 Torunn Gresdal Rønning
Torunn Gresdal Rønning Hege Enger1
Hege Enger1 Jan Egil Afset
Jan Egil Afset Christina Gabrielsen Ås
Christina Gabrielsen Ås- 1The Norwegian MRSA Reference Laboratory, Department of Medical Microbiology, Clinic of Laboratory Medicine, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
- 2Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway
- 3Department of Medical Microbiology, Clinic of Laboratory Medicine, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
Infections caused by multidrug-resistant (MDR) bacteria are recognized as a critical One Health concern which poses a significant threat to public health, leading to increased morbidity and mortality across both high- and low-income countries. In this study, we investigated the epidemiology and molecular mechanisms of multidrug-resistant methicillin-resistant Staphylococcus aureus (MDR-MRSA) strains identified in Norway from 2008 to 2020, in order to gain a better understanding of the evolution and dissemination of multidrug resistance in S. aureus. A total of 452 MDR-MRSA strains isolated from 429 individuals were analyzed from a dataset of 23,412 MRSA strains. Methods included epidemiological characterization, antimicrobial susceptibility testing (AST), and genetic analysis of a selection of strains using nanopore sequencing to identify antimicrobial resistance (AMR) genes and mutations, as well as their location on plasmids, SCCmec and other mobile genetic elements (MGEs). The study revealed an overall increasing trend in MDR-MRSA strains, with healthcare-associated strains being more prevalent among MDR-MRSA compared to the overall MRSA population. Significant heterogeneity in spa-types and clonal complexes exhibiting multidrug resistance was observed, with high resistance rates against multiple antibiotic groups, particularly erythromycin, ciprofloxacin/norfloxacin, tetracycline, gentamicin, and clindamycin in addition to cefoxitin. The predominant MDR-MRSA clones included t1476/CC8, t127/CC1, t189/CC188, and t030, t037/CC239. Among these, MRSA t1476/CC8 showed an upward trend toward the conclusion of the study period, indicating the emergence of a MDR-MRSA clone. A broad range of AMR genes and mutations were detected, linked to a wide variety of MGEs, highlighting the complex mechanisms of resistance development and dissemination within the MRSA population. This study highlights the rising challenge posed by MDR-MRSA strains, and reveals the multifactorial nature of AMR in S. aureus, thus emphasizing the importance of continued surveillance, antibiotic stewardship and infection control measures, as well as global cooperation, in order to combat the spread of these multidrug-resistant pathogens.
Introduction
Staphylococcus aureus colonizes the skin and mucosal surfaces of about 30% of the human population (Howden et al., 2023). This bacterium is however also an important human pathogen, causing a wide range of infections ranging from mild skin and soft-tissue infections to severe and invasive disease, such as endocarditis, osteomyelitis, bloodstream infection and sepsis (Cheung et al., 2021).
S. aureus is furthermore a bacterial pathogen which has the capacity to incorporate a wide variety of mobile genetic elements (MGEs) making it able to adapt to different hosts and environments (Richardson et al., 2018). These MGEs, which include plasmids, transposons, bacteriophages and staphylococcal cassette chromosome (SCC) elements, can facilitate the horizontal transfer of genes that encode important virulence factors as well as antibiotic resistance determinants providing resistance against almost all the clinically relevant groups of antibiotics.
Plasmids play a pivotal role in horizontal gene transfer, significantly contributing to the dissemination of antimicrobial resistance (AMR) among bacteria (Zhao et al., 2023). Well-known examples in S. aureus include the widely disseminated blaZ-encoding plasmids that provide resistance to penicillins (Mores et al., 2021). Furthermore, the acquisition of the staphylococcal cassette chromosome mec (SCCmec) carrying the mecA (or mecC) gene provides resistance to all beta-lactam antibiotics defining S. aureus as methicillin-resistant (MRSA) (Ito et al., 2014). SCCmec can furthermore contain additional antibiotic resistance- and virulence genes contributing to the adaptability and pathogenicity of MRSA strains (Ito et al., 1999; Katayama et al., 2001). Bacteriophages, or phages, are prevalent in the genome of most bacteria, often introducing additional genes that enhance virulence and antibiotic resistance (Leinweber et al., 2021). In human-adapted S. aureus strains, Sa3int phages are particularly significant as they carry genes that help bacteria evade the immune system, thus increasing their virulence (Leinweber et al., 2021). These examples illustrate the importance of horizontal gene transfer and MGEs in the dissemination of AMR and the evolution of bacterial pathogenicity in S. aureus.
Infections caused by multidrug-resistant (MDR) bacteria, including MRSA, are recognized as a critical One Health concern which poses a significant threat to public health (Aslam et al., 2021). These infections result in increased mortality and morbidity across both high- and low-income countries (GBD 2021 Antimicrobial Resistance Collaborators, 2024). To better understand the mechanisms driving the spread of multidrug resistance in MRSA, this study aimed to examine the epidemiology and molecular mechanisms of multidrug-resistant MRSA strains identified in Norway in the period 2008-2020.
Results
Epidemiological characteristics of multidrug-resistant MRSA in Norway 2008-2020
A subset of 452 MDR-MRSA strains isolated from 429 persons were included in the study, from a total of 23,412 MRSA strains (1.9%) (Table 1) in the study period from 2008 through 2020. Although the number of MDR-MRSA strains per year was low (ranging from 28 to 73) and with some fluctuations, we observed an overall increasing trend (Figure 1), except for the COVID-19 pandemic year 2020. This coincided with an overall increase in the total number of MRSA strains in Norway in the same period.
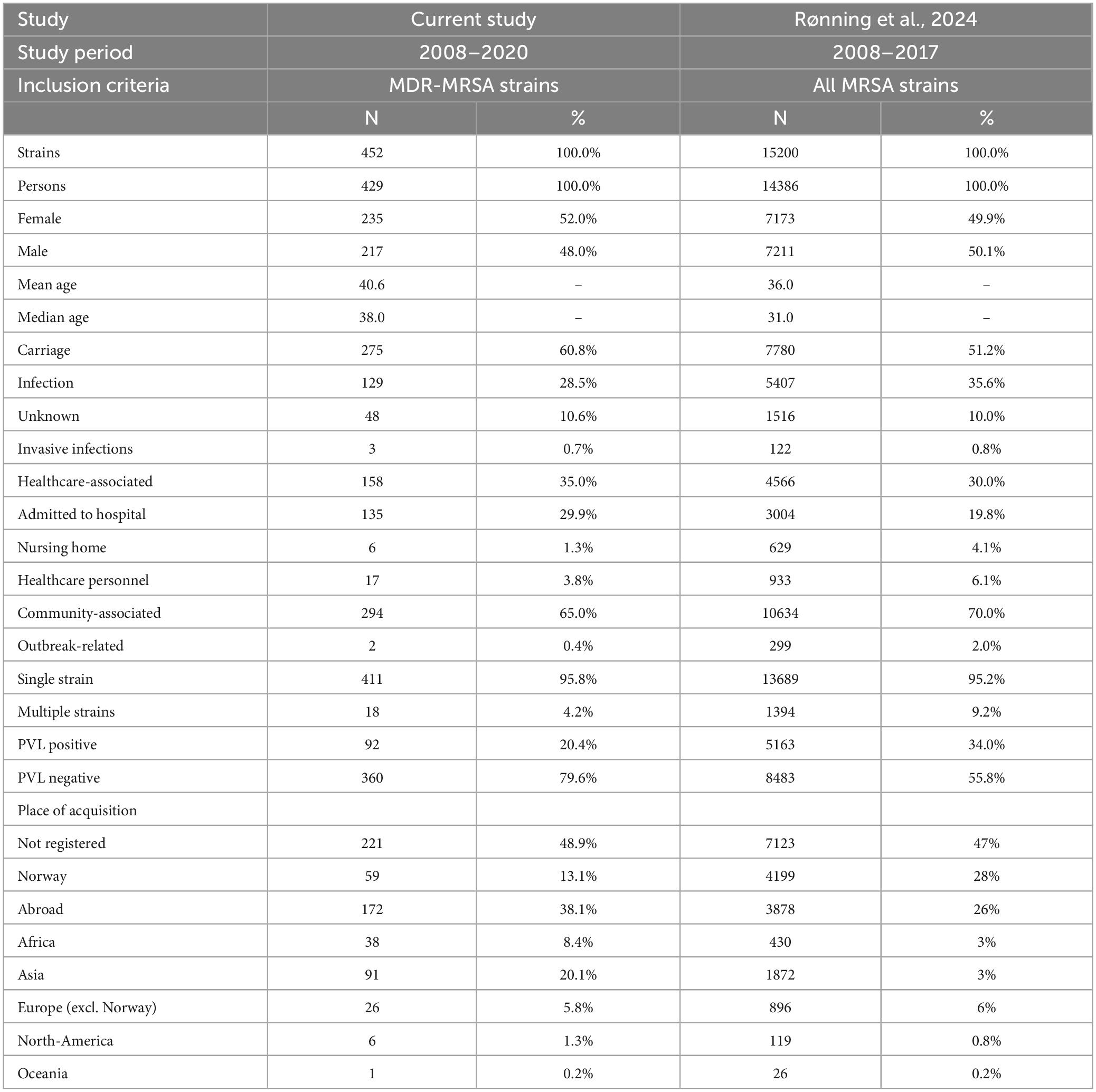
Table 1. Epidemiological and molecular characteristics of the strains included in the study, compared to data from Rønning et al. (2024).
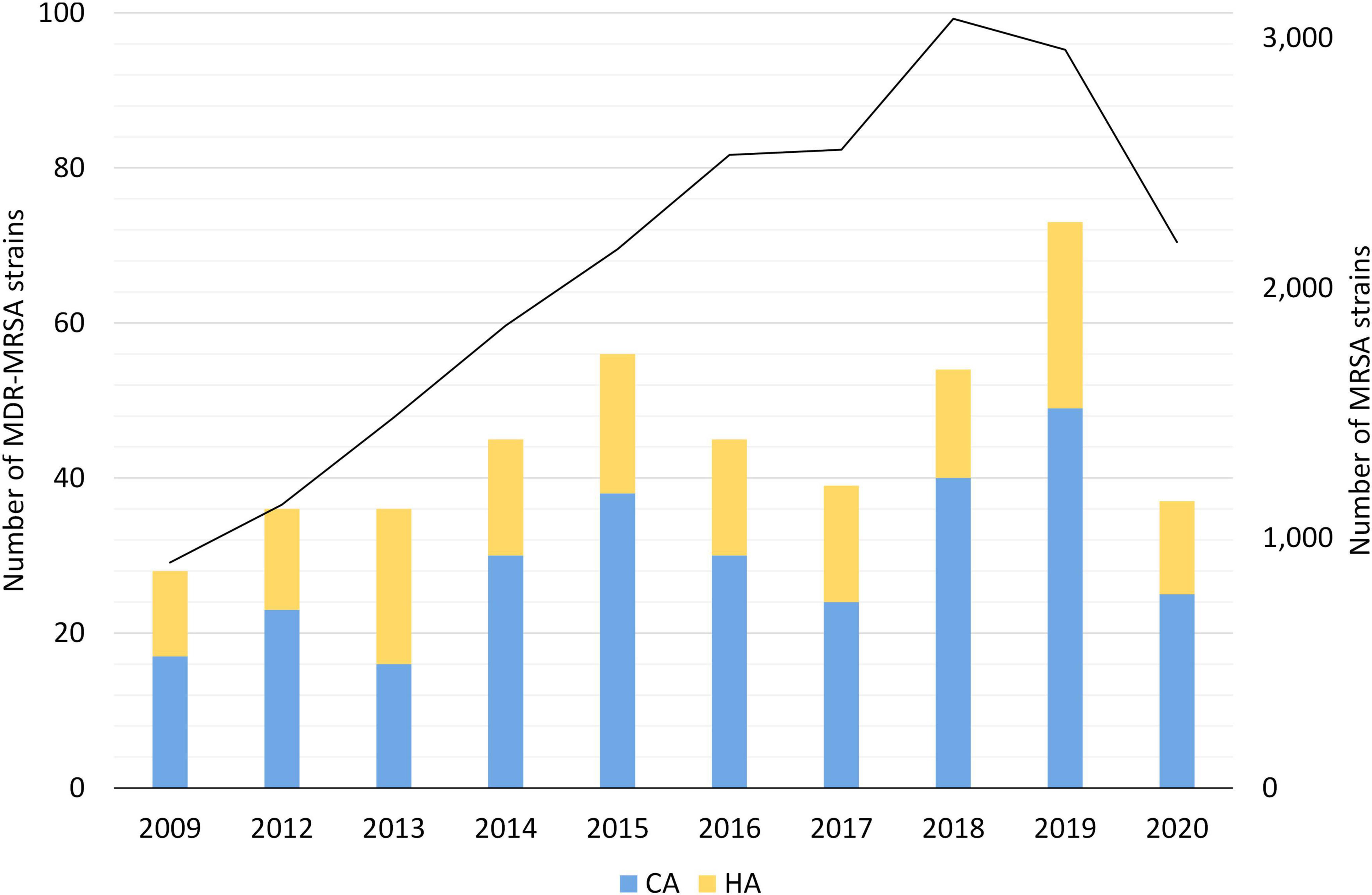
Figure 1. Yearly number of MDR-MRSA strains in Norway in 2008-2020. Strains are classified as Healthcare-Associated (HA) or Community-Associated (CA) as indicated by the colored key and scale on the left axis. Total number of MRSA strains per year indicated by black line, with scale on the right axis.
