 Ruyu Yan
Ruyu Yan Jun Ji1
Jun Ji1 Han Shen
Han Shen Xiaoli Cao
Xiaoli Cao- 1Department of Laboratory Medicine, Nanjing Drum Tower Hospital Clinical College of Nanjing University of Chinese Medicine, Nanjing, China
- 2Department of Laboratory Medicine, Nanjing Drum Tower Hospital Clinical College of Nanjing University, Nanjing, China
Objective: This study analyzes the global prevalence and distribution of vancomycin resistance genes (van) in Enterococcus faecium and examines the genetic relationship and epidemiological characteristics of strains carrying these genes.
Method: A total of 3,256 E. faecium genome sequences were downloaded, and 2,235 high-quality genomes were retained after quality filtering. The blastn tool was used to screen these genomes for van genes, and sequence types (STs) were determined using pubMLST profiles.
Result: Among the 2,235 genomes, 1,071 (47.9%) harbored van genes, with eight genotypes identified, including vanA, vanB, vanD, and vanM, accounting for 47.6%. There were 83 distinct STs among the strains carrying van genes, with ST17 being the most prevalent. Most strains carrying van genes were isolated from humans, primarily in the United States, and commonly from rectal swabs. In 2015, vanA was the most prevalent van gene, particularly in ST17 strains.
Conclusion: This study highlights the widespread distribution of van genes and their significant presence in human populations and clinical settings, emphasizing the importance of monitoring and intervening in the spread of ST17 strains.
1 Introduction
Enterococcus faecium (E. faecium) is a common opportunistic pathogen in hospital settings, frequently responsible for infections of the blood, urinary tract, and abdome (Reynolds et al., 2021). The extensive use of vancomycin and other antibiotics has facilitated the emergence and proliferation of vancomycin-resistant Enterococcus faecium (VREfm) (Ji et al., 2024), a bacterium that poses significant challenges due to its limited treatment options, prolonged hospital stays, and high mortality rate (McHugh et al., 2024). Recognizing its critical impact, the World Health Organization included VREfm in its 2024 list of pathogens that urgently require new therapeutic approaches.
Resistance to vancomycin in VREfm is primarily mediated by vancomycin resistance (van) genes, which are typically located on plasmids (Li et al., 2022). The principal van genes in Enterococcus—vanA, vanB, vanD, and vanM—encode D-alanine:D-lactate ligase, conferring high-level resistance (Geraldes et al., 2022).
Epidemiological studies have identified distinct geographical and clonal patterns of these resistance genes. For example, Elstrøm et al. (2019) reported an incidence of 7.09 per 100,000 person-years for VRE infections/colonization in Norway (Eichel et al., 2023), while in Egypt, the pooled prevalence of VRE among clinical isolates as high as 26%, with E. faecium reaching 32.5% (Azzam et al., 2023). A meta-analysis in Nigeria found that 63.1% of E. faecium strains exhibited resistance to vancomycin (Orababa et al., 2021). Moreover, different regional studies highlight the prevalence of specific sequence types (STs) associated with these resistance genes: in Brazil, the vanA gene predominantly occurs in ST21 strains (Farias et al., 2023); in Poland, nearly all carriers of the vanA gene (97.6%) are associated with the Clonal Complex CC2 and CC87 (Wardal et al., 2022); in Algeria, the vanA gene mainly appears in ST80 and ST789, part of CC17 (Benamrouche et al., 2021); in Germany, the vanB gene is most frequent, particularly in ST117 (Nürnberger et al., 2021); and in China, a study from a tertiary hospital found the vanA gene most prevalent in ST78, ST192, and ST570 (Zhou et al., 2020). However, data on the global prevalence and characteristics of van genes among E. faecium remain limited.
With the increased use of vancomycin, the number of strains carrying van genes has risen and rapidly evolved. The advancement of whole-genome sequencing (WGS) technology has also led to a significant increase in sequenced bacterial genomes, with van resistance genes being increasingly reported, potentially establishing E. faecium carrying van genes as a common pathogen (Karaman et al., 2020). However, global distribution data on E. faecium carrying van genes remain limited. In this study, we first explored the distribution of van genes in E. faecium isolates based on global databases. For strains carrying van genes, we further investigated STs, evolutionary relationships, as well as their regional and temporal distribution.
2 Materials and methods
2.1 Genome sequence download and pre-processing
E. faecium genome sequences (GenBank format) were systematically retrieved in batch format using Aspera1 as of the cutoff December 7, 2023. The GenBank files for all 3,256 E. faecium genomes were downloaded using Perl script. All 3,256 strains were annotated using Prodigal, chosen for its robustness in gene prediction, to ensure consistency across the dataset. Quality filtering was performed using CheckM v1.1.3 and Quest 5.0.2 software (Li et al., 2023; Joddha et al., 2023). We set specific parameters to ensure the selection of high-quality genomes: integrity greater than 90%, contamination rate less than 5%, a maximum of 500 contigs per genome, and an N50 of at least 40 kb (Wattanasombat and Tongjai, 2024). These stringent criteria ensured that only the most reliable and complete genomes were included for further analysis. After rigorous filtering, 2,235 high-quality genomes were obtained. These genomes met all specified quality standards and were deemed suitable for in-depth genetic and epidemiological analysis.
2.2 Identification of vancomycin resistance genes
We compiled a structured resistance gene database using van resistance gene sequences sourced from the NCBI Pathogen Resistance Gene Database. The nucleotide coding sequences for all genes in the 2,235 high-quality genomes, as annotated by Prodigal, were compared against our structured resistance gene database using blastn. This comparison was aimed at obtaining a detailed distribution of van-positive genes across all analyzed genomes. To ensure specificity and relevance of the matching results, we set stringent parameters for the blastn analysis: E-value = 1 × 10−5, identity ≥90%, coverage ≥90%, match length ≥30.
2.3 Sequence typing of van-carrying Enterococcus faecium strains
For the sequence typing of 1,071 van-carrying E. faecium strains, a self-designed tool, ST-tool was utilized. This tool was developed specifically to handle the unique demands of our study, ensuring accurate and efficient sequence typing based on established genetic markers. Seven housekeeping gene sequence files for E. faecium from the pubMLST website were acquired. These files represent the core of our sequence typing analysis, as they contain critical genetic markers used for identifying STs among different E. faecium isolates. Using blastn, we compared the housekeeping gene sequences from genomes carrying van genes against the seven housekeeping gene sequence files obtained. The criteria for this comparison were stringent, requiring 100% identity and 100% coverage to ensure that only exact matches were considered. This high threshold was set to accurately assign sequence types without ambiguity. The results from the blastn comparisons were then cross-referenced with the profile file from pubMLST to determine the STs of each strain. By matching gene sequences to known profiles, we could assign an ST to each van-carrying E. faecium strain, facilitating further epidemiological and resistance mechanism analysis.
2.4 Phylogenetic tree construction
Two phylogenetic trees were constructed, one from 915 genomes containing the vanA gene and another from 130 E. faecium genomes containing the vanB gene. Initially, genomic nucleotide sequence files were downloaded in batch from the NCBI genome database using the specified genome assembly numbers. The E. faecium genomes were annotated using Prokka version 1.14.6 (Zhao et al., 2022), and the resulting GFF files served as input for Roary version 3.13.0 (Sultana et al., 2022), which performed a metagenomic analysis to yield core gene multiple sequence alignment files. From these, single nucleotide polymorphism (SNP) sequence files were extracted using SNP-sites v2.5.1 (Coll et al., 2022). Subsequently, the optimal nucleotide substitution model, GTR + G, was determined using jModelTest 2 based on the SNP sequence files (Jacob Machado et al., 2021). A maximum likelihood phylogenetic tree was then constructed for each gene group using RAxML-NG v. 1.2.1 software (Togkousidis et al., 2023), employing the GTR + G model with 1,000 bootstrapping samples. The resulting phylogenetic trees were imported into iTOL Version 6.5.8 software (Letunic and Bork, 2024), where branches with bootstrap values less than 50 were removed. The trees were exported as SVG files, which were then enhanced visually using Adobe Illustrator. Throughout this process, 487 core gene multiple sequence alignment files were acquired from the 915 genomes containing vanA, and 1,156 core gene multiple sequence alignment files were obtained from the 130 genomes containing vanB.
