 Yizi Yang1†
Yizi Yang1† Yulin Zhang
Yulin Zhang Xiao-Hua Zhang
Xiao-Hua Zhang- 1Frontiers Science Center for Deep Ocean Multispheres and Earth System, and College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Laboratory for Marine Ecology and Environmental Science, Qingdao Marine Science and Technology Center, Qingdao, China
- 3Key Laboratory of Evolution and Marine Biodiversity (Ministry of Education), and Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao, China
The Challenger Deep of the Mariana Trench, which is the deepest site in the ocean, contains rich deposits of n-alkanes in its sediments. However, the alkane metabolic processes of the bacteria in this extreme environment were not well understood. In this study, we isolated a strain Mixta calida HXX308 (Proteobacteria) from sediment samples of the Challenger Deep (10,816 meters below sea level). HXX308 grows under pressures ranging from 0 to 40 MPa, with optimal growth at lower pressures. Additionally, it degrades approximately 20% of eicosane at both atmospheric pressure (0.1 MPa) and 20 MPa. Metabolic profiling indicated that HXX308 possesses a complete aerobic alkane metabolism pathway, along with nitrate reduction and sulfate reduction pathways, which support its adaptation to the trench’s anoxic environment. Comparative genomic studies showed that most strains in the genus Mixta contain the alkane-degrading gene LadB. Characterization of the LadB gene in HXX308 confirmed its role in the degradation of medium-to long-chain alkanes (C18-36). HXX308 is the first Mixta strain isolated from marine environment. Although this strain originated from the trench, its hydrocarbon metabolic characteristics are similar to those of cultures of terrestrial origin, suggesting that the alkanes in these sediments are likely from the terrestrial environment. Our study enhances the understanding of alkane-degrading in the phylum Proteobacteria and provides insights into the environmental adaptation of M. calida HXX308 in the Mariana Trench.
1 Introduction
The hadal zone (6,000–11,000 meters below sea level, mbsl), which is the deepest part of the ocean comprises trenches and submarine canyons, and accounts for approximately 45% of the total vertical depth of the ocean’s water mass (Jamieson et al., 2010). It is characterized by extreme environmental conditions, including complete darkness, high pressure (60 ~ 110 MPa), low temperature (1.0 ~ 2.5°C), low dissolved oxygen (156 ~ 172 μM), and relatively isolated topography, creating a unique and diverse biological community (Blankenship-Williams and Levin, 2009; Nunoura et al., 2015; Liu et al., 2023). Compared to the surrounding abyssal plains, the sedimentation in the hadal zone (>6,000 mbsl) is enhanced, which is attributed to seismically driven mass-wasting events (such as collapses and mudflows) along the slopes, and the funneling effect promoted by the “V-shaped” topography of the trench (Bao et al., 2018). Episodic downslope mass-transport events disrupt the continuous sedimentation at the deepest parts of the trench, introducing non-indigenous microorganisms that lead to the accumulation of organic carbon and alterations in redox conditions (Liu et al., 2024). These processes play a significant role in shaping the subseafloor community composition, and the erratic abundance and activity profiles with depth (Liu et al., 2024; Nunoura et al., 2015; Ichino et al., 2015).
These complex geological events, coupled with the unique environment of the trench, shaped unique microbial communities and their biogeochemical processes. It has been reported that sediments in various trenches are characterized by high organic matter content, abundant and active microbial communities, and rapid microbial carbon turnover, positioning hadal trenches as key sites for studying the degradation of deep-sea organic carbon (Wenzhöfer et al., 2016; Luo et al., 2018). Trench sediments are rich in hydrocarbons, with the average concentration of alkanes in surface sediments at a depth of approximately 10,900 m in the Mariana Trench being 2.3 μg/g dry weight, which supports the growth of hydrocarbon-degrading bacteria (Glud et al., 2013; Liu et al., 2019). Investigations have revealed that in the sediments at approximately 11,000 m of the Mariana Trench, the predominant aliphatic hydrocarbon-degrading genera including Alcanivorax, Thalassolituus, and Oleibacter (Liu et al., 2019). Additionally, the species Venatorbacter sp. C2-1 can grow with n-alkanes as the sole carbon source under in situ temperature (4°C) and pressure (58 MPa), demonstrating the adaptability of these bacteria to the extreme conditions of the hadal zone (Wang et al., 2023; Liu et al., 2023). These alkane-degrading bacteria in the Mariana Trench play multifaceted crucial roles in their habitat. Firstly, they are key participants in the carbon cycle, capable of converting complex organic carbon (like lignin, cellulose, and chitin) into simple inorganic carbon, thereby facilitating the carbon cycling within the marine ecosystem (Zhao et al., 2020; Tian et al., 2018). These bacteria reduce the accumulation of organic carbon in the sediments by degrading alkanes, maintaining the stability of the sedimentary environment. Moreover, their metabolic activities produce substances beneficial to other microorganisms, such as organic acids and amino acids, promoting the diversity and stability of microbial communities and sustaining the ecological balance of the hadal ecosystem (Wang et al., 2023). Additionally, during the process of alkane degradation, these bacteria actively participate in the nitrogen cycle, thereby influencing nitrogen transformations and cycling in the sediments. This participation is of significant importance for maintaining the balance of nitrogen cycling in the hadal environment (Yang et al., 2024; Huang et al., 2024). However, despite the progress made in research, the degradation potential and mechanisms of microorganisms in hadal sediments remain inadequately understood. Further in-depth studies of the functions and interactions of these microorganisms will aid in a better comprehension of the biogeochemical processes in trench ecosystems, as well as the adaptation and survival strategies of microorganisms in extreme environments (Arora and Panosyan, 2019).
In the sediments at the bottom of the Mariana Trench, Proteobacteria were identified as the dominant bacterial group, regardless of the method used, including pure culture techniques (Peoples et al., 2019), 16S rRNA gene amplicon sequencing (Cui et al., 2019) or metagenomic analyses (Chen et al., 2021; Liu et al., 2024). The genus Mixta belongs to the class Gammaproteobacteria, order Enterobacterales, and family Erwiniaceae. Species within this genus are characterized as facultatively anaerobic, motile, Gram-negative rods (Palmer et al., 2018). Till now, 14 species of genus Mixta were validly published,1 which were isolated from various environments, including plants, insects, food and the human body (Xia et al., 2020). The type species of genus Mixta, M. calida (type strain 1400/07T = LMG 25383T = DSM 22759T), was isolated from infant formula (Popp et al., 2010). Currently, research on various species of Mixta are focused primarily on their role as symbiotic bacteria in insects for plant disease control (Guz et al., 2020), and issues related to pathogenicity and food safety (Parra-Flores et al., 2022; Casale et al., 2023). However, the ecological functionality and importance of Mixta in natural environments remain significantly underestimated.
Total of 93 candidate hydrocarbon-degrading Proteobacteria strains were isolated from deep-sea trench sediments using a mixture of alkanes and diesel oil as the sole carbon source in our lab before. Among them, M. calida HXX308 is a representative strain of the genus Mixta, which was isolated from a marine environment for the first time and has not been previously reported to possess hydrocarbon-degrading capabilities. To explore the metabolic characteristics of strain HXX308, including its hydrocarbon degradation mechanisms and adaptation strategies to high-pressure environments, this study further investigated its metabolic network and hadal environment adaptation mechanisms based on high-pressure cultivation experiments, comparative genomic analysis, and heterologous characterization of the key hydrocarbon oxidation gene LadB. Our research provides a new perspective on how bacteria participate in the degradation of hydrocarbons to drive the material metabolic cycle in hadal environments.
2 Materials and methods
2.1 Sampling
A sediment core sample MT20-750 was collected from the Mariana Trench (11°19.904′N, 142°12.083′E) at depth of 10,816 m by the research vessel “Dongfanghong 3” in July 2020, as described in Liu et al. (2024). The sediment core, approximately 750 cm in length, is currently the deepest and longest sediment sample retrieved from the Challenger Deep. Utilizing mixed alkanes and petroleum diesel as the sole carbon sources, 93 cultures of potential hydrocarbon-degrading bacteria were isolated, belonging to 17 genera, predominantly from the class Gammaproteobacteria, followed by Alphaproteobacteria (Supplementary Figure S1). One representative strain from each of the 17 genera (Supplementary Figure S1) was selected to determine alkane degradation rates, with Alcanivorax venustensis ZYF844 used as the positive control (Supplementary Figure S2). The results showed that although M. calida HXX308 does not have as high degradation activity as some obligate alkane-degrading bacteria, it does exhibit hydrocarbon-degrading ability. Moreover, this strain is the first reported marine-derived strain in its genus, while other strains are terrestrial origin. Therefore, this study systematically investigates its metabolic characteristics and environmental adaptation mechanisms.
2.2 Genome sequencing and assembly
Mixta calida HXX308 was cultured in marine 2216E liquid medium at 28°C, 170 rpm until it reached the logarithmic phase (OD₆₀₀nm = 0.4 ~ 0.6), which took ~18 h. Cells were harvested by centrifugation at 12,000 rpm for 10 min at 4°C, then washed three times with 0.85% (w/v) saline to remove residual medium and growth metabolites (Liu et al., 2019). The cells were snap-frozen in liquid nitrogen, and sent to Guangdong Magigene Biotechnology Co., Ltd. (Magigene) for whole genome sequencing by using the PacBio Sequel IIe platform. Genomic DNA extraction and purified was performed using a proprietary method by Magigene. The integrity and purity of the DNA were assessed using 1% agarose gel electrophoresis. DNA concentration and purity were measured using Qubit 4.0 (Thermo Fisher Scientific, Waltham, USA) and NanoDrop One (Thermo Fisher Scientific, Waltham, USA).
After qualification, the genomic DNA was fragmented and end-repaired using G-tubes to prepare the SMRTbell DNA template library according to the manufacturer’s instructions. The library quality was evaluated using Qubit 4.0 (Thermo Fisher Scientific, Waltham, USA), and the average fragment size was assessed using Agilent 4200 (Agilent, Santa Clara, CA, USA). SMRT sequencing was performed on the Pacific Biosciences Sequel IIe sequencer (PacBio, Menlo Park, USA) following the standard protocol. The sequencing coverage depth was approximately 360X to ensure comprehensive genome coverage.
Prodigal v2.6.3 (Hyatt et al., 2010) was employed for coding gene prediction. The cleaning and assembly process of the raw Whole Genome Sequencing (WGS) sequences involved several steps. Initially, raw sequencing data were filtered to remove low-quality reads, adapter sequences, and reads with a high proportion of ambiguous bases (N’s). Subsequently, the cleaned reads were assembled using the Canu assembler v2.2 (Koren et al., 2017), which is designed for long-read sequencing data and is capable of producing high-quality genome assemblies. The assembly was further polished using the Arrow software to correct errors and improve the accuracy of the assembled genome.
Gene function annotations were performed by conducting whole-genome Blast searches against the following databases: NR (Non-Redundant Protein Database), Swiss-Prot, GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), and COG (Clusters of Orthologous Groups). Secretory protein predictions were made using SignalP, LipoP, TMHMM, and PSORTb. Based on the assembly results and the predicted coding genes, along with analyses of non-coding RNA, gene functional annotations were visualized using Circos software (Krzywinski et al., 2009) to generate a whole genome map.
