 Tracy L. Nicholson
Tracy L. Nicholson Sarah M. Shore
Sarah M. Shore Yihui Wang
Yihui Wang Lindsey Zimmerman2
Lindsey Zimmerman2 Tod J. Merkel
Tod J. Merkel- 1National Animal Disease Center, Agricultural Research Service, USDA, Ames, IA, United States
- 2Division of Bacterial, Parasitic and Allergenic Products, Center for Biologics Evaluation and Research, FDA, Silver Spring, MD, United States
Bordetella bronchiseptica is a highly contagious bacterial respiratory pathogen with a broad host range of wild and domesticated mammals that can cause a variety of clinical disease outcomes ranging from asymptomatic carriage to severe pneumonia. The goal of this study was to evaluate the genetic diversity of B. bronchiseptica isolates obtained from primates and evaluate the antimicrobial resistance harbored by these isolates. Two isolates were identified as belonging to B. bronchiseptica lineage II and 13 isolates represented new sequence types within B. bronchiseptica lineage I clonal complex 6. The lineage II isolates harbored the lowest sequence identity observed across all genes evaluated and did not contain several well characterized virulence and fimbrial genes. Western blotting revealed no reactivity to a lineage II strain when using antibodies generated against pertactin (PRN) from a lineage I-1 strain or antibodies generated against a domain of filamentous haemagglutinin (FHA) from a lineage I-1 strain. Isolates harbored variation within the wbm locus containing genes encoding for the expression of antigenically distinct O-antigen types and the cya operon was replaced by the ptp operon in several isolates, expanding the phylogenetic distribution of this operon replacement. Thirteen isolates exhibited phenotypic resistance to four antibiotic classes tested, however the Bordetella-specific β-lactamase was the only antimicrobial resistance gene identified. Collectively, the data in this report expands the known phylogenetic diversity and genetic variation of B. bronchiseptica isolates.
Introduction
Bordetella bronchiseptica is a highly contagious respiratory bacterial veterinary pathogen that can cause a variety of clinical disease outcomes ranging from asymptomatic carriage to severe bronchopneumonia (Mattoo and Cherry, 2005; Chambers et al., 2019; Brockmeier et al., 2019). B. bronchiseptica is closely related to Bordetella pertussis and Bordetella parapertussishu, the causative agents of whooping cough or pertussis in humans and combined are collectively referred to as the “classical bordetellae” (Goodnow, 1980; Parkhill et al., 2003; Preston et al., 2004; Diavatopoulos et al., 2005). B. parapertussis consists of two genetically distinct lineages: B. parapertussishu, which is only known to infect humans, and B. parapertussisov, which is only known to infect sheep (Porter et al., 1994). The human-restricted pathogens, B. pertussis and B. parapertussishu, are considered to have evolved independently from a B. bronchiseptica ancestor (Diavatopoulos et al., 2005; Preston et al., 2004; Parkhill et al., 2003). In addition to their close genetic relatedness, classical bordetellae harbor many of the same virulence factors, which are similarly regulated (Linz et al., 2016; Chen and Stibitz, 2019; Parkhill et al., 2003). Despite these similarities, the classical bordetellae differ in traits such as host specificity, disease severity, and duration of infection. In addition to only infecting humans, B. pertussis and B. parapertussishu also lack an animal reservoir and the ability to survive in the environment (Hewlett et al., 2014; Pinto and Merkel, 2017). In contrast, B. bronchiseptica infects a variety of mammals, often establishing chronic infections, and is capable of surviving in the environment (Goodnow, 1980; Brockmeier et al., 2019; Badhai and Das, 2021). While rare, human B. bronchiseptica infections have been reported (Barcala Salido et al., 2022; Register et al., 2012; Agarwal et al., 2022; Moore et al., 2022; Woolfrey and Moody, 1991).
A key regulatory mechanism that is shared among the classical bordetellae is that the majority of virulence gene expression is regulated by a two-component sensory transduction system encoded by the bvg locus (Chen and Stibitz, 2019; Cotter and Jones, 2003). This locus contains BvgS, a histidine kinase sensor protein, and BvgA, a DNA-binding response-regulator protein. In response to a variety of environmental cues, BvgAS controls the expression of phenotypic phases transitioning between a virulent (Bvg+) phase and a non-virulent (Bvg−) mode (Chen and Stibitz, 2019; Cotter and Jones, 2003; Nicholson et al., 2012). During the virulent Bvg+ phase, the BvgAS system is fully active and virulence-activated genes (vags), such as filamentous hemagglutinin (FHA), pertactin (PRN), fimbriae, dermonecrotic toxin (DNT), adenylate cyclase toxin (ACT), and a type III secretion system (T3SS), are fully expressed (Cotter and Jones, 2003; Nicholson et al., 2012; Nicholson, 2007; Nicholson et al., 2024).
The overwhelming majority of publicly available genomic sequencing data for the classical bordetellae is derived from clinical B. pertussis isolates due to the medical importance of whooping cough and the epidemiological surveillance of vaccine-driven selection occurring among B. pertussis isolates. The goals of the current study were to fill this gap by utilizing whole-genome sequencing (WGS) analysis to evaluate the genetic diversity of B. bronchiseptica (Bb) isolates obtained from non-human primates and assess the phenotypic and genotypic antimicrobial resistance (AMR) of these isolates. Collectively, the data in this report broadens the known phylogenetic diversity and genetic variation of Bb isolates.
Materials and methods
Isolate information
Five Bb isolates were obtained from nasopharyngeal samples collected from baboons and 14 Bb isolates were obtained from nasopharyngeal samples collected from rhesus monkeys (Supplementary Table 1). All isolates were obtained during routine screening for various pathogens, while animals were housed in large groups. Isolates M2020-2 and M2020-4 were obtained from baboons housed together and these are the only isolates obtained from animals known to be housed together. All isolates were either obtained from samples collected as part of previous studies or were obtained from samples submitted as part of field case investigations and did not require Institutional Animal Care and Use Committee (IACUC) approval. Frozen stocks (−80°C in 30% glycerol) of Bb isolates were received and then streaked onto tryptic soy agar containing 5% sheep blood (Becton, Dickinson and Co. Franklin Lakes, NJ) and incubated overnight at 37°C with 5% CO2. Classical bordetellae isolates used in the Bridel et al. (2022) study and previously characterized Bb strains RB50 and KM22, TN27 (KM22 ΔfhaB), and TN28 (KM22 Δprn) were also included in the analyses (Supplementary Table 1) (Parkhill et al., 2003; Nicholson et al., 2020; Nicholson et al., 2009).
Whole-genome sequencing, assembly, and annotation
WGS data was acquired from a variety of methods. For genomic DNA extraction from all isolates except M2020-2 and M2020-4, single colonies were inoculated into lysogeny broth (LB) and grown aerobically at 37°C overnight in a shaking incubator (250 rpm). The High Pure PCR Template Preparation Kit (Roche Diagnostics Corp., Indianapolis, IN) was used to purify total genomic DNA. The Qubit 1X dsDNA BR Assay Kit (Life Technologies, Eugene, OR) was used to assess quality standards and determine DNA concentration. WGS assemblies for isolates M2020-3, M2020-5, C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, and C2020-15 were obtained using Illumina short read data. Library preparation was performed using a custom Illumina TruSeq-style protocol (Arbor Biosciences, Ann Arbor, MI) and sequenced on an Illumina NovaSeq 6000 in 150 bp paired-end mode. The Illumina datasets were assessed for quality using FastQC1 and adapter trimming performed using BBduk.2 Assembly was performed using SPAdes v. 3.15.4 in --careful mode (Bankevich et al., 2012). Resulting assemblies were filtered to retain only contigs greater or equal to 1,500 bases in length.
For isolates M2020-2 and M2020-4, single colonies were inoculated into LB broth and grown overnight aerobically at 37°C. Bacterial cells were centrifuged at 5,000 rpm to pellet, washed in 10 mL PBS and pelleted again. The cell pellets were frozen overnight at-20°C. The pellets were resuspended in 2 mL of 25% sucrose in TE (10 mM Tris, 1 mM EDTA, pH 8.0). Fifty microliters of 100 mg/mL lysozyme (Roche, Indianapolis, IN) was added and the samples incubated for 1 h at 37°C. Next, 100 μL of 20 mg/mL Proteinase K (Roche, Indianapolis, IN), 30 μL RNase A (Roche, Indianapolis, IN), 400 μL 0.5 M EDTA (pH 8.0), and 250 μL of 10% sarkosyl were added to the samples, which were then incubated on ice for 2 h, followed by overnight incubation in a 50°C water bath. Sample volume was adjusted to 7.5 mL with TE and mixed with 5 mL phenol:chloroform:isoamyl alcohol (25:24:1), then centrifuged for 15 min at 4,000 rpm to separate the phases. The aqueous phase was transferred to a fresh tube and the phenol:chloroform:isoamyl alcohol phase separation was repeated twice. The aqueous phase was transferred to a new tube and mixed with an equal volume of chloroform:isoamyl alcohol (24:1), followed by centrifugation for 5 min at 4,000 rpm to separate the phases. The chloroform:isoamyl alcohol extraction was repeated one additional time. To the final aqueous phase, 2.5 volumes 100% ethanol was added and incubated at −20°C for 15 min to precipitate the DNA, followed by centrifugation at 4,000 rpm for 10 min. The pellet was washed with 10 mL cold 70% ethanol, air dried and resuspended in 250 μL TE. DNA concentrations were determined by UV spectrophotometry and assed for quality standards.
Complete genome assemblies for isolates M2020-2 and M2020-4 were obtained using a hybrid assembly approach combining Illumina short sequencing reads and PacBio long reads. For long read PacBio sequencing, library preparation was performed using the SMRTbell Express Template Prep Kit 2.0 (PacBio, Menlo Park, CA) and sequenced on a Sequel II system using an 8 M SMRT Cell. For short read Illumina sequencing, libraries were prepared using the KAPA Hyper Prep Kit (Roche Sequencing Solutions, Pleasanton, CA) and sequenced on an Illumina NovaSeq 6000 instrument in 150 bp paired-end mode. The Illumina data was assessed for quality using FastQC (see text footnote 1) and adapter trimming performed using BBduk (see text footnote 2). Assembly of the long read data was performed using Canu v. 2.2 (Koren et al., 2017) with default settings. Circlator v. 1.50 (Hunt et al., 2015) was used to generate a closed circular assembly with no overlaps and rotate the sequence to start at the dnaA gene. Pilon v. 1.23 (Walker et al., 2014) was used to polish the assembly using the Illumina short read data. Multiple rounds of Pilon polishing were performed, until no additional corrections were made.
Complete genome assembly of isolate C2020-8 was performed using a hybrid approach using a combination of long and short reads. Short read Illumina data was obtained using the same library preparation and sequencing as described above for the short-read only assemblies. For long-read PacBio sequencing, a HiFi Multiplex library with 10–20 kb insert size was prepared and sequenced on a Sequel II system using a SMRT Cell 8 M in CCS/HiFi mode. Long read data was assembled using Flye v. 2.9.1 with options --pacbio-hifi mode --asm-coverage 100-g 5.3 m (Kolmogorov et al., 2019), resulting in a closed, circular assembly. The Illumina data was then used to polish the assembly using Pilon v. 1.23 (Walker et al., 2014). The final assembly was rotated to start at the dnaA gene.
Isolates M2020-1 and C2020-7 were assembled using a hybrid approach combining Illumina short read data and Nanopore long read data. Short read Illumina data was obtained using the same library preparation and sequencing as described above for the short-read only assemblies. The genomic library for Nanopore sequencing was prepared with the SQK-RBK004 Rapid Barcoding Kit (Oxford Nanopore, Oxford, United Kingdom), following the manufacturer’s instructions. Sequencing was performed using a MinION Mk1C instrument with a MIN106D flow cell (version R9). The run length was 72 h and base calling was performed using Guppy v. 6.2.11 (high-accuracy mode, minimum read length of 200 bp, minimum Q score of 9).
Long read data was assembled using Flye v. 2.9.1 with settings --nano-raw-g 5.3 m (Kolmogorov et al., 2019) followed by error-correction with Medaka v. 1.11.1 (Oxford Nanopore, Oxford, United Kingdom). For both isolates, this resulted in assembly of a single, closed circular chromosome. The Illumina short read data was then used to polish the long read assemblies using Polypolish v. 0.5.0 and POLCA v. 4.1.0 as described in Wick et al. (2017) and Zimin and Salzberg (2020). The resulting assemblies were rotated to start at the dnaA gene.
Unless otherwise specified, default software settings were used. All assemblies were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v. 6.5 (all assemblies except C2020-7 and M2020-1) or v. 6.7 (C2020-7 and M2020-1) (Tatusova et al., 2016).
Comparative genomic analysis
Multi-locus sequence typing (MLST) was performed in silico utilizing the BIGSdb-Pasteur databases hosted by the Institut Pasteur.3 Average nucleotide identity (ANI) for analyzed genomes was calculated using FastANI, which uses an algorithm based on MinHash mapping to calculate pairwise genome-to-genome ANI values (Jain et al., 2018). In R, the ComplexHeatmap package (Gu et al., 2016) was used to generate a distance matrix heatmap clustered by hierarchical clustering using a complete linkage method with Euclidean distance.
The presence of the 9 bp deletion found in the complete genome assembly of M2020-4 bvgS compared to M2020-2 was verified by Sanger sequencing. Briefly, the bvgS gene was PCR amplified from 2 independent genomic DNA samples from each isolate using primers bvgSq1-for (5′-TGGTGGAACTGATAGACCTCGCCAAACGCAACAAT-3′) and bvgSq1-rev (5′-TACCGCCGCGAGATGCCGCCCAATTATCCGTAAGG-3′). PCR was performed using Platinum SuperFi II PCR Master Mix (ThermoFisher, Waltham, MA) with manufacturer’s recommended conditions. The PCR product was purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI) and sequenced using primers Sfor-3F (5′-ATCAGCGCCAGCTACTAC-3′) and SMS-5R (5′-GAGCTGCACGACGCCAAG-3′).
A custom database comprised of known Bordetella virulence-associated genes in Bb strain KM22 was used to identify corresponding homologues by BLASTN (Altschul et al., 1997) searches and the percent identity for each gene relative to the reference gene from Bb strain KM22 (accession # CP022962) was determined. Genes and locus tag information is provided in Supplementary Table 2.
Non-redundant or unique amino acid sequences of either FHA or PRN from analyzed Bordetella isolates containing a full-length nucleotide gene sequence were aligned using MAFFT (Katoh and Standley, 2013). Amino acid sequences were evaluated using hierarchical clustering based on the pairwise percent identity between isolates. Clustering was performed in R using the ComplexHeatmap package, with complete linkage and Euclidean distance as the clustering method (Gu et al., 2016). The amino acid sequences were additionally used to infer approximately-maximum-likelihood phylogenetic trees with FastTree (v2.11) and iTOL (Price et al., 2010; Letunic and Bork, 2024).
O-antigen loci were identified by a BLASTN (Altschul et al., 1997) search for the genes known to flank the O-antigen locus, using the RB50 genes BB0120 and wlbL/BB0145 (Vinogradov et al., 2010) as the query sequences. Sequences were compared to the known O-antigen Bb serotypes O1, O2, and O3 and the B. parapertussis Bpp5 O-antigen locus (Vinogradov et al., 2010; Hester et al., 2013; Hester et al., 2015) by MAFFT alignment (Katoh and Standley, 2013).
