 Chan Kyeong Lee
Chan Kyeong Lee Ho Joung Lee
Ho Joung Lee Song Hee Jeong
Song Hee Jeong Sang Jun Lee
Sang Jun Lee- Department of Systems Biotechnology, Institute of Microbiomics, Chung-Ang University, Anseong, Republic of Korea
The CRISPR-Cas system, an adaptive immune mechanism in prokaryotes against bacteriophages, has been developed into a versatile tool for recognizing and cleaving target nucleic acid sequences. In this study, we developed a model system by integrating CRISPR-Cas12a into the genome of temperate bacteriophage λ, enabling precise regulation of lysogeny and lysis in Escherichia coli. We confirmed that λ phage, armed with Cas12a nuclease and CRISPR RNA (crRNA) targeting specific sequences, could inhibit the lysogenic cycle of E. coli cells. We demonstrated that the CRISPR-Cas12a-loaded temperate λ phage mimicked a lytic phage by selectively killing cells carrying the target genomic sequence. Furthermore, by employing truncated crRNA to enhance target recognition specificity, we found that the synthetic phage could distinguish single nucleotide variations in the genomic target DNA, enabling precise targeting and selective elimination of target cells in homogeneous bacterial cultures. To further validate its specificity, we tested this system in mixed bacterial cultures, wherein Cas12a nuclease and truncated crRNA-loaded bacteriophages selectively eliminated only those cells carrying the target sequences perfectly matching the crRNA. These results highlight the potential of this approach for advancing precision microbiome modulation.
1 Introduction
The CRISPR-Cas system is an adaptive immune mechanism found in bacteria and archaea that protects the host from invading foreign genetic elements (Barrangou et al., 2007; Datsenko et al., 2012). Among these systems, the Type I-E CRISPR-Cas system in Escherichia coli is particularly well-characterized and has served as a model for studying CRISPR-Cas-mediated immunity (Makarova et al., 2011). The CRISPR-Cas system comprises a guide RNA (gRNA) that recognizes specific DNA sequences and a CRISPR-associated (Cas) nuclease that causes cleavage. By modifying the target recognition sequence (TRS) in the gRNA, various DNA targets can be specifically recognized and cleaved (Wiles et al., 2015). This system is also used for genome editing in various organisms, including microbes (Shen et al., 2019), plants (Li et al., 2020), animals (Niu et al., 2014), and humans (Gillmore et al., 2021).
Since the first demonstration of the genome editing of T7 bacteriophage using the CRISPR-Cas system, this system has evolved into an effective tool for bacteriophage genome engineering (Kiro et al., 2014). Among the various CRISPR systems, CRISPR-Cas9 has been widely used for bacteriophage genome editing owing to its high specificity and efficiency. For instance, Martel et al. utilized CRISPR-Cas9 to generate small and large deletions and point mutations and performed gene replacements in the orf of virulent phage 2972 (Martel and Moineau, 2014). Furthermore, CRISPR-Cas9 has been applied to edit the cI857 gene of the λ phage at a single-nucleotide level, enabling its conversion to cIWT (Lee et al., 2022a). Recently, CRISPR-Cas12a has emerged as a promising alternative to Cas9 for genome editing in view of its lesser off-target effects, simplified gRNA design, and ease of cellular delivery (Ha et al., 2020; Lee et al., 2018; Slaman et al., 2024). Unlike Cas9, Cas12a generates staggered DNA ends upon cleavage, which promotes homology-directed repair and enhances the efficiency of transgene integration (Huang et al., 2022; Moescheid et al., 2023). Additionally, under specific conditions, Cas12a exhibits higher cleavage efficiency than Cas9, which makes it useful for bacteriophage genome editing (Chen et al., 2024; Dong et al., 2021).
As the CRISPR-Cas technology has facilitated bacteriophage genome engineering, its applications have expanded to encompass gene function identification, bacteriophage–host interactions, and pathogen detection. For example, Tao et al. employed CRISPR-Cas9 to delete the rnlB gene or introduce an amber mutation in T4 phage, demonstrating that rnlB is not essential for phage infection (Tao et al., 2017). Similarly, Hoshiga et al. (2019) replaced the tail fiber of T2 phage with that of PP01 phage using CRISPR-Cas9, enhancing its infectivity toward E. coli O157:H7 and demonstrating the critical role of the short tail fiber of PP01 in bacterial adsorption. Moreover, reporter phages carrying the nanoluciferase (nluc) gene have been developed for detecting urinary tract infections in clinical samples (Meile et al., 2023).
Originally a prokaryotic defense mechanism against bacteriophages, the CRISPR-Cas system has been repurposed to selectively eliminate specific microorganisms based on their DNA sequences via bacteriophage-mediated delivery (Balcha and Neja, 2023). The CRISPR-Cas system can be introduced into target cells via phagemids or phage genomes and has been used to eradicate harmful bacteria carrying antibiotic resistance or virulence genes (Fagen et al., 2017). For instance, a CRISPR-Cas9 system targeting the β-lactam resistance genes blaNDM-1 or blaSHV-18 was delivered via phagemids and could effectively kill antibiotic-resistant E. coli (Citorik et al., 2014). Similarly, a λ phage carrying a CRISPR-Cas3 cascade system and a CRISPR array targeting β-lactam resistance genes successfully eliminated antibiotic resistance plasmids in E. coli, restoring its antibiotic sensitivity (Yosef et al., 2015). Selle et al. (2020) demonstrated that ϕCD24-2, a CRISPR-Cas3-armed bacteriophage, effectively eradicated Clostridioides difficile in a mouse infection model. CRISPR-Cas-armed bacteriophages have also been utilized to eliminate pathogenic bacteria, such as Staphylococcus aureus (Park et al., 2017) and E. coli (Gencay et al., 2024), which cause human diseases. However, previous studies have primarily focused on targeting bacteria at the genome or gene level to induce cell death.
In this study, we explored whether temperate λ phages equipped with the Cas12a system could control lysogenic cycle of bacterial cells. Additionally, to precisely distinguish cells with single nucleotide variations (SNVs) in the target genomic DNA, we tested Cas12a-equipped phages carrying truncated CRISPR RNA (crRNA). Using the LacZ phenotype complementation, we evaluated the phage-induced lysis and lysogeny formation in target cells under mixed bacterial culture conditions. Based on our results, we discuss the precise SNV-level control enabled by CRISPR-Cas12a-loaded phages and their potential applications in the microbiome field.
2 Materials and methods
2.1 Strains and culture conditions
The E. coli strains used in this study are listed in Supplementary Table S1. All E. coli strains were cultured in Luria-Bertani (LB) broth (LPS Solution, Cat. No. LB-05, Korea) at 30°C or 37°C with shaking at 180 rpm. E. coli DH5α was used as the cloning host for crRNA plasmid construction. When necessary, ampicillin (50 μg/mL), kanamycin (25 μg/mL), chloramphenicol (12.5 μg/mL), or spectinomycin (75 μg/mL) was added to the medium. To prepare electrocompetent cells, E. coli strains were cultured overnight at either 30°C or 37°C, depending on the experimental requirements: 30°C for strains carrying prophages or temperature-sensitive plasmids (such as pKD46 and pCJH027), and 37°C for strains containing other plasmids. The overnight culture was inoculated into fresh LB broth at 1% of the final volume and cultured until the optical density at 600 nm (OD600 nm) reached 0.4. For E. coli strains harboring pKD46, L-arabinose was added to a final concentration of 1 mM, followed by an additional 3 h incubation at 30°C. The cells were harvested and washed twice with 10% glycerol. The cells were then resuspended in 10% glycerol at 0.5% (v/v) of the culture volume, aliquoted into 50 μL, and stored at −80°C.
