral contigs were subsequently matched against the non-redundant (NR) protein database for initial virus identification. Open reading frames (ORFs) within the viral contigs were identified using NCBI ORFfinder,1 and homologous proteins were identified using HMMER v3.3 against the Pfam protein family database and the RNA-dependent RNA polymerase (RdRP) database of RNA viruses. This approach allowed for further classification and identification of novel viruses based on the latest ICTV species classification standards.
Finally, to estimate viral abundance across the libraries, the quality-controlled paired-end reads were mapped to the identified viral genomes using Bowtie2. Alignments were processed using Samtools v0.1.1 (Li et al., 2009), and RPKM and FPKM values for each contig were calculated using Salmon, providing quantitative insights into viral prevalence within the libraries.
2.2.3 Sanger sequencing
Primers specific to each identified virus were designed for Sanger sequencing. Total RNA from the six libraries obtained in section 2.2.1 was subjected to RT-PCR to synthesize cDNA, with an expected product size of 400 to 600 bp and an annealing temperature (Tm) of 55–60°C. To address potential challenges in primer design, such as complex secondary structures or low copy numbers, four primer pairs were designed for distinct regions of each viral sequence. Each primer pair was tested sequentially to achieve the desired bands; if none of the four pairs yielded a band, the virus was considered absent in that sample. Primer design was conducted using Primer-BLAST,2 and primer sequences are provided in Supplementary Table 2. Detailed reaction conditions are as follows: The Premix Taq PCR reaction system consisted of the following components with their respective volumes. Premix Taq was 12.5 μL, cDNA was 1 μL, F-primer was 1 μL, R-primer was 1 μL, and ddH2O was added to make the total volume up to 25 μL.
The PCR conditions included an initial denaturation at 95°C for 5 min, followed by 35 cycles of 30 s at 95°C, 40 s at 55°C, and 40 s at 72°C, concluding with a final extension at 72°C for 10 min. PCR products were analyzed using 1.2% agarose gel electrophoresis. If the target band was observed, it was excised from the gel, and the cDNA was purified. Purified products were sent to BGI-Shenzhen for sequencing. Sequencing results were aligned using Geneious v9.0.2, and sequences with similarity above 95% were considered indicative of the virus’s presence.
2.2.4 Phylogenetic tree construction
Phylogenetic analysis was performed by aligning conserved viral sequences identified in this study with known sequences from the same viral families using MAFFT v7.407 with the E-INS-I algorithm. Special focus was placed on RNA-dependent RNA polymerase (RdRp) sequences of RNA viruses, as these are key conserved regions useful for differentiating RNA virus types. Unaligned sequences and poorly aligned regions were removed with TrimAl v1.4 to enhance phylogenetic accuracy. The phylogenetic tree was then constructed using the maximum likelihood (ML) method in IQ-TREE v2.0.3, with bootstrap analysis conducted 1,000 times to validate the reliability of the results. The amino acid substitution model was selected based on the best-fit analysis provided by ModelTest.
2.2.5 Identification of non-viral microorganisms
Non-viral reads were classified and annotated using Kraken v2.0.7-beta 47 (Wood and Salzberg, 2014). For accurate microbial identification, rRNA reads were filtered from the dataset using SortmeRNA (Kopylova et al., 2012). The filtered rRNA reads were then compared to the nucleotide database using the NCBI BLAST suite, applying an E-value threshold of 10−3. The microorganism with the lowest E-value match was identified as the closest species.
2.3 Correlation between the detection of relevant viral diseases in cucurbits and other vegetable crops and the occurrence of thrips
2.3.1 Detection of target viruses in cucurbits and other vegetable crops
Between July 2021 and December 2022, we conducted a survey on the occurrence of viral diseases in cucurbit and other vegetable fields in Hainan, China (Supplementary Table 1). During this period, both thrips and plant samples were collected simultaneously, allowing for a direct comparison of virus presence in thrips populations with the incidence of viral symptoms in corresponding plant samples. A total of 543 leaf samples exhibiting symptoms such as dwarfism, mosaic patterns, malformation, yellow spots, or necrosis were collected and stored at −80°C for further analysis. DNA and RNA were extracted using a plasmid DNA extraction kit (Tiangen Biotech, Beijing, China) and the Trizol method, respectively.
Four target plant viruses (WSMoV, MYSV, WGMMV, TeMV), previously identified in the thrips virome, were detected in these plant tissue samples using reverse transcription and PCR. PCR products were analyzed via agarose gel electrophoresis, with target DNA bands excised and purified for sequencing. Detected plant viruses were then identified through sequence alignment with the NCBI nucleotide database. Detection rates for suspected viral samples were grouped by plant family to evaluate the prevalence of target viruses in cucurbit and other vegetable crops. These four viruses were selected as they were the only plant viruses identified in our initial virome analysis of thrips.
2.3.2 Correlation analysis
We conducted a Pearson correlation analysis to examine the detection of four plant viruses within the T. palmi and F. intonsa viromes. Specifically, we assessed the correlation between the detection rate of MYSV in cucurbit crops and the occurrence of T. palmi. Additionally, we examined the correlation between the detection rate of WSMoV in both Cucurbitaceae and Fabaceae and the abundance of T. palmi. In both analyses, virus detection in plant samples was based on presence/absence rather than incidence rates. Similarly, we analyzed the correlation between the detection rate of WGMMV in cucurbit crops and the occurrence of F. intonsa, as well as the correlation between the detection rate of TeMV in leguminous crops and the occurrence of F. intonsa.
3 Results
3.1 Occurrence of thrips in the study region
Thrips were observed on crops across seven plant families: Cucurbitaceae, Fabaceae, Solanaceae, Brassicaceae, Liliaceae, Malvaceae, and Apiaceae, with 10, 14, 9, 12, 12, 6, and 5 thrips species identified in each, respectively. Additionally, 15 thrips species were identified on other crops. Among the thrips collected throughout the survey, F. intonsa, T. palmi, and M. usitatus were the dominant species, accounting for 36.55, 32.19, and 24.84% of the total, respectively. F. intonsa and T. palmi primarily infested cucurbit crops, whereas M. usitatus was most prevalent on leguminous crops (Figure 1).