In total, 275 (60.8%) of the MDR-MRSA strains were classified as carriage strains and 129 (28.5%) were classified as infection strains. Of the infection strains, the majority were associated with wounds (74.4%), abscesses (14.7%) or pus (7.8%). In total, only three strains were from invasive infections (0.7%). No information on sampling site was available for 48 (10.6%) of the strains. In total, 158 strains (35.0%) were classified as healthcare-associated (HA), and 135 strains (29.9%) were from patients admitted to hospital. The remaining 294 strains (65.0%) were classified as community-associated (CA). Only two of the MDR-MRSA strains (0.4%) were registered as related to outbreaks.
According to the registered place of acquisition for the MDR-MRSA strains, 13.1% were acquired in Norway, and 38.1% were acquired abroad, while no information about place of acquisition was available for 48.9% of the strains. The strains that were acquired abroad, were mainly acquired in Asia (20.1%), followed by Africa (8.4%) and Europe excluding Norway (5.8%).
The overall sex distribution of the MDR-MRSA strains was even, with 235 (52.0%) from females, and 217 (48.0%) from males. The mean age of persons was 40.6 years, with a median age of 38 years. More than one strain was isolated from 18 individuals (4.2%). The strains were isolated at varying time intervals, ranging from 1 to 6 years between each collection. All strains exhibited consistent spa-types (or in one case clonal complex) between isolate one and isolates two or three from the same individual.
All MDR-MRSA strains in this study showed phenotypic resistance to cefoxitin and contained the mecA-gene, while 20.4% (n = 92) of strains contained the virulence factor and epidemiological marker Panton-Valentine leucocidin (PVL).
Successful MDR-MRSA clones and phenotypic antimicrobial susceptibility profiles
Of the 452 MDR-MRSA strains included in the study, 361 (79.9%) showed antibiotic resistance toward five different antibiotic groups, while 70 (15.5%) demonstrated resistance against six antibiotic groups. Furthermore, 17 strains (3.8%) displayed resistance against seven antibiotic groups, and four strains (0.9%) showed antibiotic resistance against eight antibiotic groups (Figure 2).
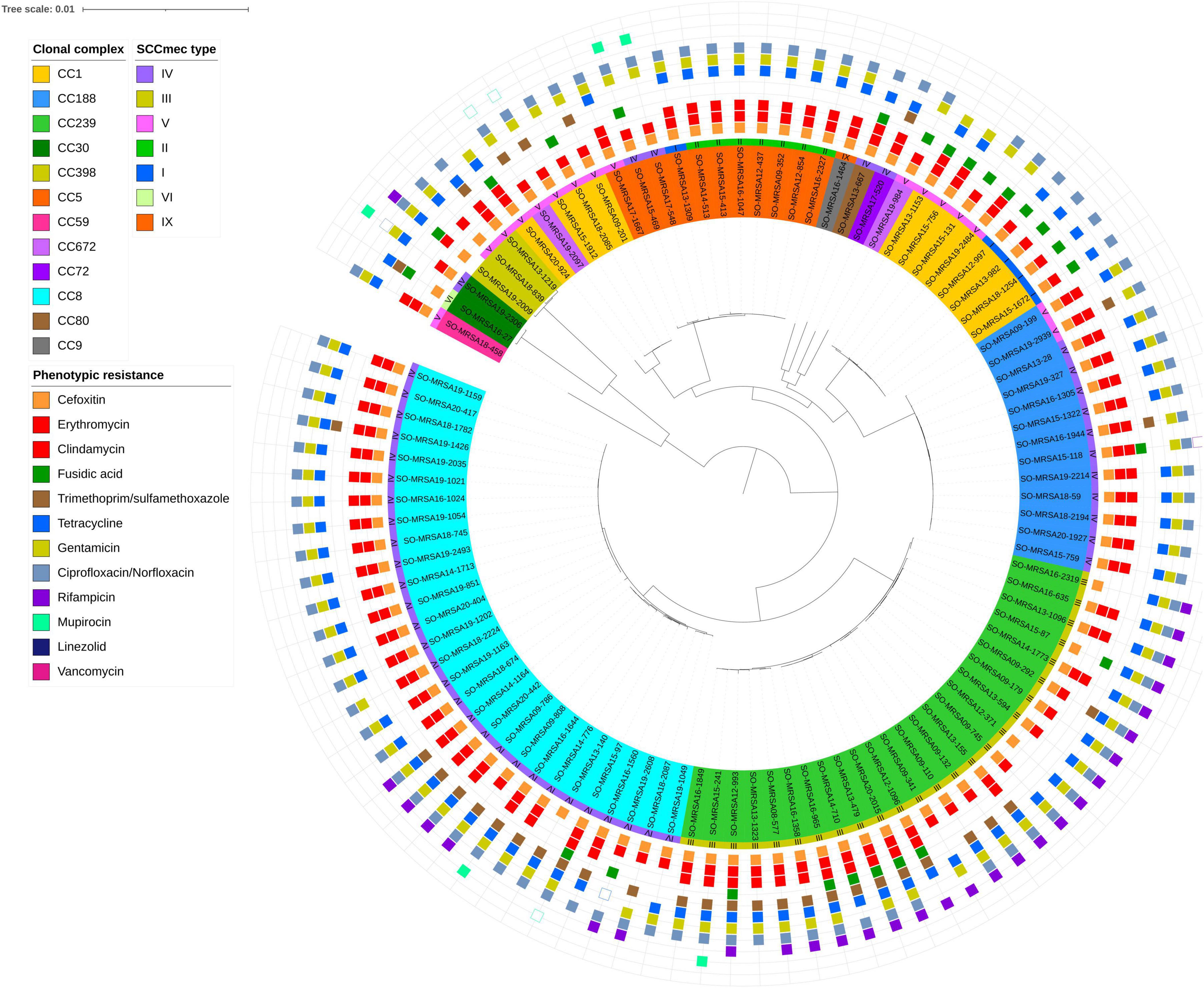
Figure 2. Core genome phylogeny of whole genome sequenced MDR-MRSA strains (n = 101). The nodes within the tree are assigned distinct colors based on clonal complex and SCCmec type. Phenotypic resistance profiles based on AST are visually represented through colored boxes. Box with fill indicates resistant, while outlined box indicates intermediate resistance (2008-2018) or susceptible increased exposure (2019-2020).
For strains showing antibiotic resistance against five or six antibiotic groups, we observed a very heterogeneous collection of spa-types. Strains resistant to five antibiotic groups belonged to more than 50 distinct spa-types of 22 different CCs, while strains resistant to six antibiotic groups belonged to 22 spa-types of 9 different CCs. Conversely, in strains showing resistance against seven or eight antibiotic groups, a very limited number of spa-types were observed. These included spa-types t008, t030, t034, t037, t064, t1476, and t451, belonging to clonal complexes 8, 239 and 398.
Overall, phenotypic antibiotic susceptibility testing revealed almost universal resistance toward erythromycin (93.1%) and ciprofloxacin/norfloxacin (92.9%) in addition to cefoxitine (100.0%) (Figure 3). High levels of resistance were also observed to tetracycline (83.9%), gentamicin (81.7%), and clindamycin (69.3% total). 175/452 (38.7%) of the strains showed constitutive resistance against clindamycin, while 140 (31.0%) showed inducible resistance against clindamycin. Moderate to low levels of resistance were observed for fusidic acid (27.8%), trimethoprim-sulfamethoxazole (19.4%), rifampicin (13.2%) and mupirocin (10.0%). No isolates were resistant toward linezolid (0.0%) or vancomycin (0.0%).
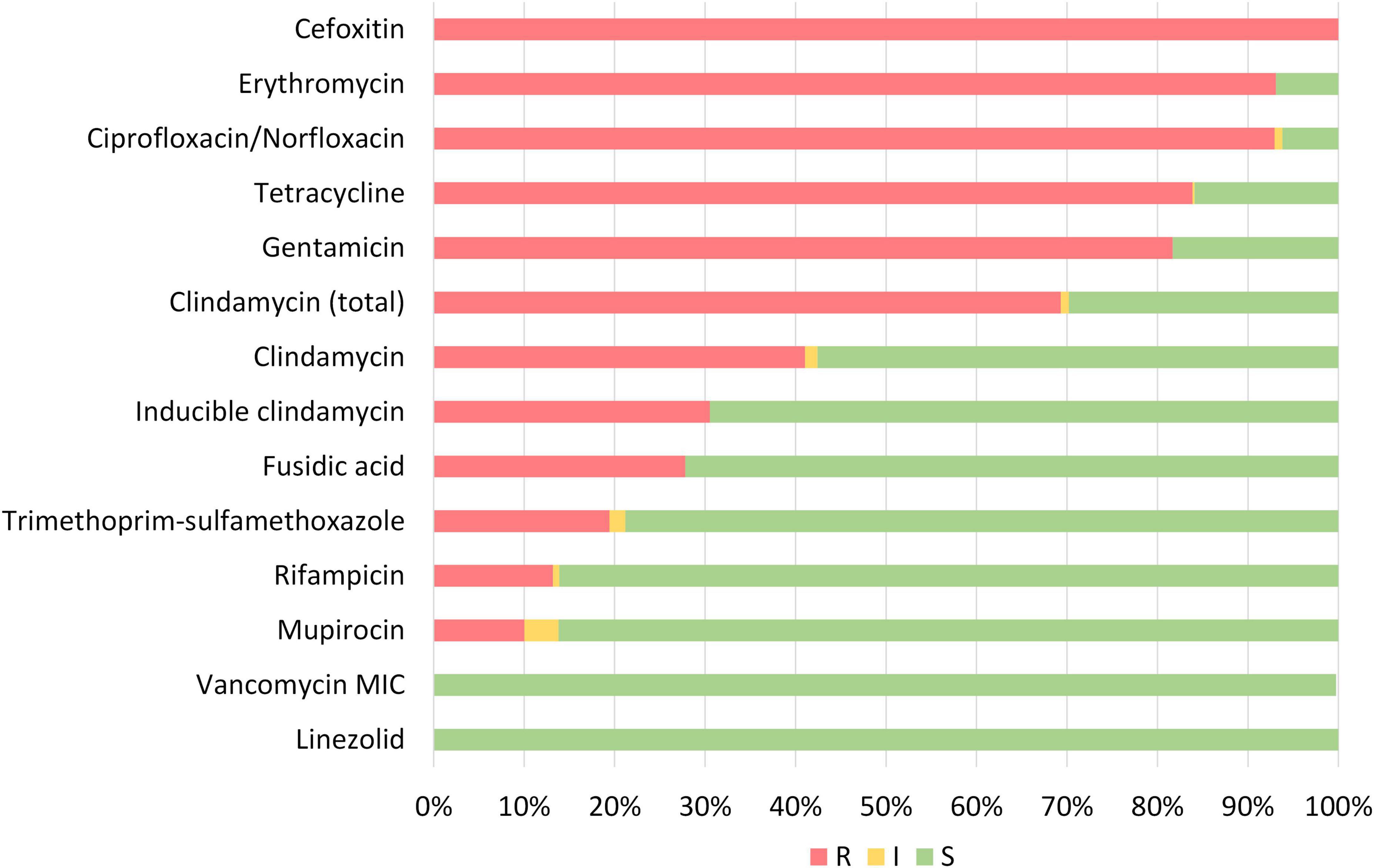
Figure 3. Phenotypic susceptibility of all MDR-MRSA strains categorized as resistant (R), intermediate/susceptible increased exposure (I) or susceptible (S) toward tested antibiotics.
Of the most successful MDR-MRSA spa-types over the course of the study period were t127/CC1, t189/CC188, t030, t037/CC239, and t1476/CC8 (Figure 4). Collectively, these spa-types accounted for 45.1% (204/452) of the strains included in this study.
