 Dominika Klimek
Dominika Klimek Olga Maria Lage
Olga Maria Lage Magdalena Calusinska
Magdalena Calusinska- 1Environmental and Industrial Biotechnology, Luxembourg Institute of Science and Technology (LIST), Esch-sur-Alzette, Luxembourg
- 2Faculty of Science, Technology and Medicine (FTSM), University of Luxembourg, Esch-sur-Alzette, Luxembourg
- 3Department of Biology, Faculty of Sciences, University of Porto, Porto, Portugal
- 4Interdisciplinary Centre of Marine and Environmental Research (CIIMAR/CIMAR), Porto, Portugal
Biomass-rich environments host diverse microbial communities that contribute to the degradation and recycling of organic matter. Understanding the community structure within these habitats is essential for elucidating the ecological roles and metabolic capacities of specific microbial groups. Here, we conducted an analysis of biomass-rich environments including diverse soil types, sediments, anaerobic digesters, termite guts, termite nests and other decaying biomasses, to explore the phylogenetic diversity and community structure of the Planctomycetota phylum, using short-read 16S rRNA gene amplicon sequencing. All sampled environments showed presence of Planctomycetota, with relative abundance ranging from nearly absent in animal manure to approximately 10% in soils. Across all samples, virtually 1,900 operational taxonomic units (OTUs) were identified, classified into diverse classes within Planctomycetota. Planctomycetotal phylogenetic diversity was highest in soils and sediments, while termite guts, exhibiting the lowest phylogenetic diversity, were dominated by a few core OTUs shared across different termite species. Notably, a single OTU, closely matching the 16S rRNA gene sequence of the Singulisphaera genus, was detected in all environments, though with relative abundance ranging from only a few reads to over 6% of the planctomycetotal community. Four environments such as soil, sediment, termite nest and decaying biomasses showed similar community structure with predominant genera such as Tepidisphaera, Telmatocola, and distantly related to Thermogutta, and Anatilimnicola. However, among these environments, weighted UniFrac analysis revealed that planctomycetotal communities in termite nests exhibited greater phylogenetic relatedness. Termite gut communities were the most divergent, followed by those in anaerobic digesters, where OTUs assigned to Anaerobaca and Anaerohalosphaera were the most abundant. Termite gut and phytoplankton bloom samples were dominated by OTUs affiliated with Pirellulales, suggesting their host-specific associations. Animal manure showed the presence of Planctomycetota, with 25% of detected OTUs not recognized by the SILVA database, possibly representing a novel, host-specific lineage distantly related to the Pirellulales order.
Introduction
Biomass can be defined as organic matter from living organisms like plants, animals or microorganisms that can be processed (Bonechi et al., 2017). Plant biomass, mainly lignocellulose, represents a vital form of renewable energy, and it has been increasingly used as a feedstock for a wide range of energy products (Bajpai, 2020). Carbohydrates make up the largest fraction of plant biomass, with cellulose and hemicellulose being the most abundant components. Cellulose microfibrils, embedded in a matrix of hemicellulose and lignin, form a highly resistant and intertwined structure that poses challenges for lignocellulose degradation and conversion processes (Chen, 2014). The particular components of plant biomass drive the structure and dynamics of microbial communities in diverse environments. Thus, studying the bacteria in environments with high organic loads, such as those rich in primary and secondary plant biomasses, is crucial for understanding their roles in biomass degradation (Lillington et al., 2020; Wang et al., 2016; Weimer, 2022). This is specifically insightful, as biomass-rich systems impose unique selective pressures that may drive bacteria along distinct evolutionary pathways, thereby fostering the emergence of diverse and potentially novel bacterial lineages (Martin and Sousa, 2016). Bacteroidota serves as an excellent example of a widespread phylum in which only certain lineages are known biomass degraders, often targeting different fractions of organic matter (Fernández-Gómez et al., 2013). For instance, marine members of Bacteroidota, such as the Flavobacteriia class, specialize in degrading high-molecular-weight organic matter from algal blooms, while Bacteroides genus is adept at degrading complex dietary polysaccharides including lignocellulose (Briliūtė et al., 2019; Kappelmann et al., 2019).
Microbial communities are pivotal in decomposing biomass and cycling nutrients across varied environments, each with distinct ecological roles. In soil, microbes break down complex plant materials, releasing essential nutrients, enhancing soil structure, and finally supporting plant growth (Žifčáková et al., 2017). Composting is an aerobic process which relies on microbial activity to decompose organic matter, producing a stable, nutrient-rich humus that enhances soil fertility and structure (Partanen et al., 2010). Built environments, such as landfills and anaerobic digestion (AD) reactors, host diverse microbial communities that play essential roles in organic matter decomposition, fermentation and methane production (Nguyen et al., 2019; Wang et al., 2017). In the animal gut, particularly in herbivores, specialized microbial community ferments fibrous biomass, producing short-chain fatty acids that supply energy to the host (Lee and Hase, 2014). In termites, this mutualistic relationship is taken to an extraordinary level – their gut functions as one of the smallest and most efficient bioreactors on the planet, housing a highly specialized microbiota capable of breaking down lignocellulosic biomass, particularly wood, which few other animals can digest (Goux et al., 2023). The lignocellulose-degrading microorganisms are also considered attractive producers of biotechnologically important enzymes due to their evolved systems for breaking down complex organic materials (Calusinska et al., 2020; Reichart et al., 2021; Sethupathy et al., 2021). The enzymes responsible for degradation of carbohydrates are called carbohydrate-active enzymes (CAZymes) and are present in virtually all living forms (Drula et al., 2022). Previous observations indicated that genomes belonging to the Planctomycetota phylum harbor a significant number of diverse CAZymes (Gharechahi et al., 2022; López-Mondéjar et al., 2022; Sun et al., 2023). For instance, Planctomycetota from AD reactors, but also peat-enriched wetlands are presumed to be well-equipped toward complex polysaccharide decomposition (Ivanova et al., 2018; Vanwonterghem et al., 2016). As expected, the genomes most likely to possess the greatest capacity for polysaccharide scavenging were primarily recovered from metagenomes of environments rich in organic matter (Klimek et al., 2024).
Since the discovery of Planctomycetota, numerous studies have documented their widespread occurrence and abundance across diverse environments. Members of this phylum have been detected in marine and brackish waters (Cabello-Yeves et al., 2021; Figueroa et al., 2021; Howe et al., 2023; Storesund et al., 2020), extreme habitats (Elshahed et al., 2007; Farias et al., 2017; Spring et al., 2015) including glaciers and permafrost (Malard and Pearce, 2018; Vipindas et al., 2020; Yang et al., 2016) and various terrestrial ecosystems (Buckley et al., 2006; Hu et al., 2022). Although primarily considered environmental bacteria, certain species, particularly within the Gemmata genus, have been linked to opportunistic infections in immunocompromised individuals (Aghnatios and Drancourt, 2016; Kaboré et al., 2020). Representatives of this phylum have also been found in caves (De Mandal et al., 2017; Shen et al., 2022) and as host-associated bacteria, including associations with phytoplankton (Bondoso et al., 2017; Kim et al., 2016; Vollmers et al., 2017), invertebrates (Eilmus and Heil, 2009; Richards et al., 2017; Sharma et al., 2014; Vargas-Albores et al., 2017), animals (Gharechahi et al., 2022; Tran et al., 2018; Yildirim et al., 2010), and within rhizosphere microbiomes (Nunes da Rocha et al., 2009; Ravinath et al., 2022). In built environments, studies of microbial communities in wastewater treatment plants frequently report the presence of Planctomycetota, particularly in relation to the anaerobic ammonium oxidation (anammox) process (Suto et al., 2017). Nevertheless, while the phylogenetic diversity of Planctomycetota inhabiting AD and peatlands were already analyzed (Dedysh and Ivanova, 2019; Klimek et al., 2025), their presence in the other biomass-rich habitats remains poorly understood.
To fill this gap, in this study, we aimed to explore and organize current knowledge on the phylogenetic diversity of Planctomycetota community across diverse biomass-rich environments. For that purpose, we compiled previously generated datasets from AD reactors, soil, and termite gut and nest samples, and complemented them with additional samples from habitats such as freshwater sediments, decaying biomass, wetlands, and rhizospheres or soil covered by cryptogams (e.g., lichens and mosses). All samples were further analyzed using 16S rRNA gene sequencing to assess the diversity of planctomycetotal operational taxonomic units (OTUs) and to examine whether these taxa are phylogenetically conserved or broadly distributed across the studied environments.
Materials and methods
Data collection and sample preparation
To assess the planctomycetotal diversity, we combined our previous amplicon sequencing data with the datasets newly generated in this study. Briefly, datasets for anaerobic digester microbial community were taken from Calusinska et al. (2018), while the samples from termite species were taken from Marynowska et al. (2020). Only a random selection of sequenced samples was used targeting between 5 and 20 samples per environment (74 in total, Supplementary Table S1). To complement the datasets with other biomass-rich environments, we additionally sampled different soil types, compost, peat-accumulated wetlands, freshwater sediments, decaying biomass and animal manure for amplicon sequencing (in total 45 more samples, Supplementary Table S1). Samples were collected using sterilized spatulas at the minimum of 3 cm depth material (e.g., soil, sediment, compost, manure) and were frozen if not processed the same day. The exact list of all the samples used in this study is detailed in Supplementary material.
16S rRNA gene amplification and sequencing
To generate 16S rRNA gene amplicons sequencing data of additionally sampled habitats, DNA of 45 samples were extracted from 0.5 g of sample material using PowerSoil kit (QIAGEN) following the manufacturer’s instructions. No technical replicates were prepared. The quality of extracted DNA was assessed using NanoDrop spectrophotometer. The 16S rRNA gene sequences were amplified using pair of custom primers (F-ACTCAAAKGAATWGACGG and R primer-ACGGGCGGTGTGTRC) targeting about 500 bp of the V6-8 region. The conditions for PCR cycling were as specified: initial denaturation 30s at 98°C followed by 22 cycles of denaturation 5 s at 98°C, annealing 30s at 58°C and 30s at 72°C of elongation. Final elongation step was performed at 72°C for 5 min. Amplicon library sequencing was prepared using Nextera XT DNA Library Preparation Kit and compatible set of indices (Nextera XT Index Kit v2), with addition of PhiX control at 5%. Paired-end sequencing was performed on an Illumina MiSeq instrument using v3 chemistry.
16S rRNA gene amplicon data processing
The newly generated amplicon sequencing data along with previous datasets were processed together. The data processing including paired reads merging, quality filtering as well as OTU clustering and construction, were undertaken using USEARCH v11.0.667. Paired-end reads were merged by using-fastq_mergepairs -relabel @ command. The primer sequences and distal bases were truncated from the merged reads with the following parameters: -fastx_truncate-stripleft 22, −stripright 15. Singletons and reads shorter than 400 bp were removed using-sortbysize and-fastq_maxee commands. Sequences which shared 97% similarity were binned into operational taxonomic unit (OTUs) with the UPARSE algorithm (−cluster_otus) using filtered reads. OTU count table was built by-otus command. Taxonomy was assigned to each OTU using “classify.seqs” command, implemented in mothur v1.48.0 by aligning to the SILVA 16S rRNA gene database v138 (Quast et al., 2013; Schloss et al., 2009). Taxonomical annotation was further used to retain all OTUs assigned to Planctomycetota (100%). Subsequently, OTU read counts were trimmed and samples with less than 100 planctomycetotal reads were removed from further analysis. To account for variation in sequence depth between sequenced datasets and samples, counts were normalized by subsampling each sample’s read counts to a median of the sample sums (median*x/sum(x)).
Phylogenetic analysis of planctomycetotal community
Retrieved planctomycetotal OTU sequences were first aligned using muscle. Phylogenetic tree was then constructed in Geneious v2019.0.3 using Neighbor-Joining method. Additionally, Faith’s Phylogenetic Diversity (PD) index was used to quantify the total branch length of a phylogenetic tree that encompasses all taxa in a sample (implemented in picante R package; Kembel et al., 2010). Using mothur (unweighted.unifrac), UniFrac distances were also computed using pre-calculated tree in Newick format (Lozupone and Knight, 2005). The planctomycetotal OTU sequences were additionally blasted against a custom database containing 16S rRNA gene sequences retrieved from all the type strains of Planctomycetota described until August 2024 (blastn -max_target_seqs 1 -evalue1e-5).
Statistical analysis of amplicon sequencing data
All analyses were carried out in R studio using the vegan package v2.6 (commands are detailed in brackets) and applied to a matrix containing the planctomycetotal normalized read counts (Oksanen et al., 2001). Alpha diversity, e.g., within-sample planctomycetotal diversity, was estimated by the number of observed distinct OTUs (richness) and the Shannon Index (diversity). Bray-Curtis and Jaccard dissimilarity indices were computed to quantify community composition differences across samples, e.g., beta-diversity (vegdist). To provide visual representation of sample clustering, the calculated dissimilarities were used for ordination analysis using non-metric multidimensional scaling, nMDS (metaMDS). The correlation between indices was assessed using Spearman’s rank correlation. To assess the significance of habitat differences, the Analysis of Similarities, ANOSIM (anosim) test was performed using Bray-Curtis distances with 1,000 permutations. Additionally, permutational multivariate analysis of variance (PERMANOVA) was applied to evaluate statistical significance of group differences (adonis2). The significance of differences between environments was assessed using pairwise PERMANOVA (pairwise.adonis package). Differences in alpha diversity indices (Shannon and Faith’s Phylogenetic Diversity) across environments were assessed using the Kruskal-Wallis test, followed by pairwise Wilcoxon rank-sum comparisons with false discovery rate correction for multiple testing. For equality of variances testing, the homogeneity of group dispersions was validated (betadisper). Finally, the Multi-Response Permutation Procedure (MRPP) was conducted to further test for significant differences between habitats (mrpp). To test for the presence of differentially abundant OTUs and genera between significantly different samples and environments, both similarity percentage (SIMPER, implemented in vegan) and Linear discriminant analysis Effect Size (Lefse) analyses were applied. Lefse was calculated using mothur (lefse) and the threshold for the linear discriminant analysis score was set to 2.0.