2.3 Phylogenetic analysis, gene annotation and metabolic pathway prediction
Twenty-one reference genomes closely related to M. calida HXX308 were obtained from the NCBI database (Sharma, 2018). The 16S rRNA gene of HXX308 were aligned with those of downloaded strains belonging to the genus Mixta. Multiple sequence alignment and trimming were performed using Muscle in MAGE 11 (Tamura et al., 2021), and a phylogenetic tree was constructed using the maximum likelihood (ML) method, with confidence values represented by bootstrap values based on 1,000 iterations.
To further determine the taxonomic position of these strains, the sequences of single-copy core genes from the whole genomes were extracted and used to develop phylogenetic trees using ML method. Specifically, Prokka v1.4.6 (Seemann, 2014) was used to functional annotate the 22 genomes, followed by the identification of homologous gene families in all genomes using Orthofinder v2.5.5 (Emms and Kelly, 2019). After obtaining the single-copy gene sequences shared by all species, multiple sequence alignment and trimming were performed using MAFFT and trimAl (Katoh and Standley, 2013; Capella-Gutiérrez et al., 2009). The core genes were concatenated, and the phylogenetic tree was constructed using IQ-TREE software (Nguyen et al., 2015), with confidence values indicated by bootstrap values based on 1,000 iterations. Finally, the phylogenetic tree was polished using the online tool Chiplot (Xie et al., 2023).
Following the method of Khot et al. (2021) hydrocarbon-degrading genes were predicted by aligning the annotated protein files with a concatenated HMM sequence file, defining sequences with an e-value < 10−50 as potential hydrocarbon-degrading proteins (Khot et al., 2021). Functional annotation of carbohydrate-active enzymes (CAZymes) was performed using the dbCAN module integrated with the CAZy database (E-value <10−30) (Lombard et al., 2013). This approach systematically identified and classified CAZyme families based on Hidden Markov Models (HMMs) to ensure accurate functional assignment. Annotation of protease families was conducted using the MEROPS database (E-value <10−5) (Rawlings et al., 2018), which provides comprehensive classification and functional information for peptidases. Additionally, the genomes were uploaded to the KEGG database2 for classification and annotation of metabolic pathways (Kanehisa et al., 2016), and a schematic of the metabolic pathways was created using Adobe Illustrator.
2.4 Bacterial cultivation and n-alkane degradation
To assess the n-alkanes degradation ability of M. calida HXX308 under high pressure conditions, ORN7a inorganic salt medium was prepared (details in Supplementary Table S1), distributed in air-tight syringes, and inoculated with freshly grown HXX308. Each syringe (ultraviolet disinfection for 30 min before use) was supplemented with 5 mL ORN7a medium with 1 mg of n-eicosane as the sole carbon source. These disposable plastic syringes were then placed in a stainless steel high-pressure vessel (380 mL, maximum pressure 60 MPa; Nantong Feiyu Oil Science and Technology Exploitation, China). Pressure was applied using water with a manual pump. Cultivation was conducted at room temperature for 30 days under four pressure conditions: 0, 20, 40 and 60 MPa.
Once the incubation was terminated, residual n-alkanes were extracted from the culture via ultrasonic extraction for three times with dichloromethane (DCM). The n-alkane content was analyzed using an Agilent 8860GC System detector, with an HP-5 chromatographic column (30 m × 320 μm × 0.25 μm) and helium as the carrier gas at a flow rate of 1.9775 mL/min. The temperature program was set with an initial temperature of 60°C, held for 1 min, and then increased to 280°C at a rate of 5°C/min, maintained for 25 min. A Flame Ionization Detector (FID) detector was used. A 10 μL syringe was used for injection, with 2 μL injected each time. n-Alkanes with different chain lengths present in the samples were determined by comparing their retention times with standards (Sigma), which included n-alkanes with chain lengths ranging from C8 to C38. The retention times of these standards were used to identify the specific n-alkanes in the samples. Concentrations were calculated based on their chromatographic responses relative to the internal standard deuterated n-tetracosane (D-24; Sigma), which was added to each sample to correct for any variations in sample injection volume and instrument response. All glassware used in this process was heated at 450°C for 6 h to remove organic contaminants.
To investigate the growth of M. calida HXX308 after cultivation under high pressure, a high-pressure point plate experiment was conducted. Before and after high-pressure cultivation, bacterial suspensions were serially diluted, and 10 μL amounts were spotted onto marine 2216E agar plates. These were incubated in a constant temperature chamber at 28°C for 24 h, and bacterial growth was subsequently observed.
2.5 Construction of the recombinant expression plasmid of the alkane degrading gene
The n-alkane oxidation gene LadB was cloned from M. calida HXX308, and the recombinant expression plasmid pRSET-A-LadB (Supplementary Figure S3) was constructed following the method of Wang et al. (2018). Briefly, the LadB gene sequence KGOCCCKL_01363, along with the two required electron transport proteins KGOCCCKL_03163 (Flavodoxin/ferredoxin-NADP reductase) and KGOCCCKL_04039 (2Fe-2S ferredoxin), were inserted into the expression plasmid pRSET-A (Bomaide Biological, Beijing). Each gene, including LadB, and two electron transport proteins, was equipped with a promoter and ribosome binding site (RBS) in its upstream region. The recombinant plasmid used specific primers for the LadB gene: LadB-F: 5′-ATGGCATTATCC GTATTC-3′; LadB-R: 5′-GCTTTGCGAGACTTTCTG-3′. The amplification products were analyzed by running agarose gel electrophoresis, and the presence of clear bands indicated successful transformation. The recombinant plasmid, verified for successful construction, was transferred into 100 μL of competent Escherichia coli BL21 cells using the heat shock method (42°C for 90 s) (Bomaide Biological, Beijing) (Froger and Hall, 2007). The transformed competent cells were then plated on LB agar plates (Haibo Biotech, Qingdao) containing ampicillin (50 μg/mL), and incubated at 37°C for ~24 h until colonies appeared. Single colonies were then transferred to 5 mL of fresh LB liquid medium supplemented with ampicillin (100 μg/mL) to prepare seed cultures for later use.
2.6 Verification of alkane degradation capability in recombinant strains
To verify the n-alkane degradation ability of the recombinant strain E. coli BL21-pRSET-A-LadB, 2 mL of overnight culture were inoculated into 150 mL of antibiotic-containing LB liquid medium, and incubated at 37°C with shaking at 170 rpm until the OD600nm reached 0.5–0.6, which took ~3 h. Then, isopropyl-β-D-1-thiogalactopyranoside (IPTG) with the final concentration of 1 mM was added to the culture, with incubation for an additional 2 h to induce plasmid expression. Alkane degradation activity was then assessed. The induced expression system was centrifuged at 6000 rpm for 10 min at 4°C, and the supernatant was discarded. The retained cells were washed with M9 medium containing glucose and antibiotics (M9-glucose), and then resuspended in 90 mL of fresh M9-glucose medium with 90 μL of 50 mg/mL ampicillin. One milliliter of the resuspended cells was added to 10 mL of mixed alkanes (C18 and C19, 2.5 mg of each alkane per 10 mL) in ORN7a medium, and the mixture was incubated at 37°C with shaking at 170 rpm for 72 h. Additionally, two control groups included E. coli BL21-pRSET-A and E. coli BL21-pRSET-A-Flavodoxin-2Fe-2S ferredoxin, with un-inoculated blank medium as a negative control. Each group consisted of three replicates. Finally, the residual n-alkane concentration in the culture was extracted to determine the alkane degradation ability of the recombinant strain E. coli BL21-pRSET-A-LadB.
3 Results
3.1 The phylogenetic characteristics of Mixta calida HXX308
The previous research in our laboratory isolated 93 potential hydrocarbon-degrading Proteobacteria from the deep-sea sediments using mixed alkanes and petroleum diesel as the sole carbon source. Among them, Cereibacter and Mixta have not been reported to have hydrocarbon-degrading capabilities. In this study, one representative strain was selected from each genus for alkane degradation capability testing (Supplementary Figure S1). It was found that different genera have different degradation preferences for chain length. Specifically, Acinetobacter lwoffi HXX052, Erythrobacter nanhaiensis HXX086, Alcanivorax dieselolei HXX443, Tritonibacter mobilis HXX101, and Advenella kashmirensis HXX265 showed weakened degradation capabilities with increasing alkane chain length. In contrast, other genera, including M. calida HXX308, exhibited stronger degradation capabilities with longer chain lengths. Moreover, this genus has not been reported to have hydrocarbon-degrading capabilities, so this paper conducts an in-depth exploration of its metabolic characteristics and environmental adaptation strategies. To clarify the evolutionary status of M. calida HXX308, we constructed 16S rRNA and core genome phylogenetic trees using sequences from M. calida HXX308 and 21 closely related strains. The 16S rRNA phylogenetic analysis revealed that M. calida HXX308 clustered with four species within the genus Mixta, namely M. calida 22759 to which it is most closely related, M. intestinalis, M. tenebrioni, and M. gaviniae (Figure 1A). The core genome phylogenetic tree, based on single-copy orthologous genes, yielded results that are essentially congruent with those of the 16S rRNA tree, further confirming that M. calida 22759 is the most closely related group to M. calida HXX308 (Figure 1B). These phylogenetic analyses provide a robust framework for understanding the evolutionary relationships within the Mixta clade, and highlight the taxonomic position of M. calida HXX308 within the phylum Proteobacteria, class Gammaproteobacteria, order Enterobacterales, and family Erwiniaceae.