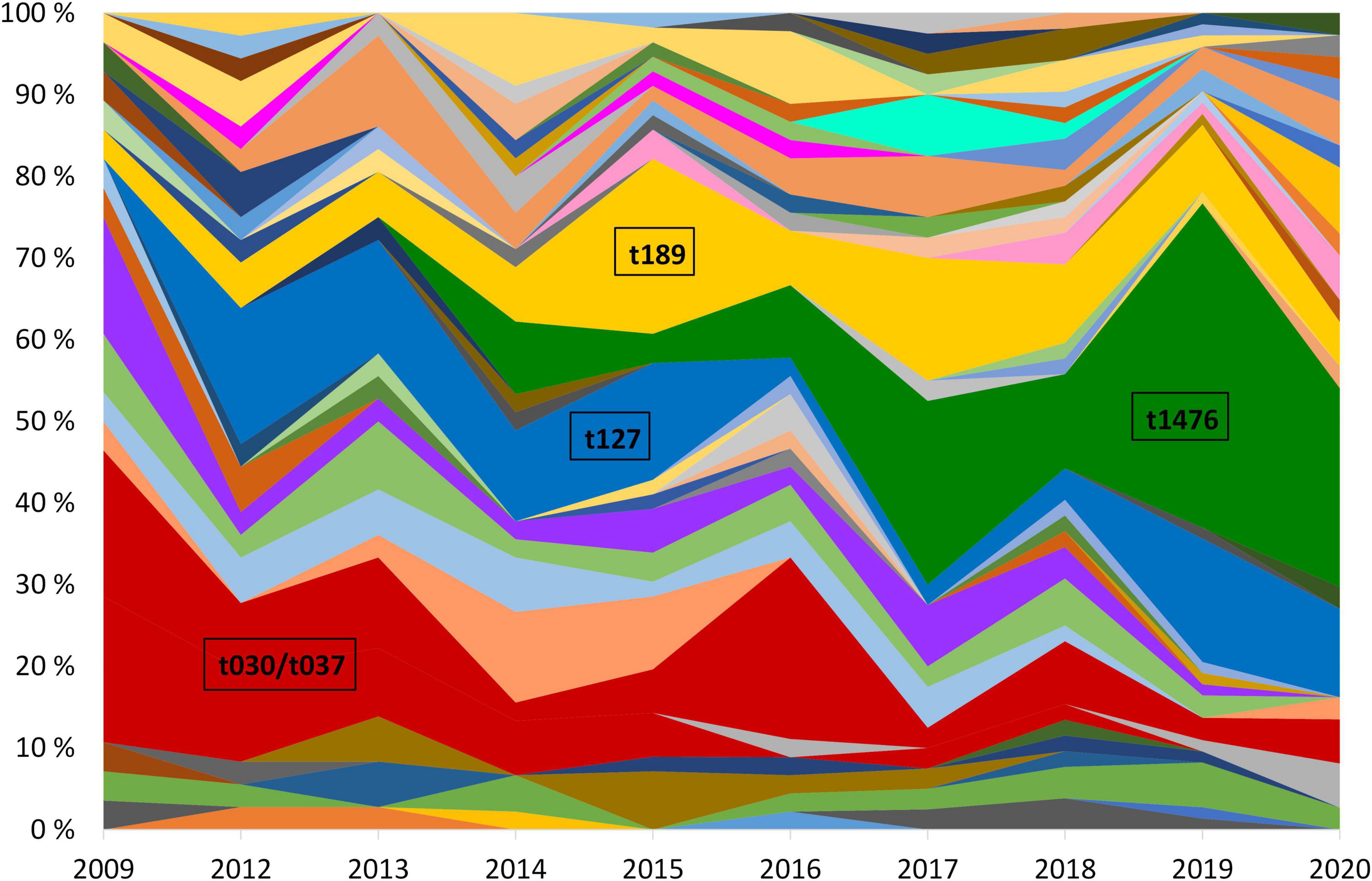
Figure 4. Yearly relative distribution of MDR-MRSA spa-types (> 5 per year) in the period 2009-2020. The years 2010 and 2011 are excluded due to missing data. Major spa-types are highlighted.
MDR-MRSA t1476/CC8 (n = 63, 13.9%) was the most successful clone in the study period (Figure 4). The number of strains which belonged to this genotype increased considerably from 2008 to 2019 (from 0 strains in 2008 to 29 strains in 2019). In this group a majority of cases were from females (65.1%), and most of the strains were from carriage (76.2%). Based on country of acquisition, 38.1% of the strains were associated with countries in Sub-Saharan Africa, while 42.9% had no record of acquisition (Figure 5). All the strains were resistant against beta-lactams, aminoglycosides, fluoroquinolones and tetracyclines. The majority were additionally resistant against macrolides (n = 62, 98.4%) (Table 2).
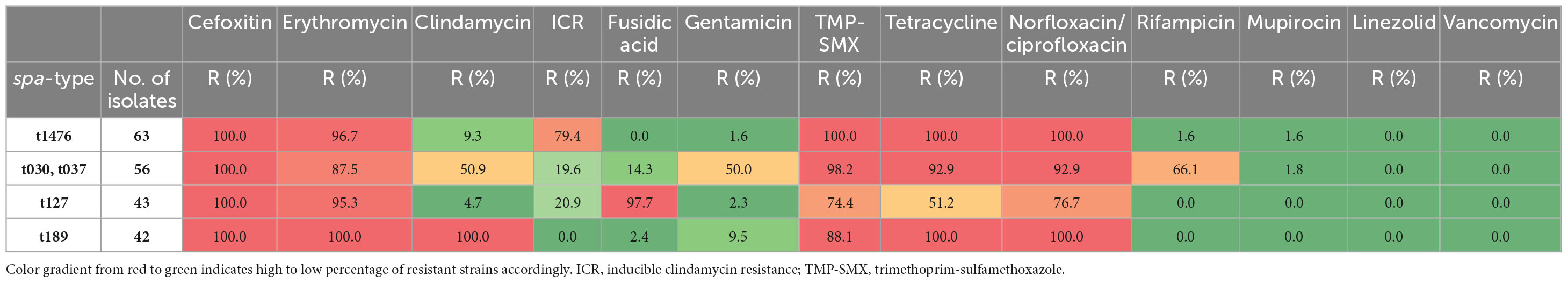
Table 2. Proportion of strains resistant to the tested antibiotics, for the four most prevalent MDR-MRSA clones.
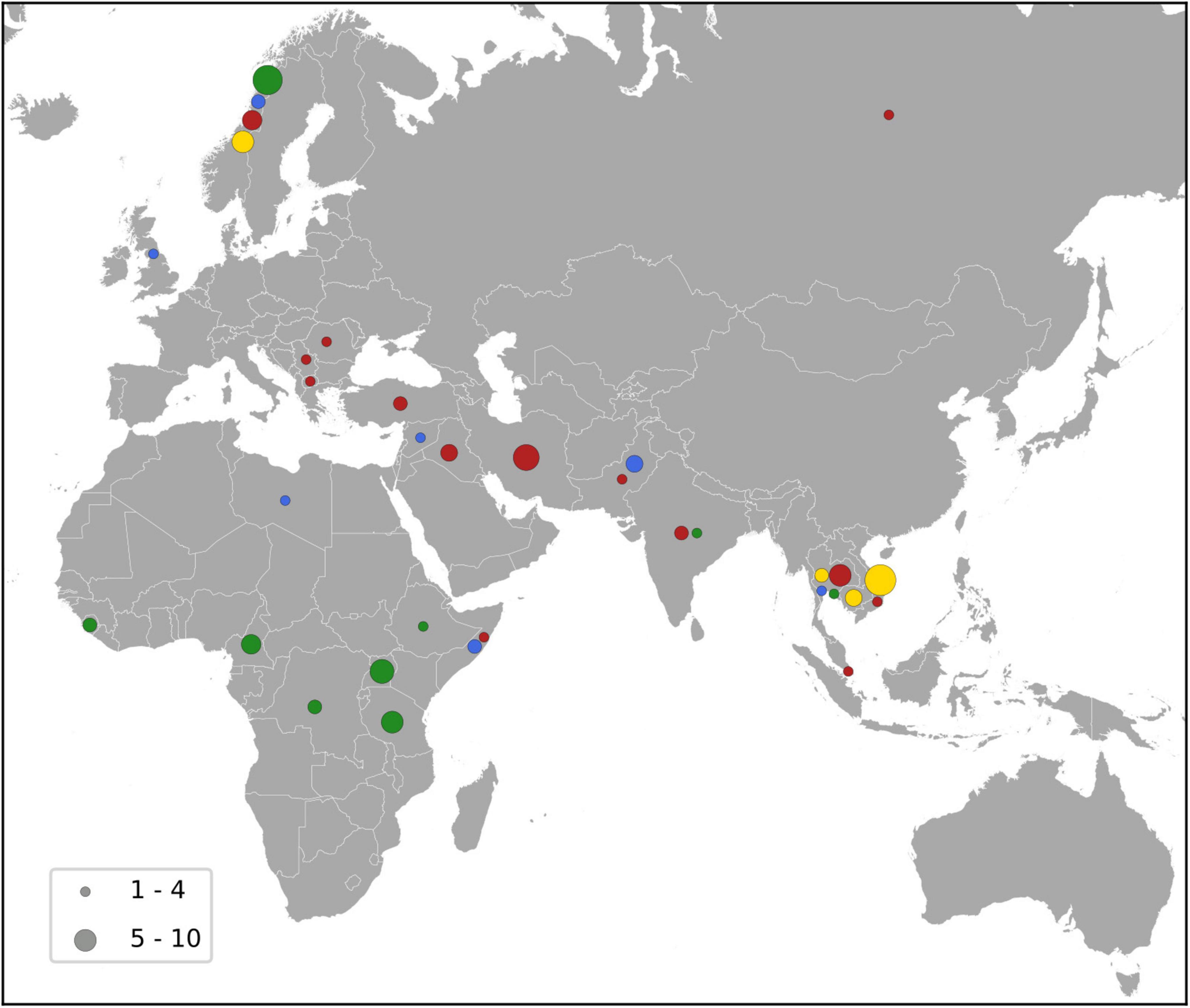
Figure 5. Map showing the country of acquisition for the major MDR-MRSA clones. The major clones include t1476 (green), t127 (blue), t189 (yellow) and t030/t037 (red), and the number of strains is indicated by the size of the circles as shown by the key.
MDR-MRSA t127/CC1 was the second most frequent spa-type in this study, accounting for 43 out of 452 strains (9.5%). All of these strains were resistant against five antibiotic groups (Table 2), the most common profile being resistance to beta-lactams, MLS, tetracyclines, fusidanes and fluoroquinolones (n = 20, 46.5%). The sex distribution was 48.8% female and 51.1% male, and the proportion of carriage (51.1%) was similar to infections (48.8%). For most of the cases from whom MDR-MRSA t127/CC1 strains were isolated, there was no record of place of acquisition (79.0%). Known countries of acquisition, however, included European as well as Asian and African countries (Figure 5).
MDR-MRSA t189/CC188 was the third most frequent spa-type in this study, accounting for 42 out of 452 strains (9.3%). These strains showed resistance against five antibiotic groups, the most common profile included resistance to beta-lactams, MLS, aminoglycosides, tetracyclines and fluoroquinolones (n = 37, 88.1%) (Table 2). The proportion of female cases was high in this group (69.0%), and carriage (57.1%) was more frequent than infection (40.5%). For 35.8% of strains, the place of acquisition were Southeast Asian countries (Figure 5).
MDR-MRSA t037/CC239 and t030/CC239 were the fourth (n = 36, 8.0%) and fifth (n = 20, 4.4%) most frequent spa-types in this study. These were most frequent at the start of the study period, while the number of strains declined in more recent years. These two spa-types belong to the same clonal complex (CC239) and share similar phenotypic antibiotic resistance patterns. A majority of strains in the two groups were resistant toward tetracyclines (98.2%), aminoglycosides (92.9%), MLS (87.5%), fluoroquinolones (92.9%), TM/S (50.0%), and ansamycins (66.1%) (Table 2). Notably, two strains (3.6%), both t037, were resistant against all the antibiotics tested except for linezolid and vancomycin. In contrast to the other successful clones, there were more men (64.3%) than women (42.9%) in this group, and a majority of strains were from carriage (62.5%). 42.9% of the strains were registered as acquired in Asian countries (Northern, Western, Southern, and South-eastern subregions) (Figure 5).
Genotypic resistance determinants associated with different groups of antibiotics
For the sequenced MDR-MRSA strains (n = 101), the presence of AMR-associated genes and mutations were predicted using the AMRFinder Plus tool and database. In total, 39 different AMR genes and 31 different AMR mutations were identified, with a median of 10 (range 7-15) different genes and 3 (range 1-10) mutations per strain. In accordance with phenotypic resistance data, the most common AMR determinants detected provided resistance against beta-lactams, MLS, fluoroqinolones, tetracyclines and aminoglycosides (Figure 2 and Table 3). The specific resistance determinants associated with each group of antibiotics are more closely described in the following subsections. Based on both phenotypic and genotypic results, there were detected no strains that could be regarded as XDR-MRSA (Magiorakos et al., 2012) in this strain collection.
