 Khansa Ejaz1*
Khansa Ejaz1* Muhammad Zakria2
Muhammad Zakria2 Peiqi Zhang3
Peiqi Zhang3 Jose Huguet Tapia3
Jose Huguet Tapia3 Muhammad Arif4
Muhammad Arif4 Frank White3
Frank White3 Sumera Yasmin1*
Sumera Yasmin1*- 1Soil and Environmental Biotechnology Division, National Institute for Biotechnology and Genetic Engineering College, Pakistan Institute for Engineering and Applied Sciences (NIBGE-C, PIEAS), Faisalabad, Punjab, Pakistan
- 2National Agricultural Research Centre, Crop Diseases Research Institute, Islamabad, Pakistan
- 3Department of Plant Pathology, University of Florida, Gainesville, FL, United States
- 4Agricultural Biotechnology Division, National Institute for Biotechnology and Genetic Engineering College, Pakistan Institute for Engineering and Applied Sciences (NIBGE-C, PIEAS), Faisalabad, Punjab, Pakistan
The emergence of hostile and novel plant pathogenic strains poses a serious threat to global food security, which renders the strategies for disease management in modern agriculture ineffective. Preventing the consequences of these emerging phytopathogens requires accurate genetic information about the pathogen population to formulate effective management strategies. Bacterial leaf blight (BLB), caused by Xanthomonas oryzae pv. oryzae (Xoo), is the foremost reason for substantial yield losses in rice crops worldwide, especially in Asia. The genetic information regarding the Pakistani Xoo population is still unexplored. To bridge this gap, two representative Pakistani Xoo isolates, namely PkXoo1 and PkXoo2, were sequenced using long-read Oxford Nanopore Technology (ONT). Both isolates were obtained from the Basmati rice-growing region of Pakistan, with substantially high virulence on certain susceptible rice varieties. The final assembly of PkXoo1 and PkXoo2 yielded a circular chromosome of approximately 4.9 MB with a G + C content of 64%. Genome annotation of both strains revealed the presence of key genes associated with hypersensitivity and virulence in Xoo. The AnnoTALE analysis showed that both strains contained 18 transcription activator-like (TAL) effectors, three of which were predicted to be pseudoTALes. A phylogenomic analysis grouped PkXoo1 and PkXoo2 with strains belonging to India and Thailand, placing them far apart from other major Asian Xoo strains. The present study revealed significant findings about the conservation of repeat variable di-residues (RVDs) in major TAL effectors and the utility of high-throughput sequencing technologies for TAL effector analysis and pathogen tracking. The complete genome sequence of Xoo isolates from Pakistan will enhance sequence resources for the global comparison of Xoo diversity across the region. This information is also of great significance for launching effective and durable breeding programs.
1 Introduction
Outbreaks of diseases caused by Xanthomonas have been reported worldwide across multiple hosts (Quezado Duval et al., 2004). The most studied and widely prevalent xanthomonads are pathovars of Xanthomonas oryzae, which cause bacterial leaf blight (BLB) and bacterial leaf streak (BLS) in rice (Timilsina et al., 2020). Bacterial Leaf Blight (BLB) is a destructive rice disease caused by Xanthomonas oryzae pv. oryzae (Xoo). It affects almost all rice-growing regions worldwide and is considered a serious yield-limiting factor in rice production. Xoo is a vascular pathogen that penetrates the host plant via hydathodes and wounds (Niño-Liu et al., 2006).
Key aspects of the mechanism underlying BLB disease have been elucidated, providing an efficient roadmap for breeding resistance. To successfully colonize the host and induce disease symptoms, Xoo secretes a series of transcription activator-like (TAL) effectors into the host through the type III secretion system (T3SS) (Boch and Bonas, 2010; Jacques et al., 2016). TAL effectors translocate to the host nucleus, where they bind to effector-specific DNA bases in the promoter region of specific genes and trigger the transcription of either susceptibility (S) or executor (E) host genes (Bogdanove et al., 2010). They bind to their target genes in a continuous, code-like manner, which is specified by the central domain or central repeat region (CRR) of an individual TAL effector. The CRR consists of nearly identical tandem repeats (typically ranging from 33 to 35 amino acid residues), which form a super-helical structure by aligning around the target DNA. However, the residues at the 12th and 13th positions of all repeats are hypervariable, commonly acknowledged as repeat variable di-residues (RVDs). Each RVD determines an exclusive interaction with a specific, single-base pair of the target DNA sequence. The succeeding RVDs in a TAL effector protein are responsible for binding to a specific DNA sequence located at the promoter region of the targeted gene, called the effector binding elements (EBEs) (Boch et al., 2009; Moscou and Bogdanove, 2009). This code-like mechanism is the key principle of TAL effector-DNA specificity, enabling the determination of TAL effector binding sites in the genome of host plants (Noël et al., 2013). Understanding this mechanism offers exciting novel opportunities for breeders and researchers to develop resistant rice varieties (Hutin et al., 2015).
Generally, activated genes are susceptibility (S) genes that help pathogens colonize the host plant. The Sugars Will Eventually be Exported Transporter (SWEET) family of sugar transporters is considered a well-characterized group of S genes, commonly known as SWEET genes (Streubel et al., 2013). SWEET genes export sucrose into the xylem vessels, which are eventually used by the bacteria for disease progression and symptom development (Carpenter et al., 2020). There are more than 20 SWEET genes present in rice, but only 3 of them, OsSWEET11a (formerly SWEET11) (Wu et al., 2022), OsSWEET13, and OsSWEET14, are stated to be upregulated by diverse TAL effectors from different Xoo strains (Oliva et al., 2019). These are all members of the Clade III of the SWEET gene family. On the other hand, TAL effectors may also activate resistance (R) genes that trigger localized host cell death to restrict host infection, ultimately leading to successful host defense (Bogdanove et al., 2010; Boch et al., 2014).
TAL effector coding genes are assumed to be highly varying and dynamic. Previous population genomic studies have also reported high-degree variations in the genomes of Xoo strains. It is crucial to understand the evolution and diversity of TAL effectors, as variations in TAL effector repeats have played a significant role in the production of novel toxins and in breaking plant resistance against pathogens (Erkes et al., 2017; Kaur et al., 2020). Due to the highly repetitive structure and sequence identity among different tal genes, assembling these genes within the genome is difficult using conventional sequencing methods, such as first- and second-generation sequencing approaches. Nowadays, the employment of high-throughput, long-read sequencing technologies, such as single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technology (ONT), is proving effective in resolving complex genomic structures, such as repetitive elements. These technologies also enable the identification of the position and organization of bacterial pathogenicity islands encoding virulence effectors, particularly the TAL effector region (Ciuffreda et al., 2021). The comprehensive knowledge gained about the evolution and diversity of the TALome will help assess the durability of resistance genes, which may depend on the conservation of certain TAL effectors. This information is of immense importance for developing more effective and well-defined means to control the pathogen population and for designing effective resistance breeding strategies (Hutin et al., 2015).
Pakistan is the ninth-largest producer and the fourth-largest exporter of premium-quality rice in the world. A substantial amount of revenue is generated from rice exports, contributing significantly to the country’s economy (USDA, 2024). Pakistani deluxe rice, widely known as Basmati, is very popular around the globe. Basmati enjoys an inimitable place in the global market due to its properties such as taste, aroma, and texture (Shahzadi et al., 2018; Akhter and Haider, 2020). In Pakistan, BLB was first reported in 1977 (Mew et al., 1989), and in recent years, it remains one of the major causes of losses in rice crops. This is mainly because information regarding the virulence genes, especially the TAL effector repertoire in the Pakistani Xoo population, is still unavailable, leading to a lack of effective control measures. Whole genome-based identification of the TAL effector repertoire in the prevalent Pakistani Xoo isolates is not only important to enlarge the geographic coverage of the pathogen’s genomic resources but also to develop information-based, durable resistant cultivars (Ejaz et al., 2022).
The identification of tal genes in the Pakistani Xoo isolates and their comparison with other Xoo strains representing East Asian lineages was the main objective of the present study. This study focused on a comparative genomic analysis to identify the TAL effector repertoire in two Xoo strains isolated from the Basmati rice-growing area of Pakistan. The knowledge gained from this study will be helpful in designing more informed host resistance strategies for the development of broad and robust resistance in rice.
2 Materials and methods
2.1 Xoo isolates used
Two Xoo isolates were collected from BLB-affected farmer fields in the Basmati rice-growing area of Punjab, Pakistan (31.1704° N, 72.7097° E), between 2017 and 2019. Two representative Xoo isolates were selected based on their higher virulence on susceptible rice varieties (IR24 and Super Basmati) and were named PkXoo1 and PkXoo2, with Pk representing Pakistan. PkXoo1 was previously reported in our study (Ejaz et al., 2023), which defined its basic genome features and was used in this study for descriptive comparison. PkXoo1 and PkXoo2 were confirmed as Xoo using species-specific (Xo3756) and pathovar-specific (Xoo80) primers in a multiplex PCR (Lu et al., 2014).
2.2 Virulence testing of PkXoo1 and PkXoo2
Preserved Xoo colonies stored at −80°C were streaked onto Wakimoto agar plates (Sucrose 2%, Peptone 0.5%, Ca(NO3)2 0.05%, FeSO4 0.005%) (Cappuccino and Sherman, 2004). The inoculum was prepared from a single, pure Xoo colony in nutrient broth with an optimal density (OD) of 1.0 at 600 nm (OD600). Seeds of two susceptible rice varieties, IR24 and Super Basmati, were first sown in plastic pots. The seedlings were transplanted into earthen pots after 15 days. The experiment was conducted in a net house under natural conditions during the rice season (June–October), with 80% relative humidity. Forty-five-day-old rice plants of each differential variety were clip-infected using the scissor dip method (Kauffman and Rao, 1972). A total of 10 rice leaves were inoculated with bacteria, and leaves cut with scissors and dipped in sterilized water were used as the control. Disease symptoms were observed and scored based on the percent Disease Leaf Area (% DLA) 21 days post-inoculation (DPI) using the following formula: %DLA = lesion length/total length × 100.