2.2 Construction of galK-targeting crRNA plasmids
The plasmids used in this study are listed in Supplementary Table S2, and the primers used for plasmid construction are listed in Supplementary Table S3. To construct a crRNA plasmid for LbCas12a, pHL027 (modified from pJYS2_crtYf; Addgene plasmid #85544) carrying the crRNA of FnCas12a was used as a template. Two DNA fragments containing the spectinomycin resistance gene and the crRNA gene were amplified using galK_LbCas12a_20_F and galK_LbCas12a_20_R primers (see Supplementary Table S3), and assembled by Gibson assembly using the HiFi DNA Assembly Master Mix (NEB, Cat. No. E2621, USA). The constructed crRNA plasmid for LbCas12a was designated as pCK055. Subsequently, crRNA plasmids targeting the galK gene with various lengths of TRS were constructed using pCK055 as a template (Supplementary Figure S1A). The construction of galK-targeting crRNA plasmids was confirmed via Sanger sequencing (Supplementary Figure S1B).
2.3 Generation of λ cas12a phage
The phages used in this study are listed in Supplementary Table S4. To replace the b2 region of the λ phage genome, which consisted of the ea59, ea31, and ea47 genes, with the Lbcas12a gene, the PrpsL-cas12a-CmR cassette was constructed. The Lbcas12a fragment was amplified from pCJH027 (a gift from Jennifer Doudna; Addgene plasmid #183074) as the template. The PrpsL and CmR fragments were individually amplified from the HL080 template using primer pairs containing homologous sequences required for recombineering. The three purified DNA fragments were ligated using Gibson assembly and subsequently amplified using PCR to create the PrpsL-cas12a-CmR cassette. The cassette was electroporated into L-arabinose-induced HL081/pKD46 competent cells using a 0.1 cm cuvette (Bio-Rad) under the conditions of 25 μF, 200 Ω, and 1.8 kV. Immediately thereafter, 950 μL of SOC medium was added, and the cells were recovered by incubation at 30°C for 2 h with shaking at 180 rpm. The recovered cells were spread onto LB agar plates supplemented with chloramphenicol and incubated at 30°C. The integration of PrpsL-cas12a-CmR into the b2 region of the λ prophage genome was confirmed using colony PCR.
2.4 Assembly of cas12a and crRNA genes in the λ phage genome
To insert a crRNA in the downstream region of the cas12a gene in the CK116 strain, a cas12a (532 bp)-PJ23119-crRNA-KmR cassette was constructed. A 532-bp fragment of this gene was amplified from CK116 as the template, and the PJ23119-crRNA fragment, with a TRS length of 20 nucleotide (nt), was amplified from pCK055. The noncoding region between cas12a and PJ23119-crRNA and the KmR fragment were separately amplified from HL081. The four DNA fragments were assembled via Gibson assembly and subsequently amplified to generate a single DNA cassette. The cas12a (532 bp)-PJ23119-crRNA-KmR cassette was electroporated into L-arabinose-induced CK116/pKD46 competent cells. Electroporation was performed as described above. The cells were spread onto LB agar plates supplemented with kanamycin and incubated at 30°C. The integration of the crRNA into CK116 strain was confirmed using colony PCR and Sanger sequencing, and the resulting strain was designated as CK120. To incorporate crRNAs with different TRS lengths into the CK116 strain, cas12a (532 bp)-PJ23119-crRNA-KmR cassettes, each containing crRNAs with varying TRS lengths, were constructed. These DNA cassettes with overhangs complementary to the crRNA gene were amplified using CK120 as a template. The subsequent steps were performed following the procedure used for the construction of CK120. The construction of these phages was confirmed using colony PCR and Sanger sequencing (Supplementary Figure S2).
2.5 Incorporation of the lacZ gene in the synthetic λ cas12a-crRNA phage
To introduce the lacZ gene into CK117 (N23) and CK124 (N16), a cas12a (532 bp)-PJ23119-crRNA-lacZ-CmR cassette was constructed. The cas12a (532 bp)-PJ23119-crRNA fragments with different TRS lengths were amplified using CK117 and CK124 as templates. The lacZ and CmR cassettes were amplified from MG1655 and HL080, respectively. The three DNA fragments were assembled via Gibson assembly and amplified to generate the cas12a (532 bp)-PJ23119-crRNA (N23 or N16)-lacZ-CmR cassette. The cassette was electroporated into L-arabinose-induced CK117 or CK124 cells harboring the pKD46 plasmid. The recovered cells were spread onto LB agar plates supplemented with chloramphenicol and incubated at 30°C. The location of the lacZ gene downstream of the crRNA was confirmed using colony PCR. The PCR primers used for the insertion of cas12a, cas12a-crRNA, and cas12a-crRNA-lacZ into the λ prophage are listed in Supplementary Table S3.
2.6 Plaque isolation and phage lysate preparation
Lysogenic cells were initially grown at 30°C and then shifted to 42°C during the log phase. Subsequently, 100 μL of the diluted culture supernatant and 150 μL of phage-free E. coli MG1655 cell culture were added to 15 mL of LB top agar containing 5 mM CaCl2, 10 mM MgSO4, and 0.75% Bacto agar (BD Difco, Cat. No. 214010, USA) and gently mixed. The mixture was then overlaid onto LB agar and incubated at 30°C for 18 h. To obtain phage lysates from the formed plaques, a single phage plaque was added to the E. coli MG1655 culture grown in LB broth supplemented with 5 mM CaCl2 and 10 mM MgSO4. The culture was incubated at 37°C until an OD600 nm of 0.3 was reached, after which it was transferred to 42°C and incubated for 4 h.
2.7 Spotting assay
To evaluate the infectivity of λ phages, a spotting assay was performed as follows: 150 μL of overnight culture of E. coli host cells was mixed with 15 mL of the LB top agar and overlaid onto an LB agar plate. If needed, X-gal (final 50 μg/mL; Bioneer, Cat. No. C-8002-1, Korea) was added for confirming the β-galactosidase activity caused by infected lacZ-incorporated phages. Phage lysates (~ 109 pfu/ml), either undiluted or serially diluted in dilution buffer (100 mM NaCl, 10 mM MgSO4, 50 mM Tris–HCl [pH 7.5], 0.01% gelatin), were spotted onto the LB top agar plates and incubated at 30°C for 18 h or at 37°C for 12 h. Clear or turbid spots were observed on a lawn of E. coli cells. As negative controls, dilution buffer, fresh LB medium, and supernatant medium collected from ΔgalK or galK WT cell cultures after centrifugation (12,000 rpm, 4°C, 10 min) were spotted on the same plates.
2.8 Phage infection in bacterial cultures
Bacterial host cells were grown at 30°C or 37°C in 50 mL of LB broth supplemented with 5 mM CaCl2 and 10 mM MgSO4 in 125 mL flask with agitation at 180 rpm. When the culture reached an OD600 nm of 0.4, phage lysate (~ 109 pfu/ml) was added at a multiplicity of infection of 0.1. The multiplicity of infection was calculated based on the assumption that 1 mL of culture at an OD600 nm of 1.0 contained 8 × 108 cells. The OD600 nm value was measured using a spectrophotometer (Biochrom Libra S70, Harvard Bioscience, Inc., MA, USA).
For mixed cell cultures, when the OD600 nm reached 0.4, galK 504A and galK WT cells were mixed at an equal ratio. Phage infection and OD600 nm measurements were performed as described above. To assess microbial control using CRISPR-Cas12a-loaded λ phages in the mixed culture, the culture was spread onto MacConkey agar (BD Difco, Cat. No. 281810, USA) plates containing either 0.5% D-galactose (CAS No. 59–23-4) or 0.5% lactose (CAS No. 63–42-3). galK 504A and galK WT cells infected with λ cas12a galK-N16-lacZ were spread on M9 minimal agar (1 × M9 minimal salts, 0.1 mM CaCl2, 1 mM MgSO4, and 1.5% agar) plates supplemented with 0.5% D-galactose. Ten colonies obtained from the M9 D-galactose plates were randomly selected and streaked on MacConkey agar containing either D-galactose or lactose. The plates were incubated at 30°C for 16 h or 72 h to evaluate phage-mediated bacterial growth and selection.
