 Linghuan Yang†
Linghuan Yang† Hilal Samut†
Hilal Samut† Leonie Kemmerling
Leonie Kemmerling Renato Hohl Orsi
Renato Hohl Orsi Martin Wiedmann
Martin Wiedmann Ruixi Chen
Ruixi Chen Cristina Resendiz-Moctezuma*
Cristina Resendiz-Moctezuma*- Department of Food Science, Cornell University, Ithaca, NY, United States
Despite the emergence of whole genome sequencing (WGS) for Salmonella characterization, serotype assignment remains important as it allows identification of Salmonella subgroups that differ in distribution, virulence, and ecology. However, it has been shown that multiple divergent lineages of the same Salmonella serovar may have evolved independently multiple times and may present distinct epidemiological characteristics. Previous studies that aimed to identify the phylogeny of certain Salmonella serovars often used isolates from specific geographical locations or outbreaks and a small number of isolates to infer the phylogeny. To address these limitations and to advance the understanding of Salmonella’s evolutionary patterns, we (i) identified the phylogenetic grouping (i.e., mono-, para-, or polyphyly) of the 100 most common Salmonella serovars analyzing 63,204 genomes available in the NCBI Pathogen Detection database, (ii) identified, for each polyphyletic serovar, the lineages that contain the majority of genomes, and (iii) inferred the antigen divergence between the five most common serovars (i.e., Salmonella Enteritidis, Typhimurium, Newport, I 4,[5],12:i:-, and Infantis) and their respective closely-related serovars. Among the 100 most common Salmonella serovars analyzed, 19 serovars are monophyletic, nine are paraphyletic, and 72 are polyphyletic. In 47 of the 72 polyphyletic serovars, one lineage contains more than 90% of the serovar’s confirmed genomes. Antigen divergence results suggest that serovars Typhimurium and I 4,[5],12:i:- (often referred to as monophasic Typhimurium) have emerged independently of each other multiple times, except for the major I 4,[5],12:i:- lineage, which emerged from the major Typhimurium lineage. Furthermore, divergence in Salmonella serovars appears to primarily occur through modifications in the H1 antigen. Hence, this study shows that (i) a much larger number of serovars than previously known are polyphyletic; (ii) serovars previously known to be polyphyletic contain more lineages than previously known; and (iii) many serovars include lineages that only have a few isolates with a given serovar. Our data suggests that, in the age of genomics, molecular serotyping should be combined with other phylogenetically informative approaches to not just assign a serovar but to also indicate the serovar lineage for polyphyletic and paraphyletic serovars.
1 Introduction
Salmonellosis, one of the most common foodborne diseases in the world, is caused by bacteria from the genus Salmonella, which has two species, enterica and bongori. Salmonella enterica is further divided into six subspecies (i.e., enterica, salamae, houtenae, arizonae, diarizonae, and indica) (Coburn et al., 2007), with at least one study suggesting 11 subspecies (Pearce et al., 2021); however, the status of these five additional subspecies still needs to be confirmed and formally accepted. Within Salmonella enterica (S.), more than 2,600 serovars have been identified; of these, nearly 1,500 serovars belong to subspecies enterica (Issenhuth-Jeanjean et al., 2014). Salmonella serovars can be further classified into two groups: typhoidal (i.e., Typhi, Paratyphi A, Paratyphi B, Paratyphi C, and Sendai), and non-typhoidal Salmonella (NTS; e.g., Typhimurium, Enteritidis, and Infantis) serovars. While the five typhoidal serovars mainly cause typhoid fever in humans, NTS are usually characterized by gastroenteritis with symptoms including acute abdominal pain, diarrhea, vomiting, and sometimes nausea (Eng et al., 2015). NTS serovars stand out as a large group of pathogens with diverse ecological niches and a notable impact on public health (Cheng et al., 2019). Approximately, 93.8 million human illnesses, and 155,000 deaths are estimated to be caused by NTS serovars annually worldwide (Murray et al., 2012), while two of the typhoidal serovars (i.e., S. Typhi and S. Paratyphi A) are estimated to cause 25.7 million human illnesses and 178,000 deaths annually worldwide (Kirk et al., 2015).
Within Salmonella serovars, some are monophyletic [e.g., S. Dublin (Chen et al., 2024)], where the isolates within a serovar cluster in a single lineage and share a common ancestor that is not shared by isolates of any other serovar. Some Salmonella serovars have been shown to be paraphyletic [e.g., S. Bredeney (Worley et al., 2018)], where the isolates within a serovar cluster in a single lineage and share a common ancestor, but this common ancestor is also shared with isolates of a different serovar. Finally, some Salmonella serovars are polyphyletic [e.g., S. Kentucky (Chen et al., 2024)], where the isolates within a serovar are spread across multiple divergent lineages and do not share a common ancestor. Multiple divergent lineages of the same Salmonella serovar that have emerged independently from each other might present very distinct phenotypic and epidemiological characteristics (Feasey et al., 2016). For example, a recent study analyzing the phylogeny of seven Salmonella serovars (i.e., Salmonella Cerro, Dublin, Enteritidis, Infantis, Kentucky, Montevideo, and Reading) associated with meat and poultry revealed that polyphyletic lineages of the same serovar might display distinct characteristics, such as two S. Reading lineages, one associated with turkey and one associated with swine (Chen et al., 2024).
Although Salmonella enterica serovars are highly diverse, they are mainly defined by the combination of 47 somatic (O) and 114 flagellar (H) antigens (McQuiston et al., 2004; Seif et al., 2019). O antigens are lipopolysaccharide (LPS) chains present on the bacterial LPS layer (Liu et al., 2014). The O antigens are encoded by the rfb gene cluster (also known as O-antigen gene cluster), which is typically located on the chromosome and consists of 3–20 genes depending on the serogroup. These genes are involved in the synthesis of sugar precursors, sugar transferases and O-unit processing and modification (Liu et al., 2014). H antigens are flagellar proteins subdivided into H1 and H2, encoded by fliC and fljB, respectively, that contribute to the motility of Salmonella (Silverman et al., 1979; Minamino et al., 2018). These flagella genes are alternatively expressed, which means H1 and H2 antigens cannot be expressed at the same time. This phenomenon is mediated by the Hin recombinase (Hughes et al., 1988) and results in phase variation.
Traditional Salmonella serotyping (White-Kauffmann-Le Minor scheme; WKL scheme) is based on serological agglutination of the O, H, and, to a lower extent, capsular (Vi) antigens. However, traditional serotyping cannot distinguish between isolates that cluster into distinct lineages of polyphyletic serovars (Wattiau et al., 2011; Achtman et al., 2012; Liu and Hsiao, 2022). Therefore, although traditional serotyping still provides a crucial historical link to epidemiological information, higher resolution in subtyping and characterization of Salmonella beyond the serovar level is required for better discrimination. With the advent of whole genome sequencing (WGS) in the last decades, WGS-based subtyping methods enabled researchers and public health agencies to obtain information with higher resolution and faster turnaround time (Ibrahim and Morin, 2018). Importantly, WGS data can also be used for the identification of lineages that independently emerged within a single serovar via phylogenetic analysis using single nucleotide polymorphisms (SNPs) or core genome multilocus sequence typing (cgMLST) (Worley et al., 2018; Yin et al., 2020; Cherchame et al., 2022; Chen et al., 2024).
Previous studies have individually characterized the phylogeny of specific serovars using different methods. For example, Yin et al. (2020) analyzed 4,498 Salmonella genomes representing 89 serovars by selecting genomes representing each sequence type (ST) within each serovar and identified seven putative polyphyletic serovars—Salmonella Montevideo, Bareilly, Saintpaul, Muenchen, Paratyphi B, Kentucky, and Newport—using MLST for phylogenetic analysis (Yin et al., 2020). Worley et al. (2018) analyzed a set of 445 Salmonella genomes representing 260 serovars and found approximately 10% of the serovars to be polyphyletic (Worley et al., 2018). Zhang et al. (2019b) used 2,258 genomes representing 107 serovars, by selecting isolates representing serovar, and found that 24 were polyphyletic using core genome alignment (Zhang et al., 2019b). However, these studies either used a limited number of genomes from each serovar or used a limited number of reference serovars for comparison. Reference serovars are a set of genomes selected to demonstrate the diversity of Salmonella enterica subspecies (i.e., enterica, salamae, houtenae, arizonae, diarizonae, and indica) in a phylogenetic analysis. Inclusion of a comprehensive number of these reference serovars in phylogenetic analysis is important as it increases the resolution of the analysis allowing for a higher confidence in the phylogenetic grouping (i.e., mono-, para-, or polyphyly) of the target serovar. In this study, 514 reference genomes representing 301, 49, 127, and 37 serovars of the subspecies enterica, arizonae, diarizonae, and houtenae were used, respectively. By analyzing 63,204 genomes across 100 serovars in the present study (92 Salmonella serovars) and two previous studies from our group [i.e., 8 Salmonella serovars, (Chen et al., 2022; Chen et each ST within each al., 2024)], we aim to bridge these gaps by employing WGS data to (i) delineate the phylogeny of the 100 most common Salmonella serovars, (ii) identify the major lineage(s) within polyphyletic serovars, and (iii) to infer antigen divergence between specific serovars and their respective closely-related Salmonella serovars. This knowledge will help shift Salmonella assessment and control strategies to a phylogeny-based approach, ultimately improving Salmonella surveillance and reducing the burden of salmonellosis.
2 Materials and methods
2.1 Identification of the 100 most common Salmonella serovars in NCBI Pathogen Detection
To identify the 100 Salmonella serovars most frequently found in NCBI Pathogen Detection (NCBI PD), metadata for all Salmonella enterica genomes (n = 552,201) available at the time was downloaded, as a CSV file, from the NCBI PD website1 on August 8, 2023. Salmonella serovar information provided in the “computed type” field in the NCBI PD was used to initially assign isolates to a serotype; isolates without a “computed type” (n = 1,409) or isolates where no antigens had been assigned (i.e., the entry was -:-:-) (n = 4,741) were identified. Among these isolates, 702 were singletons (i.e., not part of any SNP clusters) and were excluded from downstream analysis since our approach to assign the serovar of an isolate depends on the serotype assignment provided by NCBI PD (see below). Isolates belonging to SNP clusters (n = 5,448)—including those lacking computed types (n = 1,208) or antigen assignments (n = 4,240)—were still accounted for as other isolates in the same SNP clusters could have serotypes assigned by NCBI PD, and the total number of isolates in these SNP clusters were considered. Exclusion of these singletons may have affected the order or the inclusion of some serovars among the 100 most common serovars, as these 702 excluded singletons were not counted. For each serovar, we subsequently determined (i) the total number of isolates assigned to a given serotype, (ii) the total number of SNP clusters for a given serovar, and (iii) the total number of singletons (defined as isolates that do not cluster with any other isolates) found for a given serovar. Serovars were subsequently ranked based on the number of isolates available in the NCBI PD. Among the 100 most common Salmonella serovars in the NCBI PD identified with this approach, eight serovars (i.e., Salmonella Cerro, Dublin, Enteritidis, Infantis, Kentucky, Montevideo, Reading, and Saintpaul) had been previously investigated by our group using a very similar methodology as the one used here (Chen et al., 2022; Chen et al., 2024).
2.2 Retrieval of genome sequences for the most common Salmonella serovars
The workflow followed in Sections 2.2 through 2.4 has been summarized in Figure 1 and the annotated codes used in each pipeline are publicly available in https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Codes.