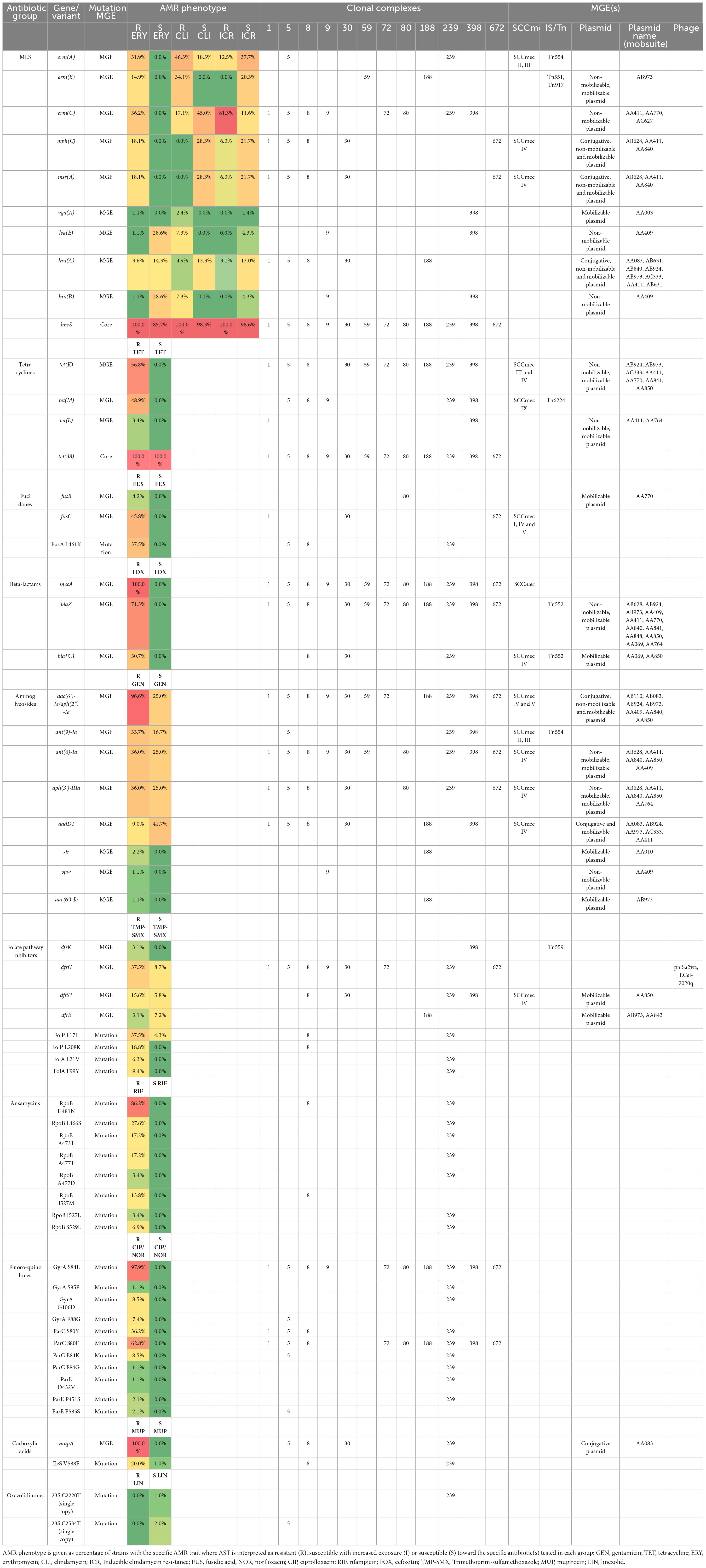
Table 3. AMR traits detected in MDR-MRSA strains in Norway, and association with clonal complex and detected MGEs.
Beta-lactam resistance
All MDR-MRSA strains included in this study (n = 452) harbored the mecA-gene. In addition, we detected penicillin resistance genes in 98.0% of the sequenced strains (Supplementary Table 1). Of these, 71.3% harbored the blaZ-gene, and 26.7% contained blaPC1. Four percent of strains had mecA, blaZ, and blaPC1 genes, while two strains (2%) solely harbored the mecA-gene.
Macrolide and lincosamide resistance
Almost all (95.0%) of the sequenced MDR-MRSA strains were phenotypically resistant against erythromycin and clindamycin (Supplementary Table 1). This was, however, linked to different genes and gene combinations. The most common gene was erm(C), detected in 35.4% of MLS-resistant strains (Table 3 and Figure 6), followed by erm(A), detected in 31.3% of MLS-resistant strains. The different genetic profiles were not associated with distinct MLS phenotypic profiles, but rather included erythromycin resistance with either constitutive or inducible resistance or susceptibility to clindamycin. The erm(B) gene was found in 14 strains (Table 3 and Supplementary Table 1), and all of these had a phenotypic profile of erythromycin resistance and constitutive clindamycin resistance.
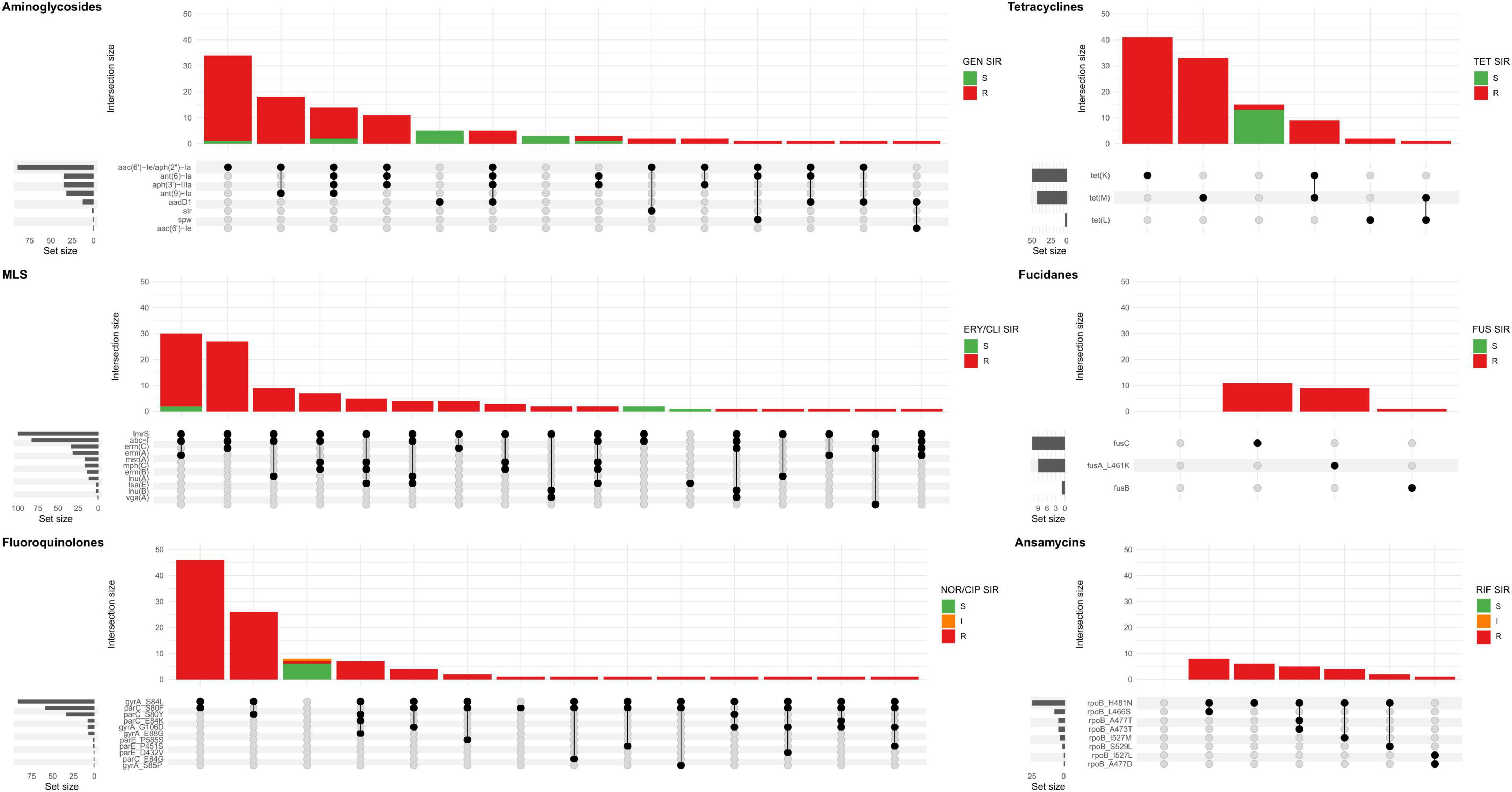
Figure 6. Upset plot showing all combinations AMR genes and mutations (set) associated with the major antibiotic groups in sequenced MDR-MRSA strains. The major antibiotic groups include aminoglycosides, tetracyclines, macrolide, lincosamide and streptogramin (MLS), fucidanes, fluoroquinolones, and ansamycins. Strains (intersection) are colored according AST, interpreted as resistant (R), susceptible with increased exposure (I) or susceptible (S) toward the specific antibiotic(s) tested in each group: GEN, gentamicin; TET, tetracycline; ERY, erythromycin; CLI, clindamycin; FUS, fusidic acid; NOR, norfloxacin; CIP, ciprofloxacin; RIF, rifampicin.
The combination of mph(C) and mrs(A) was present in the erythromycin resistant and inducible clindamycin resistant-profile, and the only erythromycin resistant profile (Figure 6). The rarest genes associated with MLS resistance were vga(A) and the combination of lsa(E) and lnu(B), with the phenotypic profile erythromycin susceptible and constitutive clindamycin resistance. The gene lmrS was present in both erythromycin resistant and susceptible strains, and did not appear to provide phenotypic resistance to erythromycin at levels sufficient for detection by the methods used in this study.
Ciprofloxacin/norfloxacin resistance
Of the sequenced MDR-MRSA strains, 94 (93.1%) exhibited phenotypic resistance to ciprofloxacin or norfloxacin. The most frequent mutations found associated with quinolone resistance were GyrA S84L (91 strains, 90.1%), ParC S80F (58 strains, 57.4%), and ParC S80Y (34 strains, 33.7%) (Table 3 and Supplementary Table 1). Combinations of quinolone-conferring mutations were found in 92 strains (91.1%). The most frequent combinations of mutations were GyrA S84L and ParC S80F (found in 46 strains, 45.5%) (Figure 6) and GyrA S84L and ParC S80Y (found in 26 strains, 25.7%). Mutations in the chromosomal gyrA, parC and parE genes were found in all quinolone-resistant strains, among multiple spa-types of different clonal complexes. This thus appears to be a quite common adaptation in the general MDR-MRSA population to acquire quinolone resistance.
Tetracycline resistance
88 (87.1%) of sequenced MDR-MRSA strains exhibited phenotypic resistance to tetracycline, having either the tet(M) (42.6%), tet(K) (49.5%) or in a few cases the tet(L) (3.0%) genes in different combinations (Table 3, Supplementary Table 1 and Figure 6). In two strains we did not detect any gene(s) likely causing tetracycline resistance. Previously described chromosomal mutations associated with tetracycline resistance in MepA (N369Y), RpsJ (Y58D) or 16S rDNA, were not identified in any of the strains. The tet(38)-gene was detected in all the sequenced strains in this study, but did not appear to cause phenotypic tetracycline resistance at levels sufficient for detection by the methods used in this study (Table 3).
Gentamicin resistance
Of the sequenced MDR-MRSA strains, 89 (88.1%) exhibited phenotypic resistance to gentamicin. The most common gene encoding aminoglycoside resistance were aac(6’)-Ie/aph(2”)-Ia, found in 86 of the 89 (96.9%) strains (Table 3 and Supplementary Table 1). 35 strains (34.7%) had the aph(3’)-IIIa-gene encoding amikacin/kanamycin resistance, 35 strains (34.7%) had ant(6)-Ia-gene encoding streptomycin resistance and 32 (31.7%) had ant(9)-Ia-gene encoding spectinomycin resistance. 13 strains (12.9%) had aadD1-gene encoding kanamycin/tobramycin resistance. Single strains had the aac(6’)-Ie-gene encoding amikacin/kanamycin/tobramycin-resistance and the aminoglycoside spw-gene, and three strains had the str-gene encoding streptomycin. The most frequently found combination of aminoglycoside-genes was aac(6’)-Ie/aph(2”)-Ia together with the ant(9)-Ia-gene, found in 18 strains (17.8%) (Figure 6).
Trimethoprim-sulfamethoxazole resistance
Overall, 34 (33.7%) of sequenced MDR-MRSA strains exhibited phenotypic resistance to TMP-SMX. As phenotypic testing is performed with the TMP-SMX combination drug, and resistance to both components separately is required for resistance, the detection and interpretation of genes and mutations contributing to TMP-SMX resistance can be complex. While we observed full concordance between phenotypic susceptibility and lack of previously described genes/mutations encoding TMP-SMX resistance, there was only a 23/34 (67.6%) concordance between phenotypic resistance and having a combination of genes/mutations previously reported to confer phenotypic TMP-SMX resistance (Table 3 and Supplementary Table 1). In the concordant cases, the drfG, dfrS1, dfrK or dfrE genes encoding trimethoprim resistance were detected, either in a single, two or three copies, together with the F17L and E208K-mutations in FolP providing sulfametoxazole resistance (Supplementary Table 1).
Rifampicin resistance
Overall, 26 (25.7%) of sequenced MDR-MRSA strains exhibited phenotypic resistance to rifampicin. Chromosomal mutations in rpoB conferring rifampicin resistance described by Guerillot et al. were found in 26 of the sequenced strains (Guérillot et al., 2018). The most common mutations were RpoB H481N alone or in combination (Table 3, Figure 6, and Supplementary Table 1). The chromosomal rpoB-mutations conferring rifampicin resistance seemed to be clonal, and were found only in strains belonging to CC8 and CC239, which included the most resistant strains in this study with resistance toward seven or eight antibiotic groups. The H481N mutation has furthermore been reported to promote the emergence of a subpopulation of small colony variants with reduced susceptibility to vancomycin and daptomycin. Although not investigated here, this raises an additional concern regarding the use of rifampicin treatment and the possible effect on emergence of MDR-MRSA strains.