To determine the presence of the cya or ptp loci, in silico PCR was performed in Geneious Prime 2023.0.14 using the primer sets described in Sukumar et al. (2014) to identify the respective operons and further analyze genomic regions encoding these loci.
Comparison of genes encoding fimbrial protein subunits within the fimNX region was performed by BLASTN search (Altschul et al., 1997) to identify the region and MAFFT alignment (Katoh and Standley, 2013) to categorize the gene families.
Phenotypic and genomic AMR analysis
Phenotypic antibiotic resistance was determined using the broth microdilution method by National Veterinary Services Laboratories (Ames, IA) following standard operating procedures. Minimum inhibitory concentrations (MICs) were determined for each isolate using the Trek BOPO7F and GNX3F plates (Thermo Fisher Scientific Inc., Oakwood Village, OH) with Escherichia coli ATCC 25922 (ATCC, Manassas, VA) serving as the quality control strain. MICs were evaluated in accordance with Clinical Laboratory Standards Institute (CLSI) recommendations based on the VET01S and M100Ed34 standards for resistance interpretations after incubation for 24 h (CLSI, 2024a; CLSI, 2024b; Pruller et al., 2015).
Result interpretation breakpoints used for Bb were values provided by the CLSI guidelines for ampicillin, clindamycin, florfenicol, tildipirosin, tulathromycin (CLSI, 2024a). Since breakpoints specific to Bb are limited, breakpoints used for treating Pseudomonas aeruginosa infections were used for amikacin, gentamicin, aztreonam, doripenem, imipenem, meropenem, piperacillin/tazobactam, ticarcillin/clavulanic acid, cefepime, cefotaxime, ceftazidime, ciprofloxacin, levofloxacin, colistin, polymixin B, doxycycline, minocycline, tetracycline, tigecycline (CLSI, 2024a; CLSI, 2024b). Breakpoints used for treating Pasteurella multocida infections were used for neomycin, spectinomycin, penicillin, ceftiofur, danofloxacin, enrofloxacin, gamithromycin, tiamulin (CLSI, 2024a). Breakpoints used for treating Mannheimia haemolytica infections were used for tilmicosin (CLSI, 2024a). Isolates were considered resistant to sulfadimethoxine when MIC was equal to or exceeded 256 μg/mL, and isolates were considered resistant to trimethoprim/sulfamethoxazole when MIC exceeded 2 μg/mL (Vilaro et al., 2023). Antimicrobial susceptibility data (AST), along with test rages and clinical breakpoints used for interpretations, for all isolates are listed in Supplementary Table 3.
An in silico search for antimicrobial resistance (AMR) genes was performed using AMRFinderPlus v. 3.12.8 with database version 2024-05-02.2 (Feldgarden et al., 2019). The input option --nucleotide was used to analyze the assembled genome FASTA sequences with default settings. The database used the blaBOR protein sequence from B. bronchiseptica RB50 as the reference for the search (accession WP_010926363.1). The RB50 blaBOR nucleotide sequence (locus_tag BB2049) was then employed to compare to the blaBOR gene sequences identified in the Bb primate isolates.
Western blots
Selected isolates were grown on Bordet-Gengou (BG) agar (Difco, Sparks, MD) + streptomycin (40 μg/μL) plates, and a single colony was inoculated into 3 mL Stainer-Scholte broth + 40 μg/μL streptomycin (SS/Sm) and grown aerobically overnight at 37°C. This overnight culture was then used to inoculate 10 mL SS/Sm broth to an OD600 of 0.05, which was grown aerobically overnight at 37°C. The cultures were harvested by centrifugation, and the cell pellets were resuspended in 1/5th volume 8 M urea. The lysates were heated to 95°C for 10 min, then cooled to room temperature. The Qubit Protein BR Kit (Thermo Fisher, Rockford, IL) was used to determine protein concentration and the lysates stored at −80°C prior to use, the lysates were diluted in 4X LDS Sample Buffer (Life Technologies, Carlsbad, CA) to 500 ng/μL or 250 ng/μL in 1X LDS + 4 M urea. SDS-PAGE was performed using NuPAGE 7% Tris-Acetate gels (Life Technologies, Carlsbad, CA) loaded and run under denaturing conditions according to the manufacturer’s recommendations. Transfer to PVDF membranes was performed using an Xcell Blot Module (Life Technologies, Carlsbad, CA) following manufacturer’s instructions. Membranes were blocked for 1 h at room temperature in 1X PBS + 0.1% Tween-20 + 5% non-fat dry milk. Primary antibody incubations were performed overnight at 4°C in 1X PBS + 0.1% Tween-20 + 5% non-fat dry milk. Membranes were washed 4 × 5 min in 1X PBS + 0.1% Tween-20. Secondary antibody incubations were for 2 h at room temperature in 1X PBS + 0.1% Tween-20 + 2.5% non-fat dry milk. Membranes were washed 4 × 5 min in 1X PBS + 0.1% Tween-20 and then incubated with SuperSignal West Pico PLUS Chemiluminescent Substrate (Life Technologies, Carlsbad, CA) per manufacturer’s protocol. Digital images were obtained using an Azure 600 imager (Azure Biosystems, Dublin, CA). KM22 FHA residues 400–1,025 were used to generate mouse polyclonal α-FHA-heparin binding domain antibodies and KM22 FHA residues 2,000–2,525 were used to generate mouse polyclonal α-FHA-mature C-terminal domain antibodies (Genscript United States, Inc., Piscataway, NJ). Primary antibodies to FHA were used at 1:500 dilutions and detected with the secondary antibody goat anti-mouse IgG-HRP 31432 (Life Technologies, Carlsbad, CA) used at a 1:10,000 dilution. The primary antibody to pertactin, α-PRN 29854, is a rabbit polyclonal antibody generated against purified PRN from a Bb swine isolate produced at the NADC and was used at a 1:5,000 dilution and detected with the secondary antibody goat anti-rabbit IgG-HRP 4030-05 (Southern Biotech, Birmingham, AL).
Data availability
The genome assemblies have been deposited at DDBJ/ENA/GenBank under the following BioProject accessions: PRJNA954421, PRJNA954729, PRJNA954420, and PRJNA954735. The sequencing data has been deposited in the Sequence Read Archive (SRA) under the following accession numbers: SRP435095, SRP435093, SRP435092, and SRP435108. Detailed information regarding BioSample, GenBank, and SRA accession numbers is provided in Supplementary Table 1.
Results
Phylogenetic analysis of Bb primate isolates
MLST was performed to begin characterizing the Bb primate isolates. One new fumC allele was identified in baboon isolates M2020-1, M2020-5, and M2020-3 and one new tyrB allele was identified in all rhesus monkey isolates (C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-8, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, and C2020-15) except for C2020-7. Baboon isolates M2020-2 and M2020-4 were determined to be ST67 and M2020-1, M2020-5, and M2020-3 were assigned to ST111, a new ST. Isolate C2020-7 was determined to be ST33, while all other rhesus monkey isolates were assigned to ST109, a new ST (Table 1).
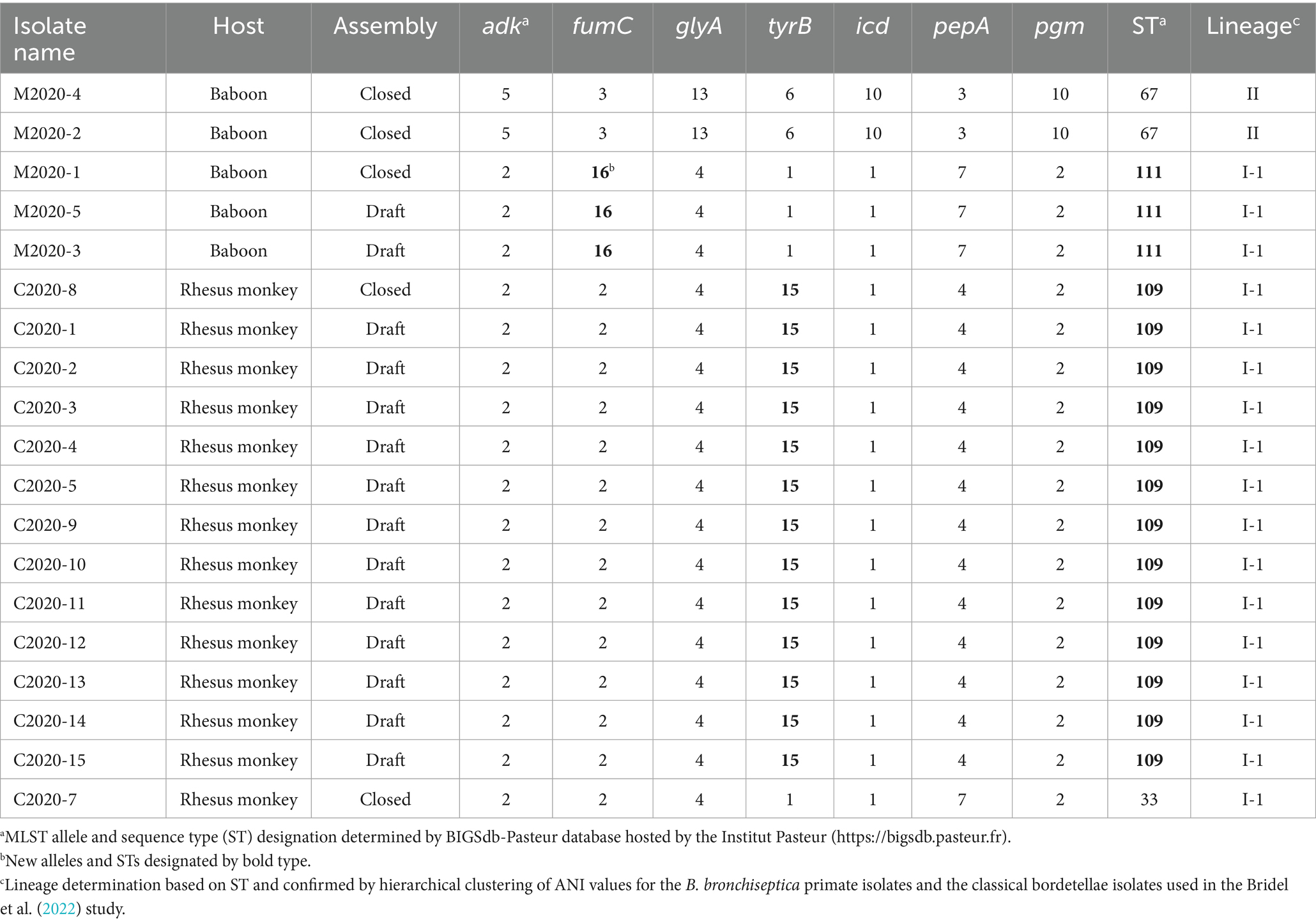
Table 1. Isolate information, MLST allele, sequence type (ST), and lineage designation.
Based on the phylogenetic tree from the cgMLST-based typing method developed by Bridel et al. (2022), baboon isolates M2020-2 and M2020-4 would group into lineage II since both were determined to be ST67, while all other isolates would group into lineage I-1 since they were ST33, ST109, and ST111 (Table 1). To further confirm the lineage for the Bb primate isolates, average nucleotide identity (ANI) values were determined for the Bb primate isolates and the classical bordetellae isolates used in the Bridel et al. (2022) study. Hierarchical clustering of ANI values demonstrated that isolates M2020-1, M2020-5, M2020-3, C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-8, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, and C2020-15 were lineage I-1 isolates and isolates M2020-2 and M2020-4 were linage II isolates (Figure 1).
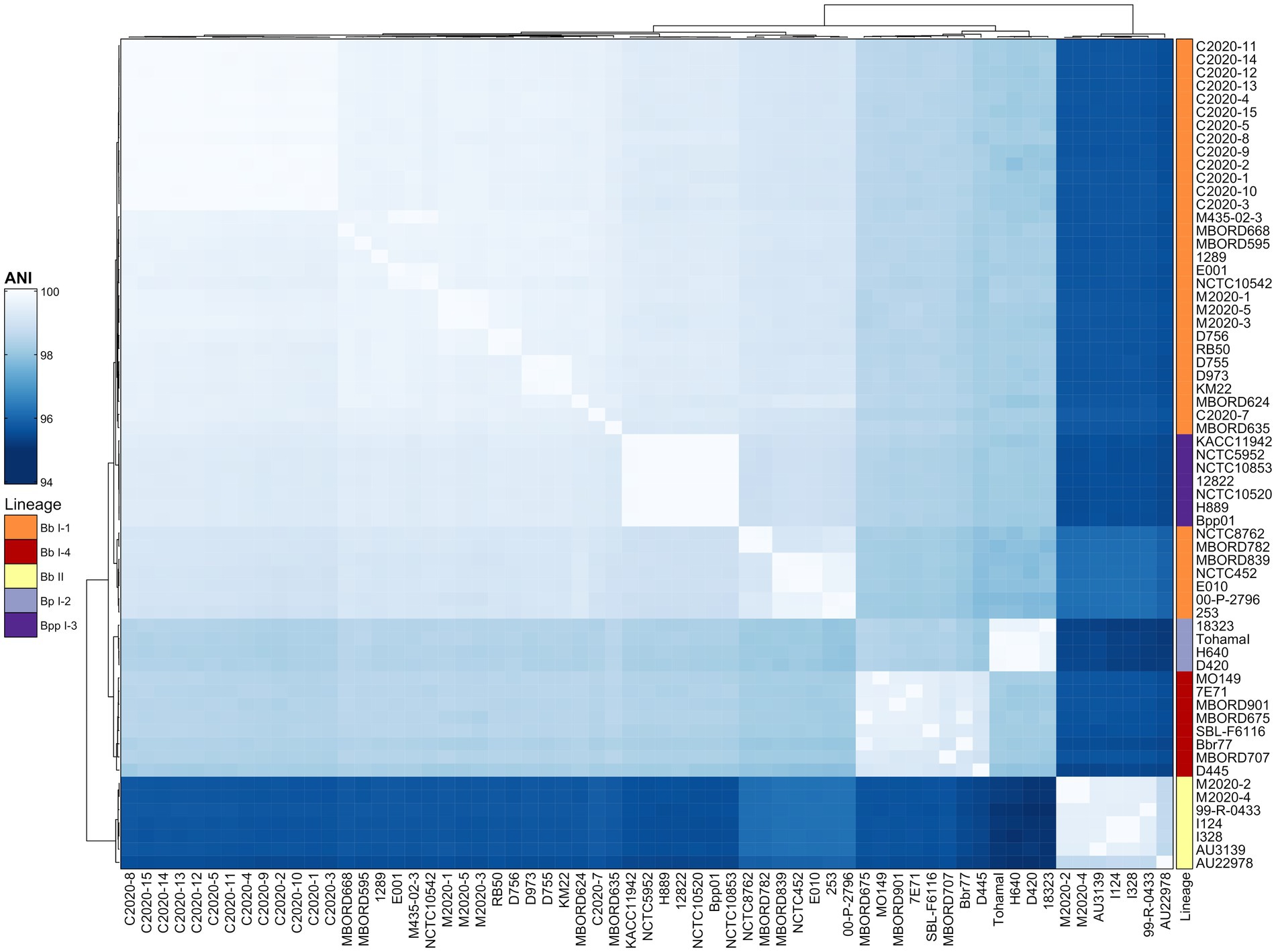
Figure 1. Hierarchically clustered heatmap displaying the relatedness of Bordetella isolates based on ANI. Pairwise ANI values for analyzed genomes were used to generate a distance matrix heatmap clustered by hierarchical clustering using a complete linkage method with Euclidean distance. Isolate names are provided at the bottom and right side of heatmap. ANI is represented using the color scale legend shown at the left of the heatmap. Dendrograms are on the left and top sides of the heat map. Lineage designations B. bronchiseptica (Bb I-1, Bb I-4, Bb II), B. parapertussis (Bpp I-3), and B. pertussis (Bp I-2) are shown alongside the isolate names on the right side of the heatmap colored according to the legend shown at the left of the heatmap.