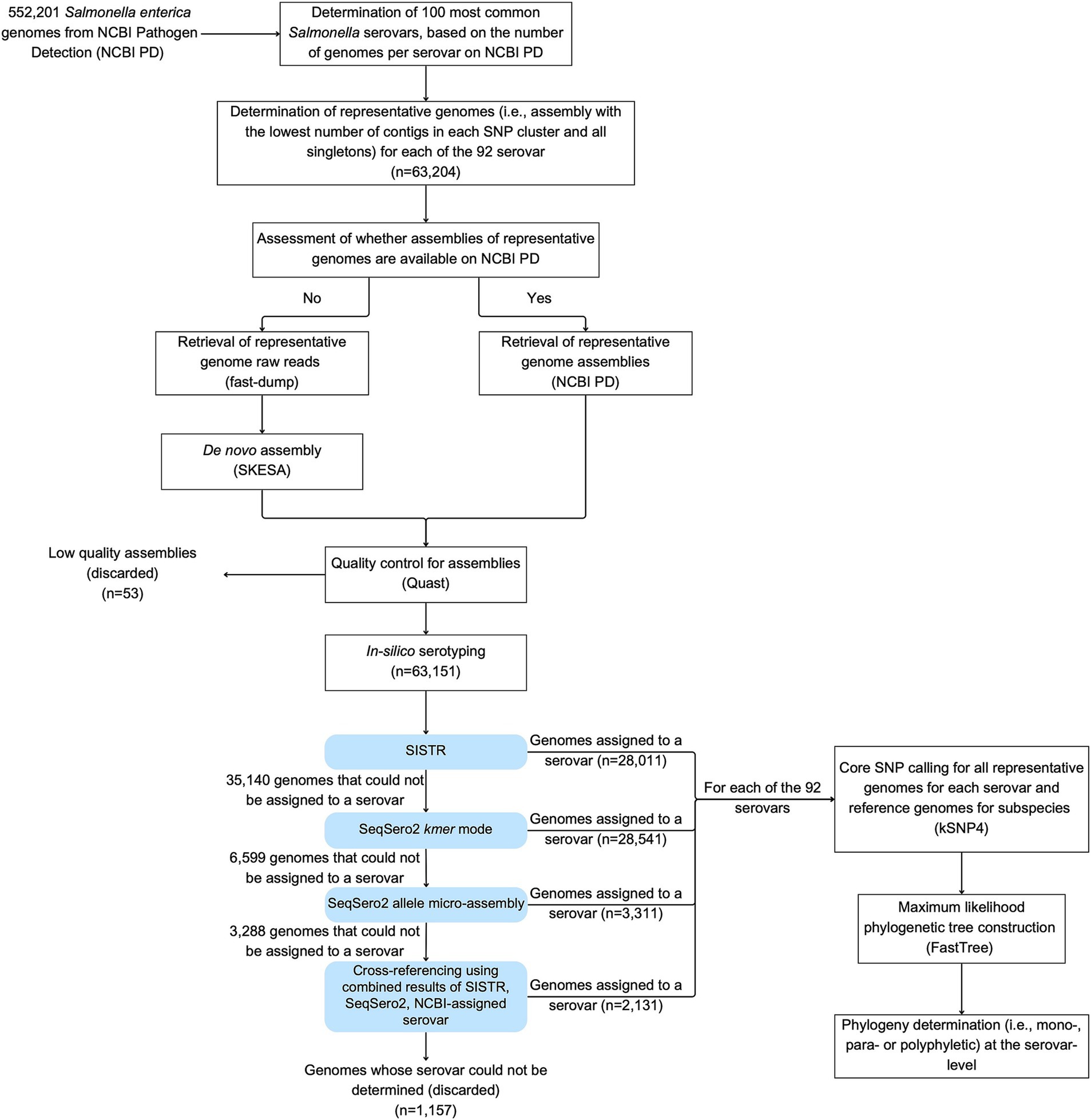
Figure 1. Workflow used in the study to identify the phylogeny of the 100 most common Salmonella serovars in NCBI PD. Representative genomes included (i) the assembly with the lowest number of contigs from each SNP cluster (i.e., best assembly), and (ii) assemblies for all singletons. Fifty-three genomes representing singleton isolates were removed because their assemblies were considered with low quality: (i) > 500 contigs, (ii) N50 < 20,000 bp, or (iii) a genome size < 4 or > 6 Mbp. For any genomes where the NCBI-assigned serovar could not be confirmed with SISTR, SeqSero2 k-mer, and SeqSero2 allele micro-assembly, serovar confirmation and assignment was carried out by cross-referencing results from multiple approaches. Specifically, in cases where the SISTR results for “qc_status” had warning messages, but the (i) SISTR results for “serovar_antigen” and “serovar_cgMLST,” and (ii) either one or both of the SeqSero2 modes predicted the same serovar, then this serovar was assigned to the genome (even if it differed from the NCBI-assigned serovar).
To perform phylogenetic analyses of 92 of the 100 most common serovars that had not been previously analyzed by our group, we identified representative genomes of each of these 92 serovars for subsequent analyses. For each serovar, representative genomes included (i) the assembly with the lowest number of contigs from each SNP cluster, and (ii) assemblies for all singletons. Where available, whole genome assemblies were downloaded from NCBI PD (see text footnote 1); otherwise, raw reads were retrieved using fastq-dump (Chen et al., 2018) and assembled de novo using SKESA (Souvorov et al., 2018). QUAST (Gurevich et al., 2013) was then used to assess the quality of each assembly and only assemblies that met the following criteria were used for subsequent analyses: (i) < 500 contigs, (ii) N50 > 20,000 bp, and (iii) a genome size between 4 and 6 Mbp. Singletons not meeting these criteria were not included in further analyses. All assemblies with the lowest number of contigs in a given SNP cluster met the assembly quality criteria.
2.3 In-silico serotyping
To confirm the serovar assignments obtained from NCBI PD, all representative genomes that passed the quality criteria described above were subjected to in-silico serotyping using SISTR (Yoshida et al., 2016). The NCBI-assigned serovar (i.e., “computed type” in the NCBI PD) was considered confirmed if (i) both the antigen prediction (“serovar_antigen”) and the cgMLST prediction (“serovar_cgmlst”) obtained from SISTR matched the NCBI-assigned serovar, and (ii) the SISTR quality control status (“qc_status”) was “PASS.” For genomes that did not meet these criteria, serovar prediction was performed using the k-mer approach implemented in SeqSero2 (Zhang et al., 2019a). If the serovars assigned by SeqSero2 k-mer and by NCBI matched, the NCBI-assigned serovar was considered confirmed. If the NCBI-assigned serovar still could not be confirmed, serovar prediction was performed using the allele micro-assembly approach implemented in SeqSero2 (Zhang et al., 2019a); if results from SeqSero2 allele micro-assembly matched the NCBI-assigned serovar, the serovar was considered confirmed. For any genomes where the NCBI-assigned serovar could not be confirmed with these steps, the serovar prediction results from all four sources (NCBI, SISTR, SeqSero2 k-mer, and SeqSero2 allele micro-assembly) were used for further serovar confirmation and assignment (“serotype assignment by cross referencing”). Specifically, in cases where the SISTR results for “qc_status” had warning messages, but the (i) SISTR results for “serovar_antigen” and “serovar_cgMLST,” and (ii) either one or both of the SeqSero2 modes predicted the same serovar, then this serovar was assigned to the genome (even if it differed from the NCBI-assigned serovar). Singletons that were assigned a different serovar than the serovar assigned by NCBI were removed from any further analyses; traditional serotyping was not carried out to resolve these inconsistencies because isolates were not readily available. When representative genomes from a given SNP cluster were assigned a different serovar than the serovar assigned by NCBI, the second-best assembly in the SNP cluster was then used for in-silico serotyping. If the second-best assembly was assigned the same serovar as the one assigned by NCBI, the second-best assembly was kept for downstream analysis.
2.4 Maximum-likelihood tree reconstruction
Among the 100 most common serovars, eight serovars (i.e., Salmonella Enteritidis, Infantis, Reading, Cerro, Dublin, Kentucky, Montevideo, and Saintpaul), for which our group had previously performed phylogenetic analyses (Chen et al., 2022; Chen et al., 2024), were not included in the phylogenetic reconstruction here, bringing the total number of serovars of interest to 92. To reconstruct the phylogeny of each of the 92 serovars analyzed here, kSNP4 was used to generate a core SNP matrix using a k-mer size of 19 nucleotides (Hall and Nisbet, 2023). Each kSNP4 analysis included assemblies for all confirmed representative SNP cluster genomes and singletons, plus 301, 49, 127, and 37 reference genomes representing Salmonella enterica subsp. enterica, arizonae, diarizonae, and houtenae serovars, respectively (available at: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Reference_Genomes). For S. subsp. enterica, we used the reference genome set reported by Chen et al. (2024), which included 285 genomes. However, this reference genome set did not include all serovars from the 100 most common serovars identified here (e.g., serovars Derby, Concord). Therefore, the serovars not included in the set reported by Chen et al. (2024) were added, for a total of 301 reference genomes in the set (Chen et al., 2024). For S. subsp. arizonae, the reference genome set described by Shariat et al. (2021) was used (Shariat et al., 2021). For S. subsp. diarizonae and houtenae, we selected representative genomes from all serovars within these subspecies, based on the isolates available on NCBI PD.
For tree construction, a S. subsp. indica (GCA_010567565.1), a subsp. diarizonae (GCA_010556605.1), a subsp. arizonae (GCA_007099465.1), and a Salmonella bongori (GCA_037109255.1) genome was used as the outgroup for serovars from subsp. enterica, subsp. houtenae, subsp. diarizonae, and subsp. arizonae, respectively. FastTree was then used to infer the maximum-likelihood phylogeny for each serovar with the General Time-Reversible (GTR)-CAT model and 1,000 bootstrap replicates (Price et al., 2010). The Interactive Tree of Life (iTOL) was used for tree visualization and annotation (Letunic and Bork, 2021).
The phylogenetic grouping definition delineated by Oosterbroek (1987) was used to determine whether a non-monophyletic serovar showed paraphyletic or polyphyletic phylogeny (Oosterbroek, 1987). Briefly, monophyletic serovars are characterized by having a most recent common ancestor (MRCA; the ancestor node of all isolates of a given serovar) that has not given rise to any other serovar. Paraphyletic serovars are characterized by having an MRCA that has given rise to at least another monophyletic serovar or a monophyletic group of other serovars. Polyphyletic serovars are characterized by having an MRCA that has given rise to other serovars that do not form a monophyletic group (Oosterbroek, 1987). Phylogeny for the eight serovars described in previous studies (Chen et al., 2022; Chen et al., 2024) was analyzed again using the criteria described above for consistency. Lineages were identified and labeled alphabetically based on the number of isolates within each lineage. For example, lineage A (e.g., Newport A) represents the lineage with the most isolates within a given serovar, while lineage B represents the lineage with the second largest number of isolates. In this study, a lineage is defined as comprising at least two isolates of the same serovar clustering together. Isolates that do not cluster with any other isolate of the same serovar were defined as “stand-alone singletons” and were not considered as lineages here; these stand-alone singletons were still included in our analyses.
2.5 Phylogenetic inference of antigenic variation linked to divergence of closely-related Salmonella serovars
To infer the divergence of closely-related serovars and their antigenic formula variations, the five most common serovars available in the NCBI PD database (Salmonella Enteritidis, Typhimurium, Newport, I 4,[5],12:i:-, and Infantis) were selected. Two of the five most common serovars analyzed in this section had been previously reported in another study by our group: S. Enteritidis and Infantis (Chen et al., 2024). To better infer the phylogeny with a statistically robust framework using a large number of genomes, the maximum likelihood phylogenetic trees for these serovars (i.e., S. Enteritidis and S. Infantis) were reconstructed using a larger set of reference serovars (n = 301; the full list available at: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Reference_Genomes) and core SNPs to determine diversification events of the five most common Salmonella serovars. Closely-related serovars were defined as all serovars that (i) share an MRCA with a lineage belonging to at least one of the five serovars analyzed in this section, and (ii) where the MRCA node has a bootstrap value of > 0.7. The antigenic formula of monophyletic and paraphyletic lineages belonging to the five serovars analyzed here were compared to the antigenic formula of their closely-related serovars; for paraphyletic lineages, the antigenic formula of a given lineage was also compared to that of serovars paraphyletically-clustered within the lineage. Lineages that share an MRCA with ≥ 5 serovars (e.g., I 4,[5],12:i:- B and D) were not included in the antigenic formula diversification analysis. Stand-alone singletons were also not included in the antigenic formula diversification analysis as they were not considered lineages. The antigenic differences between the five serovars of interest and their respective closely-related serovars were further classified as somatic (O) or flagellar (H) differences. Differences in O antigens were further divided into (i) major difference (i.e., differences that lead to O antigens that change the serogroup of the serovars) and (ii) minor differences (i.e., differences that lead to O antigens that do not change the serogroup of the serovars). Hence, major O antigen differences involve serovars that belong to different serogroups, while minor O antigen differences involve serovars that belong to the same serogroup. A total of nine categories were used to describe the overall antigen differences as (i) H1 differences only, (ii) H2 differences only, (iii) H1 and H2 differences, (iv) H2 and minor O differences, (v) H2 and major O differences, (vi) H1 and minor O differences, (vii) H1 and major O differences, (viii) H1, H2 and minor O differences, and (ix) H1, H2 and major O differences.
3 Results
3.1 The 100 most common serovars in the NCBI PD represent 94.55% of the total Salmonella enterica genomes in the NCBI PD database
A total of 552,201 genomes downloaded from NCBI PD were used to determine the 100 most common Salmonella serovars in this database (based on the serotype assignment captured in the “computed type” field). The 100 most common serovars represented S. enterica subspecies enterica (n = 96), arizonae (n = 1), diarizonae (n = 1), and houtenae (n = 2), and were classified into 23 serogroups that are B (n = 21), C1 (n = 19), C2-C3 (n = 13), D1 (n = 11), E1 (n = 8), G (n = 7), and 17 serogroups that contained less than five serovars each (n = 21). The five most common serovars were, in order, (i) S. Enteritidis, (ii) S. Typhimurium, (iii) S. Newport, (iv) S. I 4,[5],12:i:-, and (v) S. Infantis (see Supplementary Table 1 for full list). The 100 most common serovars included 93 named serovars as well as seven unnamed serovars (e.g., I 4,[5],12:i:-; the fourth most common serovar). Overall, based on the “computed type” field on NCBI PD, the 100 most common serovars represent 94.55% of all Salmonella enterica genomes in the NCBI PD database (Supplementary Figure 1).