3 Results
3.1 Validation of the activity of Cas12a nuclease-loaded λ phage
We constructed the λ cas12a phage by inserting the Lbcas12a gene into the b2 region of the λ cI857 phage genome (Figure 1A). λ cI857 is a temperature-sensitive phage with a point mutation in the cI gene that enables temperature-dependent genetic switching (Valdez-Cruz et al., 2010). At 28–32°C, the CI repressor suppressed the expression of lytic gene, allowing λ phage to remain in the lysogenic state. However, at temperatures greater than 37°C, the CI repressor became unstable, triggering a switch to the lytic cycle (Villaverde et al., 1993).
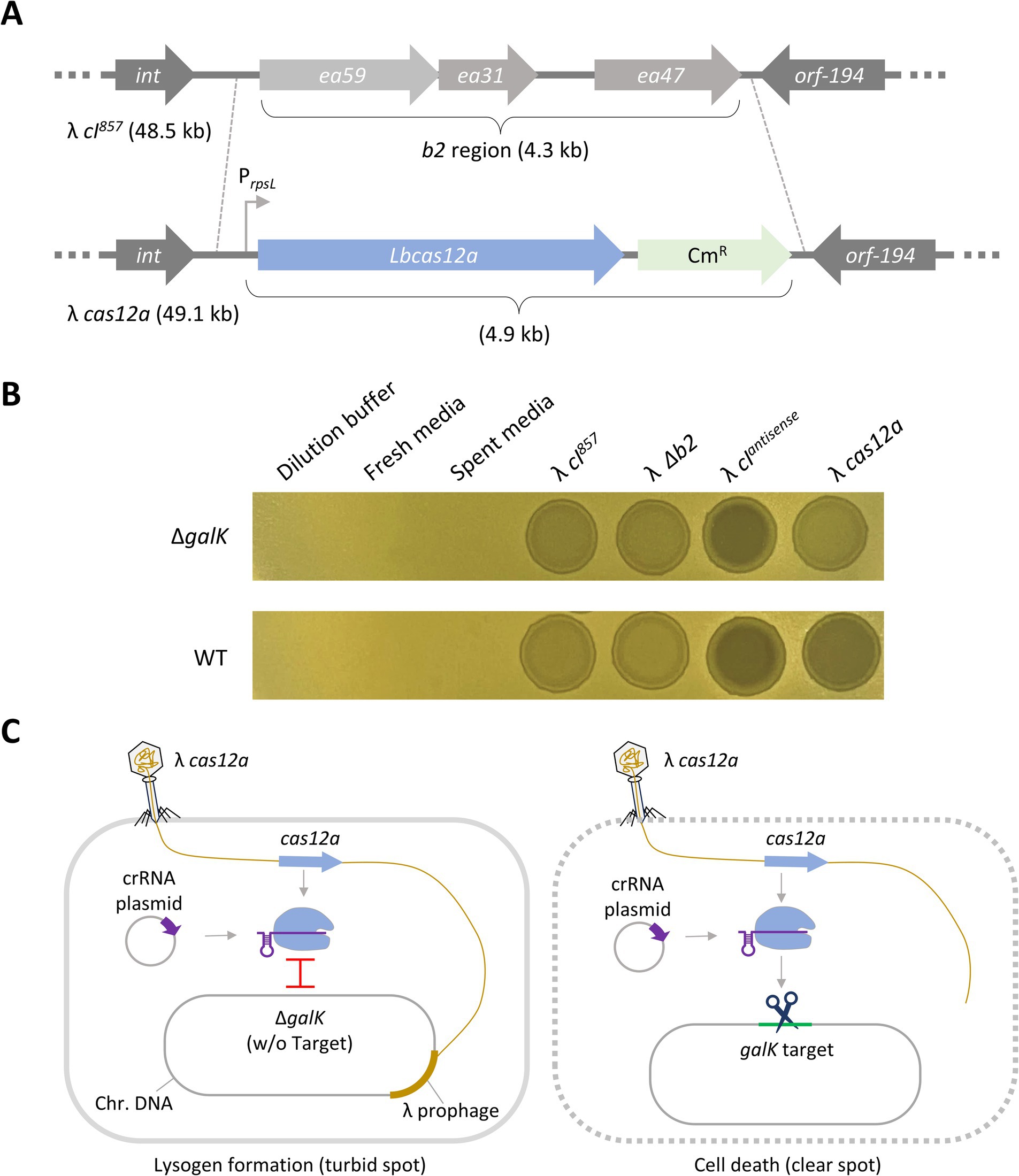
Figure 1. Death of target DNA-containing cells induced by λ phage carrying Cas12a. (A) Construction of λ cas12a phage. The cas12a gene was inserted into the b2 region of the λ cI857 prophage genome via homologous recombination. (B) Spotting assay of various synthetic λ phages on galK mutant strains. λ cI857, λ Δb2, λ cIantisense, and λ cas12a were spotted on LB top agar plates with ΔgalK or galK WT cells harboring galK-targeting crRNA plasmids. The plates were incubated at 30°C for 18 h. (C) Mechanism of cell death induced by λ cas12a. λ cas12a expresses the Cas12a nuclease and the crRNA plasmid in the host cell generates crRNA targeting the galK gene. This leads to the formation of the Cas12a–crRNA complex. If the host genome lacks the galK target, the phage remains in the prophage state without inducing cell death. Conversely, the presence of the galK target results in cell death.
To evaluate the activity of the constructed phage, we examined the turbidity of spots formed by it on LB top agar plates inoculated with either ΔgalK or galK WT cells carrying the galK-targeting crRNA plasmid (pCK058) at 30°C. Turbid spots were formed on ΔgalK cells, whereas clear spots were formed on galK WT cells (Figure 1B). Clear spots were observed even when phages (~109 pfu/ml) were diluted up to 10−3 (Supplementary Figure S3). In addition, to confirm the expression of Cas12a and crRNA in phage-infected lysogenic cells, the galK-targeting crRNA plasmid (pCK058) was transformed into λ cI857- and λ cas12a-lysogenic cells (HL051 or CK116), and the number of surviving colonies was compared (Supplementary Figure S4). These results indicate that the Cas12a nuclease and crRNA, expressed from the λ cas12a and crRNA plasmid, respectively, formed a Cas12a–crRNA complex capable of effectively recognizing and cleaving the galK target, leading to cell death. However, in the absence of the galK target, the Cas12a–crRNA complex could not cleave the target DNA, preventing cell death and resulting in the formation of turbid spots (Figure 1C). Additionally, regardless of the target gene, λ cI857 and λ cI857 Δb2 formed turbid spots on both ΔgalK and galK WT cells, attributed to their lysogenic cycle. In contrast, λ cIantisense formed clear spots, indicative of a lytic cycle, because of reduced expression of lysogenic CI repressors in the cells (Figure 1B).
3.2 Effect of truncated crRNAs on target recognition and host lysogeny
Truncated crRNAs have been reported to enhance the target specificity of Cas12a, allowing it to distinguish SNVs when used as a genome-editing tool (Lee et al., 2022b). Building on this premise, we tested whether truncated crRNAs could differentiate target DNA with single-nucleotide differences and lead to cell death. λ cas12a phage lysates were spotted on ΔgalK, galK 504A, and galK WT cells containing galK-targeting crRNA plasmids with different TRS lengths ranging from 23 nt (Δ0) to 15 nt (Δ8). When λ cas12a infected cells without crRNA or ΔgalK cells, turbid spots were formed, indicative of lysogeny. In contrast, clear spots were observed in galK 504A and galK WT cells carrying crRNA plasmids with TRS lengths ranging from 23 nt (Δ0) to 17 nt (Δ6), indicating cell death caused by the cleavage of target genomic DNA by the active Cas12a–crRNA complex (Figure 2A; Supplementary Figure S5). Notably, when using the 16 nt (Δ7) crRNA plasmid, turbid spots were observed in galK 504A cells, whereas clear spots were observed in galK WT cells. For the 15 nt (Δ8) crRNA plasmid, clear spots were not observed in either of these cell genotypes (Figure 2A). These results demonstrated that truncated 16 nt (Δ7) crRNA enabled λ phages carrying Cas12a to distinguish single-nucleotide differences in target DNA, determining host cell death or lysogeny (Figure 2B).