Results
Sampled habitats and repository data used in this study
Throughout the study, we use the term biomass-rich environments to describe habitats that we analyzed, characterized by high levels of primary and secondary organic plant matter. These environments included diverse representative niches sampled in Luxembourg, Belgium and French Guiana (Supplementary Table S1). To simplify the analysis, we split the dataset of samples into two environment type categories. Main type included AD, soil, freshwater sediment, termite nest (mound), termite gut and all the other environments. Sub-type environment categories included AD (AD-WWTP from wastewater treatment plant fed with sewage sludge and AD-farm coming from agriculture-and biowaste-fed reactors), soil (soil including arable, grassland and forest, rhizosphere, moss-lichen covered and high fens), compost, decaying biomass (algal bloom, fungus-infected trees, biological mat), freshwater sediment, termite nest and termite gut. Three animal manure samples (two cattle and one horse) were collected, but due to the low number of planctomycetotal reads, only one sample (horse manure) was included in the analysis (Figure 1a, not shown). The samples taken from wetlands were also grouped together into “high fens” category when needed (Supplementary Table S1; Figure 1). Although our study did not include technical replicates, repeated measurements of the same environment type (biological replicates) should provide sufficient consistency in the data.
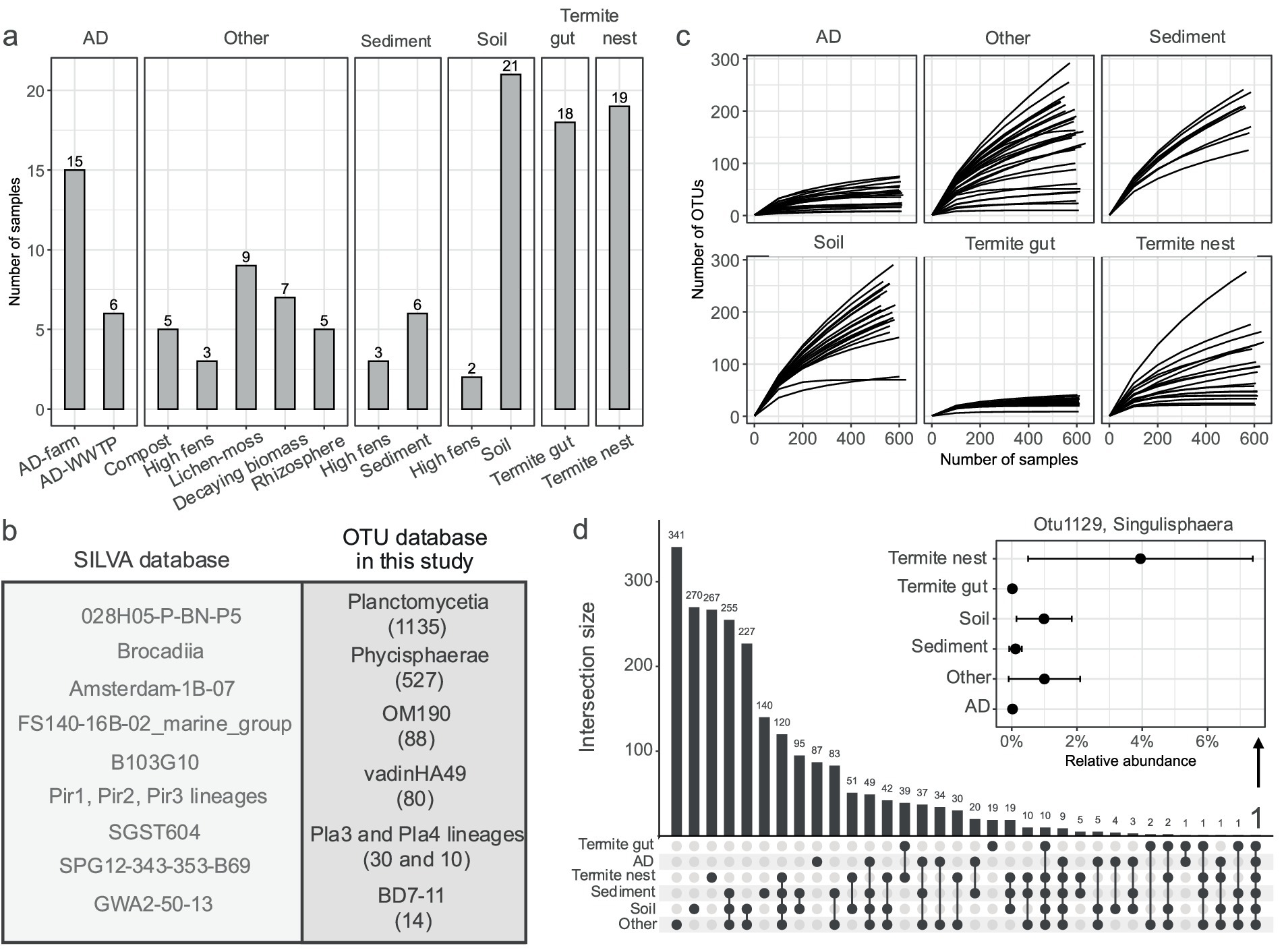
Figure 1. Studied environments and recovered OTUs of Planctomycetota. (a) Number of samples considered for the analysis, distinguished by the main type and sub-type environment categories. (b) Comparison of SILVA taxonomy of retrieved OTUs to all the other known SILVA taxonomy. Left box: taxa in SILVA not found in this study. Right box: taxa in SILVA found in this study, in brackets number of detected OTUs assigned to each class. (c) Rarefaction curves showing OTU richness across habitats, illustrating the relationship between sequencing depth and microbial diversity. (d) UpSet plot representing the intersection of shared and unique OTUs across different samples. Additionally, the relative abundance of one common OTU for all the environments (Otu1129) grouped in main type environments is depicted.
The amplicon sequencing output and SILVA taxonomy comparison
The sequenced dataset from this study included 45 samples, producing 8.7 M of read pairs. The initial quality check revealed an average read length of 484 bp, with a base quality score consistently above 30. Most of the reads passed the quality filtering (5.9 M), and we obtained a total of 1.7 M of unique, high-quality sequences remained for downstream analysis. The sequences were next combined with the reads from previous datasets yielding a final dataset of 119 samples (Supplementary Table S1; Figure 1a). OTU clustering of all the sequences with singletons removal resulted in generation of 26,578 OTUs, including a total of 1,899 OTUs that belonged to the Planctomycetota phylum, contributing to about 7% of the total bacterial diversity. At first, to assess the diversity within the studied environments, we classified retrieved OTUs against the SILVA database, comparing the detected taxonomic classes to those known in SILVA to identify the extent of overlapping class-level diversity (Figure 1b). Of all known classes and orders within the Planctomycetota phylum cataloged in the SILVA database, more than half were detected in this study, highlighting the broad diversity present in biomass-rich environments. Taxonomic classification of the identified OTUs revealed the presence of nine putative and known classes of Planctomycetota. Specifically, 1,135 OTUs were assigned to Planctomycetia, while 527 to Phycisphaerae classes. Next, putative classes such as OM190, vadinHA49 and Pla4 lineages were represented by 88, 80, and 30 OTUs, respectively. Fewer than 15 OTUs were found to be assigned to the Pla3-lineage, BD7-11, ODP123, and Ca. Brocadiia. The rarefaction curves for each sample reached a plateau only for AD, termite gut and certain other sub-type environments, indicating that sequencing depth was sufficient to capture the majority of the planctomycetotal diversity only for these systems (Figure 1c).
Shared and unique planctomycetotal OTUs among habitats
The majority of OTUs were shared between soil, sediment and other type environments, with the largest intersection of 255 OTUs observed, indicating significant phylogenetic overlap between these ecosystems (Figure 1d). In contrast, a smaller subset of 19 OTUs was uniquely found in termite gut samples, reflecting the specialized nature of the microbial community associated with this environment. The AD reactor samples also shared a considerable number of OTUs (49) with the soil, sediment and other category samples, which may be indicative of the transfer of microbes through organic waste inputs used in anaerobic digestion process. Only one OTU belonging to MSBL9-clade (Sedimentisphaerales) was found to be common between the AD and termite gut sample sets (Otu28617). The smallest intersections involved termite gut, which shared relatively few OTUs with other environments (10). Only one OTU, e.g., Otu1129, assigned to the Singulisphaera genus, was evidenced to be present in all the environments studied and it shared 99.1% sequence identity to the 16S rRNA sequence of Singulisphaera rosea S26. Importantly, the relative abundance of Otu1129 in the environments other than soil and termite nest (on average between 1.5 and 4%) was negligible, e.g., 0.2%, 0.2–0.5, and 0.5% of planctomycetotal abundance in gut, sediment and AD (Figure 1d).
Planctomycetota relative abundance and alpha diversity in studied environments
The Planctomycetota phylum was observed with abundances ranging from nearly 0% to almost 10% in the studied samples (Figure 2a). Looking at the individual samples, the highest abundances were detected in lichen-moss soil cover (9.82%) and termite nest (7.8%) samples and the lowest in horse manure specimen (Figure 2b, 0.1%). On average, soil samples contained the highest abundance of Planctomycetota (5.2% ± 1.4), followed by the sediment and other soil-types samples (Figure 2b), resulting in 4.2% (±1.1), 4.1% (±2.8), and 4.0% (±1.8), respectively. In soil samples, the higher relative abundance of Planctomycetota can be attributed to the cumulative presence of diverse OTUs at low concentrations (Supplementary Table S3). In turn, the lowest average planctomycetotal abundance was found in AD reactors (0.91% ± 0.99) and termite gut (1.1% ± 1.4) habitats.
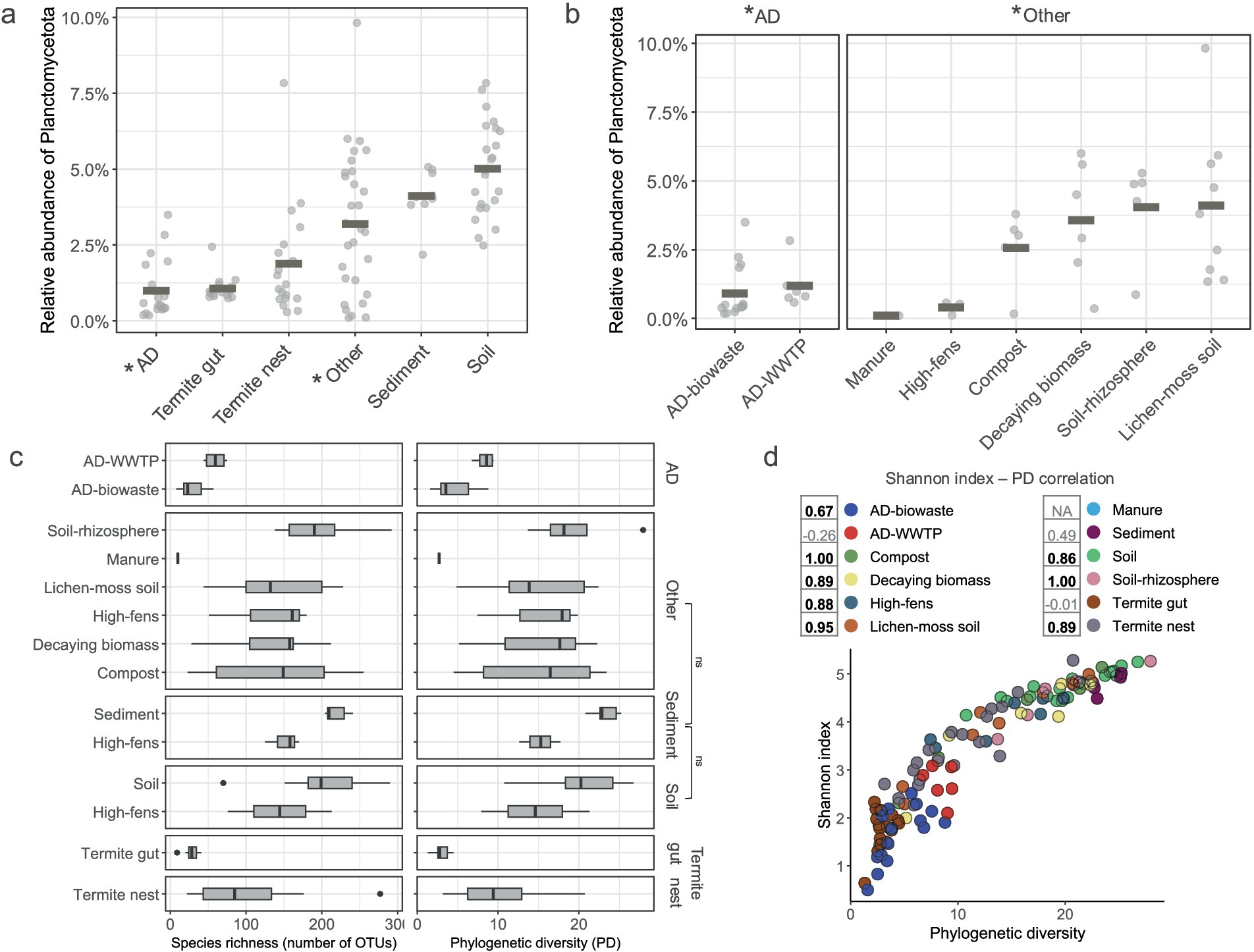
Figure 2. Alpha-diversity of Planctomycetota communities. (a,b) Relative abundance of Planctomycetota in samples grouped by (a) the main type environments (b) and sub-type environments. Gray rectangles indicate the mean abundance (c) Planctomycetotal OTU richness (number of distinct OTUs) and phylogenetic diversity (Faith’s PD) for all the sub-type environments; ns, non significant. (d) The correlation between SD and PD indices of all the studied samples, colored by sub-type environment categories. Numbers indicate Spearman rank rho correlation values and significant correlations (p < 0.05) are denoted in bold. Manure is represented by only one sample and therefore no correlation was calculated.