The genome sequences for M2020-2 and M2020-4 were almost identical. The genome sequence assembly for M2020-2 is only 6 bp larger than M2020-4. The differences between the final genome assemblies for these isolates include several nucleotide differences in intergenic regions located in polynucleotide tracts upstream of fim2, fimN, bipA, and fim3, as well as a 9-bp deletion in bvgS for M2020-4. The deletion in the bvgS for M2020-4 corresponds to bps 2,270–2,278 in the bvgS sequence for M2020-2 and results in amino acids 758, 759, and 760 missing in BvgS for M2020-4. M2020-4 displays a Bvg-colony morphology, which is likely the result of the mutations in BvgS.
Comparative analysis of genes encoding BvgAS, FHA, and PRN
While Bb strain RB50 has routinely been used as a reference strain in Bb studies, KM22 was chosen as the reference strain in this study because it shares numerous traits and characteristics that are more consistent with the vast majority of Bb isolates for which genomic data is available in GenBank. KM22 is a ST7 and ST7 is the most common Bb ST in GenBank, whereas RB50 is ST12. Similar to other Bb isolates, KM22 contains functional genes for tcfA, brkA, bapC, and putative-adhesin 2, while genes tcfA, brkA, and bapC are pseudogenes in RB50 and putative-adhesin 2 is absent from the RB50 genome. Gene content at the fimNX locus also differs between KM22 and RB50, with KM22 having four fimbrial subunit genes in the locus and RB50 having three genes. The three gene arrangement is uncommon among Bb isolates. Additionally, similar to the majority of Bb isolates, KM22 harbors genes encoding for an O-antigen serotype O2, while RB50 harbors genes encoding for an O-antigen serotype O1.
To examine genomic differences that may influence how the Bb primate isolates interact with their hosts and environment, we compared the nucleotide sequences of genes encoding well-characterized regulatory and virulence factors. Lineage II baboon isolates M2020-2 and M2020-4 harbored the lowest sequence identity for all regulatory and virulence-associated genes evaluated (Table 2; Supplementary Table 2). The nucleotide sequence identity for bvgA ranged from 95.08 to 100% and for bvgS the identity ranged from 90.25 to 99.97% (Table 2).
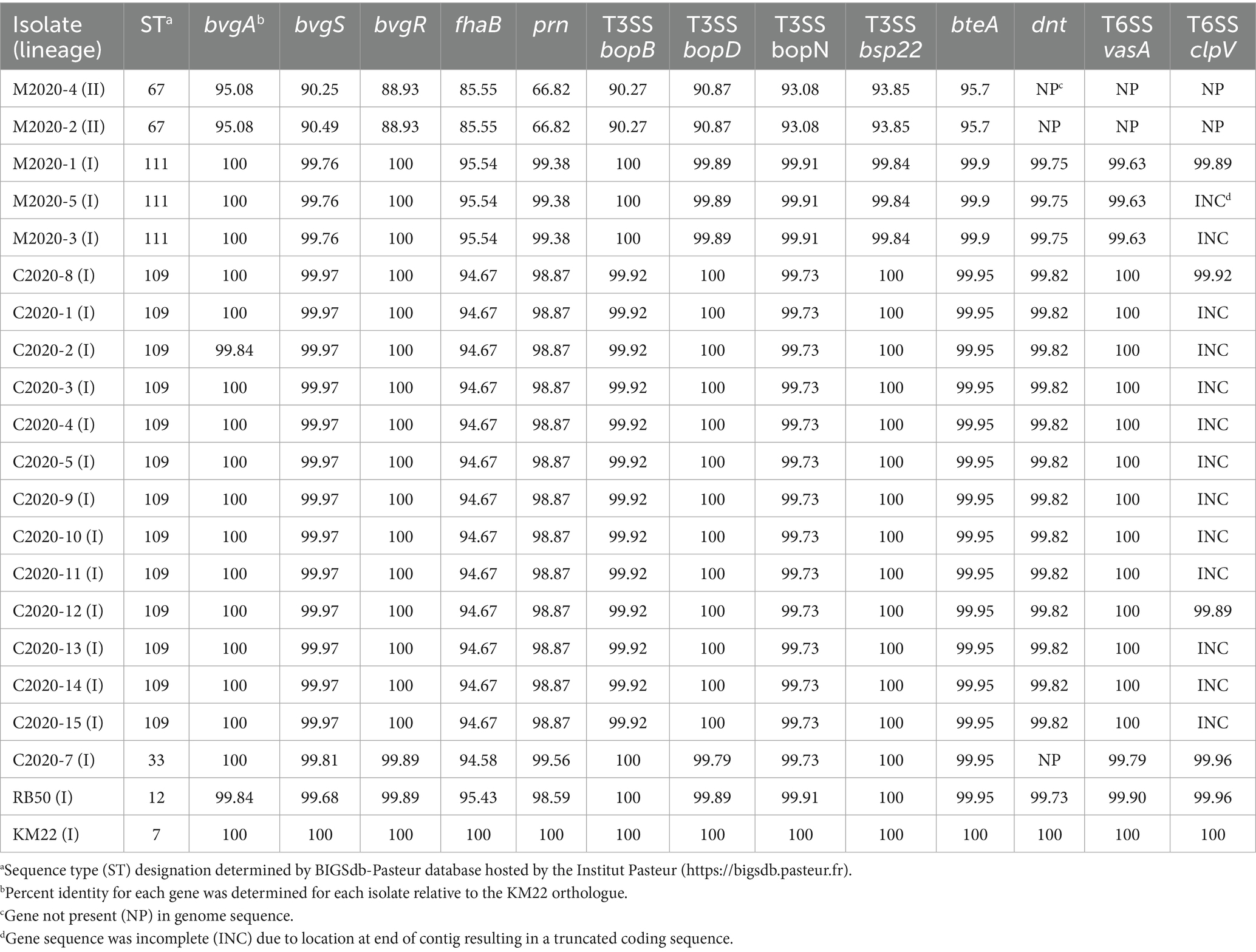
Table 2. Virulence-associated genes.
The nucleotide sequence identity relative to KM22 for fhaB, which encodes for FHA, ranged from 85.55 to 95.54% (Table 2). Primate isolates with the same ST harbored identical fhaB gene sequences. The fhaB nucleotide sequence from isolate C2020-7 (ST33) was slightly different compared to the other rhesus monkey isolates (Table 2).
To further examine the diversity of FHA, sequence alignments were used to determine pairwise amino acid (AA) identity for FHA sequences from the primate isolates as well as the classical bordetellae isolates used in the Bridel et al. (2022) study. The AA identity was then used to generate a distance matrix heatmap clustered by hierarchical clustering, which revealed a correlation between FHA AA identity and bordetellae lineage (Figure 2). Specifically, the B. parapertussis (Bpp) lineage I-3 isolates cluster together, the B. pertussis (Bp) lineage I-2 isolates cluster together, and the Bb lineage I-1 isolates cluster together. Two Bb lineage I-4 isolates cluster together and between Bpp lineage I-3 isolates and Bp lineage I-2 isolates, while the other two Bb I-4 isolates cluster together and between and Bb lineage II isolates and Bb I-1 isolates. All Bb lineage II isolates, including baboon isolates M2020-2 and M2020-4, cluster together. The FHA AA identity is conserved among the lineage II isolates and divergent from the FHA AA identity from all other isolates (Figure 2). A phylogenetic tree inferred that the FHA AA sequences resulted in an overall similar tree topology to the lineage group distribution represented in the hierarchically clustered heatmap (Supplementary Figure 1A).
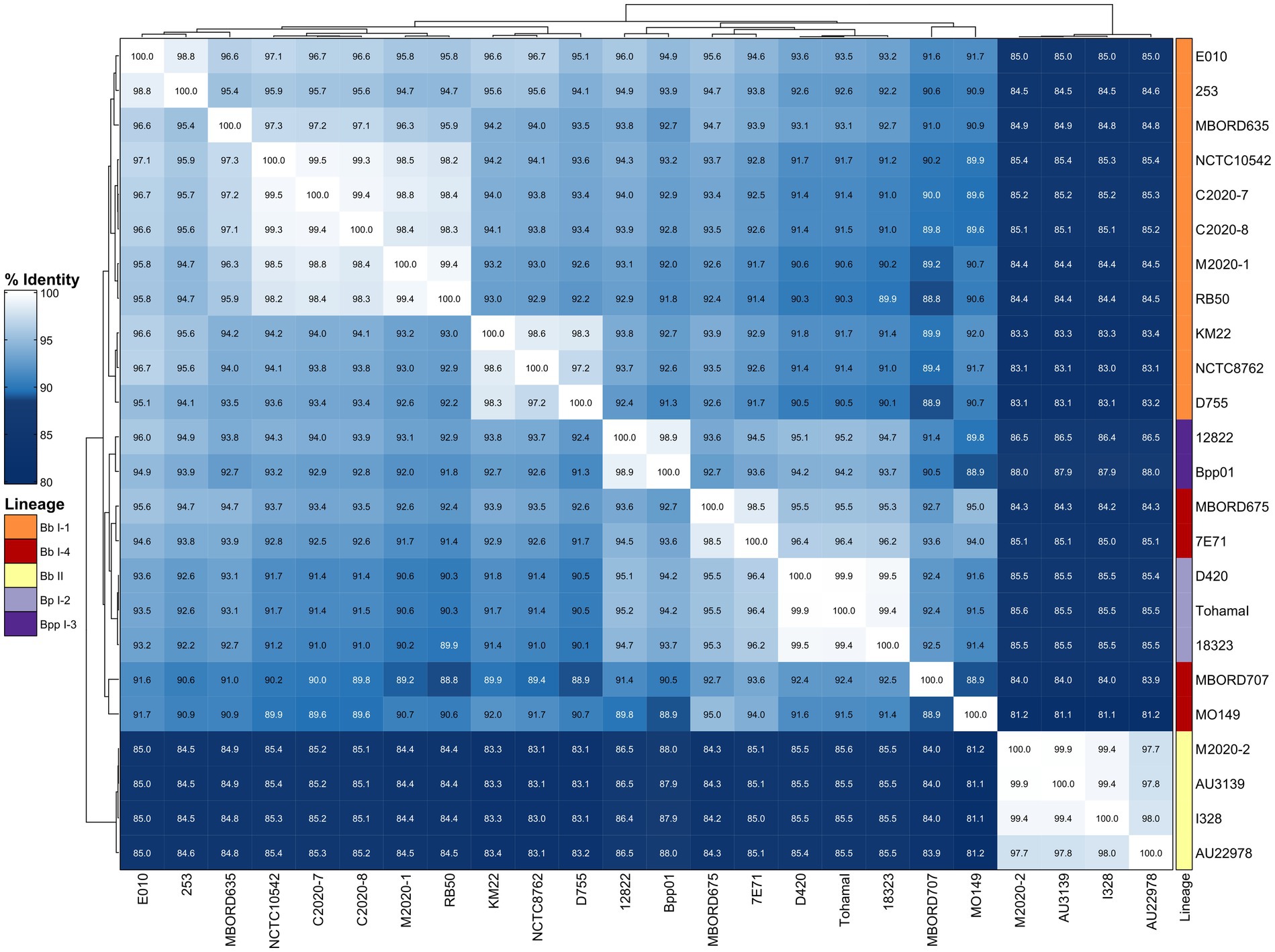
Figure 2. Hierarchically clustered heatmap displaying the relatedness of Bordetella isolates based on FHA AA identity. Pairwise AA percent identity of unique FHA sequences from isolate genomes containing a full-length fhaB gene was determined and used to generate a distance matrix heatmap clustered by hierarchical clustering using a complete linkage method with Euclidean distance. Isolate names are provided at the bottom and right side of heatmap. FHA AA identity is represented using the color scale shown left of the heatmap. Dendrograms are on the left and top sides of the heat map. Lineage designations B. bronchiseptica (Bb I-1, Bb I-4, Bb II), B. parapertussis (Bpp I-3), and B. pertussis (Bp I-2) are shown alongside the isolate names on the right side of the heatmap colored according to the legend shown left of the heatmap.
Given that primate isolates with the same ST harbored identical fhaB gene sequences, the FHA protein sequence from isolates M2020-1 (ST111), C2020-7 (ST33), C2020-8 (ST109), and M2020-2 (ST67) were used for further analyses. Protein alignments demonstrated that the AA differences compared to KM22 FHA for isolate M2020-2 (ST67), as well as all lineage II isolates, is observed throughout the whole protein sequence and not isolated to specific domains (Supplementary Figure 1B; Figure 3). While the FHA protein sequence from M2020-1 (ST111), C2020-7 (ST33), and C2020-8 (ST109) is more similar to KM22 than the FHA protein sequence from M2020-2 (ST67), there are AA differences compared to KM22 throughout the protein sequence with most located toward the C-terminal prodomain, which is removed during processing (Jungnitz et al., 1998; Coutte et al., 2001; Mazar and Cotter, 2006) (Figure 3).
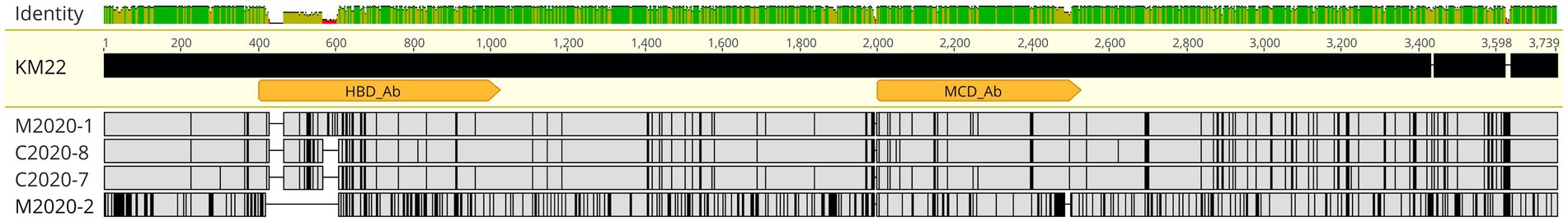
Figure 3. FHA alignment. MAFFT alignment of FHA protein sequences from Bb isolates M2020-1, C2020-8, C2020-7, and M2020-2. FHA from strain KM22 served as the reference sequence. Orange arrows indicate the FHA region corresponding to residues 400–1,025 used to generate polyclonal α-FHA antibodies that recognize the HBD and the region corresponding to residues 2,000–2,525 used to generate polyclonal α-FHA antibodies that recognize the MCD. In the aligned sequences, vertical black lines represent residues that differ from the KM22 FHA reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
Further focusing on specific domains, many AA differences compared to KM22 were observed for isolates M2020-1 (ST111), C2020-7 (ST33), C2020-8 (ST109), and M2020-2 within the Heparin Binding Domain (HBD) corresponding to residues 400–1,025 (Figure 3). Similarly, several AA differences compared to KM22 were observed for isolates M2020-1 (ST111), C2020-7 (ST33), C2020-8 (ST109), and M2020-2 (ST67) within the Mature C-terminal Domain (MCD) corresponding to residues 2,000–2,525. More AA differences compared to KM22 within both domains were observed for M2020-2 (ST67) (Figure 3). These observed AA differences led us to test whether antibodies raised against the HBD or the MCD of FHA from KM22 would recognize FHA from these isolates.
