 Shicheng Chen
Shicheng Chen Grace Agah1
Grace Agah1 Jochen Blom
Jochen Blom Edward D. Walker
Edward D. Walker- 1College of Health and Human Sciences, Northern Illinois University, DeKalb, IL, United States
- 2Bioinformatics and Systems Biology, Justus-Liebig University Giessen, Giessen, Germany
- 3Department of Microbiology, Genetics, and Immunology, Michigan State University, East Lansing, MI, United States
Introduction: Elizabethkingia miricola is a gram-negative bacterium that causes life-threatening infections in vulnerable populations. Unlike other species in the Elizabethkingia genus, E. miricola also leads to meningitis-like diseases in aquatic invertebrates such as frogs, raising concerns about its zoonotic transmission potential. Management of its infection is complicated by unclear transmission pathways and multi-drug resistance.
Methods: In this study, we analyzed three clinical strains (E. miricola Mich-1, Mich-2, and Mich-3) isolated from patients in Michigan using morphology observations, biochemical tests, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF/MS), and genome sequencing.
Results: Average Nucleotide Identity (ANI) analysis revealed that the Michigan strains were nearly identical and shared 96.52% identity with the type strain E. miricola DSM 14571, confirming their classification as E. miricola. Comprehensive comparative genomic analyses were conducted across 28 strains, including human isolates and strains from invertebrates like frogs. The strains exhibited open pan-genome characteristics. Mich-1 shared 3,199 genes (83.2%) with human isolates but fewer genes with frog-derived isolates (ranging from 3,319 to 3,375). This phylogenetic analysis highlights regional variation and the global diversity of E. miricola isolates, revealing connections between clinical and environmental strains. Antibiotic susceptibility testing revealed that the three clinical strains were resistant to 13 out of 16 tested drugs, with susceptibility only to trimethoprim/sulfamethoxazole and ciprofloxacin. The strains carried five β-lactamase-encoding genes (BlaB-10, BlaB-39, CME-1, CME-2, and GOB-25), conferring resistance to penams, cephalosporins, and carbapenems. Several virulence-associated genes were conserved across clinical and frog isolates. These genes contribute to stress adaptation, adherence, and immune modulation.
Discussion: This study underscores the evolutionary adaptability of E. miricola genomes, highlighting their capacity to acquire genetic traits that enable survival in diverse niches. This adaptability facilitates the emergence of more resistant and virulent strains, posing significant threats to both human and animal health.
1 Introduction
Elizabethkingia is a genus of gram-negative, non-fermenting, aerobic rod (Breurec et al., 2016; Coyle, 2017; Janda and Lopez, 2017). Bacteria Elizabethkingia are widely distributed in various environments such as soil, water, plants, and animals (Lin et al., 2019). At least seven species have been identified with medical importance, including E. anophelis, E. miricola, E. meningoseptica, E. occulta, E. bruuniana, E. ursingii, and E. umeracha (Nicholson et al., 2018; Hem et al., 2022). Among them, E. anophelis is more frequently isolated from human specimens, followed by E. miricola and E. meningoseptica (Coyle, 2017; Janda and Lopez, 2017; Lin et al., 2019; Hem et al., 2022; Zajmi et al., 2022). Different from other Elizabethkingia, E. miricola causes serious acute infections in both humans and aquatic invertebrate animals (Ransangan et al., 2013; Zajmi et al., 2022; Wu et al., 2024). The common clinical presentations in humans include bacteremia, pneumonia, sepsis, and meningitis (Coyle, 2017; Janda and Lopez, 2017; Lin et al., 2019; Lee and Hsueh, 2023; Wu et al., 2024). Overall, the elderly, neonates, immunosuppressed patients, and individuals with underlying chronic medical conditions are more susceptible to E. miricola infections (Lin et al., 2019; Huang et al., 2024). In anuran species, it caused meningitis-like diseases in bull frogs, northern leopard frogs (Lithobates pipiens), Chapa bug-eyed frogs (Theloderma bicolor), and Vietnamese warty toads (Bombina microdeladigitora) (Zajmi et al., 2022). Additionally, E. miricola was also isolated from Tra catfish (Pangasius hypophthalmus) filets in the industrial processing lines in Vietnam (Ransangan et al., 2013; Zajmi et al., 2022).
Managing E. miricola infections has been particularly challenging due to its multidrug resistance (MDR) (Comba et al., 2022; Wu et al., 2024). This bacterium exhibits intrinsic resistance to a wide range of important antibiotics that are used for treating infections by gram-negative bacteria (Wu et al., 2024). The MDR mechanisms remain unexplored in E. miricola. However, many reports showed that the MDR in E. anophelis is primarily mediated by chromosomally encoded determinants (Comba et al., 2022; Wu et al., 2024). Its resistance extends to nearly all β-lactam antibiotics, driven by the presence of three distinct β-lactamase genes: blaCME, an Ambler class A serine extended-spectrum β-lactamase (ESBL), and blaB and blaGOB, which encode Ambler class B metallo-β-lactamases (MBLs) (Hu et al., 2020; Yang et al., 2021; Comba et al., 2022; Andriyanov et al., 2024). The dissemination of resistance genes is largely facilitated by mobile genetic elements such as conjugative transposons and prophages, which carry genes encoding efflux pumps, enzyme-degrading proteins, and enzyme-modifying proteins, further complicating treatment options (Comba et al., 2022; Andriyanov et al., 2024; Huang et al., 2024). Large-scale outbreaks and the global distribution of this group of pathogens have been observed (Breurec et al., 2016; Perrin et al., 2017; McTaggart et al., 2019; Hu et al., 2022b; Mallinckrodt et al., 2023; Wu et al., 2024). The resistance to antimicrobials and disinfectant treatment phenotypes have been linked with the formation of biofilms (Hu et al., 2022a; Puah et al., 2022). Compounding these challenges, E. miricola is often misclassified as E. meningoseptica or E. anophelis, indicating that the infections by E. miricola have been underestimated (Andriyanov et al., 2024; Huang et al., 2024; Wu et al., 2024). Antibiograms of Elizabethkingia isolates often reveal inconsistent resistance patterns, underscoring the need for local susceptibility testing to guide effective treatment (Huang et al., 2024). Infections caused by Elizabethkingia spp. are associated with increased mortality when inappropriate antimicrobial therapy is administered (Huang et al., 2024). Accurate identification of this clinically significant pathogen in time and understanding its MDR mechanisms are particularly important for improving the management of Elizabethkingia infections (Coyle, 2017; Janda and Lopez, 2017; Huang et al., 2024).
Three E. miricola strains were detected in Michigan patients during the same period as the large Elizabethkingia outbreak that occurred in the United States between 2015 and 2016 (Perrin et al., 2017). During that time, most attention was drawn to E. anophelis (Perrin et al., 2017). Outbreaks of E. miricola infections in humans have been poorly documented, with only a small outbreak reported in intensive care units in Spain in 2021 (Soler-Iborte et al., 2024). So far, there were at least 9 available genomes of E. miricola deposited in the GenBank with most isolated from Midwest regions in United States. The transmission pathways of E. miricola remain unclear, though current research suggests several potential routes (Hu et al., 2020; Zajmi et al., 2022; Huang et al., 2024; Wu et al., 2024). Healthcare-associated transmission is considered the most plausible, supported by its detection in hospital settings and its similarity to E. anophelis and E. meningoseptica, which have been linked to outbreaks via contaminated medical devices and inadequate sterilization practices (Coyle, 2017; Janda and Lopez, 2017; Lin et al., 2019; Hu et al., 2022a). However, many affected patients have neither been hospitalized nor lived in long-term healthcare facilities, indicating alternative routes (Coyle, 2017; Janda and Lopez, 2017; Lin et al., 2019; Mallinckrodt et al., 2023). Environmental exposure is another possibility, as E. miricola has been isolated from freshwater, soil, animals, and plants, highlighting its resilience in both natural and built environments (Lin et al., 2019; Kadi et al., 2022; Zajmi et al., 2022). Zoonotic transmission is also suggested by its presence in diseased animals, such as cultured fish and frogs, pointing to potential environmental or direct animal-to-human transmission (Lin et al., 2019; Puah et al., 2022; Zajmi et al., 2022). While direct evidence of person-to-person transmission is lacking, outbreaks of related species in healthcare settings suggest that such transmission, possibly involving healthcare workers, could occur, especially among immunocompromised individuals (Lin et al., 2019; Puah et al., 2022; Zajmi et al., 2022).