To characterize the planctomycetotal diversity within each sample, we assessed the Faith’s phylogenetic diversity (PD) and richness (observed OTUs) across habitats (Figures 2c,d). Both indices varied significantly among the studied environments (p < 0.01). However, pairwise comparisons between habitats revealed that soil, sediment, and “other” types of environments exhibited similar SD values, as indicated by non-significant pairwise comparisons (Figure 2c). Soils and sediments exhibited the highest PD (23.3 ± 1.7 and 20.3 ± 4.2, respectively) indicating a broad evolutionary span of the planctomycetotal community. In contrast, termite guts showed the lowest PD (3 ± 0.8), suggesting more evolutionarily narrow community. AD samples showed higher discrepancy of planctomycetotal diversity, and AD-WWTP had higher PD (8.4 ± 1.1) than AD-farm (4.5 ± 2.2). Similar patterns occurred when analyzing the species richness, with the highest number of observed OTUs in soil and sediment samples. We next calculated the Shannon diversity index (SD) and compared it with the PD value for each sample (Figure 2d). Spearman correlation analysis between these indices indicated a significant positive correlation (p < 0.05) for most of the habitats, suggesting a higher number of observed OTUs and the evolutionary breadth of planctomycetotal communities. In contrast, the habitats with lower PD, such as termite guts and ADs, were also characterized by lower species richness. The comparison between PD and SD showed that, while AD-WWTP had higher species diversity, the negative correlation between these two indices suggests that the taxa may be more closely related, despite their OTU abundance. Taken together, this indicates that the microbial communities in both AD-WWTP and the termite guts are more specialized.
Beta diversity of planctomycetotal communities among studied environments
Subsequently, we examined differences in the planctomycetotal community composition between studied environments. nMDS plot using Bray–Curtis dissimilarity revealed distinct clustering patterns based on the sampled habitat, especially visible for planctomycetotal communities in anaerobic digesters and termite guts (Figure 3a). The nMDS analysis yielded a stress value of 0.0998, indicating a good representation of the data. The PERMANOVA test confirmed that these differences in community composition were statistically significant (Figure 3a), with pairwise PERMANOVA revealing significant differences between all habitat types except among sediment, soil, and the “other” category environments. However, the significant betadisper result suggests that part of the difference detected by PERMANOVA could also be influenced by differences in how dispersed the community compositions are within each habitat. We also confirmed that the clustering patterns indicated only termite gut as a more homogenous community (delta of 0.34), while all the other environments showed higher variability (delta > 0.66). It is important to note that delta values may vary depending on multiple factors; therefore, these thresholds should be interpreted within the context of this dataset and not as universal cutoffs. Overall similarity index scored to 0.316 supporting a moderate to strong agreement within groups, meaning that the samples within each habitat were more similar to each other than what would be expected randomly (Figure 3a). Across all samples, not surprisingly, the most abundant classes were Planctomycetia and Phycisphaerae, constituting at least one third of total sequences (Figure 3b). Representatives of other putative planctomycetotal classes such as OM190 (proposed Saltatorellus) and vadinHA49 were also found to be in higher abundances in sediments and termite gut samples. Certain Planctomycetota, such as Pla3 (UBA8108 in GTDB) and other yet-unknown lineages, were detected almost exclusively in AD-WWTP and manure samples, suggesting a possible niche specialization (Figure 3b).
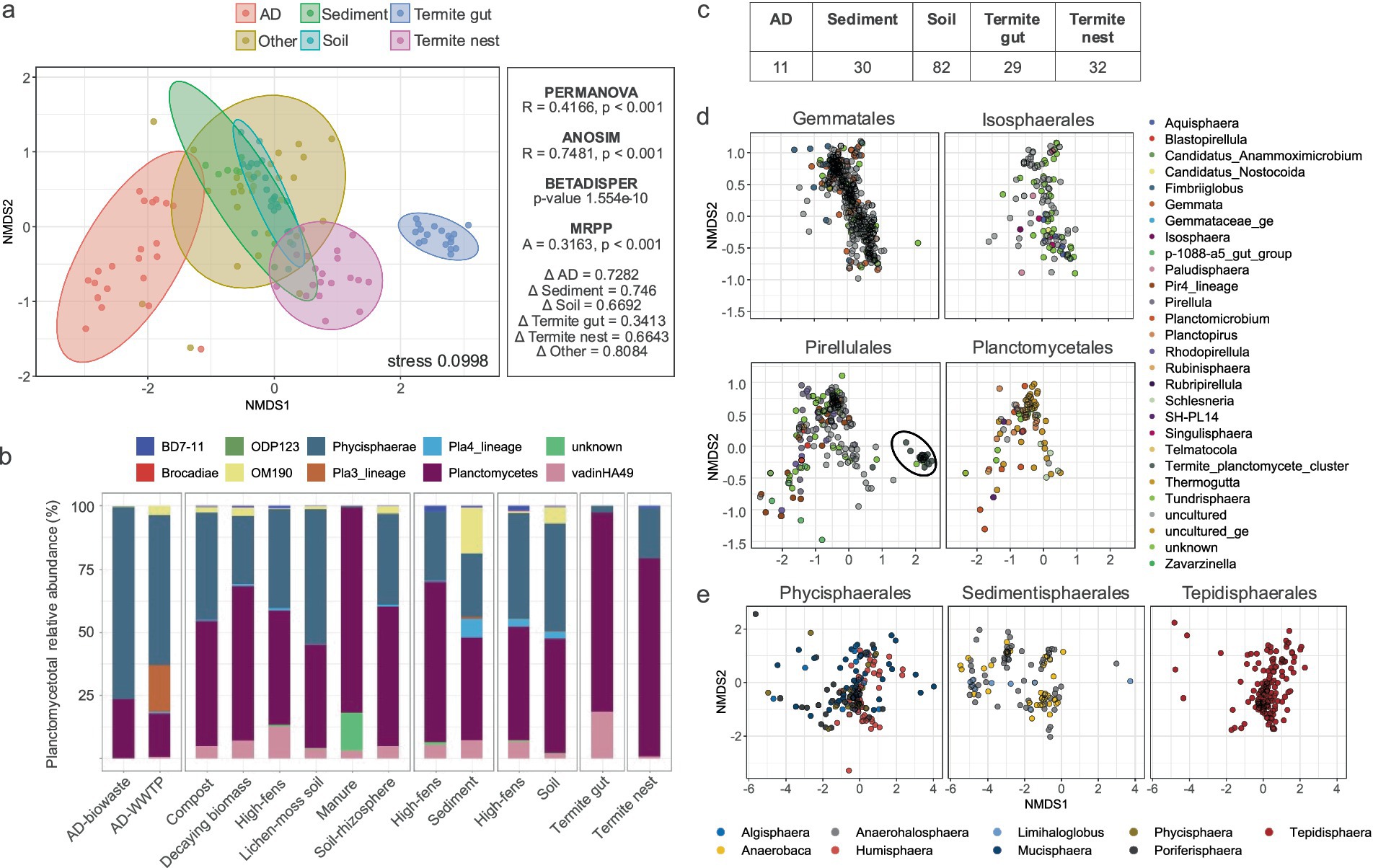
Figure 3. Beta diversity of Planctomycetota communities. (a) nMDS plot illustrating planctomycetotal community composition across diverse environments based on Bray-Curtis dissimilarities. Right panel presents the results of beta diversity statistical tests. (b) Barplot represents the planctomycetotal community for class level as assigned against SILVA database. (c) Number of differentially abundant OTUs in each of the environments. (d) nMDS plot for the orders within the Planctomycetia class, colored by genera; in black ellipse the termite cluster is highlighted. (e) nMDS plot for the orders within the Phycisphaerae class, colored by genera.
To identify taxa that were differentially abundant between habitats, SIMPER and LEfSe analyses were employed (Figure 3c). As a result, we identified 178 OTUs significantly different among all main type habitats, belonging primarily to 50 genera. The highest number of differentially abundant OTUs was found in soil (82), but representing only 7.8% of all the OTUs discovered. On the other hand, 40% of all the OTUs found in termite guts were differentially abundant. We next tried to visualize potential co-occurrence patterns among taxa and performed nMDS based on taxonomic distributions. As a result, we observed that Gemmatales and Isosphaerales tended to overlap, as did Planctomycetales and Pirellulales (Figure 3d). Additional, clear grouping of termite-derived Pirellulales can also be observed (Figure 3d) Among Phycisphaerae, certain genera of Phycisphaerales and Tepidisphaerales tended to collocate, while Sedimentisphaerales showed clearer groupings (Figure 3e). In the following section we evaluated the genera that contribute the most to the patterns observed.
The closest relatives of OTUs to known species
To gain a deeper ecological understanding of Planctomycetota, we compared their OTUs against a custom database containing formally described type species. The dataset was then stratified to identify potential OTUs matching known species, genera, and families, using standard identity thresholds of 98.7, 94.5, and 86.5% (Figure 4a). The majority of the retrieved OTUs in this study, e.g., between 22 and 60%, represent potential new genera within known families, depending on the lineage and its environmental origin (Supplementary Table S2). In turn, 49 OTUs exhibited high identity, potentially representing known species (Supplementary Table S2). Among them, four were commonly found in soils, termite nests and sediments: Caulifigura coniformis Pan44 (Otu774) (Kallscheuer et al., 2020b), Schlesneria paludicola MPL7 (Otu3995) (Kulichevskaya et al., 2007), Paludisphaera soli JC670 (Otu12607) (Kaushik et al., 2020) and Singulisphaera rosea S26 (Otu1129) (Kulichevskaya et al., 2012). The other typical soil-sediment OTUs with high identities were: Urbifossiella limnaea ETA_A1 (Otu5302), Humisphaera borealis M1803 (Otu2624) (Dedysh et al., 2021) or Anatilimnocola aggregata ETA_A8 (Otu3323) (Kallscheuer et al., 2021). In turn, in AD samples with high identities to known species were Posidoniimonas polymericola Pla123a (Otu5525) and Planctomyces bekefii (Otu3705) (Dedysh et al., 2020). Subsequently, we applied the Kruskal-Wallis test to identify genera with significant abundance changes across main environmental types, revealing a panel of 28 genera (Figure 4b). Most of these genera were not highly abundant in the studied environments, contributing between 0 and 25% to the planctomycetotal community. Only Aeoliella-assigned OTUs were highly abundant in manure sample representing >75% of the planctomycetotal community, while Anaerobaca and Anaerohalosphaera, both retrieved from ADs, were moderately abundant in these environments (between 50 and 75%). Tepidisphaera was also dominant Planctomycetota in diverse soils including rhizosphere and lichen-moss covered soils. Ca. Brocadiia-, Thermostilla-and Mucisphaera-related OTUs were identified with a high portion of distantly related 16S rRNA gene sequences. Frigoriglobus seemed to be a hallmark genus for the termite nest samples, and about 30% of its sequences expressed high identity to Frigoriglobus tundricola PL17 (Kulichevskaya et al., 2020). However, termite nest is most abundantly inhabited by OTUs potentially attributed to Telmatocola-related representatives (50 and 75%). Thermogutta-related OTUs were widespread in different habitats, however, they were also largely represented by within-family sequences. In turn, both Singulisphaera-and Schlesneria-assigned OTUs were represented by a high fraction of sequences with higher identities, e.g., passing genus threshold.