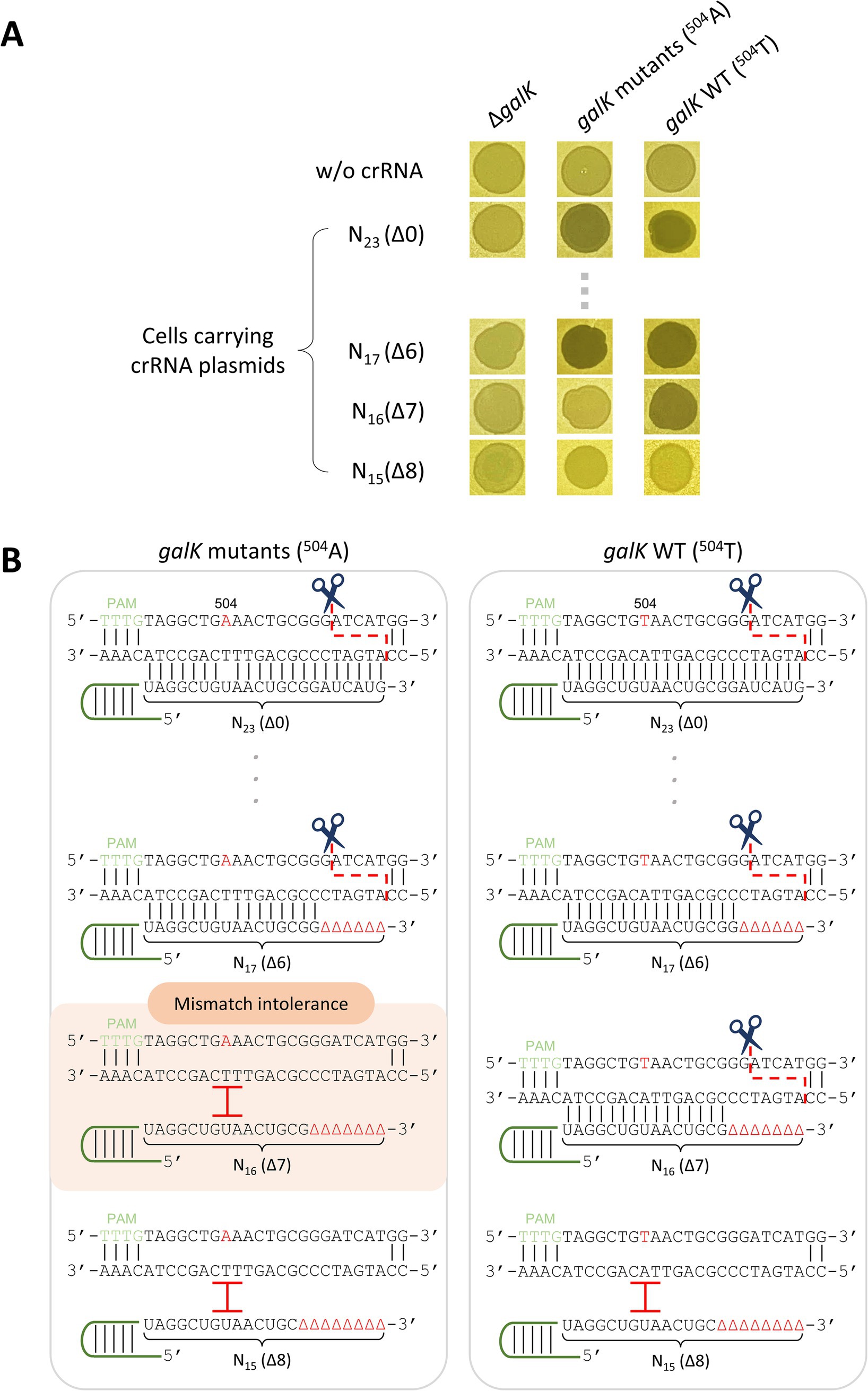
Figure 2. Discrimination of single-nucleotide variations using Cas12a and truncated crRNA. (A) Spotting assay of λ cas12a on various galK strains at 30°C. N23 and N17-N15 represent the length of the target recognition sequence (TRS) in the crRNA, and Δ in the parentheses indicates the number of truncated nucleotides at the 3′-end of the crRNA. (B) Mismatch intolerance. “504” represents the 504th nucleotide position in the galK gene of the E. coli genome. The nucleotides A and T in red represent the variant and original sequences, respectively. The black lines indicate base pairing. The scissors icon and red, dashed line show the position where the Cas12a–crRNA complex recognizes and cleaves the target DNA sequence. The red I indicates that the Cas12a–crRNA complex fails to recognize and cleave the target DNA.
3.3 Control of host viability by λ phages armed with truncated crRNAs and Cas12a nuclease
We investigated whether the results observed for cells carrying crRNA plasmids were due to multiple copies of the crRNA gene or if similar results could be achieved by loading a single copy of crRNA into the λ cas12a phage. For this, λ cas12a-crRNA phages were constructed by inserting galK-targeting crRNAs with various TRS lengths ranging from 23 nt (N23) to 15 nt (N15) into λ cas12a (Figure 3A). Spotting assays were then performed to evaluate whether the Cas12a–crRNA complex expressed from the λ cas12a-crRNA phages could control cell death by distinguishing single-nucleotide differences in the target gene within the host genome.