Epidemiological studies, genomic analyses, and expanded environmental sampling are essential to better understand the transmission pathways, MDR mechanisms, and strategies for disease management of E. miricola (Hem et al., 2022; Huang et al., 2024). In this study, we sequenced three E. miricola strains collected during a cluster outbreak in Michigan. A detailed phylogenetic analysis was conducted to compare genetic variations between strains isolated from amphibians and those from clinical settings. Furthermore, we investigated the molecular mechanisms underlying drug resistance in this emerging pathogen. Our research on the geographical distribution, phylogenetic structure, and MDR mechanisms of E. miricola provides valuable insights into its drug resistance and virulence factors, as well as predictions regarding host-pathogen interactions and host-environment responses.
2 Materials and methods
2.1 Culture
Three strains of E. miricola (Mich-1, Mich-2, and Mich-3) were isolated from patients in Michigan (see Table 1). Strain Mich-1 was obtained from the whole blood of a female patient on February 22, 2016, while strains Mich-2 and Mich-3 were isolated from whole blood samples of different male patients on November 10, 2015. The E. miricola strains were cultured aerobically in tryptic soy broth (TSB) at 30°C. When using tryptic soy agar (TSA), Bacto agar (Difco, Detroit, MI) was added to TSB at a final concentration of 20 g/liter. Sheep blood agar (SBA) was from Thermo Scientific (Waltham, MA). Streptococcus pneumoniae, Streptococcus pyogenes, and Enterococcus faecalis were employed as controls for alpha, beta, and gamma hemolysis, respectively. Hemolysis patterns were identified after the tested strains were cultured on blood agar plates at 37°C for 48 h. To characterize the biochemical properties of Mich-1, we inoculated 150 μL of the bacterial suspension into a Biolog GEN III microplate (Biolog Inc., Hayward CA) and incubated it at 30°C. Color changes in plate wells were analyzed according to the manufacturer’s instructions.
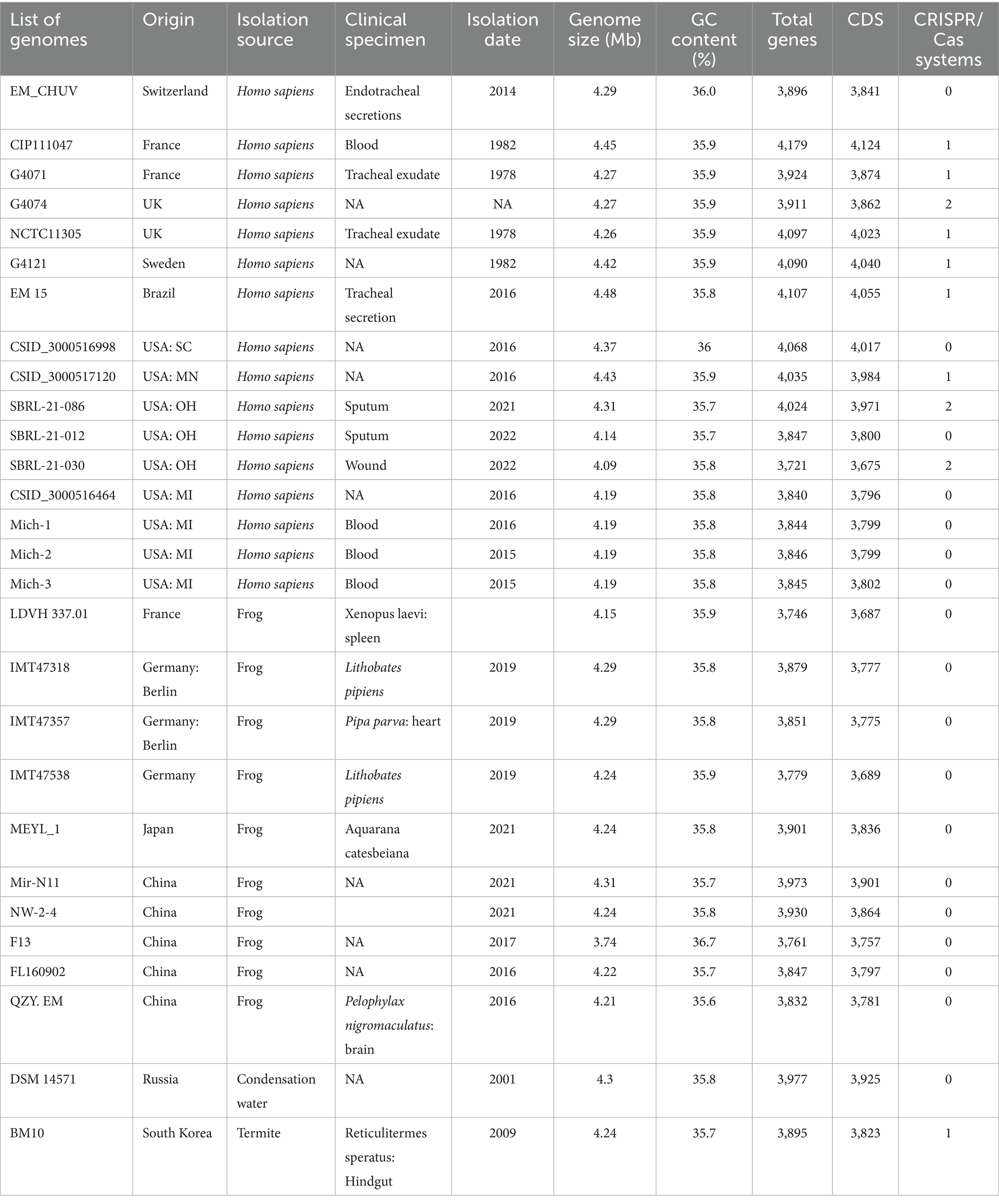
Table 1. Genomic features in selected Elizabethkingia species.
2.2 MALDI-ToF MS analyses
Pure strains of E. miricola were grown on sheep blood agar plates at 35.5°C for 24 h. A single colony was then smeared onto a metal target plate. Following this, 1 μL of α-cyano-4-hydroxycinnamic acid matrix solution was applied to the smeared area and allowed to dry. The target plate was subsequently placed into the VITEK mass spectrometer, a MALDI-TOF/MS system (BioMérieux, Durham, NC, United States). The resulting spectra, covering a mass range of 2 to 20 kDa, were analyzed and compared to the reference spectra of known species in the VITEK MS MS-ID database (version 2.0) for accurate identification.
2.3 Antibiotic susceptibility testing
A 0.5 McFarland bacterial suspension was prepared using a 24-h culture and transferred into an AST-GN69 card, which was then loaded into a VITEK 2 system (BioMérieux, Durham, NC, United States). The following antimicrobials were tested: piperacillin, piperacillin/tazobactam, trimethoprim/sulfamethoxazole, ampicillin, ampicillin/sulbactam, meropenem, aztreonam, Cefotaxime, ceftriaxone, cefazolin, amikacin, gentamicin, tetracycline, tigecycline, ciprofloxacin, and nitrofurantoin. The interpretation of results was based on standards recommended by the Clinical and Laboratory Standards Institute (CLSI) for non-Enterobacteriaceae.
2.4 Genomic DNA extraction, genome sequencing, assembly, and annotation
DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison). The concentration of genomic DNA was quantified with a Nanodrop2000 UV–Vis Spectrophotometer (Thermo Scientific) and a Qubit DNA assay kit. DNA integrity was assessed via a 1.5% (w/v) agarose gel assay. Next-generation sequencing (NGS) libraries were prepared using the Illumina TruSeq Nano DNA Library Preparation Kit. The completed libraries were evaluated with Qubit dsDNA HS, Caliper LabChipGX HS DNA, and Kapa Illumina Library Quantification qPCR assays. The libraries were pooled for multiplexed sequencing and loaded onto a single standard MiSeq flow cell (v2). Sequencing was conducted in a 2 × 250 bp paired-end format using a v2 500-cycle reagent cartridge. NGS libraries were sequenced using Illumina MiSeq paired-end sequencing technology at the Research Technology Support Facility (RTSF) of Michigan State University. The sequencing reads were assembled using the CLC Genomics Workbench v. 23.05. The assembled genome sequences for Mich-1, Mich-2 and Mich-3 were submitted to the Prokaryotic Genome Automatic Annotation Pipeline (PGAAP 3.3) available in National Center for Biotechnology Information (NCBI) for annotation (Tatusova et al., 2016). The predicted CDSs were translated and analyzed against the NCBI non-redundant database, Pfam, TIGRfam, InterPro, KEGG and COG (Tatusova et al., 2016).