Western blot analysis of whole-cell lysates of KM22, TN27 (KM22 containing an in-frame deletion of the fhaB gene) (Nicholson et al., 2009), C2020-7, M2020-1, M2020-2, and C2020-8 revealed the presence of several large ~250 kDa polypeptides indicative of multiple processed FHA proteins (Delisse-Gathoye et al., 1990; Domenighini et al., 1990; Coutte et al., 2001; Mazar and Cotter, 2006; Jurnecka et al., 2018) in all isolates except TN27, when using α-FHA antibodies generated against the MCD of KM22 (Figure 4A). In contrast, western blot analysis using α-FHA antibodies generated against the HBD of KM22 revealed the presence of several processed FHA proteins of ~250 kDa only in whole-cell lysates of KM22, C2020-7, M2020-1, and C2020-8 and no FHA proteins of ~250 kDa in whole-cell lysates of TN27 and M2020-2 (Figure 4B). These data demonstrate that antibodies raised against the MCD of FHA from KM22 recognize FHA from these isolates despite AA differences compared to KM22, while antibodies raised against the HBD of FHA from KM22 do not recognize FHA from M2020-2.
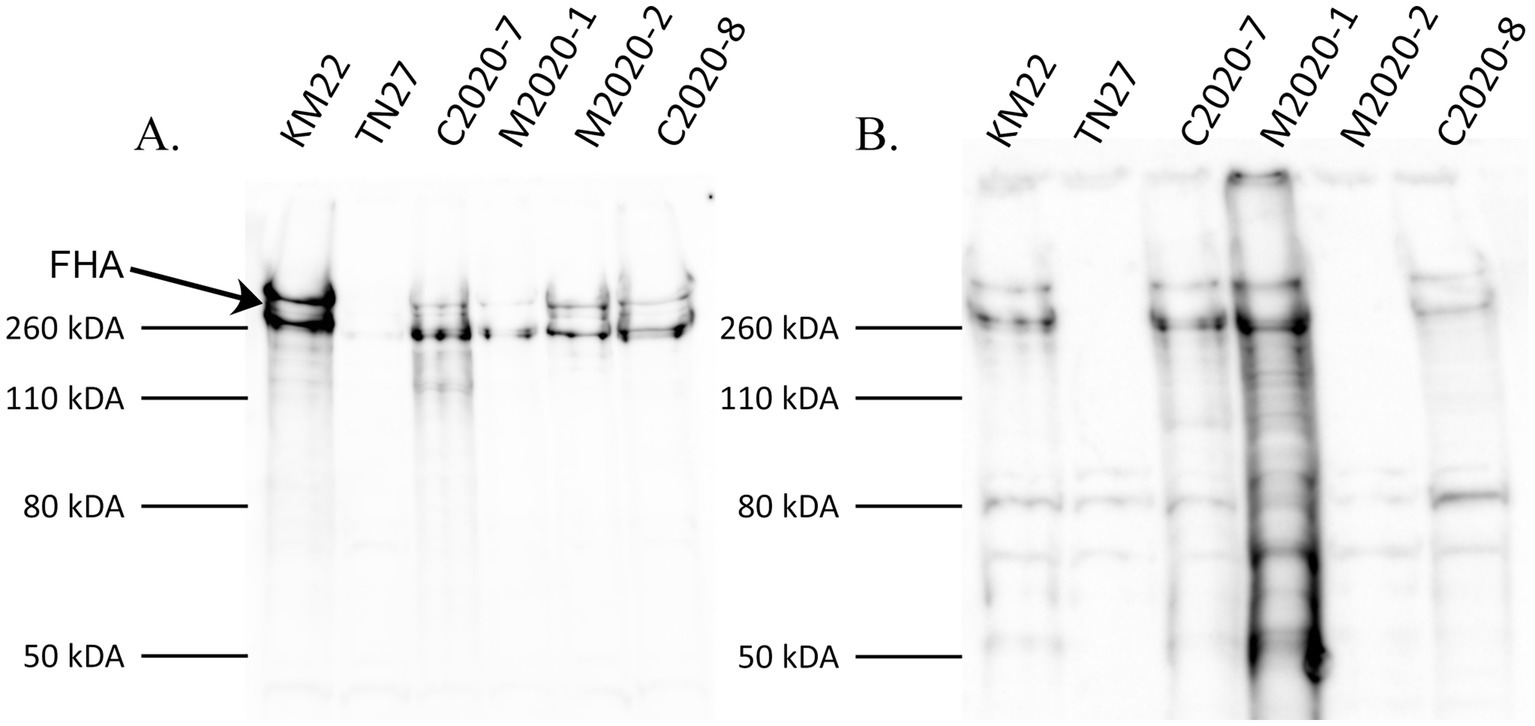
Figure 4. Western blot analysis using α-MCD and α-HBD KM22 antibodies. Western blot analysis of whole-cell bacterial lysates from isolates KM22, TN27 (ΔfhaB), C2020-7, M2020-1, M2020-2, and C2020-8. Membranes were probed with α-MCD (A) and α-HBD (B) antibodies. Sizes are indicated in kilodaltons on the left and the large ~250 kDa polypeptides indicative of multiple processed FHA proteins are indicated.
The nucleotide sequence identity for prn, which encodes for PRN, ranged from 66.82 to 99.56%, and similar to fhaB sequences, primate isolates with the same ST harbored identical prn gene sequences (Table 2). The prn nucleotide sequences from lineage II baboon isolates M2020-2 and M2020-4 (both ST67) were identical and had the lowest observed identity compared to KM22 at 66.82%. The low prn gene sequence identity observed for the baboon lineage II isolates M2020-2 and M2020-4 suggests that the gene identified as prn in these isolates might not be a pertactin orthologue. Given that the gene identified as prn in these isolates was the only potential prn gene identified by BLASTN search methods, it was included in this and subsequent analyses. The prn nucleotide sequences from the 13 ST109 rhesus monkey isolates were identical to each other. The prn nucleotide sequence from rhesus monkey isolate C2020-7 (ST33) was slightly different compared to the other ST109 rhesus monkey isolates (Table 2).
Similar to the correlation between FHA AA identity and bordetellae lineage, hierarchical clustering of primate and classical bordetellae isolates based on pairwise PRN AA identity, indicated a correlation between PRN AA identity and bordetellae lineage. The PRN AA identity is conserved among the Bb lineage II isolates and divergent from the PRN AA identity from all other isolates (Figure 5). A phylogenetic tree inferred from the PRN AA sequences resulted in an overall tree topology that correlated with the lineage group distribution represented in the hierarchically clustered heatmap (Supplementary Figure 2A).
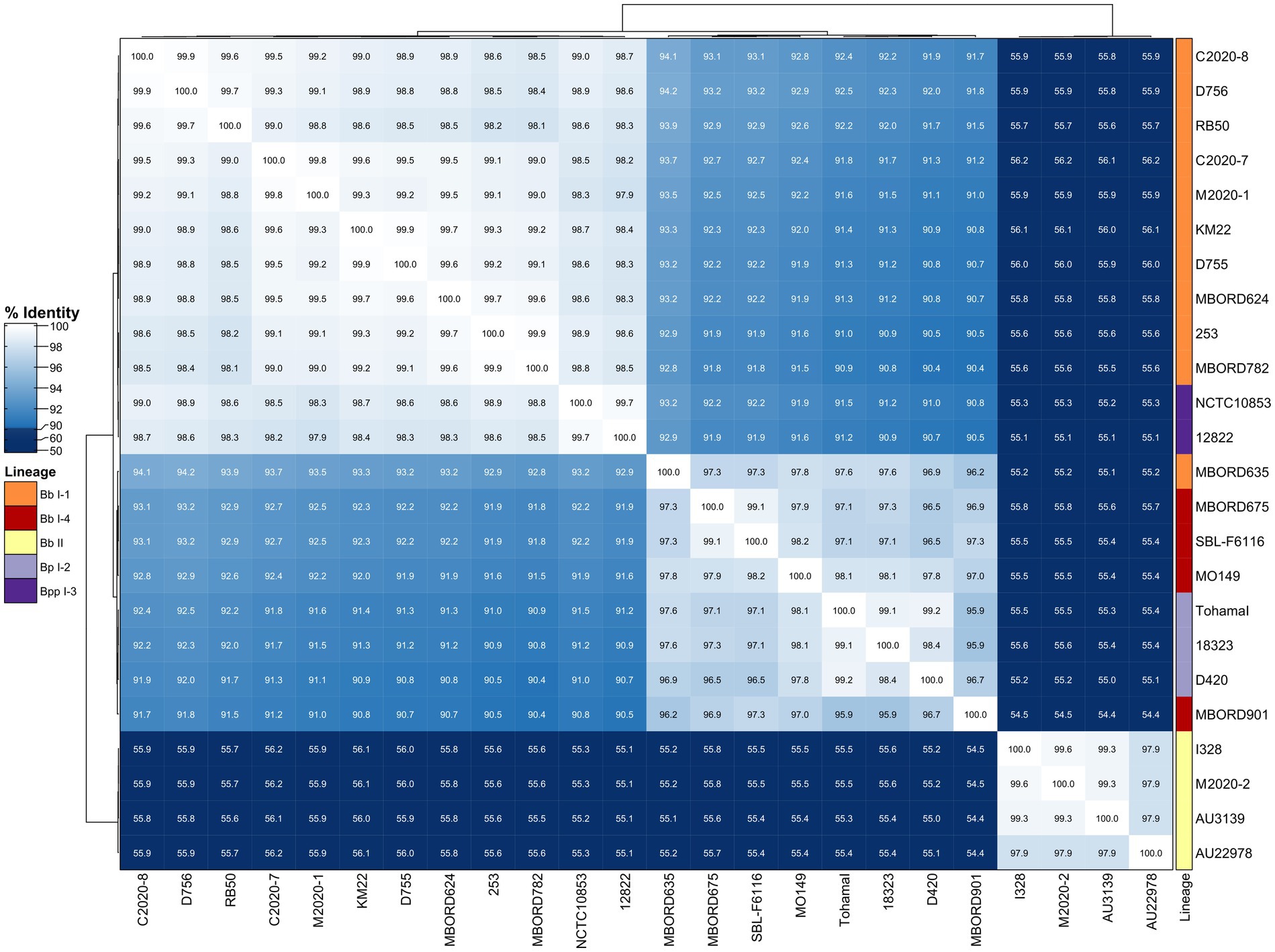
Figure 5. Hierarchically clustered heatmap displaying the relatedness of Bordetella isolates based on PRN AA identity. Pairwise AA percent identity of unique PRN sequences from isolate genomes containing a full-length prn gene was determined and used to generate a distance matrix heatmap clustered by hierarchical clustering using a complete linkage method with Euclidean distance. Isolate names are provided at the bottom and right side of heatmap. PRN AA identity is represented using the color scale shown left of heatmap. Dendrograms are on the left and top sides of the heat map. Lineage designations B. bronchiseptica (Bb I-1, Bb I-4, Bb II), B. parapertussis (Bpp I-3), and B. pertussis (Bp I-2) are shown alongside the isolate names on the right side of the heatmap colored according to the legend shown left of heatmap.
PRN sequences from isolates M2020-1 (ST111), C2020-7 (ST33), C2020-8 (ST109), and M2020-2 (ST67) were used in alignments and demonstrated that while AA differences compared to KM22 PRN were not located within specific domains, greater sequence diversity was observed within the first two-thirds of the protein and more sequence conservation was observed toward the C-terminal for isolate M2020-2 (ST67), as well as all lineage II isolates (Supplementary Figure 2B; Figure 6).
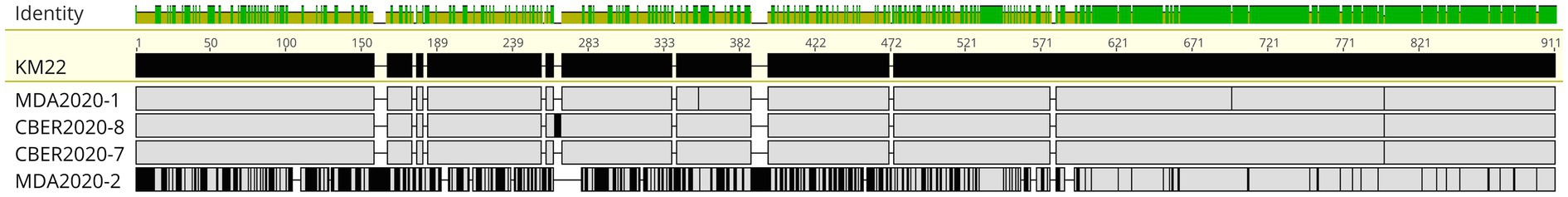
Figure 6. PRN alignment. MAFFT alignment of PRN protein sequences from Bb isolates M2020-1, C2020-8, C2020-7, and M2020-2. FHA from KM22 served as the reference sequence. In the aligned sequences, vertical black lines represent residues that differ from the KM22 FHA reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
Further evaluation of the PRN AA alignment between KM22 and M2020-2 showed that PRN from M2020-2 contains two RGD domains similar to KM22 (data not shown). However, the first RGD domain PRN from M2020-2 is located closer to the N-terminal and does not align with the first RGD domain in PRN from KM22 (data not shown). The second RGD domain PRN from M2020-2 does align with the second RGD domain in PRN from KM22 (data not shown). The second RGD domain for both proteins is located in the more conserved C-terminal end, which is not present in the mature form of PRN (Gotto et al., 1993; Charles et al., 1994). In contrast, PRN protein sequences from M2020-1 (ST111), C2020-7 (ST33), and C2020-8 (ST109) were highly similar to KM22 (Figure 6). The observed AA differences led us to test whether antibodies raised against PRN would recognize PRN from M2020-2 (ST67).
Western blot analysis of whole-cell lysates of KM22, TN28 (KM22 containing an in-frame deletion of the prn gene) (Nicholson et al., 2009), C2020-7, M2020-1, M2020-2, and C2020-8 revealed the presence of a ~69 kDa polypeptide indicative of PRN only in whole-cell lysates of KM22, C2020-7, M2020-1, and C2020-8 when using α-PRN antibodies generated against the purified PRN from a Bb swine isolate (Figure 7). Contrastingly, no PRN was observed in whole-cell lysates of TN28 and M2020-2 when using α-PRN antibodies (Figure 7). These data demonstrate that antibodies raised against PRN from a Bb swine isolate recognize PRN from KM22, C2020-7, M2020-1, and C2020-8 and do not recognize PRN from M2020-2.

Figure 7. Western blot analysis using α-PRN antibodies. Western blot analysis of whole-cell bacterial lysates from isolates KM22, TN28 (Δprn), C2020-7, M2020-1, M2020-2, and C2020-8. Membranes were probed with α-PRN antibodies. Sizes are indicated in kilodaltons on the right.