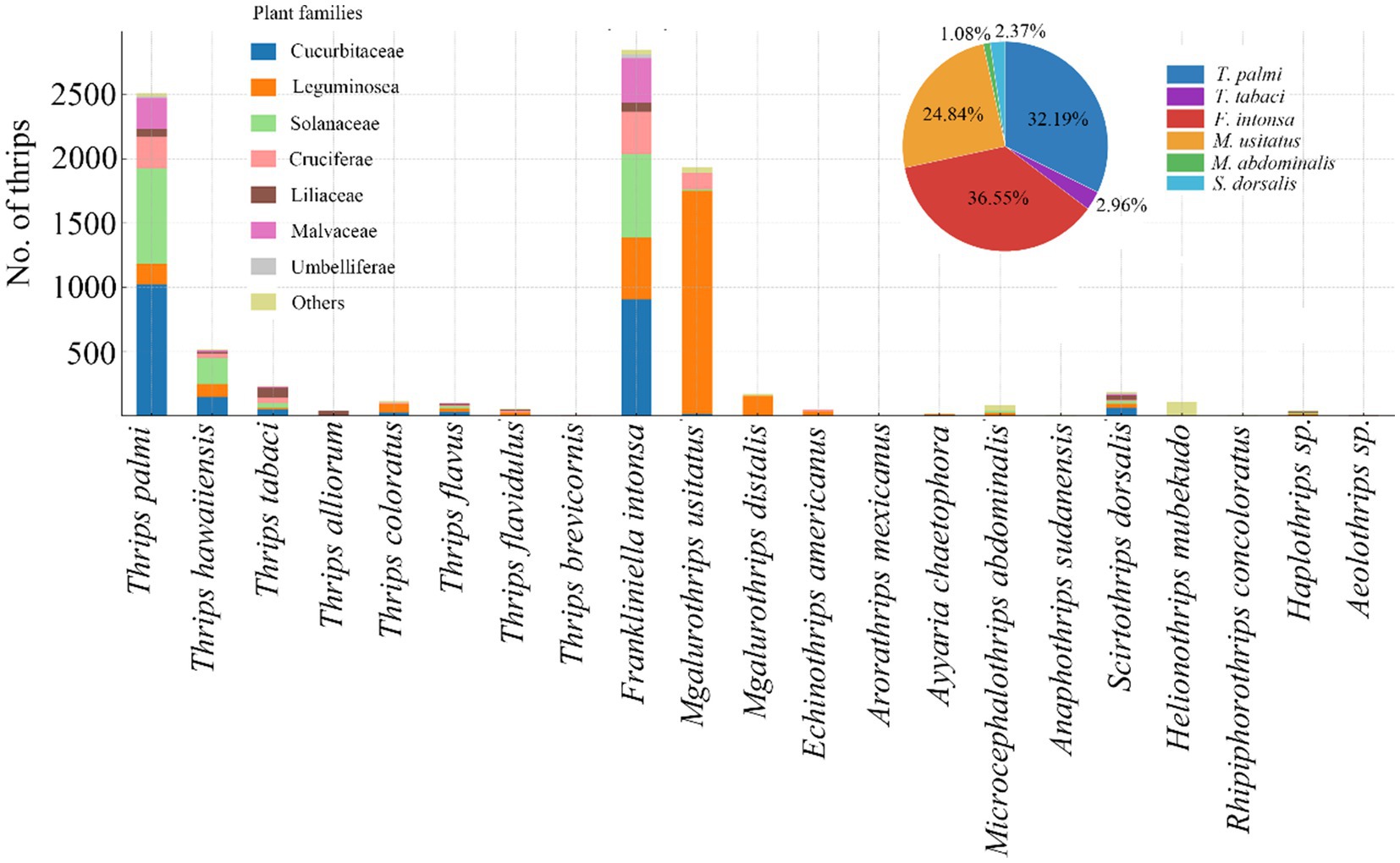
Figure 1. Major thrips species collected in survey on Hainan Island, southern China (pie chart) and their distribution on cucurbits and other vegetables (by family, see bar chart).
3.2 Analysis of thrips viromes
3.2.1 Virus identification
From each of the six thrips sample libraries, a total of 12.88 to 13.80 GB of data were generated, reflecting the data range across different species. After filtering out low-quality reads, 65,532,884 clean reads were retained. Through de novo assembly, reference genome alignment, and BLASTx searches, we identified 36,269 initial viral contigs, with lengths ranging from 200 to 20,390 nt and an average length of 502 nt. Following iterative extension using Bowtie2 and Samtools, and after rigorous screening and quality control, we obtained 30 high-quality viral sequences from the six sample pools, identifying 19 distinct viruses. These viruses were categorized into nine viral families, with three unclassified viral groups comprising one double-stranded RNA virus (Totiviridae), 11 positive-sense single-stranded RNA viruses, six negative-sense single-stranded RNA viruses, and one unclassified RNA virus (Supplementary Table 3). Among the identified viruses, negative-sense single-stranded RNA viruses were the most prevalent. Picornavirales accounted for 35.63% of total viral reads, followed by Amarillovirales (27.80%) and Martellivirales (16.06%) (Table 1).