Fusidic acid resistance
Of the sequenced MDR-MRSA strains, 25 (24.8%) exhibited phenotypic resistance to fusidic acid. The most common gene that was associated with fusidic acid resistance was fusC, found in 11 strains (Table 3 and Supplementary Table 1). The fusB-gene was present in one strain (t044, CC80). The fusA L461K mutation was found in nine strains, were spa-type t037 was the most frequent spa-type. For three strains, we did not detect any genetic determinants likely causing phenotypic fusidic acid resistance. These strains however had disk diffusion inhibition zones close to the susceptibility breakpoint (S > 24 mm). There was 100% concordance for phenotypic susceptibility to fusidic acid and having no genetic determinants detected.
Mupirocin resistance
Of the sequenced MDR-MRSA strains, five (5.0%) exhibited phenotypic resistance to mupirocin (Table 3 and Supplementary Table 1). All of these strains contained the mupA-gene. One strain had two copies of the gene, and one strain furthermore had the chromosomal IleS V588F mutation in addition to the mupA-gene. The V588F IleS-mutation was however also detected in one mupirocin susceptible strain.
Linezolid resistance
No phenotypic resistance to linezolid was observed in this study, and no genes associated with linezolid resistance (cfr, optrA, poxtA) were detected in the sequenced MDR-MRSA strains. Linezolid resistance may also be caused by mutations in copies of the 23S rDNA genes (7). We identified two mutations associated with linezolid resistance in three MDR-MRSA strains; C2192T (n = 1) and C2534T (n = 2) (Howe et al., 2003; Liakopoulos et al., 2010) (Table 3). However, neither of the strains demonstrated phenotypic resistance when exposed to linezolid, as all had mutations in only a single copy of the gene.
Mobile genetic elements associated with antibiotic resistance determinants
For the sequenced MDR-MRSA strains (n = 101), the presence of AMR genes within specific types of MGEs were investigated. The specific resistance determinants associated with each type of MGE are more closely described in the following subsections.
Plasmids
In the present study, a total of 191 plasmids were identified, in 83.2% of the sequenced MDR-MRSA strains. The number of plasmids per strain varied from 1 to 6, with a median of 2 plasmids per strain, and a median size of 10,318 bp. The mobilizable group of plasmids included both the largest and some of the smallest plasmids, ranging in size from 1,299 to 98,879 bp (Supplementary Figure 1). A majority of the plasmids were previously described in S. aureus (86.4%) or other Staphylococci (10.5%). However, plasmids previously described in Escherichia coli (2.1%), Lactobacillus pentosus (0.5%), and Salmonella enterica 0.5%) were also detected.
The plasmids were clustered into 29 sub communities using Pling, while two of the nodes were excluded due to likely being partial plasmids or transposons. Among these sub communities, 15 comprised of multiple plasmids while 17 were singletons. The largest sub community included 117 plasmids, indicating that a majority of the plasmids in this study were related. The largest sub community included plasmids from strains of 25 different spa-types and 11 different sequence types. These plasmids encoded various combinations of 21 different AMR-genes, covering resistance against beta-lactams, MLS, aminoglycosides, trimethoprim, tetracyclines, fosfomycins and fusidanes. The second largest sub community included 11 plasmids, from strains with five different spa-types, and four different sequence types. These plasmids contained combinations of five different AMR-genes, covering resistance against beta-lactams, aminoglycosides, lincosamide and mupirocin.
The majority of plasmids were convergent plasmids (48.4%), carrying combinations of AMR-, stress and virulence-genes (e.g., AA411) (Supplementary Table 2). Some of the detected plasmids were however strictly AMR- (32.3%), stress- (12.9%) or virulence-plasmids (6.5%), carrying genes of only one specific category (e.g., AC627, AA851, and AA379, respectively). In total, 112 plasmids (58.6%) carried one or several AMR genes, with a median of 4 AMR genes per plasmid (ranging from 1 to 14). The AMR genes most commonly found on plasmids were blaZ (n = 45), aac(6’)-Ie/aph(2”)-Ia (n = 30), erm(C) (n = 26) and tet(K) (n = 24). The blaZ gene was found on 15 different plasmids, the aac(6’)-Ie/aph(2”)-Ia on nine different plasmids, the erm(C) gene on three different plasmids and the tet(K) gene on eight different plasmids (Figure 7). Other genes were only found on specific plasmids; e.g., mup(A) was only found in the conjugative plasmid AA083. The most common AMR plasmids identified in the MDR-MRSA strains were the non-mobilizable plasmid AC627 (n = 20), and the mobilizable plasmids AA411, AB973, and AC333 (n = 13). Conjugative AMR plasmids included the AA083 (n = 4) and the AB110 (n = 3) plasmids.
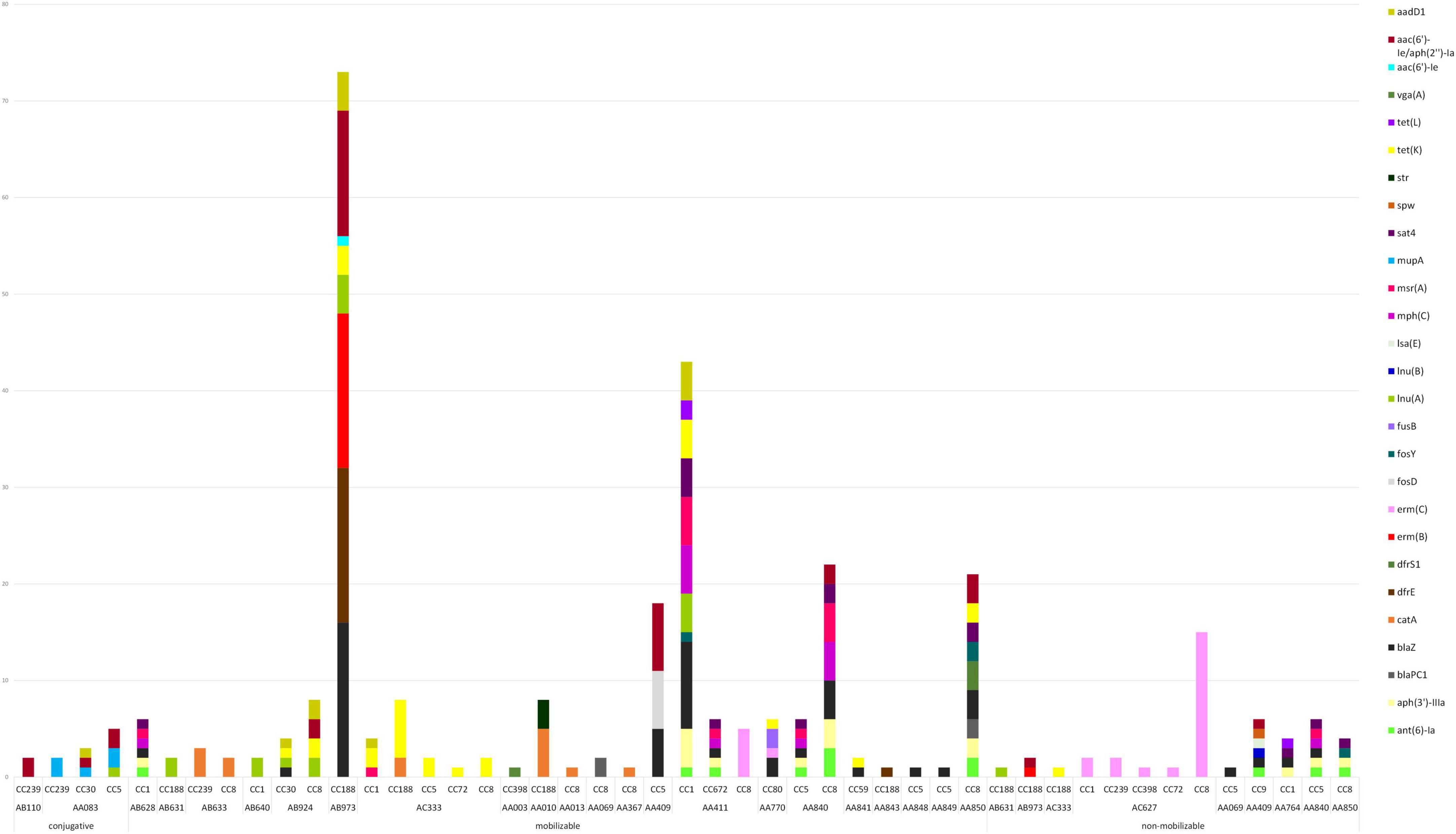
Figure 7. Plasmid type, predicted mobility and AMR genes found within plasmids in sequenced MDR-MRSA strains from Norway.
The small non-mobilizable plasmid AC627 (Figure 7 and Supplementary Table 2), which only holds the erm(C)-gene, was found in multiple strains belonging to CC1 (t127), CC239 (t037, and t632), CC398 (t011), CC72 (t3092), and CC8 (t064, t1476, t1952, and t451). For seven strains where we initially detected no genetic cause of macrolide resistance, it was upon further inspection detected that this plasmid was present, but had not been assembled correctly. Previous studies have also reported challenges in the detection of small plasmids using long read assemblers (Johnson et al., 2023). Consequently, this is an important aspect to consider for analysis of small plasmids when employing this methodological approach.
Most of the detected plasmids (76.0%) were found in a single clonal complex (Figure 7), and thus appeared to be relatively stably maintained within a specific genetic background while not being disseminated to other clones to any large extent. Consequently, these plasmids likely have diminished capacity for disseminating AMR traits across the more widely distributed MRSA clones. For instance, the medium sized mobilizable plasmid AB973, holding the AMR genes erm(B), blaZ, blaR1, blaI, dfrE, lnu(A), tet(K), aac(6’)-Ie/aph(2”)-Ia and aadD1, was only found in strains belonging to CC188 (Figure 7 and Supplementary Table 2). A few groups of AMR-plasmids (18.8%) however appeared to be spreading more successfully to diverse MRSA backgrounds. The medium sized conjugative plasmid AA083, holding mup(A), lnu(A), qacC, aac(6’)-Ie/aph(2”)-Ia, and aadD1, was found in CC239 (t037), CC30 (t665) as well as CC5 (t067 and t9408). Furthermore, the small plasmids AC333 and AC627, the small to medium-sized plasmids AA411, AB924, and AA840, were found in multiple clonal backgrounds, and thus demonstrate the highest potential for horizontal transfer between different MRSA clones.
Staphylococcal chromosome cassette mec (SCCmec)
SCCmec was detected in all but one (99.0%) of the sequenced MDR-MRSA strains. The lengths of SCCmec chromosome cassettes were quite uniform, with a median length of 45,587 bp. The smallest and largest SCCmec elements detected were both SCCmec type IV, of length 24,060 and 83,838 bp, respectively. The most prevalent SCCmec types identified in the sequenced strains were type IV (n = 47, 46.5%) followed by type III (n = 25, 24.8%). Both SCCmec types contained a wide variety of AMR genes (Figure 8). Type IV was typically detected in strains belonging to CC8 and CC188. The most commonly detected AMR genes in this SCCmec, in addition to mecA, were aac(6’)-Ie/aph(2”)-Ia, drfS1 (n = 17), and tet(K) (n = 16). SCCmec type III was only detected in strains belonging to CC239. The most predominant AMR genes found within this type, aside from mecA, were ant(9)-Ia and erm(A) (n = 14).