2.5 Metadata extraction and integration
We extracted meta information from the GenBank files of 2,235 genomes using custom Perl scripts. This metadata included critical epidemiological and clinical details such as the isolation time, country of origin, host, and sample source of each strain. The extracted metadata was then integrated with STs obtained from the sequence typing analysis. To facilitate this integration, both datasets were compiled into a single table, allowing for a comprehensive dataset that combines genetic and epidemiological data.
3 Results
3.1 A high prevalence of the van gene in global Enterococcus faecium populations
Among the 2,235 strains of E. faecium analyzed, 1,071 (47.9%) were found to carry the van gene. In all E. faecium, vanA was the most prevalent, identified in 915 strains (40.9%), followed by vanB in 130 strains (5.8%). Additionally, vanD was detected in 15 strains (0.7%), vanM in 12 strains (0.5%), and vanP in four strains (0.2%). The vanN gene was identified in two strains (0.1%).
3.2 Multiple sequence types serve as hosts for van genes
In total, 83 different STs were identified among the 1,071 E. faecium strains carrying van gene. Among them, ST17 was the most common, found in 87 (8.1%) strains, followed by ST117 (6.1%) in 66 strains, ST78 in 61 (5.7%) strains, ST203 in 48 (4.5%) strains, and ST736 in 40 (3.7%) strains, the distribution of the major van genotypes such as vanA, vanB, vanD, and vanM, along with STs, is shown in Figure 1.
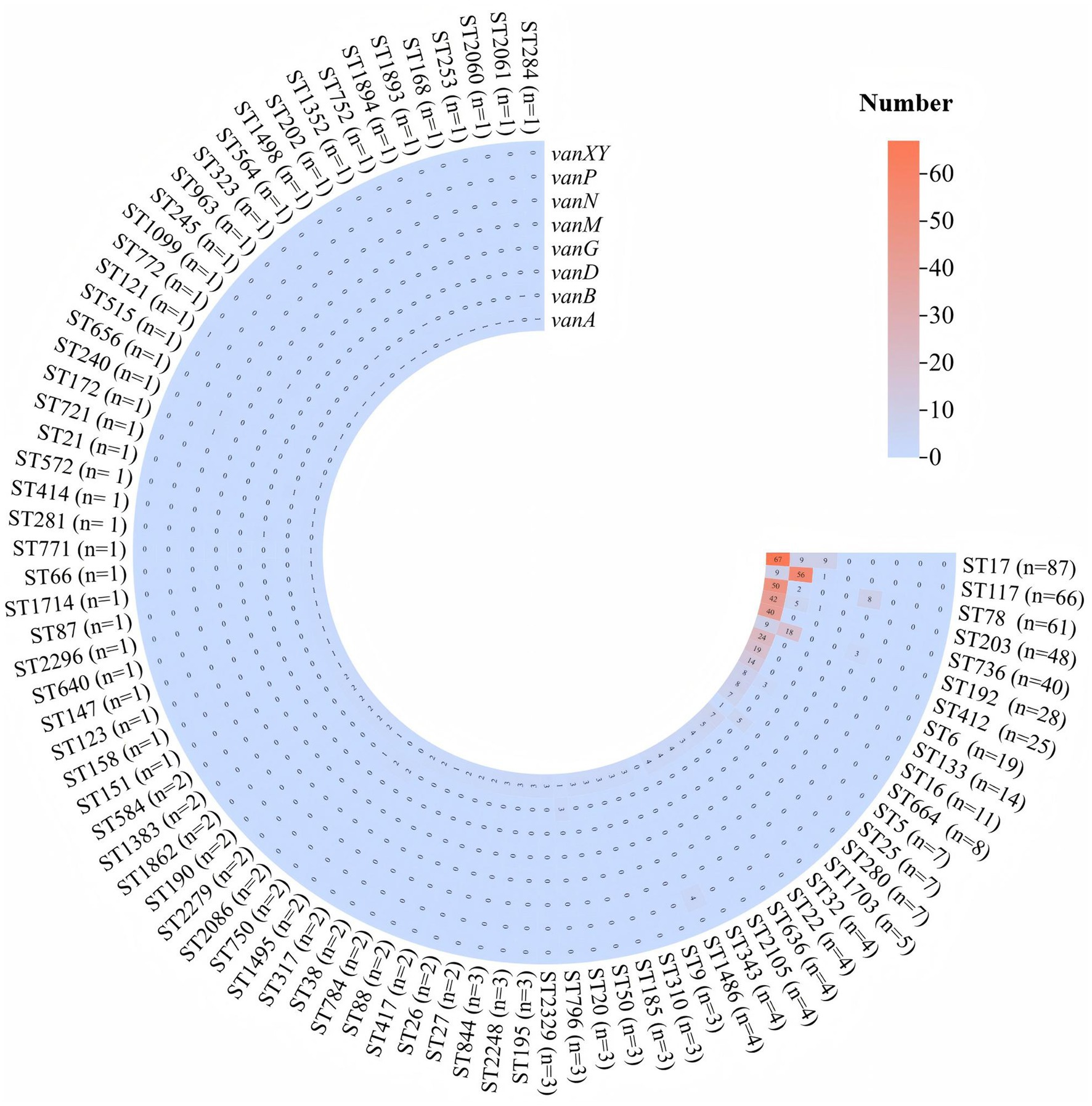
Figure 1. Distribution of vancomycin resistance genes among the predominant sequence types of Enterococcus faecium. The heatmap displays the presence of six vancomycin resistance genes: vanA, vanB, vanD, vanG, vanN, vanP, and vanXY. Each row represents a specific ST, with the number of isolates indicated in parentheses. The color gradient, from blue to red, indicates the number of isolates carrying each resistance gene, with higher numbers shown in red and lower numbers in blue.
4 Genetic relationship
4.1 Phylogenetic tree for strains carrying vanA gene
A total of 915 strains carrying vanA gene were analyzed through whole-genome phylogenetic reconstruction, revealing distinct genetic clusters alongside geographic and ST information, as shown in Figure 2. The innermost ring of the circular phylogenetic tree indicated that the majority of isolates originated from Europe, followed by Asia and North America, with fewer contributions from Africa, Oceania, and South America. Some isolates lacked geographic metadata and were labeled as NA. Country-level data, represented in the middle ring, showed high proportions of isolates from the United Kingdom, Germany, the USA, France, the Netherlands, and Australia, with additional contributions from countries such as China, Japan, India, and Saudi Arabia. These isolates formed both geographically concentrated clusters—suggesting local outbreaks—and mixed-country branches, indicating potential international transmission. The outermost ring showed a wide diversity of STs, with frequent representation of ST17 and ST78 across multiple regions. These STs often formed dense clusters, implying both local clonal expansion and the global spread of dominant vanA-carrying lineages. Overall, the phylogenetic structure highlighted a combination of localized hospital-associated outbreaks and widespread dissemination of high-risk clones across continents.