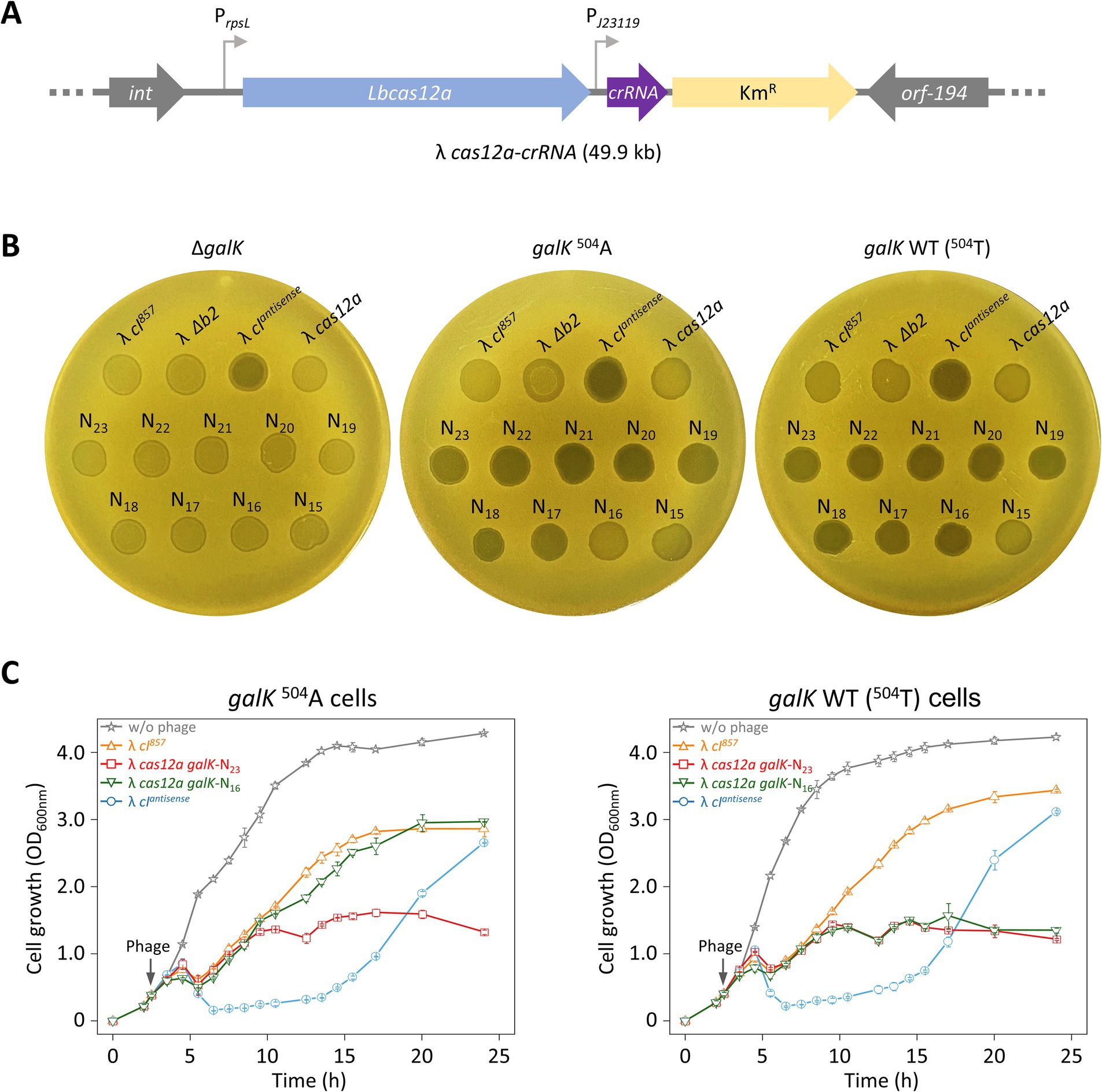
Figure 3. Sequence-specific target recognition and cell death control using λ phage carrying Cas12a and crRNA. (A) Construction of λ cas12a-crRNA phage. The cas12a gene and crRNA gene targeting the galK gene were inserted into the b2 region of the λ cI857 prophage genome. (B) Spotting assay of various synthetic λ phages including λ cas12a-crRNA at 30°C. λ cI857, λ Δb2, λ cIantisense, λ cas12a, and λ cas12a-crRNA were spotted on LB top agar plates containing ΔgalK, galK 504A, or galK WT cells, and incubated at 30°C for 18 h. N23-N15 represent λ cas12a-crRNA with crRNA TRS lengths ranging from 23 nt (Δ0) to 15 nt (Δ8). (C) Growth curves of galK 504A and galK WT strains either uninfected or infected with λ cI857, λ cIantisense, λ cas12a galK-N23, and λ cas12a galK-N16, respectively, at 30°C. The gray arrow indicates the time point of phage infection. OD600 nm values represent the mean obtained from three independent cultures.
λ cI857, λ cI857 Δb2, and λ cas12a phages produced turbid spots, whereas λ cIantisense produced clear spots, regardless of the cellular genotype (ΔgalK, galK 504A, or galK WT) (Figure 3B). As expected, all λ cas12a–crRNA phages (N23–N15) formed turbid spots in ΔgalK cells, as the Cas12a–crRNA complex fails to cleave the genome, thereby preventing cell death and allowing lysogeny, which leads to turbid spot formation. In the case of galK 504A or galK WT cells, clear spots were observed with λ cas12a-crRNA phages (N23 –N17). However, λ cas12a-crRNA phages (N16) formed turbid spots in galK 504A cells, whereas clear spots were observed in galK WT cells (Figure 3B). These results indicated that Cas12a and crRNA expressed from λ cas12a-crRNA form an active complex, which effectively recognizes and cleaves the galK target, enabling precise control of target cell death. When incubated at 37°C, the cI857 gene no longer supports the lysogenic cycle, resulting in clear spots on all LB top agar plates regardless of the phage or host cell type (Supplementary Figure S6A).
In view of the results obtained using top agar plates, we investigated whether similar effects would be observed in liquid culture. We monitored the growth of galK 504A and galK WT cells infected with λ cI857, λ cIantisense, λ cas12a galK-N23, or λ cas12a galK-N16. When infected with lysogenic λ cI857, cells showed rapid growth recovery after an initial lysis phase between 4 and 6 h. In contrast, when infected with lytic λ cIantisense, cell growth was suppressed up to 15 h, followed by a period of rapid recovery. The growth patterns associated with λ cI857 and λ cIantisense were consistent for both the cell types (galK 504A or galK WT). The recovery growth following phage infection could result either from lysogeny or as a consequence of the emergence of phage-insensitive mutants, which is described in the next section.
Cells infected with λ cas12a galK-N23 showed no significant growth recovery after initial lysis, with OD600 nm remaining stable between 1 and 1.5 for up to 24 h in both galK 504A and galK WT cells at 30°C. In contrast, λ cas12a galK-N16 exhibited distinct effects depending on the host cell type—in galK 504A cells, the growth pattern resembled that of cells infected with lysogenic λ cI857, suggesting that Cas12a failed to recognize and cleave the galK 504A target; however, in galK WT cells, initial growth recovery after lysis was followed by inhibition, closely mimicking the growth suppression pattern observed for λ cas12a galK-N23-infected cells at 30°C (Figure 3C). At 37°C, ΔgalK, galK 504A, and galK WT cells infected with various phages, including the lytic λ cIantisense, exhibited similar growth patterns regardless of the phage type (Supplementary Figure S6B). These results indicated that synthetic phages carrying Cas12a and truncated crRNAs can effectively suppress lysogeny by precisely controlling cell death and distinguishing SNVs within the target DNA.
3.4 Removal of genetic variations in mixed bacterial cultures using synthetic phages
As mentioned in the results described above, the regrowth observed after phage infection could have resulted either from lysogeny or from the emergence of phage-insensitive mutants. Previous studies have used colony color changes to identify lysogenic cells (Chaib et al., 2019). However, such changes were not observed with λ phages in our study. To overcome this limitation, we engineered the λ phage genome by inserting the lacZ gene, enabling colony color to serve as an indicator of lysogeny. A complementation assay was then performed to confirm that lacZ was successfully delivered to ΔlacZ target cells during lysogeny. We introduced the lacZ gene into λ cI857, λ cas12a-galK N23, and λ cas12a-galK N16 to make λ cI857-lacZ, λ cas12a-galK N23-lacZ, and λ cas12a-galK N16-lacZ (Figure 4A). Lysates of these lacZ-incorporated phages were spotted on LB top agar containing X-gal and seeded with galK WT ΔlacZ cells. Blue spots were observed for the lacZ-incorporated phages (Supplementary Figure S7A). λ cas12a-galK N23-lacZ produced clear spots on both galK 504A and galK WT cells, with no distinction between them. However, λ cas12a-galK N16-lacZ formed clear spots only on galK WT cells (Supplementary Figure S7B). These results indicated that the presence of the lacZ gene did not affect the formation of turbid or clear spots on LB top agar plates containing the target cells.