3.2 In-silico serotyping of the representative genomes for the 100 most common Salmonella serovars showed that only 1.52% of them do not match the serotype assigned by the NCBI PD
In total, 63,204 representative genomes were initially selected for our analyses based on the NCBI PD’s “computed types” prediction (Table 1). However, 53 genomes were discarded as either their genome data were no longer available on NCBI, or the genome assemblies did not meet the assembly quality criteria. These 53 genomes were singletons, and none of the SNP clusters were discarded during quality control. In-silico serotyping approaches, including SISTR, SeqSero2 k-mer, and SeqSero2 micro-assembly were used to confirm or revise the NCBI PD serovar assignments for the 63,151 high-quality genomes representing among the 100 most common Salmonella serovars (referred as “target serovars” hereon) on NCBI PD. As each genome represent a distinct isolate, we will use the term “isolate” going forward. The 63,151 isolates included (i) 20,497 isolates representing each a SNP cluster and confirmed as belonging to the target serovar, (ii) 41,497 singleton isolates confirmed as the target serovar, and (iii) 1,157 isolates that were not confirmed as the target serovar. The 1,157 isolates that were not confirmed as the target serovar listed on NCBI PD included (i) 957 isolates (1.52%) where the in-silico approaches used here identified a different serovar than the serovar assigned on NCBI PD, (ii) 171 isolates (0.27%) for which the genome data were contaminated with multiple serovars as determined by SeqSero2 micro-assembly results, and (iii) 29 isolates (0.05%) where the in-silico approaches used here did not identify an O antigen.
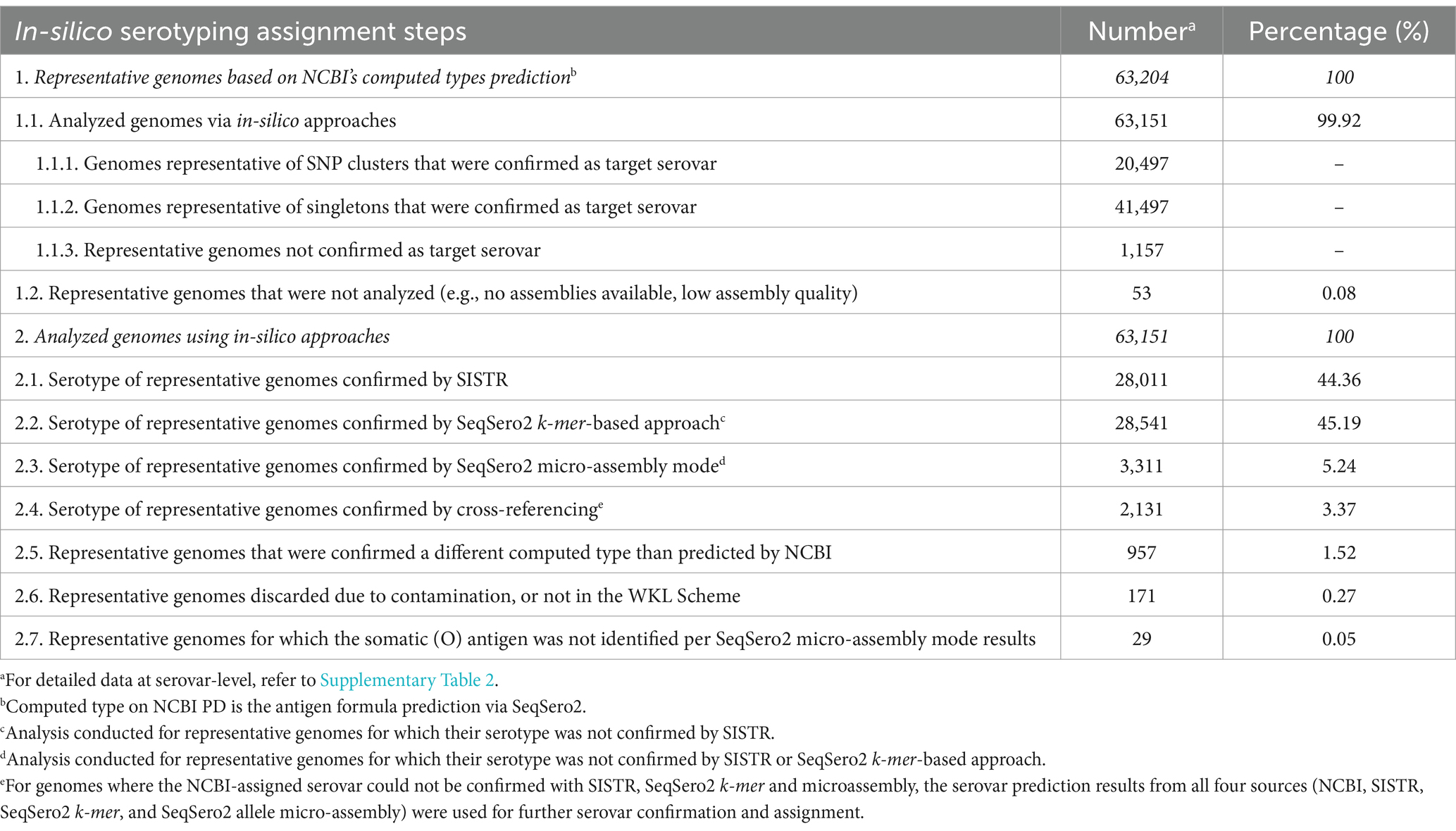
Table 1. Serovar identification of representative genomes via in-silico serotyping tools for the 100 most common Salmonella serovars available on NCBI PD.
Using SISTR alone, out of the 63,151 representative isolates, the serotype for 28,011 isolates was confirmed. However, we observed that the serotype of 32,552 representative isolates belonging to 43 target serovars could not be confirmed via SISTR (e.g., Salmonella Newport, Javiana, and Muenchen; Supplementary Table 2). The SeqSero2 k-mer approach confirmed the serotype of 28,541 out of the 35,140 representative isolates not confirmed by SISTR, and the SeqSero2 micro-assembly approach confirmed the serotype of 3,311 out of 6,599 representative isolates that were not confirmed by either SISTR or SeqSero2 k-mer approaches. Lastly, cross-referencing was used to confirm the serotype of 2,131 out of 3,288 representative isolates not confirmed by the individual approaches.
Serovars Montevideo, I 4,[5],12:b:-, and IV 48:g,z51:- had the largest number of isolates for which the results from our in-silico serotyping analyses did not match the NCBI-assigned serovar. Serovar Montevideo (antigenic formula 6,7,14,[54]:g,m,[p],s:[1,2,7]) had 136 isolates that were confirmed a different serovar than the NCBI-assigned serovar (i.e., Montevideo). Based on our in-silico analyses, these 136 isolates were either identified as serovar Bareilly (n = 25; antigenic formula 6,7,14:y:1,5) or lacked sufficient information for an unambiguous serovar assignment (n = 111). Serovar I 4,[5],12:b:- had 114 isolates that did not match the NCBI-assigned serovar (i.e., I 4,[5],12:b:-), these 114 isolates were identified as the serovar Paratyphi B (antigenic formula 1,4,[5],12:b:1,2). Finally, serovar IV 48:g,z51:- had 110 isolates that did not match the NCBI-assigned serovar (i.e., IV 48:g,z51:-), these isolates were identified as IIIa 48:g,z51:-.
3.3 Phylogenetic analysis of the 100 most common Salmonella serovars showed that 19 are monophyletic, 9 are paraphyletic, and 72 are polyphyletic
Maximum-likelihood trees (Supplementary Figures 2–95, including the trees of the 92 serovars analyzed here, and the re-analyzed trees of the serovars S. Enteritidis and S. Montevideo) were analyzed at the serovar level to identify serovars that were mono-, poly-, or paraphyletic; further analyses were performed to identify monophyletic and paraphyletic lineages within the 72 polyphyletic serovars. Among the 100 most common Salmonella serovars, which include 92 serovars analyzed here (see Supplementary Figures 2–27, 29–64, and 66–95 for the maximum-likelihood phylogenetic trees at https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Supplementary_Figures) and 8 serovars analyzed in two previous studies (Chen et al., 2022; Chen et al., 2024), our results showed that (i) 19% of the serovars are monophyletic, (ii) 9% are paraphyletic, and (iii) 72% are polyphyletic (Figure 2); polyphyletic serovars presented either more than one lineage, or one lineage and at least one stand-alone singleton. Across all 100 serovars, we identified 252 lineages (i.e., monophyletic or paraphyletic lineages); the maximum number of lineages was found for serovar Oranienburg, which included 10 lineages. Among polyphyletic serovars with more than one lineage, 215 lineages were identified including (i) 165 monophyletic, and (ii) 50 paraphyletic lineages.
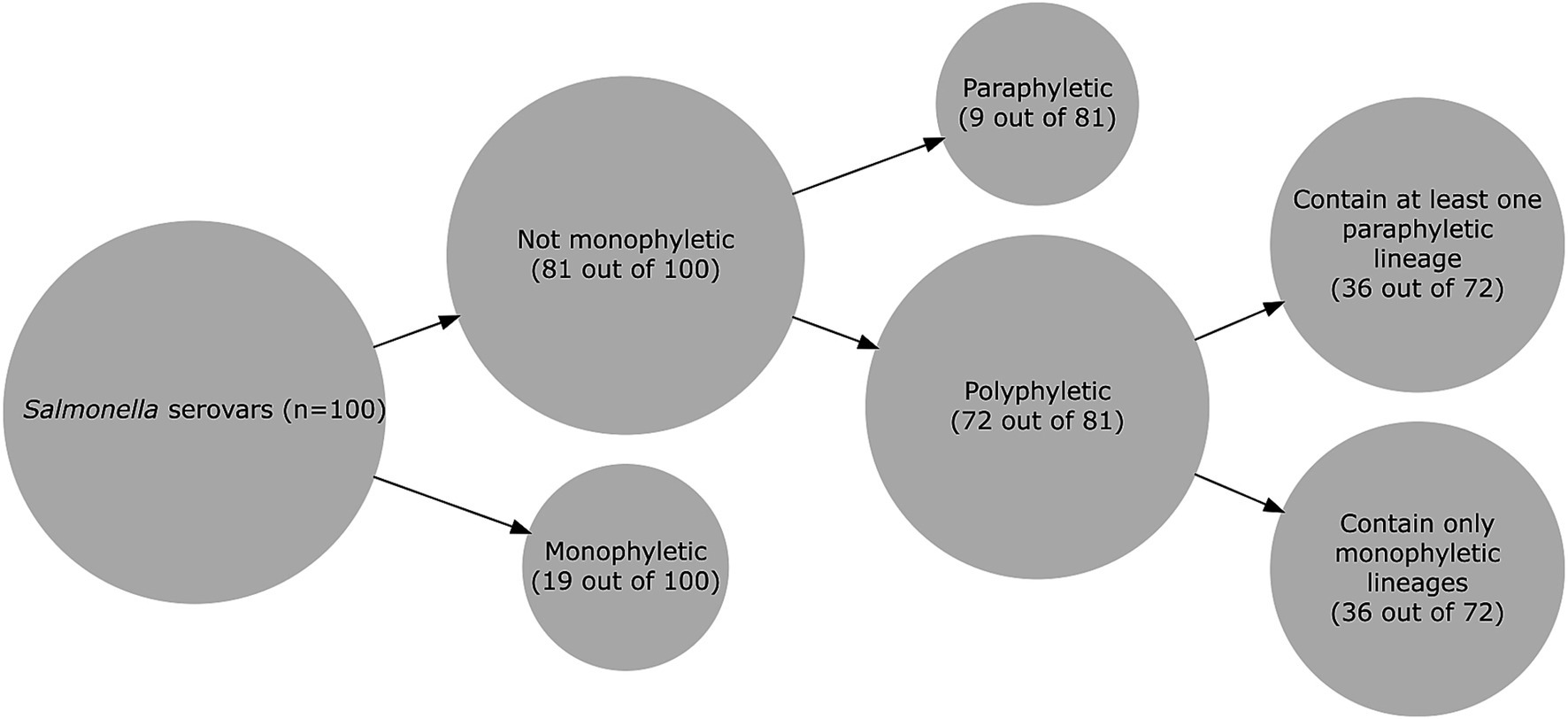
Figure 2. Phylogenetic classification of the 100 most common Salmonella serovars available on the NCBI PD database. The size of the circle is proportional to the number of Salmonella serovars in a given group.
Among the 19 monophyletic Salmonella serovars identified here (see Table 2 for a full list), Salmonella Javiana, Typhi, and Braenderup were the three most common monophyletic serovars with 16,637, 13,735, and 9,943 confirmed isolates, respectively. Salmonella Haifa was the least common monophyletic serovar with 353 confirmed isolates (Supplementary Table 1).

Table 2. Overall phylogenetic grouping (i.e., mono-, para-, or polyphyly) of the 100 most common Salmonella serovars on NCBI PD (in alphabetical order).
Nine of the 100 Salmonella serovars analyzed in this study were classified as paraphyletic. These serovars have only one lineage which includes all isolates assigned to the target serovar. However, within each given lineage, we have identified at least one isolate that belongs to a different serovar. For example, serovar Poona has one paraphyletic lineage that includes 3,908 confirmed isolates, as well as serovar Bristol. Serovar Agbeni has 1,851 confirmed isolates clustered within one lineage that also includes serovars Limete and Ituri. Finally, serovar Johannesburg has 1,730 confirmed isolates clustered within one lineage that also includes serovar Urbana.