2.3 DNA extraction and long-read sequencing of the Xoo isolates using Oxford Nanopore Technology
For nanopore sequencing, bacterial cultures from a single colony were cultivated in nutrient broth at 28 ± 2°C for 48 h at 100 rpm until the OD reached 0.8 at 600 nm. The culture (1 mL) was then harvested in a bio-grade centrifuge tube for 5 min at 13,000 rpm. DNA was extracted using the Thermo Fisher Scientific GeneJet Genomic DNA Purification Kit (Massachusetts, United States) following the manufacturer’s protocol. The samples were sequenced using ONT (Oxford, United Kingdom). The extracted DNA was cleaned up with 0.6 volume of AMPure XP beads (Beckman-Coulter, California, United States) to attain the recommended concentration of DNA for Nanopore library preparation and to remove small DNA fragments. This step was also repeated during library preparation after fragmentation. DNA quantity and quality were measured using a NanoDropTm One/OneC Microvolume UV–Vis Spectrophotometer (Thermo Fisher Scientific, Massachusetts, United States) to determine the A260/280 and A260/230 ratios, as well as a Qubit 4 Fluorometer (Thermo Fisher Scientific, Massachusetts, United States). For long-read sequencing of each strain, 2–3 μg of genomic DNA was used to prepare the sequencing library. The library was prepared using the PCR-free Rapid Sequencing Kit (RAD004) and Rapid Barcoding Kit (RBK004) following the manufacturer’s protocol (ONT, Oxford, United Kingdom). Oxford Nanopore sequencing was performed at the Department of Plant Pathology, University of Florida, FL, United States. A total of 12 μL of the prepared library from each sample was loaded onto the flow cell according to the manufacturer’s protocol. Multiplexed (barcoded) samples were sequenced using a MinION Flow Cell (R9.4.1) (ONT, United Kingdom) for 72 h. Single samples were also sequenced using a Flongle Flow Cell (R 9.4.1) for 24 h. Sequencing progress was monitored in real-time through the MinKNOW (version: 21.06.13 and 22.05.5) graphic user interface with user-defined parameters. The reads obtained were saved automatically in the system, and the data were retrieved in the form of Fast5 files. The Fast5 reads were base-called using Guppy (version 4.4.1) (Wick et al., 2019). Guppy converted the Fast5 reads into FASTQ format.
2.4 Genome assembly and post-finishing
The sequenced reads obtained after base-calling were chopped using Porechop1 to cleave adapters. Trimmed reads were filtered to remove reads less than 1,000 bp using the Filtlong tool (v0.2.1).2 Reads with a quality higher than Q10 and a length greater than 1Kb were de novo assembled using Flye (v2.9) (Kolmogorov et al., 2019). The genome assembly derived from Flye was further corrected and refined using Racon (v1.4.3) (Vaser et al., 2017) and Medaka (v1.7.0).3 Assembly completeness scores were calculated using Benchmarking Universal Single-Copy Orthologs (BUSCO) (v5.4.3) (Simão et al., 2015) with the xanthomonadales_odb10 dataset, which consists of 1,152 single-copy orthologs. Although the BUSCO matrices of the polished assembly derived after Medaka were quite significant, manual assessment exposed significant problems with homopolymeric nucleotides, resulting in frequent frameshifts and a large number of apparent pseudogenes. The assembly was further rectified using Homopolish (v0.0.1) (Huang et al., 2021). The Circlator (Hunt et al., 2015) was used to fix the start position and to circularize the assembly. Contamination from any other genome was checked using ContEST16S (Lee et al., 2017).
2.5 Genome annotation
Genome annotation was performed using Prokka (v1.14.5) (Seemann, 2014) and GeneMarkS-2 + (Lomsadze et al., 2018), as implemented in the NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016). Gene function and pathway analyses were performed using BLASTP searches against functional databases, including the Clusters of Orthologous Groups (COGs) database (Tatusov et al., 2003), the Gene Ontology (GO),4 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa, 2002).
2.6 Prediction of TAL effectors
The TAL effector repertoire of the two Xoo genomes was predicted using the “TALE Prediction” tool from the AnnoTALE suite (v1.4.1) (Grau et al., 2016). The final circular assembly was used as input for the prediction of the TAL effector genes. The DNA sequences of the predicted TAL effectors were used as input to identify RVDs. The AnnoTALE class builder with class_definitin_29–07-22 (downloaded in November 2022) was used to assign classes to the TAL effectors based on their RVD patterns. DisTAL, from the QueTAL suite (Perez-Quintero et al., 2015), was used to calculate similarities based on RVDs between the TAL effectors of available reference Xoo genomes (PXO99A, PXO86) in the database. The amino acid sequences of the predicted TAL effectors were used as input for DisTAL.
2.7 Phylogenetic and comparative genomic analysis of the Pakistani Xoo isolates with other Xoo strains representing east Asian lineages
A set of Xanthomonas genomes was downloaded from the NCBI Assembly database, and complete genomes were aligned using Parsnp (v1.6.2) (Treangen et al., 2014) for phylogenomic analysis. The tree was visualized using FigTree.5 Genome-wide pairwise alignments were calculated using Parsnp, and a phylogenetic tree was constructed with RAxML-ng (v1.1.0) (Kozlov et al., 2019) using the GTR = G model and bootstrap with 100 replications. For structural comparison, the complete genomes of PkXoo1 and PkXoo2 were aligned with reference strains (IXO-280, SK2-3, MAFF311018, PXO71, and PXO99A) using progressive Mauve (Darling et al., 2010) with standard preset settings.
2.8 Prediction of effector binding elements
After the TAL sequences were extracted, the TALE-NT 2.0 Target Finder tool (Doyle et al., 2012) was used to predict the targets or effector binding elements (EBEs) of some important TAL effectors. The RVD sequence was used as input. Predictions were carried out using the standard preset settings (score cutoff = 3.0; upstream base of the binding site = T). Predictions were made on the promoterome (defined as the 5′ UTR plus 1,000 base pairs upstream of the transcriptional start site of each transcript) for the clade III SWEET genes, using the MSU Rice Genome Annotation Project Release 7. The FindM tool from the signal search analysis (SSA) server (Ambrosini et al., 2003) was used with default parameters to determine the distance between the binding site and the TATA box.
3 Results
3.1 Virulence analysis of the two Xoo isolates, PkXoo1 and PkXoo2, collected from the basmati-growing region of Pakistan
The artificially inoculated leaves of the susceptible rice variety Super Basmati showed BLB symptoms, and the percent diseased leaf area (DLA) for both isolates was calculated. On the basis of molecular identification and phenotypic characteristics, both Xoo isolates seemed quite similar. The % DLA for both isolates ranged from 50 to 70% on the susceptible varieties, which confirmed the aggressive behavior of both Xoo isolates. In the pathotype analysis, PkXoo1 and PkXoo2 showed virulent interactions with rice NILs carrying single BLB-R genes (IRBB3, IRBB4, and IRBB5) (Shah et al., 2025).
3.2 Genome characteristics of PkXoo1 and PkXoo2
Two Xoo isolates, PkXoo1 and PkXoo2, collected from Basmati-growing areas of Punjab, Pakistan, were selected to compare the Xoo genomic information from the Pakistan region, which is currently not available. Oxford Nanopore sequencing with ultra-long reads was used for the complete genome sequencing of the two strains. The total number of bases retrieved after the sequencing of PkXoo1 and PkXoo2 was 636,943,704.00 and 1,315,318,885.00, respectively. The number of reads generated was 193,312.0 and 239,360.0, respectively. Other sequencing statistics are provided in Table 1. The sequenced reads were de novo assembled using Flye, resulting in a single circular contig for each strain. Racon and Medaka were used to polish the assembly, and Homopolish was used to give the assembly a final look, which significantly improved the assembly’s BUSCO completeness (Table 2). After circularizing and adjusting the starting site, the final assembly yielded a single circular chromosome consisting of 4,930,582 base pairs (bp) and 4,941,434 bp, with 133X and 274X genome coverage, respectively. No plasmids were found in either Xoo strains. The BUSCO results revealed the completeness of the assembly up to 98.8 and 99%, respectively, although no contamination was found in the two genomes, as confirmed by ContEst16. The genome characteristics of PkXoo1 and PkXoo2 and other reference strains are listed in Table 3. The genomes of PkXoo1 and PkXoo2 encoded 749 and 770 pseudogenes, respectively, while in comparison, PXO99A has 759 and MAFF 311018 has 747 pseudogenes (Ochiai et al., 2005; Salzberg et al., 2008). These results revealed that the quality of the ultralong sequencing platform was relatively high.