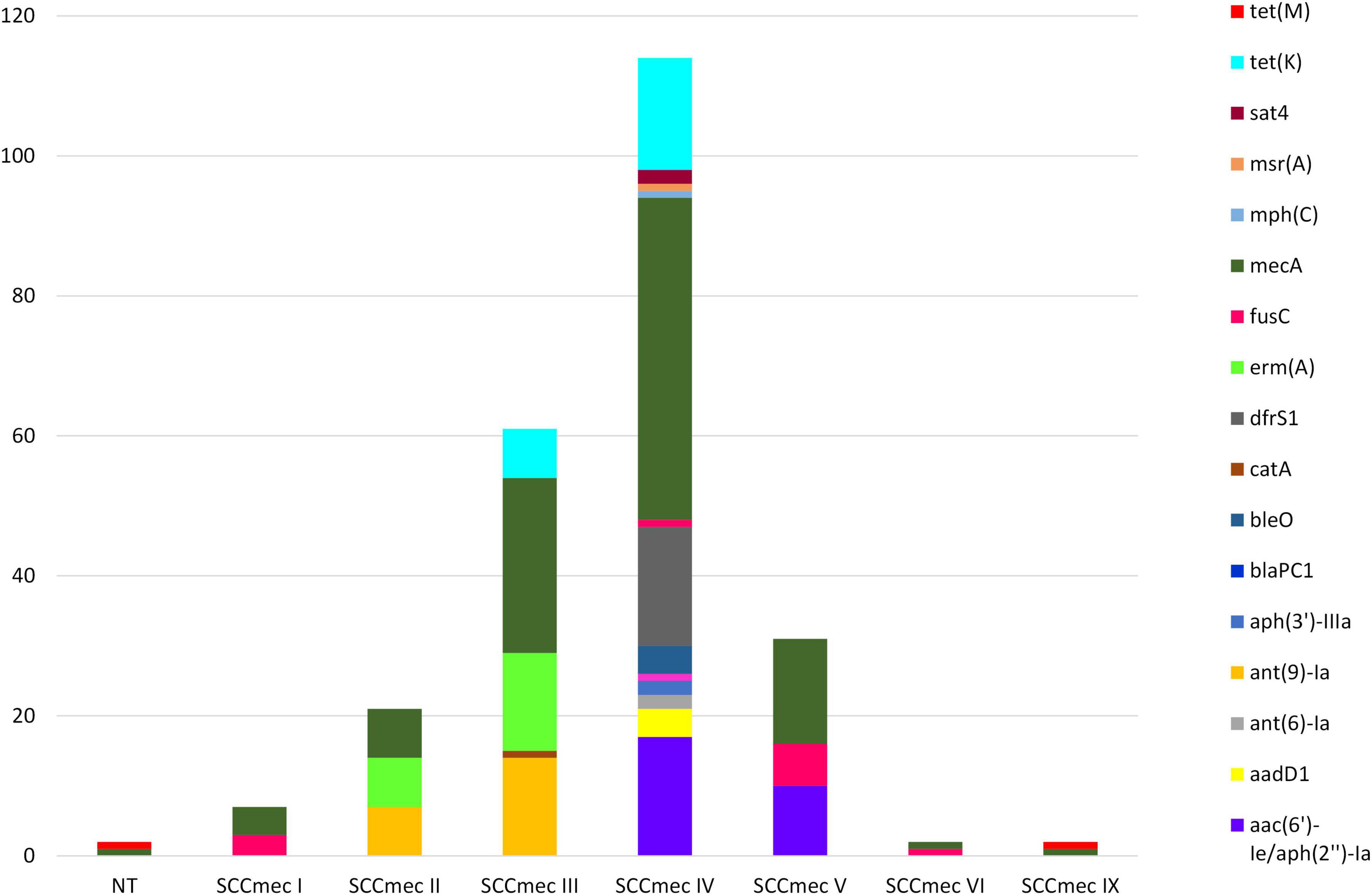
Figure 8. AMR genes identified in SCCmec elements in sequenced MDR-MRSA strains from Norway. NT, non-typeable.
Prophages
A total of 256 prophages harboring virulence and/or AMR-genes were identified in 86 (85.1%) of the sequenced MDR-MRSA strains. Strains which harbored prophages had on average three prophages per genome (range 1-7) with a mean length of 25,151 bp. Only three strains (3.0%) had prophages encoding an AMR gene (Figure 9), specifically the dfrG gene providing trimethoprim resistance. This gene was detected, in single or multiple copies, in the Staphylococcus phages ECel-2020q and phiSa2wa-st72 accordingly. The prophages identified were however associated with several known virulence factors. This included the well-known PVL toxin (encoded by lukF-PV and lukS-PV), δ-hemolysin (hld) and the enterotoxin-encoding genes sea, sec2, sek, sel, sep, and seq. Prophages encoding the human immune evasion cluster were also detected, encoding the staphylokinase gene (sak) and the staphylococcal complement inhibitor (scn). The prophages harboring this cluster were phage 23MRA, ECel-2020g, P630, phiNM3, phiSAa119, SA1014ru, Sa2wa, SA345ru, SA7, SA780ru, SAP090B, and tp310-3, which were present in 79 (78.2%) of the sequenced MDR-MRSA strains.
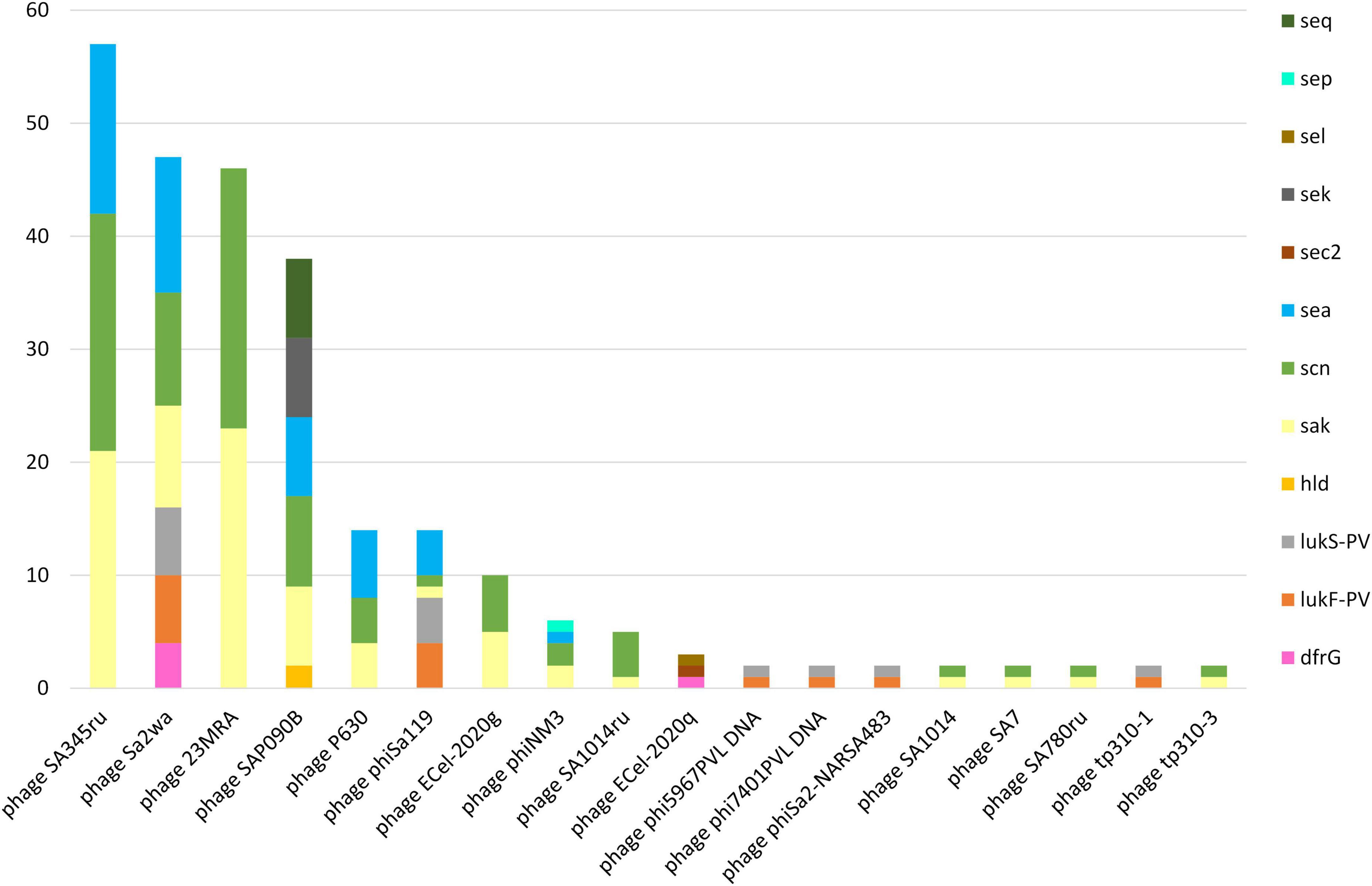
Figure 9. AMR and virulence genes in identified prophages in sequenced MDR-MRSA strains from Norway.
Composite transposons
A total of 207 composite transposons containing AMR-genes were identified in 76 (75.2%) of the sequenced MDR-MRSA strains. Within these strains, we detected a median of one transposon per genome, with a mean length of 6,032 bp. All of these transposons contained AMR gene(s). The Tn5405, Tn551, and Tn552 were found in both the chromosome and on plasmids (Figure 10). The Tn554, Tn558, Tn559, Tn6224, and Tn917 were only found in the chromosomes of the analyzed strains.
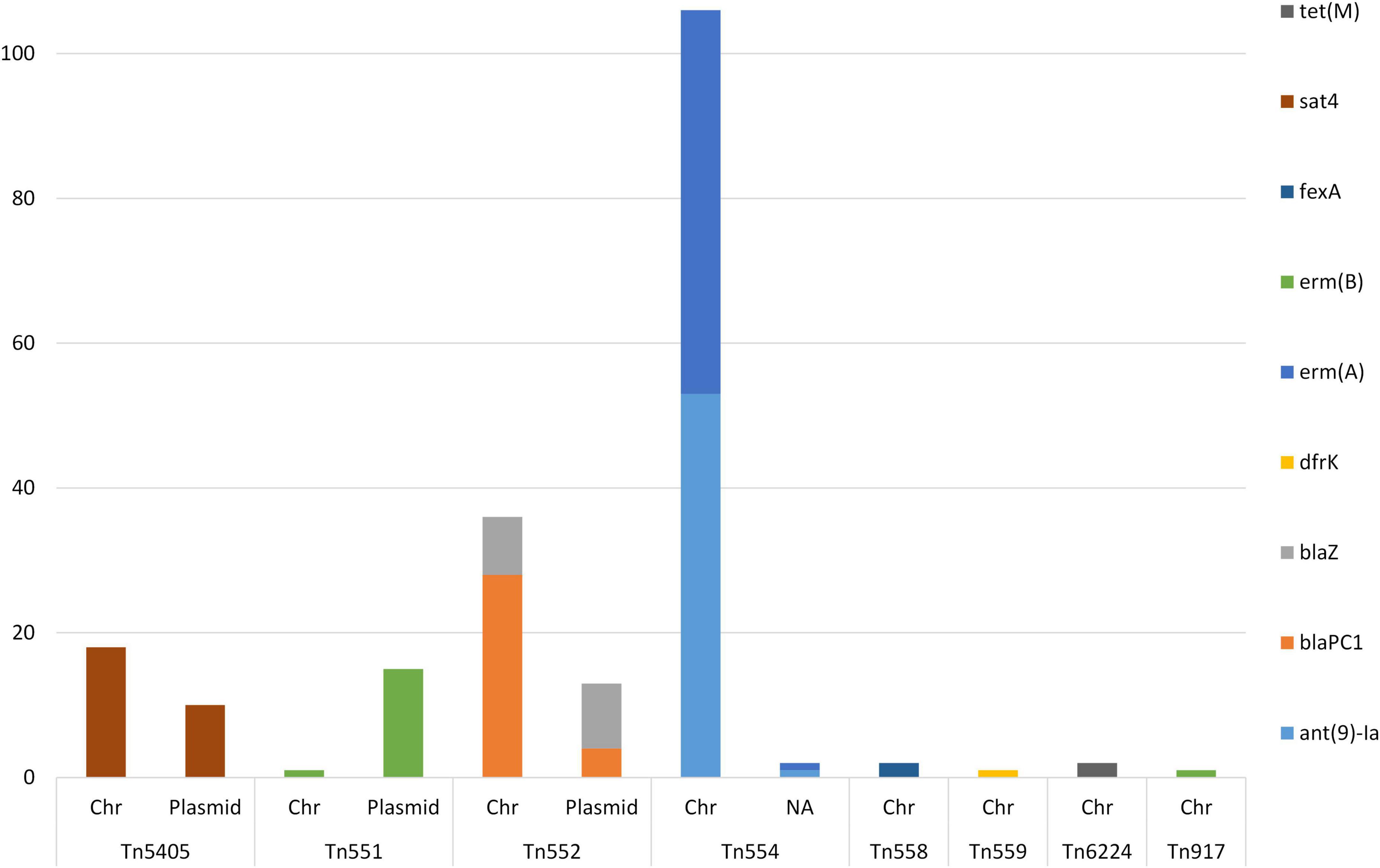
Figure 10. AMR genes identified in composite transposons in sequenced MDR-MRSA strains from Norway. Genomic location indicated by chromosome (Chr) or plasmid.
The AMR genes predominantly found within composite transposons were the aminoglycoside resistance gene ant(9)-Ia and the macrolide resistance gene erm(A) (n = 54) (Figure 10). Additionally, the beta-lactam resistance genes blaPC1 (n = 32) and blaZ (n = 17) were found in transposons, as well as the MLS gene erm(B) (n = 17). Genes found more rarely in transposons were tet(M) (n = 2) and dfr(K) (n = 1).
Tn554-transposons (carrying the ant(9)-Ia- and erm(A) AMR genes), were present in one to four copies per strain. The Tn551 and the Tn552- transposons was detected in a single or two copies when present in the strains. The Tn664-, Tn5405-, Tn558-, Tn559, and Tn917-transposons, were present in a single copy per strain.
Discussion
This study included all MDR-MRSA strains (n = 452) detected in Norway during the study period 2008-2020, from a total of 23,412 MRSA strains. This project was made possible by the continued Norwegian MRSA surveillance effort that has been ongoing since 2008, with the aim of keeping multidrug-resistant pathogens from becoming endemic in healthcare institutions in Norway. Although the overall and yearly proportion of MDR-MRSA was relatively low, we observed an increase in the number of MDR-MRSA strains over time, as well as major changes in molecular epidemiology, during the study period. Specifically, we observed temporal shifts and predominance of certain genotypes (spa-types t1476, t127, t188, and t030/t037), indicating that these were successfully established and disseminating MDR-MRSA clones.