Overall, among the 72 polyphyletic serovars, 36 serovars contain at least one paraphyletic lineage (e.g., Salmonella Enteritidis, Typhimurium), and 27 serovars contain only monophyletic lineages (e.g., Salmonella Newport, Kentucky). The remaining nine polyphyletic serovars (e.g., Salmonella Agona, Mbandaka, Indiana) include only one monophyletic/paraphyletic lineage and one or two stand-alone singletons; for example, S. Agona had 7,810 confirmed isolates, with 7,809 of the isolates clustering in a monophyletic lineage, and one isolate as a stand-alone singleton. Salmonella Enteritidis, Typhimurium, Newport, I 4,[5],12:i:-, and Infantis were the most common polyphyletic serovars.
3.4 For most polyphyletic serovars, more than 90% of the isolates represent a single lineage
The 72 polyphyletic serovars identified in this study included between one and 10 lineages (Table 3). Polyphyletic serovars with more than one lineage (n = 63) most commonly included two, three, or four lineages (25, 13, and 16 serovars, respectively). Among the 63 serovars with more than one lineage, 47 serovars had only one lineage including > 10% of the isolates. Among these 47 serovars, 42 serovars had > 90% of the isolates grouped in a single lineage. For example, S. Typhimurium had 99.9% of the isolates included in a single lineage (Typhimurium A; see Table 3). The remaining 16 of the 63 polyphyletic serovars with at least two lineages identified in this study, had multiple lineages with > 10% of the isolates. For example, S. Newport had 38,377 isolates distributed in four lineages: Newport A contained 61.6% (n = 23,633 isolates), Newport B contained 37.0% (n = 14,230 isolates), while the remaining 1.4% (n = 514 isolates) were distributed in Newport C and D (Table 3).
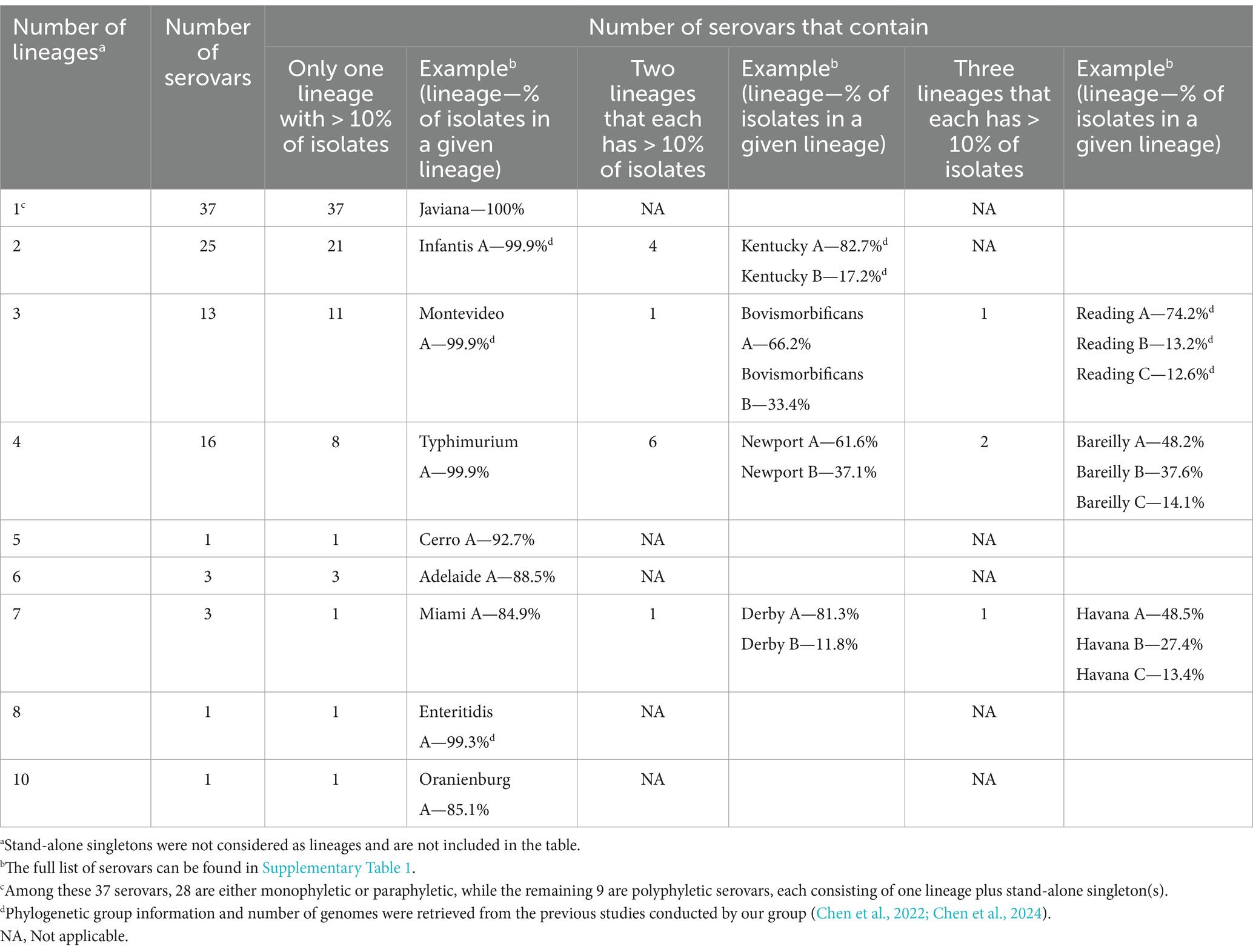
Table 3. Distribution of the 100 most common Salmonella serovars available on NCBI PD by number of lineages and the number of serovars that have one, two, or three lineages containing more than 10% of all genomes within each serovar.
3.5 Closely-related serovars diverged primarily through changes in the H1 antigen
To better understand the diversification that ultimately leads to the emergence of different Salmonella serovars, we characterized the antigenic variations of the five most common Salmonella serovars (i.e., Salmonella Enteritidis, Typhimurium, Newport, I 4,[5],12:i:-, and Infantis) and their closely-related serovars (i.e., serovars clustered within paraphyletic lineages or serovars that share an MRCA supported by a > 0.7 bootstrap value). These five serovars included 14 monophyletic and eight paraphyletic lineages. Amongst the 14 monophyletic lineages, five were not compared to any other serovar because they did not share an MRCA with five or less serovars supported by a bootstrap > 0.7. Thus, nine out of the 14 monophyletic lineages were compared to 17 closely-related serovars (e.g., the monophyletic Typhimurium D was compared with Salmonella Stanley, Schleissheim, and Paratyphi B as these 4 serovars cluster with a bootstrap value of 0.95, Figure 3). The eight paraphyletic lineages were compared to 16 serovars that fell within the lineages of interest (e.g., the paraphyletic Typhimurium B and S. Heidelberg were compared as S. Heidelberg clustered within Typhimurium B, Figure 3). Four of these eight paraphyletic lineages were also compared to seven serovars that shared an MRCA supported by a bootstrap value > 0.7 with a given paraphyletic lineages (e.g., paraphyletic lineage I 4,[5],12:i:- A was compared to Salmonella Worb and Baildon, Supplementary Figure 98). In total, 40 antigenic diversification events were identified, which included (i) differences in flagellar (H) antigens only (n = 13) and (ii) differences in flagellar (H) and somatic (O) antigens (n = 27). The differences in flagellar antigens were classified as (i) H1 differences only (n = 8), (ii) H2 differences only (n = 2), and (iii) H1 and H2 differences (n = 3). The differences in flagellar and somatic antigens were classified as (i) H2 and minor O differences (n = 2), (ii) H2 and major O differences (n = 2), (iii) H1 and minor O differences (n = 2), (iv) H1 and major O differences (n = 4), (v) H1, H2 and minor O differences (n = 6), and (vi) H1, H2 and major O differences (n = 11). We identified no diversification events that included only O antigen differences (Tables 4, 5). In summary, of the 40 diversification events we found, 34, 27, and 26 were associated with differences in the H1, O, and H2 antigens, respectively. All 5 serovars assessed here (i.e., Enteritidis, Typhimurium, Newport, I 4,[5],12:i:-, and Infantis) showed similar patterns of diversification, with most events involving the lineage A of a given serovar being associated with variations in flagella antigens only.
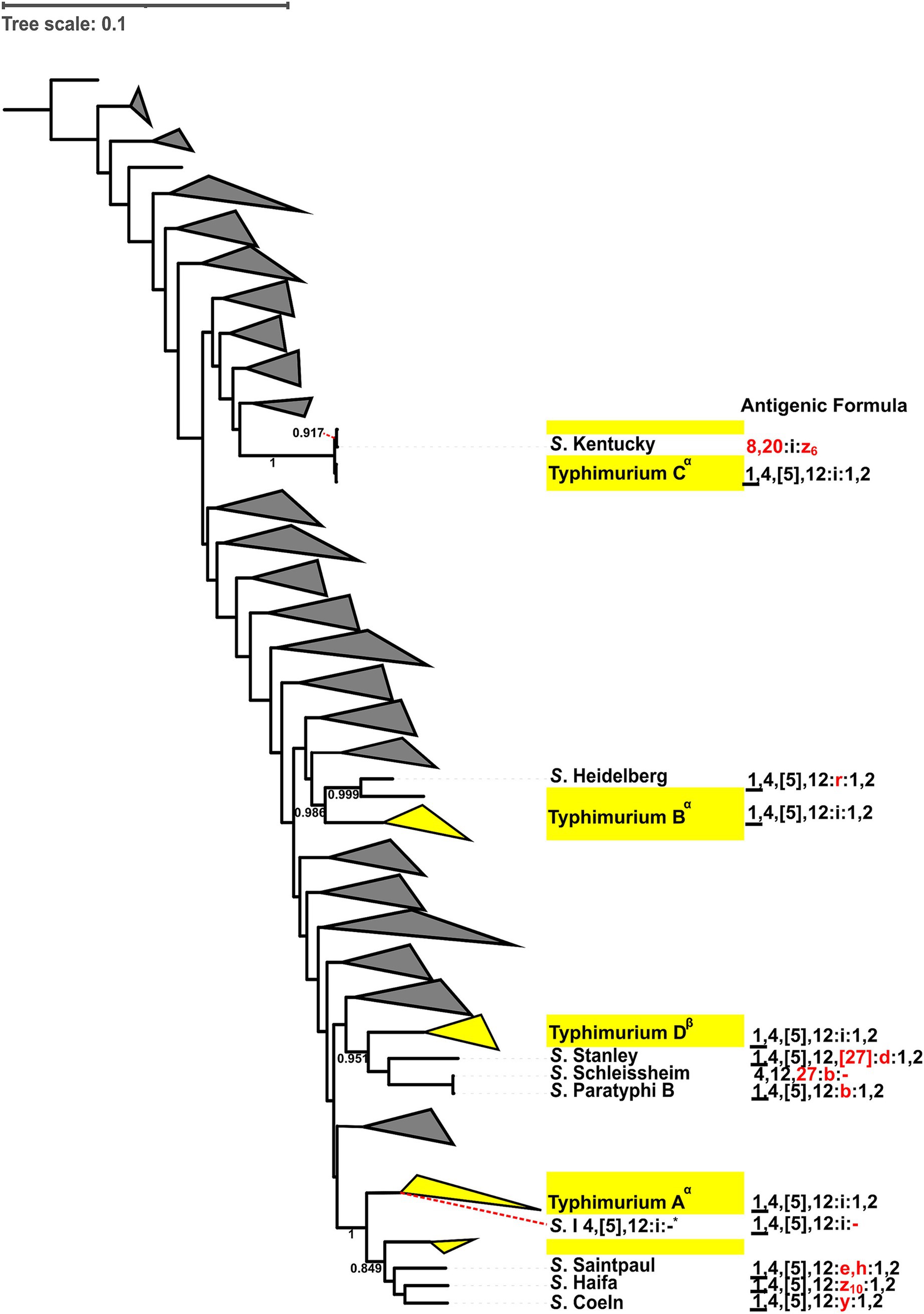
Figure 3. Antigenic formula diversification comparisons of the four lineages of S. Typhimurium (i.e., Typhimurium A-D) with corresponding closely-related serovars. Closely-related serovars share the same most recent common ancestor as the S. Typhimurium lineages. Differences in somatic (O) and (H) flagellar antigens between closely-related serovars and S. Typhimurium lineages are indicated in red. Bootstrap values are provided for the ancestral nodes. Antigenic formula comparisons were made if the bootstrap value of ancestral nodes between Typhimurium lineages and closely-related serovar was >0.7. Closely-related serovars and their antigenic formulas are shown explicitly. Yellow triangles represent genomes from the target serovar (i.e., S. Typhimurium), while branches representing non-Typhimurium serovars (distantly related, at least one node away, and not sharing an MRCA) are collapsed to form grey triangles. Superscripts were added to the phylogenetic lineages to differentiate those that are paraphyletic (α) from those that are monophyletic (β). The S. I 4,[5],12:i:- reference isolate (*) clusters within Typhimurium A. Antigenic formulas: (i) underlined O factors (_)—determined by phage conversion and the genome should be lysogenized with the specific converting phage; (ii) curly brackets ({ })—O antigens represented in curly brackets cannot coexist with the other antigens in curly brackets; (iii) square brackets ([ ])—O or H antigens that are present or absent with no relation to phage conversion; (iv) brackets (( ))—O or H antigens that are weakly agglutinable.