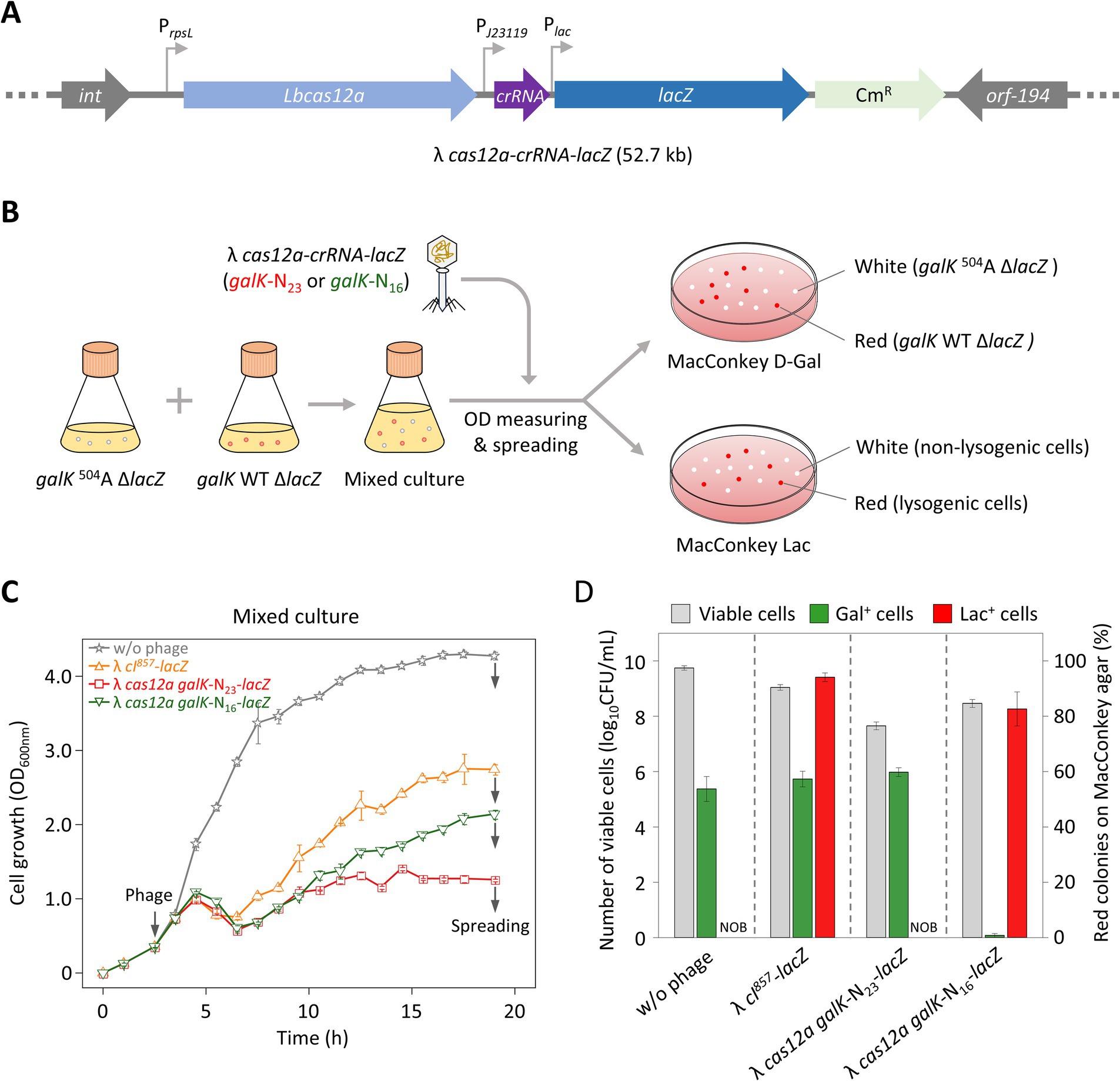
Figure 4. Control of cell death in mixed cell cultures with different galK phenotypes using λ phages carrying the CRISPR-Cas system and lacZ. (A) Construction of the λ cas12a-crRNA-lacZ phage. The cas12a gene, galK-targeting crRNA, and lacZ gene were inserted into the b2 region of the λ cI857 prophage genome. (B) Verification of cell death and lysogen formation in galK 504A ΔlacZ and galK WT ΔlacZ strains on MacConkey agar plates. galK 504A ΔlacZ and galK WT ΔlacZ cells were cultured to early exponential phase, mixed at equal ratio, and either left uninfected or were infected with the phage. After 19 h of incubation, the cultures were spread on MacConkey D-Gal and MacConkey Lac plates. On the MacConkey D-Gal plate, white and red colonies represent galK 504A ΔlacZ and galK WT ΔlacZ cells, respectively. On the MacConkey Lac plate, white and red colonies represent non-lysogenic and lysogenic cells, respectively. (C) Growth curves of mixed cultures containing galK 504A ΔlacZ and galK WT ΔlacZ strains, either uninfected or infected with various synthetic λ phages at 30°C. The gray arrows on the left indicate the time point of phage infection, whereas gray arrows on the right indicate the time point of spreading. Each OD600 nm value represents the mean of values for three independent cultures. (D) Proportions of galK 504A ΔlacZ and galK WT ΔlacZ strains, as well as lysogenic cells, observed on MacConkey agar plates. The gray bars represent the total number of viable cells (white colonies + red colonies) observed on the plates. The green bars indicate the proportion of galK WT ΔlacZ cells among the viable cells, whereas the red bars represent the proportion of lysogenic cells among the viable cells. The data are presented as mean ± standard deviations based on three independent experiments. NOB, Not observed.
To investigate whether λ cas12a galK-N23-lacZ or λ cas12a galK-N16-lacZ selectively eliminated target cells while regulating lysogen formation in liquid culture, we treated mixed cell cultures containing strains with a ΔlacZ background with lacZ-loaded phages. At 19 h after the start of incubation, the culture was spread on MacConkey agar plates containing D-galactose or lactose. The galK 504A ΔlacZ strain, carrying a nonsense mutation in the galK gene and unable to metabolize galactose, formed white colonies on MacConkey D-galactose plates, whereas the galK WT ΔlacZ strain formed red colonies. Furthermore, lysogen host cells infected with lacZ-incorporated phages produced red colonies on MacConkey lactose plates, whereas ΔlacZ cells that did not acquire the phage genetic material continued to form white colonies (Figure 4B).
Infection with λ cI857-lacZ triggered a rapid recovery of the growth curve in mixed cells following initial cell lysis. In contrast, λ cas12a galK-N23-lacZ infection resulted in only minimal growth recovery. The growth pattern observed with λ cas12a galK-N16-lacZ infection was intermediate between those of λ cI857-lacZ and λ cas12a galK-N23-lacZ infections (Figure 4C). On MacConkey D-galactose plates, infections with either λ cI857-lacZ or λ cas12a galK-N23-lacZ produced a similar ratio of red to white colonies, comparable with that in untreated cultures, indicating that these phages did not distinguish between galK 504A and galK WT cells (Figure 4D). However, λ cas12a galK-N16-lacZ infection resulted in less than 1% of the colonies being red, which indicated that this phage selectively eliminated galK WT ΔlacZ cells while sparing galK 504A ΔlacZ cells in the mixed culture.
On MacConkey lactose plates, neither the untreated cultures nor those infected with λ cas12a galK-N23-lacZ produced red colonies, indicating that λ cas12a galK-N23-lacZ effectively inhibited lysogen formation. In contrast, infections with λ cI857-lacZ and λ cas12a galK-N16-lacZ resulted in high proportions of red colonies, at 94 and 83%, respectively. To determine whether the red colonies observed on MacConkey galactose plates following λ cas12a galK-N16-lacZ infection were lysogenic, ten colonies randomly selected from M9 D-galactose plates were streaked on either MacConkey D-galactose or MacConkey lactose plates. All red colonies on MacConkey D-galactose plates appeared as white colonies on the MacConkey lactose plates, indicating the gal+ cells were not lysogenic (Supplementary Figure S8). These results indicated that λ cI857-lacZ forms lysogens regardless of the cell strain, whereas λ cas12a galK-N16-lacZ fails to eliminate galK 504A cells, allowing them to remain present as lysogens.
4 Discussion
Bacteriophage λ is a temperate phage capable of switching between the lytic and lysogenic cycles upon host cell infection. In the lytic cycle, the host cell undergoes lysis, releasing newly assembled phages, whereas in the lysogenic cycle, the phage genome integrates into the host chromosome and is passively replicated (Court et al., 2007). Lysogeny enables prophages to acquire virulence factors and vertically transmit them across generations (Canchaya et al., 2004; Fan and Kan, 2015; Mokrousov, 2009; Naidoo and Zishiri, 2023). Host cells carrying a prophage exhibit superinfection immunity, preventing reinfection by similar phages and potentially limiting bacteriophage-based strategies for microbial control targeting antibiotic-resistant bacteria or pathogens. To overcome this limitation, we engineered a CRISPR-Cas-based λ phage system capable of precise DNA recognition and regulated lysogen formation.