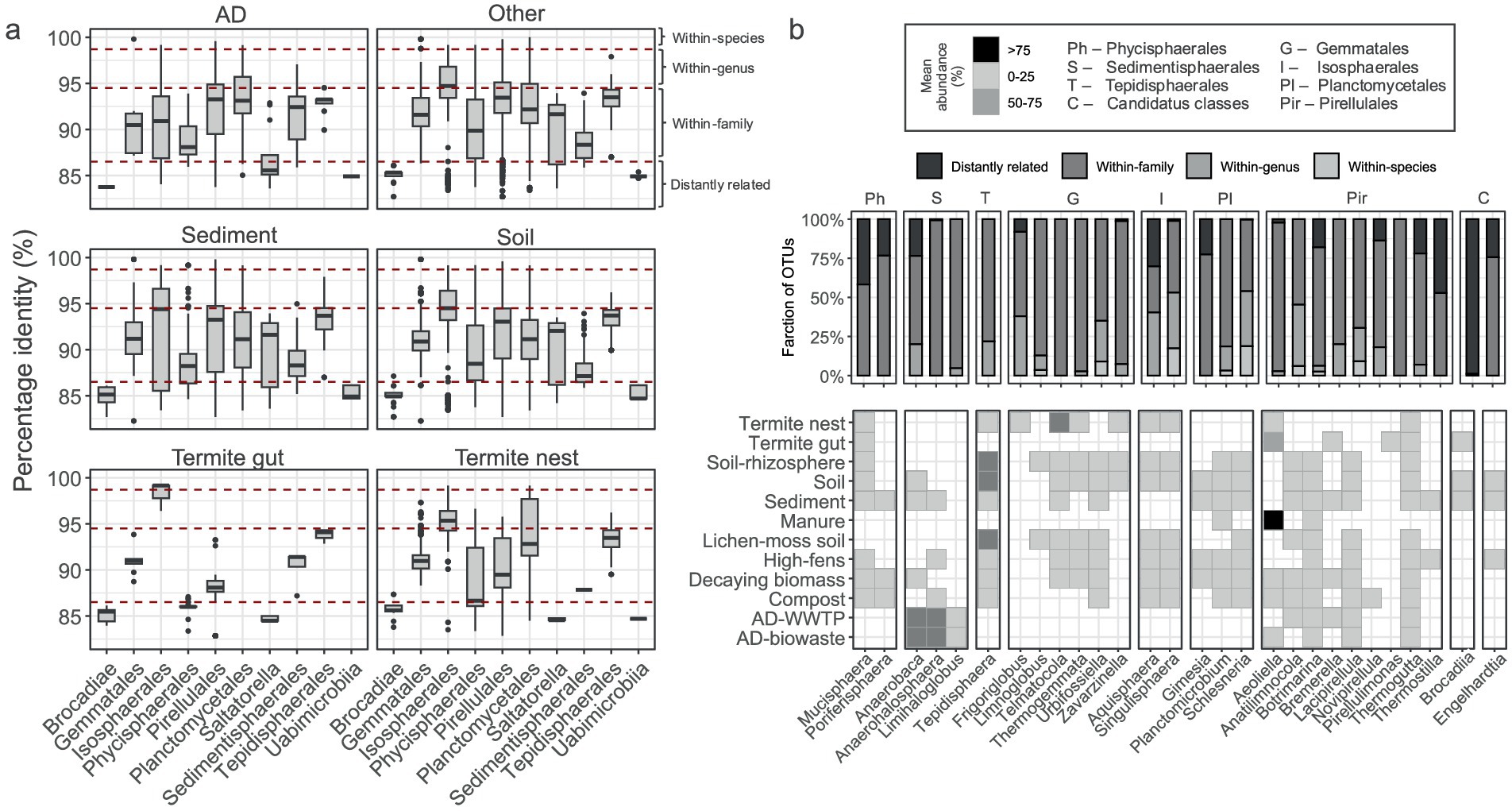
Figure 4. Comparison of Planctomycetota OTU sequences to the formally described type strains. (a) Percentage identity (%) for individual OTUs gathered at the order level; red lines limit the set thresholds for species, genus, family delineation. (b) The significantly different genera among all the studied environments. Barplot represents fraction of OTUs assigned to each thresholds category. Heatmap represents planctomycetotal average abundance range in each habitat (legend in the top box).
Planctomycetotal shared community
The phylogenetic tree based on UniFrac distances showed that samples from termite guts, termite nests and AD (except two WWTP samples) formed three separate clusters, indicating strong planctomycetotal community structures specific to these environments (Figure 5). In contrast, samples from diverse soil types, sediments, composts and decaying biomasses showed mixed clustering patterns, often more influenced by specific local conditions (e.g., High-fens region) rather than habitat type. The statistical differences between these environments were indeed not significant, further strengthening the observation of similar community structure. Regardless, three clusters on the tree are visible: (I) comprising diverse soils, decaying biomass and sediment samples, (II) encompassing forest soil and High fens samples and (III) mostly consisting of soil samples. In cluster I, Pirellulales dominated the community, but sediment samples also comprised Saltatorellus and Sedimentisphaerales. Cluster II was mainly abundant in Planctomycetales, and Gemmatales, while cluster III was dominated by Tepidisphaerales. Pond bloom sample diverged remarkably from all the others, exhibiting a highly distinct community dominated by Pirellulales OTUs with approximately 80% of total relative abundance (Figure 5). Next, we identified a panel of shared OTUs that significantly contributed to the observed community structure of soil, sediment and other type habitats, reflecting the presence of mixed planctomycetotal communities. A total of 29 shared OTUs was assigned to Tepidisphaera, while other OTUs were assigned to Telmatocola (19), Thermogutta (18), Anatilimnocola (16), Engelhardtia (12), Mucisphaera and Anaerobaca (both 11) and Humisphaera (10).
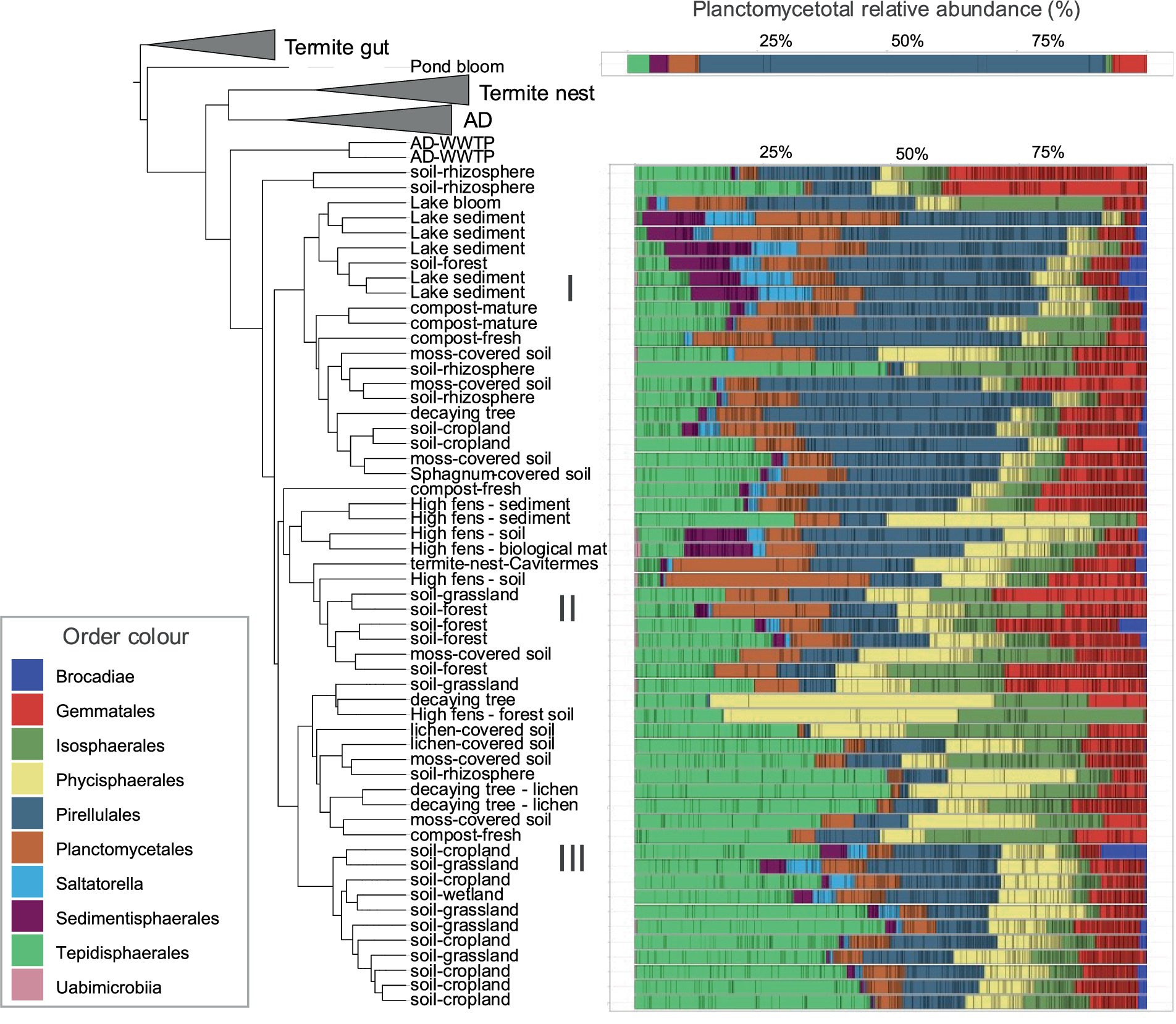
Figure 5. Shared Planctomycetota phylogeny for soil, sediment and other environment type samples. Weighted UniFrac phylogenetic tree illustrates the relationships between samples based on their planctomycetotal community composition, alongside corresponding barplots show the relative abundance of taxa at the order level.
Common and specific lineages in different environmental stages
In compost samples the community changed from early stage to more digested matter (Figure 6a). The vadinHA49 class was more commonly found in later stages of composting, e.g., in well-digested material. The vadinHA49 clones retrieved in this study were distantly related to other known, described Planctomycetota with average percentage identity below 85%. Blasted 16S rRNA gene sequences of vadinHA49 against deposited sequences in GTDB revealed that this group represents SZUA-567 putative class. Comparison of two different freshwater blooms showed significant differences in planctomycetotal communities (Figure 6a). Even though both samples taken from freshwater cyanobacterial blooms were sampled during its density peak leading to decomposition, pond sample was abundant in Pirellulales (Figure 6a), while bloom occurred on lake was more diverse, including other bacteria such as vadinHA49. Samples from decaying trees also did not show strong common patterns in relative abundance of Planctomycetota, however, Tepidisphaerales and Pirellulales seemed to be the dominant groups in these ecosystems. Except freshwater bloom samples, all the sampled environments seemed to be dominated by the same orders such as Tepidisphaerales, Pirellulales and Planctomycetales.
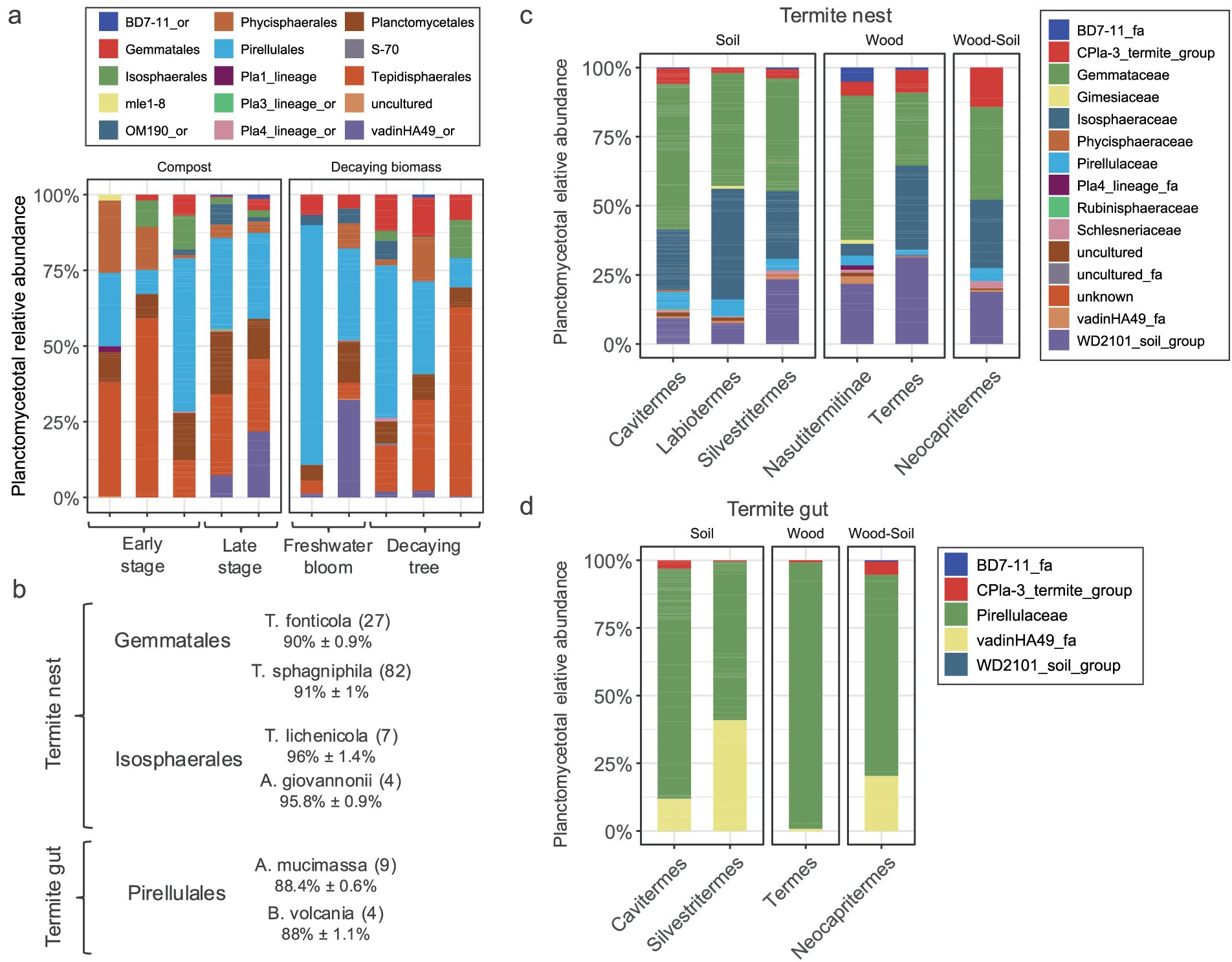
Figure 6. Phylogenetic affiliation of Planctomycetota for selected environments. (a) Relative abundance of Planctomycetota in actively degrading biomasses such as compost, freshwater blooms and decaying trees. (b) Percentage identity of the most abundant OTUs assigned to the main Planctomycetota orders: Gemmatales and Isosphaerales in termite nests and Pirellulales in termite guts. Shortcuts: T. fonticola (Thermogemmata fonticola 2918), T. sphagnifila (Telmatocola sphagniphila SP2), T. lichenicola (Tundrisphaera lichenicola P12), A. giovannonii (Aquisphaera giovannonii OJF2), A. mucimassa (Aeoliella mucimassa Pan181) and B. volcania (Bremerella volcania Pan97). In brackets the number of OTUs assigned to these strains are indicated. (c,d) Termite nest (c) and gut (d) planctomycetotal community. Relative abundance of different families of Planctomycetota are gathered for each species of termites.