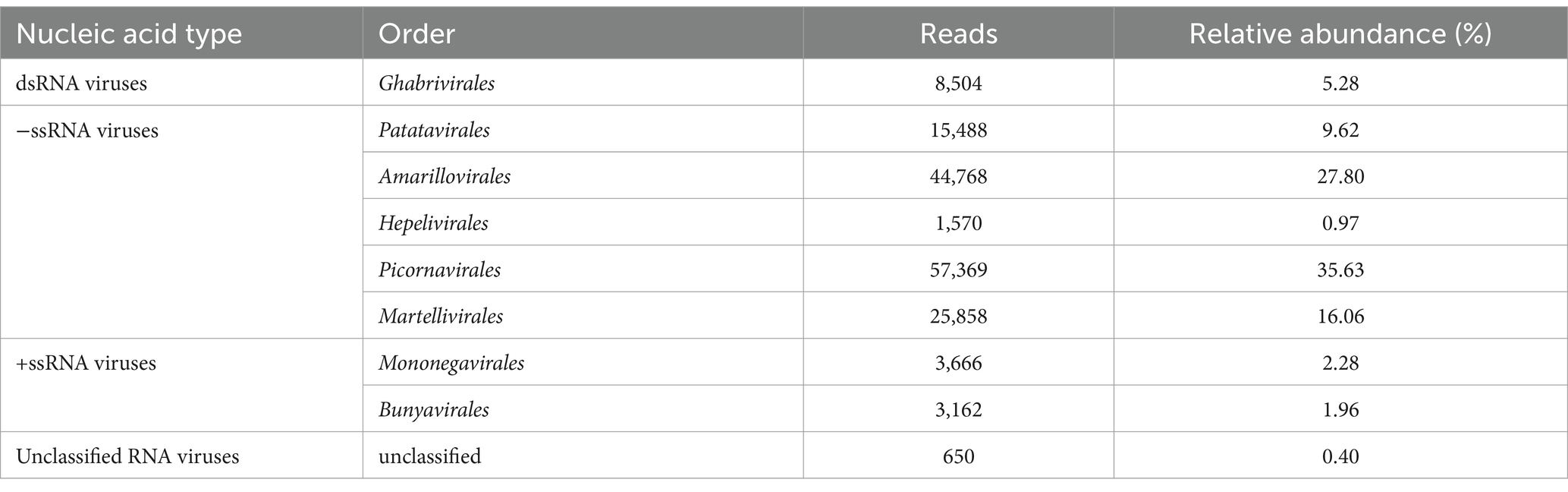
Table 1. Relative abundance of viral classifications in libraries.
3.2.2 Viral community composition
Based on host categories, the viruses carried by thrips were classified as fungal viruses (one species), plant viruses (four species), arthropod viruses (13 species), and vertebrate viruses (one species) (Supplementary Table 3). The four plant viruses identified were MYSV, WSMoV, TeMV, and WGMMV, with MYSV and WSMoV detected in T. palmi, and TeMV and WGMMV in F. intonsa (Figure 2A).
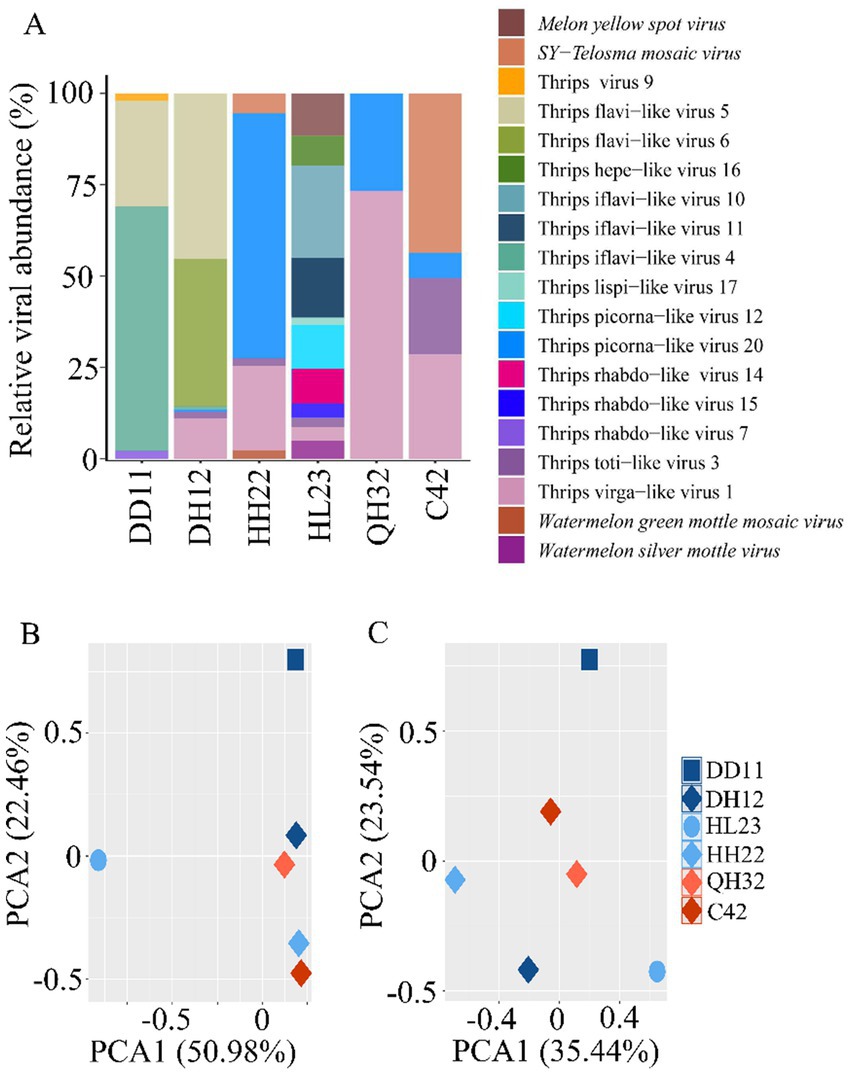
Figure 2. Comparison of viral and bacterial communities across different thrips samples. (A) Comparison of viral species and abundance across different thrips sample libraries. Principal component analysis (PCA) of the viral (B) and bacterial (C) communities in the different thrips samples.
Analysis of the viral community composition in the three thrips samples revealed significant differences in virus species and abundance among samples (p < 0.05). In the T. palmi sample (HL23), 11 viruses were detected, with Thrips iflavi-like virus 10 being the most abundant, accounting for 25.26% of the total. In the F. intonsa samples (DH12, HH22, QH32, C42), between 2 and 6 viruses were detected, each dominated by a different virus: Thrips flavilike viruses 5 and 6 in DH12, Thrips picorna-like virus 20 in HH22, Thrips virga-like virus 1 in QH32, and SY-Telosma mosaic virus in C42. In the M. usitatus sample (DD11), four viruses were identified, with Thrips iflavi-like virus 4 being the most prevalent, comprising 66.84% of the total (Figure 2A).