PRN repeat region typing is a discriminatory method used for genotyping Bb isolates based on the frequency of GGXXP and PQP repeat regions (Register, 2004; Register, 2001; Boursaux-Eude and Guiso, 2000). The regions containing GGXXP and PQP repeats could not be amplified in silico using PCR typing schemes described by Register (2001) and Register (2004) for lineage II isolates M2020-2 and M2020-4 due to lack of sequence conservation indicating that attempts to obtain PCR amplicons of these regions would likely be unsuccessful. Further in silico analysis of these regions found no GGXXP in the PRN sequence in any lineage II isolates (M2020-2, M2020-4, I328, I124, AU3139, or AU22978) and PQP was found only once in the PRN of these isolates. In addition to having a divergent prn gene sequence, the prn gene is located in a different genomic region in lineage II isolates compared to lineage I Bb, Bp, and Bpp isolates (Figure 8). As depicted in Figure 8, the prn gene is located in between predicted siroheme synthase (cobA /cysG) and uracil phosphoribosyltransferase (upp) genes for lineage I Bb isolates KM22 (lineage I-1), MO149 (lineage I-4), Bp D420 (lineage I-2), and Bpp 12,822 (lineage I-3) (Figure 8A). Bp D420 (lineage I-2) harbors a predicted transposase between prn and the upp gene (Figure 8A). There is no predicted gene located between these genes or within the similar genomic region in lineage II isolates I328 and M2020-2 (Figure 8B), with the flanking genes located directly adjacent to each other. In contrast, the prn gene is located in between two genes predicted to encode for enoyl-CoA hydratase for lineage II isolates I328 and I328 and M2020-2 (Figure 8B). There is no predicted gene located between the two genes predicted to encode for enoyl-CoA hydratase or within the similar genomic region in lineage I isolates (Figure 8A); the enoyl-CoA hydratase genes are directly adjacent to one another in lineage I isolates.

Figure 8. Genomic organization and alignment of the region containing the prn gene. MAFFT alignment of the region containing the prn gene for isolates KM22, MO149, D420, and 12,822 (A) and I328 and M2020-2 (B). Bb isolate and (lineage) shown at left. In the aligned sequences, the prn gene (yellow) and flanking genes (blue) are shown with vertical black lines that represent residues that differ from the KM22 reference. Base pair numbers are displayed at top. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
Comparative analysis of type III and type VI secretion systems
Focusing on type III secretion system (T3SS) genes, nucleotide sequence identity for bopB ranged from 90.27 to 100%, bopD ranged from 90.87 to 100%, bopN ranged from 93.08 to 99.91%, bsp22 ranged from 93.85 to 100%, bteA ranged from 95.27 to 99.95% (Table 2). Primate isolates with the same ST harbored identical bopB, bopD, bopN, and bteA gene sequences. However, the bsp22 nucleotide sequences from all rhesus monkey isolates were identical to the reference KM22 sequence and baboon isolates with the same ST harbored identical bsp22 gene sequences (Table 2).
The entire type VI secretion system (T6SS) locus was absent in the genomes of lineage II baboon isolates M2020-2, M2020-4; however, genes within the T6SS locus were highly conserved in all other lineage I primate isolates. The T6SS genes vasA and clpV listed in Table 2 reflect the observed conservation among all T6SS genes (Supplementary Table 2). The nucleotide sequence identity compared to KM22 for vasA ranged from 99.63 to 100% % and the nucleotide sequence identity for clpV ranged from 99.89 to 99.96% relative to KM22 among the genomes in which a complete clpV gene sequence could be compared (Table 2). The clpV gene was truncated at the end of contig for several genomes thus limiting comparative analysis. Lineage I primate isolates with the same ST harbored identical vasA gene sequences (Table 2). Additionally, lineage I primate isolates with the same ST harbored identical gene sequences for all T6SS genes for which gene sequences could be compared (Supplementary Table 2). T6SS-4 and T6SS-6 were the most conserved T6SS genes among all isolates and were 100% identical to the KM22 reference orthologue (Supplementary Table 2).
Diversity among the fimbriae and adhesin genes
Next, we investigated the nucleotide diversity of predicted fimbriae and adhesin genes among the primate isolates. The nucleotide sequence identity for each isolate relative to the KM22 orthologue is shown in Table 3. Isolates M2020-2 and M2020-4 did not harbor genes fimN, fimbrial protein 1, bcfA/putative adhesin 1, putative adhesin 2 (Table 3 and Supplementary Table 2). All other predicted fimbriae and adhesin genes harbored by isolates M2020-2 and M2020-4 had the lowest sequence identity observed (Table 3; Supplementary Table 2). Genes fim2-like and putative-adhesin 2 were absent from RB50. Overall, a high degree of conservation was observed for predicted fimbriae and adhesin genes among lineage I baboon isolates M2020-1, M2020-5, and M2020-3 and rhesus monkey isolates C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-8, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, and C2020-15. The nucleotide sequence for genes fim2, fim3, fimC, fimD, fimbrial protein 2, and putative-adhesin 2 for rhesus monkey isolate C2020-7 were slightly different compared to the other lineage I baboon and rhesus monkey isolates (Table 3; Supplementary Table 2).
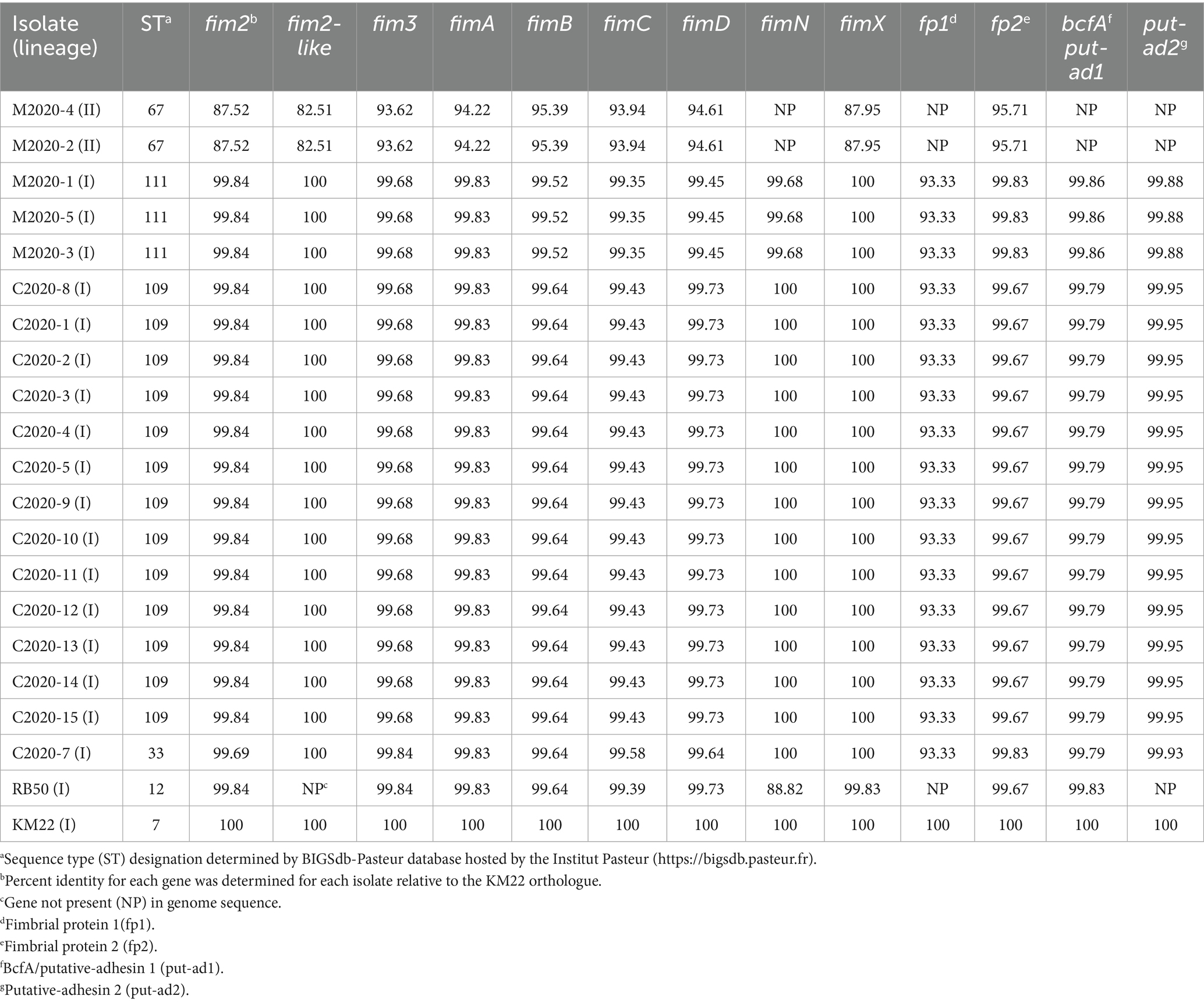
Table 3. Fimbriae and adhesin genes.
Focusing on the fimNX locus, all lineage I baboon isolates M2020-1, M2020-5, and M2020-3 and all lineage I rhesus monkey isolates C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-8, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, C2020-15, and C2020-7 harbored five predicted fimbrial genes (Figure 9). These predicted fimbrial genes include fimbrial protein 3, fimbrial protein 1, fimN, fim2-like, and fimX (Figure 9A). In contrast, lineage II baboon isolates M2020-4 and M2020-2 harbored only two predicted fimbrial genes fim2-like and fimX (Figure 9B). Despite the diversity in the number of genes harbored, the fimNX locus was located in the same genomic location flanked by tripartite ATP-independent periplasmic (TRAP) transporter and phenylacetate-CoA ligase gene (paaK) genes in all primate isolates, as well as in RB50 and KM22 (Figures 9A,B and data not shown).
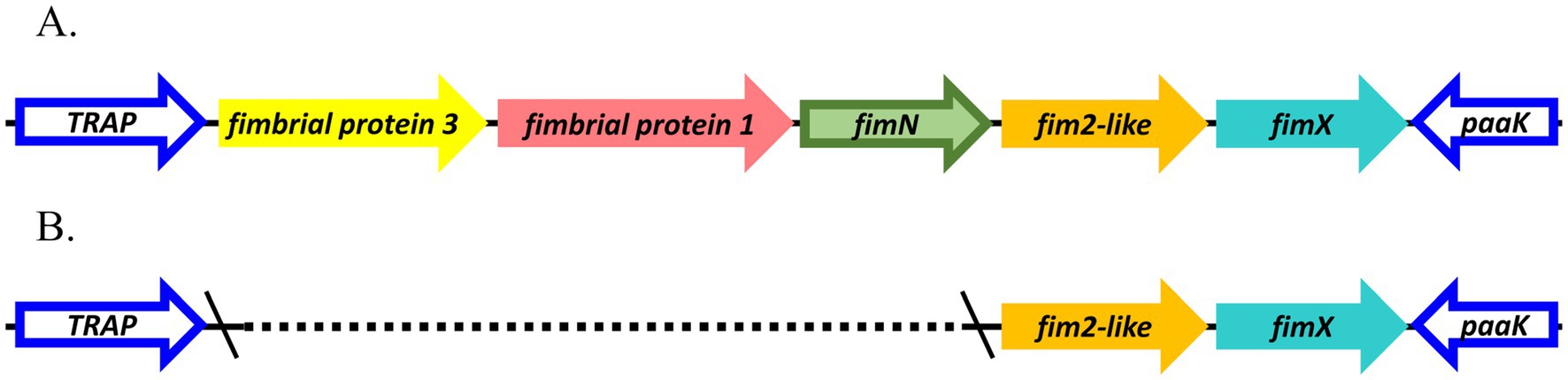
Figure 9. Organization of fimNX locus. Predicted fimbrial protein genes are represented as arrows; gene names refer to names used in Table 3 and are color-coded by nucleotide sequence identity. fimNX locus for isolates (A) M2020-1, M2020-5, M2020-3, C2020-8, C2020-1, C2020-2, C2020-3, C2020-4, C2020-5, C2020-9, C2020-10, C2020-11, C2020-12, C2020-13, C2020-14, C2020-15, C2020-7 and (B) M2020-4 and M2020-2.
Comparative analysis of the cya, ptp, and loci
Genes cyaA, cyaB, cyaC, cyaD, and cyaE of the cya operon were absent from the genomes of lineage II baboon isolates M2020-2 and M2020-4, however, these genes were highly conserved among all other lineage I primate isolates. Additionally, the cyaX gene was present and highly conserved in all primate isolates (Table 4). It has been demonstrated that the cya operon, which contains genes that encode, activate, and secrete Adenylate cyclase toxin, was replaced by an operon predicted to encode peptide transport (ptp) proteins in Bb ST37 isolates (Buboltz et al., 2008). The ptp operon was flanked by gabD and cyaX genes in Bb ST37 isolates, and the genes were reported as highly conserved among ST37 isolates (Buboltz et al., 2008). Similarly, genes within the cya operon were replaced by the ptp operon, flanked by gabD and cyaX genes, in lineage II isolates M2020-4, M2020-2, 99-R-0433, and AU3139 (Table 5). Neither the cya operon or the ptp operon was harbored by lineage II isolates I124 and I328. Lineage II isolate AU3139 harbored both the cya operon or the ptp operon, however, the cyaA gene was predicted to be non-functional due to a frameshift (Table 5).
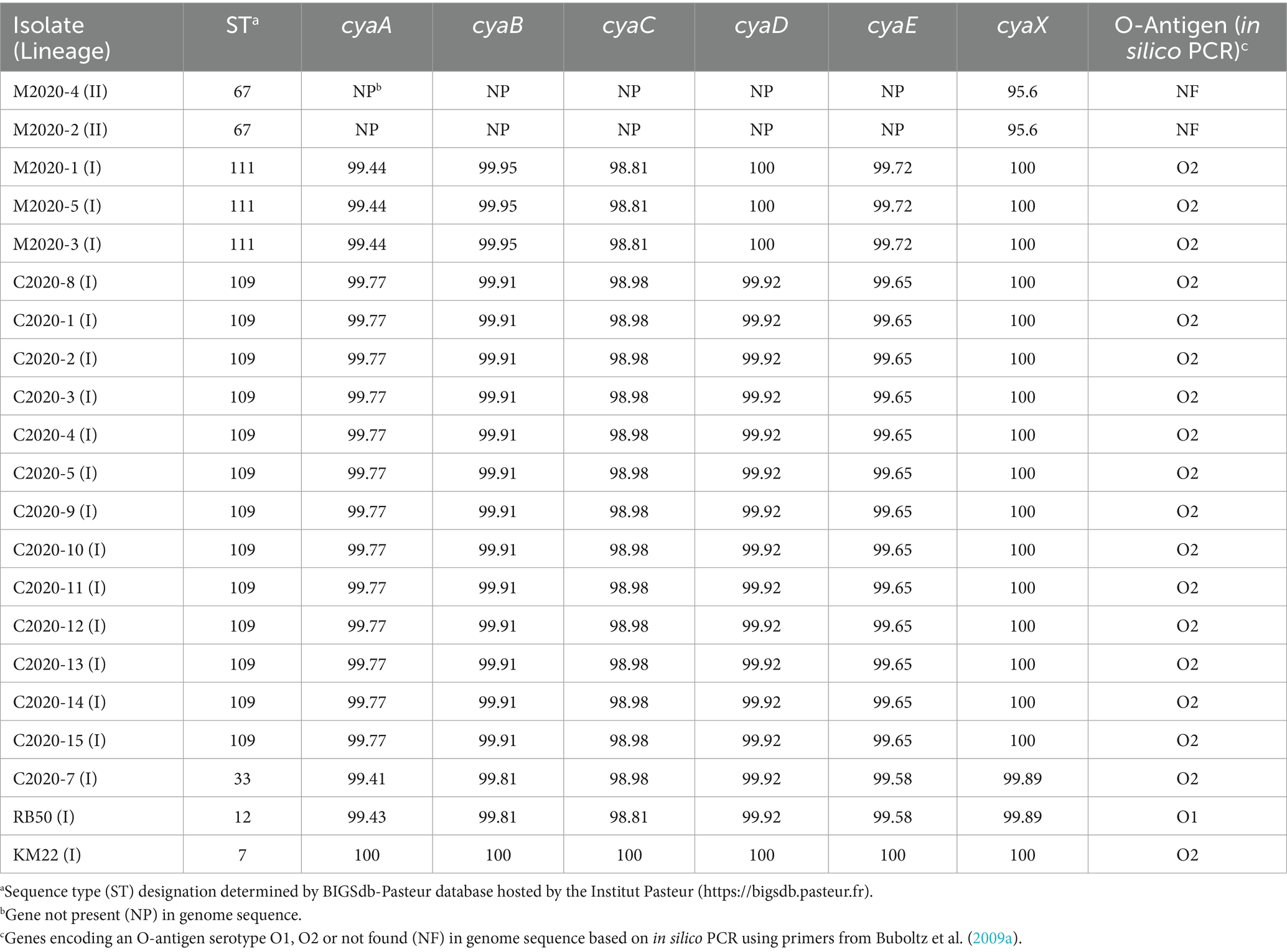
Table 4. cyaA operon and the O-antigen serotype.