This study was limited by the small total number of MDR-MRSA strains, the absence of antimicrobial susceptibility testing (AST) for all antibiotic groups, and the lack of comprehensive epidemiological data for all strains. Information on place of acquisition for the MDR-MRSA strains is also inherently limited by some uncertainty. However, it is interesting to note that only 13.1% of strains were registered as acquired in Norway, while a majority were registered as acquired abroad. Compared to the overall MRSA population (Rønning et al., 2024), this suggests import to be responsible for a comparatively larger proportion of MDR-MRSA strains, and similarly that Asia and Africa are the most frequent routes of transmission. This is also supported by the fact that we detect specific clones that have previously been reported as endemic or prevalent in these specific regions and countries (Lawal et al., 2022; Mohamad Farook et al., 2022). These findings highlight the importance of global cooperation, surveillance, antibiotic stewardship and infection control efforts that can limit both the emergence and global dissemination of these important pathogens.
A high proportion of the MDR-MRSA strains were healthcare-associated, mostly isolated from patients during hospital admission. The proportion of healthcare-associated strains was markedly higher among the MDR-MRSA strain collection than that reported in our surveillance study of the whole Norwegian MRSA population (Rønning et al., 2024). This suggests that the most highly resistant MRSA clones are more often hospital-associated than less resistant MRSA clones, although they have become more common in the community setting as well. This may among other factors be due to the high selection pressures and competition between MRSA clones provided by high antibiotic exposure (de Vos et al., 2021). MRSA are also endemic in many hospital environments wordwide (Stefani et al., 2012), thus providing reservoirs for efficient spread into a vulnerable populations, especially in resource-limited settings with inadequate infection control (Christopher et al., 2011). A limitation to this study is the lack of temporal data on hospital and nursing home admissions, and thus that we had to rely on a broad definition of healthcare-associated MRSA. With this in mind, it is possible that the number of HA- and specifically hospital-associated cases are overestimated. However, the same definition of HA-MRSA was used in the surveillance study of the whole Norwegian MRSA population, in support of the relative differences observed in this study.
Interestingly, there was a larger proportion of MDR-MRSA strains from carriage than from clinical infections, compared to the corresponding numbers for the overall MRSA population in Norway (Rønning et al., 2024). This finding may reflect that a majority of these strains have been acquired abroad, and are thus (in Norway) mainly detected from asymptomatic carriers, e.g., due to screening upon contact with the healthcare system and differing screening practices. While it is possible that some MDR-MRSA strains have reduced virulence due to e.g., the physiological costs of maintaining resistance mechanisms, this can vary significantly between different strains and settings (Rao et al., 2022; Rudkin et al., 2012; Taglialegna et al., 2019). Furthermore, it is possible that MDR-MRSA strains have acquired virulence factors that enhance their ability to colonize and persist in the host (Jiang et al., 2023). The prevalence of PVL-positive strains was 20.4%, which was much lower than for the whole Norwegian MRSA strain population (Rønning et al., 2024). This also likely reflects the low number of infection compared to carriage strains, as PVL positive strains have typically been linked to clinical infections, whereas PVL-negative strains are more commonly associated with asymptomatic carriage (Boyle-Vavra and Daum, 2007).
In the MDR-MRSA strain collection, we observed almost universal resistance to antibiotics such as erythromycin and ciprofloxacin/norfloxacin in addition to cefoxitine. High levels of resistance were also observed for tetracycline, gentamicin, and clindamycin. Thus, in this group of strains, the choice of antibiotic treatment for potential infections is alarmingly and severely restricted. Resistance to mupirocin is, however, low, meaning that this is still an option for eradication/decolonization of a majority of these strains. Vancomycin and linezolid remain as treatment options for all strains, and there were detected no strains that could be regarded as XDR-MRSA in this strain collection.
We observed a very large heterogeneity of spa-types and clonal complexes that were resistant toward five or six groups of antibiotics, indicating that multi-drug resistance is indeed a major challenge within the general MRSA population, not only in a few specific clones. The limited number of genotypes that were resistant to seven or eight groups of antibiotics, however, included well-known clones like MRSA CC239, that have been evolving antimicrobial resistance in high selection pressure environments for many decades, given their continuous presence and global spread since the large hospital outbreaks of the 1970s (Monecke et al., 2018; Shanson, 1981). This underscores the critical importance of preventing the introduction of MRSA into hospitals and other healthcare facilities, especially in low-prevalence countries, and furthermore the importance of antibiotic stewardship and infection control in mitigating the risk of developing highly resistant strains within these environments.
Overall, we observed significant concordance between phenotypic (AST) and genotypic antimicrobial resistance profiles. In almost all cases, previously described AMR genes or mutations were detected, that could explain the observed phenotypes. Additionally, we noticed high clonality of resistance profiles, which corresponds well with the apparent clonality of specific MGEs carrying these AMR genes. Consequently, one can reasonably anticipate an antimicrobial profile based on genotyping results, although we do not suggest that antimicrobial susceptibility testing should be bypassed. On the other hand, certain genes and mutations which do not appear to confer phenotypic resistance in S. aureus, at least not to an extent which provides resistance according to clinical breakpoints, continue to be reported as resistance genes in the major AMR databases. Furthermore, especially for some groups of antibiotics, the genetics underlying the resistance phenotypes are still not well enough understood and/or are difficult to interpret. This underscores the necessity for further investigation into the mechanisms underlying bacterial resistance as well as the importance of continuous curation and updating of AMR databases.
Nanopore sequencing technology was used to investigate the specific MGEs associated with AMR genes and mutations. Long-read sequencing technologies such as Oxford Nanopore Technologies (8) have facilitated a much more comprehensive investigation of MGEs, which can often be problematic to resolve by short-read sequencing (9). Consequently, our investigation revealed that AMR genes in MDR-MRSA strains are predominantly located within plasmids, as well as within staphylococcal cassette chromosome mec (SCCmec) elements and transposons, with a limited presence on prophages. The most common AMR genes were associated with wide range of MGEs, including different SCCmec types and multiple transposons and plasmids. Some MGEs were clonal, while others were widely distributed, indicating high potential for spread within the MRSA population. A majority of the plasmids were furthermore closely related, although differing in AMR traits, thus indicating high plasticity. The most prevalent AMR phenotypes were furthermore caused by multiple AMR genes and/or mutations. As an example, we observed eight different genes or gene combinations in strains with MLS resistance. This large variation is likely a consequence of high selection pressure and thus multiple adaptations of MRSA clones over a long period of time, which now serves as an arsenal of genes available for providing resistance against different MLS antibiotics. This high diversity both in AMR genes and in acquisition mechanisms thus provide a significant advantage for dissemination of antibiotic resistance within the MRSA population. These findings highlight the complexity of resistance gene dissemination and the adaptive strategies employed by S. aureus in response to antimicrobial pressures.
Materials and methods
Study design and population
This study is based on MRSA strains submitted to the Norwegian MRSA reference laboratory from 2008 to 2020. Inclusion was based on phenotypic resistance to five or more of the following antibiotic/resistance groups, with the antibiotic tested provided in parentheses; beta-lactams (cefoxitin), macrolide/lincosamide/streptogramin B (MLS) (erythromycin and clindamycin), aminoglycosides (gentamicin), tetracyclines (tetracycline), fucidanes (fusidic acid), fluoroquinolones (ciprofloxacin or norfloxacin), folate pathway inhibitors (trimethoprim-sulfamethoxazole, TMP-SMX), ansamycins (rifampicin), oxazolidinones (linezolid), and glycopeptides (vancomycin). For lack of a more informative terminology, we refer to this strain collection as MDR-MRSA (Magiorakos et al., 2012) throughout this manuscript.
Clinical and epidemiological data
Clinical and epidemiological data on all cases was collected from the Norwegian Surveillance System for Communicable Diseases (MSIS) and request forms from the referring laboratory or treating physician available from the laboratory information system (LIMS) at the national reference laboratory. The Information from MSIS included age, sex, admission to hospital or nursing home, place of acquisition and if it was part of a known outbreak (data from the Norwegian outbreak rapid alert system Vesuv) (Leinweber et al., 2021). The data obtained from the LIMS included sample date, sampling site/type of sample and laboratory results. All MDR-MRSA cases were categorized as carriage, infection, invasive infection or unknown based on sampling site/type of sample. Age groups were defined according to Aslam et al. (2021). Due to lack of temporal data for hospitalized patients and nursing home stays, a case was classified as healthcare-associated (HA) if it was diagnosed during a hospital or nursing home stay, or if MDR-MRSA was detected in healthcare workers MLS (HCWs). Conversely, all other cases were classified as community-acquired (CA).
Genotyping and antimicrobial susceptibility testing
Culturing, DNA extraction, spa-typing and assignment of spa-types to clonal complexes (CC) was performed as described previously (Rønning et al., 2024). The strains included in this study had previously undergone phenotypic antimicrobial susceptibility testing (AST) toward the following antibiotics: cefoxitin, erythromycin, clindamycin, fusidic acid, trimethoprim -sulfamethoxalate, tetracycline, gentamicin, ciprofloxacin/norfloxacin, rifampicin, mupirocin, linezolid, and vancomycin. Susceptibility testing was performed as previously described (Enger et al., 2022) on all strains using the EUCAST (European Committee on Antimicrobial Susceptibility Testing) disk diffusion method and categorized as either susceptible, intermediate/susceptible increased exposure, or resistant according to the current EUCAST breakpoints at the time of testing. For clindamycin, inducible resistance was recorded as described in the EUCAST expert rules (EUCAST, 2023). For vancomycin, the gradient strip test from BioMeriéux (2008-2013) or Liofilchem (2014-2020) was used.
Whole genome sequencing
A selection of strains (n = 101) were subjected to whole genome sequencing (WGS) by nanopore methodology, hereafter referred to as sequencing. These included all strains resistant to seven or more antibiotic groups (n = 21), and a randomized selection among the remaining strains (n = 80). Bacterial cells were first treated with proteinase K (2 mg/mL) and lysostaphin (0.1 mg/mL) for 15 min with shaking at 37°C, before heating for 15 min at 65°C. Genomic DNA was then isolated using the EZ1 DNA tissue kit with an EZ1 Advanced XL instrument (Qiagen). Sequencing libraries were prepared and multiplexed using the Rapid Barcoding Sequencing kit (SQK-RBK004), according to the RBK_9054_v2_revJ_14Aug2019 protocol. Sequencing libraries were loaded onto a R9.4.1 SpotON flow cell (FLO-MIN106D) and sequenced on a MinION Mk1B sequencer (Oxford Nanopore Technologies).
Bioinformatic analyses
Dorado (Samarakoon et al., 2023) v.0.4.2 was used for basecalling (SUP v3.6 model) and demultiplexing. Assembly was performed using Flye v.2.9.2 (Kolmogorov et al., 2019; Lin et al., 2016). Racon and Medaka (SUP v5.0.7 model) were used for polishing. Additionally, Homopolish (Huang et al., 2021) was used to remove possible systematic errors from the nanopore sequencing. The sequences are available from GenBank under BioProject ID PRJNA1186082. Annotation was performed with Prokka v.1.14.6 (Seemann, 2014). Definition of the core genome and phylogeny were performed using Roary v3.13.0 with default settings (Page et al., 2015) and FastTree with the GTR model (Price et al., 2009).
Plasmid classification was performed by MOB-suite v.3.1.8 (Robertson and Nash, 2018), with the typing and clustering modules. Mashtree (Katz et al., 2019) was then used to construct a plasmid phylogeny. Plasmids were furthermore merged into communities/subcommunities using the Pling bioinformatic tool (Frolova et al., 2024).
Putative prophages were detected using Phastest (Wishart et al., 2023) including only intact phages, and identified using nBLAST against reference S. aureus phages in the GenomeNet Virus-Host DataBase (VHDB) (Mihara et al., 2016). Transposons and IS-elements were detected using the MobileElementFinder (Johansson et al., 2020). SCCmec chromosome cassettes were typed using SCCmecFinder (Kaya et al., 2018). Whole SCCmec elements were extracted by in silico PCR with SeqKit (Shen et al., 2016), using modified primers from Ito et al. (2001).
For whole genomes as well as for specific MGEs, AMR genes and point mutations were detected using AMRFinderPlus v3.10.30 with organism-specific settings for S. aureus, and cut-off of 90% on protein identity and 50% on coverage (Feldgarden et al., 2021).
Visualization
Upset plots were created using R studio v4.3.3 with the ggplot2 (Wickham, 2016), ComplexUpset (Krassowski, 2020; Lex et al., 2014) and cowplot (Wilke, 2024) packages. Visualization of the core genome tree with phenotypic and genotypic traits was performed with iTol v.5 (Letunic and Bork, 2021). The world map was created using GeoPandas (Jordahl et al., 2020) and Matplotlib (Hunter, 2007).
Data availability statement
Sequence data are available from GenBank under BioProject ID PRJNA1186082. There are ethical restrictions to the sharing of epidemiological metadata, due to Norwegian legislation on medical and health research. With ethical approval from the Regional Committees for Medical and Health Research in Norway, the underlying metadata can however be made available by application to the Norwegian Surveillance System for Communicable Diseases (MSIS).