To effectively engineer bacteriophages, it is crucial to identify the nonessential regions of the genome that can be removed and to determine the size limits of foreign genes that can be introduced. The b2 region of the λ phage is a nonessential gene, and its removal has no impact on the survival or replication of the phage (Casjens and Hendrix, 2015). We, therefore, inserted the cas12a gene and crRNAs into the b2 region of λ cI857 (Figure 1A). Regarding size limits, the λ phage is known to accommodate up to 110% of the wild-type genome length (53.4 kb) while remaining infectious. However, a λ phage with a 53.4 kb genome exhibits a 1,000-fold reduction in infectivity compared with that of the wild-type phage (Nurmemmedov et al., 2012). To address this, we inserted a cas12a-crRNA-lacZ cassette into the λ phage genome (48.5 kb) to create λ cas12a-crRNA-lacZ (52.7 kb), which effectively suppressed lysogen formation and selectively eliminated target cells (Figure 4A).
The CRISPR-Cas system exhibits mismatch tolerance, allowing it to recognize and cleave target DNA even when 1–2 mismatches exist between the gRNA and target DNA (Fu et al., 2013). To overcome this issue, previous studies have successfully performed single-nucleotide editing within the target DNA using truncated gRNA (Lee et al., 2023; Lee et al., 2022b). Therefore, in this study, we designed crRNA plasmids with various TRS lengths to evaluate whether truncated crRNA could distinguish host cells with SNVs and regulate cell lysis (Figure 2A; Supplementary Figure S2). We observed that LbCas12a requires a crRNA with a TRS length of 16 nt (Δ7) to selectively recognize and cleave target DNA at a single-nucleotide resolution, thereby inducing cell lysis. This result is consistent with the minimum TRS length required for FnCas12a to recognize and cleave target DNA (Lee et al., 2022b).
When treated with most phages in this study, cells in liquid culture eventually recovered their growth within 24 h, reaching an OD of 3.0, regardless of whether they were infected by lysogenic λ cI857 or lytic λ cIantisense (Figure 3C). This growth recovery was likely due to either lysogen formation or the emergence of phage-insensitive mutants. To confirm the actual reason, we constructed lacZ-incorporated phages and examined whether the LacZ phenotype was transferred to ΔlacZ cells in mixed cultures (Figures 4C,D).
Li et al. reported that the primary mechanism of resistance to phages in E. coli is the inability of phages to recognize and bind to specific receptors on the bacterial surface (Li et al., 2019). In particular, a missense mutation in LamB, an essential receptor for λ phage infection, can block the attachment of phages to the host receptor, thereby preventing infection (Charbit et al., 1988; Werts et al., 1994). When λ cas12a galK-N23-lacZ was applied, the viable cell population remained approximately 60% Gal+, indicating that the mixture of galK 504A and galK WT cells was maintained and that the cells remained LacZ− (Figure 4D). This suggests that the surviving cells were likely phage-insensitive mutants. We assume that the growth of phage-infected cells was initially inhibited, but it was later restored due to the emergence of phage-resistant mutants, such as those with receptor mutations. In contrast, when cells were treated with λ cas12a galK-N16-lacZ, all surviving cells exhibited the Gal− LacZ+ phenotype, indicating the formation of lysogens in galK 504A cells (Figure 4D). This observation is consistent with previous studies showing that temperate λ phages initially induce lysis upon infecting E. coli, but the cell growth subsequently recovers as the lysogen population increases (Maynard et al., 2010; Maynard et al., 2012).
In conclusion, we demonstrated that a temperate phage carrying a Cas12a-truncated crRNA can discriminate host bacteria based on a SNV in the target genome. Guided by the designed truncated crRNA sequence, the synthetic phage precisely modulated lysogeny in the host. Moreover, in a mixed-culture model, the synthetic temperate phage selectively targeted the SNV-containing strain and exhibited lytic activity. These findings offer important insights into the potential application of this strategy as a precision microbiome engineering tool capable of selectively eliminating undesirable bacteria at the SNV level while preserving non-target strains.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
CL: Conceptualization, Formal analysis, Investigation, Methodology, Writing – original draft. HL: Formal analysis, Investigation, Writing – review & editing. SJ: Formal analysis, Investigation, Writing – review & editing. SL: Conceptualization, Formal analysis, Funding acquisition, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MSIT) (RS-2024-00342735). This research was also supported by the Chung-Ang University Graduate Research Scholarship in 2024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1575339/full#supplementary-material
References
Balcha, F. B., and Neja, S. A. (2023). CRISPR-Cas9 mediated phage therapy as an alternative to antibiotics. Anim. Dis. 3, 1–12. doi: 10.1186/s44149-023-00065-z
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Canchaya, C., Fournous, G., and Brussow, H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. doi: 10.1111/j.1365-2958.2004.04113.x
Casjens, S. R., and Hendrix, R. W. (2015). Bacteriophage lambda: early pioneer and still relevant. Virology 479-480, 310–330. doi: 10.1016/j.virol.2015.02.010
Chaib, A., Philippe, C., Jaomanjaka, F., Claisse, O., Jourdes, M., Lucas, P., et al. (2019). Lysogeny in the lactic acid bacterium is responsible for modified Colony morphology on red grape juice agar. Appl. Environ. Microbiol. 85, e00997-19. doi: 10.1128/AEM.00997-19
Charbit, A., Gehring, K., Nikaido, H., Ferenci, T., and Hofnung, M. (1988). Maltose transport and starch binding in phage-resistant point mutants of maltoporin: functional and topological implications. J. Mol. Biol. 201, 487–493. doi: 10.1016/0022-2836(88)90630-4
Chen, Y., Yan, B., Chen, W., Zhang, X., Liu, Z., Zhang, Q., et al. (2024). Development of the CRISPR-Cas12a system for editing of Pseudomonas aeruginosa phages. iScience 27:110210. doi: 10.1016/j.isci.2024.110210
Citorik, R. J., Mimee, M., and Lu, T. K. (2014). Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141–1145. doi: 10.1038/nbt.3011
Court, D. L., Oppertheim, A. B., and Adhya, S. L. (2007). A new look at bacteriophage λ genetic networks. J. Bacteriol. 189, 298–304. doi: 10.1128/JB.01215-06
Datsenko, K. A., Pougach, K., Tikhonov, A., Wanner, B. L., Severinov, K., and Semenova, E. (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3:945. doi: 10.1038/ncomms1937
Dong, J., Chen, C., Liu, Y., Zhu, J., Li, M., Rao, V. B., et al. (2021). Engineering T4 bacteriophage for in vivo display by type V CRISPR-Cas genome editing. ACS Synth. Biol. 10, 2639–2648. doi: 10.1021/acssynbio.1c00251
Fagen, J. R., Collias, D., Singh, A. K., and Beisel, C. L. (2017). Advancing the design and delivery of CRISPR antimicrobials. Curr. Opin. Biomed. Eng. 4, 57–64. doi: 10.1016/j.cobme.2017.10.001
Fan, F., and Kan, B. (2015). Survival and proliferation of the lysogenic bacteriophage CTXPhi in Vibrio cholerae. Virol. Sin. 30, 19–25. doi: 10.1007/s12250-014-3550-7