Planctomycetotal community in termite bodies and nests
The Planctomycetota community in termite nests was distinct from soil samples, although there were overlaps, mainly driven by the presence of specific OTUs distantly related to Gemmatales and Isosphaerales (Figure 6b). Overall, the main distinction between the soil and termite nest planctomycetotal communities lies in the presence of termite-specific microbes, which shape the community structure and make it distinct from the typical soil members (Figures 5, 6c). Looking at the different feeding styles of termites, the wood-feeding termite nests showed higher richness (158 ± 128 OTUs), compared to nests of termites feeding on soil humus (84 ± 44 OTUs) and mixed humus-wood feeders (40 OTUs). Conversely, OTU richness in termite guts was the highest in humus-feeders (30 ± 6 OTUs), rather than in wood-and mixed-feeders (20 and 9 OTUs, respectively). Termite guts were chiefly dominated by OTUs assigned to Pirellulaceae and vadinHA49 families, except a single sample of wood-feeder, consisting of only 9 Pirellulales OTUs (Figure 6d). Otu993 seemed to be a core termite gut representing Termite_planctomycete_cluster in SILVA database, being present in all the termite species studied. Otu993 displayed the highest 16S rRNA gene similarity to Aeoliella mucimassa Pan181 of 88.5% suggesting the potentially novel sister family to Lacipirellulaceae (Wiegand et al., 2020) (Figure 6b).
Discussion
In the context of biomass degradation, microbial community analyses are essential not only for revealing taxonomic diversity but also for providing insights into possible ecological functions and guiding future microbial isolation efforts. Therefore, the aim of this study was to characterize the microbial community of the Planctomycetota phylum based on 16S rRNA gene sequencing, providing insights into their diversity and composition across diverse natural and built biomass-rich systems. We revealed significant variations in Planctomycetota community composition that reflect common patterns found in microbial communities globally. The OTU rarefaction curves showed contrasts in Planctomycetota richness across environments (Figure 1) and disclosed OTU diversity across habitat types that was further supported by alpha diversity metrics (Figure 2). Commonly, microbial communities from soil, followed by sediment, show the highest alpha diversities (Walters and Martiny, 2020), and this is well reflected by Planctomycetota richness and diversity in these environments. Microbes from habitats such as anaerobic digesters or termite guts are predicted to exhibit narrow OTU diversity, as both communities are highly specialized for the specific anaerobic niches present in these environments (Goux et al., 2023). Consequently, the observation of a limited number of planctomycetotal OTUs is not unexpected. In some environments, the total planctomycetotal abundance is negligible, approaching levels typically associated with the rare biosphere (below 1% or even lower), while in other environments, the greater diversity of species parallels the higher abundance. Despite this variability, in both cases, Planctomycetota could serve as potential genetic reservoirs, harboring unique genetic diversity that may contribute to maintaining the functional resilience of the entire ecosystem.
The species delineation and estimation are still challenging due to the limited resolution of 16S rRNA gene amplicon sequencing or influence of primer selection, which can lead to misclassification and inaccuracies in estimating microbial diversity (Abellan-Schneyder et al., 2021; Johnson et al., 2019). In this study, OTU clustering was performed at the 97% sequence similarity threshold, a widely used approximation for species-level diversity. While higher-resolution approaches, such as 99% OTU clustering or amplicon sequence variants, are proposed as the current standard (Callahan et al., 2019; Edgar, 2018), 97% OTU clustering still offers certain advantages. In our study, such approach allowed comparability across datasets originating from different sequencing runs, helped reduce the impact of sequencing noise or artifacts and importantly, lowered the risk of subdividing taxonomic units into poorly supported taxa, resulting in more conservative and robust taxonomic assignments (Schloss, 2021). Still, future work employing full-length 16S rRNA gene sequencing may offer a more accurate view of Planctomycetota diversity.
Surprisingly, one OTU with high identity to Singulisphaera rosea S26 belonging to the Isosphaeraceae family (Isosphaerales order) was shared among all the studied habitats, but only abundant in diverse soil types, termite nests and other type environments including composts or high fens (Figure 1). Previously, Planctomycetota, including Isosphaerales, were indeed found to represent a significant fraction of the microbial communities in lichen-dominated areas, contributing to 8–13% of total bacterial sequences (Ivanova et al., 2016). Isolated Singulisphaera strains from these habitats were also shown to utilize complex polysaccharides including a characteristic component of lichen biomass (lichenan), but overall Singuliphaera spp. strains are capable of utilizing a wide range of polysaccharides including cellulose and chitin (Kulichevskaya et al., 2012; Ivanova et al., 2024). The widespread presence of Singulisphaera OTUs suggests its potential metabolic versatility, making it a compelling subject for further study. This is particularly important in light of its metabolic plasticity as members of the Isosphaerales order are also known to harbor plasmids (Coronel et al., 2024; Ivanova et al., 2017).
The high relative abundance of Pirellulales and Tepidisphaerales in several habitats including soils, sediments, freshwater blooms and animal guts suggests that these microbes play substantial roles in the studied environments, including putative breakdown of biomass. This degradation process is likely driven by fermentative and hydrolytic pathways, as described species from both orders are heterotrophic bacteria possessing multiple CAZymes, which certainly facilitate the breakdown of complex organic matter (Klimek et al., 2024). For instance, previous study indicated that Tepidisphaera was involved in degradation of extrapolymeric substances in biochar-assisted bioremediation reactors (Jiang et al., 2020). The planctomycetotal community in the pond bloom sample was dominated by three Pirellulales OTUs accounting for approximately 80% of the total relative abundance (Figure 5). The OTUs identified had the highest similarities of 95.3% to Aureliella helgolandensis and 95.1 and 94.3% to Bremerella alba FF15. Both strains were isolated from host-associated environments as they derived from jellyfish and macroalgae ecosystems (Godinho et al., 2021; Kallscheuer et al., 2020a). This highly specialized community aligns well with previous observations, as members of the Pirellulales were shown to proliferate during cyanobacterial blooms in freshwater environment, where they became highly abundant, albeit not particularly transcriptionally active (Kallscheuer et al., 2021). In turn, Planctomycetota assigned to Pirellulales, were also evidenced as the most transcriptionally active group of microbes in the hydrothermal plume, involved in organic matter scavenging (Li et al., 2016).
Based on the set thresholds for taxonomic delineation, the OTUs recovered in this study were primarily associated with already known bacterial families (Figure 4). These families are generally represented by cultured type species, suggesting that most discoveries in biomass-rich environments will likely involve new genera within the established higher ranks. However, some yet-unknown lineages likely remain undescribed. All OTUs distantly related to Brocadiia suggest existence of new class-level lineages of Planctomycetota specific to soils, sediments, but also termite guts. One of the lineages of putative class status is vadinHA49 (in SILVA, SZUA-567 in GTDB). This group has been previously identified in various environments, including anaerobic digesters, the intestinal tracts of insects, peat bogs, and sulfur spring sediments (Ivanova et al., 2018; Sabree and Moran, 2014; Youssef et al., 2012). It was also evidenced that vadinHA49 strongly correlated with the higher availability of organic matter, suggesting its involvement in biomass degradation during AD (Liu et al., 2020). In addition to that, vadinHA49 was also identified as one of the bacterial groups able to persist the cadmium pollution (Song et al., 2019). However, studies on vadinHA49 are scarce, and its potential metabolic functions remain of high interest, though they are largely unexplored. Thermogutta exhibited differential abundance across the studied environments, simultaneously with many OTUs related to this genus shared among soil, sediment, and other habitat types. Additionally, their lower sequence similarities to members of the Thermoguttaceae family suggest that they represent a novel lineage within this family. Thermoguttaceae representatives inhabit terrestrial, subsurface or anthropogenic ecosystems with elevated temperatures (Slobodkina et al., 2015, 2016); yet our analysis clearly shows their widespread occurrence in other ecosystems. Previously, we identified Thermoguttaceae as putative biomass degraders due to the presence of multiple CAZymes (Klimek et al., 2024). Therefore, the wide distribution of these novel lineages across the studied biomass-rich environments is not unexpected.
Soil, sediment and other type environments shared certain widespread taxa (Figure 5) distantly affiliated to Tepidisphaera (Tepidisphaerales), Telmatocola (Gemmatales), Thermogutta, and Anatilimnicola (both Pirellulales). As previously noted for the Isosphaera genus, the genera listed may also exhibit metabolic versatility that enables adaptation to diverse environmental conditions. Despite pronounced physicochemical differences among habitats, these planctomycetotal groups may fulfill similar ecological functions, contributing to a convergence in core community composition. Future comparative genomic analyses of representative genomes from these genera will be instrumental in elucidating their metabolic capabilities and ecological roles. Planctomycetota are important and diverse components of soil microbial communities, and their diversity correlates strongly with environmental heterogeneity, particularly in relation to organic and nitrate levels as well as land use history (Buckley et al., 2006). Phylotypes belonging to Gemmata were previously linked to N2O reduction in soils (Xiao et al., 2025) and were also found to be the most abundant genera in the microbial community derived from garden soil that was actively removing the heavy metal pollutions (Singh et al., 2022). As also evidenced in our study, Gemmatales were previously listed among abundant orders in termite nests and suggested as an indicator of elevated phosphorus content in soil (González Plaza and Hradecký, 2023). Still, the abundance of Planctomycetota in the termite nest are usually lower than in the surrounding soil (Chen et al., 2021).
Interestingly, two trends of planctomycetotal beta diversity could be observed (Figure 3). Both Gemmatales and Isosphaerales tended to overlap in their community distribution, likewise Planctomycetales and Pirellulales, most likely due to their preferred environmental niches. The former two are more frequently associated with terrestrial and freshwater systems, in contrast to Planctomycetales and Pirellulales, which representative strains were typically isolated from marine, brackish or other aquatic environments (Vitorino and Lage, 2022). This is, however, more likely a result of their close phylogenetic relatedness, as phylogeny can exhibit a strong signal for habitat preferences, though this does not necessarily imply phylogenetic niche conservatism (Barberán et al., 2014). Based on the 16S rRNA gene phylogeny, Isosphaerales and Gemmatales are positioned on separate branch, indicating they are more distantly related to Pirellulales and Planctomycetales (Vitorino and Lage, 2022).