Ethics statement
The project was approved, with exemption from informed consent of participants, by the Norwegian Regional Committees for Medical and Health Research Ethics (REC) South-East with reference number 352380. A Data Protection Impact Assessment was performed and approved by the responsible institution, the Clinic of Laboratory Medicine at St. Olavs Hospital, Trondheim University Hospital.
Author contributions
TR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. HE: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. JA: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. CÅ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project was supported by funds from the Clinic of Laboratory Medicine, St. Olavs University Hospital and from The Norwegian Surveillance Programme for Antimicrobial Resistance (NORM).
Acknowledgments
We would like to acknowledge the contribution of all submitting medical microbiology laboratories to the continued MRSA surveillance effort in Norway, as well as the laboratory personnel who have contributed to the analysis of these strains at the Norwegian MRSA reference laboratory at St. Olavs Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1564943/full#supplementary-material
References
Aslam, B., Khurshid, M., Arshad, M., Muzammil, S., Rasool, M., Yasmeen, N., et al. (2021). Antibiotic resistance: One health one world outlook. Front. Cell. Infect. Microbiol. 11:771510. doi: 10.3389/fcimb.2021.771510
Boyle-Vavra, S., and Daum, R. (2007). Community-acquired methicillin-resistant Staphylococcus aureus: The role of Panton-Valentine leukocidin. Lab. Invest. 87, 3–9. doi: 10.1038/labinvest.3700501
Cheung, G., Bae, J., and Otto, M. (2021). Pathogenicity and virulence of Staphylococcus aureus. Virulence 12, 547–569. doi: 10.1080/21505594.2021.1878688
Christopher, S., Verghis, R., Antonisamy, B., Sowmyanarayanan, T., Brahmadathan, K., Kang, G., et al. (2011). Transmission dynamics of methicillin-resistant Staphylococcus aureus in a medical intensive care unit in India. PLoS One 6:e20604. doi: 10.1371/journal.pone.0020604
de Vos, A., de Vlas, S., Lindsay, J., Kretzschmar, M., and Knight, G. (2021). Understanding MRSA clonal competition within a UK hospital; the possible importance of density dependence. Epidemics 37:100511. doi: 10.1016/j.epidem.2021.100511
Enger, H., Larssen, K., Damås, E., Aamot, H., Blomfeldt, A., Elstrøm, P., et al. (2022). A tale of two STs: Molecular and clinical epidemiology of MRSA t304 in Norway 2008-2016. Eur. J. Clin. Microbiol. Infect. Dis. 41, 209–218. doi: 10.1007/s10096-021-04353-9
EUCAST. (2023). The European Committee on Antimicrobial Susceptibility Testing Expert Rules v 3.2. Vaxjo: EUCAST.
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J., Haendiges, J., Haft, D., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Frolova, D., Lima, L., Roberts, L., Bohnenkämper, L., Wittler, R., Stoye, J., et al. (2024). Applying rearrangement distances to enable plasmid epidemiology with pling. Microb. Genom. 10:001300. doi: 10.1099/mgen.0.001300
GBD 2021 Antimicrobial Resistance Collaborators. (2024). Global burden of bacterial antimicrobial resistance 1990-2021: A systematic analysis with forecasts to 2050. Lancet 404, 1199–1226. doi: 10.1016/S0140-6736(24)01867-1
Guérillot, R., Gonçalves da Silva, A., Monk, I., Giulieri, S., Tomita, T., Alison, E., et al. (2018). Convergent evolution driven by rifampin exacerbates the global burden of drug-resistant Staphylococcus aureus. mSphere 3:e00550-17. doi: 10.1128/mSphere.00550-17
Howden, B., Giulieri, S., Wong Fok Lung, T., Baines, S. L., Sharkey, L. K., Lee, J. Y. H., et al. (2023). Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 21, 380–395. doi: 10.1038/s41579-023-00852-y
Howe, R., Wootton, M., Noel, A., Bowker, K., Walsh, T., and MacGowan, A. (2003). Activity of AZD2563, a novel oxazolidinone, against Staphylococcus aureus strains with reduced susceptibility to vancomycin or linezolid. Antimicrob. Agents Chemother. 47, 3651–3652. doi: 10.1128/AAC.47.11.3651-3652.2003
Huang, Y., Liu, P., and Shih, P. (2021). Homopolish: A method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22:95. doi: 10.1186/s13059-021-02282-6
Hunter, J. (2007). Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95. doi: 10.1109/MCSE.2007.55
Ito, T., Katayama, Y., and Hiramatsu, K. (1999). Cloning and nucleotide sequence determination of the entire mec DNA of pre-methicillin-resistant Staphylococcus aureus N315. Antimicrob. Agents Chemother. 43, 1449–1458. doi: 10.1128/AAC.43.6.1449
Ito, T., Katayama, Y., Asada, K., Mori, N., Tsutsumimoto, K., Tiensasitorn, C., et al. (2001). Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1323–1336. doi: 10.1128/AAC.45.5.1323-1336.2001
Ito, T., Kuwahara-Arai, K., Katayama, Y., Uehara, Y., Han, X., Kondo, Y., et al. (2014). Staphylococcal cassette chromosome mec (SCCmec) analysis of MRSA. Methods Mol. Biol. 1085, 131–148. doi: 10.1007/978-1-62703-664-1_8
Jiang, J., Cameron, D., Nethercott, C., Aires-de-Sousa, M., and Peleg, A. (2023). Virulence attributes of successful methicillin-resistant Staphylococcus aureus lineages. Clin. Microbiol. Rev. 36:e0014822. doi: 10.1128/cmr.00148-22
Johansson, M., Bortolaia, V., Tansirichaiya, S., Aarestrup, F., Roberts, A., and Petersen, T. (2020). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: Mobileelementfinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/jac/dkaa390
Johnson, J., Soehnlen, M., and Blankenship, H. (2023). Long read genome assemblers struggle with small plasmids. Microb. Genom. 9:mgen001024. doi: 10.1099/mgen.0.001024
Jordahl, K., Bossche, J. V. D., Fleischmann, M., Wasserman, J., McBride, J., Tratner, J. G., et al. (2020). geopandas/geopandas: v0.8.1. Zenodo.
Katayama, Y., Ito, T., and Hiramatsu, K. (2001). Genetic organization of the chromosome region surrounding mecA in clinical staphylococcal strains: Role of IS431-mediated mecI deletion in expression of resistance in mecA-carrying, low-level methicillin-resistant Staphylococcus haemolyticus. Antimicrob. Agents Chemother. 45, 1955–1963. doi: 10.1128/AAC.45.7.1955-1963.2001
Katz, L., Griswold, T., Morrison, S., Caravas, J., Zhang, S., den Bakker, H., et al. (2019). Mashtree: A rapid comparison of whole genome sequence files. J. Open Source Softw. 4:01762. doi: 10.21105/joss.01762
Kaya, H., Hasman, H., Larsen, J., Stegger, M., Johannesen, T., Allesøe, R., et al. (2018). SCC mec finder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere 3:e00612-17. doi: 10.1128/mSphere.00612-17
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Lawal, O., Ayobami, O., Abouelfetouh, A., Mourabit, N., Kaba, M., Egyir, B., et al. (2022). A 6-year update on the diversity of methicillin-resistant Staphylococcus aureus clones in Africa: A systematic review. Front. Microbiol. 13:860436. doi: 10.3389/fmicb.2022.860436
Leinweber, H., Sieber, R., Larsen, J., Stegger, M., and Ingmer, H. (2021). Staphylococcal phages adapt to new hosts by extensive attachment site variability. mBio 12:e0225921. doi: 10.1128/mBio.02259-21
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R., and Pfister, H. (2014). UpSet: Visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 20, 1983–1992. doi: 10.1109/TVCG.2014.2346248
Liakopoulos, A., Spiliopoulou, I., Damani, A., Kanellopoulou, M., Schoina, S., Papafragas, E., et al. (2010). Dissemination of two international linezolid-resistant Staphylococcus epidermidis clones in Greek hospitals. J. Antimicrob. Chemother. 65, 1070–1071. doi: 10.1093/jac/dkq065
Lin, Y., Yuan, J., Kolmogorov, M., Shen, M., Chaisson, M., and Pevzner, P. (2016). Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. U S A. 113, E8396–E8405. doi: 10.1073/pnas.1604560113
Magiorakos, A., Srinivasan, A., Carey, R., Carmeli, Y., Falagas, M., Giske, C., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Mihara, T., Nishimura, Y., Shimizu, Y., Nishiyama, H., Yoshikawa, G., Uehara, H., et al. (2016). Linking virus genomes with host taxonomy. Viruses 8:66. doi: 10.3390/v8030066
Mohamad Farook, N., Argimón, S., Abdul Samat, M., Salleh, S., Sulaiman, S., Tan, T., et al. (2022). Diversity and dissemination of methicillin-resistant Staphylococcus aureus (MRSA) genotypes in Southeast Asia. Trop. Med. Infect. Dis. 7:438. doi: 10.3390/tropicalmed7120438
Monecke, S., Slickers, P., Gawlik, D., Müller, E., Reissig, A., Ruppelt-Lorz, A., et al. (2018). Molecular Typing of ST239-MRSA-III From diverse geographic locations and the evolution of the SCC mec III element during its intercontinental spread. Front. Microbiol. 9:1436. doi: 10.3389/fmicb.2018.01436
Mores, C., Montelongo, C., Putonti, C., Wolfe, A., and Abouelfetouh, A. (2021). Investigation of plasmids among clinical staphylococcus aureus and staphylococcus haemolyticus isolates from egypt. Front. Microbiol. 12:659116. doi: 10.3389/fmicb.2021.659116
Page, A., Cummins, C., Hunt, M., Wong, V., Reuter, S., Holden, M., et al. (2015). Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Price, M., Dehal, P., and Arkin, A. (2009). FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rao, Y., Peng, H., Shang, W., Hu, Z., Yang, Y., Tan, L., et al. (2022). A vancomycin resistance-associated WalK(S221P) mutation attenuates the virulence of vancomycin-intermediate Staphylococcus aureus. J. Adv. Res. 40, 167–178. doi: 10.1016/j.jare.2021.11.015
Richardson, E., Bacigalupe, R., Harrison, E., Weinert, L., Lycett, S., Vrieling, M., et al. (2018). Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2, 1468–1478. doi: 10.1038/s41559-018-0617-0
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4:e000206. doi: 10.1099/mgen.0.000206
Rønning, T., Enger, H., Afset, J., and Ås, C. (2024). Insights from a decade of surveillance: Molecular epidemiology of methicillin-resistant Staphylococcus aureus in Norway from 2008 to 2017. PLoS One 19:e0297333. doi: 10.1371/journal.pone.0297333
Rudkin, J., Edwards, A., Bowden, M., Brown, E., Pozzi, C., Waters, E., et al. (2012). Methicillin resistance reduces the virulence of healthcare-associated methicillin-resistant Staphylococcus aureus by interfering with the agr quorum sensing system. J. Infect. Dis. 205, 798–806. doi: 10.1093/infdis/jir845
Samarakoon, H., Ferguson, J., Gamaarachchi, H., and Deveson, I. (2023). Accelerated nanopore basecalling with SLOW5 data format. Bioinformatics 39:btad352. doi: 10.1093/bioinformatics/btad352
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shanson, D. (1981). Antibiotic-resistant Staphylococcus aureus. J. Hosp. Infect. 2, 11–36. doi: 10.1016/0195-6701(81)90003-7
Shen, W., Le, S., Li, Y., and Hu, F. (2016). SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. doi: 10.1371/journal.pone.0163962
Stefani, S., Chung, D., Lindsay, J., Friedrich, A., Kearns, A., Westh, H., et al. (2012). Meticillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 39, 273–282. doi: 10.1016/j.ijantimicag.2011.09.030
Taglialegna, A., Varela, M., Rosato, R., and Rosato, A. (2019). VraSR and virulence trait modulation during daptomycin resistance in methicillin-resistant Staphylococcus aureus Infection. mSphere 4:e00557-18. doi: 10.1128/mSphere.00557-18
Wilke, C. (2024). cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’. R package version 1.1.3.
Wishart, D., Han, S., Saha, S., Oler, E., Peters, H., Grant, J., et al. (2023). PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 51, W443–W450. doi: 10.1093/nar/gkad382
Keywords: Staphylococcus aureus, MRSA, multidrug resistance, AMR, epidemiology, Norway
Citation: Rønning TG, Enger H, Afset JE and Ås CG (2025) Trends and characteristics of multidrug-resistant MRSA in Norway 2008-2020. Front. Microbiol. 16:1564943. doi: 10.3389/fmicb.2025.1564943
Received: 22 January 2025; Accepted: 11 April 2025;
Published: 09 May 2025.
Edited by:
Hazem Ramadan, Mansoura University, EgyptReviewed by:
Okon Okwong Kenneth, Federal University, Wukari, NigeriaXiao-Ming Lu, Fujian University of Technology, China
Copyright © 2025 Rønning, Enger, Afset and Ås. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina Gabrielsen Ås, Y2hyaXN0aW5hLmdhYnJpZWxzZW5Ac3RvbGF2Lm5v