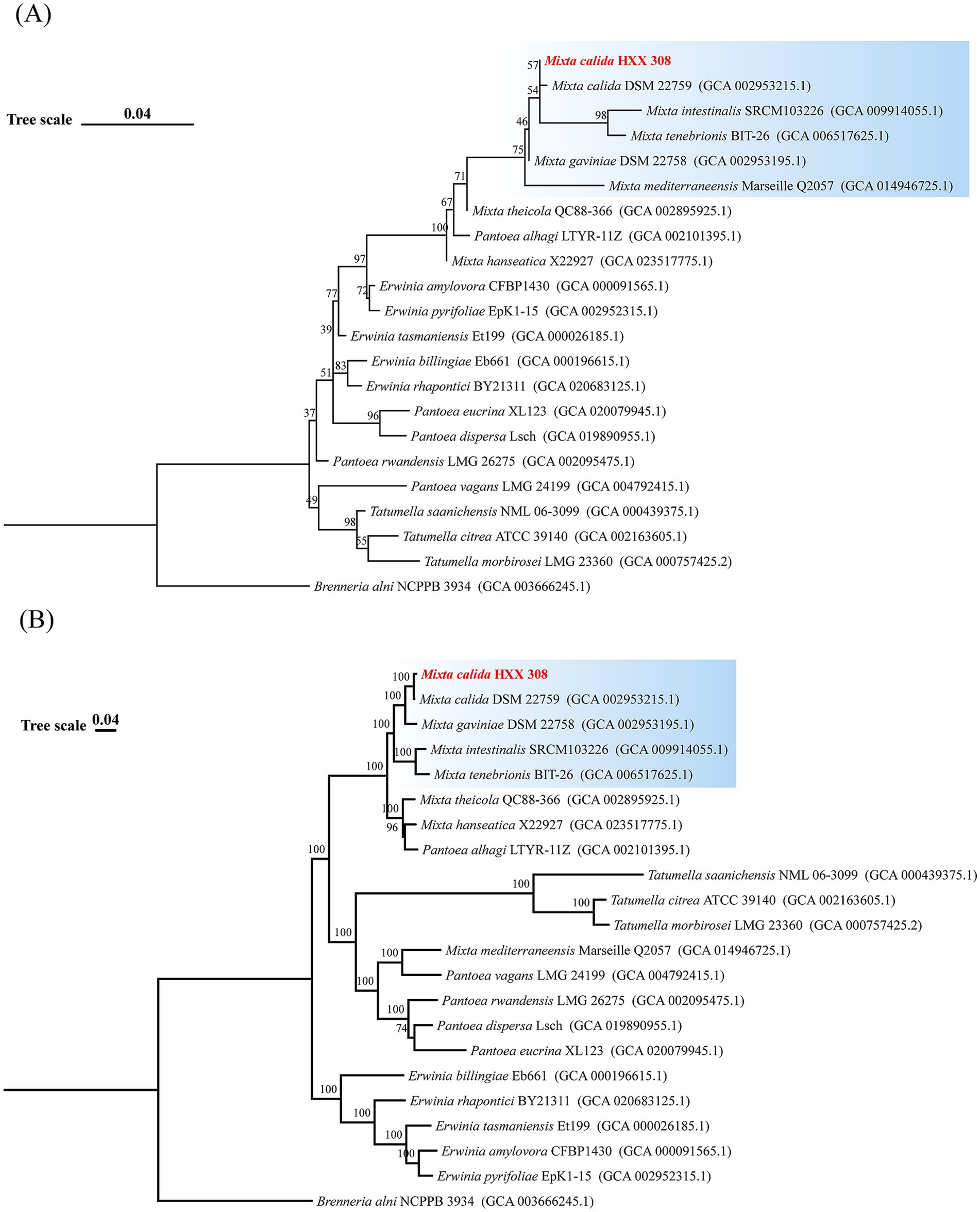
Figure 1. Phylogenetic tree of M. calida HXX308 and its related strains. (A) Phylogenetic tree constructed based on the 16S rRNA gene sequences of 22 strains. (B) Phylogenetic tree constructed based on the sequences of 1,226 single-copy core genes. The values at the nodes represent the bootstrap values based on 1,000 replicates. The scale bar indicates the substitution frequency per site.
3.2 Genomic characteristics of Mixta calida HXX308 and its reference strains
The circular genomic map of M. calida HXX308 (BioSample Accession: SAMN46867115) shows that it consists of a single chromosome containing 4,218,089 bp base pairs (Supplementary Tables S2–S4), with a GC content of 59.6% (Supplementary Figure S4). This genome includes 86 tRNA genes, eight 5S rRNA genes, seven 16S rRNA genes and seven 23S rRNA genes. Comparing the genomic characteristics of M. calida HXX308 with its reference strains (Table 1), it is evident that the HXX308 genome closely resembles that of the type strain of this genus (M. calida DSM22759), namely regarding genome size and GC content. However, the number of genes and encoded proteins in HXX308 are higher than those in M. calida DSM22759. Compared with other strains within the genus Mixta, HXX308 exhibits a smaller genome size (4.2 Mbp) and a higher GC percentage (56.9%) (Table 1). For example, the type strain M. calida DSM22759 has a genome size of 4.3 Mbp and a GC content of 57.0%. However, M. calida HXX308 was isolated from a marine environment, whereas other strains of Mixta were isolated from terrestrial habitats, including plants, insects, and food sources.
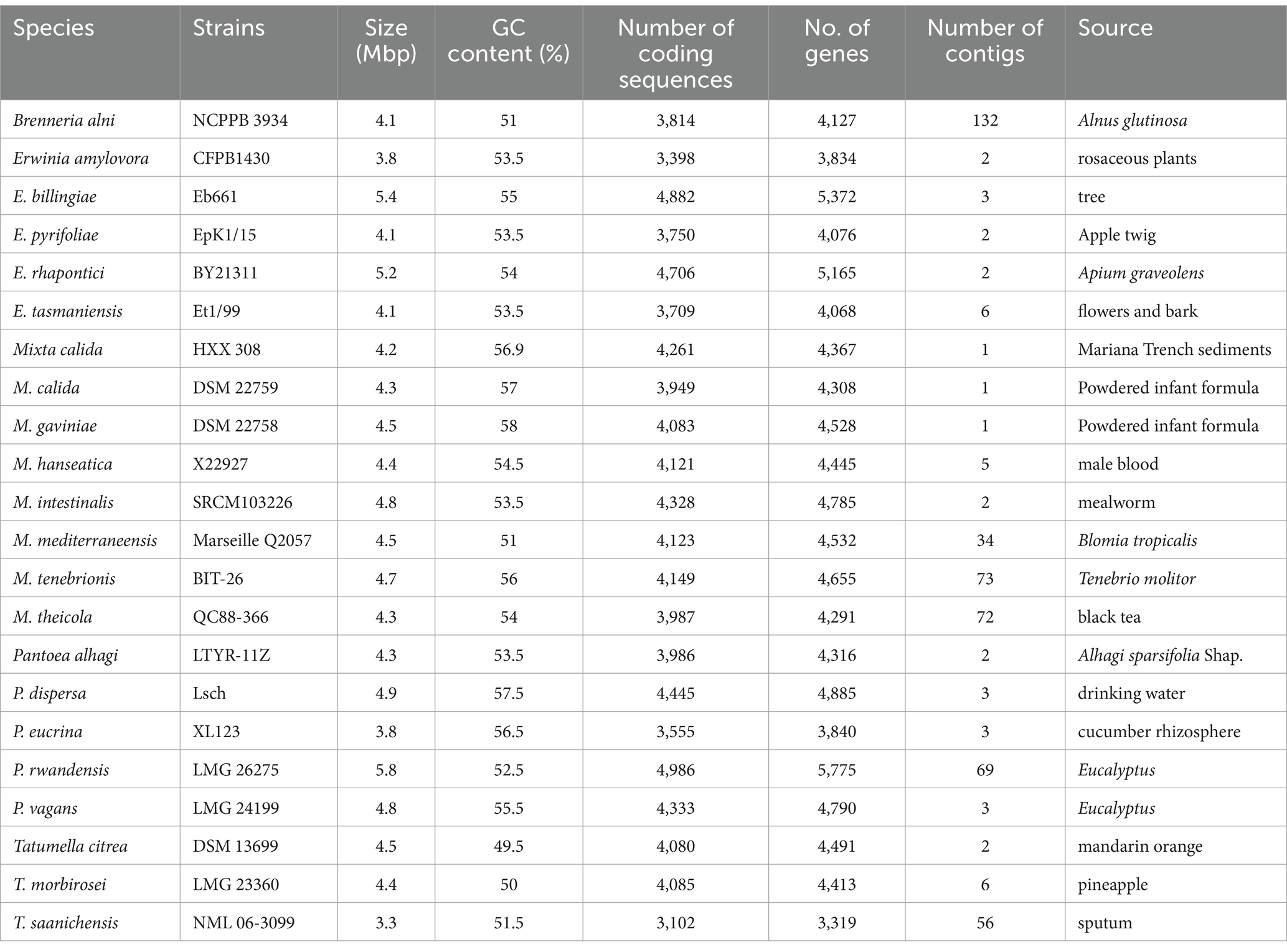
Table 1. The genomic characteristics of M. calida HXX308 with reference strains.
3.3 Metabolic network reconstruction of Mixta calida HXX308
A comparison of alkane degradation related genes between HXX308 and reference strains revealed that the former encodes aerobic long-chain alkane degradation gene LadB, which is capable of degrading C15-36 n-alkanes. Additionally, it encoded genes, specifically AhyA or AssA, for anaerobic alkane degradation, and the genes, including AbcA or K27540, for anaerobic aromatic hydrocarbon-degrading (Figure 2). Our analysis indicates that HXX308 contains genes involved in aerobic alkane oxidation (LadB), anaerobic alkane oxidation (AhyA and AssA), and anaerobic aromatic hydrocarbon oxidation (AbcA and K27540). However, it is essential to clarify that while we have focused on key genes known to participate in these pathways, our analysis does not cover all possible genes that may also play a role. Therefore, we should avoid claiming that the pathway is ‘relatively complete’ without additional genetic evidence.
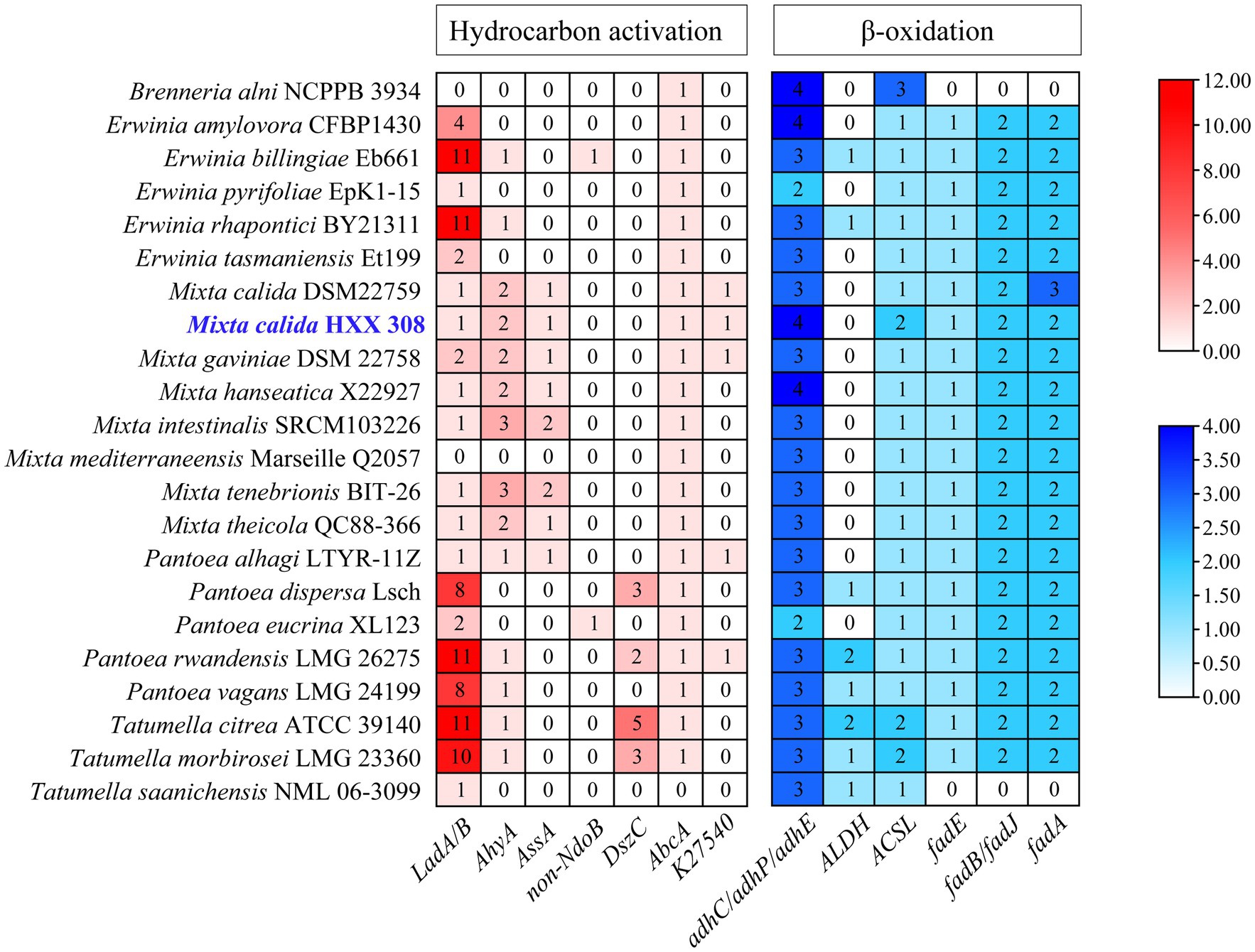
Figure 2. The hydrocarbon-degrading genes encoded by M. calida HXX308 and its reference strains. The depth of the legend color represents the gene copy number.