2.5 Bioinformatics
Genomes of 28 E. miricola strains and two other clinically important Elizabethkingia species, E. anophelis and E. meningoseptica, were downloaded from GenBank (NCBI) and reannotated using Prokka, a rapid prokaryotic genome annotation tool (Seemann, 2014). The multi-drug resistance genes were predicted in the CARD database (Alcock et al., 2023). Prophage and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) were predicted using CRISPRfinder (Grissa et al., 2007). The virulence factors of Elizabethkingia species were predicted using the VFDB (Chen et al., 2016). For genome similarity assessment, average nucleotide identity (ANI) and digital DNA–DNA hybridization (dDDH) values were computed using the web tools ANI calculator (Yoon et al., 2017) and GGDC 3.0 (Meier-Kolthoff et al., 2022), respectively. For quantification and type of prokaryotic regulatory system proteins, web tool P2RP was used (Barakat et al., 2013). The pan-genome, core genome, and specific genes of Michigan isolates were analyzed by comparison with other representative Elizabethkingia using EDGAR 3.2 (Dieckmann et al., 2021). The sizes of pan-genome and core genomes were approximated using the core/pan development feature. The Elizabethkingia pangenome was further calculated using Roary v3.13.0 (Page et al., 2015) built in Galaxy1 and visualized using Phandango v1.3.1 (Hadfield et al., 2018).
2.6 Accession of genome sequences
Data from the whole-genome shotgun projects were deposited at DDBJ/ENA/GenBank for E. miricola Mich-1, Mich-2, and Mich-3 under accession numbers JBEUGN000000000, JBEUGO000000000, and JBEUGP000000000, respectively. BioProject numbers are PRJNA1125339, PRJNA1125343, and PRJNA 746122 and the BioSample accession numbers are SAMN41894042, SAMN41894043, and SAMN 20181758, respectively.
3 Results
3.1 Physiological and biochemical characteristics of Elizabethkingia miricola Mich-1, Mich-2, and Mich-3
The three clinical isolates E. miricola Mich-1, Mich-2, and Mich-3 exhibited similar morphological characteristics: colonies were medium-sized, creamy, slightly mucoid, and round. Strain Mich-1 was selected as the representative for further identification. Mich-1 cells showed a straight rod with a smooth surface and defined cell borders and had a diameter of 0.4 μm and length of 15.0 μm (Figure 1A). It showed gamma hemolysis (no hemolysis) after incubation on sheep blood agar at 35°C for 24 h (see Figure 1B) while S. pyogenesis, E. faecalis and S. pneumoniae demonstrated beta, gamma and alpha hemolysis, respectively (Figures 1C–E). We assessed the ability of Mich-1 to utilize various carbon and nitrogen sources, as well as its tolerance to salt, pH, and surfactants, using Biolog GEN III microplates (Supplementary Table S1). The results demonstrated that Mich-1 metabolized a range of carbohydrates, including dextrin, d-maltose, d-trehalose, gentibiose, d-lactose, d-melibiose, d-glucose, N-acetyl-d-glucosamine, d-mannose, d-galactose, d-fucose, l-fucose, d-fructose, d-mannitol, d-arabitol, and d-glycerol. In addition, it utilized various nitrogen sources, such as gelatin, glycyl-l-proline, l-alanine, l-arginine, l-aspartic acid, l-glutamic acid, l-histidine, l-pyroglutamic acid, l-serine, pectin, d-galacturonic acid, l-galactonic acid lactone, and d-glucuronic acid. However, its growth was inhibited by 4% NaCl, the surfactant Niaproof 4, or pH levels below 5.0. These findings suggest that Mich-1 is capable of surviving in diverse environments. However, all three strains Mich-1, Mich-2 and Mich-3 were misidentified as E. meningosepticum by the MALDI-TOF/MS analysis (see Supplementary Figure S1).
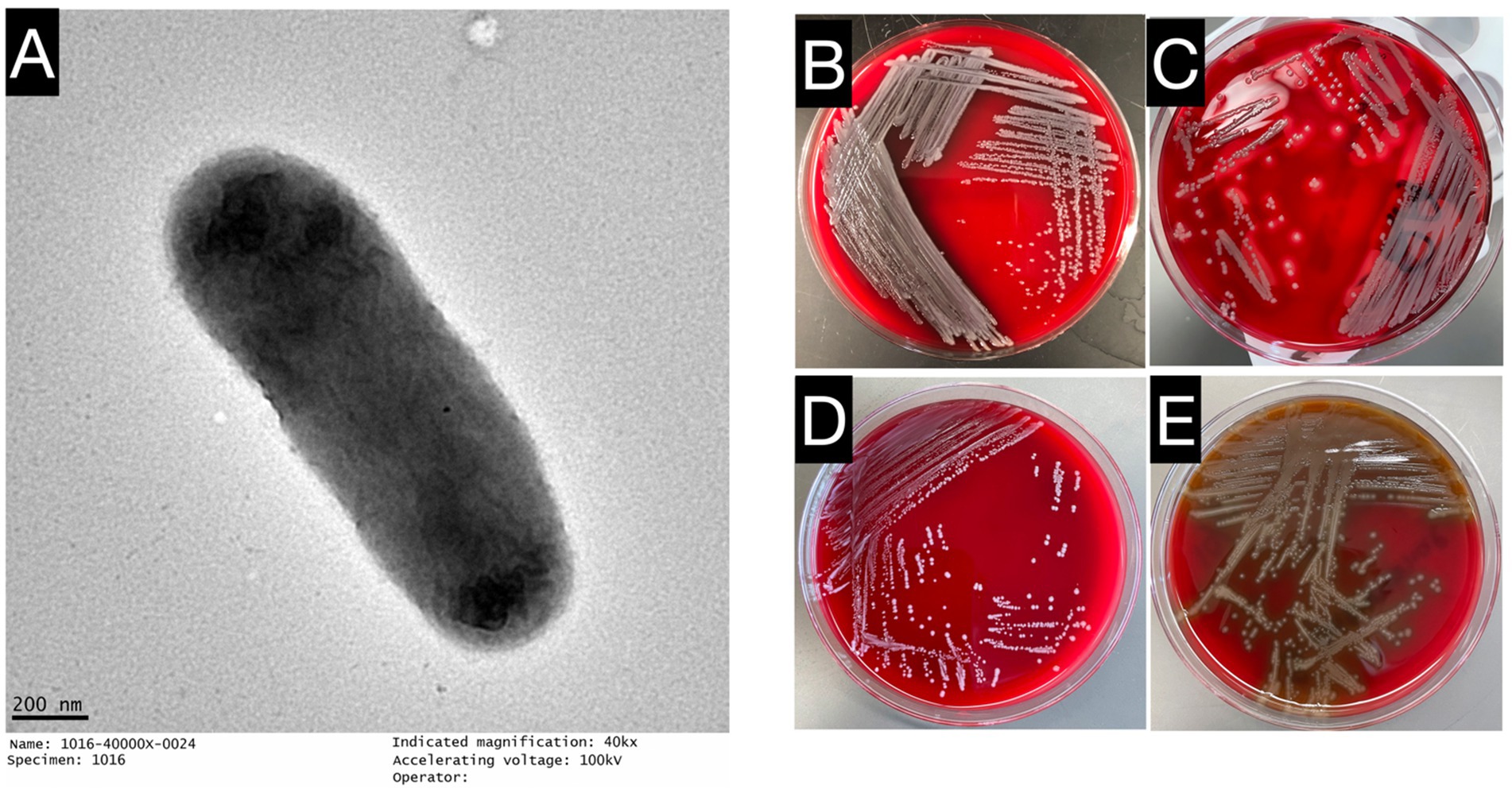
Figure 1. Growth characteristics and microscopic observations of E. miricola Mich-1. (A) Electron microscopy image of E. miricola Mich-1, visualized with a negative stain to highlight bacterial morphology. (B) Growth of E. miricola Mich-1 on sheep blood agar, demonstrating gamma hemolysis (non-hemolytic). (C) S. pyogenes on sheep blood agar, showing complete hemolysis, used as a positive control for beta-hemolysis. (D) E. faecalis on sheep blood agar, demonstrating gamma hemolysis (non-hemolytic), used as a non-hemolytic control. (E) S. pneumoniae on sheep blood agar, exhibiting alpha hemolysis (partial hemolysis), used as a control for partial hemolysis.
3.2 Genomic features and phylogenetic inferences
The genomic characteristics of the Elizabethkingia strains are summarized in Table 1. The assemblies of Mich-1, Mich-2, and Mich-3 contained 10, 14, and 11 contigs, respectively, with similar genome sizes of 4.19 Mb (Table 1). Among the 28 selected E. miricola genomes, sizes ranged from 3.74 Mb to 4.48 Mb, with an average size of 4.25 Mb. Notably, the average genome size of isolates from U. S. patients (4.23 Mb) was slightly smaller than those from European and Brazilian patients (4.34 Mb), a difference that was statistically significant (p < 0.05). However, no significant difference in genome size was observed between isolates from humans and frogs (p > 0.05). A similar trend was observed in the total predicted gene numbers across these genomes. The genomes of Mich-1, Mich-2, and Mich-3 contained 3,799, 3,799, and 3,802 coding sequences (CDSs), respectively. The average GC content of the three E. miricola strains isolated from Michigan patients was 35.8%, consistent with the average GC content of isolates from U. S. patients (35.81%), but slightly lower than that of European and Brazilian isolates (35.9%; p < 0.05). Additionally, the three Michigan isolates lacked predicted CRISPR elements (Table 1). Overall, the genomes of clinical isolates showed high similarity to those from frog isolates (Figure 2).