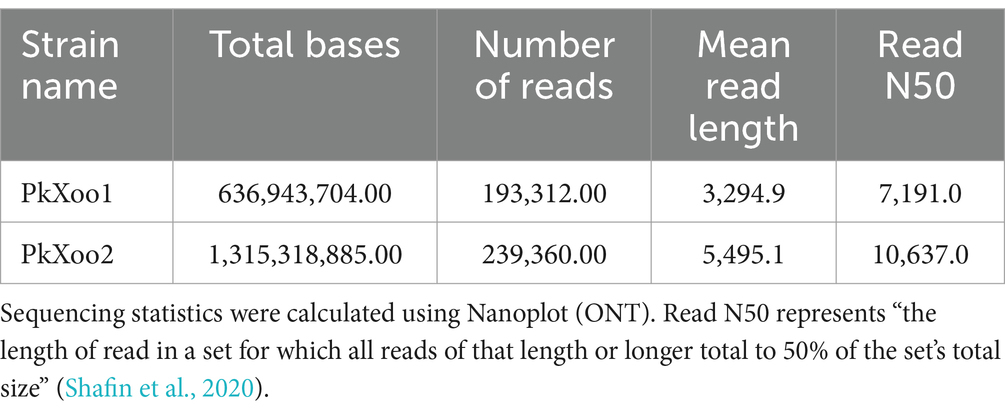
Table 1. Nanoplot results for PkXoo1 and PkXoo2.
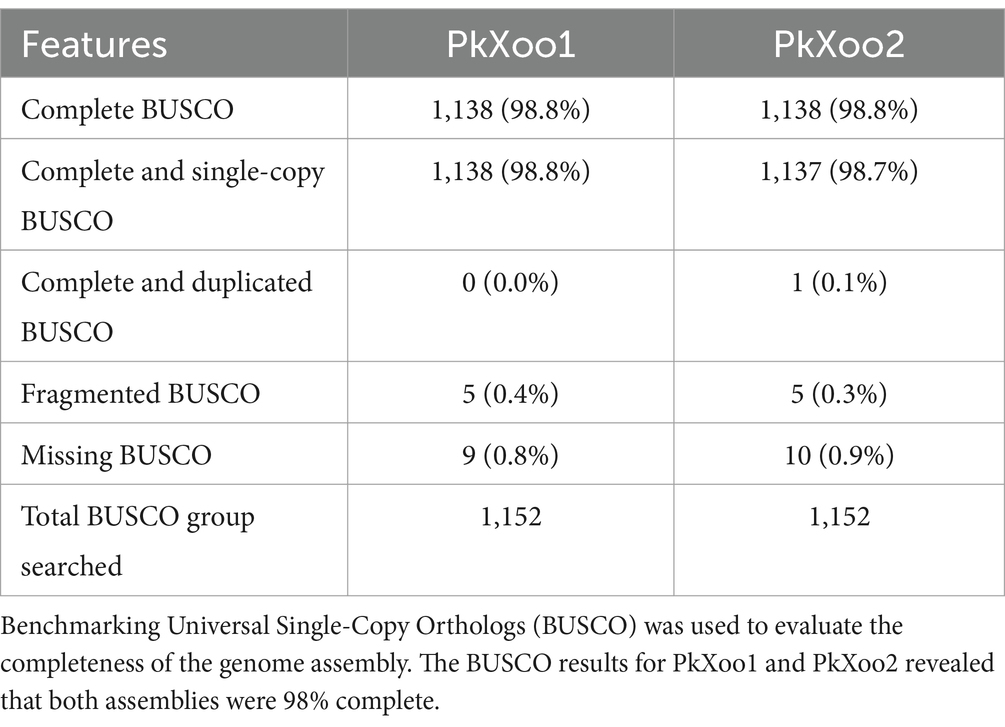
Table 2. BUSCO results for PkXoo1 and PkXoo2.
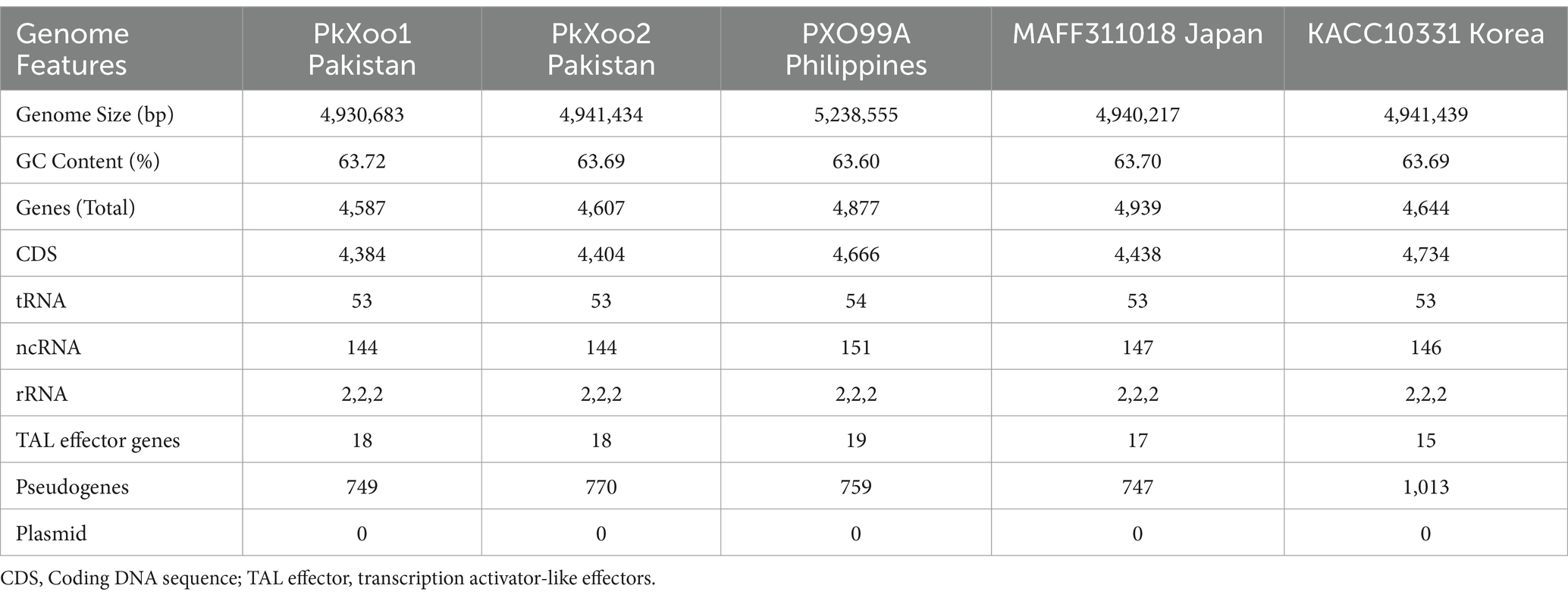
Table 3. Genome features of PkXoo1 and PkXoo2 in comparison with reference Xoo strains.
3.3 Genome annotation
Initially, the annotation was performed using the NCBI automated pipeline, and then, Prokka was also used for the annotation of the two Xoo isolates. The results revealed the presence of 4,384 and 4,404 protein-encoding genes (CDS) in PkXoo1 and PkXoo2, respectively (Table 3). Like other Xanthomonas genomes, both Xoo isolates typically encoded pathogenicity/virulence genes, including extracellular hydrolases, extracellular polysaccharides, adhesins, the type 2 secretion system, and type 3 secretion effectors, specifically TAL effectors. Gene functions and pathway analysis were performed using BLASTP against functional databases.
3.4 TAL effectors of PkXoo1 and PkXoo2
To interact with the host and promote proliferation, virulence, and dispersal, Xoo exploits TAL effectors. The TAL effector repertoires of PkXoo1 and PkXoo2 were annotated using the AnnoTALE suite, which assigned classes based on the RVDs (Grau et al., 2016). The number of predicted TAL effectors and other features is presented in Tables 4, 5. The AnnoTALE analysis revealed the presence of 15 TAL effectors, along with 3 pseudoTALes, in both isolates. Almost two or three pseudoTALes are found alongside the typical tal genes in the genome of Xoo. TAL effectors encoded by pseudogenes were defined as the presence of an early stop codon, a frameshift mutation, or the insertion of a large fragment that led to the absence of one or all matches to the nuclear localization signal (NLS) consensus or the absence of a string of 35 amino acids with at least 80% sequence similarity to the acidic activation domain sequence (Van Den Ackerveken et al., 1996).