Animal gut habitats exhibited unique planctomycetotal assemblages predominantly affiliated with the Pirellulales order (Figure 5). The low abundance of Planctomycetota in the studied manure sample is consistent with previous reports on their presence in horse gut (Mach et al., 2022). However, 20% of OTUs reconstructed in this study could not be classified within any group in the SILVA database. Their percentage identity to Aeoliella mucimassa Pan181 (Lacipirellulaceae; Pirellulales) was oscillating around 88.3% ± 0.8%, indicating the presence of potentially novel lineages that may be specifically associated with the gastrointestinal tract of animals. Although our study lacked sufficient Planctomycetota reads in cattle manure samples, there is substantial evidence supporting their presence and diversity in the cattle rumen and ruminants overall. This is demonstrated by the abundance of assembled genomes from gut microbiome studies and their sporadic detection in various scientific reports (Hagey et al., 2019; Kim et al., 2014; Mao et al., 2012; Stewart et al., 2019). The relative abundance of Planctomycetota in the rumen is generally low, typically below 0.1%; however, studies have shown a significant increase in their abundance in grass-fed cattle and older animals (Liu et al., 2022; Wang et al., 2021). Samples collected from a diverse range of animals, including mammals, birds, fish, and insects, are predominantly inhabited by Pirellulales, suggesting a highly adaptable, host-associated planctomycetotal group (Medina-Silva et al., 2018). The ecological role and distribution of this bacterial group within the human gut microbiome remain unresolved. To date, Planctomycetota were only identified in a single 16S rRNA gene-based study, derived from stool samples of individuals in France and Senegal (Cayrou et al., 2013). Additionally, a limited number of genomes have been recovered from gut samples of hunter-gatherer populations (Carter et al., 2023). Previously, we noticed that Pirellulales express the potential to degrade mucosal layer components (mucins) from animal tissues such as the gastrointestinal tract (Klimek et al., 2025), presenting a promising avenue for future research. In addition to potential of Pirellulales for dietary and host polysaccharide degradation, they may also play other ecological roles within the gut ecosystem. Pirellulaceae-related lineages increased their abundance in cattle fed with grass infected with toxic endophyte Epichloë coenophialum, likely due to their ability to degrade main alkaloid (ergovaline) produced by this fungus (Mote et al., 2019). Pirellulaceae have the potential for degradation of hydrocarbon, and are resistant to pollutants (de Araujo et al., 2021; Flores et al., 2014), which may allow them to contribute to bioremediation processes. In general, other Planctomycetota from yet unidentified lineages have also been suggested as potential heavy metal detoxifiers, as their abundance was previously found to positively correlate with elevated cadmium and lead concentrations (Li et al., 2022).
Another interesting gut microbiome is that of termites. Termites play essential roles in ecosystems by decomposing plant material and contributing to nutrient cycling, ultimately returning organic matter to the soil (Ohkuma and Brune, 2011). Termite microbial communities are often inhabited by host-specific lineages (Arora et al., 2022). So-called lower termites are mainly wood-feeders that help in breaking down dead wood, while higher termites, mainly belonging to the Termitidae family, are characterized with wider food repertoire (Brune, 2014). Still, most of the higher termites ingest humus-rich soil, which contains decayed organic material, plant debris, and minerals. Lower termites represent the ancestral lineages that rely on protozoa for cellulose digestion, in contrast to higher termites, which have evolved a fully bacterial gut with a complex hindgut (Marynowska et al., 2023). The presence of Planctomycetota MAGs across a wide range of termite species supports the notion that this phylum constitutes a core component of the termite gut microbiome (Hervé et al., 2020), despite their lower abundancies. The ecological role of Planctomycetota in this environment remains purely hypothetical and is primarily inferred from genomic data. So far, Planctomycetota were suggested to increase the solubility of humic substances or to be involved in the digestion of microbial polymers (Köhler et al., 2008). Still, many plantomycetotal MAGs recovered from metagenomes from both lower and higher termites were found to be abundant in genes coding for pectinases (Salgado et al., 2024), implying they are likely involved in the degradation of plant organic matter. The current understanding of Planctomycetota abundance across different gut regions remains fragmentary. Their higher abundancies were found in the alkaline posterior compartments of hindgut in the soil-feeding termites belonging to the Cubitermes ugandensis and Ophiotermes sp. Ox79b (Köhler et al., 2008; Mikaelyan et al., 2017). In our study, the entire gut was taken for analysis, without separating it into compartments. Despite this, our findings, along with prior studies, indicate that Planctomycetota are not exclusively linked to higher termites nor alkaline conditions (Abdul Rahman et al., 2015). For instance, Planctomycetota were also found abundant in the foregut of the lower termites like the wood-feeding Coptotermes formosanus (Dar et al., 2024). Further investigation into the exact differentiation between planctomycetotal species in lower and higher termites would help elucidate their roles in either early-stage carbohydrate metabolism to lignocellulose degradation and fermentation. Counterintuitively, we observed higher richness and diversity of Planctomycetota in humus-feeders than in wood-feeders, indicating presence of a few, but specialized microbes for targeting lignocellulose. These novel, termite-specific bacterial lineages are uniquely adapted to the environment, reflecting observed tight clustering of termite gut samples. Termite gut and ADs systems are indeed characterized by distinct microbial communities, yet much of the encoded CAZyme diversity is shared among their microbes (Goux et al., 2023). Overall, future comparative genomic analysis of termite gut with gastrointestinal systems of ruminants will help identify specialized Planctomycetota essential for lignocellulose breakdown.
Data availability statement
All the results presented in this study are included in the article and its related Supplementary material. The raw sequencing reads used in this study were obtained from previously published datasets, as referenced in the Materials and methods (Data collection and sample preparation section). The processed data, including raw count tables and FASTA files of generated OTUs, are available on Figshare, https://doi.org/10.6084/m9.figshare.28847258.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
DK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Funding acquisition. OL: Conceptualization, Supervision, Writing – review & editing. MC: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Our research was supported by the Luxembourg National Research Fund (AFR grant, ref. 14583934).
Acknowledgments
We would like to thank Xavier Goux for his help in reviewing our manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1579219/full#supplementary-material
References
Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K., Warnecke, F., et al. (2015). A molecular survey of Australian and north American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome 3:5. doi: 10.1186/s40168-015-0067-8
Abellan-Schneyder, I., Matchado, M. S., Reitmeier, S., Sommer, A., Sewald, Z., Baumbach, J., et al. (2021). Primer, pipelines, parameters: issues in 16S rRNA gene sequencing. mSphere 6:10.1128/msphere.01202-20. doi: 10.1128/msphere.01202-20
Aghnatios, R., and Drancourt, M. (2016). Gemmata species: Planctomycetes of medical interest. Future Microbiol. 11, 659–667. doi: 10.2217/fmb-2015-0001
Arora, J., Kinjo, Y., Šobotník, J., Buček, A., Clitheroe, C., Stiblik, P., et al. (2022). The functional evolution of termite gut microbiota. Microbiome 10:78. doi: 10.1186/s40168-022-01258-3
Bajpai, P. (2020). “Chapter 7- advantages and disadvantages of biomass utilization” in Biomass to energy conversion technologies. ed. P. Bajpai (Elsevier), 169–173.
Barberán, A., Ramirez, K. S., Leff, J. W., Bradford, M. A., Wall, D. H., and Fierer, N. (2014). Why are some microbes more ubiquitous than others? Predicting the habitat breadth of soil bacteria. Ecol. Lett. 17, 794–802. doi: 10.1111/ele.12282
Bondoso, J., Godoy-Vitorino, F., Balagué, V., Gasol, J. M., Harder, J., and Lage, O. M. (2017). Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol. Ecol. 93:fiw255. doi: 10.1093/femsec/fiw255
Bonechi, C., Consumi, M., Donati, A., Leone, G., Magnani, A., Tamasi, G., et al. (2017). “Biomass” in Bioenergy Systems for the Future. Eds, F. Dalena, A. Basile, C. Rossi (Woodhead Publishing), 3–42.
Briliūtė, J., Urbanowicz, P. A., Luis, A. S., Baslé, A., Paterson, N., Rebello, O., et al. (2019). Complex N-glycan breakdown by gut Bacteroides involves an extensive enzymatic apparatus encoded by multiple co-regulated genetic loci. Nat. Microbiol. 4, 1571–1581. doi: 10.1038/s41564-019-0466-x
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Buckley, D. H., Huangyutitham, V., Nelson, T. A., Rumberger, A., and Thies, J. E. (2006). Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. AEM 72, 4522–4531. doi: 10.1128/AEM.00149-06
Cabello-Yeves, P. J., Callieri, C., Picazo, A., Mehrshad, M., Haro-Moreno, J. M., Roda-Garcia, J. J., et al. (2021). The microbiome of the Black Sea water column analyzed by shotgun and genome centric metagenomics. Environ. Microbiome 16:5. doi: 10.1186/s40793-021-00374-1
Callahan, B. J., Wong, J., Heiner, C., Oh, S., Theriot, C. M., Gulati, A. S., et al. (2019). High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 47:e103. doi: 10.1093/nar/gkz569
Calusinska, M., Goux, X., Fossépré, M., Muller, E. E. L., Wilmes, P., and Delfosse, P. (2018). A year of monitoring 20 mesophilic full-scale bioreactors reveals the existence of stable but different core microbiomes in bio-waste and wastewater anaerobic digestion systems. Biotechnol. Biofuels 11:196. doi: 10.1186/s13068-018-1195-8
Calusinska, M., Marynowska, M., Bertucci, M., Untereiner, B., Klimek, D., Goux, X., et al. (2020). Integrative omics analysis of the termite gut system adaptation to Miscanthus diet identifies lignocellulose degradation enzymes. Commun. Biol. 3, 275–212. doi: 10.1038/s42003-020-1004-3
Carter, M. M., Olm, M. R., Merrill, B. D., Dahan, D., Tripathi, S., Spencer, S. P., et al. (2023). Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell 186, 3111–3124.e13. doi: 10.1016/j.cell.2023.05.046
Cayrou, C., Sambe, B., Armougom, F., Raoult, D., and Drancourt, M. (2013). Molecular diversity of the Planctomycetes in the human gut microbiota in France and Senegal. APMIS 121, 1082–1090. doi: 10.1111/apm.12087
Chen, H. (2014). Chemical composition and structure of natural lignocellulose, in: Biotechnology of lignocellulose. Dordrecht: Springer Netherlands, 25–71.
Chen, Q., Hu, H., Yan, Z., Li, C., Nguyen, B. T., Zheng, Y., et al. (2021). Termite mounds reduce soil microbial diversity by filtering rare microbial taxa. Environ. Microbiol. 23, 2659–2668. doi: 10.1111/1462-2920.15507
Coronel, M. D. M. Q., Devos, D. P., and Garcillán-Barcia, M. P. (2024). Specificities and commonalities of the Planctomycetes plasmidome. Environ. Microbiol. 26:e16638. doi: 10.1101/2024.01.22.576637
Dar, M. A., Xie, R., Jing, L., Qing, X., Ali, S., Pandit, R. S., et al. (2024). Elucidating the structure, and composition of bacterial symbionts in the gut regions of wood-feeding termite, Coptotermes formosanus and their functional profile towards lignocellulolytic systems. Front. Microbiol. 15:1395568. doi: 10.3389/fmicb.2024.1395568
de Araujo, J. E., Taketani, R. G., Pylro, V. S., Leite, L. R., Silva, M. C. P. E., Lemos, L. N., et al. (2021). Genomic analysis reveals the potential for hydrocarbon degradation of Rhodopirellula sp. MGV isolated from a polluted Brazilian mangrove. Braz. J. Microbiol. 52, 1397–1404. doi: 10.1007/s42770-021-00483-6
Dedysh, S. N., Beletsky, A. V., Ivanova, A. A., Kulichevskaya, I. S., Suzina, N. E., Philippov, D. A., et al. (2021). Wide distribution of Phycisphaera-like planctomycetes from WD2101 soil group in peatlands and genome analysis of the first cultivated representative. Environ Microbiol. 23, 1510–1526. doi: 10.1111/1462-2920.15360
Dedysh, S. N., Henke, P., Ivanova, A. A., Kulichevskaya, I. S., Philippov, D. A., Meier-Kolthoff, J. P., et al. (2020). 100-year-old enigma solved: identification, genomic characterization and biogeography of the yet uncultured Planctomyces bekefii. Environ Microbiol. 22, 198–211. doi: 10.1111/1462-2920.14838
Dedysh, S. N., and Ivanova, A. A. (2019). Planctomycetes in boreal and subarctic wetlands: diversity patterns and potential ecological functions. FEMS Microbiol. Ecol. 95:fiy227. doi: 10.1093/femsec/fiy227
De Mandal, S., Chatterjee, R., and Kumar, N. S. (2017). Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol. 17:90. doi: 10.1186/s12866-017-1002-x
Drula, E., Garron, M.-L., Dogan, S., Lombard, V., Henrissat, B., and Terrapon, N. (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. doi: 10.1093/nar/gkab1045
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. doi: 10.1093/bioinformatics/bty113
Eilmus, S., and Heil, M. (2009). Bacterial Associates of Arboreal Ants and Their Putative Functions in an obligate ant-plant mutualism. AEM 75, 4324–4332. doi: 10.1128/AEM.00455-09
Elshahed, M. S., Youssef, N. H., Luo, Q., Najar, F. Z., Roe, B. A., Sisk, T. M., et al. (2007). Phylogenetic and metabolic diversity of Planctomycetes from anaerobic, sulfide-and sulfur-rich Zodletone Spring. Oklahoma. Appl. Environ. Microbiol. 73, 4707–4716. doi: 10.1128/AEM.00591-07
Farias, M. E., Rasuk, M. C., Gallagher, K. L., Contreras, M., Kurth, D., Fernandez, A. B., et al. (2017). Prokaryotic diversity and biogeochemical characteristics of benthic microbial ecosystems at La Brava, a hypersaline lake at Salar de Atacama, Chile. PLoS ONE 12:e0186867. doi: 10.1371/journal.pone.0186867
Fernández-Gómez, B., Richter, M., Schüler, M., Pinhassi, J., Acinas, S. G., González, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Figueroa, D., Capo, E., Lindh, M. V., Rowe, O. F., Paczkowska, J., Pinhassi, J., et al. (2021). Terrestrial dissolved organic matter inflow drives temporal dynamics of the bacterial community of a subarctic estuary (northern Baltic Sea). Environ. Microbiol. 23, 4200–4213. doi: 10.1111/1462-2920.15597
Flores, C., Catita, J. A. M., and Lage, O. M. (2014). Assessment of planctomycetes cell viability after pollutants exposure. Antonie Van Leeuwenhoek 106, 399–411. doi: 10.1007/s10482-014-0206-4
Gharechahi, J., Sarikhan, S., Han, J.-L., Ding, X.-Z., and Salekdeh, G. H. (2022). Functional and phylogenetic analyses of camel rumen microbiota associated with different lignocellulosic substrates. npj Biofilms Microbiomes 8:46. doi: 10.1038/s41522-022-00309-9
Godinho, O., Botelho, R., Albuquerque, L., Wiegand, S., Kallscheuer, N., da Costa, M. S., et al. (2021). Bremerella alba sp. nov., a novel planctomycete isolated from the surface of the macroalga Fucus spiralis. Syst. Appl. Microbiol. 44:126189. doi: 10.1016/j.syapm.2021.126189
González Plaza, J. J., and Hradecký, J. (2023). The tropical cookbook: termite diet and phylogenetics—over geographical origin—drive the microbiome and functional genetic structure of nests. Front. Microbiol. 14:1089525. doi: 10.3389/fmicb.2023.1089525
Goux, X., Liu, T., Westerholm, M., and Calusinska, M. (2023). “Microbial degradation of lignocellulose in natural and engineered systems – from the smallest to the biggest bioreactor” in Microbial fermentations in nature and as designed processes (Hoboken, United States: John Wiley & Sons, Ltd), 167–205.