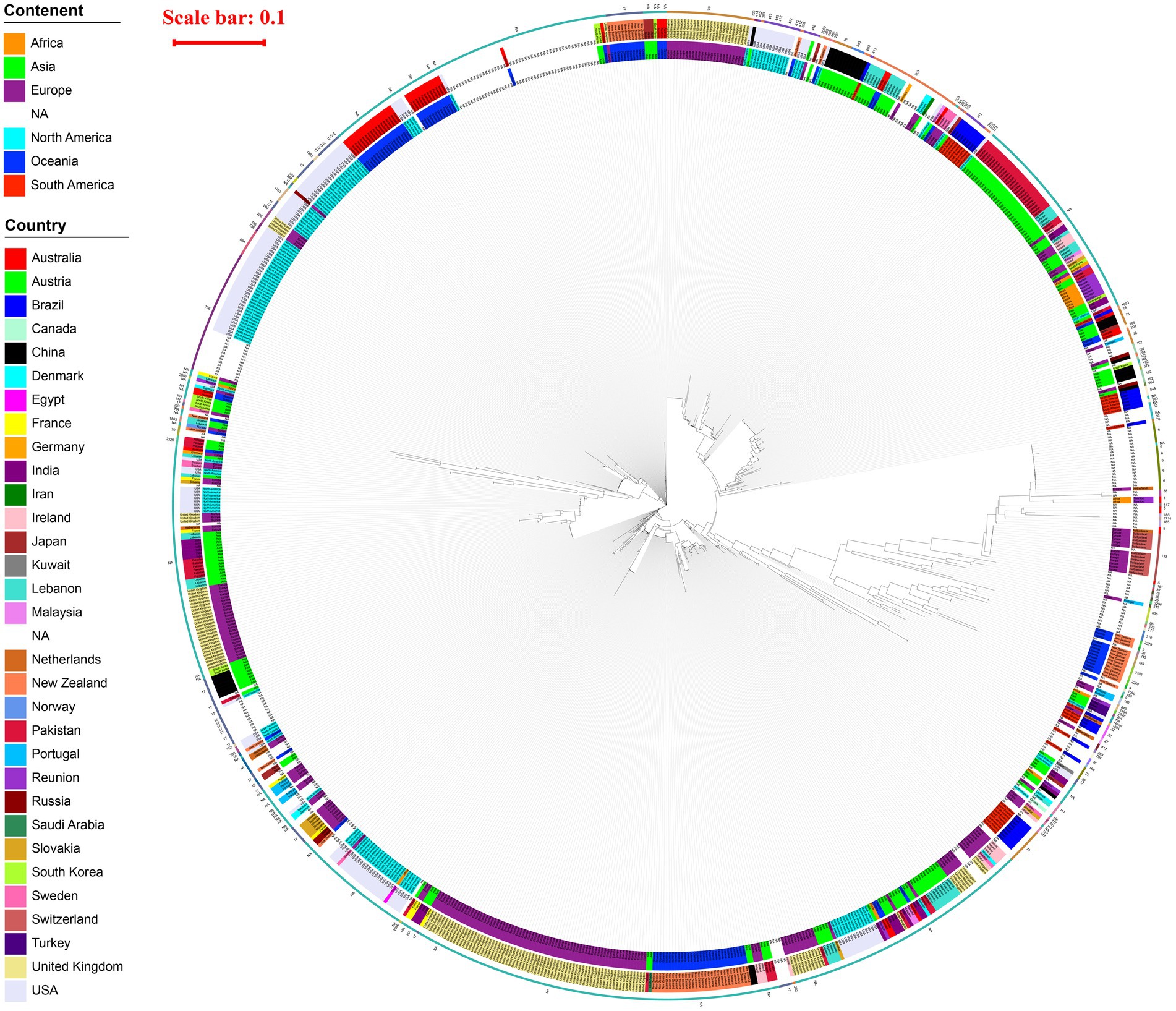
Figure 2. Phylogenetic tree of 915 vanA-carrying Enterococcus isolates circular maximum likelihood phylogenetic tree of 915 vanA-carrying Enterococcus isolates. The innermost ring indicates the continent of origin, the middle ring shows the country, and the outermost ring represents the MLST sequence type (ST) of each isolate. A red scale bar (0.1) indicates genetic distance. The tree reveals extensive geographic and genetic diversity, with multiple clades corresponding to both regional clusters and widespread international lineages. STs such as ST17, ST78, and ST203 are frequently observed and often associated with specific geographic regions.
4.2 Phylogenetic tree for strains carrying vanB gene
In the phylogenetic tree of 130 strains carrying vanB gene, we found that the innermost ring of the circular phylogenetic tree showed that most isolates originated from Europe, with additional contributions from Asia, North America, and Oceania; several were labeled as NA due to missing metadata. At the country level, Germany accounted for the largest number of isolates, forming a prominent clonal cluster, while other countries such as the United Kingdom, USA, France, Australia, Japan, and Lebanon were also represented. The outermost ring indicated a strong dominance of ST117, which formed a large, closely related clade, primarily composed of isolates from Germany and other European countries. Other STs, including ST17, ST203, and ST192, were present in smaller numbers and often clustered geographically. These findings suggest a major clonal expansion of ST117 within Europe—particularly in Germany—alongside evidence of both localized outbreaks and international dissemination of vanB-carrying lineages, as shown in Figure 3.
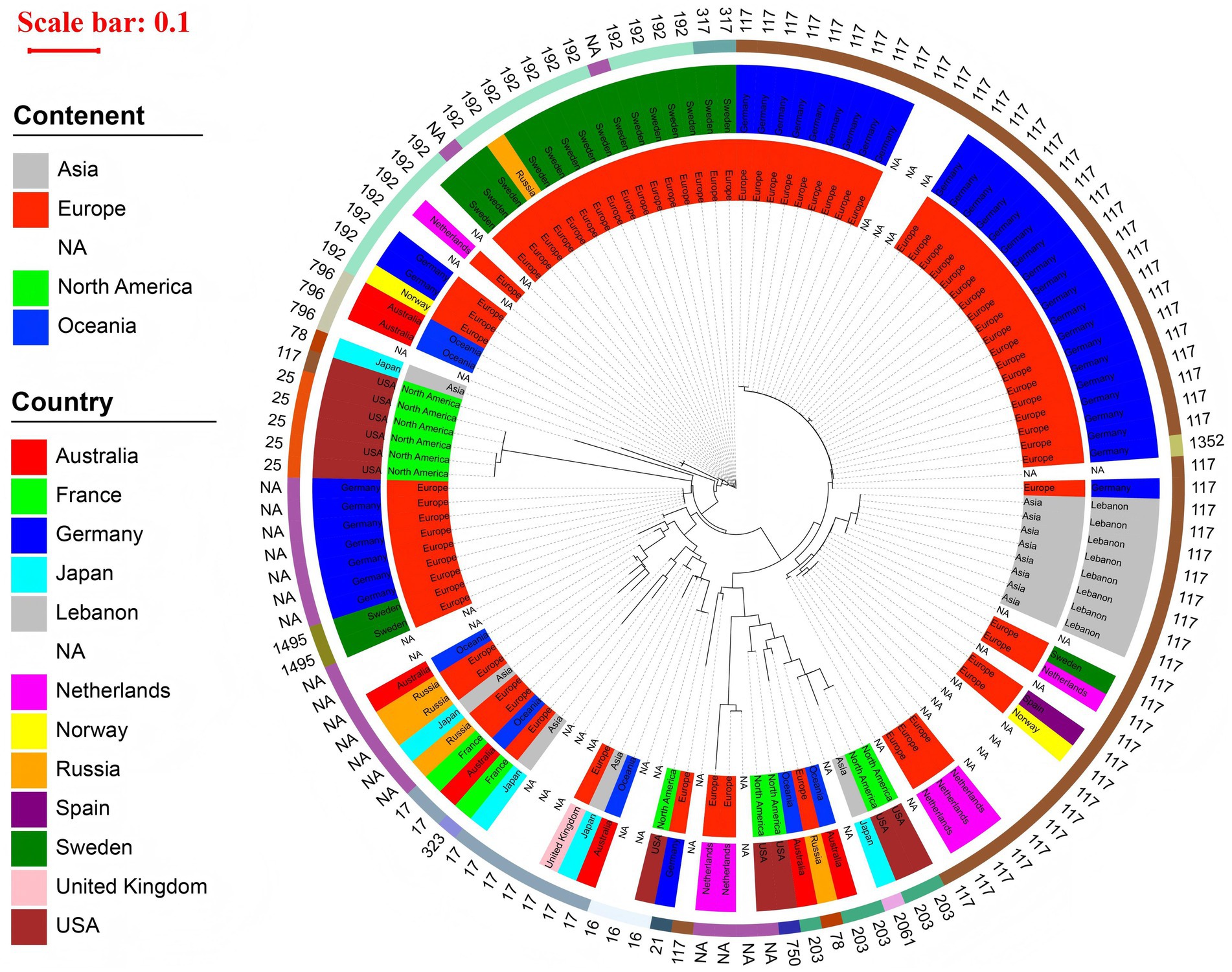
Figure 3. Phylogenetic tree of 130 vanB-carrying Enterococcus isolates circular maximum likelihood phylogenetic tree of 130 vanB-carrying Enterococcus isolates. The innermost ring represents the continent, the middle ring indicates the country, and the outermost ring displays the MLST sequence type (ST) of each isolate. A red scale bar (0.1) shows the genetic distance. The tree is dominated by a large ST117 clade composed primarily of isolates from Germany and other European countries. Additional STs—including ST203, ST17, and ST192—form smaller, geographically associated clusters. The tree highlights both localized clonal expansion and international dissemination of vanB-carrying strains.