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi: 10.1038/nbt.2623
Gencay, Y. E., Jasinskyte, D., Robert, C., Semsey, S., Martinez, V., Petersen, A. O., et al. (2024). Engineered phage with antibacterial CRISPR-Cas selectively reduce E. coli burden in mice. Nat. Biotechnol. 42, 265–274. doi: 10.1038/s41587-023-01759-y
Gillmore, J. D., Maitland, M. L., and Lebwohl, D. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502. doi: 10.1056/NEJMoa2107454
Ha, D. I., Lee, J. M., Lee, N. E., Kim, D., Ko, J. H., and Kim, Y. S. (2020). Highly efficient and safe genome editing by CRISPR-Cas12a using CRISPR RNA with a ribosyl-2′-O-methylated uridinylate-rich 3′-overhang in mouse zygotes. Exp. Mol. Med. 52, 1823–1830. doi: 10.1038/s12276-020-00521-7
Hoshiga, F., Yoshizaki, K., Takao, N., Miyanaga, K., and Tanji, Y. (2019). Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 366:fnz041. doi: 10.1093/femsle/fnz041
Huang, J., Zhou, Y., Li, J., Lu, A., and Liang, C. (2022). CRISPR/Cas systems: delivery and application in gene therapy. Front. Bioeng. Biotechnol. 10:942325. doi: 10.3389/fbioe.2022.942325
Kiro, R., Shitrit, D., and Qimron, U. (2014). Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 11, 42–44. doi: 10.4161/rna.27766
Lee, H. J., Kim, H. J., and Lee, S. J. (2022a). Control of λ lysogenic Escherichia coli cells by synthetic λ phage carrying cIantisense. ACS Synth. Biol. 11, 3829–3835. doi: 10.1021/acssynbio.2c00409
Lee, H. J., Kim, H. J., and Lee, S. J. (2023). Miniature CRISPR-Cas12f1-mediated single-nucleotide microbial genome editing using 3′-truncated sgRNA. CRISPR J. 6, 52–61. doi: 10.1089/crispr.2022.0071
Lee, H. J., Kim, H. J., Park, Y. J., and Lee, S. J. (2022b). Efficient single-nucleotide microbial genome editing achieved using CRISPR/Cpf1 with maximally 3′-end-truncated crRNAs. ACS Synth. Biol. 11, 2134–2143. doi: 10.1021/acssynbio.2c00054
Lee, K., Zhang, Y. X., Kleinstiver, B. P., Guo, J. A., Aryee, M. J., Miller, J., et al. (2018). Activities and specificities of CRISPR/Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol. J. 17, 362–372. doi: 10.1111/pbi.12982
Li, C., Li, W., Zhou, Z., Chen, H., Xie, C., and Lin, Y. (2020). A new rice breeding method: CRISPR/Cas9 system editing of the Xa13 promoter to cultivate transgene-free bacterial blight-resistant rice. Plant Biotechnol. J. 18, 313–315. doi: 10.1111/pbi.13217
Li, P., Lin, H., Mi, Z., Xing, S., Tong, Y., and Wang, J. (2019). Screening of polyvalent phage-resistant Escherichia coli strains based on phage receptor analysis. Front. Microbiol. 10:850. doi: 10.3389/fmicb.2019.00850
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Martel, B., and Moineau, S. (2014). CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 42, 9504–9513. doi: 10.1093/nar/gku628
Maynard, N. D., Birch, E. W., Sanghvi, J. C., Chen, L., Gutschow, M. V., and Covert, M. W. (2010). A forward-genetic screen and dynamic analysis of lambda phage host-dependencies reveals an extensive interaction network and a new anti-viral strategy. PLoS Genet. 6:e1001017. doi: 10.1371/journal.pgen.1001017
Maynard, N. D., Macklin, D. N., Kirkegaard, K., and Covert, M. W. (2012). Competing pathways control host resistance to virus via tRNA modification and programmed ribosomal frameshifting. Mol. Syst. Biol. 8:567. doi: 10.1038/msb.2011.101
Meile, S., Du, J., Staubli, S., Grossmann, S., Koliwer-Brandl, H., Piffaretti, P., et al. (2023). Engineered reporter phages for detection of Escherichia coli, Enterococcus, and Klebsiella in urine. Nat. Commun. 14:4336. doi: 10.1038/s41467-023-39863-x
Moescheid, M. F., Wisitphongpun, P., Mann, V. H., Quack, T., Grunau, C., Grevelding, C. G., et al. (2023). Enhanced efficiency of RNA-guided Cas12a versus Cas9 transgene knock-in and activity at a Schistosoma mansoni genome safe harbor. bioRxiv. doi: 10.1101/2023.09.12.557428
Mokrousov, I. (2009). Corynebacterium diphtheriae: genome diversity, population structure and genotyping perspectives. Infect. Genet. Evol. 9, 1–15. doi: 10.1016/j.meegid.2008.09.011
Naidoo, N., and Zishiri, O. T. (2023). Comparative genomics analysis and characterization of Shiga toxin-producing Escherichia coli O157:H7 strains reveal virulence genes, resistance genes, prophages and plasmids. BMC Genomics 24:791. doi: 10.1186/s12864-023-09902-4
Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J., Wang, L., et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843. doi: 10.1016/j.cell.2014.01.027
Nurmemmedov, E., Castelnovo, M., Medina, E., Catalano, C. E., and Evilevitch, A. (2012). Challenging packaging limits and infectivity of phage λ. J. Mol. Biol. 415, 263–273. doi: 10.1016/j.jmb.2011.11.015
Park, J. Y., Moon, B. Y., Park, J. W., Thornton, J. A., Park, Y. H., and Seo, K. S. (2017). Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci. Rep. 7:44929. doi: 10.1038/srep44929
Selle, K., Fletcher, J. R., Tuson, H., Schmitt, D. S., McMillan, L., Vridhambal, G. S., et al. (2020). In vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 antimicrobials. mBio 11, e00019-20. doi: 10.1128/mBio.00019-20
Shen, W., Zhang, J., Geng, B., Qiu, M., Hu, M., Yang, Q., et al. (2019). Establishment and application of a CRISPR-Cas12a assisted genome-editing system in Zymomonas mobilis. Microb. Cell Fact. 18:162. doi: 10.1186/s12934-019-1219-5
Slaman, E., Kottenhagen, L., de Martines, W., Angenent, G. C., and de Maagd, R. A. (2024). Comparison of Cas12a and Cas9-mediated mutagenesis in tomato cells. Sci. Rep. 14:4508. doi: 10.1038/s41598-024-55088-4
Tao, P., Wu, X., Tang, W. C., Zhu, J., and Rao, V. (2017). Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth. Biol. 6, 1952–1961. doi: 10.1021/acssynbio.7b00179
Valdez-Cruz, N. A., Caspeta, L., Perez, N. O., Ramirez, O. T., and Trujillo-Roldan, M. A. (2010). Production of recombinant proteins in E. coli by the heat inducible expression system based on the phage lambda pL and/or pR promoters. Microb. Cell Fact. 9:18. doi: 10.1186/1475-2859-9-18
Villaverde, A., Benito, A., Viaplana, E., and Cubarsi, R. (1993). Fine regulation of cI857-controlled gene expression in continuous culture of recombinant Escherichia coli by temperature. Appl. Environ. Microbiol. 59, 3485–3487. doi: 10.1128/aem.59.10.3485-3487.1993
Werts, C., Michel, V., Hofnung, M., and Charbit, A. (1994). Adsorption of bacteriophage lambda on the LamB protein of Escherichia coli K-12: point mutations in gene J of lambda responsible for extended host range. J. Bacteriol. 176, 941–947. doi: 10.1128/jb.176.4.941-947.1994
Wiles, M. V., Qin, W., Cheng, A. W., and Wang, H. (2015). CRISPR-Cas9-mediated genome editing and guide RNA design. Mamm. Genome 26, 501–510. doi: 10.1007/s00335-015-9565-z
Keywords: CRISPR-Cas12a, bacteriophage, lambda, lysogen, Escherichia coli
Citation: Lee CK, Lee HJ, Jeong SH and Lee SJ (2025) Precision targeting of genetic variations in mixed bacterial cultures using CRISPR-Cas12a-programmed λ phages. Front. Microbiol. 16:1575339. doi: 10.3389/fmicb.2025.1575339
Edited by:
Karen Fong, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Cécile Philippe, Institut Français de Recherche pour l’Exploitation de la Mer (IFREMER), FranceJasna Rakonjac, Massey University, New Zealand
Amita Pandey, Amity University, India
Copyright © 2025 Lee, Lee, Jeong and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang Jun Lee, c2FuZ2psZWVAY2F1LmFjLmty