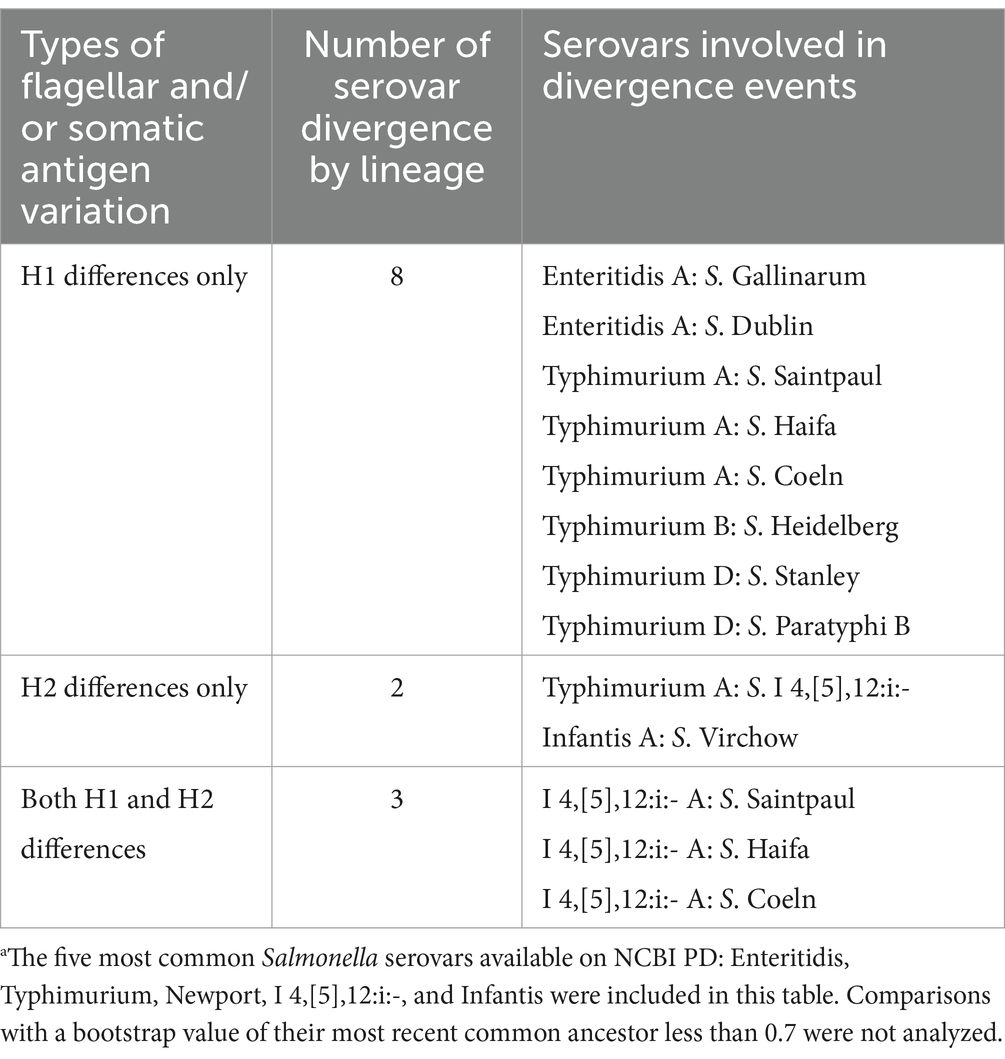
Table 4. Inference of divergence among the top five most common Salmonella serovars available on NCBI PDa by variations in flagellar (H) antigens (n = 13).
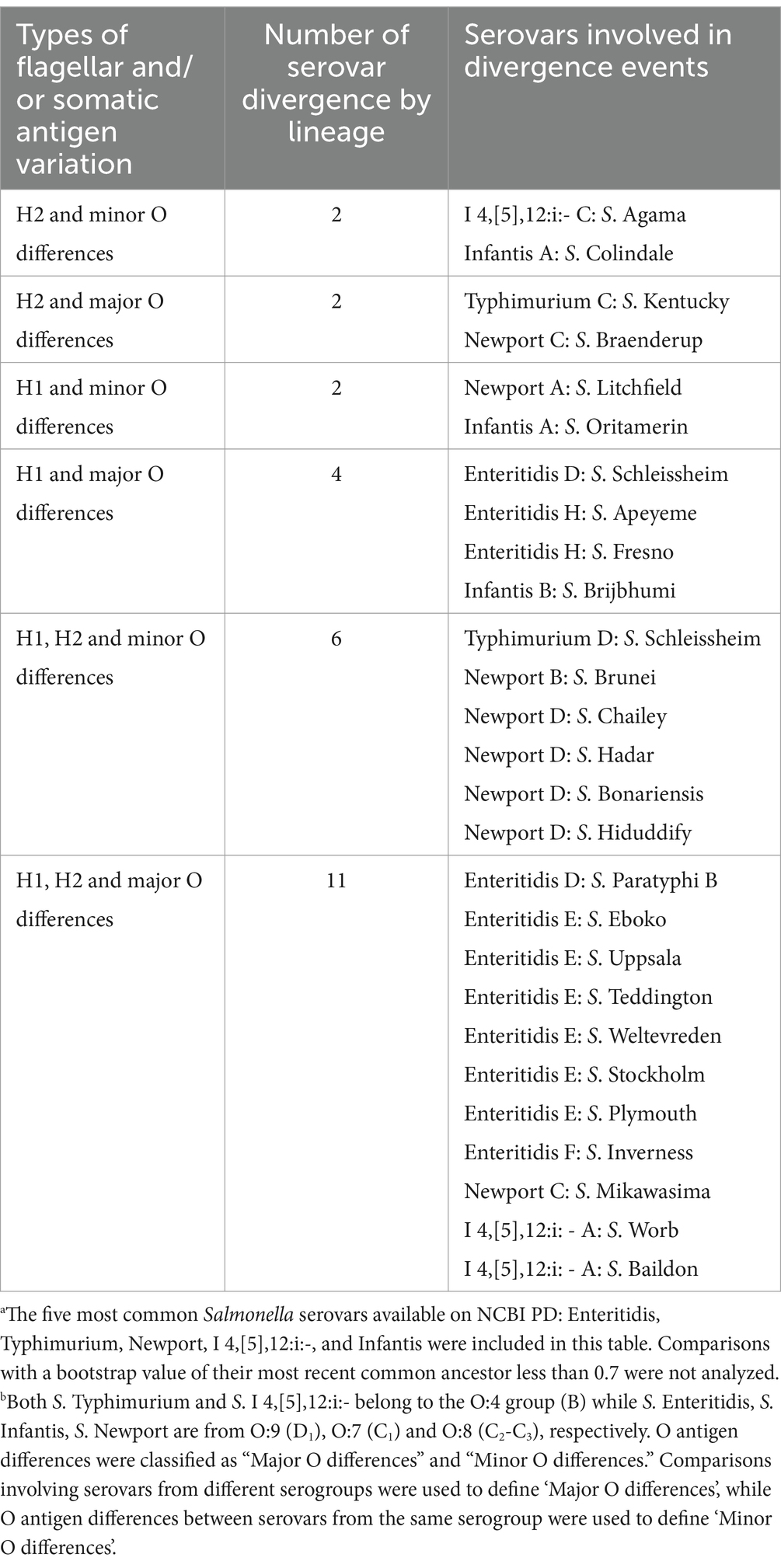
Table 5. Inference of divergence among the top five most common Salmonella serovars available on NCBI PDa by variations in flagellar (H) and somatic (O) antigensb (n = 27).
Specifically, out of the 13 diversification events involving five of the eight S. Enteritidis lineages, two events (both involving Enteritidis A) involved differences in the H1 antigen only, three events involved differences in the H1 and major O antigens, and eight events involved differences in the H1, H2 and major O antigens (Tables 4, 5 and Supplementary Figure 96). Out of the nine diversification events involving the four S. Typhimurium lineages, six events (including three events involving Typhimurium A) involved differences in the H1 antigen only, one event (involving Typhimurium A) involved differences in the H2 antigen only, one event involved differences in the H2 and major O antigens, and one event involved differences in the H1, H2 and minor O antigens (Figure 3; Tables 4, 5). Out of the eight diversification events involving the four S. Newport lineages, one event (involving Newport A) involved differences in the H1 and minor O antigens, one event involved differences in the H2 and major O antigens, five events involved differences in the H1, H2 and minor O antigens, and one event involved differences in the H1, H2 and major O antigens (Tables 4, 5 and Supplementary Figure 97). Out of the seven diversification events involving two of the four I 4,[5],12:i:- lineages, one event (involving I 4,[5],12:i:- A) involved differences in the H2 antigen only, three events (all of which involved I 4,[5],12:i:- A) involved differences in the H1 and H2 antigens, one event involved differences in the H2 and minor O antigens, and two events (both involving I 4,[5],12:i:- A) involved differences in the H1, H2 and major O antigens (Supplementary Figure 98 and Tables 4, 5). Out of the four diversification events involving the two Infantis lineages, one event (involving Infantis A) involved differences in the H2 antigen only, one event (involving Infantis A) involved differences in the H2 and minor O antigens, one event (involving Infantis A) involved differences in the H1 and minor O antigens, and one event involved differences in the H1 and major O antigens (Supplementary Figure 99 and Tables 4, 5).
3.6 Salmonella I 4,[5],12:i:- has emerged multiple times independently from Salmonella Typhimurium
To better understand the evolution of S. I 4,[5],12:i:- (often referred to as monophasic Typhimurium) and its relationship to S. Typhimurium, a phylogenetic analysis of all representative S. I 4,[5],12:i:- isolates and at least one representative S. Typhimurium isolate from each S. Typhimurium lineage was performed (Figure 4). While I 4,[5],12:i:- lineage A shares an MRCA with S. Typhimurium, I 4,[5],12:i:- lineages B, C, and D, and the stand-alone singleton I 4,[5],12:i:- S1 do not. Hence, our results suggest that S. I 4,[5],12:i:- has emerged multiple times independently from S. Typhimurium.
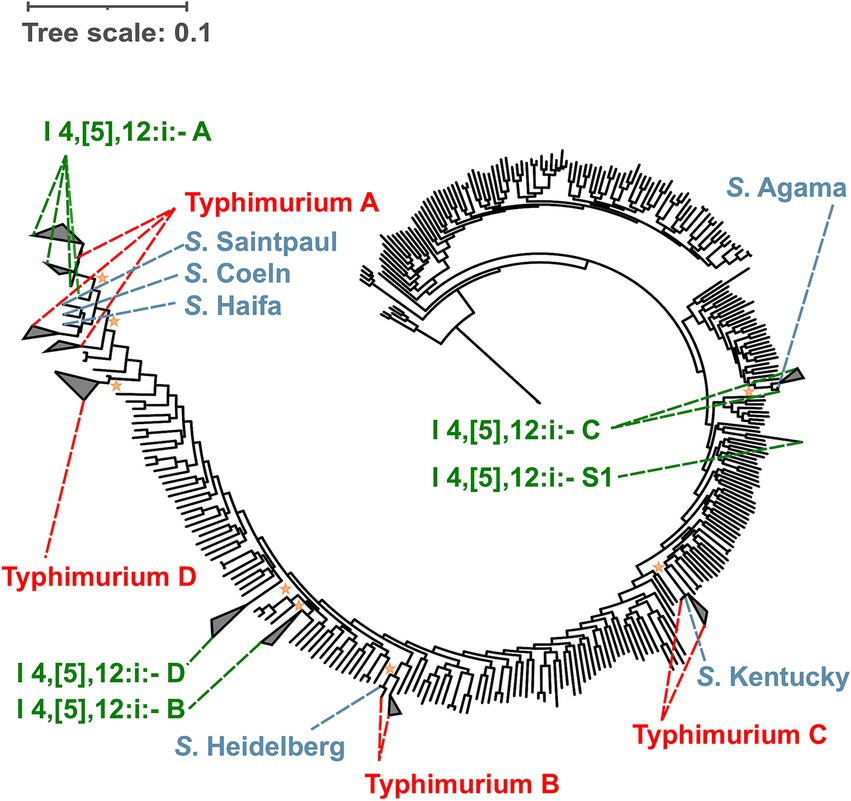
Figure 4. Maximum likelihood phylogenetic tree for S. I 4,[5],12:i:- suggests that S. I 4,[5],12:i:- has diverged multiple times independently without sharing an MRCA with S. Typhimurium. Both Salmonella Typhimurium and I 4,[5],12:i:- are polyphyletic serovars. The four lineages belonging to S. I 4,[5],12:i:- (i.e., I 4,[5],12:i:- A, B, C, and D) and one stand-alone singleton (i.e., S. I 4,[5],12:i:- S1) are shown in green. The four lineages belonging to S. Typhimurium (i.e., Typhimurium A, B, C, and D) are shown in red. Reference serovars that clustered within the paraphyletic lineages of Salmonella I 4,[5],12:i:- (i.e., I 4,[5],12:i:- A, and C) and Typhimurium (i.e., Typhimurium A, B, and C) are shown in blue. The major I 4,[5],12:i:- lineage (i.e., I 4,[5],12:i:- A) shares an MRCA with S. Typhimurium, while the other three S. I 4,[5],12:i:- lineages (i.e., I 4,[5],12:i:- B, C, and D) emerged independently without sharing an MRCA with S. Typhimurium. In addition to the 301 reference genomes representing Salmonella subsp. enterica used to reconstruct the phylogenetic trees, reference genomes were selected from Typhimurium A (n = 3), B (n = 2), C (n = 2), and D (n = 1) lineages. The average pairwise number of nucleotide substitutions per site was used to report branch lengths. For rooting the tree, one genome belonging to Salmonella enterica subsp. indica was used as the outgroup. The Shimodaira-Hasegawa test with 1,000 resamples was used to assess the clustering confidence. The orange stars represent the most recent common ancestor of each I 4,[5],12:i:- lineage with a bootstrap value higher than 0.7. For individual phylogenetic trees for Salmonella I 4,[5],12:i:- and Typhimurium refer to Supplementary Figures 39 and 90, respectively.