Instead, we can state that HXX308 has the genetic potential to engage in both aerobic and anaerobic alkane oxidation pathways, as well as in anaerobic aromatic hydrocarbon oxidation. This suggests a versatile metabolic capability. To ascertain whether HXX308 shares similar hydrocarbon-degrading genes with other strains, we conducted a comparative analysis using KEGG and CANT-HYD annotations, which involved comparing the presence and quantity of specific genes. Comparative analysis indicated that HXX308 shares identical hydrocarbon-degrading genes and β-oxidation genes with the model strain M. calida DSM 22759. In terms of aerobic alkane degradation, both HXX308 and the reference strains encode only the aerobic alkane degradation gene LadB. It is predicted that these strains can solely degrade long-chain alkanes under aerobic conditions.
To further investigate the metabolic potential of these strains, particularly regarding carbohydrate degradation or synthesis and macromolecular proteins utilization, the CAZy and MEROPS databases were used to annotate CAZymes and peptidase families of these genomes. For carbohydrate metabolism, Mixta strains exhibited similar CAZymes copy numbers, with glycoside hydrolases (GHs) being the most abundant, followed by glycosyl transferases (GT), and the lowest copy numbers of carbohydrate-binding modules (CBM) and polysaccharide lyases (PL) (Figure 3A). M. calida HXX308 encodes 99 carbohydrate-active enzyme genes, including 47 GHs, 37 GTs, 5 CEs, 4 AAs, 3 CBMs, and 3 PLs (Figure 3A and Supplementary Table S6). This is similar to the type strain M. calida DSM 22759, indicating the similar potential of carbohydrate utilization. We annotated peptidases against the MEROPS database (Figure 3B and Supplementary Table S7) and, by comparing the number of peptidase families among different genera, found that the genus Mixta possesses a greater number of serine peptidases (S) and unclassified peptidases (U). For all genomes examined, the dominant peptidase families were serine proteases (S) and metalloproteases (M), with no representation of aspartic proteases (P) or glutamic acid proteases (G). Detailed analysis of metalloproteases and serine proteases across the genomes (Figures 4A,B and Supplementary Table S5) revealed that S09 (prolyl oligopeptidase) and M20 (glutamate carboxypeptidase) were the predominant families. In the genome of M. calida HXX308, the abundance of peptidases in family M20 was lower than that in other strains within the genus Mixta, whereas S11 (D-Ala-D-Ala carboxypeptidase A) was more abundant. Additionally, HXX308 possessed enzymes M1 (aminopeptidase N) and M16 (pitrilysin), which were absent in other reference strains.
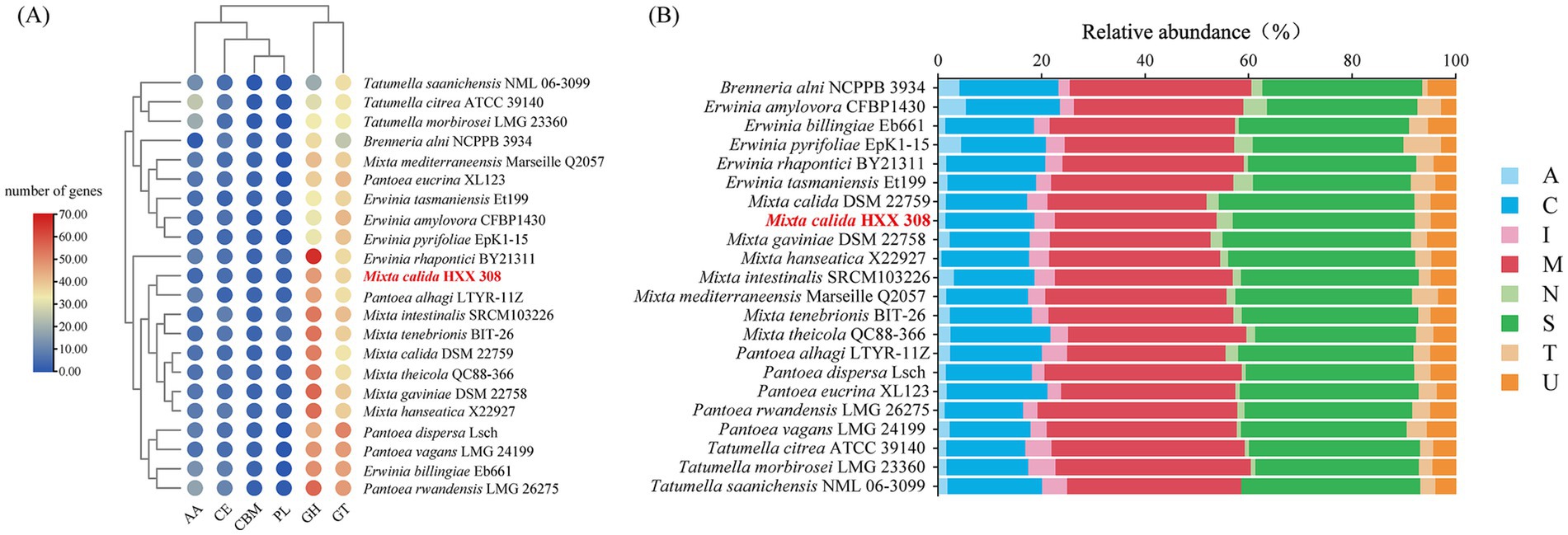
Figure 3. Comparative analysis of enzymes involved in carbohydrate metabolism and macromolecular protein utilization in M. calida HXX 308 and its reference strains. (A) Composition of carbohydrate-active enzymes in strain HXX 308 and its reference strains; (B) Functional categories of peptidases in strain HXX 308 and its reference strains.

Figure 4. The absolute abundance of metalloprotease (A) and serine protease (B) encoded genes in the genomes of M. calida HXX 308 and its reference strains.
To further elucidate the metabolic characteristics of HXX308, its metabolic pathway was reconstructed (Figure 5 and Supplementary Table S8), revealing the ability to utilize various carbon sources, including carbohydrates (fructose, sucrose, and glycerate), amino acids, oligopeptides, and phospholipids. This strain possesses a complete alkane metabolic pathway, enabling the entire process from alkane oxidation to acetyl-CoA, which is integrated into the TCA cycle. Additionally, HXX308 encodes 11 copies of the methyl-accepting chemotaxis protein (MCP), which aids in the bacterial perception of alkanes in the surrounding environment (Liu et al., 2015). MCP facilitates substrate degradation by attracting cells, whereas the transport of long-chain alkanes from the extracellular to the intracellular environment relies on outer membrane transport proteins, FadL (Zhou et al., 2017). Moreover, HXX308 possesses key genes that encode for assimilatory sulfate reduction and dissimilatory nitrate reduction, including nitrate reductase (narGHI) and nitrite reductase (nirBD). These pathways indicate the potential of HXX308 to utilize sulfate and nitrate found in sediments.
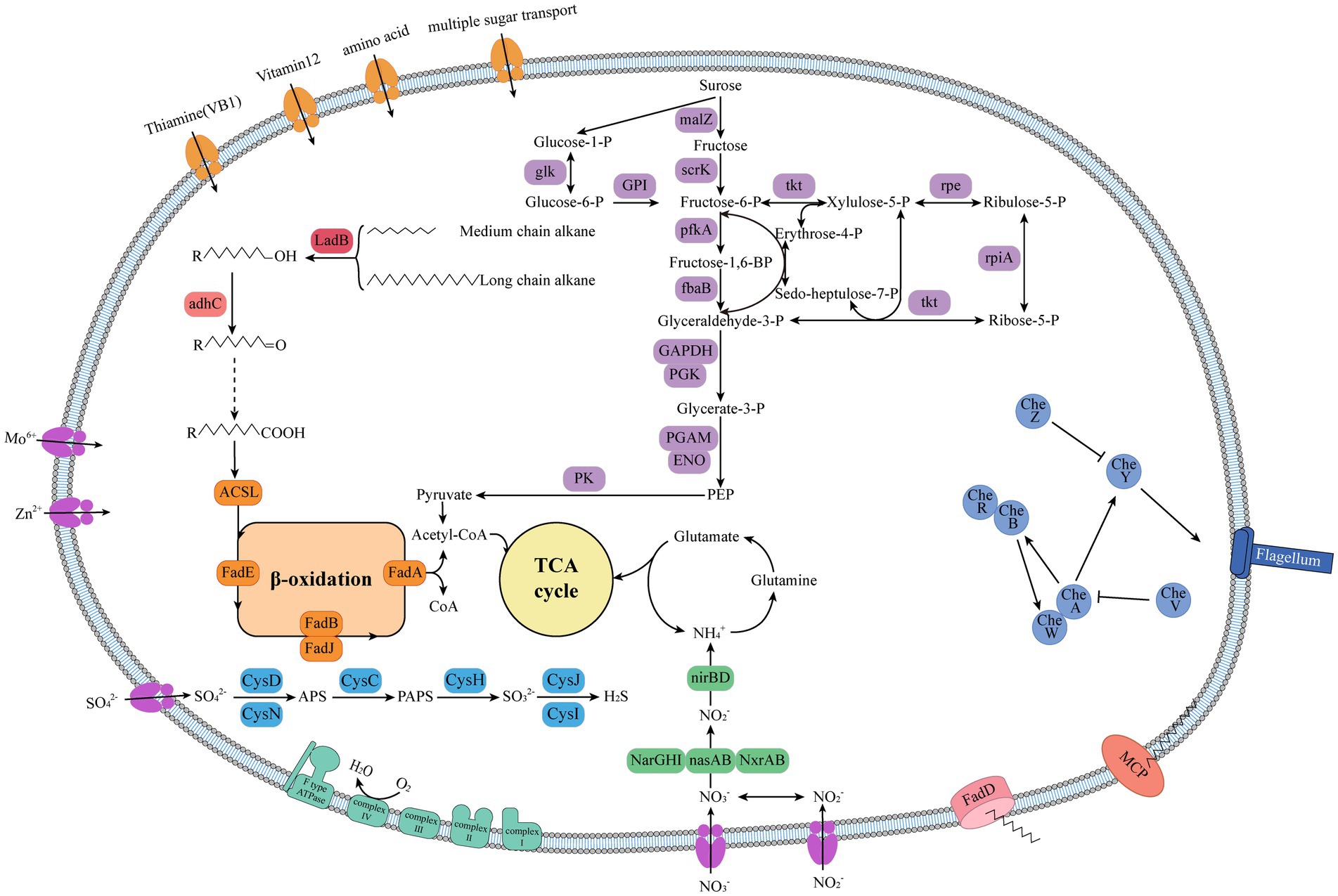
Figure 5. The metabolic pathway reconstruction of M. calida HXX 308.