5 Human was the predominant host for Enterococcus faecium carrying van gene
Among the 1,071 E. faecium strains carrying van gene analyzed, human sources accounted for the highest proportion, comprising 61.3% (n = 657) of all isolates. The most common human-derived samples included stool (n = 288), blood (n = 118), and urine (n = 33). Animal and dairy-related samples constituted 2.5% (n = 27) of the isolates, with the Gallus gallus domesticus being the most prevalent (n = 14), followed by pigs (n = 6), dogs (n = 2), and cats (n = 2), as detailed in Table 1.

Table 1. The hosts and sample types of van-positive Enterococcus faecium isolates with known origin.
6 Distributional characteristics of strains carrying van genes
6.1 Geographical distribution of strains carrying van genes
From the 2,235 genomes of E. faecium, a total of 1,071 E. faecium carrying van gene were obtained, with their geographical distribution detailed in (Supplementary Table S1). Notably, the United States recorded the highest number of isolates at 160, followed by the United Kingdom with 148 isolates, and New Zealand with 62 isolates. In the United States, the most common STs were ST736 (n = 27), ST17 (n = 22), and ST412 (n = 14), the predominant van is vanA (91.9%), followed by vanB (6.9%). In the United Kingdom, ST78 was the most prevalent (n = 25), followed by ST280 (n = 4) and ST17 (n = 1), with vanA being the predominant van gene (98.6%). Meanwhile, in New Zealand, ST17 was the most frequently found (n = 12), followed by ST2105 (n = 4) and ST195 (n = 3), with vanA predominating as the most common van gene (98.4%) as shown in Figure 4.
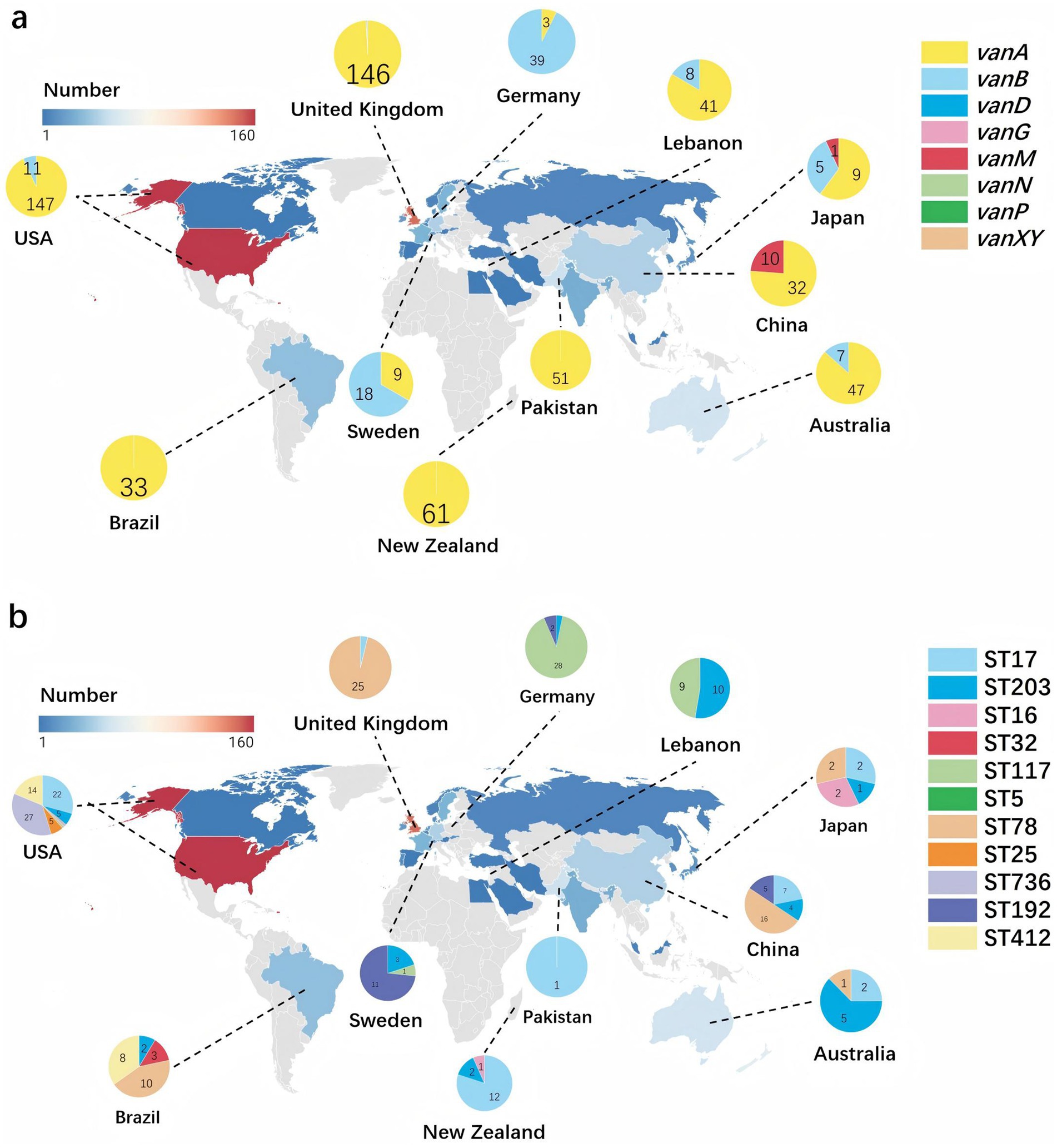
Figure 4. Global distribution of Enterococcus faecium dominant sequence types and vancomycin resistance genes. The color gradient indicates gene frequency, with red representing higher counts and blue representing lower counts. The geographic distribution of all strains included in the study is covered. (a) Highlights the distribution of eight different van gene subtypes, focusing on the 11 countries with the highest van gene frequencies. Each segment of the pie chart is color-coded according to the vancomycin resistance gene, with the legend on the right indicating the corresponding colors for vanA, vanB, vanD, vanG, vanM, vanN, vanP, and vanXY. (b) Uses pie charts to show the distribution of the 10 most prevalent STs, displaying only the 11 countries with the highest van gene frequencies on the map. Each segment of the pie chart is color-coded according to the ST, with the legend on the right indicating the corresponding colors for ST17, ST203, ST16, ST32, ST117, ST5, ST78, ST25, ST736, ST192, and ST412.