4 Discussion
Salmonella subtyping and classification heavily relies on serovar identification. However, previous studies have shown that strains of the same serovar might have different phenotypic characteristics because they belong to lineages that emerged independently from each other. For example, different lineages of Salmonella Newport and Kentucky have been suggested to demonstrate varying virulence capabilities, appear to have adapted to different hosts, and appear to exhibit distinct geographic distributions (Cao et al., 2013; Haley et al., 2016). Therefore, identifying distinct lineages within Salmonella serovars can aid traceback and outbreak investigations as some of these lineages may show different associations to geographic locations, commodities, and hosts. Although the phylogeny of the most clinically relevant serovars has been well studied (Yin et al., 2020), many other Salmonella serovars remain understudied. Hence, this study used the 100 most common Salmonella serovars in the NCBI PD database to assess their phylogeny in a standardized manner. With the WGS-based phylogenetic analysis of the 100 most common Salmonella serovars in the NCBI PD database, we showed that (i) different in-silico serotyping tools (i.e., SISTR, SeqSero2 k-mer-based, and SeqSero2 allele micro-assembly approaches) may produce different predictions for some isolates representing certain serovars, (ii) polyphyly is common among Salmonella serovars, and (iii) divergence of closely related Salmonella serovars is driven primarily by changes in the H1 flagellar antigens, and to a lesser extent by changes in the H2 flagellar and somatic antigens. While our results are consistent with previous studies that showed clustering of Salmonella genome sequences by serovar and STs (Cherchame et al., 2022), we showed that (i) a larger proportion of serovars than previously known are polyphyletic; (ii) serovars previously known to be polyphyletic contain more lineages than previously known; and (iii) many serovars include lineages that only have a few isolates with a given serovar. For example, we report for the first time that serovar Hadar, which is commonly found in turkey, is polyphyletic and we also report that serovar Oranienburg include at least 10 distinct lineages [a prior study (Cherchame et al., 2022) only reported 3 lineages for this serovar].
4.1 Strategies regarding sampling and WGS data-sharing might bias the geographical representation of Salmonella isolates in public databases
The NCBI PD database includes WGS data for more than 550,000 Salmonella genomes from across the world, with 57.9% of these genomes originating from the United States (US) (as of August 2023). In the US, regulatory agencies put considerable effort into environmental monitoring of certain food commodities (Crandall et al., 2024) and multiple US federal departments, including but not limited to the US Centers for Disease Control and Prevention (CDC), the US Department of Agriculture (USDA), the Food and Drug Administration (FDA) perform WGS on Salmonella isolates and contribute sequence data to NCBI PD. For example, the USDA Food Safety and Inspection Service samples poultry plants several times a year (USDA, 2021), contributing a substantial number of sequence data for US poultry isolates to NCBI PD. Similarly, all human clinical isolates of Salmonella obtained in the US are whole genome sequenced and have their WGS data added to NCBI PD. Also, Public Health England (PHE) deposits a substantial number of Salmonella genomes sequences in NCBI PD. However, NCBI PD does not reflect the number of reported human cases of Salmonella in other countries. For example, the rate of confirmed salmonellosis cases in the Czech Republic (71.9 cases per 100,000 population) and Slovakia (67.5) is high (ECDC, 2024), however, only 21 Salmonella isolates from these locations are represented in the NCBI PD database. Therefore, the classification of the 100 most common Salmonella serovars used in this study greatly reflects the prevalence of Salmonella serovars in the US (and hence may be biased towards serovars with high prevalence in the US), with a few exceptions; for example, S. Napoli is among the top 5 serovars causing infection in Italy (Graziani et al., 2015; Sabbatucci et al., 2018), but it is rare in the US. Initiatives, such as that of the Chinese Academy of Science (CAS), which sequenced almost 8,000 Salmonella genomes from 22 Chinese provinces, making them publicly available in the Chinese Local Salmonella Genome Database version 2 (CLSGDB v2) (Wang et al., 2023b), and also through NCBI, are important to increase the representation of non-US Salmonella isolates in NCBI PD.
4.2 Different in-silico serotyping tools produced different predictions for some isolates of certain serovars
Our findings here are consistent with previous studies that have shown that different in-silico serotyping approaches provide results that vary between in-silico methods and from those obtained by traditional serotyping (Uelze et al., 2020), as each of these approaches use different databases and algorithms to predict the serotype. It is well known that in-silico serotyping is inexpensive and time-efficient compared to traditional serotyping (Banerji et al., 2020). However, constant improvements are still required for robust serotyping. SISTR and SeqSero2 may give ambiguous predictions for some isolates of certain serovars, thus requiring biochemical tests for full characterization when (i) the antigenic formula cannot be fully identified (e.g., missing O antigen genes), and (ii) different serovars share the same antigenic formula in the WKL scheme (e.g., Salmonella Choleraesuis, Paratyphi C, and Typhisuis share the same antigenic formula: 6,7:c:1,5). For example, for several serovars, such as Salmonella Agona, Enteritidis, and Infantis, Uelze et al. (2020) reported that SeqSero2 was repeatedly unable to identify the O antigen of the analyzed genomes (Uelze et al., 2020). When serovars share the same antigenic formula, identification of phenotypic traits based on genetic markers, if available, could result in more accurate serovar prediction with higher discriminatory power (Yoshida et al., 2016). SeqSero2 already implemented such additional genetic markers (e.g., identifying SNPs in specific genes) for several serovars, like Salmonella Enteritidis and Paratyphi B pathotypes. However, implementation of genetic markers for other serovars that share the same antigenic formula (e.g., Salmonella Choleraesuis, Paratyphi C, and Typhisuis) would increase the robustness of serovar identification. Lastly, these tools need to be regularly updated with recently identified serovars [e.g., S. Lubbock emerged from S. Mbandaka by acquiring fliC operon of S. Montevideo; (Bugarel et al., 2019)] to provide up-to-date serovar identification across serotyping methods. The 1.52% inconsistency between the serotypes assigned by NCBI PD and our approach suggests that the NCBI PD assignment methodology could be improved and that it may be valuable to independently confirm serovar assignments. We observed several isolates from the same NCBI PD SNP cluster (therefore these isolates were all genetically closely related as assessed by SNP differences) that were assigned different serovars. For example, the largest S. Enteritidis SNP cluster on NCBI PD includes isolates classified as serovar Hillingdon, and other unnamed serovars (e.g., I 9,46:g,m:1,2) with an antigenic formula different from that of S. Enteritidis (i.e., I 1,9,12:g,m:-).
4.3 Polyphyly is common among Salmonella serovars
Our comprehensive phylogenetic analysis of the 100 most common Salmonella serovars, covering 94.55% of the total number of Salmonella genomes available in the NCBI PD database, revealed that 72% of the Salmonella serovars are polyphyletic. This finding is noteworthy as it contrasts with previous studies that reported different frequencies of polyphyly among Salmonella serovars. For instance, Worley et al. (2018) found approximately 10% of Salmonella serovars to be polyphyletic, among 445 isolates that represented 260 serovars analyzed in their study (Worley et al., 2018). These authors assessed the phylogeny of Salmonella serovars using WGS data of a limited number of isolates from each serovar. Another study used pangenome WGS analysis of 219 genomes and observed polyphyly in 43.1% of the 58 serovars frequently isolated from both the US and Europe (Cherchame et al., 2022). Furthermore, analysis of the 20 most frequently reported serovars associated with human clinical cases in the US suggested that 35% (7/20) of the serovars were polyphyletic based on a 7-gene MLST analysis (Yin et al., 2020). The discrepancy in these findings and the findings reported in the current study highlights the influence of methodological differences in phylogeny inference. The present study represents a robust analysis of the phylogeny of the 100 most common Salmonella serovars since we identified genomes to represent each SNP cluster and every singleton within each specific serovar, which resulted in the representation of 63,151 genomes. While previous studies (Yin et al., 2020; Cherchame et al., 2022) showed clustering of Salmonella genome sequences by serovar and STs, our analyses included 514 reference genomes representing 301, 49, 127, and 37 serovars of the subspecies enterica, arizonae, diarizonae, and houtenae, respectively, in order to identify additional polyphyletic linages; this analysis across a wide range of serovars was needed in order to reliably identify different (polyphyletic) lineages and to identify the closest neighbors of these lineages.
Several factors could lead to differences in the phylogenetic grouping of Salmonella serovars. Using an appropriate set of reference serovars to construct phylogenetic trees and including all available isolates of interest during phylogeny construction is crucial. For example, previous studies suggested that Salmonella Typhimurium, Enteritidis, Havana, and Montevideo, appeared to be monophyletic (Sangal et al., 2010; Cherchame et al., 2022). However, our results and other studies (D'Alessandro et al., 2018; Zhang et al., 2019b; Yin et al., 2020; Chen et al., 2024) found that these serovars are polyphyletic. With the exception of S. Havana, we observed that these serovars have one lineage that includes > 99% of the isolates. This could explain the discrepancy between our results and those from previous studies. Results from previous studies may have reported different findings regarding the phylogeny of these serovars because (i) they selected a limited number of isolates from the target serovar, and (ii) they did not include a large enough number of reference serovars representing subspecies enterica to capture the true nature of their phylogeny. This also affects the number of lineages identified in polyphyletic serovars. For example, our analysis of S. Oranienburg (using 2,427 Oranienburg genomes, that represent both SNP clusters and singletons) resulted in 10 lineages. However, studies conducted by Cherchame et al. (2022) and Zhang et al. (2019b) using a limited number of representative S. Oranienburg isolates only identified three and four lineages, based on 24 and 29 isolates, respectively (Zhang et al., 2019b; Cherchame et al., 2022).
4.4 Associations of lineages or clades within Salmonella serovars with specific geolocations, commodities, and hosts can aid traceback and outbreak investigations
Our findings revealed that Salmonella Typhi is among the most common monophyletic serovars. S. Typhi, which causes typhoid fever in humans (Yap and Thong, 2017), shows minimal genetic variation and strong geographical clustering, which can help with epidemiological tracking (Wong et al., 2015; Yap and Thong, 2017). Similarly, S. Dublin, another monophyletic serovar primarily associated with cattle, also shows a strong phylogeographical association (Fenske et al., 2019; Kudirkiene et al., 2020).
Several polyphyletic serovars also show association with specific regions at the lineage level. For example, S. Newport (Sangal et al., 2010), and S. Mississippi (Cheng et al., 2021) have geographically distinct lineages. Specifically, our phylogenetic analysis of S. Kentucky revealed two main lineages, and two stand-alone singletons. This is consistent with previous findings that have shown that most S. Kentucky isolates fall into two main evolutionary lineages: (i) Kentucky-I (ST152), mostly associated with poultry in the US, and (ii) Kentucky-II (ST198), frequently linked to cattle and human cases in Europe (Tate et al., 2022; Richards et al., 2023). In contrast, S. Newport has been linked to many human gastroenteritis cases in the US and Europe (Sangal et al., 2010). According to our analysis, S. Newport has four distinct lineages, with two main lineages covering nearly 60 and 37% of the S. Newport isolates. Similar to these findings, previous studies suggested that S. Newport isolates fall into three distinct lineages, which show geographic patterns; Lineage I (Newport C in this study) was mostly comprised of European isolates, while Lineages II (Newport A in this study) and III (Newport B in this study) were mostly linked to North America (Sangal et al., 2010). S. Derby, which our analysis revealed to have seven lineages, was previously reported to show lineage associated host specificities, with isolates in distinct lineages being associated with poultry or swine (Sevellec et al., 2018). In addition, previous studies have also identified STs that are associated with specific reservoirs and human hosts. For example, a Chinese study reported that S. Typhimurium ST34 isolates, which are associated with swine, and S. Typhimurium ST19 isolates, which are associated with chickens, were found to mainly cause gastro-infection in children and adults, respectively (Wang et al., 2023a). These findings further support that lineage specific characteristics may be valuable in outbreak and traceback investigations.