A comparative genomic analysis between HXX308 and DSM22759 (Table 2) revealed that the former harbors more genes related to oligopeptide transport, alkane chemotaxis, carbon fixation, and sugar metabolism. In contrast to HXX308, DSM22759 contains a higher number of genes with unknown functions.
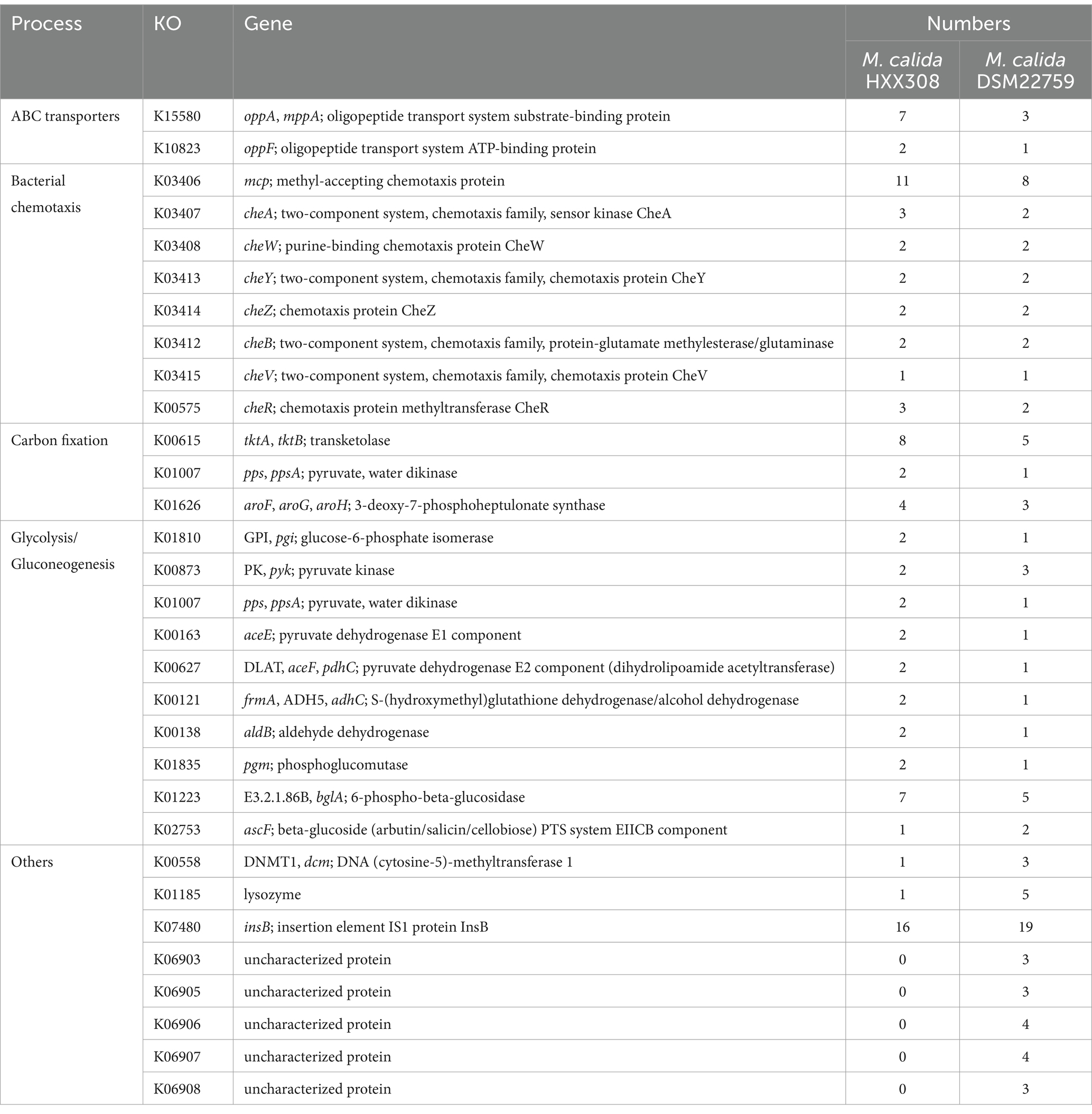
Table 2. The list of KEGG Orthologs (KO) and gene numbers involved in certain metabolic processes in M. calida HXX308 and M. calida DSM22759.
3.4 The growth and alkane degradation capability of Mixta calida HXX308 under high pressure
To assess the growth of HXX308 under different pressure conditions, a high-pressure processing was conducted at 0, 20, 40 and 60 MPa. The results indicated that HXX308 was able to grow under pressures ranging from 0 to 40 MPa, with growth progressively diminished as the pressure increased. Notably, no growth was observed after 30 days of incubation at 60 MPa (Figures 6A,B). The alkane degradation capacity of HXX308 under high pressure was also evaluated. Thus, HXX308 was able to degrade ~20% of C20 under both atmospheric (0.1 Mpa) and 20 MPa pressures (Figure 6C).
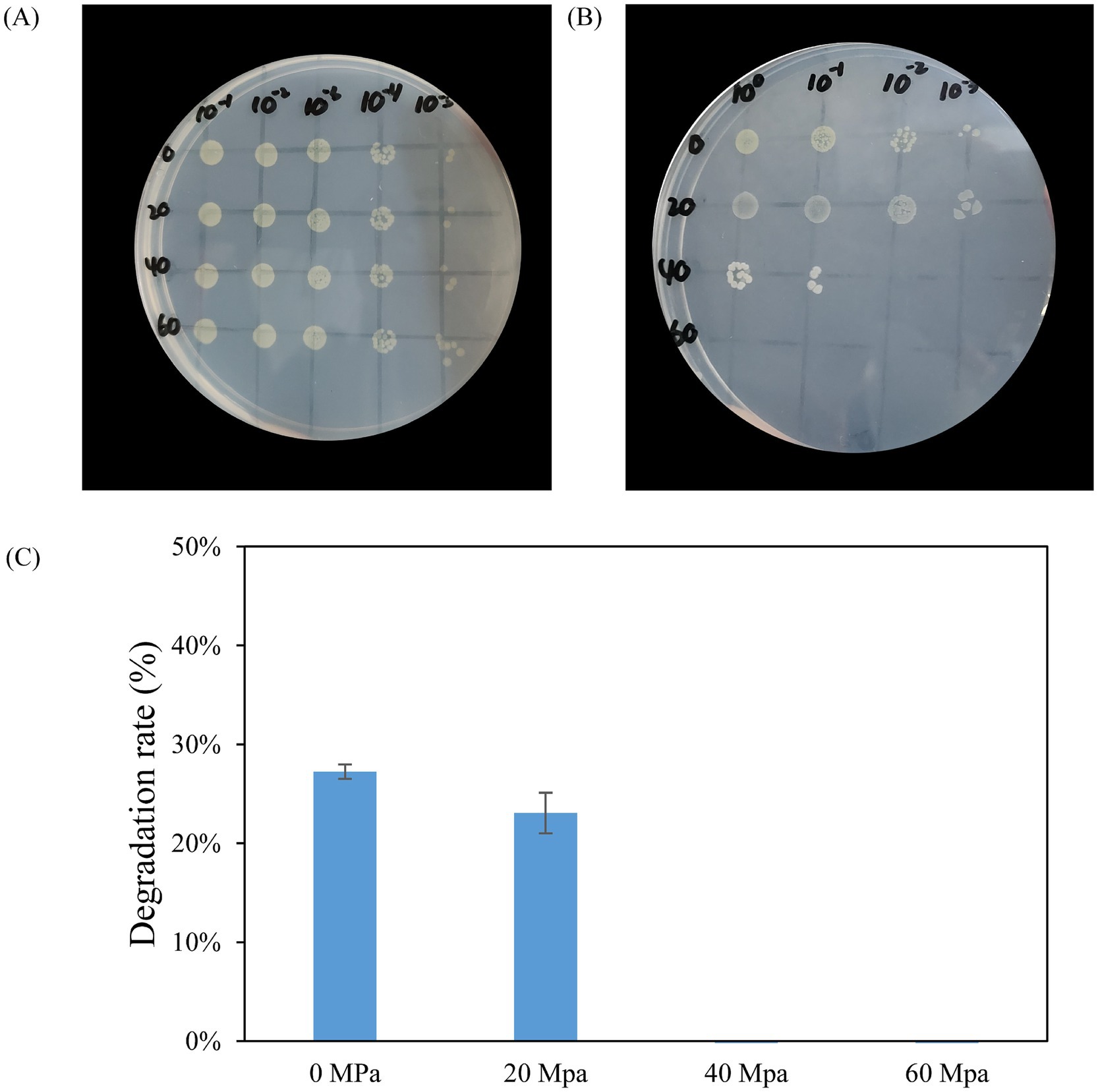
Figure 6. Verification of HXX308’s growth capacity under various pressures (A) Initial results from the high-pressure dot plate assay before high-pressure cultivation; (B) Results from the high-pressure dot plate assay following high-pressure cultivation; (C) Degradation rates after 30 days of incubation using C20 as the sole carbon source under various pressures. Error bars represent the standard deviation from triplicate measurements.
3.5 Alkanes degrading ability of LadB in Mixta calida HXX308
Based on the annotation in the CANT-HYD database, HXX308 possesses the alkane-degrading gene LadB. The protein sequence analysis indicated that the sequence of LadB (KGOCCCKL_01363) from HXX308 showed the highest similarity (100%) to the FMNH2-dependent alkanesulfonate monooxygenase [Mixta calida] (NCBI GenBank ID: WP_312393135.1). LadB is an alkane monooxygenase that belongs to the LadA-type alkane monooxygenase family, and is capable of degrading long-chain alkanes ranging from C11 to C32 (Boonmak et al., 2014). However, the specific sequence and functional characteristics of LadB in HXX308 have not been extensively reported, suggesting that further investigation of this gene may provide novel insights into the mechanisms of alkane degradation. To verify the alkane-degrading activity of LadB in HXX308, the LadB gene was characterized. The recombinant expression strain E. coli BL21-pRSET-A-Flavodoxin-2Fe-2S ferredoxin-LadB was found to degrade 5.96% of C18 and 4.27% of C19 within 72 h (Figure 7 and Supplementary Figure S5), thereby confirming the alkane-degrading activity of the LadB gene in HXX308.