Hagey, J. V., Bhatnagar, S., Heguy, J. M., Karle, B. M., Price, P. L., Meyer, D., et al. (2019). Fecal microbial communities in a large representative cohort of California dairy cows. Front. Microbiol. 10:1093. doi: 10.3389/fmicb.2019.01093
Hervé, V., Liu, P., Dietrich, C., Sillam-Dussès, D., Stiblik, P., Šobotník, J., et al. (2020). Phylogenomic analysis of 589 metagenome-assembled genomes encompassing all major prokaryotic lineages from the gut of higher termites. PeerJ 8:e8614. doi: 10.7717/peerj.8614
Howe, K. L., Seitz, K. W., Campbell, L. G., Baker, B. J., Thrash, J. C., Rabalais, N. N., et al. (2023). Metagenomics and metatranscriptomics reveal broadly distributed, active, novel methanotrophs in the Gulf of Mexico hypoxic zone and in the marine water column. FEMS Microbiol. Ecol. 99:fiac153. doi: 10.1093/femsec/fiac153
Hu, X., Gu, H., Liu, J., Wei, D., Zhu, P., Cui, X., et al. (2022). Metagenomics reveals divergent functional profiles of soil carbon and nitrogen cycling under long-term addition of chemical and organic fertilizers in the black soil region. Geoderma 418:115846. doi: 10.1016/j.geoderma.2022.115846
Ivanova, A. A., Kulichevskaya, I. S., Merkel, A. Y., Toshchakov, S. V., and Dedysh, S. N. (2016). High diversity of Planctomycetes in soils of two lichen-dominated sub-Arctic ecosystems of northwestern Siberia. Front. Microbiol. 7:2065. doi: 10.3389/fmicb.2016.02065
Ivanova, A. A., Naumoff, D. G., Kulichevskaya, I. S., Rakitin, A. L., Mardanov, A. V., Ravin, N. V., et al. (2024). Planctomycetes of the genus Singulisphaera possess Chitinolytic capabilities. Microorganisms 12:1266. doi: 10.3390/microorganisms12071266
Ivanova, A. A., Naumoff, D. G., Miroshnikov, K. K., Liesack, W., and Dedysh, S. N. (2017). Comparative genomics of four Isosphaeraceae Planctomycetes: a common Pool of plasmids and glycoside hydrolase genes shared by Paludisphaera borealis PX4T, Isosphaera pallida IS1BT, Singulisphaera acidiphila DSM 18658T, and strain SH-PL62. Front. Microbiol. 8:412. doi: 10.3389/fmicb.2017.00412
Ivanova, A. A., Wegner, C.-E., Kim, Y., Liesack, W., and Dedysh, S. N. (2018). Metatranscriptomics reveals the hydrolytic potential of peat-inhabiting Planctomycetes. Antonie Van Leeuwenhoek 111, 801–809. doi: 10.1007/s10482-017-0973-9
Jiang, B., Zeng, Q., Liu, J., Hou, Y., Xu, J., Li, H., et al. (2020). Enhanced treatment performance of phenol wastewater and membrane antifouling by biochar-assisted EMBR. Bioresour. Technol. 306:123147. doi: 10.1016/j.biortech.2020.123147
Johnson, J. S., Spakowicz, D. J., Hong, B.-Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-1
Kaboré, O. D., Godreuil, S., and Drancourt, M. (2020). Planctomycetes as host-associated Bacteria: a perspective that holds promise for their future isolations, by mimicking their native environmental niches in clinical microbiology laboratories. Front. Cell. Infect. Microbiol. 10:519301. doi: 10.3389/fcimb.2020.519301
Kallscheuer, N., Rast, P., Jogler, M., Wiegand, S., Kohn, T., Boedeker, C., et al. (2021). Analysis of bacterial communities in a municipal duck pond during a phytoplankton bloom and isolation of Anatilimnocola aggregata gen. Nov., sp. nov., Lacipirellula limnantheis sp. nov. and Urbifossiella limnaea gen. Nov., sp. nov. belonging to the phylum Planctomycetes. Environ. Microbiol. 23, 1379–1396. doi: 10.1111/1462-2920.15341
Kallscheuer, N., Wiegand, S., Boedeker, C., Peeters, S. H., Jogler, M., Heuer, A., et al. (2020b). Caulifigura coniformis gen. nov., sp. nov., a novel member of the family Planctomycetaceae isolated from a red biofilm sampled in a hydrothermal area. Antonie Van Leeuwenhoek. 113, 1927–1937. doi: 10.1007/s10482-020-01439-w
Kallscheuer, N., Wiegand, S., Boedeker, C., Peeters, S. H., Jogler, M., Rast, P., et al. (2020a). Aureliella helgolandensis gen. Nov., sp. nov., a novel Planctomycete isolated from a jellyfish at the shore of the island Helgoland. Antonie Van Leeuwenhoek 113, 1839–1849. doi: 10.1007/s10482-020-01403-8
Kappelmann, L., Krüger, K., Hehemann, J.-H., Harder, J., Markert, S., Unfried, F., et al. (2019). Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13, 76–91. doi: 10.1038/s41396-018-0242-6
Kaushik, R., Sharma, M., Gaurav, K., Jagadeeshwari, U., Shabbir, A., Sasikala, C., et al. (2020). Paludisphaera soli sp. nov., a new member of the family Isosphaeraceae isolated from high altitude soil in the Western Himalaya. Antonie Van Leeuwenhoek. 113, 1663–1674. doi: 10.1007/s10482-020-01471-w
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kim, J. W., Brawley, S. H., Prochnik, S., Chovatia, M., Grimwood, J., Jenkins, J., et al. (2016). Genome analysis of Planctomycetes inhabiting blades of the red alga Porphyra umbilicalis. PLoS One 11:e0151883. doi: 10.1371/journal.pone.0151883
Kim, M., Kim, J., Kuehn, L. A., Bono, J. L., Berry, E. D., Kalchayanand, N., et al. (2014). Investigation of bacterial diversity in the feces of cattle fed different diets1. J. Anim. Sci. 92, 683–694. doi: 10.2527/jas.2013-6841
Klimek, D., Herold, M., and Calusinska, M. (2024). Comparative genomic analysis of Planctomycetota potential for polysaccharide degradation identifies biotechnologically relevant microbes. BMC Genomics 25:523. doi: 10.1186/s12864-024-10413-z
Klimek, D., Herold, M., Vitorino, I. R., Dedova, Z., Lemaigre, S., Roussel, J., et al. (2025). Insights into the phylogenetic and metabolic diversity of Planctomycetota in anaerobic digesters and the isolation of novel Thermoguttaceae species. FEMS Microbiol. Ecol. 101:fiaf025. doi: 10.1093/femsec/fiaf025
Köhler, T., Stingl, U., Meuser, K., and Brune, A. (2008). Novel lineages of Planctomycetes densely colonize the alkaline gut of soil-feeding termites (Cubitermes spp.). Environ. Microbiol. 10, 1260–1270. doi: 10.1111/j.1462-2920.2007.01540.x
Kulichevskaya, I. S., Detkova, E. N., Bodelier, P. L. E., Rijpstra, W. I. C., Sinninghe Damsté, J. S., and Dedysh, S. N. (2012). Singulisphaera rosea sp. nov., a planctomycete from acidic Sphagnum peat, and emended description of the genus Singulisphaera. Int. J. Syst. Evol. Microbiol. 62, 118–123. doi: 10.1099/ijs.0.025924-0
Kulichevskaya, I. S., Ivanova, A. O., Belova, S. E., Baulina, O. I., Bodelier, P. L. E., Rijpstra, W. I. C., et al. (2007). Schlesneria paludicola gen. nov., sp. nov., the first acidophilic member of the order Planctomycetales, from Sphagnum-dominated boreal wetlands. Int J Syst Evol Microbiol. 57, 2680–2687. doi: 10.1099/ijs.0.65157-0
Kulichevskaya, I. S., Ivanova, A. A., Naumoff, D. G., Beletsky, A. V., Rijpstra, W. I. C., Sinninghe Damsté, J. S., et al. (2020). Frigoriglobus tundricola gen. nov., sp. nov., a psychrotolerant cellulolytic planctomycete of the family Gemmataceae from a littoral tundra wetland. Syst Appl Microbiol. 43:126129. doi: 10.1016/j.syapm.2020.126129
Lee, W.-J., and Hase, K. (2014). Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 10, 416–424. doi: 10.1038/nchembio.1535
Li, D., Chen, J., Zhang, X., Shi, W., and Li, J. (2022). Structural and functional characteristics of soil microbial communities in response to different ecological risk levels of heavy metals. Front. Microbiol. 13:1072389. doi: 10.3389/fmicb.2022.1072389
Li, M., Jain, S., and Dick, G. J. (2016). Genomic and transcriptomic resolution of organic matter utilization among Deep-Sea Bacteria in Guaymas Basin hydrothermal plumes. Front. Microbiol. 7:1125. doi: 10.3389/fmicb.2016.01125
Lillington, S. P., Leggieri, P. A., Heom, K. A., and O’Malley, M. A. (2020). Nature’s recyclers: anaerobic microbial communities drive crude biomass deconstruction. Curr. Opin. Biotechnol. 62, 38–47. doi: 10.1016/j.copbio.2019.08.015
Liu, J., Bai, Y., Liu, F., Kohn, R. A., Tadesse, D. A., Sarria, S., et al. (2022). Rumen microbial predictors for short-chain fatty acid levels and the grass-fed regimen in Angus cattle. Animals 12:2995. doi: 10.3390/ani12212995
Liu, T., Schnürer, A., Björkmalm, J., Willquist, K., and Kreuger, E. (2020). Diversity and abundance of microbial communities in UASB reactors during methane production from hydrolyzed wheat straw and Lucerne. Microorganisms 8:1394. doi: 10.3390/microorganisms8091394
López-Mondéjar, R., Tláskal, V., da Rocha, U. N., and Baldrian, P. (2022). Global distribution of carbohydrate utilization potential in the prokaryotic tree of life. mSystems 7:e0082922. doi: 10.1128/msystems.00829-22
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Mach, N., Midoux, C., Leclercq, S., Pennarun, S., Le Moyec, L., Rué, O., et al. (2022). Mining the equine gut metagenome: poorly-characterized taxa associated with cardiovascular fitness in endurance athletes. Commun. Biol. 5, 1–15. doi: 10.1038/s42003-022-03977-7
Malard, L. A., and Pearce, D. A. (2018). Microbial diversity and biogeography in Arctic soils. Environ. Microbiol. Rep. 10, 611–625. doi: 10.1111/1758-2229.12680
Mao, S., Zhang, R., Wang, D., and Zhu, W. (2012). The diversity of the fecal bacterial community and its relationship with the concentration of volatile fatty acids in the feces during subacute rumen acidosis in dairy cows. BMC Vet. Res. 8:237. doi: 10.1186/1746-6148-8-237
Martin, W. F., and Sousa, F. L. (2016). Early microbial evolution: the age of anaerobes. Cold Spring Harb. Perspect. Biol. 8:a018127. doi: 10.1101/cshperspect.a018127
Marynowska, M., Goux, X., Sillam-Dussès, D., Rouland-Lefèvre, C., Halder, R., Wilmes, P., et al. (2020). Compositional and functional characterisation of biomass-degrading microbial communities in guts of plant fibre-and soil-feeding higher termites. Microbiome 8:96. doi: 10.1186/s40168-020-00872-3
Marynowska, M., Sillam-Dussès, D., Untereiner, B., Klimek, D., Goux, X., Gawron, P., et al. (2023). A holobiont approach towards polysaccharide degradation by the highly compartmentalised gut system of the soil-feeding higher termite Labiotermes labralis. BMC Genomics 24:115. doi: 10.1186/s12864-023-09224-5
Medina-Silva, R., Oliveira, R. R., Trindade, F. J., Borges, L. G. A., Lopes Simão, T. L., Augustin, A. H., et al. (2018). Microbiota associated with tubes of Escarpia sp. from cold seeps in the southwestern Atlantic Ocean constitutes a community distinct from that of surrounding marine sediment and water. Antonie Van Leeuwenhoek 111, 533–550. doi: 10.1007/s10482-017-0975-7
Mikaelyan, A., Meuser, K., and Brune, A. (2017). Microenvironmental heterogeneity of gut compartments drives bacterial community structure in wood-and humus-feeding higher termites. FEMS Microbiol. Ecol. 93:fiw210. doi: 10.1093/femsec/fiw210
Mote, R. S., Hill, N. S., Skarlupka, J. H., Turner, Z. B., Sanders, Z. P., Jones, D. P., et al. (2019). Response of beef cattle fecal microbiota to grazing on toxic tall fescue. Appl. Environ. Microbiol. 85, e00032–e00019. doi: 10.1128/AEM.00032-19
Nguyen, L. N., Nguyen, A. Q., and Nghiem, L. D. (2019). “Microbial Community in Anaerobic Digestion System: progression in microbial ecology” in Water and wastewater treatment technologies. eds. X.-T. Bui, C. Chiemchaisri, T. Fujioka, and S. Varjani (Singapore: Springer), 331–355.