4.5 Divergence of closely related Salmonella serovars involves variations in somatic and flagellar antigens, but primarily through changes in the H1 antigen
Our results showed that antigen diversification of the five most common Salmonella serovars was primarily associated with differences in the H1 and H2 antigens, compared to differences in the O antigens. This might be related to the complexity of the genetic material underlying the expression of different antigens. H1 and H2 antigens are encoded by single genes, while O antigens are encoded by the rfb gene cluster (Silverman and Simon, 1980; Liu et al., 2014; Horvath et al., 2019). Differences in the H1 antigen are likely to be associated with fewer evolutionary changes, ultimately following the principal of parsimony (Kannan and Wheeler, 2012). The O and H antigens are both involved in pathogenesis and are immunogenic, leading to the production of antibodies by the host (Reeves, 1993). The O antigen plays a role in allowing the pathogen to evade phagocytosis by immune cells (Farhana and Khan, 2024). More specifically, variations in the length of the O-antigen chain can obstruct complement-mediated killing and bacterial defenses (Thiriard et al., 2018). It has been suggested that long O-antigen chains are associated with enhanced fitness during Salmonella-induced colitis via increased bile resistance (Crawford et al., 2012). On the other hand, Hayashi et al. (2001) concluded that the FliC protein (H1 antigen) will bind to IpaF intracellular receptors and cell surface TLR-5 receptors, which triggers a pro-inflammatory signaling pathway activating the host immune system to remove bacterial infections (Hayashi et al., 2001). Therefore, it is possible that Salmonella shows antigen divergence to maintain its ability to colonize hosts, adapt to different environments, or to evade host’s immune response (Liu et al., 2014).
Genomic rearrangement, gene deletion, gene disruption, and horizontal gene transfer followed by homologous recombination are possible mechanisms resulting in emergence of multiple antigenic phenotypes, ultimately leading to divergence of serovars (Vink et al., 2012; Burki et al., 2015; Tanner and Kingsley, 2018; Yin et al., 2020). Previous findings suggest that S. I 4,[5],12:i:- emerged multiple times from S. Typhimurium (Switt et al., 2009; Arrieta-Gisasola et al., 2020; Wang et al., 2023c). This divergence has been suggested to have occurred primarily through interruptions of the flagellar coding region (Ingle et al., 2021). This genetic alteration typically involves the integration of an insertion sequence into fljB, which disrupts the expression of the H2 antigen in S. I 4,[5],12:i:- (Arrieta-Gisasola et al., 2020). As a result, S. I 4,[5],12:i:- emerged expressing only the H1 antigen (encoded by fliC), lacking the ability to switch between two flagellar phases. S. I 4,[5],12:i:- is characterized by high multidrug resistance, especially in mcr-carrying S. I 4,[5],12:i:- ST34, which has been implicated in several outbreaks worldwide (Biswas et al., 2019; Laisnez et al., 2025; Wu et al., 2025). Additionally, the emergence of S. I 4,[5],12:i:- ST34 has been associated with increased phage resistance through lysogenic conversion, which may have allowed S. I 4,[5],12:i:- ST34 to spread rapidly across multiple host species and become a major concern to public health (Charity et al., 2022). Although it has been clear that S. I 4,[5],12:i:- have emerged multiple times, it remained unclear whether all S. I 4,[5],12:i:- isolates emerged from S. Typhimurium. Our results further increase the understanding of S. I 4,[5],12:i:- and S. Typhimurium by inferring the overall phylogeny of these serovars and their respective lineages. Noticeably, we observed that only one S. I 4,[5],12:i:- lineage shares an MRCA with a S. Typhimurium lineage, while the other three S. I 4,[5],12:i:- lineages do not, thus suggesting that these three S. I 4,[5],12:i:- lineages have emerged independently from S. Typhimurium.
Our findings emphasize the importance of using WGS for accurate phylogenetic analysis and lineage identification. While traditional serotype designations are still valuable to maintain a link with historical epidemiological data and to resolve ambiguous in-silico results, classification systems (such as serotyping) that heavily relies on phenotypic traits are unlikely to fully capture the genetic diversity and evolutionary relationships within Salmonella serovars (Banerji et al., 2020). Serotype-based nomenclature can be useful for monophyletic and paraphyletic serovars, however, for polyphyletic serovars, serotyping does not provide enough discriminatory power to identify different lineages within the serovar. The prevalence of polyphyly observed in our study highlights the limitations of serotyping and the need for genomic approaches to discern lineages of a given serotype. WGS-based phylogenetic analysis provides a more comprehensive understanding of the phylogenetic history of Salmonella serovars, which could enable more effective root-cause analysis, as well as traceback and outbreak investigations. Hence, a nomenclature that incorporates both classical serotype information and the phylogenetic lineages (a “hybrid nomenclature”) (e.g., Salmonella Kentucky A, Salmonella Kentucky B) would be beneficial for researchers, industry, regulators and public health professionals, as it would provide for improved isolate characterization and classification, which can be linked to virulence potential as well as likely sources and geographic origins. While Salmonella strain designations based on either solely MLST-derived STs (Achtman et al., 2012) or based on ST and serovar (Chattaway et al., 2021) have previously been proposed, these approaches would not allow stakeholders to easily identify distinct polyphyletic lineages [e.g., as a given phylogenetic lineage may include several STs (Chen et al., 2024)]. Integration of ST into proposed “hybrid nomenclature” would however be easy (e.g., Salmonella Kentucky A ST152). Importantly, phylogenetic-based “hybrid nomenclature” is already currently used by the Genome Taxonomy Database (GTDB) project (Parks et al., 2018), in which polyphyletic genera (e.g., Bacillus) are named with alphabetical suffixes added to the genus name (e.g., Bacillus_A, Bacillus_B) to differentiate each polyphyletic lineage, supporting the feasibility of this approach.
Data availability statement
The metadata, reference genomes, codes, and supplementary figures presented in this study can be found in online repositories. Metadata: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Metadata; Reference genomes: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Reference_Genomes; Codes: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Codes; and Supplementary figures: https://github.com/FSL-MQIP/USDA_Salmonella_Phylogeny_Project/tree/main/Supplementary_Figures.
Author contributions
LY: Data curation, Formal analysis, Investigation, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing, Methodology, Resources. HS: Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. LK: Data curation, Formal analysis, Methodology, Writing – review & editing. RO: Data curation, Methodology, Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft. MW: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. RC: Visualization, Writing – review & editing, Data curation, Methodology. CR-M: Data curation, Formal analysis, Investigation, Project administration, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Food Safety and Defense (A1332) grant number 2021-67017 33830/project accession no. 1024163 from the USDA National Institute of Food and Agriculture.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1547190/full#supplementary-material
Footnotes
References
Achtman, M., Wain, J., Weill, F. X., Nair, S., Zhou, Z., Sangal, V., et al. (2012). Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 8:e1002776. doi: 10.1371/journal.ppat.1002776
Arrieta-Gisasola, A., Atxaerandio-Landa, A., Garrido, V., Grillo, M. J., Martinez-Ballesteros, I., Laorden, L., et al. (2020). Genotyping study of Salmonella 4,[5],12:i:- monophasic variant of serovar Typhimurium and characterization of the second-phase flagellar deletion by whole genome sequencing. Microorganisms 8:2049. doi: 10.3390/microorganisms8122049
Banerji, S., Simon, S., Tille, A., Fruth, A., and Flieger, A. (2020). Genome-based Salmonella serotyping as the new gold standard. Sci. Rep. 10:10776. doi: 10.1038/s41598-020-67917-3
Biswas, S., Li, Y., Elbediwi, M., and Yue, M. (2019). Emergence and dissemination of mcr-carrying clinically relevant Salmonella Typhimurium monophasic clone ST34. Microorganisms 7:298. doi: 10.3390/microorganisms7090298
Bugarel, M., Cook, P. W., den Bakker, H. C., Harhay, D., Nightingale, K. K., and Loneragan, G. H. (2019). Complete genome sequences of four Salmonella enterica strains (Including those of serotypes Montevideo, Mbandaka, and Lubbock) isolated from peripheral lymph nodes of healthy cattle. Microbiol. Resour. Announc. 8:e01450-18. doi: 10.1128/MRA.01450-18
Burki, S., Frey, J., and Pilo, P. (2015). Virulence, persistence and dissemination of Mycoplasma bovis. Vet. Microbiol. 179, 15–22. doi: 10.1016/j.vetmic.2015.02.024
Cao, G., Meng, J., Strain, E., Stones, R., Pettengill, J., Zhao, S., et al. (2013). Phylogenetics and differentiation of Salmonella Newport lineages by whole genome sequencing. PLoS One 8:e55687. doi: 10.1371/journal.pone.0055687
Charity, O. J., Acton, L., Bawn, M., Tassinari, E., Thilliez, G., Chattaway, M. A., et al. (2022). Increased phage resistance through lysogenic conversion accompanying emergence of monophasic Salmonella Typhimurium ST34 pandemic strain. Microb. Genom. 8:mgen000897. doi: 10.1099/mgen.0.000897
Chattaway, M. A., Langridge, G. C., and Wain, J. (2021). Salmonella nomenclature in the genomic era: A time for change. Sci. Rep. 11:7494. doi: 10.1038/s41598-021-86243-w
Chen, R., Cheng, R. A., Wiedmann, M., and Orsi, R. H. (2022). Development of a genomics-based approach to identify putative hypervirulent nontyphoidal Salmonella isolates: Salmonella enterica Serovar Saintpaul as a model. mSphere 7, e00730–e00721. doi: 10.1128/msphere.00730-21
Chen, R., Yang, L., Pajor, M. S., Wiedmann, M., and Orsi, R. H. (2024). Salmonella associated with agricultural animals exhibit diverse evolutionary rates and show evidence of recent clonal expansion. MBio 15:e0191324. doi: 10.1128/mbio.01913-24
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cheng, R. A., Eade, C. R., and Wiedmann, M. (2019). Embracing diversity: differences in virulence mechanisms, disease severity, and host adaptations contribute to the success of nontyphoidal Salmonella as a foodborne pathogen. Front. Microbiol. 10:1368. doi: 10.3389/fmicb.2019.01368
Cheng, R. A., Orsi, R. H., and Wiedmann, M. (2021). Phylogeographic clustering suggests that distinct clades of Salmonella enterica serovar Mississippi are endemic in Australia, the United Kingdom, and the United States. mSphere 6:e0048521. doi: 10.1128/mSphere.00485-21
Cherchame, E., Ilango, G., Noel, V., and Cadel-Six, S. (2022). Polyphyly in widespread Salmonella enterica serovars and using genomic proximity to choose the best reference genome for bioinformatics analyses. Front. Public Health 10:963188. doi: 10.3389/fpubh.2022.963188
Coburn, B., Grassl, G. A., and Finlay, B. (2007). Salmonella, the host and disease: a brief review. Immunol. Cell Biol. 85, 112–118. doi: 10.1038/sj.icb.7100007
Crandall, P. G., O’Bryan, C. A., Wang, D., Gibson, K. E., and Obe, T. (2024). Environmental monitoring in food manufacturing: Current perspectives and emerging frontiers. Food Control 159:110269. doi: 10.1016/j.foodcont.2023.110269
Crawford, R. W., Keestra, A. M., Winter, S. E., Xavier, M. N., Tsolis, R. M., Tolstikov, V., et al. (2012). Very long O-antigen chains enhance fitness during Salmonella-induced colitis by increasing bile resistance. PLoS Pathog. 8:e1002918. doi: 10.1371/journal.ppat.1002918
D'Alessandro, B., Perez Escanda, V., Balestrazzi, L., Iriarte, A., Pickard, D., Yim, L., et al. (2018). A novel prophage identified in strains from Salmonella enterica serovar Enteritidis is a phylogenetic signature of the lineage ST-1974. Microb. Genom. 4:e000161. doi: 10.1099/mgen.0.000161
ECDC (2024). Salmonellosis - Annual epidemiological report for 2022. Stockholm: European Centre for Disease Prevention and Control.
Eng, S.-K., Pusparajah, P., Ab Mutalib, N.-S., Ser, H.-L., Chan, K.-G., and Lee, L.-H. (2015). Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front. Life Sci. 8, 284–293. doi: 10.1080/21553769.2015.1051243
Farhana, A., and Khan, Y. S. (2024). Biochemistry, lipopolysaccharide. Treasure Island (FL): StatPearls.