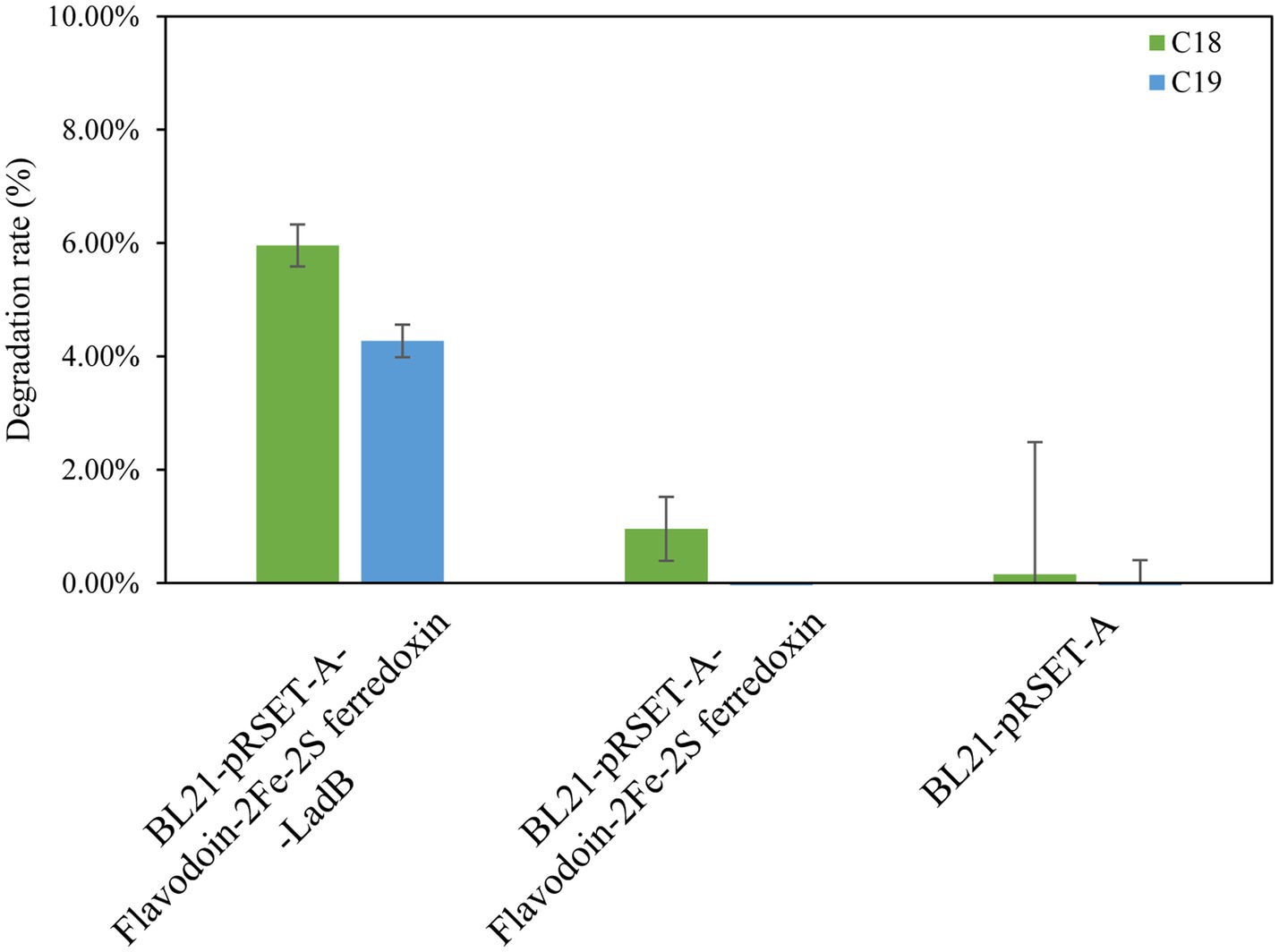
Figure 7. Cloning and expression of LadB gene in M. calida HXX308 and validation of alkane-degrading ability in the recombinant strain E. coli BL21-pRSET-A-Flavodoxin-2Fe-2S ferredoxin-LadB. Determination of the alkane degradation ability of the recombinant strains. The error bars are based on triplicate measurements.
4 Discussion
4.1 The genus Mixta is generally characterized by its ability to degrade hydrocarbons
Proteobacteria is a dominant group in hadal trench sediments. In the Atacama Trench, Proteobacteria is one of the most abundant bacterial phyla, with its highest relative abundance observed in the oxic zone, particularly with Gammaproteobacteria comprising 20.9% of the total microbial community (Schauberger et al., 2021). Similarly, metagenomic sequencing and taxonomic analysis conducted by Zhang and Jing (2024) revealed that Proteobacteria is the dominant phylum in the surface sediments (0–4 cm) of the Challenger Deep in the Mariana Trench. Proteobacteria have been reported as dominant hydrocarbon-degrading taxa in hadal environments. In the Mariana Trench, the proportion of hydrocarbon-degrading bacteria at depths greater than 10,400 m is the deepest observed in any natural environment on Earth. These bacteria are primarily from the genera Oleibacter, Thalassolituus and Alcanivorax, which are known to consume aliphatic hydrocarbons (Liu et al., 2019; Teramoto et al., 2011; Wei et al., 2022; Kasai et al., 2002).
Alcanivorax is one of the most extensively studied genera for alkane degradation, particularly in marine environments, with its alkane metabolism mechanisms being thoroughly investigated (Gregson et al., 2019). However, the role of the genus Mixta in hydrocarbon-degrading in hadal environments remains unexplored. This study reports isolate M. calida HXX308, which was recovered from abyssal sediments at 487–490 cmbsf, and represents the first investigation of its alkane degradation processes.
The alkane degradation pathway is a complex regulatory network involving alkane sensing, chemotaxis, uptake, transport and metabolism (Rojo, 2009). In Alcanivorax dieselolei B5, alkane presence is sensed by OmpS, which activates a chemotaxis complex (comprising mcp, cheR, and cheW) to direct bacterial movement toward higher alkane concentrations (Wang and Shao, 2014). Genomic analysis of HXX308 revealed the presence of alkane chemotaxis protein genes (mcp) and a diverse set of chemotaxis-related genes (cheR, cheA, cheB, cheW, cheV, cheY, cheZ). Here, cheR and cheB regulate the methylation level of receptors to modulate cheA activity, which in turn phosphorylates cheY to regulate flagellar motion. cheW and cheV assist in signal transduction, whereas cheZ terminates signaling by dephosphorylating cheY (Xu et al., 2021).
In terms of alkane transport, A. dieselolei B5 utilizes selective outer membrane transport proteins (OmpT-1, OmpT-2, and OmpT-3) to transport alkanes of varying chain lengths into the cell (Wang and Shao, 2014). In contrast, HXX308 lacks OmpT proteins and instead employs FadL for the transport of medium-and long-chain alkanes across the outer membrane (van den Berg et al., 2004). While OmpT proteins can selectively transport short-, medium-and long-chain alkanes, FadL is specialized for medium-and long-chain alkanes (Liu et al., 2022).
Regarding alkane degradation, A. dieselolei B5 initiates the process via AlmA, which oxidizes alkanes to primary alcohols. These are subsequently oxidized to fatty acids by AdhA and AldA, followed by β-oxidation to generate acetyl-CoA, which enters the tricarboxylic acid cycle to produce carbon dioxide (Wang and Shao, 2014). HXX308 follows a different pathway, using LadB to oxidize alkanes to primary alcohols, which are then converted to fatty acids by AdhC. These fatty acids undergo β-oxidation to acetyl-CoA, ultimately entering the tricarboxylic acid cycle to generate carbon dioxide. In terms of substrate specificity, AlmA targets long-chain alkanes (C26–C38) (Chen et al., 2025), whereas LadB exhibits higher activity toward medium-and long-chain alkanes (C15–C36) (Feng et al., 2007; Wentzel et al., 2007).
In this study, we observed that M. calida HXX308 can grow under pressures ranging from 0 to 40 MPa in the laboratory, which does not encompass the pressure range of its native environment in the Mariana Trench (60–110 MPa). This discrepancy raises questions about the adaptability of HXX308 to the extreme conditions of the hadal zone. We further discuss possible reasons for this observation: (1) the lack of specific protective mechanisms in the laboratory setting, such as pressure-buffering substances or the synergistic effects of microbial communities; (2) the potential adaptation of HXX308 to lower pressure conditions during its cultivation in the laboratory; and (3) the possibility that the pressure in the native environment is the result of a long-term adaptation process that cannot be fully replicated in short-term experiments.
4.2 Adaptation of Mixta calida HXX308 to the trench environment
As a representative of extreme marine environments, hadal trenches are characterized by high pressure, low temperature and darkness (Wang et al., 2019). Microbes inhabiting these environments have evolved unique metabolic features to adapt to these extreme conditions. For example, in this study, M. calida HXX308 was found to possess genomic traits related to deep-sea adaptation, including a streamlined genome with a higher ratio of proteins to genes compared to other strains of Mixta (Liang et al., 2022). This streamlined genome facilitates adaptation to oligotrophic environments. Similar genome streamlining has been observed in Parcubacteria transitioning from near-surface soils to groundwater habitats (Chaudhari et al., 2024). Additionally, streamlined strains of Bacillus subtilis have been shown to exhibit high resistance to DNA-damaging agents (Dervyn et al., 2023).
Moreover, M. calida HXX308 has metabolic traits that enable it to adapt to hypoxic environments. Sediments in the Mariana Trench typically have lower dissolved oxygen content compared to open water, with oxygen concentrations decreasing with depth due to microbial respiration and organic matter mineralization (Li et al., 2019; Xiao et al., 2024). To survive in these low-oxygen conditions, microorganisms often adjust their metabolic pathways, utilizing alternative respiratory chain acceptors or anaerobic respiration. For example, Pseudomonas alcaliphila strain MBR uses an electrode as an electron donor for dissimilatory nitrate reduction to ammonium and denitrification (Su et al., 2012). Similarly, heterotrophic denitrifiers oxidize sulfide to mitigate toxicity and support complete denitrification, providing a competitive advantage in low-oxygen environments (Shao et al., 2025).
M. calida HXX308 possesses key genes that encode for the assimilatory sulfate reduction pathway and the F-type ATPase, indicating its ability to utilize sulfate reduction—a primary anaerobic respiration pathway—in hypoxic environments. The expression of narGHI under low-oxygen conditions further suggests that HXX308 may use nitrate as an electron acceptor, reducing it to nitrite and then to ammonium, consistent with the low-oxygen environment of Mariana Trench sediments (Mareen et al., 2021).