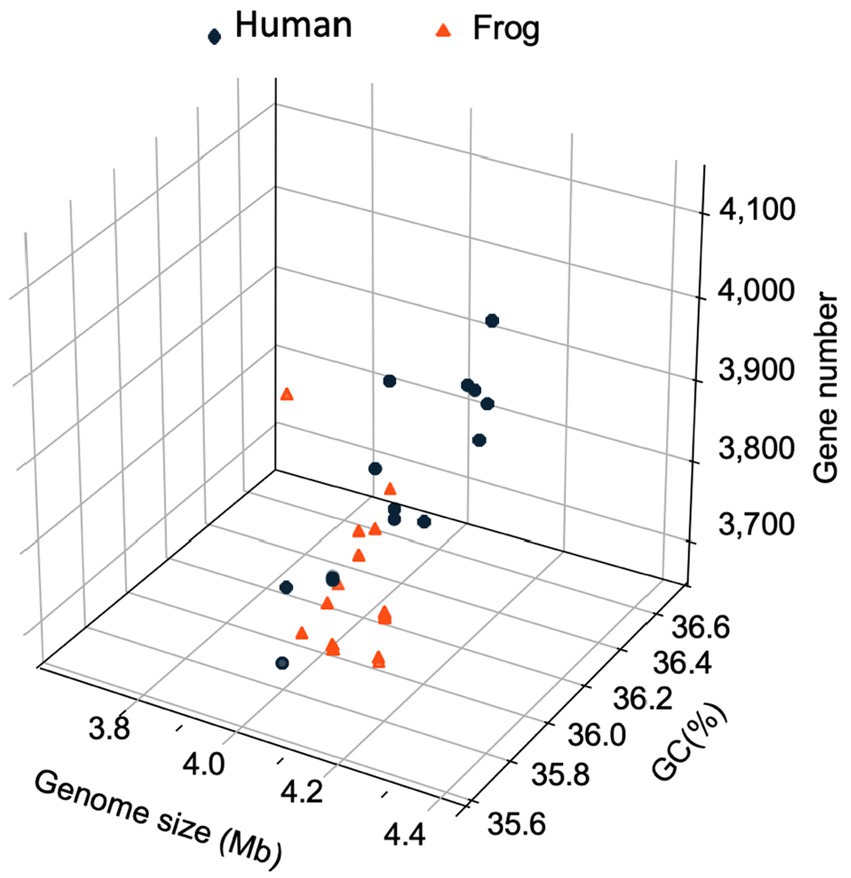
Figure 2. Three-dimensional plots of genome feature in selected E. miricola isolates. A 3D scatter plot illustrating the relationship between genome size (x-axis), coding sequence (CDS) count (y-axis), and GC content (z-axis) across the selected E. miricola isolates.
The genomes of E. miricola Mich-1, Mich-2, Mich-3, and other selected Elizabethkingia species were analyzed using ANI and digital DNA–DNA hybridization (dDDH) (Supplementary Table S2). ANI analysis confirmed that the three isolates belonged to the E. miricola species, as they shared 96.38% identity with the type strain E. miricola DSM 14571 (Figure 3; Supplementary Table S2), exceeding the 95% threshold for species delineation. Furthermore, the ANI values among the three isolates were 100%, indicating they are identical at the genomic level. For comparison, strain CSID_3000516464, also isolated from a Michigan patient in 2016, showed high identity with these strains. Interestingly, isolates from frogs and condensation water exhibited high ANI values with clinical isolates from patients, indicating significant genomic similarity. In contrast, strain BM10, isolated from termites, showed a lower ANI value of 95%, precisely at the species cutoff threshold. ANI values between E. miricola and other Elizabethkingia species, such as E. anophelis and E. meningoseptica, were below 86%, confirming their distinction as separate species. The dDDH analysis further supported the ANI findings. The dDDH values for Mich-1, Mich-2, and Mich-3 relative to the type strain E. miricola DSM 14571 were 73.5%, exceeding the 70% threshold for species definition. Overall, dDDH results were consistent with the ANI analysis (Supplementary Table S2), reinforcing the classification of these isolates within the E. miricola species.
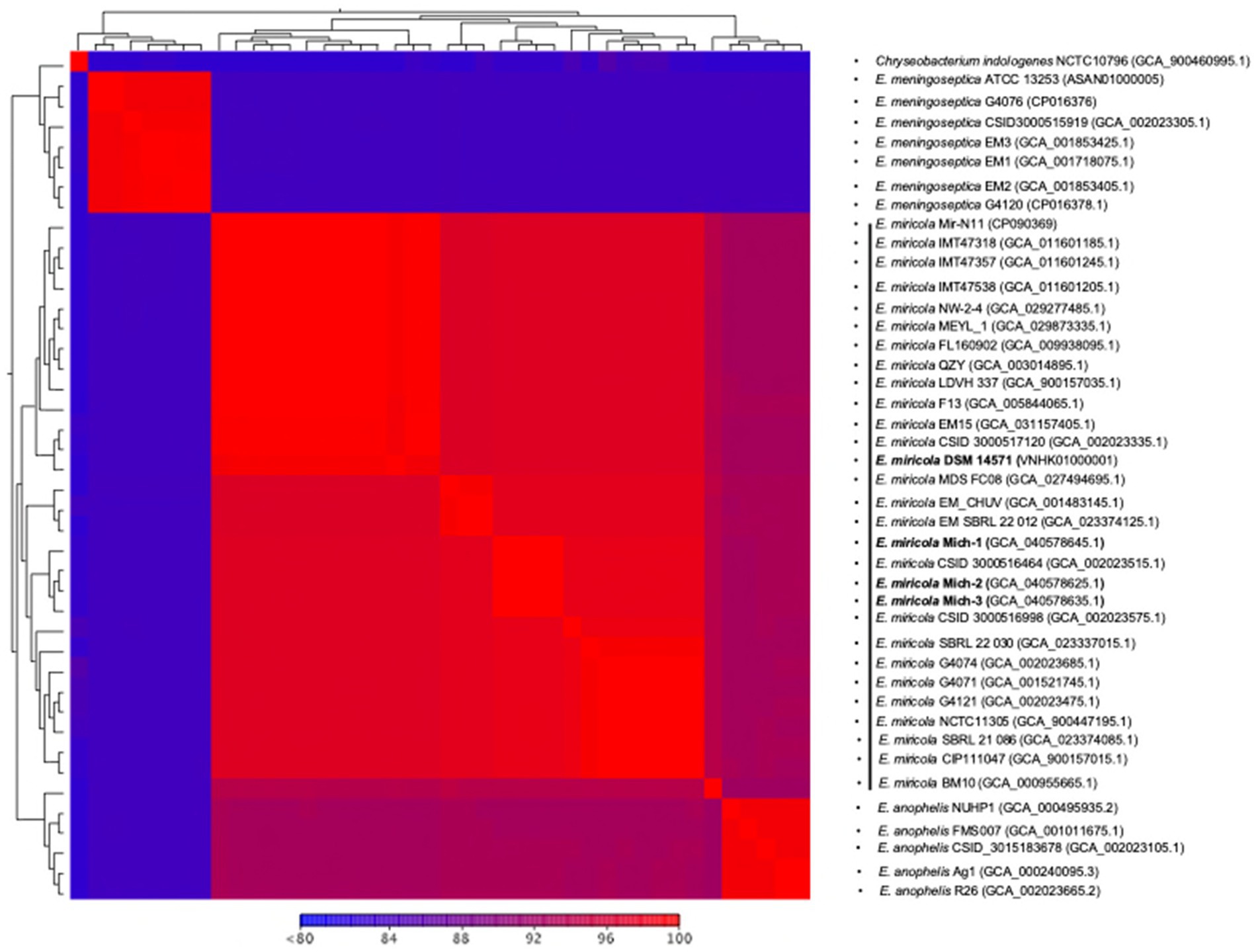
Figure 3. Heatmap of pairwise ANI values of selected Elizabethkingia genomes. Heatmap showing the pairwise Average Nucleotide Identity (ANI) values between the selected Elizabethkingia genomes. The scale ranges from 80 to 100% similarity, with values less than 80% depicted in blue and 100% similarity in red. This heatmap illustrates the genetic relatedness and diversity among the different Elizabethkingia strains, highlighting the degree of genomic similarity between clinical and environmental isolates. The heatmap was generated using ANI analysis.