Principal component analysis (PCA) demonstrated that viral community compositions differed significantly among the three thrips species (p < 0.05), while viral composition was similar among thrips on the same host plant family. This finding suggests that viral communities are primarily influenced by the host plant (Figures 2B,C).
3.2.3 Identification and analysis of dsRNA viruses
In three F. intonsa sample libraries (C42, DH12, HH22) and one T. palmi sample library (HL23), we identified a novel double-stranded RNA (dsRNA) virus in the Totiviridae family, tentatively named Thrips toti-like virus 3 (TTV3). The full genome of TTV3 is 4,858 nucleotides long and contains two open reading frames (ORFs). This specific ORF arrangement in the TTV3 genome is significant as it provides the basis for further exploring the virus’s replication mechanism and its interaction with the host thrips. The first ORF encodes a protein of unknown function with a molecular mass of 81.072 kDa, while the second ORF encodes an RNA-dependent RNA polymerase (RdRp) (Figure 3A). Phylogenetic analysis shows that TTV3 forms a distinct evolutionary branch within the Totivirus genus, exhibiting 36.83% amino acid sequence similarity to Camponotus nipponicus virus, a virus found in tree-dwelling ants (Figure 3B).
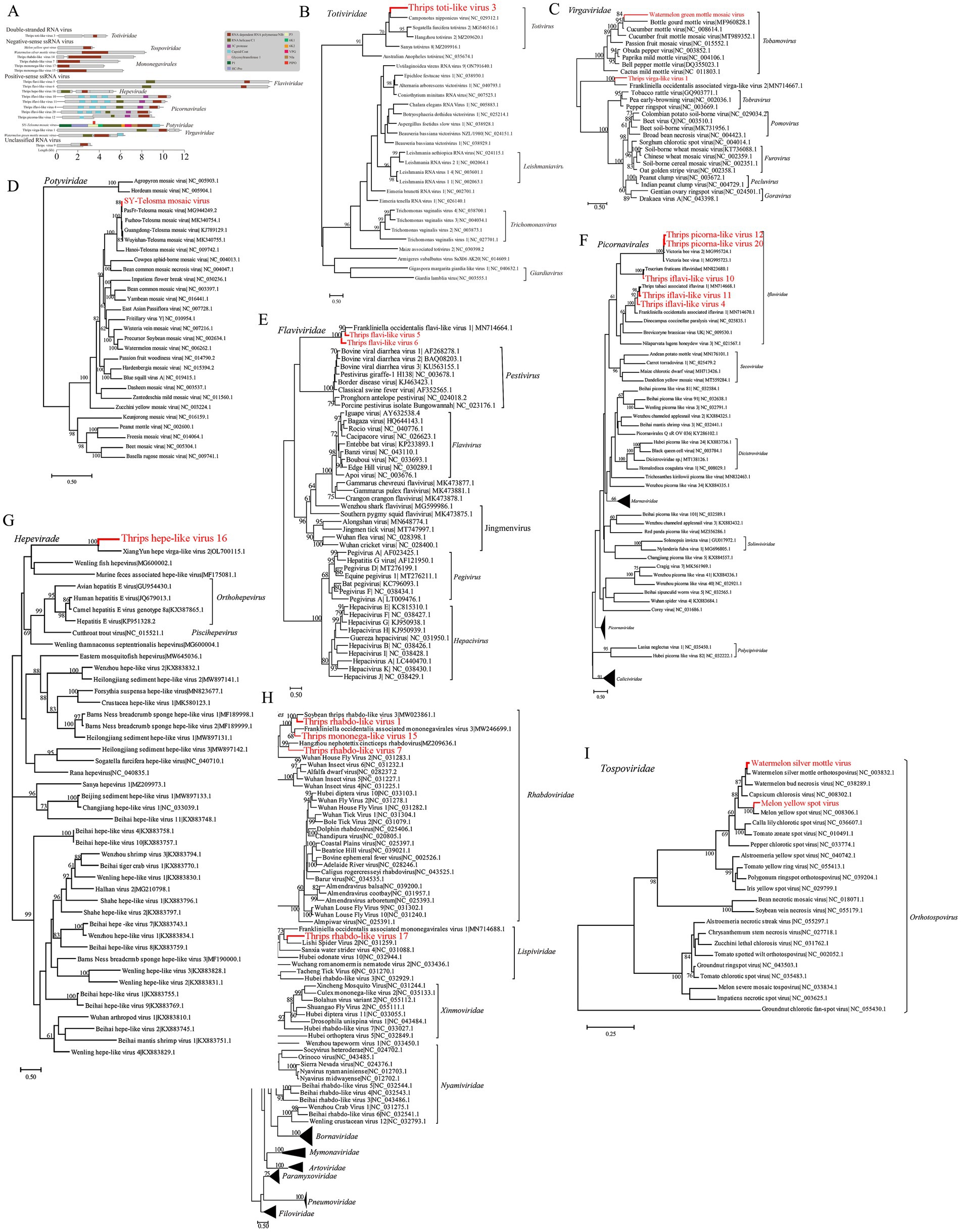
Figure 3. Presentation of the genomic structure of double-stranded RNA viruses, and the phylogenetic relationships of viruses from multiple families (including Virgaviridae, etc.), negative-sense single-stranded RNA viruses, and Tospoviridae viruses identified in this study with known counterparts. Schematic of viral genome structure and phylogenetic relationships of double-stranded RNA viruses (A,B). (A) Schematic representation of the viral genome structure. (B) Phylogenetic relationships of the double-stranded RNA viruses identified in this study (marked in red) with other known viruses in the Totiviridae family. Phylogenetic relationships of viruses identified in this study with known viruses across multiple families (C–G). (C) Phylogenetic relationships of the Virgaviridae viruses identified in this study (marked in red) with other known viruses in the Virgaviridae family. (D) Phylogenetic relationships of the Potyviridae viruses identified in this study (marked in red) with other known viruses in the Potyviridae family. (E) Phylogenetic relationships of the Flaviviridae viruses identified in this study (marked in red) with other known viruses in the Flaviviridae family. (F) Phylogenetic relationships of the Picornavirales viruses identified in this study (marked in red) with other known viruses in the Picornavirales order. (G) Phylogenetic relationships of the Hepeviridae viruses identified in this study (marked in red) with other known viruses in the Hepeviridae family. Phylogenetic relationships of negative-sense single-stranded RNA viruses and Tospoviridae viruses identified in this study (H,I). (H) Phylogenetic relationships of the negative-sense single-stranded RNA viruses identified in this study (marked in red) with other known viruses in the Mononegavirales order. (I) Phylogenetic relationships of the Tospoviridae viruses identified in this study (marked in red) with other known viruses in the Tospoviridae family.