4.3 Mixta calida HXX308 possesses more genes adapted to the trench environment than the reference strain Mixta calida DSM22759
Currently, known strains of the genus Mixta have been isolated from terrestrial environments, such as plants, insects, food and the human body (Xia et al., 2020). However, this study isolated M. calida HXX308 from sediment at a depth of 10,816 m in the Mariana Trench, marking the first marine-sourced strain of the genus. Despite its origin in the trench, HXX308 exhibits hydrocarbon metabolic characteristics similar to those of the type species M. calida DSM22759.
Detailed analysis revealed that HXX308 possesses more genes encoding the oligopeptide transport system substrate-binding protein (OppA) compared to M. calida DSM22759 (Table 2). OppA is a key component of the oligopeptide transport system, responsible for peptide uptake and providing bacteria with nutrients and environmental status information (Monnet, 2003). In nutrient-poor environments, like the trench, the expression of OppA is upregulated to enhance nutrient acquisition (Urbanowski et al., 2000). This suggests that HXX308 has adapted to the oligotrophic conditions of the trench by increasing its capacity for peptide transport.
Additionally, HXX308 has more copies of genes including tktA, aroG, and pps, involved in aromatic compound synthesis. These genes are crucial for the production of 3-deoxy-D-arabinoheptulosonate-7-phosphate (DAHP), a key intermediate in aromatic compound biosynthesis (Patnaik et al., 1995; Vimala and Harinarayanan, 2016; Shaw et al., 2018). The increased presence of these genes likely enhances HXX308’s ability to fix carbon and survive in nutrient-limited environments.
In the glycolysis/gluconeogenesis pathway, HXX308 exhibits a greater number of related genes, particularly bglA. The enzyme encoded by bglA, 6-phospho-β-glucosidase, catalyzes the hydrolysis of 6-phospho-β-glucosides to produce glucose-6-phosphate, facilitating cellulose breakdown and sugar metabolism (Desai et al., 2010; Veldman et al., 2020). This suggests that HXX308 has a stronger capacity for sugar metabolism compared to terrestrial strains, such as M. calida DSM22759.
Overall, the increased number of genes related to peptide transport, aromatic compound synthesis, and sugar metabolism in HXX308 highlights its adaptation to the oligotrophic and extreme conditions of the Mariana Trench.
5 Conclusion
In this study, we investigated the alkane degradation Proteobacteria strain M. calida HXX308, which was isolated from deep-sea sediment in the Mariana Trench. High-pressure alkane degradation assays and high-pressure processing indicated that strain HXX308 could grow at pressures ranging from 0 to 40 MPa, with reduced growth at higher pressures. It could degrade approximately 20% of eicosane at atmospheric pressure (0.1 MPa) and 20 MPa. Phylogenetic analysis revealed that HXX308 is most closely related to M. calida DSM 22759. HXX308 is capable of extensively participating in the processes of alkane metabolism, and nitrogen and sulfur cycling within sediments. Compared to terrestrial strains of the Mixta genus, HXX308 exhibits a greater number of adaptations to the Hadal Trench environment. Specifically, it encodes a higher abundance of genes associated with peptide transport, aromatic compound synthesis and carbohydrate metabolism. Collectively, these genetic features enhance its capacity for nutrient acquisition and metabolic versatility in the hypoxic and oligotrophic conditions of the trench.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SAMN46867115.
Author contributions
YY: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. XH: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. YZ: Methodology, Validation, Visualization, Writing – review & editing. X-HZ: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Laoshan Laboratory (LSKJ202203206 and LSKJ202203201), National Natural Science Foundation of China (32370118), and the Fundamental Research Funds for the Central Universities (202172002).
Acknowledgments
We thank all the scientists and crews for their assistance with sampling during the cruise conducted by Dongfanghong 3 of Ocean University of China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1579612/full#supplementary-material
Footnotes
References
Arora, N. K., and Panosyan, H. (2019). Extremophiles: applications and roles in environmental sustainability. Environ. Sustain. 2, 217–218. doi: 10.1007/s42398-019-00082-0
Bao, R., Strasser, M., McNichol, A. P., Haghipour, N., McIntyre, C., Wefer, G., et al. (2018). Tectonically-triggered sediment and carbon export to the Hadal zone. Nat. Commun. 9:121. doi: 10.1038/s41467-017-02504-1
Blankenship-Williams, L. E., and Levin, L. A. (2009). Living deep: a synopsis of Hadal trench ecology. Mar. Technol. Soc. J. 43, 137–143. doi: 10.4031/mtsj.43.5.23
Boonmak, C., Takahashi, Y., and Morikawa, M. (2014). Cloning and expression of three ladA-type alkane monooxygenase genes from an extremely thermophilic alkane-degrading bacterium Geobacillus thermoleovorans B23. Extremophiles 18, 515–523. doi: 10.1007/s00792-014-0636-y
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Casale, R., Boattini, M., Bianco, G., Comini, S., Corcione, S., Garazzino, S., et al. (2023). Bloodstream infections by Pantoea species: clinical and microbiological findings from a retrospective study, Italy, 2018–2023. Antibiotics 12:1723. doi: 10.3390/antibiotics12121723
Chaudhari, N. M., Pérez-Carrascal, O. M., Overholt, W. A., Totsche, K. U., and Küsel, K. (2024). Genome streamlining in Parcubacteria transitioning from soil to groundwater. Environ. Microbiol. 19:41. doi: 10.1186/s40793-024-00581-6
Chen, S., Cao, L., Lv, T., Liu, J., Gao, G., Li, M., et al. (2025). Regulation mechanism of the long-chain n-alkane monooxygenase gene almA in Acinetobacter venetianus RAG-1. Appl. Environ. Microbiol. 91:e0205024. doi: 10.1128/aem.02050-24
Chen, P., Zhou, H., Huang, Y., Xie, Z., Zhang, M., Wei, Y., et al. (2021). Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the challenger deep. Genome Biol. 22:207. doi: 10.1186/s13059-021-02408-w
Cui, G., Li, J., Gao, Z., and Wang, Y. (2019). Spatial variations of microbial communities in abyssal and hadal sediments across the challenger deep. PeerJ 7:e6961. doi: 10.7717/peerj.6961
Dervyn, E., Planson, A., Tanaka, K., Chubukov, V., Guerin, C., Derozier, S., et al. (2023). Greedy reduction of Bacillus subtilis genome yields emergent phenotypes of high resistance to a DNA damaging agent and low evolvability. Nucleic Acids Res. 51, 2974–2992. doi: 10.1093/nar/gkad145
Desai, S. K., Nandimath, K., and Mahadevan, S. (2010). Diverse pathways for salicin utilization in Shigella sonnei and Escherichia coli carrying an impaired Bgl operon. Arch. Microbiol. 192, 821–833. doi: 10.1007/s00203-010-0610-8
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Feng, L., Wang, W., Cheng, J., Ren, Y., Zhao, G., Gao, C., et al. (2007). Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc. Natl. Acad. Sci. USA 104, 5602–5607. doi: 10.1073/pnas.0609650104
Froger, A., and Hall, J. E. (2007). Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 6:253. doi: 10.3791/253
Glud, R. N., Fer, F. W., Middelboe, M., Oguri, K., Turnewitsch, R., Canfield, D. E., et al. (2013). High rates of microbial carbon turnover in sediments in the deepest oceanic trench on earth. Nat. Geosci. 6, 284–288. doi: 10.1038/ngeo1773
Gregson, B. H., Metodieva, G., Metodiev, M. V., and McKew, B. A. (2019). Differential protein expression during growth on linear versus branched alkanes in the obligate marine hydrocarbon-degrading bacterium Alcanivorax borkumensis SK2T. Environ. Microbiol. 21, 2347–2359. doi: 10.1111/1462-2920.14620
Guz, N., Arshad, M., Cagatay, N. S., and Dageri, A. (2020). High prevalence of Pantoea in Diaphorina Citri (hemiptera: Liviidae): vector of citrus huanglongbing disease. Curr. Microbiol. 77, 1525–1531. doi: 10.1007/s00284-020-01969-6
Huang, Y., Zhang, X., Xin, Y., Tian, J., and Li, M. (2024). Distinct microbial nitrogen cycling processes in the deepest part of the ocean. mSystems 9:e0024324. doi: 10.1128/msystems.00243-24
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11:119. doi: 10.1186/1471-2105-11-119
Ichino, M. C., Clark, M. R., Drazen, J. C., Jamieson, A., Jones, D. O. B., Martin, A. P., et al. (2015). The distribution of benthic biomass in hadal trenches: a modelling approach to investigate the effect of vertical and lateral organic matter transport to the seafloor. Deep-Sea Res. Pt. I 100, 21–33. doi: 10.1016/j.dsr.2015.01.010
Jamieson, A. J., Fujii, T., Mayor, D. J., Solan, M., Priede, I. G., and Glud, R. N. (2010). Hadal trenches: the ecology of the deepest places on earth. Trends Ecol. Evol. 25, 190–197. doi: 10.1016/j.tree.2009.09.009
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kasai, Y., Kishira, H., Sasaki, T., Syutsubo, K., Watanabe, K., and Harayama, S. (2002). Predominant growth of Alcanivorax strains in oil-contaminated and nutrient-supplemented sea water. Environ. Microbiol. 4, 141–147. doi: 10.1046/j.1462-2920.2002.00275.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khot, V., Zorz, J., Gittins, D. A., Chakraborty, A., Bell, E., Bautista, M. A., et al. (2021). CANT-HYD: a curated database of phylogeny-derived hidden markov models for annotation of marker genes involved in hydrocarbon degradation. Front. Microbiol. 12:764058. doi: 10.3389/fmicb.2021.764058
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Li, W., Huang, J., Zhang, P., Cui, G., Wei, Z., Wu, Y., et al. (2019). Periodic and spatial spreading of alkanes and Alcanivorax bacteria in deep waters of the Mariana trench. Appl. Environ. Microbiol. 85:e02089-18. doi: 10.1128/AEM.02089-18
Liang, J., Liu, J., Wang, X., Sun, H., Zhang, Y., Ju, F., et al. (2022). Genomic analysis reveals adaptation of Vibrio campbellii to the hadal ocean. Appl. Environ. Microbiol. 88:e0057522. doi: 10.1128/aem.00575-22
Liu, Y., Chen, S., Xie, Z., Zhang, L., Wang, J., and Fang, J. (2023). Influence of extremely high pressure and oxygen on hydrocarbon-enriched microbial communities in sediments from the challenger deep, Mariana Trench. Microorganisms 11:630. doi: 10.3390/microorganisms11030630