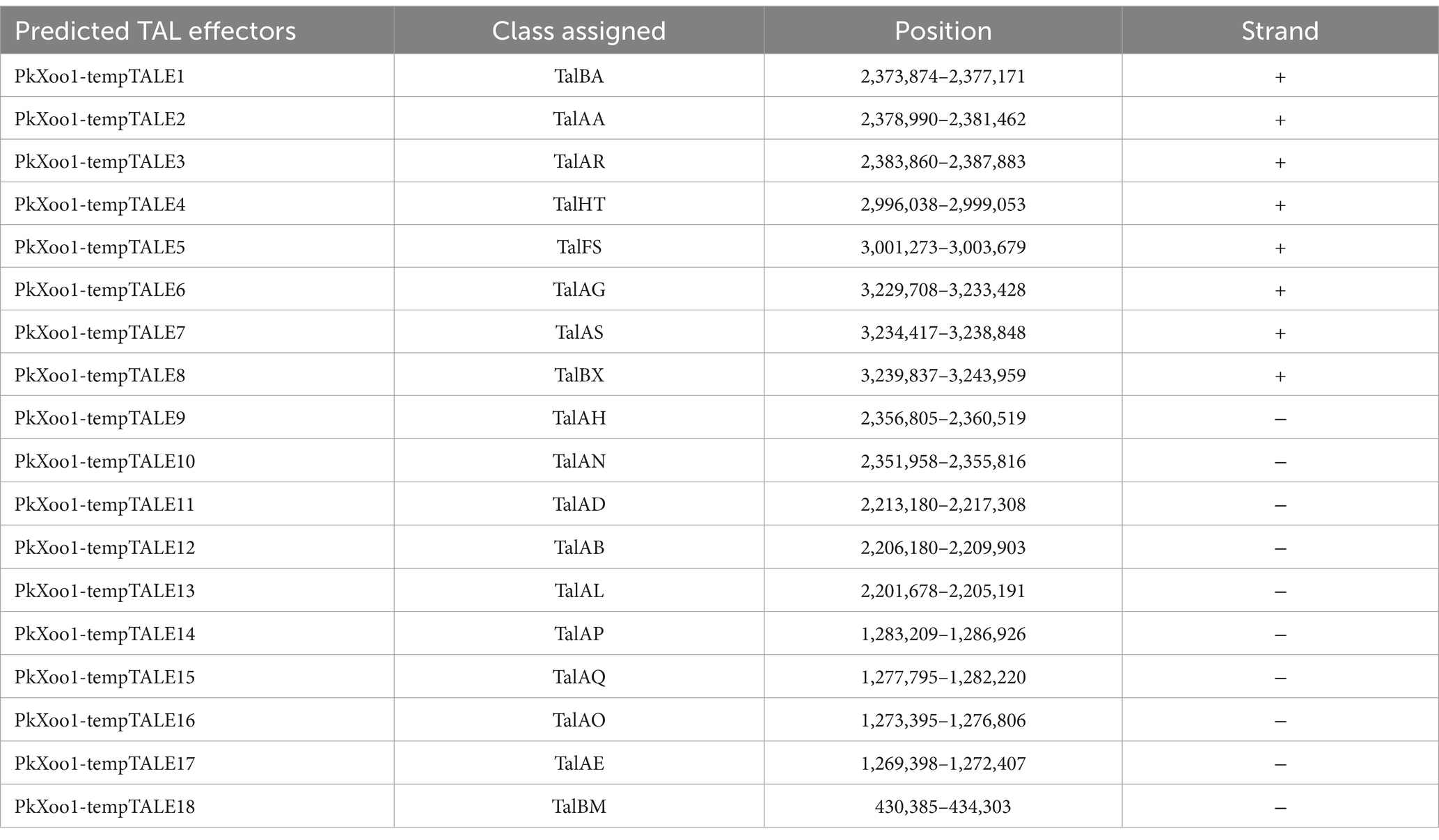
Table 4. Predicted TAL effectors in PkXoo1.
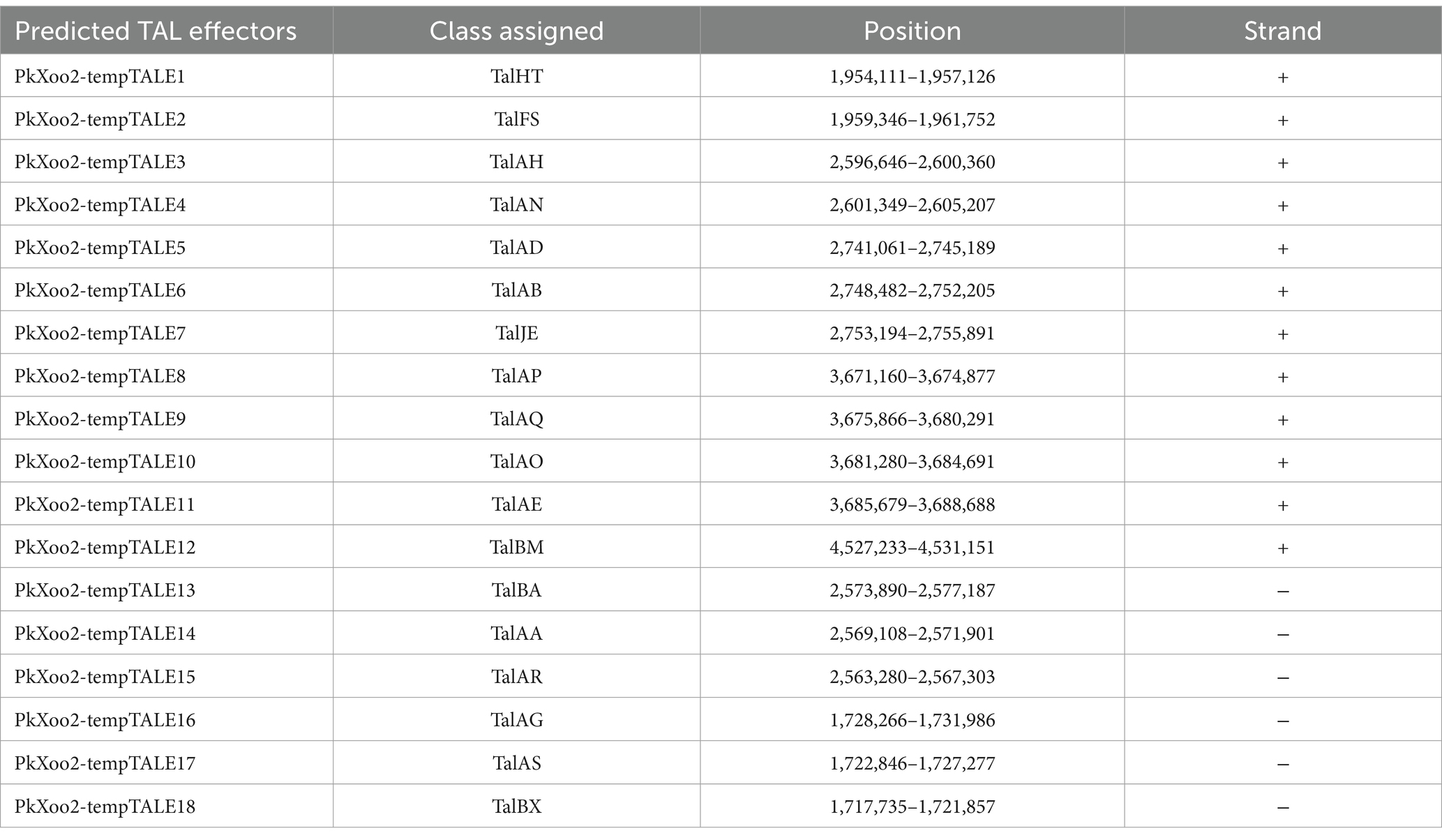
Table 5. Predicted TAL effectors in PkXoo2.
A total of 15 classic TAL effectors present in both isolates were similar to those in the genomes of other Xoo strains, specifically PXO99A and PXO86 (Salzberg et al., 2008). Both strains PkXoo1 and PkXoo2 shared almost the same TAL repertoire, except for the presence of one truncTal in PkXoo2, which was truncated at the 10th repeat. TruncTal is a TAL effector variant with modified N- and C-termini, which may undermine the resistance facilitated by definite non-executor R genes of the host plant (Ji et al., 2016; Read et al., 2016). The shared Tal effectors of both Xoo isolates were assigned to the classes TalBM, TalAE, TalAO, TalAQ, TalAP, TalAB, TalAD, TalAN, TalAH, TalBA, TalAA, TalAR, TalHT, TalFS, TalAG, TalAS, and TalBX. Furthermore, truncTal of PkXoo2 was assigned to the class TalJE, whereas its counterpart, which was present in full form in PkXoo1, was assigned to the class TalAL. The tal genes were also designated numerically based on the method described by Salzberg et al. (2008). The tal genes were positioned at eight loci on the genome. The positions of the tal genes in the genome showed rearrangements in both genomes relative to one another (Figure 1).
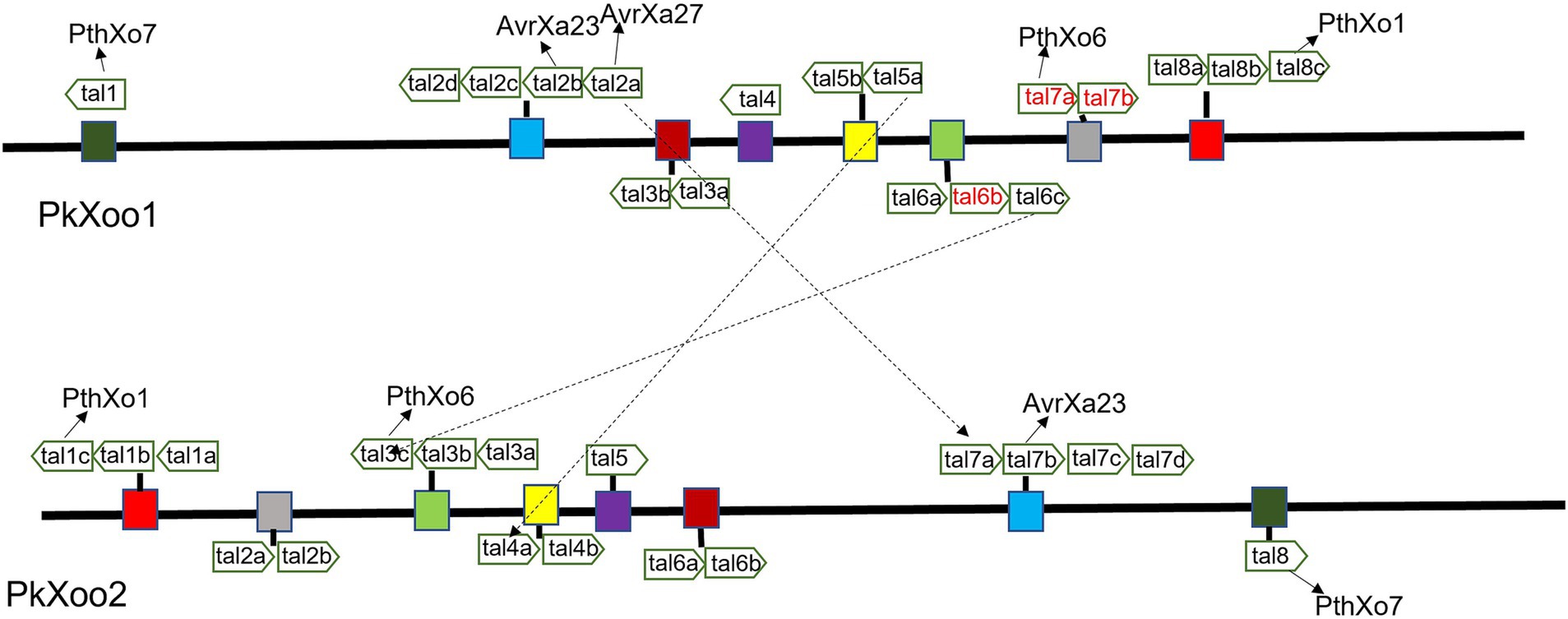
Figure 1. Map of tal genes on genomes of PkXoo1 and PkXoo2. The dotted lines represent the TAL effectors with slight differences in RVDs. The same color boxes represent homologous TAL effectors on the genomes of PkXoo1 and PkXoo2.