The four clinical isolates from Michigan patients (E. miricola Mich-1, Mich-2, Mich-3, and CSID_3000516464) formed a single clade (Figure 4). Strains SBRL 22-012, SBRL 21-086, and SBRL 22-030, isolated from Ohio between 2011 and 2012, showed distinct phylogenetic placements. Among these, SBRL 21-086 and SBRL 22-030 formed a separate clade with several clinical strains from European countries, while SBRL 22-012 clustered with E. miricola EM_CHUV, a strain isolated in Switzerland. Strain E. miricola CSID_3000516998 was phylogenetically close to the clade formed by European clinical strains. Interestingly, strain CSID_3000517120, isolated from a Minnesota patient, formed a clade with EM15, a strain from Brazil, within the broader clade of isolates obtained from frogs.
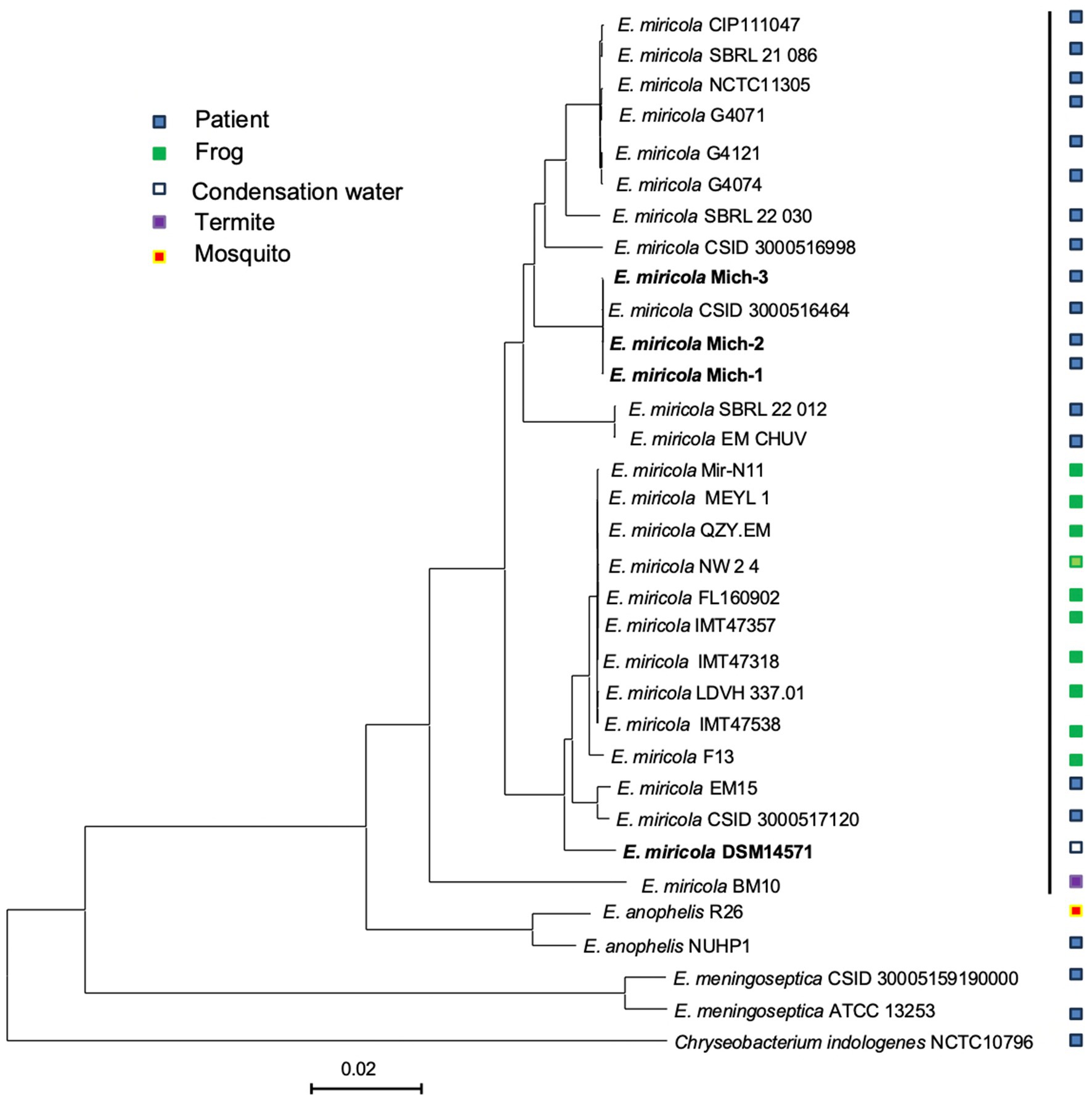
Figure 4. Genome BLAST distance phylogeny tree of selected Elizabethkingia genomes. The GBDP tree was constructed based on whole genome sequencing data. The branch lengths represent genetic distances, and the numbers above the branches indicate GBDP pseudo-bootstrap support values (percentages), with values greater than 60% based on 100 replications. The average branch support across the tree is 43.3%. The tree is rooted at the midpoint, providing a phylogenetic overview of the selected Elizabethkingia strains and their relatedness.
3.3 Gene repertoire of Elizabethkingia miricola
The core and pan-genomes of the selected 28 E. miricola genomes were analyzed to examine their gene repertoire (Figure 5). The core genome, shared by all the genomes, is typically associated with essential housekeeping functions (Supplementary Table S3). It is further categorized into hard-core genes, present in more than 99% of genomes, and soft-core genes, present in 95 to 99% of genomes. The accessory genome, shared by a subset of genomes, is often linked to pathogenicity or environmental adaptation. This category is further divided into shell genes, present in 15 to 95% of genomes, and cloud genes, present in 0 to 15%. Cloud genes include those unique to individual genomes. The pan-genome of the 28 E. miricola isolates comprises a total of 10,944 genes, with 2,201 core genes and 8,743 accessory genes. Within the core genome, 2,025 genes (9%) are hard-core, and 176 are soft-core. The accessory genome is further divided into 2,467 shell genes and 6,100 cloud genes. This genomic organization highlights the balance between conserved elements essential for survival and the diverse accessory components that enable adaptation and pathogenicity.
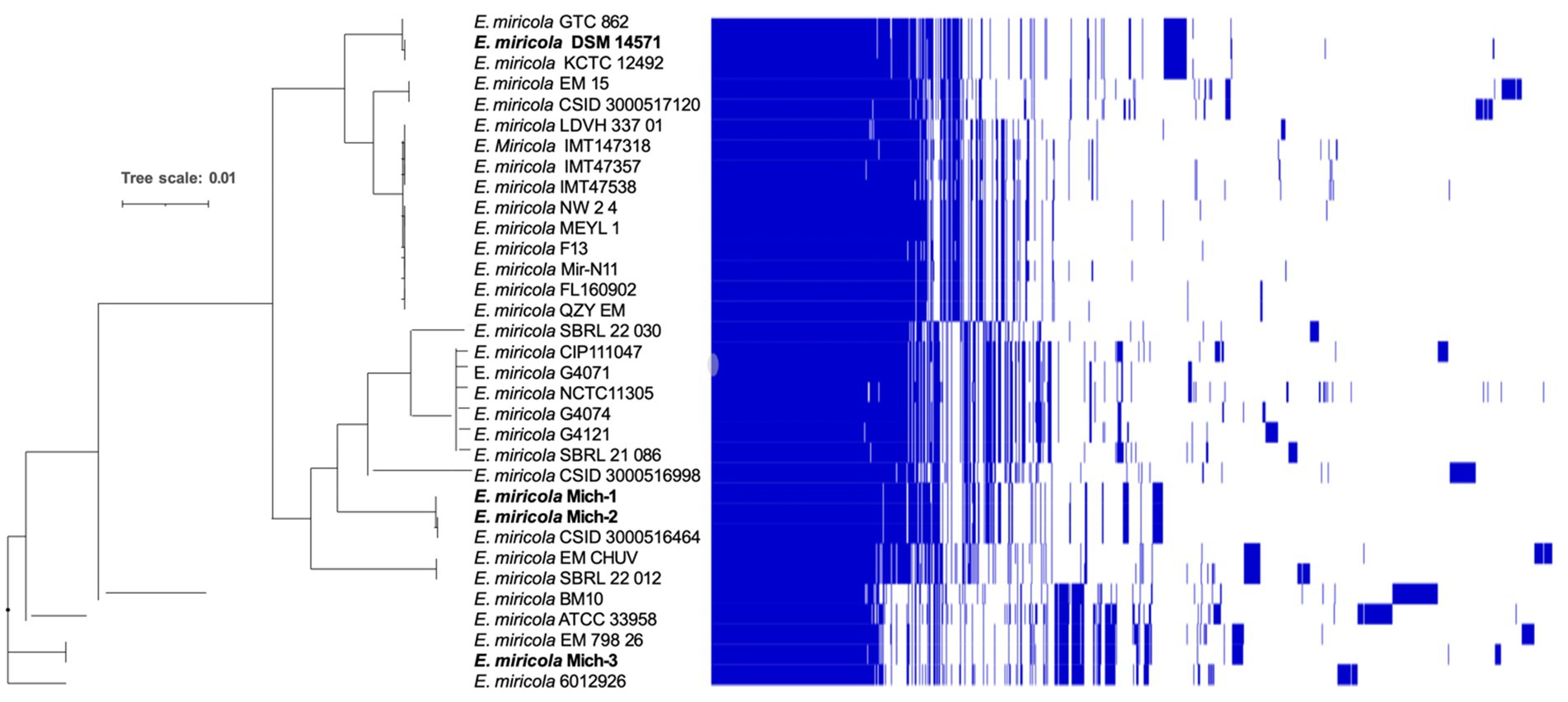
Figure 5. Pangenome analysis of E. miricola. Pan-genome presence and absence heatmap combined with a Maximum Likelihood (ML) tree. The genomes of the strains were clustered based on the presence (blue) and absence (white) of genes. This analysis provides a comprehensive view of the core and accessory genes within the E. miricola strains. The pangenome visualization was generated using Phandango, offering insights into gene distribution and the genomic diversity of the strains (Page et al., 2015; Hadfield et al., 2018).