3.2.4 Identification and analysis of +ssRNA viruses
Twelve positive-sense single-stranded RNA (+ssRNA) viruses were identified, representing various families, including Virgaviridae, Potyviridae, Flaviviridae, Picornavirales, and Hepeviridae. In one F. intonsa sample (HH22), a sequence closely matching Watermelon green mottle mosaic virus (WGMMV) was detected, with 99% amino acid homology (Figure 3C). Additionally, a novel virus related to Virgaviridae, identified in multiple thrips samples (DH12, HH22, QH32, C42, HL23), was named Thrips virga-like virus 1 (TVV1). The TVV1 genome is approximately 11,938 nucleotides in length and contains three open reading frames (ORFs), one of which includes both Hel and RdRp domains. TVV1 shares 42.8% amino acid similarity with F. intonsa-associated virga-like virus 2 (FAVV2) and forms a distinct evolutionary branch with FAVV2 (Figure 3C).
In two F. intonsa samples (C42 and HH22), sequences of Telopea mosaic virus (TeMV) from the Potyvirus genus were identified and designated SY-TeMV. The SY-TeMV genome is 10,057 nucleotides long, with a 9,630-nucleotide ORF encoding 3,209 amino acids. Phylogenetic analysis reveals that SY-TeMV forms a monophyletic branch with other Chinese isolates (Figure 3D).
Furthermore, two novel Flavivirus sequences were isolated from F. intonsa and M. usitatus samples, designated Thrips flavi-like virus 5 (TFV5) and Thrips flavi-like virus 6 (TFV6), respectively. The genomes of these viruses are notably longer than those of other members in the Flaviviridae family (Figure 3C). Phylogenetic analysis suggests that TFV5 and TFV6 are distantly related to other insect-specific flaviviruses (Figure 3E).
In the Picornavirales order, Thrips iflavi-like virus 4 (TIV4) and Thrips picorna-like virus 20 (TPV20) were detected in M. usitatus and F. intonsa samples, respectively. Additionally, four novel viruses were isolated from the T. palmi sample, including Thrips iflavi-like virus 10 (TIV10), Thrips iflavi-like virus 11 (TIV11), and Thrips picorna-like virus 12 (TPV12). These viruses belong to the Iflaviridae family but occupy distinct evolutionary branches (Figure 3F).
Lastly, a novel Hepeviridae virus, named Thrips hepe-like virus 16 (THV16), was identified in the T. palmi sample. The THV16 genome is 5,802 nucleotides long and contains three ORFs. Phylogenetic analysis indicates that THV16 is distantly related to other hepatitis viruses (Figure 3G).
3.2.5 Identification and analysis of −ssRNA viruses
Four novel virus sequences, suspected to belong to the negative-sense single-stranded RNA (−ssRNA) viruses, were identified. Among these, one virus was detected in a T. palmi sample (HL23) and was named Thrips mononega-like virus 17 (TMV17), potentially a new member of the Lispiviridae family. The other three viruses were detected in M. usitatus (DD11) and T. palmi (HL23) samples, showing amino acid sequence homology with viruses from the Rhabdoviridae family, ranging from 42.36 to 52.62%. These viruses were named Thrips rhabdo-like virus 7 (TRV7), Thrips rhabdo-like virus 14 (TRV14), and Thrips mononega-like virus 15 (TMV15), respectively. Due to limited sequencing depth, their full genome structures remain unresolved, and they were identified as vOTUs (virus operational taxonomic units) based solely on the conserved sequence of RNA-dependent RNA polymerase (RdRp).
Phylogenetic analysis revealed that TMV17 is most closely related to Lishi Spider Virus 2 and F. intonsa associated mononegavirales virus 1, with nucleotide and amino acid sequence similarities of 29.71 and 29.73%, respectively. TRV7, TRV14, and TMV15 clustered with other arthropod-hosted Rhabdoviridae members, distinct from viruses infecting vertebrate and plant hosts. Notably, TRV14 is most closely related to Soybean Thrips rhabdo-like virus 3, which infects soybean thrips, with 49.42% amino acid similarity. TMV15 shows 41.12% amino acid similarity to F. intonsa associated mononegavirales virus 3, which infects F. intonsa, suggesting that these thrips-associated viruses may represent a new evolutionary branch (Figure 3H).
Additionally, two full-length sequences with high similarity to the LRNA segment of Orthotospovirus were obtained from the T. palmi sample (HL23). These sequences were most closely related to WSMoV and MYSV, with nucleotide and amino acid sequence similarities of 95.06 and 96.42%, respectively (Figure 3I).
3.3 Detection of target viruses in cucurbits and other vegetables and their correlation with thrips occurrence
MYSV, WSMoV, and WGMMV viruses were detected exclusively in cucurbit crops, with detection rates of 32.77, 9.36, and 36.00%, respectively, while WSMoV and TeMV were detected solely in leguminous crops, with detection rates of 2.24 and 2.38%, respectively (Figure 4A). These findings suggest that these viruses may exhibit specific host preferences. The detection rate of MYSV showed a weak positive correlation with the presence of T. palmi (r2 = 0.39, p = 0.055), with a detection rate of 59% in cucumber (Figure 4B). In contrast, the detection rate of WSMoV was significantly and positively correlated with T. palmi occurrence (r2 = 0.58, p < 0.05), with the highest detection rate in watermelon at 61%, and a 6% detection rate in cowpea (Figure 4C). The detection rate of WGMMV did not show a significant correlation with the presence of F. intonsa (r2 = 0.04, p = 0.61) (Figure 4D). However, TeMV displayed a significant positive correlation with F. intonsa occurrence (r2 = 0.92, p < 0.05), with a 4% detection rate in cowpea (Figure 4E). These results suggest that these viruses may exhibit host specificity in particular crops and that their prevalence appears to be correlated with the occurrence of specific thrips populations.