TAL effector sequences were highly conserved among the Xoo isolates, except for the slight difference in the repeats and RVD sequences (Perez-Quintero et al., 2023). PkXoo1 and PkXoo2 had the same RVD pattern for their shared TAL effectors, except for tal2a, tal5a, and tal6c of PkXoo1, which were homologous to tal7a, tal4a, and tal3c of PkXoo2, respectively (Tables 4, 5). These three TAL effectors of both isolates were homologous to PthXo6, tal9a, and tal6a in PXO99A. The number of RVDs in the TAL repertoire of both isolates varied from 14 to 27 (Tables 6, 7). tal8c of PkXoo1 and talc of PkXoo2 were orthologs of PthXo1 in PXO99A. PthXo1 in PkXoo1 and PkXoo2 had RVD patterns identical to those of PthXo1 in PXO99A.

Table 6. Repeat variable di-residue (RVD) pattern of the Xanthomonas oryzae pv. oryzae strain PkXoo1.
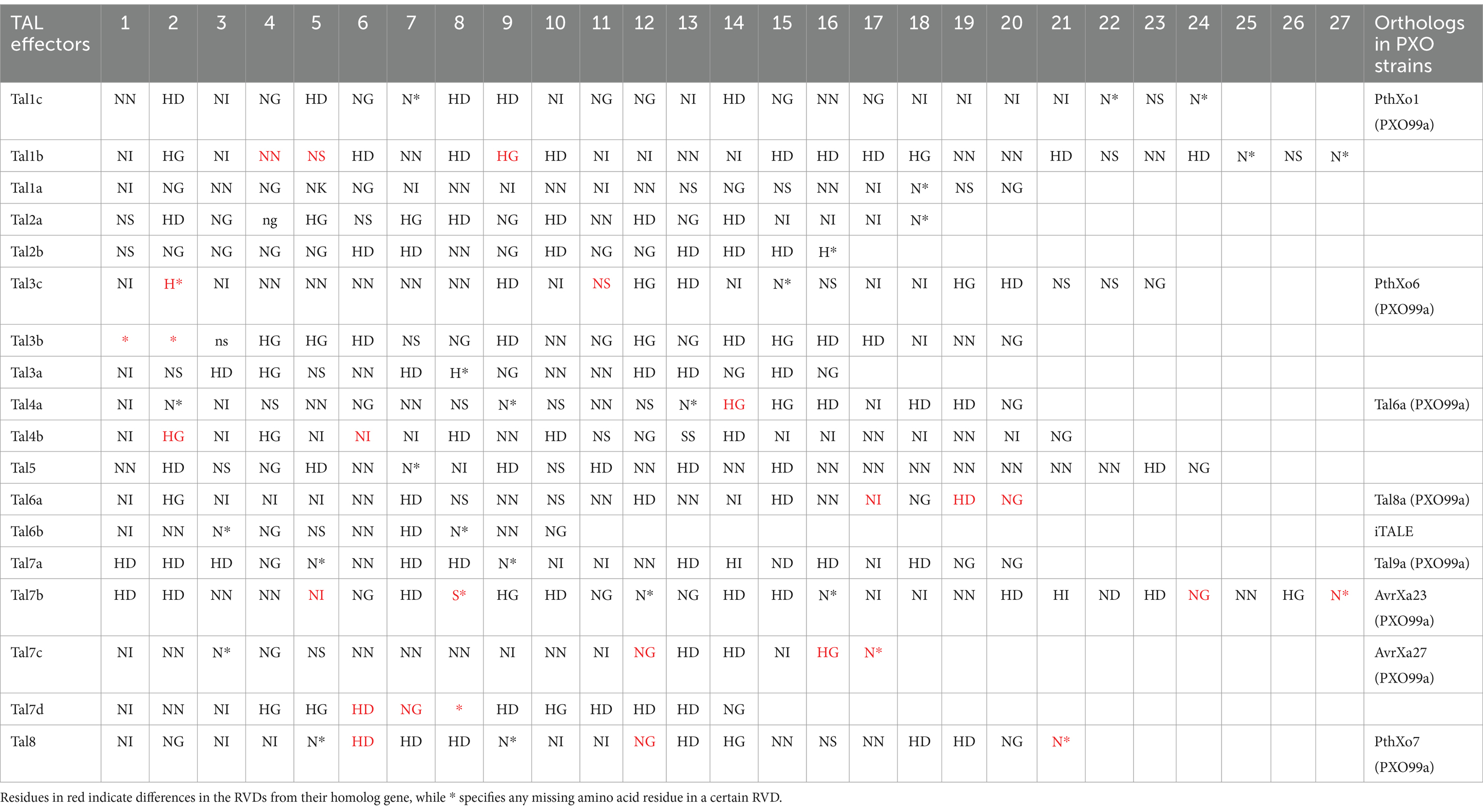
Table 7. Repeat variable di-residue (RVD) pattern of the Xanthomonas oryzae pv. oryzae strain PkXoo2.
3.5 Phylogenetic and genome comparison
Phylogeny based on whole genome alignment showed that both Pakistani isolates shared a strong relationship with isolates from neighboring Asian countries—Xoo IX-280 from India and Xoo SK2-3 from Thailand. To determine the relationship between the two isolates, a pairwise genome comparison was conducted using Mauve. The results showed that the chromosomes of PkXoo1 and PkXoo2 somehow exhibited synteny (Figure 2), but PkXoo1 showed genomic rearrangements relative to PkXoo2. The genome alignment results indicated that the genomes of the two Pakistani isolates were conserved in gene content and allelic sites. Although the two isolates were genetically identical, genomic rearrangements were observed (Figure 2).
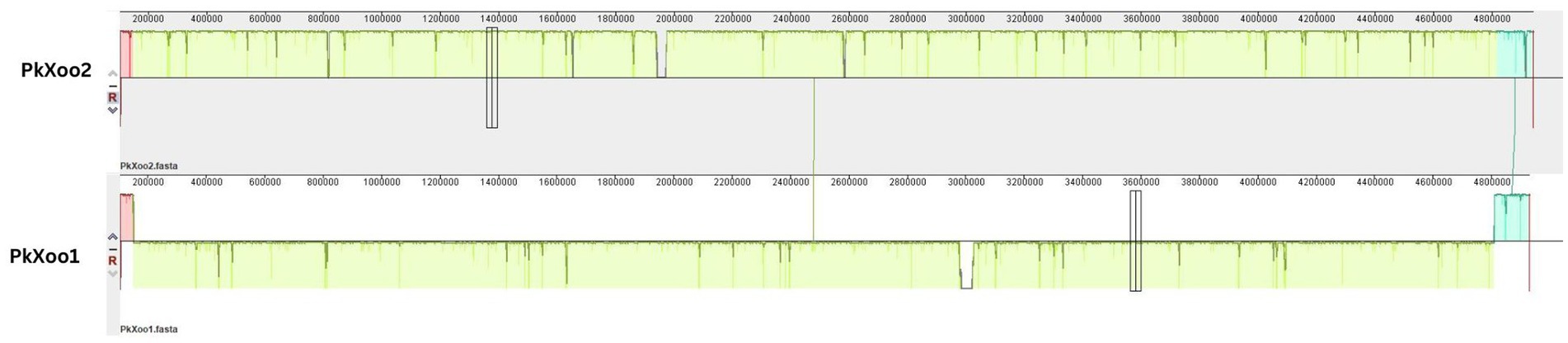
Figure 2. Comparison of the genome structure of PkXoo1 and PkXoo2. The colors represent locally colinear blocks (LCBs), which are regions that show no rearrangement across all the compared genome sequences. Synteny is displayed by a line that links orthologous LCBs. LCBs found on the bottom part represent regions that are found in reverse orientation.
To compare the genome structures of PkXoo1 and PkXoo2 with the genomes of the other Xoo strains representing East Asian lineages (Quibod et al., 2016), a pairwise genome comparison was performed using Mauve. The alignment showed that the genome structure of PkXoo2 was similar to that of the Indian strains IX-280 and SK2-3 from Thailand. While PkXoo1 showed genomic rearrangements, its genome structure was similar to that of other closely related Xoo strains (Figure 3).