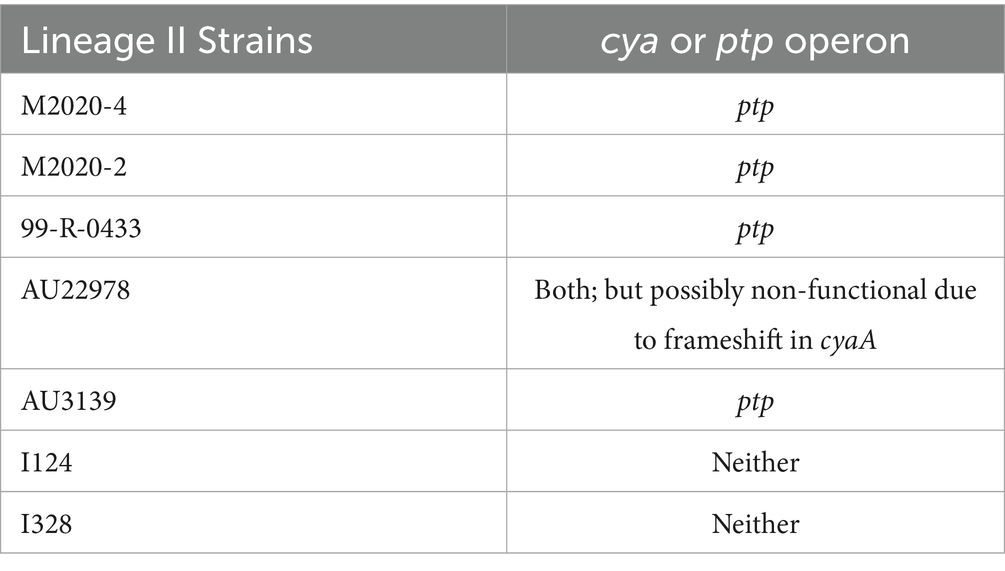
Table 5. cyaA or ptp operon.
Diversity among the wbm loci encoding O-antigen serotypes
The wbm locus contains genes required for expression of one of three antigenically distinct O-antigen types defined as O1, O2, or O3 serotype (Vinogradov et al., 2010; Preston et al., 1999; Hester et al., 2013; Buboltz et al., 2009a). An in silico PCR test based on PCR typing schemes described by Buboltz et al. (2009a) was used to screen the genomes of all primate isolates, RB50, and KM22. The O-antigen type O2 was observed in all lineage I primate isolates and KM22, while the O-antigen type O1 was observed for RB50. Neither the O-antigen type O1 or O2 were found for lineage II baboon isolates M2020-4 and M2020-2 using in silico PCR (Table 4).
Further examination of the wbm locus for all rhesus monkey isolates revealed that while the 3′ and 5′ ends of the wbm locus were conserved and highly similar to a wbm locus encoding genes for O-antigen serotype O2, a high degree of nucleotide divergence was observed for centrally located wbm genes wbmP—wbmI and wbmD (Figure 10). As shown in Figure 10, the wbm locus of C2020-8, representing all rhesus monkey isolates, was more similar to the wbm locus of Bppov Bpp5 than to the KM22 O-antigen serotype O2 locus (Figures 10A,B). Bppov strain Bpp5 does not produce an O-antigen molecule due to frameshift mutations in the wbmO and wbmI genes (Hester et al., 2015). Similar to Bppov Bpp5, wbmE is missing in the wbm locus of C2020-8 and wbmK is replaced by a unique gene, which encodes a methyltransferase (Figure 10B) (Hester et al., 2015). However, unlike the wbm locus of Bppov Bpp5, wbmO and wbmI genes within the wbm locus for rhesus monkey isolates are predicted to be functional as they do not contain frameshift mutations (Figure 10B) (Hester et al., 2015).
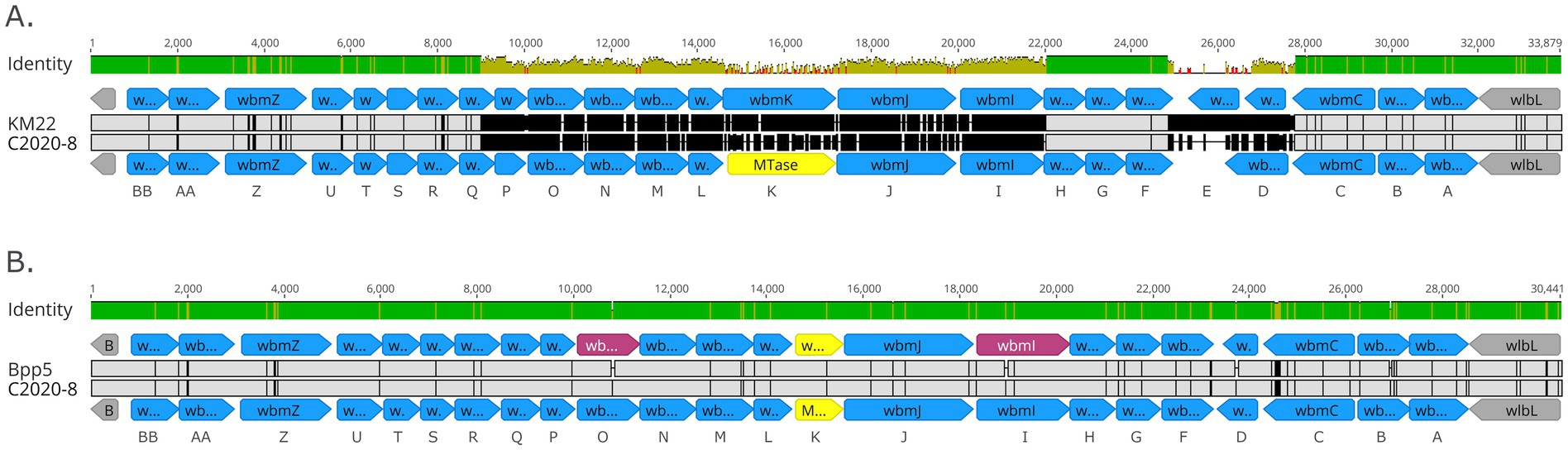
Figure 10. Organization of wbm locus from rhesus monkey Bb isolates. Alignment of the wbm locus of C2020-8 to KM22 serving as reference O-antigen serotype O2 (A) and alignment of the wbm locus of C2020-8 to Bppov strain Bpp5 serving as reference (B). wbm gene name designations are provided at bottom of each alignment. Predicted genes within wbm locus are represented as blue arrows and genes flanking the wbm locus are indicated by grey arrows. Unique gene, which is not present in O-antigen serotype O2, is indicated by yellow arrows. Pseudogenes are indicated by pink arrows. Base pair numbers are displayed above black bar at top. In the aligned sequences, vertical black lines represent residues that differ from the reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
A wbm locus was identified in lineage II baboon isolates M2020-2 and M2020-4; however, this wbm locus has very low sequence identity compared to wbm loci encoding for O-antigen serotypes O1, O2, or O3 (Supplementary Figure 3). The wbm locus identified in lineage II baboon isolates M2020-2 and M2020-4 was observed to be novel and highly similar to the wbm locus harbored by other lineage II isolates AU3139, I124, and I128 (Figure 11). The wbm locus harbored by these isolates contain 16 predicted genes that appear to encode for a novel O-antigen serotype (Figure 11). This predicted novel O-antigen locus was not harbored by all lineage II isolates evaluated. Lineage II isolate 99-R-0433 harbored a wbm locus with a high similarity (88%) to the wbm locus of strain MO149 encoding for an O-antigen serotype O3 (Vinogradov et al., 2010) and lineage II isolate AU22978 harbored a wbm locus with a high similarity (88%) to the wbm locus of strain RB50 encoding for an O-antigen serotype O1 (Buboltz et al., 2009a).
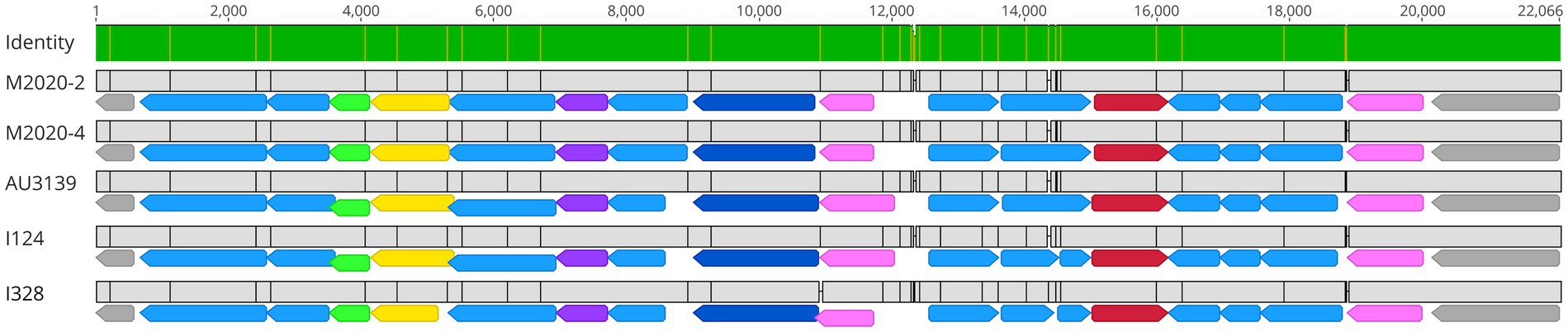
Figure 11. M2020-2 and M2020-4 harbor a novel O-antigen serotype. Alignment of the wbm locus for isolates M2020-2, M2020-4, AU3139I124, and I328. The wbm locus for these isolates includes 16 predicted genes, with functional annotations including predicted formyltransferase genes (green arrows), predicted aminotransferase genes (yellow arrows), predicted methyltransferase genes (purple arrows), predicted asparagine synthase genes (dark blue arrows), predicted glyceroltransferase genes (pink arrows), predicted sugar amidotransferase genes (red arrows), and other genes of undetermined function (light blue arrows). Genes flanking the wbm locus are indicated by grey arrows. Base pair numbers are displayed above black bar at top. In the aligned sequences, vertical black lines represent residues that differ between sequences. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical and yellow is 30 - <100% identical.
AMR distribution
Phenotypic AMR to 10 antibiotic classes was determined, and all isolates were resistant to specific antibiotics within the β-lactam class (Table 6; Supplementary Table 3). Additionally, a high prevalence of resistance was observed for macrolide/lincosamide/streptogramin (MLSb) (95%, n = 18), pleuromutilin (95%, n = 18), and fluroquinolone (69%, n = 13) antibiotic classes (Table 6; Supplementary Table 3). In contrast, no isolates exhibited resistance to aminoglycoside, tetracycline, phenicol, polymyxin, sulfonamide, or glycylcycline antibiotic classes (Table 6; Supplementary Table 3). Thirteen isolates exhibited phenotypic resistance to four of the 10 antibiotic classes tested and five isolates exhibited phenotypic resistance to three of the 10 antibiotic classes tested (Supplementary Table 3).
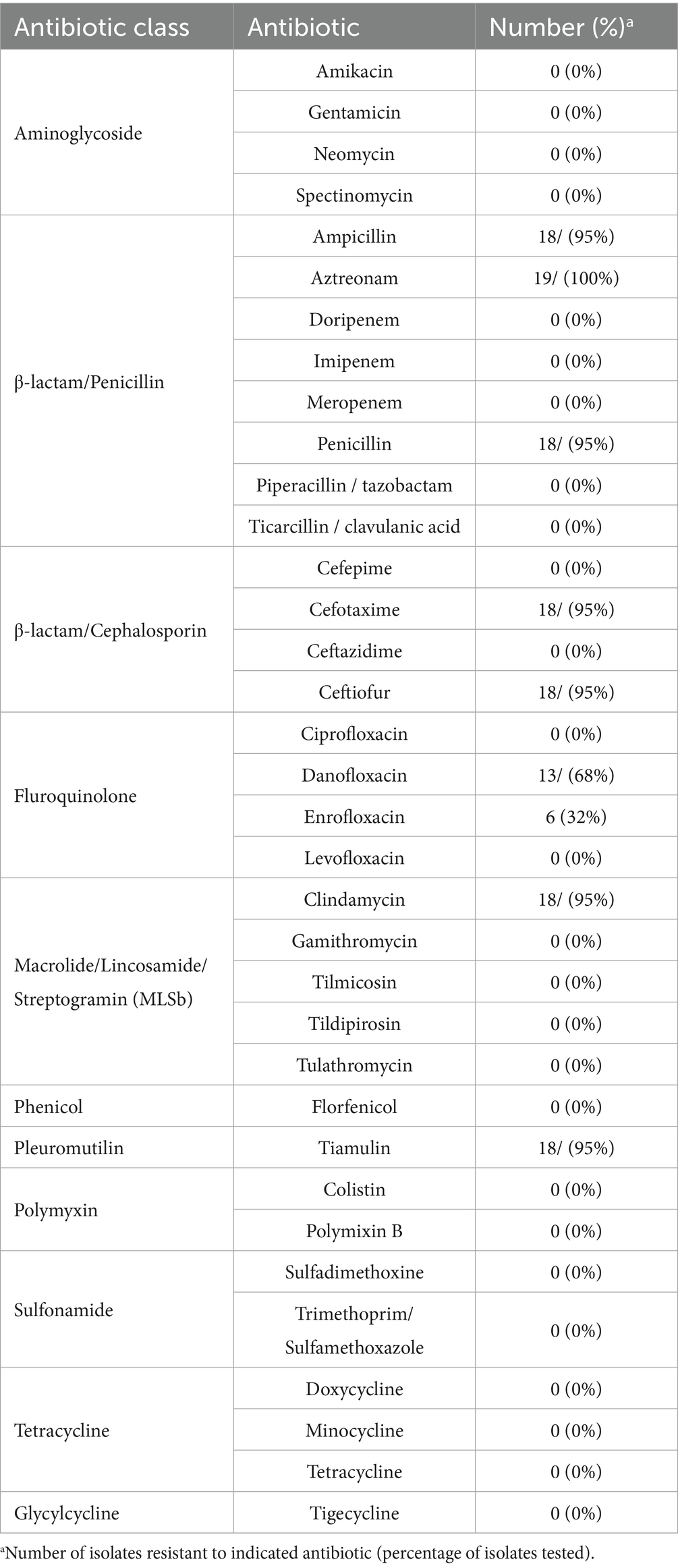
Table 6. Phenotypic AMR prevalence among primate B. bronchiseptica isolates.