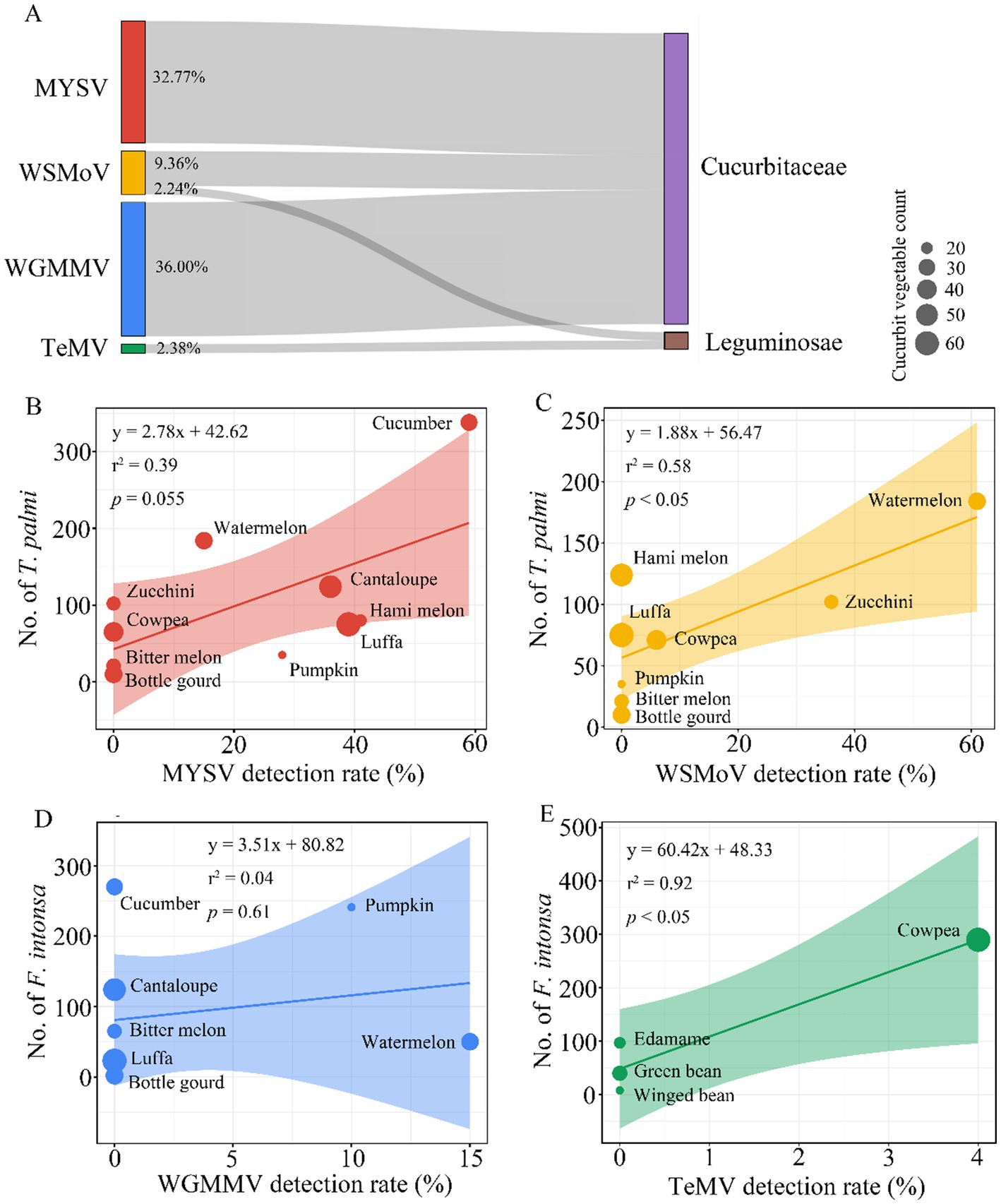
Figure 4. Correlation analysis between thrips-borne plant viruses and different families of cucurbit and leguminous crops. (A) Distribution of four plant viruses in cucurbit and leguminous crops. (B–E) Correlation between the detection rates of four plant viruses in cucurbit and leguminous crops and the occurrence of thrips on these crops. The size of the scatter points represents the number of samples tested, and the shaded areas indicate the 95% confidence intervals. (B) Correlation between the detection rate of MYSV and the abundance of T. palmi on cucurbit crops. (C) Correlation between the detection rate of WSMoV and the abundance of T. palmi on cucurbit and leguminous crops. (D) Correlation between the detection rate of WGMMV and the abundance of F. intonsa on cucurbit crops. (E) Correlation between the detection rate of TeMV and the abundance of F. intonsa on leguminous crops.
4 Discussion
In this study, we investigated the occurrence of thrips on crops from seven plant families in the tropical region of Hainan, focusing on the three dominant thrips species: F. intonsa, M. usitatus, and T. palmi. Virome analysis of six thrips sample libraries revealed 19 viruses spanning nine viral families and three unclassified viral groups, including double-stranded RNA viruses, positive-sense single-stranded RNA viruses, and negative-sense single-stranded RNA viruses. Our study also identified several novel viruses, including Ghabrivirales (Totiviridae), a double-stranded RNA virus and multiple positive- and negative-sense single-stranded RNA viruses. Additionally, detection rates of four plant viruses (MYSV, WSMoV, WGMMV, and TeMV) were significantly correlated with thrips abundance, suggesting a potential association between thrips density and the epidemiology of viral diseases in cucurbits and other vegetables.
We identified a total of 19 viruses in the thrips virome, spanning plant, arthropod, fungal, and vertebrate viruses. This finding aligns with previous studies in other arthropods, such as bees, mosquitoes, and locusts, further supporting the role of invertebrates, particularly arthropods, as significant hosts for RNA viruses (Chiapello et al., 2021; Thekke-Veetil et al., 2020; Xu et al., 2022). Additionally, we discovered several novel viruses, including two new members of the Flaviviridae family, TFV5 and TFV6, which form distinct evolutionary branches in the phylogenetic tree and may represent new viral lineages within Thysanoptera (Tree et al., 2016). These novel findings underscore the complexity of thrips as virus vectors and highlight their potential impacts on ecosystems.