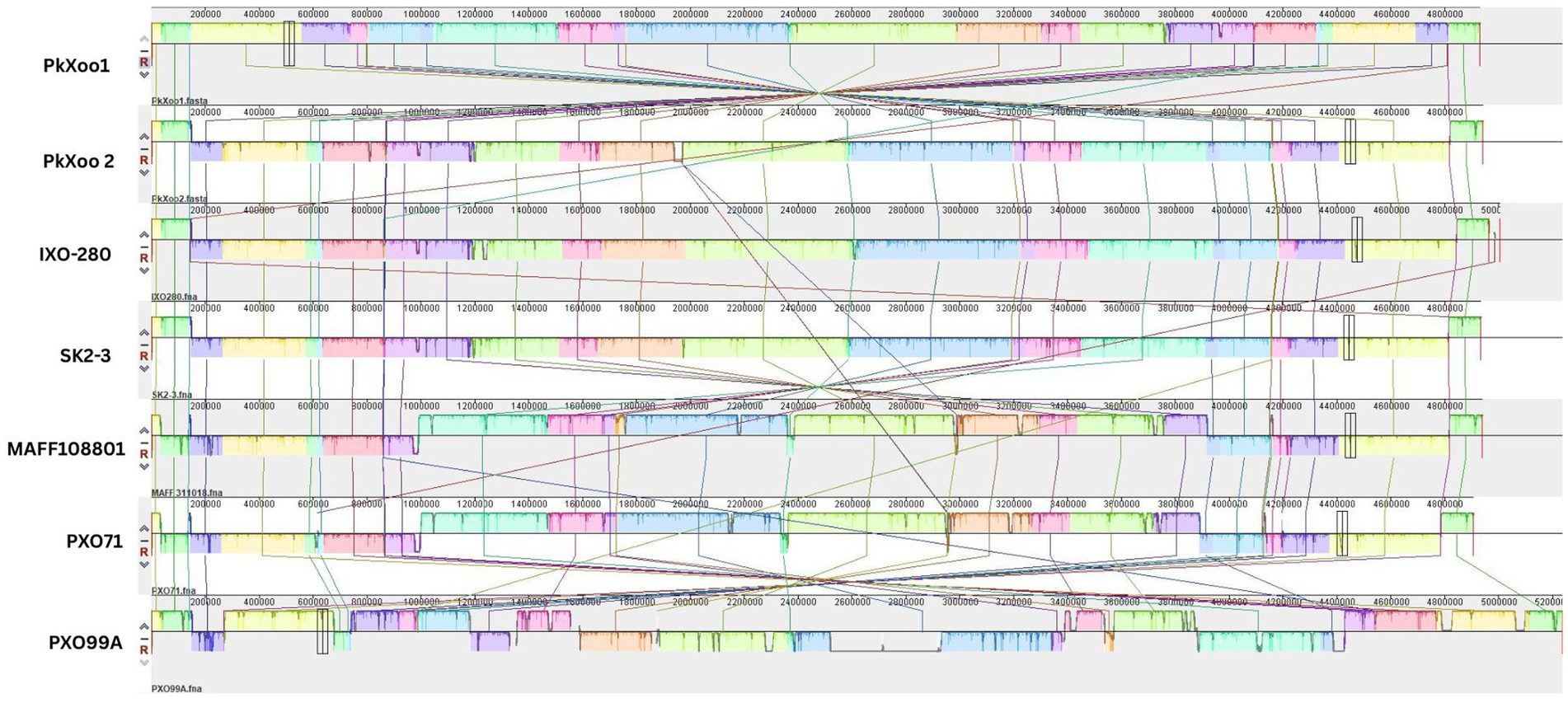
Figure 3. Pairwise comparisons of the chromosome structure of PkXoo1 and PkXoo2 using available complete genome sequences of reference Xoo strains from other Asian countries.
3.6 TAL effector EBE prediction
In the present study, we assessed the target EBEs of important TAL effectors present in the Pakistani Xoo isolates using an in silico target prediction tool. TalBX (homologous to PthXo1) and TalBM (homologous to PthXo7) were found in both Pakistani isolates. Strains that can cope with xa5-mediated resistance showed the presence of either TalBX, which corresponds to the PthXo1 TAL effector, or TalBM, which corresponds to the PthXo7 TAL effector of Xoo. For PthXo1 or TalBX, EBEs were predicted to be present in the promoter region upstream of locus LOC_Os08g42350.1, which corresponds to the OsSWEET11 gene. Similarly, for TalBM, EBEs were predicted in the promoter region upstream of locus LOC_Os01g73890, which corresponds to the OsTFIIAγ1 gene.
4 Discussion
BLB is a primary threat to rice crops, caused by Xanthomonas oryzae pv. oryzae (Xoo). Resistance to the BLB pathogen includes a durable, race-specific element conferred either by disease resistance genes or by engineered variations in the effector-binding sites of susceptibility genes. To date, 46 resistance (Xa) genes have been discovered and widely incorporated into BLB-resistant varieties (Tian et al., 2014).
Pakistan, being one of the largest rice-producing countries, exports fine-quality rice all over the world. However, to date, BLB remains one of the major constraints to achieving significant rice production in Pakistan. Outbreaks of BLB have occurred repeatedly in Pakistan, specifically in the Kallar belt, which is famous for producing high-quality Basmati rice. Recent surveys have indicated a high occurrence of BLB in Punjab province (~30–55%) (Waheed et al., 2009; Ullah et al., 2022). The knowledge regarding the virulence factors of the Pakistani Xoo isolates is still unexplored, which hinders breeding efforts to overcome this disease (Ejaz et al., 2022). In the present study, we sequenced two Pakistani Xoo isolates, PkXoo1 and PkXoo2, collected from the Kaller belt region of Punjab province, Pakistan, using the Oxford Nanopore MinION sequencer. The emergence of cost-effective, long-read, next-generation sequencing technologies has facilitated the study of whole genomes and the identification of identical/nearly identical repetitive elements and genes encoding proteins, such as TAL effectors. The potential and power of the Oxford MinION sequencer in the investigation of the Xanthomonas genus, including Xoo strains, have also been reported in previous studies (Bansal et al., 2018; Kaur et al., 2019; Fernandes et al., 2021; Erkes et al., 2023). This study can provide valuable insights into the diversity of TAL effectors in Pakistani Xoo and assist in the discovery of key TAL effectors essential for virulence.
Both Xoo isolates had a homolog of PthXo1, namely tal8c in PkXoo1 and tal1c in PkXoo2. Both shared the same RVD pattern as PthXo1 in PXO99. Previously, Carpenter et al. (2020) reported a difference of one RVD in the PthXo1 ortholog of IX-280 and SK2-3 compared to PXO99. PthXo1 is the prominent virulent determinant that binds directly to the SWEET11a promoter in rice, delivering the mechanism behind gene-for-gene recessive resistance and susceptibility (Chen et al., 2010; Römer et al., 2010). The function of PthXo1 is interrupted in rice by the recessive blight resistance gene xa13 (Salzberg et al., 2008).
The Pakistani Xoo isolates, PkXoo1 and PkXoo2, did not encompass any other major TAL effectors predicted to trigger the expression of any other SWEET gene from clade III. Remarkably, an identical copy of PthXo7, the PXO99A TAL effector that targets TFIIAγ1, was also present in both strains (tal1 in PkXoo1 and tal8 in PkXoo2). PkXoo1 and PkXoo2 also had orthologs of the PXO99A TAL effector PthXo6, homologous to tal3b in PkXoo1 and tal3c in PkXoo2. PthXo6 triggers the expression of the bZIP transcription factor TFX1. AvrXa23 and AvrXa27, which are considered avirulent determinants and trigger the response of Xa23 and Xa27 resistance genes in rice, were also present in both Pakistani Xoo isolates. The RVD pattern of AvrXa23 is different from its ortholog in PXO99A but is identical to the AvrXa23D present in PXO61, PXO71, Sk2-3, and IX-280, as reported recently by Xu et al. (2022). Carpenter et al. (2020) also reported the harmony of IX-280 and SK2-3 with xa5.
The phylogenomic analysis and genome comparison indicated that the Pakistani isolates PkXoo1 and PkXoo2 are closely related but show rearrangements and inversions compared to one another. Further analysis revealed the presence of a large number of transposases and insertion sequences in the genome of both Xoo isolates, which are likely to contribute to genome plasticity. Such a high number of insertion sequences within the genome facilitates chromosomal recombination, horizontal gene transfer, and gene duplication (Vandecraen et al., 2017). Previously, a relatively large number of insertion sequences were reported in Xoo, as these mobile elements were involved in race differentiation. Insertion elements can also lead to the loss of gene function, and virulence can develop from the inactivation of avirulence genes (Ochiai et al., 2005; Goettelmann et al., 2023).
Both Xoo isolates PkXoo1 and PkXoo2 were highly identical to the Indian Xoo strain IX-280 and a Xoo strain from Thailand, SK2-3. Both strains belong to the young and relatively high clonal lineage L-1, which is predominant in India (Midha et al., 2017; Carpenter et al., 2020). The genome structure of PkXoo2, IX-280, and SK2-3 is comparable to each other, despite the three strains belonging to different regions. In contrast, the Xoo strains from the Philippines, PXO86, PXO71, and PXO99A, have experienced genomic rearrangements relative to one another, although they belong to the same region (Carpenter et al., 2020).
Although geographically separated, PkXoo2 shares a striking genomic similarity with IX-280 and SK2-3. This has prompted us to investigate the relationship between these isolates and other Xoo strains. The clonality of PkXoo2 with IX-280 and SK2-3 suggested that immigration played a significant role in the evolution of local Xoo populations (Carpenter et al., 2020). The results highlight the necessity of developing local varieties with distinct or even better stacked resistance genes for rapid deployment.