Feasey, N. A., Hadfield, J., Keddy, K. H., Dallman, T. J., Jacobs, J., Deng, X., et al. (2016). Distinct Salmonella Enteritidis lineages associated with enterocolitis in high-income settings and invasive disease in low-income settings. Nat. Genet. 48, 1211–1217. doi: 10.1038/ng.3644
Fenske, G. J., Thachil, A., McDonough, P. L., Glaser, A., and Scaria, J. (2019). Geography shapes the population genomics of Salmonella enterica Dublin. Genome Biol. Evol. 11, 2220–2231. doi: 10.1093/gbe/evz158
Graziani, C., Luzzi, I., Owczarek, S., Dionisi, A. M., and Busani, L. (2015). Salmonella enterica serovar Napoli infection in Italy from 2000 to 2013: Spatial and spatio-temporal analysis of cases distribution and the effect of human and animal density on the risk of infection. PLoS One 10:e0142419. doi: 10.1371/journal.pone.0142419
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haley, B. J., Kim, S. W., Pettengill, J., Luo, Y., Karns, J. S., and Van Kessel, J. A. (2016). Genomic and evolutionary analysis of two Salmonella enterica serovar Kentucky sequence types isolated from bovine and poultry sources in North America. PLoS One 11:e0161225. doi: 10.1371/journal.pone.0161225
Hall, B. G., and Nisbet, J. (2023). Building phylogenetic trees from genome sequences with kSNP4. Mol. Biol. Evol. 40:msad235. doi: 10.1093/molbev/msad235
Hayashi, F., Smith, K. D., Ozinsky, A., Hawn, T. R., Yi, E. C., Goodlett, D. R., et al. (2001). The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103. doi: 10.1038/35074106
Horvath, P., Kato, T., Miyata, T., and Namba, K. (2019). Structure of Salmonella flagellar hook reveals intermolecular domain interactions for the universal joint function. Biomol. Ther. 9:462. doi: 10.3390/biom9090462
Hughes, K. T., Youderian, P., and Simon, M. I. (1988). Phase variation in Salmonella: analysis of Hin recombinase and hix recombination site interaction in vivo. Genes Dev. 2, 937–948. doi: 10.1101/gad.2.8.937
Ibrahim, G. M., and Morin, P. M. (2018). Salmonella serotyping using whole genome sequencing. Front. Microbiol. 9:2993. doi: 10.3389/fmicb.2018.02993
Ingle, D. J., Ambrose, R. L., Baines, S. L., Duchene, S., Goncalves da Silva, A., Lee, D. Y. J., et al. (2021). Evolutionary dynamics of multidrug resistant Salmonella enterica serovar 4,[5],12:i:- in Australia. Nat. Commun. 12:4786. doi: 10.1038/s41467-021-25073-w
Issenhuth-Jeanjean, S., Roggentin, P., Mikoleit, M., Guibourdenche, M., de Pinna, E., Nair, S., et al. (2014). Supplement 2008-2010 (no. 48) to the White-Kauffmann-Le Minor scheme. Res. Microbiol. 165, 526–530. doi: 10.1016/j.resmic.2014.07.004
Kannan, L., and Wheeler, W. C. (2012). Maximum parsimony on phylogenetic networks. Algorithms Mol. Biol. 7:9. doi: 10.1186/1748-7188-7-9
Kirk, M. D., Pires, S. M., Black, R. E., Caipo, M., Crump, J. A., Devleesschauwer, B., et al. (2015). World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med. 12:e1001921. doi: 10.1371/journal.pmed.1001921
Kudirkiene, E., Sorensen, G., Torpdahl, M., de Knegt, L. V., Nielsen, L. R., Rattenborg, E., et al. (2020). Epidemiology of Salmonella enterica serovar Dublin in cattle and humans in Denmark, 1996 to 2016: a retrospective whole-genome-based study. Appl. Environ. Microbiol. 86:e01894-19. doi: 10.1128/AEM.01894-19
Laisnez, V., Vusirikala, A., Nielsen, C. S., Cantaert, V., Delbrassinne, L., Mattheus, W., et al. (2025). Key role of whole genome sequencing in resolving an international outbreak of monophasic Salmonella Typhimurium linked to chocolate products. BMC Infect. Dis. 25:242. doi: 10.1186/s12879-025-10629-8
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, C. C., and Hsiao, W. W. L. (2022). Large-scale comparative genomics to refine the organization of the global Salmonella enterica population structure. Microb. Genom. 8:mgen000906. doi: 10.1099/mgen.0.000906
Liu, B., Knirel, Y. A., Feng, L., Perepelov, A. V., Senchenkova, S. N., Reeves, P. R., et al. (2014). Structural diversity in Salmonella O antigens and its genetic basis. FEMS Microbiol. Rev. 38, 56–89. doi: 10.1111/1574-6976.12034
McQuiston, J. R., Parrenas, R., Ortiz-Rivera, M., Gheesling, L., Brenner, F., and Fields, P. I. (2004). Sequencing and comparative analysis of flagellin genes fliC, fljB, and flpA from Salmonella. J. Clin. Microbiol. 42, 1923–1932. doi: 10.1128/JCM.42.5.1923-1932.2004
Minamino, T., Morimoto, Y. V., Kawamoto, A., Terashima, H., and Imada, K. (2018). “"Salmonella" flagellum” in Salmonella - A Re-emerging Pathogen. ed. M. T. Mascellino (Sine Loco: IntechOpen).
Murray, C. J., Vos, T., Lozano, R., Naghavi, M., Flaxman, A. D., Michaud, C., et al. (2012). Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2197–2223. doi: 10.1016/S0140-6736(12)61689-4
Oosterbroek, P. (1987). More appropriate definitions of paraphyly and polyphyly, with a comment on the Farris 1974 model. Syst. Zool. 36, 103–108. doi: 10.2307/2413263
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Pearce, M. E., Langridge, G. C., Lauer, A. C., Grant, K., Maiden, M. C. J., and Chattaway, M. A. (2021). An evaluation of the species and subspecies of the genus Salmonella with whole genome sequence data: Proposal of type strains and epithets for novel S. enterica subspecies VII, VIII, IX, X and XI. Genomics 113, 3152–3162. doi: 10.1016/j.ygeno.2021.07.003
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Reeves, P. (1993). Evolution of Salmonella O antigen variation by interspecific gene transfer on a large scale. Trends Genet. 9, 17–22. doi: 10.1016/0168-9525(93)90067-R
Richards, A. K., Kue, S., Norris, C. G., and Shariat, N. W. (2023). Genomic and phenotypic characterization of Salmonella enterica serovar Kentucky. Microb. Genom. 9:001089. doi: 10.1099/mgen.0.001089
Sabbatucci, M., Dionisi, A. M., Pezzotti, P., Lucarelli, C., Barco, L., Mancin, M., et al. (2018). Molecular and epidemiologic analysis of reemergent Salmonella enterica Serovar Napoli, Italy, 2011-2015. Emerg. Infect. Dis. 24, 562–565. doi: 10.3201/eid2403.171178
Sangal, V., Harbottle, H., Mazzoni, C. J., Helmuth, R., Guerra, B., Didelot, X., et al. (2010). Evolution and population structure of Salmonella enterica serovar Newport. J. Bacteriol. 192, 6465–6476. doi: 10.1128/JB.00969-10
Seif, Y., Monk, J. M., Machado, H., Kavvas, E., and Palsson, B. O. (2019). Systems biology and pangenome of Salmonella O-Antigens. MBio 10:e01247-19. doi: 10.1128/mBio.01247-19
Sevellec, Y., Vignaud, M. L., Granier, S. A., Lailler, R., Feurer, C., Le Hello, S., et al. (2018). Polyphyletic nature of Salmonella enterica serotype Derby and lineage-specific host-association revealed by genome-wide analysis. Front. Microbiol. 9:891. doi: 10.3389/fmicb.2018.00891
Shariat, N. W., Timme, R. E., and Walters, A. T. (2021). Phylogeny of Salmonella enterica subspecies arizonae by whole-genome sequencing reveals high incidence of polyphyly and low phase 1 H antigen variability. Microb. Genom. 7:000522. doi: 10.1099/mgen.0.000522
Silverman, M., and Simon, M. (1980). Phase variation: genetic analysis of switching mutants. Cell 19, 845–854. doi: 10.1016/0092-8674(80)90075-6
Silverman, M., Zieg, J., Hilmen, M., and Simon, M. (1979). Phase variation in Salmonella: genetic analysis of a recombinational switch. Proc. Natl. Acad. Sci. USA 76, 391–395. doi: 10.1073/pnas.76.1.391
Souvorov, A., Agarwala, R., and Lipman, D. J. (2018). SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 19:153. doi: 10.1186/s13059-018-1540-z
Switt, A. I., Soyer, Y., Warnick, L. D., and Wiedmann, M. (2009). Emergence, distribution, and molecular and phenotypic characteristics of Salmonella enterica serotype 4,5,12:i. Foodborne Pathog. Dis. 6, 407–415. doi: 10.1089/fpd.2008.0213
Tanner, J. R., and Kingsley, R. A. (2018). Evolution of Salmonella within hosts. Trends Microbiol. 26, 986–998. doi: 10.1016/j.tim.2018.06.001
Tate, H., Hsu, C. H., Chen, J. C., Han, J., Foley, S. L., Folster, J. P., et al. (2022). Genomic diversity, antimicrobial resistance, and virulence gene profiles of Salmonella serovar Kentucky isolated from humans, food, and animal ceca content sources in the United States. Foodborne Pathog. Dis. 19, 509–521. doi: 10.1089/fpd.2022.0005
Thiriard, A., Raze, D., and Locht, C. (2018). Diversion of complement-mediated killing by Bordetella. Microbes Infect. 20, 512–520. doi: 10.1016/j.micinf.2018.02.002
Uelze, L., Borowiak, M., Deneke, C., Szabo, I., Fischer, J., Tausch, S. H., et al. (2020). Performance and accuracy of four open-source tools for in silico serotyping of Salmonella spp. based on whole-genome short-read sequencing data. Appl. Environ. Microbiol. 86:e02265-19. doi: 10.1128/AEM.02265-19
USDA (2021). Food Safety and Inspection Service Annual Sampling Summary Report. Washington, DC: United States Department of Agriculture.
Vink, C., Rudenko, G., and Seifert, H. S. (2012). Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol. Rev. 36, 917–948. doi: 10.1111/j.1574-6976.2011.00321.x
Wang, Z., Gu, D., Hong, Y., Hu, Y., Gu, J., Tang, Y., et al. (2023c). Microevolution of Salmonella 4,[5],12:i:- derived from Salmonella enterica serovar Typhimurium through complicated transpositions. Cell Rep. 42:113227. doi: 10.1016/j.celrep.2023.113227
Wang, Y., Liu, Y., Lyu, N., Li, Z., Ma, S., Cao, D., et al. (2023a). The temporal dynamics of antimicrobial-resistant Salmonella enterica and predominant serovars in China. Natl. Sci. Rev. 10:nwac269. doi: 10.1093/nsr/nwac269
Wang, Y., Xu, X., Zhu, B., Lyu, N., Liu, Y., Ma, S., et al. (2023b). Genomic analysis of almost 8,000 Salmonella genomes reveals drivers and landscape of antimicrobial resistance in China. Microbiol. Spectr. 11:e0208023. doi: 10.1128/spectrum.02080-23
Wattiau, P., Boland, C., and Bertrand, S. (2011). Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl. Environ. Microbiol. 77, 7877–7885. doi: 10.1128/AEM.05527-11
Wong, V. K., Baker, S., Pickard, D. J., Parkhill, J., Page, A. J., Feasey, N. A., et al. (2015). Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet. 47, 632–639. doi: 10.1038/ng.3281
Worley, J., Meng, J., Allard, M. W., Brown, E. W., and Timme, R. E. (2018). Salmonella enterica phylogeny based on whole-genome sequencing reveals two new clades and novel patterns of horizontally acquired genetic elements. MBio 9:e02303-18. doi: 10.1128/mBio.02303-18
Wu, Y., Du, J., Zhou, X., Peng, X., Jia, C., Wang, B., et al. (2025). Genomic epidemiology and public health implications of zoonotic monophasic Salmonella Typhimurium ST34. Front. Cell. Infect. Microbiol. 15:1490183. doi: 10.3389/fcimb.2025.1490183
Yap, K.-P., and Thong, K. L. (2017). Salmonella Typhi genomics: envisaging the future of typhoid eradication. Trop. Med. Int. Health 22, 918–925. doi: 10.1111/tmi.12899
Yin, Z., Liu, J., Du, B., Ruan, H. H., Huo, Y. X., Du, Y., et al. (2020). Whole-genome-based survey for polyphyletic serovars of Salmonella enterica subsp. enterica provides new insights into public health surveillance. Int. J. Mol. Sci. 21:5226. doi: 10.3390/ijms21155226
Yoshida, C. E., Kruczkiewicz, P., Laing, C. R., Lingohr, E. J., Gannon, V. P., Nash, J. H., et al. (2016). The Salmonella in silico typing resource (SISTR): An open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One 11:e0147101. doi: 10.1371/journal.pone.0147101
Zhang, S., den Bakker, H. C., Li, S., Chen, J., Dinsmore, B. A., Lane, C., et al. (2019a). SeqSero2: Rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl. Environ. Microbiol. 85:e01746-19. doi: 10.1128/AEM.01746-19
Keywords: Salmonella enterica, serovar, phylogeny, evolution, divergence, whole-genome sequence, antigen, polyphyletic
Citation: Yang L, Samut H, Kemmerling L, Orsi RH, Wiedmann M, Chen R and Resendiz-Moctezuma C (2025) Phylogeny and divergence of the 100 most common Salmonella serovars available in the NCBI Pathogen Detection database. Front. Microbiol. 16:1547190. doi: 10.3389/fmicb.2025.1547190
Edited by:
Malgorzata Ziarno, Warsaw University of Life Sciences, PolandReviewed by:
Min Yue, University of Chinese Academy of Science, ChinaYanan Wang, Henan Agricultural University, China
Jingqiu Liao, Virginia Tech, United States
Luit Barkalita, Assam Agricultural University, India
Copyright © 2025 Yang, Samut, Kemmerling, Orsi, Wiedmann, Chen and Resendiz-Moctezuma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Resendiz-Moctezuma, Y3I2MzhAY29ybmVsbC5lZHU=
†These authors have contributed equally to this work and share first authorship