6.2 Annual distribution of strains carrying van genes
The initial isolations of E. faecium strains ST17 and ST117 occurred in 1998 and 2001, respectively, while strains ST203 and ST78 were first identified in 2005. Starting in 2008, the dissemination rate of ST17 began to increase significantly, exhibiting fluctuations but maintaining a consistently high presence in subsequent years. By 2015, the ST78 clone had gradually emerged as the dominant clone. However, a notable increase in the dissemination rate of ST117 was observed between 2018 and 2019, leading to its establishment as the most prevalent clone during that period, as shown in Figure 5.
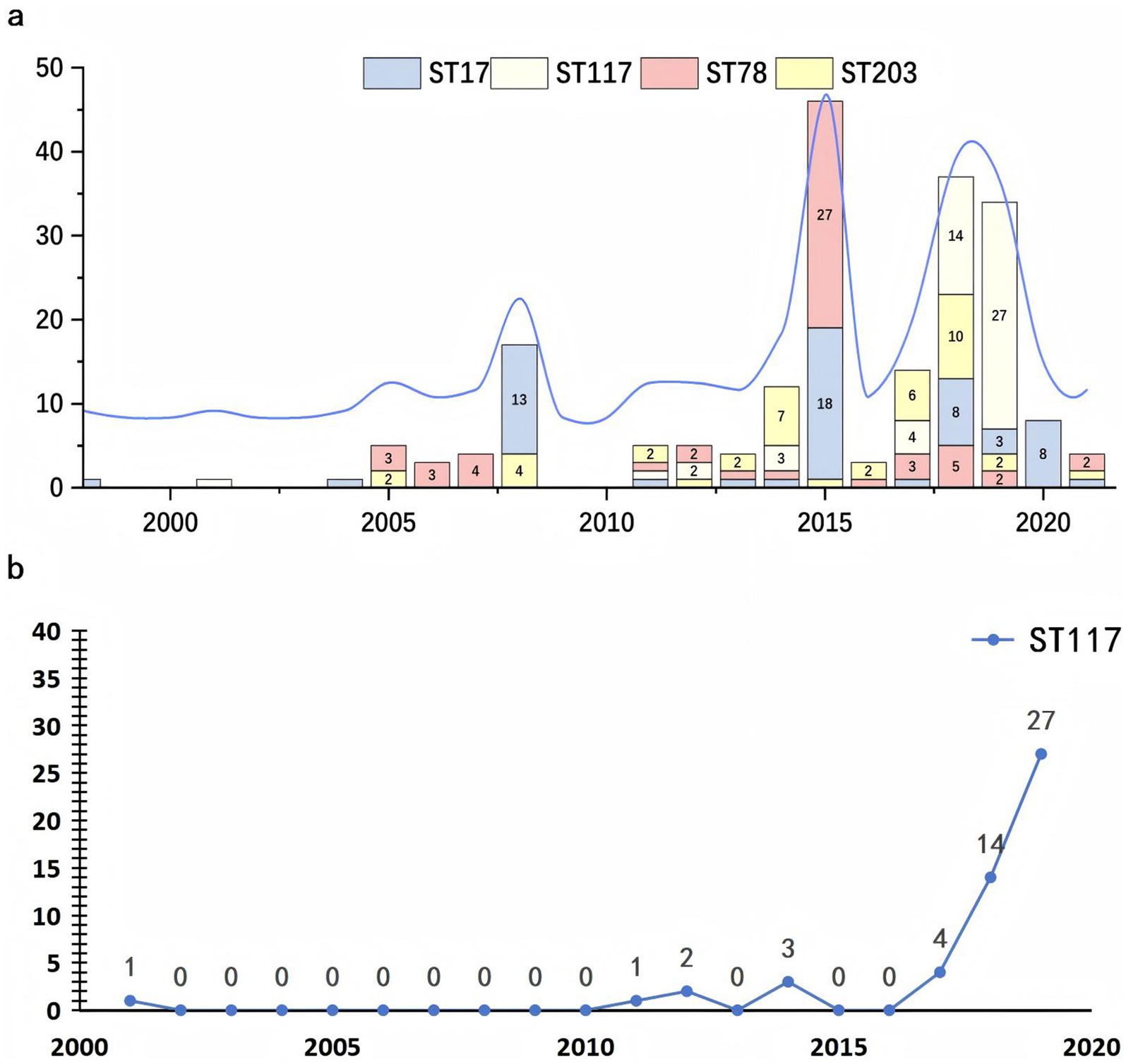
Figure 5. Temporal distribution of major sequence types of Enterococcus faecium from 2000 to 2020. (a) The stacked bar chart illustrating the annual number of isolates for each sequence type (ST), with distinct colors representing ST17 (blue), ST117 (yellow), ST78 (red), and ST203 (gray). An overlaid blue line indicates the total number of isolates per year. (b) The trend chart depicting the yearly distribution of ST117 E. faecium carrying van genes.
7 Discussion
Vancomycin remains a critical antimicrobial agent against multidrug-resistant E. faecium infections. However, resistance to this vital drug presents substantial challenges in clinical settings. This study provides valuable insights into the epidemiological distribution and characteristics of vancomycin resistance, which are crucial for developing clinical treatment strategies and preventive measures. Understanding these patterns can help healthcare providers better manage and prevent vancomycin-resistant infections.
Our analysis identified a 47.9% prevalence rate of van genes in E. faecium, with vanA being the most frequent variant (40.9%), based on global data collected from 1995 to 2022. When comparing with other studies, however, it is critical to consider differences in sample origin, geographic scope, detection method, and study objectives. For instance, our vanA prevalence is lower than the 44.7% observed in municipal wastewater samples from Germany (Valenza et al., 2024), but higher than rates from clinical isolates in Toronto (10.2%), Australia (13.7%) (Coombs et al., 2024), and Japan (0.9%) (Kohler et al., 2018). In Beijing hospitals, vanA and vanM prevalence were reported at 24.4 and 4.7% respectively (Yan et al., 2023), while all VREfm strains in a Turkish study carried vanA (Erdem et al., 2020). These variations highlight the importance of interpreting prevalence differences with caution, as biases may arise from inconsistent sampling contexts (e.g., wastewater vs. clinical), database submission trends, and genomic screening criteria. Additional van genes detected in our dataset include vanB (5.8%), vanD (0.7%), vanM (0.5%), vanP (0.2%), and vanN (0.1%). These frequencies differ from localized hospital-based reports—for example, vanB prevalence of 36.8–47.6% in a German hospital (Jochim-Vukosavic et al., 2024), vanM at 16.7% in a tertiary Chinese hospital (Zhou et al., 2020), and vanD at 27.8% in the Netherlands (Flipse et al., 2019). Again, such discrepancies underscore the limitations of direct cross-study comparison without careful consideration of sample origin, sequencing depth, and local epidemiology. Importantly, the presence of multiple van gene variants—each conferring resistance through different mechanisms such as inducible ligase systems (VanA/VanB) or intrinsic modifications (VanC)—indicates a high potential for heterogeneity in clinical outcomes and detection sensitivity (Karukappadath et al., 2023).