The relatedness of the Pakistani isolates to IX-280 and SK2-3 suggests that migration events, such as the exchange of seeds and the import and export of agricultural commodities, may be involved in the evolution of local Xoo populations (Carpenter et al., 2020). The presence of an ortholog of PthXo1 in these strains underlines the effectiveness of a complete genome sequence study using long-read sequencing technologies and TAL effector analysis-based surveillance to comprehend the breakdown of resistance genes. The results also emphasize the need for the development of local cultivars with diverse single or, preferably, stacked BLB resistance genes for the expeditious deployment of BLB-resistant cultivars.
5 Conclusion
The present study reports the TAL effector repertoire in two Xanthomonas oryzae pv. oryzae (Xoo) isolates from Pakistan. The whole genome sequencing using long-read sequencing technology provided a deep understanding of diversity, genome plasticity, and geographical relationships of the Xoo isolates. The findings of this study will be helpful to enhance sequence resources for the global comparison of pathogen diversity across the region. The presence of 17 distinct TAL effectors in both Xoo isolates unravels their virulence potential. Phylogenetic analysis surprisingly unveiled the relatedness of the Pakistani Xoo isolates with the Indian and Thai strains, suggesting the potential role of migration events in shaping the pathogen population. In the future, to uncover the population structure and TALome diversity in Pakistan, we aim to focus on sequencing more Xoo isolates. This will not only be helpful for disease surveillance in real time but will lead to sustainable and targeted disease management in rice crops.
Data availability statement
The complete genome sequence of PkXoo1 and PkXoo2 has been submitted to NCBI Gen Bank in Bio-project PRJNA861287 under accession number CP101721.2 and CP140752, respectively.
Author contributions
KE: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. MZ: Writing – review & editing, Methodology. PZ: Formal analysis, Writing – review & editing, Supervision, Software. JH: Software, Supervision, Writing – review & editing, Formal analysis. MA: Supervision, Writing – review & editing, Project administration. FW: Supervision, Validation, Writing – review & editing, Project administration. SY: Writing – review & editing, Investigation, Conceptualization, Supervision, Formal analysis, Resources, Project administration, Validation.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are deeply thankful to Zahid Iqbal (Principal Scientific Assistant), M. Sarwar (Scientific Assistant) from the Plant-Microbe Interactions Lab, and Qaiser Zaman (Scientific Assistant) from the Agricultural Biotechnology Division for their technical assistance in the lab and net house experiments. We are also grateful to the Higher Education Commission (HEC), Pakistan, for the provision of the IRSIP fellowship to Khansa Ejaz at the Department of Plant Pathology, University of Florida.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://github.com/rrwick/Porechop
2. ^https://github.com/rrwick/Filtlong
3. ^https://github.com/nanoporetech/medaka
References
Akhter, M., and Haider, Z. (2020). Basmati rice production and research in Pakistan. Sustain Agric. Res. 39, 119–136. doi: 10.1007/978-3-030-38881-2_5
Ambrosini, G., Praz, V., Jagannathan, V., and Bucher, P. (2003). Signal search analysis server. Nucleic Acid. Res. 31, 3618–3620. doi: 10.1093/nar/gkg611
Bansal, K., Kumar, S., and Patil, P. B. (2018). Complete genome sequence reveals evolutionary dynamics of an emerging and variant pathovar of Xanthomonas euvesicatoria. Genome Biol. Evol. 10, 3104–3109. doi: 10.1093/gbe/evy238
Boch, J., and Bonas, U. (2010). Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 48, 419–436. doi: 10.1146/annurev-phyto-080508-081936
Boch, J., Bonas, U., and Lahaye, T. (2014). TAL effectors–pathogen strategies and plant resistance engineering. New Phytol. 204, 823–832. doi: 10.1111/nph.13015
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi: 10.1126/science.1178811
Bogdanove, A. J., Schornack, S., and Lahaye, T. (2010). TAL effectors: finding plant genes for disease and defense. Curr. Opin. Plant Biol. 13, 394–401. doi: 10.1016/j.pbi.2010.04.010
Cappuccino, J. G., and Sherman, N. (2004). Microbiology, laboratory manual. New Delhi: Pearson Education Inc.
Carpenter, S. C., Mishra, P., Ghoshal, C., Dash, P. K., Wang, L., Midha, S., et al. (2020). An xa5 resistance gene-breaking Indian strain of the rice bacterial blight pathogen Xanthomonas oryzae pv. Oryzae is nearly identical to a Thai strain. Front. Microbiol. 11:579504. doi: 10.3389/fmicb.2020.579504
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X. Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606
Ciuffreda, L., Rodríguez-Pérez, H., and Flores, C. (2021). Nanopore sequencing and its application to the study of microbial communities. Comput. Struct. Biotechnol. J. 19, 1497–1511. doi: 10.1016/j.csbj.2021.02.020
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:11147. doi: 10.1371/journal.pone.0011147
Dossa, G. S., Sparks, A., Cruz, C. V., and Oliva, R. (2015). Decision tools for bacterial blight resistance gene deployment in rice-based agricultural ecosystems. Front. Plant Sci. 6:305. doi: 10.3389/fpls.2015.00305
Doyle, E. L., Booher, N. J., Standage, D. S., Voytas, D. F., Brendel, V. P., VanDyk, J. K., et al. (2012). TAL effector-nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acid. Res. 40, W117–W122. doi: 10.1093/nar/gks608
Ejaz, K., Faiq, A., Asif, M., Zaka, A., Nguyen, M. H., Cruz, C. V., et al. (2022). Molecular characterization and screening of Xanthomonas oryzae pv. Oryzae, isolated from Pakistan for prediction of bacterial leaf blight-resistant basmati rice. Physiol. Mol. Plant Pathol. 121:101858. doi: 10.1016/j.pmpp.2022.101858
Ejaz, K., Zakria, M., Tapia, J. H., Zhang, P., Faiq, A., Ahsan, R., et al. (2023). Genome sequence resource for Xanthomonas oryzae pv. Oryzae (causal agent of bacterial leaf blight of rice) isolated from Pakistan. PhytoFront 3, 731–733. doi: 10.1094/PHYTOFR-12-22-0148-A
Erkes, A., Grove, R. P., Žarković, M., Krautwurst, S., Koebnik, R., Morgan, R. D., et al. (2023). Assembling highly repetitive Xanthomonas TALomes using Oxford Nanopore sequencing. BMC Genomics 24, 1–18. doi: 10.1186/s12864-023-09228-1
Erkes, A., Reschke, M., Boch, J., and Grau, J. (2017). Evolution of transcription activator-like effectors in Xanthomonas oryzae. Genome Biol. Evol. 9, 1599–1615. doi: 10.1093/gbe/evx108
Fernandes, C., Martins, L., Teixeira, M., Blom, J., Pothier, J. F., Fonseca, N. A., et al. (2021). Comparative genomics of Xanthomonas euroxanthea and Xanthomonas arboricola pv. Juglandis strains isolated from a single walnut host tree. Microorganisms 9:624. doi: 10.3390/microorganisms9030624
Goettelmann, F., Koebnik, R., Roman-Reyna, V., Studer, B., and Koelliker, R. (2023). High genomic plasticity and unique features of Xanthomonas translucens pv. Graminis revealed through comparative analysis of complete genome sequences. bioRxiv.
Grau, J., Reschke, M., Erkes, A., Streubel, J., Morgan, R. D., Wilson, G. G., et al. (2016). AnnoTALE: bioinformatics tools for identification, annotation and nomenclature of TALEs from Xanthomonas genomic sequences. Sci. Rep. 6:21077. doi: 10.1038/srep21077
Huang, Y. T., Liu, P. Y., and Shih, P. W. (2021). Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 1–17. doi: 10.1186/s13059-021-02282-6
Hunt, M., Silva, N. D., Otto, T. D., Parkhill Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16, 1–10. doi: 10.1186/s13059-015-0849-0
Hutin, M., Pérez-Quintero, A. L., Lopez, C., and Szurek, B. (2015). MorTAL Kombat: the story of defense against TAL effectors through loss-of-susceptibility. Front. Plant Sci. 6:535. doi: 10.3389/fpls.2015.00535
Jacques, M. A., Arlat, M., Boulanger, A., Boureau, T., Carrère, S., Cesbron, S., et al. (2016). Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annu. Rev. Phytopathol. 54, 163–187. doi: 10.1146/annurev-phyto-080615-100147
Ji, Z., Ji, C., Liu, B., Zou, L., Chen, G., and Yang, B. (2016). Interfering TAL effectors of Xanthomonas oryzae neutralize R-gene-mediated plant disease resistance. Nat. Commun. 7:13435. doi: 10.1038/ncomms13435
Kanehisa, M. (2002). “The KEGG database” in ‘In silico’ simulation of biological processes: novartis foundation symposium (Chichester: John Wiley & Sons, Ltd), 91–103.
Kauffman, H., and Rao, P. (1972). Resistant to bacterial leaf blight–India, Rice breeding. Los Banos, CA: International Rice Research Institute.