The significant differences in viral read abundance across thrips samples may stem from varying infection rates at different collection stages, as well as the possibility that some low-abundance viruses are not actively replicating within thrips but are passively acquired from feeding on virus-contaminated plants (Neri et al., 2022). The high detection rate of abundant viruses, such as Picornavirales, suggests these viruses likely complete their life cycle within thrips, indicating active replication (Neri et al., 2022). Conversely, the low abundance of certain viruses may result from their integration into the thrips genome as endogenous viral elements, a phenomenon also observed in other arthropods (Neri et al., 2022). The discovery of these new viral species not only broadens our understanding of the diversity within the thrips virome but also underscores the complex interactions between thrips and the viruses they harbor. These findings offer valuable insights for future research on viral evolution, particularly in understanding virus adaptation to insect hosts and its potential implications for virus transmission dynamics in agricultural ecosystems.
The study results indicate that the composition of thrips viral communities is primarily influenced by their host plants, with viral diversity in different thrips populations closely linked to host plant diversity (Thongsripong et al., 2021). This finding supports the theory of virus-host co-evolution, suggesting that viruses establish specific ecological relationships with their hosts through long-term evolutionary processes (Brooks et al., 2017). The greater diversity and abundance of viruses carried by T. palmi compared to F. intonsa and M. usitatus may be attributed to T. palmi’s broader range of host plants, providing increased exposure to diverse viral species. Additionally, T. palmi may possess biological or ecological traits—such as a longer lifespan, higher mobility, or more frequent feeding on varied plant species—that enhance its efficiency as a virus vector, thus increasing its likelihood of acquiring and transmitting a broader array of viruses. These findings hold significant implications for pest management. The higher viral diversity associated with T. palmi suggests that this species should be a primary focus for monitoring and control efforts, particularly in crops where it is prevalent. Targeting T. palmi populations could help reduce the transmission of multiple plant viruses, thereby lowering the overall viral burden in agricultural ecosystems.
Using high-throughput sequencing, we detected four plant viruses—MYSV, WSMoV, WGMMV, and TeMV—in T. palmi and F. intonsa samples. Previous studies have demonstrated that MYSV and WSMoV are effectively transmitted by T. palmi, confirming the natural transmission of these viruses (Mou et al., 2021). However, the transmission mechanisms of WGMMV and TeMV remain unclear. While studies suggest that a close relative of WGMMV, CGMMV, is seed-transmitted (Yang et al., 2018), no definitive transmission pathway has been reported for WGMMV or TeMV. This study is the first to detect WGMMV and TeMV in F. intonsa samples, suggesting that F. intonsa may have come into contact with these emerging viruses. However, detecting these viruses in thrips does not necessarily imply that thrips can transmit them. Thrips may acquire viral particles by feeding on infected plants, but this does not confirm their ability to transmit the viruses to new hosts. Further research is needed to explore the specific transmission mechanisms and efficiency of these viruses, including whether F. intonsa can actively transmit WGMMV and TeMV, to better understand their transmission dynamics in the field.
Insects, particularly thrips, are significant vectors of plant viruses, with strong transmission capabilities and widespread distribution, profoundly impacting the field epidemiology and spread of viruses (Jones, 2005). This study demonstrates that the field prevalence of MYSV is significantly and positively correlated with the population size of T. palmi, suggesting that T. palmi may contribute to the transmission of MYSV in cucurbits and other vegetables. However, virus transmission is influenced by various factors, including virus acquisition efficiency, environmental conditions, and the presence of additional vectors. Further research is necessary to clarify the role of T. palmi in this process. Similarly, the correlation between WSMoV spread and T. palmi is weaker, potentially due to factors such as initial infection sources, host plant resistance, and other ecological variables. Additionally, the detection of WGMMV and TeMV in F. intonsa samples suggests that F. intonsa may be involved in their transmission, but further studies are required to elucidate the specific mechanisms.
This study enhances our understanding of the diverse range of viruses carried by thrips and reveals significant correlations between thrips populations and virus prevalence in cucurbits and other vegetables. While these correlations do not confirm causation, they offer valuable insights into the potential ecological interactions between thrips and plant viruses. These findings provide a foundation for developing targeted pest management strategies focused on key thrips species and the conditions that influence viral transmission. Future research should seek to elucidate the specific transmission mechanisms and ecological dynamics of these viruses, which will be essential for improving virus control in agricultural ecosystems.
Data availability statement
All raw sequence data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1242210. This BioProject includes six SRA datasets related to thrips collected in the tropical region of Hainan Island, southern China. The data are publicly available and can be accessed at the following link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1242210.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
QZ: Writing – original draft, Data curation. CC: Data curation, Methodology, Writing – original draft. BL: Data curation, Supervision, Writing – original draft, Writing – review & editing. YG: Data curation, Writing – review & editing. LC: Resources, Writing – review & editing. LP: Investigation, Writing – review & editing. ZY: Investigation, Writing – review & editing. HL: Methodology, Writing – review & editing. JT: Resources, Writing – review & editing. BJ: Project administration, Writing – review & editing. SL: Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Ministry of Agriculture and Rural Affairs of China (JDZX-2024) and the Central Public-Interest Scientific Institution Basal Research Fund (No. 1630042023007).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1540883/full#supplementary-material
Footnotes
References
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2017). Correction: Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 15:e1002587. doi: 10.1371/journal.pbio.1002587
Chiapello, M., Bosco, L., Ciuffo, M., Ottati, S., Salem, N., Rosa, C., et al. (2021). Complexity and local specificity of the virome associated with tospovirus-transmitting thrips species. J. Virol. 95:e0059721. doi: 10.1128/JVI.00597-21
Cunniffe, N. J., Taylor, N. P., Hamelin, F. M., and Jeger, M. J. (2021). Epidemiological and ecological consequences of virus manipulation of host and vector in plant virus transmission. PLoS Comput. Biol. 17:e1009759. doi: 10.1371/journal.pcbi.1009759
Ertunç, F. (2020). “Physiology of virus-infected plants” in Applied plant virology: advances, detection, and antiviral strategies (London: Academic Press), 199–205.