Beyond human isolates, our metadata analysis identified a subset of strains derived from non-human hosts, including poultry, pigs, dogs, and environmental sources (e.g., wastewater, surface water). Although these occurrences were infrequent, the detection of vanA- and vanB-carrying strains from animal and environmental sources suggests potential reservoirs for resistance genes. These findings align with previous studies identifying zoonotic and ecological reservoirs for VREfm (Leinweber et al., 2018), and emphasize the need for a One Health approach to resistance surveillance that integrates human, veterinary, and environmental domains.
Our multilocus sequence typing (MLST) analysis revealed that ST17, ST117, and ST78 were the most prevalent STs among vanA-carrying isolates. Notably, ST117 has gained increasing attention due to its growing presence in recent years. In our phylogenetic tree, ST117 formed several well-supported clades—especially in isolates from Europe, North America, and Oceania—suggesting clonal expansion. However, we caution against overinterpreting this trend: the apparent increase in ST117 may reflect temporal bias in genome submissions rather than true epidemiological replacement of ST17. To mitigate this, we normalized ST frequencies by total genome counts per year, and observed a consistent proportional rise in ST117 from 2010 onwards. Still, we interpret this as a potential trend rather than a definitive lineage shift, as longitudinal genomic data and in vitro fitness validation are lacking.
The biological success of ST117 may be multifactorial. Prior studies have identified several adaptive traits in ST117 strains, including plasmid-mediated metabolic flexibility (e.g., PTS sugar transporters), virulence factors (e.g., pili, secretion systems), and enhanced colonization abilities under antibiotic pressure (Stege et al., 2024). ST117 is frequently associated with Inc18 and RepA_N plasmids that stably harbor vanA, potentially facilitating its expansion in hospital environments (Al Rubaye et al., 2023). Nevertheless, in the absence of experimental evidence on growth dynamics or transmission efficiency, we can only hypothesize that ST117’s rise is driven by both fitness advantage and selection pressure in healthcare settings.
These high-risk clones formed well-supported clusters that were either geographically restricted—suggesting localized outbreaks and sustained transmission within specific healthcare systems—or dispersed across countries and continents, indicative of clonal spread via inter-hospital or international transfer of patients (Permana et al., 2023). Particularly, the wide geographic reach of ST17 supports the role as pandemic clones capable of persisting and disseminating under strong antibiotic selection pressure (Willems et al., 2011; van Hal et al., 2021). The application of Roary allowed us to identify core-genome-based phylogenetic structure (Page et al., 2015), while RAxML-NG enabled robust tree construction, revealing both deeply rooted lineages and evidence of recent diversification (Kozlov et al., 2019). Some inconsistencies between phylogeny and geography, especially in mixed-country branches, may reflect recombination events or horizontal acquisition of resistance genes, including vanA, which is commonly located on transmissible plasmids and transposons (e.g., Tn1546) (Gouliouris et al., 2018). The potential for plasmid-mediated spread of vanA among different STs within shared environments underscores the complexity of resistance dissemination beyond clonal inheritance (Gorrie et al., 2019).
In parallel, the phylogenetic reconstruction of 130 vanB-carrying isolates demonstrated a marked clonal dominance of ST117, especially among German and other European strains. This suggests not only a successful regional expansion of a healthcare-adapted lineage, but also potential plasmid stabilization of vanB within this genetic background (Lisotto et al., 2021; Falgenhauer et al., 2019). Other STs—such as ST203, ST17, and ST192—formed smaller, geographically constrained clades, suggesting independent introduction events and localized outbreaks (Lehmkuhl et al., 2024; Hughes et al., 2019; Lam et al., 2013). The ST117-dominant clade exhibited very short branch lengths, indicative of recent expansion likely driven by clonal spread within hospitals or healthcare networks under antimicrobial pressure (Xanthopoulou et al., 2020). The detection of genetically similar vanB-carrying isolates in Asia, North America, and Oceania suggests that international dissemination is also occurring, possibly via mobile genetic elements and intercontinental transfer of colonized patients (Wei et al., 2024; Santona et al., 2023; Bender et al., 2022). However, due to the relatively small number of vanB isolates, further analysis—especially of the accessory genome and plasmid content—would be necessary to confirm the role of mobile elements and recombination in their spread (Mills et al., 2025).
Together, these findings underscore the importance of integrating phylogenomic, epidemiological, and mobile genetic element data to fully understand the transmission dynamics of vanA and vanB resistance genes. The observed patterns of clade formation and geographic clustering, supported by core-genome phylogenies, highlight both long-standing epidemic lineages and ongoing regional transmission events, reinforcing the need for real-time genomic surveillance and infection control efforts, particularly targeting high-risk clones such as ST117 (Hourigan et al., 2024).
Our study has several limitations. First, the representation of E. faecium strains in the NCBI database may not be comprehensive, introducing potential selection bias and limiting the generalizability of our findings. Second, the lack of susceptibility data restricts our ability to correlate genotypes with phenotypes accurately, hindering a full understanding of how genetic variations affect clinical outcomes. Further research should focus on including data from underrepresented regions and exploring the interactions between genetic diversity and resistance mechanisms.
8 Conclusion
Using whole-genome analysis of 2,235 strains, we identified major epidemic clones such as ST17, ST78, and ST117, with ST117 showing recent expansion, especially in vanB-carrying populations. The observed clonal structure, combined with evidence of recombination and potential plasmid-mediated gene transfer, underscores the complex transmission dynamics of vancomycin resistance. Our findings emphasize the dual role of local hospital-associated outbreaks and global dissemination in shaping the current landscape of VREfm. These insights reinforce the urgent need for real-time genomic surveillance, coordinated international monitoring, and targeted infection control measures—particularly in healthcare settings—to curb the spread of high-risk resistant clones.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
RY: Visualization, Writing – original draft, Methodology, Software. JJ: Data curation, Methodology, Writing – original draft. HS: Supervision, Writing – review & editing. XC: Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81902124).