Liu, J., Chen, S., Zhao, B., Li, G., and Ma, T. (2022). A novel fadL homolog, altL, mediates transport of long-chain alkanes and fatty acids in Acinetobacter venetianus RAG-1. Appl. Environ. Microbiol. 88:e0129422. doi: 10.1128/aem.01294-22
Liu, J., Li, D., He, X., Liu, R., Cheng, H., Su, C., et al. (2024). A unique subseafloor microbiosphere in the Mariana trench driven by episodic sedimentation. Mar. Life Sci. Technol. 6, 168–181. doi: 10.1007/s42995-023-00212-y
Liu, H., Sun, W. B., Liang, R. B., Huang, L., Hou, J. L., and Liu, J. H. (2015). iTRAQ-based quantitative proteomic analysis of Pseudomonas aeruginosa SJTD-1: a global response to n-octadecane-induced stress. J. Proteome 123, 14–28. doi: 10.1016/j.jprot.2015.03.034
Liu, J., Zheng, Y., Lin, H., Wang, X., Li, M., Liu, Y., et al. (2019). Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana trench. Microbiome 7:47. doi: 10.1186/s40168-019-0652-3
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2013). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Luo, M., Glud, R. N., Pan, B., Wenzhöfer, F., Xu, Y., Lin, G., et al. (2018). Benthic carbon mineralization in hadal trenches: insights from in situ determination of benthic oxygen consumption. Geophys. Res. Lett. 45, 2752–2760. doi: 10.1002/2017GL076232
Mareen, H., Alina, B., Stephanie, F. C. L. D., Katharina, R., Philipp, B., Chantal, T., et al. (2021). Evaluation of an oxygen-dependent self-inducible surfactin synthesis in B. subtilis by substitution of native promoter PsrfA by anaerobically active PnarG and PnasD. AMB Express 11:57. doi: 10.1186/s13568-021-01218-4
Monnet, V. (2003). Bacterial oligopeptide-binding proteins. Cell. Mol. Life Sci. 60, 2100–2114. doi: 10.1007/s00018-003-3054-3
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nunoura, T., Takaki, Y., Hirai, M., Shimamura, S., Makabe, A., Koide, O., et al. (2015). Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on earth. Proc. Natl. Acad. Sci. U. S. A. 112, E1230–E1236. doi: 10.1073/pnas.1421816112
Palmer, M., Steenkamp, E. T., Coetzee, M. P. A., Avontuur, J. R., Chan, W., van Zyl, E., et al. (2018). Mixta gen. nov., a new genus in the Erwiniaceae. Int. J. Syst. Evol. Microbiol. 68, 1396–1407. doi: 10.1099/ijsem.0.002540
Parra-Flores, J., Cabal-Rosel, A., Daza-Prieto, B., Chavarria, P., Maury-Sintjago, E., Rodriguez-Fernández, A., et al. (2022). Are Enterobacteriaceae and Enterococcus isolated from powdered infant formula a hazard for infants? A genomic analysis. Foods 11:3556. doi: 10.3390/foods11223556
Patnaik, R., Spitzer, R. G., and Liao, J. C. (1995). Pathway engineering for production of aromatics in Escherichia coli: confirmation of stoichiometric analysis by independent modulation of AroG, TktA, and Pps activities. Biotechnol. Bioeng. 46, 361–370. doi: 10.1002/bit.260460409
Peoples, L. M., Grammatopoulou, E., Pombrol, M., Xu, X., Osuntokun, O., Blanton, J., et al. (2019). Microbial community diversity within sediments from two geographically separated hadal trenches. Front. Microbiol. 10:347. doi: 10.3389/fmicb.2019.00347
Popp, A., Cleenwerck, I., Iversen, C., De Vos, P., and Stephan, R. (2010). Pantoea gaviniae sp. nov. and Pantoea calida sp. nov., isolated from infant formula and an infant formula production environment. Int. J. Syst. Evol. Microbiol. 60, 2786–2792. doi: 10.1099/ijs.0.019430-0
Rawlings, N. D., Barrett, A. J., Thomas, P. D., Huang, X., Bateman, A., and Finn, R. D. (2018). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632. doi: 10.1093/nar/gkx1134
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Schauberger, C., Glud, R. N., Hausmann, B., Trouche, B., Maignien, L., Poulain, J., et al. (2021). Microbial community structure in hadal sediments: high similarity along trench axes and strong changes along redox gradients. ISME J. 15, 3455–3467. doi: 10.1038/s41396-021-01021-w
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shao, B., Xie, Y., Zhang, L., Ruan, Y., Liang, B., Zhang, R., et al. (2025). Versatile nitrate-respiring heterotrophs are previously concealed contributors to sulfur cycle. Nat. Commun. 16:1202. doi: 10.1038/s41467-025-56588-1
Sharma, S. C. S. S. (2018). The NCBI bio collections database. Database 2018:bay006. doi: 10.1093/database/bay006
Shaw, J. A., Henard, C. A., Liu, L., Dieckman, L. M., Vázquez-Torres, A., and Bourret, T. J. (2018). Salmonella enterica serovar typhimurium has three transketolase enzymes contributing to the pentose phosphate pathway. J. Biol. Chem. 293, 11271–11282. doi: 10.1074/jbc.ra118.003661
Su, W., Zhang, L., Li, D., Zhan, G., Qian, J., and Tao, Y. (2012). Dissimilatory nitrate reduction by Pseudomonas alcaliphila with an electrode as the sole electron donor. Biotechnol. Bioeng. 109, 2904–2910. doi: 10.1002/bit.24554
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Teramoto, M., Ohuchi, M., Hatmanti, A., Darmayati, Y., Widyastuti, Y., Harayama, S., et al. (2011). Oleibacter marinus gen. nov., sp. nov., a bacterium that degrades petroleum aliphatic hydrocarbons in a tropical marine environment. Int. J. Syst. Evol. Microbiol. 61, 375–380. doi: 10.1099/ijs.0.018671-0
Tian, J., Fan, L., Liu, H., Liu, J., Li, Y., Qin, Q., et al. (2018). A nearly uniform distributional pattern of heterotrophic bacteria in the Mariana trench interior. Deep-Sea Res. Pt. I 142, 116–126. doi: 10.1016/j.dsr.2018.10.002
Urbanowski, M. L., Stauffer, L. T., and Stauffer, G. V. (2000). The Gcvb gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol. Microbiol. 37, 856–868. doi: 10.1046/j.1365-2958.2000.02051.x
van den Berg, B., Black, P. N., Clemons, W. M. Jr., and Rapoport, T. A. (2004). Crystal structure of the long-chain fatty acid transporter FadL. Science 304, 1506–1509. doi: 10.1126/science.1097524
Veldman, W., Liberato, M. V., Almeida, V. M., Souza, V. P., Frutuoso, M. A., Marana, S. R., et al. (2020). X-ray structure, bioinformatics analysis, and substrate specificity of a 6-Phospho-β-glucosidase glycoside hydrolase 1 enzyme from Bacillusl licheniformis. J. Chem. Inf. Model. 60, 6392–6407. doi: 10.1021/acs.jcim.0c00759
Vimala, A., and Harinarayanan, R. (2016). Transketolase activity modulates glycerol-3-phosphate levels in Escherichia coli. Mol. Microbiol. 100, 263–277. doi: 10.1111/mmi.13317
Wang, W., and Shao, Z. (2014). The long-chain alkane metabolism network of Alcanivorax Dieselolei. Nat. Commun. 5:5755. doi: 10.1038/ncomms6755
Wang, K., Shen, Y., Yang, Y., Gan, X., Liu, G., Hu, K., et al. (2019). Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nat. Ecol. Evol. 3, 823–833. doi: 10.1038/s41559-019-0864-8
Wang, W., Wang, L., and Shao, Z. (2018). Polycyclic aromatic hydrocarbon (PAH) degradation pathways of the obligate marine PAH degrader Cycloclasticus sp. strain P1. Appl. Environ. Microbiol. 84, e01261–e01218. doi: 10.1128/AEM.01261-18
Wang, J., Zhang, Y., Liu, Y., Xie, Z., Cao, J., Zhang, H., et al. (2023). The phylogeny and metabolic potentials of an n-alkane-degrading Venatorbacter bacterium isolated from deep-sea sediment of the Mariana trench. Front. Microbiol. 14:1108651. doi: 10.3389/fmicb.2023.1108651
Wei, T. T., He, S., and Quan, Z. X. (2022). Thalassolituus alkanivorans sp. nov., a hydrocarbon-utilizing bacterium isolated from the Mariana trench. Int. J. Syst. Evol. Microbiol. 72:5404. doi: 10.1099/ijsem.0.005404
Wentzel, A., Ellingsen, T. E., Kotlar, H. K., Zotchev, S. B., and Throne-Holst, M. (2007). Bacterial metabolism of long-chain n-alkanes. Appl. Microbiol. Biotechnol. 76, 1209–1221. doi: 10.1007/s00253-007-1119-1
Wenzhöfer, F., Oguri, K., Middelboe, M., Turnewitsch, R., Toyofuku, T., Kitazato, H., et al. (2016). Benthic carbon mineralization in hadal trenches: assessment by in situ O2 microprofile measurements. Deep-Sea Res. Pt. I 116, 276–286. doi: 10.1016/j.dsr.2016.08.013
Xia, M., Wang, J., Huo, Y., and Yang, Y. (2020). Mixta tenebrionis sp. nov., isolated from the gut of the plastic-eating mealworm Tenebrio molitor L. Int. J. Syst. Evol. Microbiol. 70, 790–796. doi: 10.1099/ijsem.0.003826
Xiao, W., Xu, Y., Canfield, D. E., Wenzhoefer, F., Zhang, C., et al. (2024). Strong linkage between benthic oxygen uptake and bacterial tetraether lipids in deep-sea trench regions. Nat. Commun. 15:3439. doi: 10.1038/s41467-024-47660-3
Xie, J., Chen, Y., Cai, G., Cai, R., Hu, Z., and Wang, H. (2023). Tree visualization by one table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. doi: 10.1093/nar/gkad359
Xu, X., Li, H., Qi, X., Chen, Y., Qin, Y., Zheng, J., et al. (2021). cheA, cheB, cheR, cheV, and cheY are involved in regulating the adhesion of Vibrio harveyi. Front. Cell. Infect. Microbiol. 10:591751. doi: 10.3389/fcimb.2020.591751
Yang, N., Lv, Y., Ji, M., Wu, S., and Zhang, Y. (2024). High hydrostatic pressure stimulates microbial nitrate reduction in hadal trench sediments under oxic conditions. Nat. Commun. 15:2473. doi: 10.1038/s41467-024-46897-2
Zhang, Y., and Jing, H. (2024). Metagenome sequencing and 107 microbial genomes from seamount sediments along the yap and Mariana trenches. Sci. Data 11:887. doi: 10.1038/s41597-024-03762-7
Zhao, X., Liu, J., Zhou, S., Zheng, Y., Wu, Y., Kogure, K., et al. (2020). Diversity of culturable heterotrophic bacteria from the Mariana trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2, 181–193. doi: 10.1007/s42995-020-00027-1
Keywords: the Mariana Trench, Mixta calida, Proteobacteria, alkane degradation, comparative genomic analysis
Citation: Yang Y, He X, Zhang Y and Zhang X-H (2025) Alkane degradation mechanism of Mixta calida HXX308 isolated from sediment of the Mariana Trench. Front. Microbiol. 16:1579612. doi: 10.3389/fmicb.2025.1579612
Edited by:
Harold J. Schreier, University of Maryland, Baltimore County, United StatesReviewed by:
Nastasia Johana Freyria, McGill University, CanadaBinbin Wang, University of Nebraska-Lincoln, United States
Copyright © 2025 Yang, He, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Hua Zhang, eGh6aGFuZ0BvdWMuZWR1LmNu
†These authors have contributed equally to this work