He, S., and Creasey Krainer, K. M. (2020). Pandemics of people and plants: which is the greater threat to food security? Mol. Plant 13, 933–934. doi: 10.1016/j.molp.2020.06.007
He, Z., Guo, J.-F., Reitz, S. R., Lei, Z.-R., and Wu, S. Y. (2020). A global invasion by the thrip, Frankliniella occidentalis: current virus vector status and its management. Insect Sci. 27, 626–645. doi: 10.1111/1744-7917.12721
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H., and Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A. 101, 14812–14817. doi: 10.1073/pnas.0406166101
Jones, D. R. (2005). Plant viruses transmitted by thrips. Eur. J. Plant Pathol. 113, 119–157. doi: 10.1007/s10658-005-2334-1
Jones, R. A. C. (2021). Global plant virus disease pandemics and epidemics. Plan. Theory 10:233. doi: 10.3390/plants10020233
Karthikeyan, G., Nagendran, K., and Kumari, S. (2021). Tospovirus diseases of ornamental plants: characterization, identification, diagnosis and management. Singapore: Springer.
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Mou, D.-F., Chen, W.-T., Li, W.-H., Chen, T.-C., Tseng, C.-H., Huang, L.-H., et al. (2021). Transmission mode of watermelon silver mottle virus by Thrips palmi. PLoS One 16:e0247500. doi: 10.1371/journal.pone.0247500
Mound, L. A., and Morris, D. C. (2007). The insect order Thysanoptera: classification versus systematics. Zootaxa 1668, 395–411. doi: 10.11646/zootaxa.1668.1.21
Neri, U., Wolf, Y. I., Roux, S., Camargo, A. P., Lee, B., Kazlauskas, D., et al. (2022). Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell 185, 4023–4037.e18. doi: 10.1016/j.cell.2022.08.023
Nichol, S. T., Beaty, B. J., and Elliott, R. M. (2005). “Virus taxonomy: classification and nomenclature of viruses” in Satellites and other virus dependent nucleic acids (Moscow: International Committee on Taxonomy of Viruses), 1211–1219.
Rotenberg, D., Jacobson, A. L., Schneweis, D. J., and Whitfield, A. E. (2015). Thrips transmission of tospoviruses. Curr. Opin. Virol. 15, 80–89. doi: 10.1016/j.coviro.2015.08.003
Sánchez-Sánchez, M., Vargas-Arispuro, I., Tovar-Pedraza, J. M., González-Pérez, C. J., Aispuro-Hernández, E., Quintana-Obregón, E. A., et al. (2024). Mixed viral infections in vegetable crops: biochemical and molecular aspects. Mex. J. Phytopathol. 42:30. doi: 10.18781/R.MEX.FIT.2404-3
Sastry, K. S., and Zitter, T. A. (2014). Plant virus and viroid diseases in the tropics. Dordrecht: Springer.
Thekke-Veetil, T., Lagos-Kutz, D., McCoppin, N. K., Hartman, G. L., Ju, H.-K., Lim, H.-S., et al. (2020). Soybean thrips (Thysanoptera: Thripidae) harbor highly diverse populations of arthropod, fungal, and plant viruses. Viruses 12:1376. doi: 10.3390/v12121376
Thongsripong, P., Chandler, J. A., Kittayapong, P., Wilcox, B. A., Kapan, D. D., and Bennett, S. N. (2021). Metagenomic shotgun sequencing reveals host species as an important driver of virome composition in mosquitoes. Sci. Rep. 11:8448. doi: 10.1038/s41598-021-87122-0
Tree, M. O., McKellar, D. R., Kieft, K. J., Watson, A. M., Ryman, K. D., and Conway, M. J. (2016). Insect-specific flavivirus infection is restricted by innate immunity in the vertebrate host. Virology 497, 81–91. doi: 10.1016/j.virol.2016.07.005
Whitfield, A. E., Ullman, D. E., and German, T. L. (2005). Tospovirus-thrips interactions. Annu. Rev. Phytopathol. 43, 459–489. doi: 10.1146/annurev.phyto.43.040204.140017
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Wu, S., Tang, L., Zhang, X., Xing, Z., Lei, Z., and Gao, Y. (2018). A decade of a thrips invasion in China: lessons learned. Ecotoxicology 27, 1032–1038. doi: 10.1007/s10646-017-1864-6
Xu, Y., Jiang, J., Lin, X., Shi, W., and Cao, C. (2022). Identification of diverse viruses associated with grasshoppers unveils the parallel relationship between host phylogeny and virome composition. Virus Evol. 8:veac057. doi: 10.1093/ve/veac057
Yang, K., Yan, H., Song, L., Jin, P., Miao, W., and Cui, H. (2018). Analysis of the complete genome sequence of a potyvirus from passion fruit suggests its taxonomic classification as a member of a new species. Arch. Virol. 163, 2583–2586. doi: 10.1007/s00705-018-3885-8
Keywords: diversity, ecological relationships, high-throughput sequencing, plant virus transmission, thrips vectors, virome
Citation: Zhang Q, Cheng C, Lyu B, Gao Y, Chen L, Peng L, Yue Z, Lu H, Tang J, Jiao B and Liu S (2025) Thrips and plant viruses: a comprehensive virome analysis in tropical agriculture. Front. Microbiol. 16:1540883. doi: 10.3389/fmicb.2025.1540883
Edited by:
Susheel Kumar, National Botanical Research Institute (CSIR), IndiaReviewed by:
Vijayanandraj Selvaraj, National Botanical Research Institute (CSIR), IndiaAnna Taglienti, Council for Agricultural Research and Agricultural Economy Analysis (CREA), Italy
Copyright © 2025 Zhang, Cheng, Lyu, Gao, Chen, Peng, Yue, Lu, Tang, Jiao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoqian Lyu, bHl1YmFvcWlhbkBxcS5jb20=
†These authors have contributed equally to this work