Acknowledgments
The authors are very grateful to Tianshe Li for providing technical help on data sorting and bioinformatic analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1578903/full#supplementary-material
Footnotes
References
Al Rubaye, M., Janice, J., Bjørnholt, J. V., Löhr, I. H., Sundsfjord, A., and Hegstad, K. (2023). The population structure of vancomycin-resistant and -susceptible Enterococcus faecium in a low-prevalence antimicrobial resistance setting is highly influenced by circulating global hospital-associated clones. Microb. Genom. 9:001160. doi: 10.1099/mgen.0.001160
Azzam, A., Elkafas, H., Khaled, H., Ashraf, A., Yousef, M., and Elkashef, A. A. (2023). Prevalence of vancomycin-resistant enterococci (VRE) in Egypt (2010–2022): a systematic review and meta-analysis. J. Egypt. Public Health Assoc. 98:8. doi: 10.1186/s42506-023-00133-9
Benamrouche, N., Guettou, B., Henniche, F. Z., Assaous, F., Laouar, H., Ziane, H., et al. (2021). Vancomycin-resistant Enterococcus faecium in Algeria: phenotypic and genotypic characterization of clinical isolates. J. Infect. Dev. Ctries. 15, 95–101. doi: 10.3855/jidc.12482
Bender, J. K., Hermes, J., Zabel, L. T., Haller, S., Mürter, N., Blank, H. P., et al. (2022). Controlling an unprecedented outbreak with vancomycin-resistant Enterococcus faecium in Germany, October 2015 to November 2019. Microorganisms 10:1603. doi: 10.3390/microorganisms10081603
Coll, F., Gouliouris, T., Bruchmann, S., Phelan, J., Raven, K. E., Clark, T. G., et al. (2022). PowerBacGWAS: a computational pipeline to perform power calculations for bacterial genome-wide association studies. Commun. Biol. 45, 54–58. doi: 10.1071/MA24018
Coombs, G. W., Daley, D. A., Bell, J. M., Mowlaboccus, S., and Iredell, J. R.Australian Group on Antimicrobial Resistance (AGAR) (2024). The Australian Group on Antimicrobial Resistance. Microbiol. Aust. 44. doi: 10.33321/cdi.2020.44.72
Eichel, V. M., Last, K., Brühwasser, C., von Baum, H., Dettenkofer, M., Götting, T., et al. (2023). Epidemiology and outcomes of vancomycin-resistant Enterococcus infections: a systematic review and meta-analysis. J. Hosp. Infect. 141, 119–128. doi: 10.1016/j.jhin.2023.09.008
Elstrøm, P., Astrup, E., Hegstad, K., Samuelsen, Ø., Enger, H., and Kacelnik, O. (2019). The fight to keep resistance at bay, epidemiology of carbapenemase producing organisms (CPOs), vancomycin resistant enterococci (VRE) and methicillin resistant Staphylococcus aureus (MRSA) in Norway, 2006-2017. PLOS ONE 14:e0211741. doi: 10.1371/journal.pone.0211741
Erdem, F., Kayacan, C., Oncul, O., Karagoz, A., and Aktas, Z. (2020). Clonal distribution of vancomycin-resistant Enterococcus faecium in Turkey and the new singleton ST733. J. Clin. Lab. Anal. 34:e23541. doi: 10.1002/jcla.23541
Falgenhauer, L., Fritzenwanker, M., Imirzalioglu, C., Steul, K., Scherer, M., Heudorf, U., et al. (2019). Near-ubiquitous presence of a vancomycin-resistant Enterococcus faecium ST117/CT71/vanB -clone in the Rhine-Main metropolitan area of Germany. Antimicrob. Resist. Infect. Control 8:128. doi: 10.1186/s13756-019-0573-8
Farias, B. O., Montenegro, K. S., Nascimento, A. P. A., Magaldi, M., Gonçalves-Brito, A. S., Flores, C., et al. (2023). First report of a wastewater treatment-adapted Enterococcus faecalis ST21 harboring vanA gene in Brazil. Curr. Microbiol. 80:313. doi: 10.1007/s00284-023-03418-6
Flipse, J., von Wintersdorff, C. J. H., van Niekerk, J. M., Jamin, C., van Tiel, F. H., Hasman, H., et al. (2019). Appearance of vanD-positive Enterococcus faecium in a tertiary hospital in the Netherlands: prevalence of vanC and vanD in hospitalized patients. Sci. Rep. 9:6949. doi: 10.1038/s41598-019-42824-4
Geraldes, C., Tavares, L., Gil, S., and Oliveira, M. (2022). Enterococcus virulence and resistant traits associated with its permanence in the hospital environment. Antibiotics 11:857. doi: 10.3390/antibiotics11070857
Gorrie, C., Higgs, C., Carter, G., Stinear, T. P., and Howden, B. (2019). Genomics of vancomycin-resistant Enterococcus faecium. Microb. Genom. 5:e000283. doi: 10.1099/mgen.0.000283
Gouliouris, T., Raven, K. E., Ludden, C., Blane, B., Corander, J., Horner, C. S., et al. (2018). Genomic surveillance of Enterococcus faecium reveals limited sharing of strains and resistance genes between livestock and humans in the United Kingdom. mBio 9:e01780-18. doi: 10.1128/mBio.01780-18
Hourigan, D., Stefanovic, E., Hill, C., and Ross, R. P. (2024). Promiscuous, persistent and problematic: insights into current enterococcal genomics to guide therapeutic strategy. BMC Microbiol. 24:103. doi: 10.1186/s12866-024-03243-2
Hughes, A., Ballard, S., Sullivan, S., and Marshall, C. (2019). An outbreak of vanA vancomycin-resistant Enterococcus faecium in a hospital with endemic vanB VRE. Infect. Dis. Health 24, 82–91. doi: 10.1016/j.idh.2018.12.002
Jacob Machado, D., Castroviejo-Fisher, S., and Grant, T. (2021). Evidence of absence treated as absence of evidence: the effects of variation in the number and distribution of gaps treated as missing data on the results of standard maximum likelihood analysis. Mol. Phylogenet. Evol. 154:106966. doi: 10.1016/j.ympev.2020.106966
Ji, T., Wang, W., Wang, L., Gao, Y., Wang, Y., and Gao, X. (2024). Development and application of a rapid visual detection technique for VanA gene in vancomycin-resistant Enterococcus faecium. mSphere 9:e0066624. doi: 10.1128/msphere.00666-24
Jochim-Vukosavic, A., Schwab, F., Knegendorf, L., Schlüter, D., Bange, F. C., Ebadi, E., et al. (2024). Epidemiology and infection control of vancomycin-resistant enterococci at a German university hospital: a three-year retrospective cohort study. PLoS One 19:e0297866. doi: 10.1371/journal.pone.0297866
Joddha, H. B., Mathakiya, R. A., Joshi, K. V., Khant, R. B., Golaviya, A. V., Hinsu, A. T., et al. (2023). Profiling of antimicrobial resistance genes and integron from Escherichia coli isolates using whole genome sequencing. Genes 14:1212. doi: 10.3390/genes14061212
Karaman, R., Jubeh, B., and Breijyeh, Z. (2020). Resistance of gram-positive bacteria to current antibacterial agents and overcoming approaches. Molecules 25:2888. doi: 10.3390/molecules25122888
Karukappadath, R. M., Sirbu, D., and Zaky, A. (2023). Drug-resistant bacteria in the critically ill: patterns and mechanisms of resistance and potential remedies. Front. Antibiot. 2:1145190. doi: 10.3389/frabi.2023.1145190
Kohler, P., Eshaghi, A., Kim, H. C., Plevneshi, A., Green, K., Willey, B. M., et al. (2018). Prevalence of vancomycin-variable Enterococcus faecium (VVE) among vanA-positive sterile site isolates and patient factors associated with VVE bacteremia. PLoS One 13:e0193926. doi: 10.1371/journal.pone.0193926
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lam, M. M. C., Seemann, T., Tobias, N. J., Chen, H., Haring, V., Moore, R. J., et al. (2013). Comparative analysis of the complete genome of an epidemic hospital sequence type 203 clone of vancomycin-resistant Enterococcus faecium. BMC Genomics 14:595. doi: 10.1186/1471-2164-14-595
Lehmkuhl, J., Schneider, J. S., Werth, K. L., Scherff, N., Mellmann, A., and Kampmeier, S. (2024). Role of membrane vesicles in the transmission of vancomycin resistance in Enterococcus faecium. Sci. Rep. 14:1895. doi: 10.1038/s41598-024-52310-1