Kaur, A., Bansal, K., Kumar, S., Sonti, R. V., and Patil, P. B. (2019). Complete genome dynamics of a dominant-lineage strain of Xanthomonas oryzae pv. Oryzae harbouring a novel plasmid encoding a type IV secretion system. Access Microbiol. 1:9. doi: 10.1099/acmi.0.000063
Kaur, A., Bansal, K., and Patil, P. B. (2020). Extensive genomic rearrangements along with distinct mobilome and TALome are associated with extreme pathotypes of a rice pathogen. Genome Biol. Evol. 12, 3951–3956. doi: 10.1093/gbe/evaa025
Kolmogorov, M., Yuan, J., and Lin Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lee, I., Chalita, M., Ha, S. M., Na, S. I., Yoon, S. H., and Chun, J. (2017). ContEst16S: an algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 67, 2053–2057. doi: 10.1099/ijsem.0.001872
Li, T., Li, Y., Ma, X., Dan, X., Huang, X., Li, Q., et al. (2022). Comparative genomic analysis of two Xanthomonas oryzae pv. Oryzae strains isolated from low land and High Mountain paddies in Guangxi, China. Front. Microbiol. 13:867633. doi: 10.3389/fmicb.2022.867633
Lomsadze, A., Gemayel, K., Tang, S., and Borodovsky, M. (2018). Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 28, 1079–1089. doi: 10.1101/gr.230615.117
Lu, W., Pan, L., Zhao, H., Jia, Y., Wang, Y., Yu, X., et al. (2014). Molecular detection of Xanthomonas oryzae pv. Oryzae, Xanthomonas oryzae pv. Oryzicola, and Burkholderia glumae in infected rice seeds and leaves. Crop. J. 2, 398–406. doi: 10.1016/j.cj.2014.06.005
Mew, T. W., Reyes, R. C., and Vera Cruz, C. M. (1989). Screening for bacterial blight resistance in rice. Methods. Phytobacteriol., 338–341.
Midha, S., Bansal, K., Kumar, S., Girija, A. M., Mishra, D., Brahma, K., et al. (2017). Population genomic insights into variation and evolution of Xanthomonas oryzae pv. Oryzae. Sci. Rep. 7:40694. doi: 10.1038/srep40694
Moscou, M., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. doi: 10.1126/science.1178817
Niño-Liu, D. O., Ronald, P. C., and Bogdanove, A. J. (2006). Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol. Plant Pathol. 7, 303–324. doi: 10.1111/j.1364-3703.2006.00344.x
Noël, L. D., Denancé, N., and Szurek, B. (2013). Predicting promoters targeted by TAL effectors in plant genomes: from dream to reality. Front. Plant Sci. 4:333. doi: 10.3389/fpls.2013.00333
Ochiai, H., Inoue, Y., Takeya, M., Sasaki, A., and Kaku, H. (2005). Genome sequence of Xanthomonas oryzae pv. Oryzae suggests contribution of large numbers of effector genes and insertion sequences to its race diversity. Jpn. Agr. Res. Q. 39, 275–287. doi: 10.6090/jarq.39.275
Oliva, R., Ji, C., Atienza-Grande, G., Huguet-Tapia, J. C., Perez-Quintero, A., Li, T., et al. (2019). Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotech. 37, 1344–1350. doi: 10.1038/s41587-019-0267-z
Perez-Quintero, A. L., Lamy, L., Gordon, J. L., Escalon, A., Cunnac, S., Szurek, B., et al. (2015). QueTAL: a suite of tools to classify and compare TAL effectors functionally and phylogenetically. Front. Plant Sci. 6:545–1393. doi: 10.3389/fpls.2015.00545
Perez-Quintero, A. L., Rodriguez-R, L. M., Cuesta-Morrondo, S., Hakalová, E., Betancurt-Anzola, D., Camelo Valera, L. C., et al. (2023). Comparative genomics identifies conserved and variable TAL effectors in African strains of the cotton pathogen Xanthomonas citri pv. Malvacearum. Phytopathology 113, 1387–1393. doi: 10.1094/PHYTO-12-22-0477-SC
Quezado Duval, A. M., Leite, R. P. Jr., Truffi, D., and Camargo, L. E. (2004). Outbreaks of bacterial spot caused by Xanthomonas gardneri on processing tomato in central-West Brazil. Plant Dis. 88, 157–161. doi: 10.1094/PDIS.2004.88.2.157
Quibod, I. L., Perez-Quintero, A., Booher, N. J., Dossa, G. S., Grande, G., and Szurek, B. (2016). Effector diversification contributes to Xanthomonas oryzae pv. Oryzae phenotypic adaptation in a semi-isolated environment. Sci. Rep. 6:34137. doi: 10.1038/srep34137
Read, A. C., Rinaldi, F. C., Hutin, M., He, Y. Q., Triplett, L. R., and Bogdanove, A. J. (2016). Suppression of Xo1-mediated disease resistance in rice by a truncated, non-DNA-binding TAL effector of Xanthomonas oryzae. Front. Plant Sci. 7:1516. doi: 10.3389/fpls.2016.01516
Römer, P., Recht, S., Strauß, T., Elsaesser, J., Schornack, S., Boch, J., et al. (2010). Promoter elements of rice susceptibility genes are bound and activated by specific TAL effectors from the bacterial blight pathogen, Xanthomonas oryzae pv. Oryzae. New Phytol. 187, 1048–1057. doi: 10.1111/j.1469-8137.2010.03217.x
Salzberg, S. L., Sommer, D. D., Schatz, M. C., Phillippy, A. M., Rabinowicz, P. D., Tsuge, S., et al. (2008). Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. Oryzae PXO99A. BMC Genomics 9, 1–16. doi: 10.1186/1471-2164-9-204
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shafin, K., Pesout, T., Lorig-Roach, R., Haukness, M., Olsen, H. E., Bosworth, C., et al. (2020). Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotech. 38, 1044–1053. doi: 10.1038/s41587-020-0503-6
Shah, S. M. A., Ahsan, R., Liu, L., Li, Y., Wang, Q., Wang, Y., et al. (2025). TALome and phenotypic analysis of Pakistani Xanthomonas oryzae pv. Oryzae population revealed novel virulent TALEs contributing to bacterial blight of rice. Phytopathol. Res. 7:5. doi: 10.1186/s42483-024-00292-3
Shahzadi, N., Akhter, M., Ali, M., and Misbah, R. (2018). Economic aspects of basmati rice in Pakistan. J. Rice. Res. 6, 192–197. doi: 10.4172/2375-4338.1000192
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Streubel, J., Pesce, C., Hutin, M., Koebnik, R., Boch, J., and Szurek, B. (2013). Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. Oryzae. New Phytol. 200, 808–819. doi: 10.1111/nph.12411
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC. Bioinformatics 4, 1–14. doi: 10.1186/1471-2105-4-41
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tian, D., Wang, J., Zeng, X., Gu, K., Qiu, C., and Yang, X. (2014). The rice TAL effector–dependent resistance protein Xa10 triggers cell death and calcium depletion in the endoplasmic reticulum. Plant Cell 26, 497–515. doi: 10.1105/tpc.113.119255
Timilsina, S., Potnis, N., Newberry, E. A., Liyanapathiranage, P., Iruegas-Bocardo, F., White, F. F., et al. (2020). Xanthomonas diversity, virulence and plant–pathogen interactions. Nat. Rev. Microbiol. 18, 415–427. doi: 10.1038/s41579-020-0361-8
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 1–15. doi: 10.1186/s13059-014-0524-x
Ullah, I., Ali, H., Mahmood, T., Khan, M. N., Haris, M., Shah, H., et al. (2022). Pyramiding of four broad spectrum bacterial blight resistance genes in cross breeds of basmati rice. Plan. Theory 12:46. doi: 10.3390/plants12010046
USDA (2024). Grain and feed update, Report Number: PK2024-0022. Global Agriculture Information Network (GAIN).
Van den Ackerveken, G., Marois, E., and Bonas, U. (1996). Recognition of the bacterial avirulence protein AvrBs3 occurs inside the host plant cell. Cell 87, 1307–1316. doi: 10.1016/S0092-8674(00)81825-5
Vandecraen, J., Chandler, M., Aertsen, A., and Van Houdt, R. (2017). The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 43, 709–730. doi: 10.1080/1040841X.2017.1303661
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Waheed, M. A., Inamullah, A. H., Sirajuddin, A. H., Khan, A. Q., and Khan, A. (2009). Evaluation of rice genotypes for resistance against bacterial leaf blight. Pak. J. Bot. 41, 329–335.
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 1–10. doi: 10.1186/s13059-019-1727-y
Wu, L. B., Eom, J. S., Isoda, R., Li, C., Char, S. N., Luo, D., et al. (2022). OsSWEET11b, a potential sixth leaf blight susceptibility gene involved in sugar transport-dependent male fertility. New Phytol. 234, 975–989. doi: 10.1111/nph.18054
Keywords: rice, bacterial leaf blight, TAL effectors, RVDs, Oxford Nanopore Sequencing, type III secretion system
Citation: Ejaz K, Zakria M, Zhang P, Huguet Tapia J, Arif M, White F and Yasmin S (2025) Comparative genomics using long-read sequencing identifies nearly identical TAL effector regions in two Xanthomonas oryzae pv. Oryzae isolates collected from the basmati rice-growing region of Pakistan. Front. Microbiol. 16:1560969. doi: 10.3389/fmicb.2025.1560969
Edited by:
Febri Doni, Padjadjaran University, IndonesiaReviewed by:
Kumrop Ratanasut, Naresuan University, ThailandKalpana Singh, Guru Angad Dev Veterinary and Animal Sciences University, India
Copyright © 2025 Ejaz, Zakria, Zhang, Huguet Tapia, Arif White and Yasmin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khansa Ejaz, bWlzc2VqYXo0NDNAZ21haWwuY29tSumera Yasmin, c3VtZXJhaW1yYW4yMDEyQGdtYWlsLmNvbQ==