Focusing on specific antibiotics tested, isolates primarily exhibited resistance to ampicillin, aztreonam, and/or penicillin within the penicillin subclass of the β-lactam antibiotic class and cefotaxime and/or ceftiofur within the cephalosporin subclass of the β-lactam antibiotic class (Table 6; Supplementary Table 3). Resistance to the MLSb class was due to resistance to clindamycin (Table 6; Supplementary Table 3). The observed resistance to the fluroquinolone class was primarily due to resistance to danofloxacin (Table 6; Supplementary Table 3). While not exhibiting complete resistance to tilmicosin, within the MLSb class, 18 isolates exhibited intermediate resistance to tilmicosin (Supplementary Table 3). Similarly, while no isolates were observed to exhibit complete resistance to florfenicol, within the phenicol class, 17 isolates exhibited intermediate resistance to florfenicol (Supplementary Table 3).
Genomes were screened for chromosomal mutations and genes conferring AMR and only the Bordetella-specific blaBOR β-lactamase gene (Lartigue et al., 2005) was identified (Supplementary Table 3). All isolates harbored the blaBOR β-lactamase gene (Supplementary Table 3). The blaBOR β-lactamase gene from 13 rhesus monkey isolates were 100% identical to each other and 99.78% identical to the reference RB50 sequence blaBOR β-lactamase gene (Supplementary Table 3). The rhesus monkey isolate C2020-7 differed from the other rhesus monkey isolates and was 99.67% identical to the reference RB50 sequence (Supplementary Table 3). The lineage I baboon isolates (M2020-1, M2020-3, and M2020-5) were 100% identical to each other and 99.89% identical to the reference RB50 sequence (Supplementary Table 3). The blaBOR β-lactamase gene from the lineage II baboon isolates (M2020-2, M2020-4) were 100% identical to each other and 93.36% identical to the reference RB50 sequence (Supplementary Table 3). No other AMR elements were identified.
Discussion
There is very little whole-genome data available for Bb isolates obtained from non-human primate hosts. To our knowledge, Bb strain 1,289 obtained from a monkey host, is the only publicly available genome sequence available for a Bb strain obtained from a non-human primate species (Buboltz et al., 2009b). In this report we use whole genome sequencing to evaluate the genetic diversity of Bb isolates obtained from non-human primates, five obtained from baboons and 14 from rhesus monkeys. Most of the isolates evaluated in this report represented one of the two new sequence types (STs) within Bb lineage I-1 clonal complex 6 that were identified by this study. Two baboon isolates, M2020-2 and M2020-4, were ST67 and determined to be Bb lineage II isolates. Given that the genome sequences for M2020-2 and M2020-4 were almost identical and both isolates were obtained from baboons housed together, they are likely the same isolate transmitted between the baboons. While M2020-2 and M2020-4 were isolated from the only animals known to be cohoused together prior to inclusion in this study, the possibility that other isolates were obtained from animals housed together in large groups cannot be excluded.
It has been previously demonstrated that genes encoding virulence factors and vaccine antigens are highly variable and unique to Bb lineage II isolates (Bridel et al., 2022). The data for lineage II baboon isolates M2020-2 and M2020-4 agree with these previous observations. Specially, these isolates harbored the lowest sequence identity observed across all regulatory and virulence-associated genes evaluated. Additionally, these isolates do not have the dnt gene, T6SS locus genes, the cya operon, and fimbrial genes fimN, fimbrial protein 1, bcfA/putative adhesin 1, and putative adhesin 2.
Both FHA and PRN are Bp vaccine antigens and extensive genomic diversity has been documented within the genes encoding these antigens as well as throughout the whole genome among Bp isolates (Komatsu et al., 2010; Bart et al., 2014; Weigand et al., 2017a; Weigand et al., 2017b). Hierarchical clustering of ANI values among the primate isolates evaluated in this study and additional classical bordetellae isolates highly correlated with the phylogenetic group distribution of the cgMLST-based tree reported by Bridel et al. (2022). The similar correlation between AA identity and classical bordetellae lineage for both FHA and PRN indicate that the genes encoding these proteins are core genes in which allele variation reflects phylogenetic group distribution, which is in agreement to other reports (Bridel et al., 2022; Diavatopoulos et al., 2005; Linz et al., 2016; Park et al., 2012).
While AA differences of FHA between KM22 and M2020-2, as well as all lineage II isolates, were observed throughout the whole protein sequence and not located within specific domains, antibodies raised against the MCD of FHA from KM22 recognized FHA from M2020-2, whereas antibodies raised against the HBD of FHA from KM22 did not recognize FHA from M2020-2. This is the first report demonstrating antibodies generated against a specific domain of FHA from one Bb isolate not recognizing FHA from another isolate. Due to the high AA similarity among the lineage II isolates, the data suggests that antibodies generated against FHA from a lineage I isolate could result in decreased cross-protective immunity toward FHA from a lineage II isolate.
The prn gene sequence identity to the KM22 prn was 86.16% for the baboon lineage II isolates M2020-2 and M2020-4. The low nucleotide and AA identity observed for M2020-2 and M2020-4 was observed among all the Bb lineage II isolates. Additionally, the prn gene was located in a different genomic region in lineage II isolates compared to lineage I Bb, Bp, and Bpp isolates. Due to the low AA identity of M2020-2 compared to KM22, and other lineage I Bb isolates, antibodies raised against PRN from a Bb swine isolate did not recognize PRN from M2020-2. While the Bb swine isolate used for the purification of PRN and antibody generation was not KM22, all swine Bb isolates studied to date are Bb lineage I and harbor extreme genomic similarity (Nicholson and Shore, 2024). Due to the high AA similarity among the lineage II isolates, the data suggests that antibodies generated against PRN from a lineage I isolate would result in no cross-protective immunity toward PRN from a lineage II isolate.
PRN belongs to the type V autotransporter protein family and contains two RGD domains (Emsley et al., 1996; Hijnen et al., 2004). Bridel et al. (2022) reported Bb lineage II isolates do not harbor a prn gene sequence. The prn gene was identified in all isolates using BLASTN searches. The gene identified and evaluated as the prn gene in the lineage II isolates M2020-2 and M2020-4 was the only gene hit identified in these isolates, despite the low sequence identity. However, the possibility that the gene identified as prn might not be prn but instead another gene within type V autotransporter family cannot be excluded given the low prn gene sequence identity and the observation that the gene was located in a different genomic region compared to lineage I Bb, Bpp, and Bp isolates.
Buboltz et al. (2008) demonstrated that the cya operon was replaced by the ptp operon in Bb lineage I ST37 isolates. We observed the cya operon was replaced by the ptp operon in several lineage II isolates expanding the phylogenetic distribution of this operon replacement. Furthermore, we observed heterogeneity within this locus among lineage II isolates. Specifically, both the cya and the ptp operons were absent in some lineage II isolates and one lineage II isolate harbored both the cya operon and the ptp operon. The data reported here greatly expands knowledge regarding phylogenetic diversity and genetic variation within this locus among Bb isolates.
Recombination-mediated horizontal gene transfer events involving O-antigen genes contribute to O-antigen variation and previous studies have characterized different O-antigen types among the classical bordetellae subspecies (Hester et al., 2013). Additionally, the genetic variation within the wbm locus enables Bordetella strains to escape immunity to different O-antigen types, thereby allowing for the circulation of different Bordetella strains within the same host population (Hester et al., 2013). Building on the previously reported variation within the wbm locus, the O-antigen locus harbored by all rhesus monkey isolates was similar to the O-antigen locus reported for Bppov Bpp5 (Hester et al., 2015), with the exception of wbmO and wbmI genes, which were predicted to be functional as they do not contain frameshift mutations. Additionally, the wbm locus harbored by lineage II baboon isolates M2020-2 and M2020-4, as well as other lineage II isolates AU3139, I124, and I128, was observed to encode a novel unreported O-antigen. Collectively, the data reported here broadens the described variation within the wbm locus containing genes encoding for the expression of antigenically distinct O-antigen types.
Bb isolates are well known to be generally resistant to β-lactams and macrolides (Woolfrey and Moody, 1991; Kadlec and Schwarz, 2018). In fact, macrolide resistance was previously an important clinical treatment distinction between Bb and Bp before the recent occurrence of macrolide-resistant isolates of Bp (Woolfrey and Moody, 1991; Mattoo and Cherry, 2005; Ivaska et al., 2022). The phenotypic AMR observed among the primate isolates agrees with these reports, with all of the isolates exhibiting resistance to β-lactams. The high prevalence of resistance observed for the macrolide/lincosamide/streptogramin (MLSb) class was due to resistance to clindamycin, a lincosamide antibiotic. While structurally distinct, lincosamide and macrolide antibiotics are grouped together in the same class due to a shared mechanism of action (Canu and Leclercq, 2009). A high prevalence of resistance was observed for pleuromutilin and fluroquinolone antibiotic classes, which has not been previously reported for Bb isolates. Tetracyclines are commonly used to treat Bb veterinary infections due to a high level of sensitivity reported (Kadlec and Schwarz, 2018). Consistent with these previous reports, no isolates exhibited resistance to tetracycline. The Bordetella-specific blaBOR β-lactamase gene (Lartigue et al., 2005), harbored by all primate Bb isolates, was the only AMR gene identified. Unfortunately, with the exception of the blaBOR β-lactamase gene, the occurrence of elevated MICs among the Bb isolates toward other antibiotic classes could not be clearly associated with specific chromosomal mutations or AMR genes, which is consistent with prior reports (Kadlec and Schwarz, 2018; Pruller et al., 2015).
In conclusion, this study presents genomic and phenotypic data for several non-human primate Bb isolates. Combined, the data in this report expands the known diversity of Bb isolates and highlights distinctive traits of Bb isolates between different lineages as well as distinctive traits among Bb isolates within the same lineage.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
All isolates were either obtained from samples collected as part of previous studies or were obtained from samples submitted as part of field case investigations and did not require Institutional Animal Care and Use Committee (IACUC) approval.
Author contributions
TN: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SS: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. YW: Methodology, Resources, Writing – review & editing. LZ: Methodology, Resources, Writing – review & editing. TM: Investigation, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the United States Department of Agriculture, Agriculture Research Service Project Number 5030-32000-237-00-D. This research used resources provided by the SCINet Project of the USDA Agricultural Research Service, ARS Project Number 0201-88888-003-000D.
Acknowledgments
The authors would like to thank Ashley Belinfante and Janel Kolar for their excellent technical assistance. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1571660/full#supplementary-material
SUPPLEMENTARY FIGURE 1 | FHA phylogenetic tree and alignment. (A) Phylogenetic tree inferred from unique FHA AA sequences from analyzed Bordetella isolates containing a full-length fhaB gene used to build a maximum-likelihood phylogenetic tree with FastTree (v2.11) and iTOL (Letunic and Bork, 2024; Price et al., 2010). Support values are shown on each branch, and isolate names are color coded according to legend shown left of the heatmap. (B) MAFFT alignment of the unique FHA AA sequences from analyzed Bordetella isolates containing a full-length fhaB gene ordered by lineage. FHA from Bb strain KM22 served as the reference sequence. In the aligned sequences, vertical black lines represent residues that differ from the KM22 FHA reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
SUPPLEMENTARY FIGURE 2 | PRN alignment and phylogenetic tree. (A) Phylogenetic tree inferred from non-redundant PRN AA sequences from analyzed Bordetella isolates containing a full-length prn gene used to build a maximum-likelihood phylogenetic tree with FastTree (v2.11) and iTOL (Letunic and Bork, 2024; Price et al., 2010). Support values are shown on each branch, and isolate names are color coded according to legend shown left of the heatmap. (B) MAFFT alignment of the unique PRN AA sequences from analyzed Bordetella isolates containing a full-length prn gene ordered by lineage. PRN from Bb strain KM22 served as the reference sequence. In the aligned sequences, vertical black lines represent residues that differ from the KM22 FHA reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
SUPPLEMENTARY FIGURE 3 | M2020-2 and M2020-4 do not harbor O-antigen serotype O1, O2, or O3. Alignment of the wbm locus M2020-2 to RB50 serving as reference O-antigen serotype O1 (A), wbm locus M2020-2 to KM22 serving as reference O-antigen serotype O2 (B), and wbm locus M2020-2 to MO149 serving as reference O-antigen serotype O3 (C). M2020-2 and M2020-4 sequences are 100% identical over the region therefore only M2020-2 sequence is used in alignments. Predicted genes within the wbm locus are represented as blue arrows and genes flanking the wbm locus are indicated by grey arrows. Base pair numbers are displayed above black bar at top. In the aligned sequences, vertical black lines represent residues that differ from the reference. The top bar represents mean pairwise identity over all pairs in that column of the alignment; green is 100% identical, yellow is 30 - <100% identical, and red is <30% identical. The height of the bar represents conservation of sequence at that position; a lower height indicates low sequence conservation at that position.
Footnotes
1. ^https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
References
Agarwal, L., Singh, H., Jani, C., Banankhah, P., Abdalla, M., Kurman, J. S., et al. (2022). A wolf in sheep’s clothing: dogs confer an unrecognized risk for their immunocompromised master. Respir. Med. Case. Rep. 38:101672. doi: 10.1016/j.rmcr.2022.101672
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Badhai, J., and Das, S. K. (2021). Genomic plasticity and antibody response of Bordetella bronchiseptica strain HT200, a natural variant from a thermal spring. FEMS Microbiol. Lett. 368:fnab035. doi: 10.1093/femsle/fnab035
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barcala Salido, J. M., Mora-Delgado, J., and Lojo-Cruz, C. (2022). Bordetella bronchiseptica pneumonia in an immunocompetent pig farmer. IDCases 27:e01435. doi: 10.1016/j.idcr.2022.e01435
Bart, M. J., Harris, S. R., Advani, A., Arakawa, Y., Bottero, D., Bouchez, V., et al. (2014). Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. mBio 5:e01074. doi: 10.1128/mBio.01074-14
Boursaux-Eude, C., and Guiso, N. (2000). Polymorphism of repeated regions of pertactin in Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica. Infect. Immun. 68, 4815–4817. doi: 10.1128/IAI.68.8.4815-4817.2000
Bridel, S., Bouchez, V., Brancotte, B., Hauck, S., Armatys, N., Landier, A., et al. (2022). A comprehensive resource for Bordetella genomic epidemiology and biodiversity studies. Nat. Commun. 13:3807. doi: 10.1038/s41467-022-31517-8
Brockmeier, S. L., Register, K. B., Nicholson, T. L., and Loving, C. L. (2019). “Bordetellosis” in Diseases of swine. eds. J. J. Zimmerman, L. A. Karriker, A. Ramirez, G. W. Stevenson, and J. Zhang. 11th ed (Hoboken, NJ: John Wiley & Sons, Inc).