Elizabethkingia miricola Mich-1, Mich-2, Mich-3, and CSID_3000516464 shared at least 3,837 genes, indicating high genomic similarity among the Michigan isolates (Figure 6A). These strains had only 1 to 5 unique genes, further supporting their close relationship (Figure 6A). However, the genomic content of the Michigan isolates differed significantly from that of isolates from other Midwest regions, such as Ohio and Minnesota. For instance, E. miricola Mich-1 shared 3,505, 3,368, 3,558, and 3,427 genes with SBRL-21-030, SBRL-21-012, SBRL-21-086, and CSID_3000517120, respectively, accounting for 91.1, 87.6, 92.5, and 89.1% of its genome. Collectively, Mich-1 and these four strains shared 3,199 genes (83.2%; Figure 6A). Interestingly, Mich-1 shared more genes with frog isolates (3,319 to 3,375 genes) than with some human clinical isolates from the Midwest (Figure 6B). To explore the pan-genome characteristics of the 28 E. miricola strains, pan-genome and core-genome curves were generated (Figures 7A,B). The results revealed that the pan-genome expanded significantly as more genomes were included, confirming that E. miricola possesses an open pan-genome. Conversely, the core genome size decreased as the number of genomes increased.
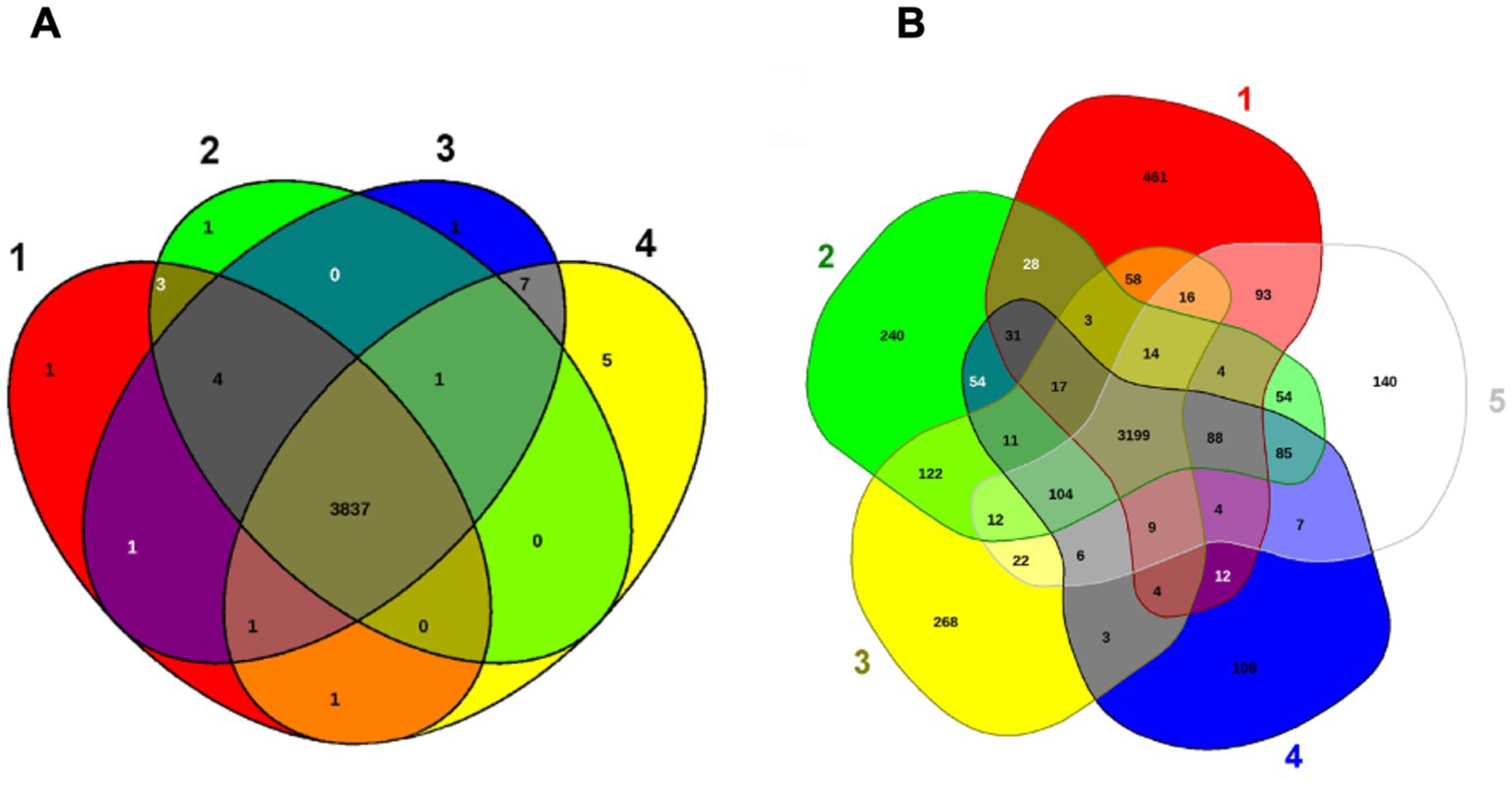
Figure 6. Venn diagram of shared and unique genes of selected Elizabethkingia genomes. The unique and shared genome among the selected strains was determined by a dual cutoff of 30% or greater amino acid identity and sequence length coverage of more than 70%. EDGAR was used for Venn diagrams. (A) 1: E. miricola CSID_3000516464; 2: E. miricola Mich-1; 3: E. miricola Mich-2; 4: E. miricola Mich-3. (B) 1: E. miricola CSID_3000517120, 2: E. miricola SBRL-21-086; 3: E. miricola SBRL-21-012 and E. miricola SBRL-21-030, 5: E. miricola Mich-1.