Leinweber, H., Alotaibi, S. M. I., Overballe-Petersen, S., Hansen, F., Hasman, H., Bortolaia, V., et al. (2018). Vancomycin resistance in Enterococcus faecium isolated from Danish chicken meat is located on a pVEF4-like plasmid persisting in poultry for 18 years. Int. J. Antimicrob. Agents 52, 283–286. doi: 10.1016/j.ijantimicag.2018.03.019
Letunic, I., and Bork, P. (2024). Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/nar/gkae268
Li, Q., Chen, S., Zhu, K., Huang, X., Huang, Y., Shen, Z., et al. (2022). Collateral sensitivity to pleuromutilins in vancomycin-resistant Enterococcus faecium. Nat. Commun. 13:1888. doi: 10.1038/s41467-022-29493-0
Li, S., Mosier, D., Dong, X., Kouris, A., Ji, G., Strous, M., et al. (2023). Frequency of change determines effectiveness of microbial response strategies. ISME J. 17, 2047–2057. doi: 10.1038/s41396-023-01515-9
Lisotto, P., Couto, N., Rosema, S., Lokate, M., Zhou, X., Bathoorn, E., et al. (2021). Molecular characterisation of vancomycin-resistant Enterococcus faecium isolates belonging to the lineage ST117/CT24 causing hospital outbreaks. Front. Microbiol. 12:728356. doi: 10.3389/fmicb.2021.728356
McHugh, M. P., Pettigrew, K. A., Taori, S., Evans, T. J., Leanord, A., Gillespie, S. H., et al. (2024). Consideration of within-patient diversity highlights transmission pathways and antimicrobial resistance gene variability in vancomycin-resistant Enterococcus faecium. J. Antimicrob. Chemother. 79, 656–668. doi: 10.1093/jac/dkae023
Mills, E. G., Hewlett, K., Smith, A. B., Griffith, M. P., Pless, L., Sundermann, A. J., et al. (2025). Bacteriocin production facilitates nosocomial emergence of vancomycin-resistant Enterococcus faecium. Nat. Microbiol. 10, 871–881. doi: 10.1038/s41564-025-01958-0
Nürnberger, L., Schmidt, D., Szumlanski, T., Kirchhoff, L., Ross, B., Steinmann, J., et al. (2021). Molecular characterization of vancomycin-resistant Enterococcus faecium isolates from two German hospitals. GMS Hyg. Infect. Control 16:Doc13. doi: 10.3205/dgkh000384
Orababa, O. Q., Soriwei, J. D., Akinsuyi, S. O., Essiet, U. U., and Solesi, O. M. (2021). A systematic review and meta-analysis on the prevalence of vancomycin-resistant enterococci (VRE) among Nigerians. Porto Biomed. J. 6:e125. doi: 10.1097/j.pbj.0000000000000125
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Permana, B., Harris, P. N. A., Runnegar, N., Lindsay, M., Henderson, B. C., Playford, E. G., et al. (2023). Using genomics to investigate an outbreak of vancomycin-resistant Enterococcus faecium ST78 at a large tertiary hospital in Queensland. Microbiol. Spectr. 11:e0420422. doi: 10.1128/spectrum.04204-22
Reynolds, J. L., Trudeau, R. E., Seville, M. T., and Chan, L. (2021). Impact of a vancomycin-resistant Enterococcus (VRE) screening result on appropriateness of antibiotic therapy. Antimicrob. Steward. Healthc. Epidemiol. 1:e41. doi: 10.1017/ash.2021.215
Santona, A., Taviani, E., Fiamma, M., Deligios, M., Hoang, H., Sanna, S., et al. (2023). Occult vancomycin-resistant Enterococcus faecium ST117 displaying a highly mutated vanB2 operon. Antibiotics 12:476. doi: 10.3390/antibiotics12030476
Stege, P. B., Beekman, J. M., Hendrickx, A. P. A., van Eijk, L., Rogers, M. R. C., Suen, S. W. F., et al. (2024). Colonization of vancomycin-resistant Enterococcus faecium in human-derived colonic epithelium: unraveling the transcriptional dynamics of host-enterococcal interactions. FEMS Microbes 5:xtae014. doi: 10.1093/femsmc/xtae014
Sultana, S., Crompton, M. E., Meurer, K., Jankiewicz, O., Morales, G. H., Johnson, C., et al. (2022). Redox-mediated inactivation of the transcriptional repressor RcrR is responsible for uropathogenic Escherichia coli's increased resistance to reactive chlorine species. mBio 13:e0192622. doi: 10.1128/mbio.01926-22
Togkousidis, A., Kozlov, O. M., Haag, J., Hohler, D., and Stamatakis, A. (2023). Adaptive RAxML-NG: accelerating phylogenetic inference under maximum likelihood using dataset difficulty. Mol. Biol. Evol. 40:msad227. doi: 10.1093/molbev/msad227
Valenza, G., Eisenberger, D., Esse, J., Held, J., Lehner-Reindl, V., Plaumann, P. L., et al. (2024). High prevalence of the recently identified clonal lineage ST1299/CT3109 vanA among vancomycin-resistant Enterococcus faecium strains isolated from municipal wastewater. mSphere 9:e0039624. doi: 10.1128/msphere.00396-24
van Hal, S. J., RJL, W., Gouliouris, T., Ballard, S. A., Coque, T. M., Hammerum, A. M., et al. (2021). The global dissemination of hospital clones of Enterococcus faecium. Genome Med. 13:52. doi: 10.1186/s13073-021-00868-0
Wardal, E., Zabicka, D., Hryniewicz, W., and Sadowy, E. (2022). VanA-Enterococcus faecalis in Poland: hospital population clonal structure and vanA mobilome. Eur. J. Clin. Microbiol. Infect. Dis. 41, 1245–1261. doi: 10.1007/s10096-022-04479-4
Wattanasombat, S., and Tongjai, S. (2024). Easing genomic surveillance: a comprehensive performance evaluation of long-read assemblers across multi-strain mixture data of HIV-1 and other pathogenic viruses for constructing a user-friendly bioinformatic pipeline. F1000Res. 13:556. doi: 10.12688/f1000research.149577.1
Wei, Y., Palacios Araya, D., and Palmer, K. L. (2024). Enterococcus faecium: evolution, adaptation, pathogenesis and emerging therapeutics. Nat. Rev. Microbiol. 22, 705–721. doi: 10.1038/s41579-024-01058-6
Willems, R. J., Hanage, W. P., Bessen, D. E., and Feil, E. J. (2011). Population biology of Gram-positive pathogens: high-risk clones for dissemination of antibiotic resistance. FEMS Microbiol. Rev. 35, 872–900. doi: 10.1111/j.1574-6976.2011.00284.x
Xanthopoulou, K., Peter, S., Tobys, D., Behnke, M., Dinkelacker, A. G., Eisenbeis, S., et al. (2020). Vancomycin-resistant Enterococcus faecium colonizing patients on hospital admission in Germany: prevalence and molecular epidemiology. J. Antimicrob. Chemother. 75, 2743–2751. doi: 10.1093/jac/dkaa271
Yan, M. Y., He, Y. H., Ruan, G. J., Xue, F., Zheng, B., and Lv, Y. (2023). The prevalence and molecular epidemiology of vancomycin-resistant Enterococcus (VRE) carriage in patients admitted to intensive care units in Beijing, China. J. Microbiol. Immunol. Infect. 56, 351–357. doi: 10.1016/j.jmii.2022.07.001
Zhao, H., He, Z., Li, Y., and Sun, B. (2022). Epidemiology of carbapenem-resistant Klebsiella pneumoniae ST15 of producing KPC-2, SHV-106 and CTX-M-15 in Anhui, China. BMC Microbiol. 22:262. doi: 10.1186/s12866-022-02672-1
Keywords: Enterococcus faecium, van, sequence types, phylogenetic tree, prevalence characteristics
Citation: Yan R, Ji J, Shen H and Cao X (2025) Deciphering vancomycin resistance in Enterococcus faecium: gene distribution, sequence typing, and global phylogenetic analysis. Front. Microbiol. 16:1578903. doi: 10.3389/fmicb.2025.1578903
Edited by:
Yasser Mahmmod, Long Island University, United StatesReviewed by:
Muhammed Duman, Bursa Uludağ University, TürkiyeMarta Laranjo, University of Evora, Portugal
Copyright © 2025 Yan, Ji, Shen and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Cao, Y2FvLXhpYW8tbGlAMTYzLmNvbQ==