Nunes da Rocha, U., Van Overbeek, L., and Van Elsas, J. D. (2009). Exploration of hitherto-uncultured bacteria from the rhizosphere. FEMS Microbiol. Ecol. 69, 313–328. doi: 10.1111/j.1574-6941.2009.00702.x
Ohkuma, M., and Brune, A. (2011). “Diversity, structure, and evolution of the termite gut microbial community” in Biology of termites: A modern synthesis. eds. D. E. Bignell, Y. Roisin, and N. Lo (Dordrecht: Springer), 413–438.
Oksanen, J., Simpson, G. L., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., et al. (2001). Vegan: Community ecology package. doi: 10.32614/CRAN.package.vegan
Partanen, P., Hultman, J., Paulin, L., Auvinen, P., and Romantschuk, M. (2010). Bacterial diversity at different stages of the composting process. BMC Microbiol. 10:94. doi: 10.1186/1471-2180-10-94
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ravinath, R., Das, A. J., Usha, T., Ramesh, N., and Middha, S. K. (2022). Targeted metagenome sequencing reveals the abundance of Planctomycetes and Bacteroidetes in the rhizosphere of pomegranate. Arch. Microbiol. 204:481. doi: 10.1007/s00203-022-03100-8
Reichart, N. J., Bowers, R. M., Woyke, T., and Hatzenpichler, R. (2021). High potential for biomass-degrading enzymes revealed by hot Spring metagenomics. Front. Microbiol. 12:668238. doi: 10.3389/fmicb.2021.668238
Richards, C., Otani, S., Mikaelyan, A., and Poulsen, M. (2017). Pycnoscelus surinamensis cockroach gut microbiota respond consistently to a fungal diet without mirroring those of fungus-farming termites. PLoS One 12:e0185745. doi: 10.1371/journal.pone.0185745
Sabree, Z. L., and Moran, N. A. (2014). Host-specific assemblages typify gut microbial communities of related insect species. Springerplus 3:138. doi: 10.1186/2193-1801-3-138
Salgado, J. F. M., Hervé, V., Vera, M. A. G., Tokuda, G., and Brune, A. (2024). Unveiling lignocellulolytic potential: a genomic exploration of bacterial lineages within the termite gut. Microbiome 12:201. doi: 10.1186/s40168-024-01917-7
Schloss, P. D. (2021). Amplicon sequence variants artificially Split bacterial genomes into separate clusters. mSphere 6:e0019121. doi: 10.1128/msphere.00191-21
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sethupathy, S., Morales, G. M., Li, Y., Wang, Y., Jiang, J., Sun, J., et al. (2021). Harnessing microbial wealth for lignocellulose biomass valorization through secretomics: a review. Biotechnol. Biofuels 14:154. doi: 10.1186/s13068-021-02006-9
Sharma, P., Sharma, S., Maurya, R. K., De, T. D., Thomas, T., Lata, S., et al. (2014). Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit. Vectors 7:235. doi: 10.1186/1756-3305-7-235
Shen, J., Smith, A. C., Barnett, M. J., Morgan, A., and Wynn, P. M. (2022). Distinct microbial communities in the soils, waters, and speleothems of a Hyperalkaline cave system. Journal of geophysical research. Biogeosciences 127:e2022JG006866. doi: 10.1029/2022JG006866
Singh, N., Singh, V., Rai, S. N., Vamanu, E., and Singh, M. P. (2022). Metagenomic analysis of garden soil-derived microbial consortia and unveiling their metabolic potential in mitigating toxic hexavalent chromium. Life 12:2094. doi: 10.3390/life12122094
Slobodkina, G. B., Kovaleva, O. L., Miroshnichenko, M. L., Slobodkin, A. I., Kolganova, T. V., Novikov, A. A., et al. (2015). Thermogutta terrifontis gen. Nov., sp. nov. and Thermogutta hypogea sp. nov., thermophilic anaerobic representatives of the phylum Planctomycetes. Int. J. Syst. Evol. Microbiol. 65, 760–765. doi: 10.1099/ijs.0.000009
Slobodkina, G. B., Panteleeva, A. N., Beskorovaynaya, D. A., Bonch-Osmolovskaya, E. A., and Slobodkin, A. I. (2016). Thermostilla marina gen. Nov., sp. nov., a thermophilic, facultatively anaerobic planctomycete isolated from a shallow submarine hydrothermal vent. Int. J. Syst. Evol. Microbiol. 66, 633–638. doi: 10.1099/ijsem.0.000767
Song, H., Peng, L., Li, Z., Deng, X., Shao, J., and Gu, J. (2019). Metal distribution and biological diversity of crusts in paddy fields polluted with different levels of cadmium. Ecotoxicol. Environ. Saf. 184:109620. doi: 10.1016/j.ecoenv.2019.109620
Spring, S., Brinkmann, N., Murrja, M., Spröer, C., Reitner, J., and Klenk, H.-P. (2015). High diversity of Culturable prokaryotes in a lithifying hypersaline microbial mat. Geomicrobiol J. 32, 332–346. doi: 10.1080/01490451.2014.913095
Stewart, R. D., Auffret, M. D., Warr, A., Walker, A. W., Roehe, R., and Watson, M. (2019). Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961. doi: 10.1038/s41587-019-0202-3
Storesund, J. E., Lanzèn, A., Nordmann, E.-L., Armo, H. R., Lage, O. M., and Øvreås, L. (2020). Planctomycetes as a vital constituent of the microbial communities inhabiting different layers of the meromictic Lake Sælenvannet (Norway). Microorganisms 8:1150. doi: 10.3390/microorganisms8081150
Sun, C.-C., Zhao, W.-J., Yue, W.-Z., Cheng, H., Sun, F.-L., Wang, Y.-T., et al. (2023). Polymeric carbohydrates utilization separates microbiomes into niches: Insights into the diversity of microbial carbohydrate-active enzymes in the inner shelf of the Pearl River Estuary, China. Front. Microbiol. 14:1180321. doi: 10.3389/fmicb.2023.1180321
Suto, R., Ishimoto, C., Chikyu, M., Aihara, Y., Matsumoto, T., Uenishi, H., et al. (2017). Anammox biofilm in activated sludge swine wastewater treatment plants. Chemosphere 167, 300–307. doi: 10.1016/j.chemosphere.2016.09.121
Tran, N. T., Zhang, J., Xiong, F., Wang, G.-T., Li, W.-X., and Wu, S.-G. (2018). Altered gut microbiota associated with intestinal disease in grass carp (Ctenopharyngodon idellus). World J. Microbiol. Biotechnol. 34:71. doi: 10.1007/s11274-018-2447-2
Vanwonterghem, I., Jensen, P. D., Rabaey, K., and Tyson, G. W. (2016). Genome-centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion. Environ. Microbiol. 18, 3144–3158. doi: 10.1111/1462-2920.13382
Vargas-Albores, F., Porchas-Cornejo, M. A., Martínez-Porchas, M., Villalpando-Canchola, E., Gollas-Galván, T., and Martínez-Córdova, L. R. (2017). Bacterial biota of shrimp intestine is significantly modified by the use of a probiotic mixture: a high throughput sequencing approach. Helgol. Mar. Res. 71:5. doi: 10.1186/s10152-017-0485-z
Vipindas, P. V., Krishnan, K. P., Rehitha, T. V., Jabir, T., and Dinesh, S. L. (2020). Diversity of sediment associated Planctomycetes and its related phyla with special reference to anammox bacterial community in a high Arctic fjord. World J. Microbiol. Biotechnol. 36:107. doi: 10.1007/s11274-020-02886-3
Vitorino, I. R., and Lage, O. M. (2022). The Planctomycetia: an overview of the currently largest class within the phylum Planctomycetes. Antonie Van Leeuwenhoek 115, 169–201. doi: 10.1007/s10482-021-01699-0
Vollmers, J., Frentrup, M., Rast, P., Jogler, C., and Kaster, A.-K. (2017). Untangling genomes of novel Planctomycetal and Verrucomicrobial species from Monterey Bay kelp Forest metagenomes by refined binning. Front. Microbiol. 8:472. doi: 10.3389/fmicb.2017.00472
Walters, K. E., and Martiny, J. B. H. (2020). Alpha-, beta-, and gamma-diversity of bacteria varies across habitats. PLoS One 15:e0233872. doi: 10.1371/journal.pone.0233872
Wang, X., Cao, A., Zhao, G., Zhou, C., and Xu, R. (2017). Microbial community structure and diversity in a municipal solid waste landfill. Waste Manag. 66, 79–87. doi: 10.1016/j.wasman.2017.04.023
Wang, C., Dong, D., Wang, H., Müller, K., Qin, Y., Wang, H., et al. (2016). Metagenomic analysis of microbial consortia enriched from compost: new insights into the role of Actinobacteria in lignocellulose decomposition. Biotechnol. Biofuels 9:22. doi: 10.1186/s13068-016-0440-2
Wang, Y., Fu, Y., He, Y., Kulyar, M. F.-A., Iqbal, M., Li, K., et al. (2021). Longitudinal characterization of the gut bacterial and fungal communities in yaks. J. Fungi 7:559. doi: 10.3390/jof7070559
Wiegand, S., Jogler, M., Boedeker, C., Heuer, A., Peeters, S. H., Kallscheuer, N., et al. (2020). Updates to the recently introduced family Lacipirellulaceae in the phylum Planctomycetes: isolation of strains belonging to the novel genera Aeoliella, Botrimarina, Pirellulimonas and Pseudobythopirellula and the novel species Bythopirellula polymerisocia and Posidoniimonas corsicana. Antonie Van Leeuwenhoek. 113, 1979–1997. doi: 10.1007/s10482-020-01486-3
Weimer, P. J. (2022). Degradation of cellulose and hemicellulose by ruminal microorganisms. Microorganisms 10:2345. doi: 10.3390/microorganisms10122345
Xiao, H., Yu, H., Wang, J., He, L., Wang, Z., Fu, Y., et al. (2025). Reforestation practices have varied the resilience of nosZ-type denitrifier communities: a 40-year soil chronosequence study. Appl. Soil Ecol. 206:105877. doi: 10.1016/j.apsoil.2025.105877
Yang, G. L., Hou, S. G., Le Baoge, R., Li, Z. G., Xu, H., Liu, Y. P., et al. (2016). Differences in bacterial diversity and communities between glacial snow and glacial soil on the Chongce ice cap, West Kunlun Mountains. Sci. Rep. 6:36548. doi: 10.1038/srep36548
Yildirim, S., Yeoman, C. J., Sipos, M., Torralba, M., Wilson, B. A., Goldberg, T. L., et al. (2010). Characterization of the fecal microbiome from non-human wild Primates reveals species specific microbial communities. PLoS One 5:e13963. doi: 10.1371/journal.pone.0013963
Youssef, N., Steidley, B. L., and Elshahed, M. S. (2012). Novel high-rank phylogenetic lineages within a sulfur Spring (Zodletone Spring, Oklahoma), revealed using a combined pyrosequencing-sanger approach. Appl. Environ. Microbiol. 78, 2677–2688. doi: 10.1128/AEM.00002-12
Keywords: Planctomycetota, planctomycetes diversity, taxonomic profiling, 16S rRNA gene sequence analysis, microbial community structure, biomass-rich environments, anaerobic digester, termite gut
Citation: Klimek D, Lage OM and Calusinska M (2025) Phylogenetic diversity and community structure of Planctomycetota from plant biomass-rich environments. Front. Microbiol. 16:1579219. doi: 10.3389/fmicb.2025.1579219
Edited by:
Mariusz Cycoń, Medical University of Silesia, PolandReviewed by:
Mudasir A. Dar, Jiangsu University, ChinaSławomir Sułowicz, University of Silesia in Katowice, Poland
Copyright © 2025 Klimek, Lage and Calusinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominika Klimek, ZG9taW5pa2Eua2xpbWVrQGxpc3QubHU=; Magdalena Calusinska, bWFnZGFsZW5hLmNhbHVzaW5za2FAbGlzdC5sdQ==