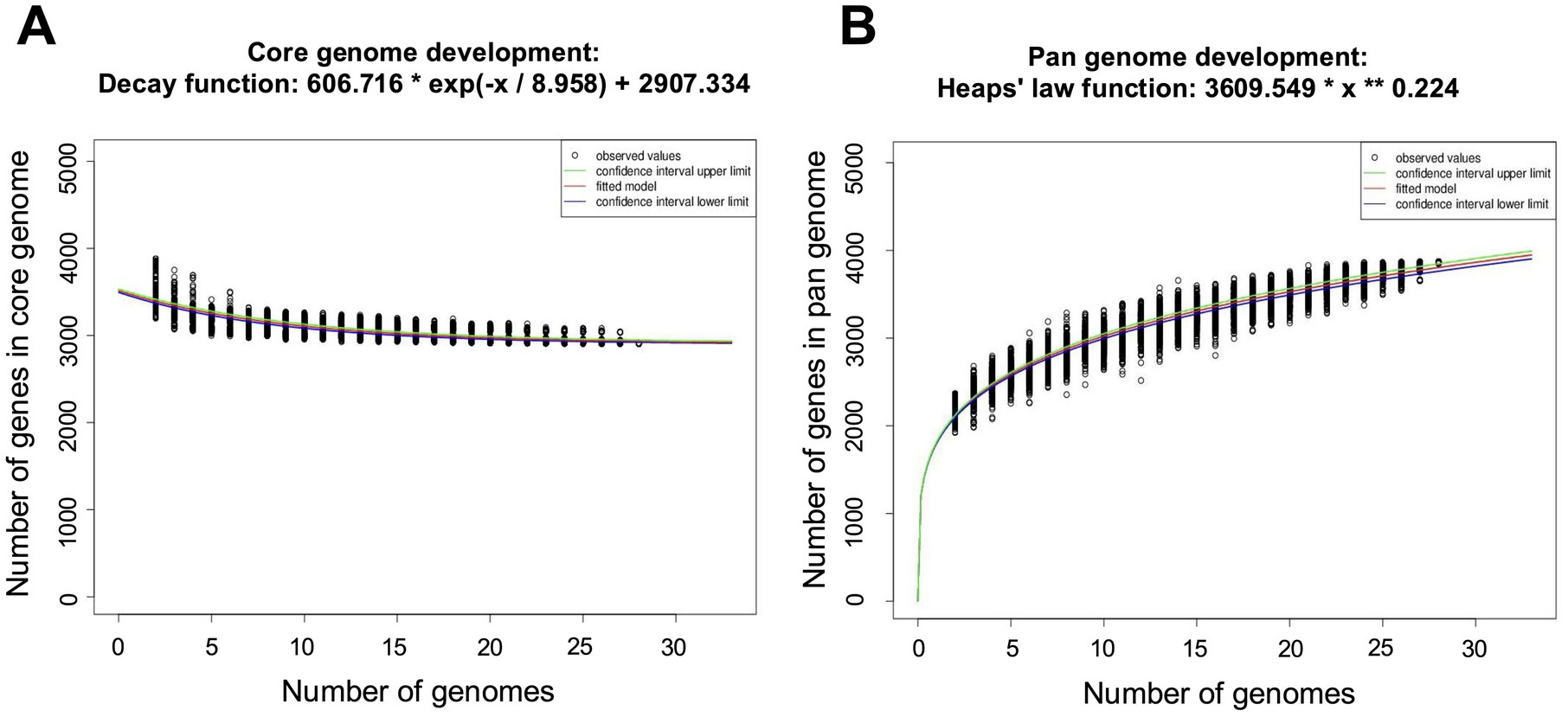
Figure 7. Pan and core genome plots of the 28 selected Elizabethkingia genomes. (A) Number of core genomes for a given number of genomes sequentially added. (B) Number of pan genomes as a function of the number of genomes sequentially added.
3.4 Regulatory system proteins
The genome of E. miricola Mich-1, isolated from Michigan patients, contained genes encoding 63 two-component system proteins, 209 transcription factor proteins, and 10 other DNA-binding proteins, for a total of 282 regulatory proteins (Table 2). Identical counts were observed for Mich-2 and Mich-3. The average total number of regulatory proteins in the Michigan isolates (282) was lower than those from other regions (296) and frog isolates (291) (Table 2). Although the total number of regulatory proteins did not differ significantly between clinical and frog isolates (p > 0.05), notable variations were observed in the numbers of response regulators (RR) and sigma factors (SF). These numbers were lower in clinical isolates compared to frog isolates (Table 2; Figure 8A). Principal component analysis (PCA) of regulatory proteins in clinically relevant isolates further revealed a separation between frog-associated isolates and those from human patients (Figure 8B). This distinction highlights potential differences in regulatory adaptations between environmental and clinical E. miricola strains.
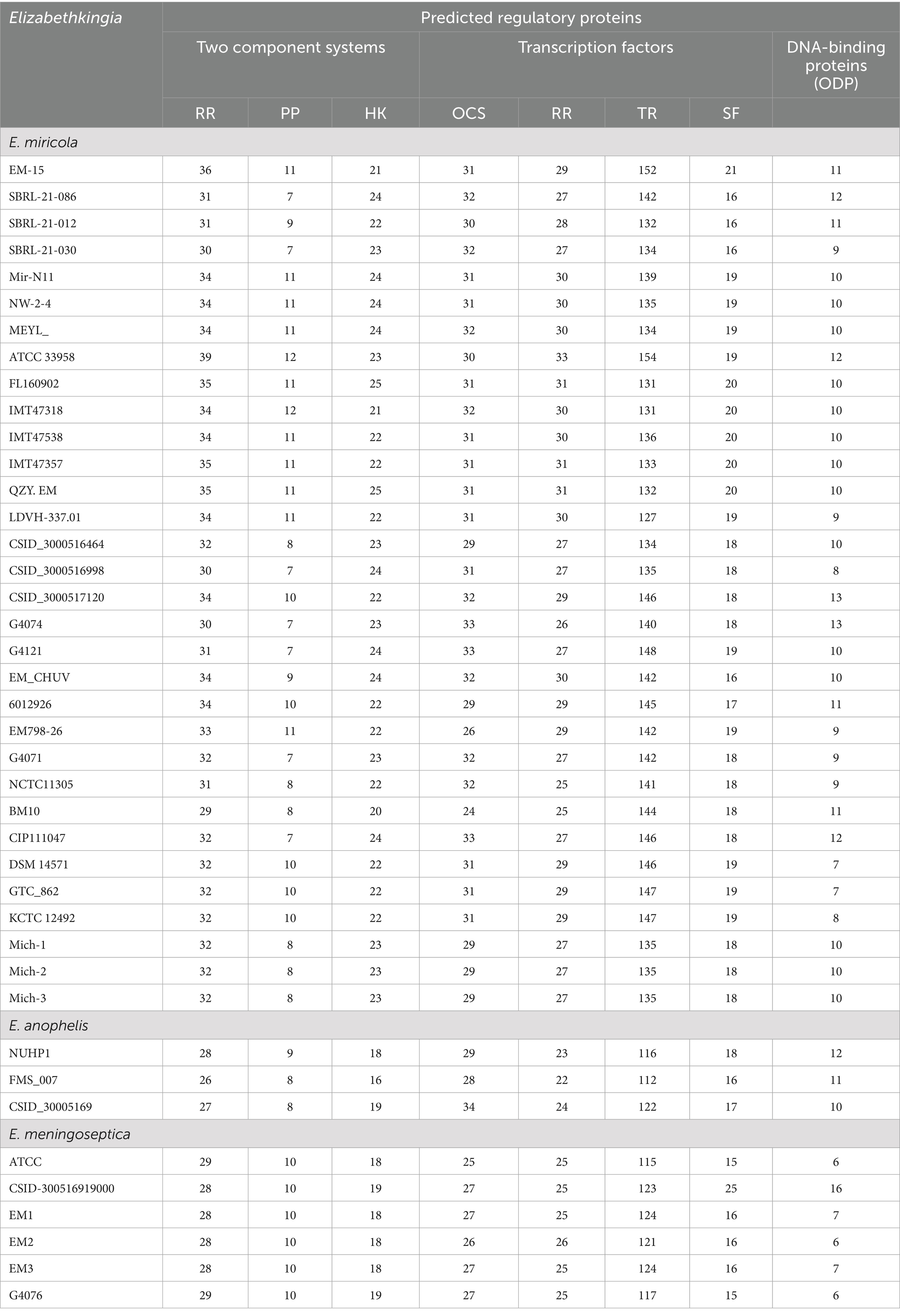
Table 2. Predicted regulatory proteins of selected Elizabethkingia genomes.