Buboltz, A. M., Nicholson, T. L., Karanikas, A. T., Preston, A., and Harvill, E. T. (2009a). Evidence for horizontal gene transfer of two antigenically distinct O antigens in Bordetella bronchiseptica. Infect. Immun. 77, 3249–3257. doi: 10.1128/IAI.01448-08
Buboltz, A. M., Nicholson, T. L., Parette, M. R., Hester, S. E., Parkhill, J., and Harvill, E. T. (2008). Replacement of adenylate cyclase toxin in a lineage of Bordetella bronchiseptica. J. Bacteriol. 190, 5502–5511. doi: 10.1128/JB.00226-08
Buboltz, A. M., Nicholson, T. L., Weyrich, L. S., and Harvill, E. T. (2009b). Role of the type III secretion system in a hypervirulent lineage of Bordetella bronchiseptica. Infect. Immun. 77, 3969–3977. doi: 10.1128/IAI.01362-08
Canu, M., and Leclercq, R. (2009). “Macrolides and lincosamides” in Antimicrobial drug resistance. eds. D. L. Mayers, J. D. Sobel, M. Ouellette, K. S. Kaye, and D. Marchaim (New York: Humana Press), 211–221.
Chambers, J. K., Matsumoto, I., Shibahara, T., Haritani, M., Nakayama, H., and Uchida, K. (2019). An outbreak of fatal Bordetella bronchiseptica bronchopneumonia in puppies. J. Comp. Pathol. 167, 41–45. doi: 10.1016/j.jcpa.2018.12.002
Charles, I., Fairweather, N., Pickard, D., Beesley, J., Anderson, R., Dougan, G., et al. (1994). Expression of the Bordetella pertussis P.69 pertactin adhesin in Escherichia coli: fate of the carboxy-terminal domain. Microbiology 140, 3301–3308. doi: 10.1099/13500872-140-12-3301
Chen, Q., and Stibitz, S. (2019). The BvgASR virulence regulon of Bordetella pertussis. Curr. Opin. Microbiol. 47, 74–81. doi: 10.1016/j.mib.2019.01.002
CLSI (2024a). “Performance standards for antimicrobial disk and dilution susceptibility tests for bacteria isolated from animals” in CLSI supplement VET01S. 11th ed (Wayne, PA. USA: Clinical and Laboratory Standards Institute).
CLSI (2024b). “Performance standards for antimicrobial susceptibility testing” in CLSI supplement M100Ed34. 34th ed (Wayne, PA. USA: Clinical and Laboratory Standards Institute).
Cotter, P. A., and Jones, A. M. (2003). Phosphorelay control of virulence gene expression in Bordetella. Trends Microbiol. 11, 367–373. doi: 10.1016/S0966-842X(03)00156-2
Coutte, L., Antoine, R., Drobecq, H., Locht, C., and Jacob-Dubuisson, F. (2001). Subtilisin-like autotransporter serves as maturation protease in a bacterial secretion pathway. EMBO J. 20, 5040–8.
Delisse-Gathoye, A. M., Locht, C., Jacob, F., Raaschou-Nielsen, M., Heron, I., Ruelle, J. L., et al. (1990). Cloning, partial sequence, expression, and antigenic analysis of the filamentous hemagglutinin gene of Bordetella pertussis. Infect Immun. 58, 2895–905.
Diavatopoulos, D. A., Cummings, C. A., Schouls, L. M., Brinig, M. M., Relman, D. A., and Mooi, F. R. (2005). Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 1:e45. doi: 10.1371/journal.ppat.0010045
Domenighini, M., Relman, D., Capiau, C., Falkow, S., Prugnola, A., Scarlato, V., et al. (1990). Genetic characterization of Bordetella pertussis filamentous haemagglutinin: a protein processed from an unusually large precursor. Mol Microbiol. 4, 787–800.
Emsley, P., Charles, I. G., Fairweather, N. F., and Isaacs, N. W. (1996). Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381, 90–92. doi: 10.1038/381090a0
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63:e00483-19. doi: 10.1128/AAC.00483-19
Goodnow, R. A. (1980). Biology of Bordetella bronchiseptica. Microbiol. Rev. 44, 722–738. doi: 10.1128/mr.44.4.722-738.1980
Gotto, J. W., Eckhardt, T., Reilly, P. A., Scott, J. V., Cowell, J. L., Metcalf, T. N. 3rd, et al. (1993). Biochemical and immunological properties of two forms of pertactin, the 69,000-molecular-weight outer membrane protein of Bordetella pertussis. Infect. Immun. 61, 2211–2215. doi: 10.1128/iai.61.5.2211-2215.1993
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Hester, S. E., Goodfield, L. L., Park, J., Feaga, H. A., Ivanov, Y. V., Bendor, L., et al. (2015). Host specificity of ovine Bordetella parapertussis and the role of complement. PLoS One 10:e0130964. doi: 10.1371/journal.pone.0130964
Hester, S. E., Park, J., Goodfield, L. L., Feaga, H. A., Preston, A., and Harvill, E. T. (2013). Horizontally acquired divergent O-antigen contributes to escape from cross-immunity in the classical bordetellae. BMC Evol. Biol. 13:209. doi: 10.1186/1471-2148-13-209
Hewlett, E. L., Burns, D. L., Cotter, P. A., Harvill, E. T., Merkel, T. J., Quinn, C. P., et al. (2014). Pertussis pathogenesis—what we know and what we don't know. J. Infect. Dis. 209, 982–985. doi: 10.1093/infdis/jit639
Hijnen, M., Mooi, F. R., van Gageldonk, P. G., Hoogerhout, P., King, A. J., and Berbers, G. A. (2004). Epitope structure of the Bordetella pertussis protein P.69 pertactin, a major vaccine component and protective antigen. Infect. Immun. 72, 3716–3723. doi: 10.1128/IAI.72.7.3716-3723.2004
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Ivaska, L., Barkoff, A. M., Mertsola, J., and He, Q. (2022). Macrolide resistance in Bordetella pertussis: current situation and future challenges. Antibiotics 11:1570. doi: 10.3390/antibiotics11111570
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jungnitz, H., West, N. P., Walker, M. J., Chhatwal, G. S., and Guzman, C. A. (1998). A second two-component regulatory system of Bordetella bronchiseptica required for bacterial resistance to oxidative stress, production of acid phosphatase, and in vivo persistence. Infect Immun. 66, 4640–50.
Jurnecka, D., Man, P., Sebo, P., and Bumba, L. (2018). Bordetella pertussis and Bordetella bronchiseptica filamentous hemagglutinins are processed at different sites. FEBS Open Bio. 8, 1256–1266.
Kadlec, K., and Schwarz, S. (2018). Antimicrobial resistance in Bordetella bronchiseptica. Microbiol. Spectr. 6, 24–35. doi: 10.1128/microbiolspec.ARBA-0024-2017
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Komatsu, E., Yamaguchi, F., Abe, A., Weiss, A. A., and Watanabe, M. (2010). Synergic effect of genotype changes in pertussis toxin and pertactin on adaptation to an acellular pertussis vaccine in the murine intranasal challenge model. Clin. Vaccine Immunol. 17, 807–812. doi: 10.1128/CVI.00449-09
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Lartigue, M. F., Poirel, L., Fortineau, N., and Nordmann, P. (2005). Chromosome-borne class a BOR-1 beta-lactamase of Bordetella bronchiseptica and Bordetella parapertussis. Antimicrob. Agents Chemother. 49, 2565–2567. doi: 10.1128/AAC.49.6.2565-2567.2005
Letunic, I., and Bork, P. (2024). Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/nar/gkae268
Linz, B., Ivanov, Y. V., Preston, A., Brinkac, L., Parkhill, J., Kim, M., et al. (2016). Acquisition and loss of virulence-associated factors during genome evolution and speciation in three clades of Bordetella species. BMC Genomics 17:767. doi: 10.1186/s12864-016-3112-5
Mattoo, S., and Cherry, J. D. (2005). Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 18, 326–382. doi: 10.1128/CMR.18.2.326-382.2005
Mazar, J., and Cotter, P. A. (2006). Topology and maturation of filamentous haemagglutinin suggest a new model for two-partner secretion. Mol Microbiol. 62, 641–54.
Moore, J. E., Rendall, J. C., and Millar, B. C. (2022). A doggy tale: risk of zoonotic infection with Bordetella bronchiseptica for cystic fibrosis (CF) patients from live licenced bacterial veterinary vaccines for cats and dogs. J. Clin. Pharm. Ther. 47, 139–145. doi: 10.1111/jcpt.13492
Nicholson, T. L. (2007). Construction and validation of a first-generation Bordetella bronchiseptica long-oligonulceotide microarray by transcriptional profiling the Bvg regulon. BMC Genomics 8:220. doi: 10.1186/1471-2164-8-220
Nicholson, T. L., Bayles, D. O., and Shore, S. M. (2020). Complete genome sequence of Bordetella bronchiseptica strain KM22. Microbiol. Resour. Announc. 9:e01207-19. doi: 10.1128/MRA.01207-19
Nicholson, T. L., Brockmeier, S. L., and Loving, C. L. (2009). Contribution of Bordetella bronchiseptica filamentous hemagglutinin and pertactin to respiratory disease in swine. Infect. Immun. 77, 2136–2146. doi: 10.1128/IAI.01379-08
Nicholson, T. L., Brockmeier, S. L., Loving, C. L., Register, K. B., Kehrli, M. E. Jr., Stibitz, S. E., et al. (2012). Phenotypic modulation of the virulent Bvg phase is not required for pathogenesis and transmission of Bordetella bronchiseptica in swine. Infect. Immun. 80, 1025–1036. doi: 10.1128/IAI.06016-11
Nicholson, T. L., and Shore, S. M. (2024). Comparative analysis of antimicrobial resistance and genetic diversity of Bordetella bronchiseptica isolates obtained from swine within the United States. Front. Microbiol. 15:1501373. doi: 10.3389/fmicb.2024.1501373
Nicholson, T. L., Waack, U., Fleming, D. S., Chen, Q., Miller, L. C., Merkel, T. J., et al. (2024). The contribution of BvgR, RisA, and RisS to global gene regulation, intracellular cyclic-di-GMP levels, motility, and biofilm formation in Bordetella bronchiseptica. Front. Microbiol. 15:1305097. doi: 10.3389/fmicb.2024.1305097
Parkhill, J., Sebaihia, M., Preston, A., Murphy, L. D., Thomson, N., Harris, D. E., et al. (2003). Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35, 32–40. doi: 10.1038/ng1227
Park, J., Zhang, Y., Buboltz, A. M., Zhang, X., Schuster, S. C., Ahuja, U., et al. (2012). Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genomics 13:545. doi: 10.1186/1471-2164-13-545
Pinto, M. V., and Merkel, T. J. (2017). Pertussis disease and transmission and host responses: insights from the baboon model of pertussis. J. Infect. 74, S114–S119. doi: 10.1016/S0163-4453(17)30201-3
Porter, J. F., Connor, K., and Donachie, W. (1994). Isolation and characterization of Bordetella parapertussis-like bacteria from ovine lungs. Microbiology 140, 255–261. doi: 10.1099/13500872-140-2-255
Preston, A., Allen, A. G., Cadisch, J., Thomas, R., Stevens, K., Churcher, C. M., et al. (1999). Genetic basis for lipopolysaccharide O-antigen biosynthesis in bordetellae. Infect. Immun. 67, 3763–3767. doi: 10.1128/IAI.67.8.3763-3767.1999
Preston, A., Parkhill, J., and Maskell, D. J. (2004). The bordetellae: lessons from genomics. Nat. Rev. Microbiol. 2, 379–390. doi: 10.1038/nrmicro886
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Pruller, S., Rensch, U., Meemken, D., Kaspar, H., Kopp, P. A., Klein, G., et al. (2015). Antimicrobial susceptibility of Bordetella bronchiseptica isolates from swine and companion animals and detection of resistance genes. PLoS One 10:e0135703. doi: 10.1371/journal.pone.0135703
Register, K. B. (2001). Novel genetic and phenotypic heterogeneity in Bordetella bronchiseptica pertactin. Infect. Immun. 69, 1917–1921. doi: 10.1128/IAI.69.3.1917-1921.2001
Register, K. B. (2004). Comparative sequence analysis of Bordetella bronchiseptica pertactin gene (prn) repeat region variants in swine vaccines and field isolates. Vaccine 23, 48–57. doi: 10.1016/j.vaccine.2004.07.020
Register, K. B., Sukumar, N., Palavecino, E. L., Rubin, B. K., and Deora, R. (2012). Bordetella bronchiseptica in a paediatric cystic fibrosis patient: possible transmission from a household cat. Zoonoses Public Health 59, 246–250. doi: 10.1111/j.1863-2378.2011.01446.x
Sukumar, N., Nicholson, T. L., Conover, M. S., Ganguly, T., and Deora, R. (2014). Comparative analyses of a cystic fibrosis isolate of Bordetella bronchiseptica reveal differences in important pathogenic phenotypes. Infect. Immun. 82, 1627–1637. doi: 10.1128/IAI.01453-13
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Vilaro, A., Novell, E., Enrique-Tarancon, V., Baliellas, J., and Fraile, L. (2023). Susceptibility trends of swine respiratory pathogens from 2019 to 2022 to antimicrobials commonly used in Spain. Porcine Health Manag. 9:47. doi: 10.1186/s40813-023-00341-x
Vinogradov, E., King, J. D., Pathak, A. K., Harvill, E. T., and Preston, A. (2010). Antigenic variation among Bordetella: Bordetella bronchiseptica strain MO149 expresses a novel o chain that is poorly immunogenic. J. Biol. Chem. 285, 26869–26877. doi: 10.1074/jbc.M110.115121
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Weigand, M. R., Peng, Y., Cassiday, P. K., Loparev, V. N., Johnson, T., Juieng, P., et al. (2017a). Complete genome sequences of Bordetella pertussis isolates with novel pertactin-deficient deletions. Genome Announc. 5:e00973-17. doi: 10.1128/genomeA.00973-17
Weigand, M. R., Peng, Y., Loparev, V., Batra, D., Bowden, K. E., Burroughs, M., et al. (2017b). The history of Bordetella pertussis genome evolution includes structural rearrangement. J. Bacteriol. 199:e00806-16. doi: 10.1128/JB.00806-16
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Woolfrey, B. F., and Moody, J. A. (1991). Human infections associated with Bordetella bronchiseptica. Clin. Microbiol. Rev. 4, 243–255. doi: 10.1128/CMR.4.3.243
Keywords: Bordetella bronchiseptica, whole-genome sequencing (WGS), comparative genomics, virulence factor, antimicrobial resistance (AMR), primate
Citation: Nicholson TL, Shore SM, Wang Y, Zimmerman L and Merkel TJ (2025) Genetic diversity of Bordetella bronchiseptica isolates obtained from primates. Front. Microbiol. 16:1571660. doi: 10.3389/fmicb.2025.1571660
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Hieu Tran-Van, Vietnam National University, Ho Chi Minh City, VietnamJhasketan Badhai, University of South Florida, United States
Andrei Giacchetto Felice, Universidade Federal do Triângulo Mineiro, Brazil
Copyright © 2025 Nicholson, Shore, Wang, Zimmerman and Merkel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy L. Nicholson, dHJhY3kubmljaG9sc29uQHVzZGEuZ292