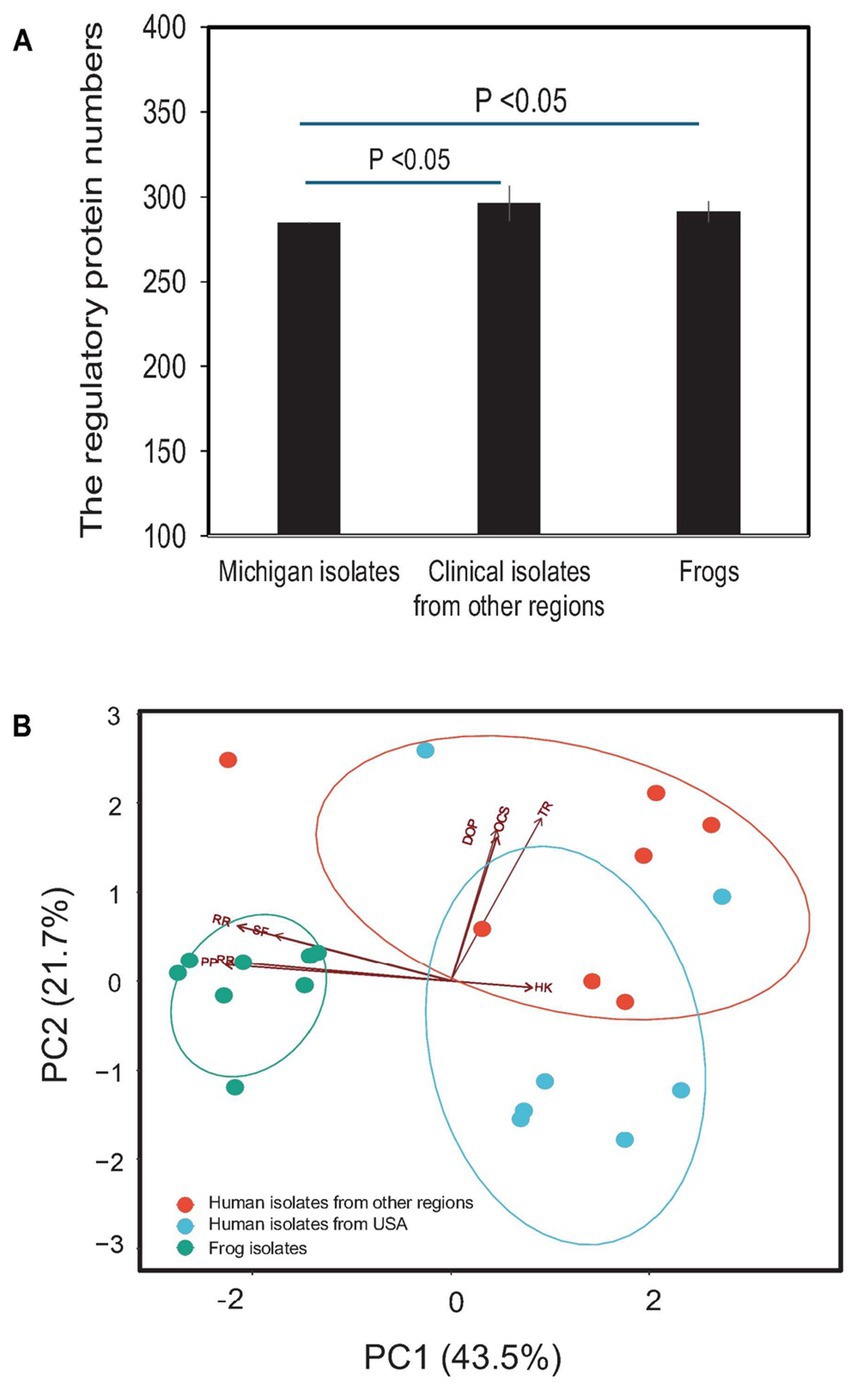
Figure 8. Comparison of predicted regulatory proteins in various E. miricola isolates. (A) Difference of total regulatory protein numbers between Michigan isolates, other clinical isolates, and frog isolates. (B) PCA analysis of various regulatory proteins including response regulators, phosphotransferase proteins, histidine kinases, one-component systems, transcriptional regulators, sigma factors, and other DNA-binding proteins.
3.5 Resistome analysis
The three Michigan isolates exhibited resistance to at least three distinct categories of antibiotics, including aminoglycosides, nitrofurans, and β-lactams. Notably, even β-lactam/inhibitor combinations, such as ampicillin/sulbactam and piperacillin/tazobactam, failed to enhance drug sensitivity. Among the antibiotics tested, the isolates demonstrated susceptibility only to trimethoprim/sulfamethoxazole (a sulfonamide) and ciprofloxacin (a quinolone), while showing intermediate susceptibility to tetracycline (Table 3). Overall, these isolates displayed multi-drug resistance profiles consistent with those previously reported in E. meningoseptica strains isolated from Michigan (Chen et al., 2017, 2019).
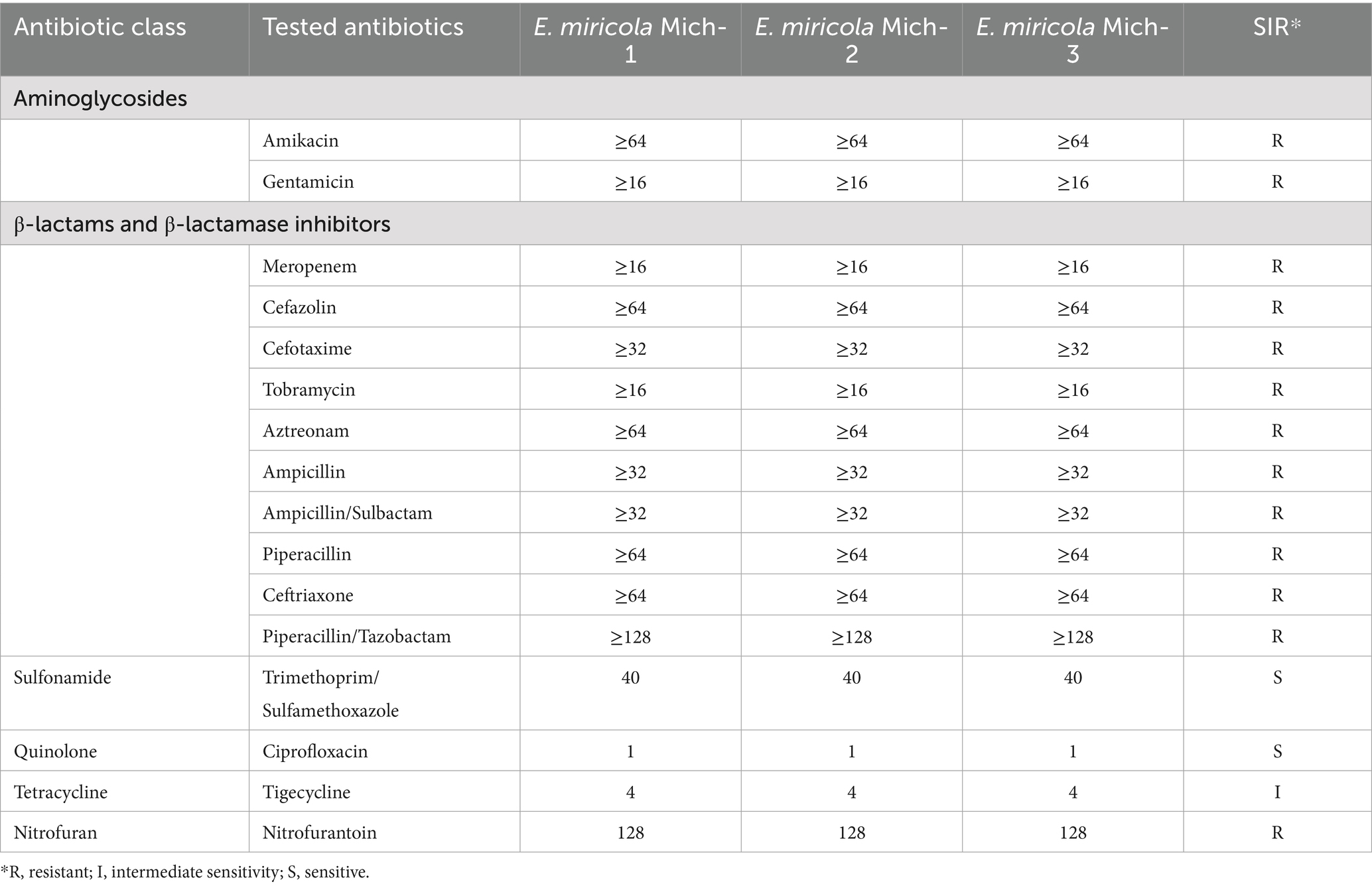
Table 3. Antibiotic susceptibility tests of Michigan isolates.
To correlate the relationship between multidrug resistance phenotypes and their possible genetic determinants, we analyzed the resistance gene profiles in the selected E. miricola strains (Figure 9). There was no difference in the distribution of drug-resistance genes among Michigan isolates (Figure 9; Supplementary Table S4). At least five different β-lactamase genes (BlaB-10, BlaB-39, CME-1, CME-2, GOB-25) were found, which may confer their resistance to cephalosporins, penams, and carbapenems. The aminoglycoside resistance gene aadS can explain its resistance to aminoglycosides. The presence of tet(X4) found in three E. miricola genomes and others may partially account for its intermediate sensitivity to tigecycline. Genes encoding several efflux pumps were also discovered, which may confer resistance to nitrofurantoin and other tested drugs in this study (Supplementary Table S4; Table 3). Collectively, certain subtypes of the antimicrobial resistance (AMR) genes were more specifically associated with isolates from either frogs or humans (Supplementary Table S4). For example, GOB-25 (metallo-β-lactamase, subclass B3) was absent in frog isolates. Instead, BlaB-16 was only found in the frog isolates (Supplementary Table S4). However, there were also some shared among these E. miricola. Most E. miricola strains carried CME-2 (class A β-lactamase) while some carried both of CME-1 and CME-2 (Supplementary Table S4; Figure 9).
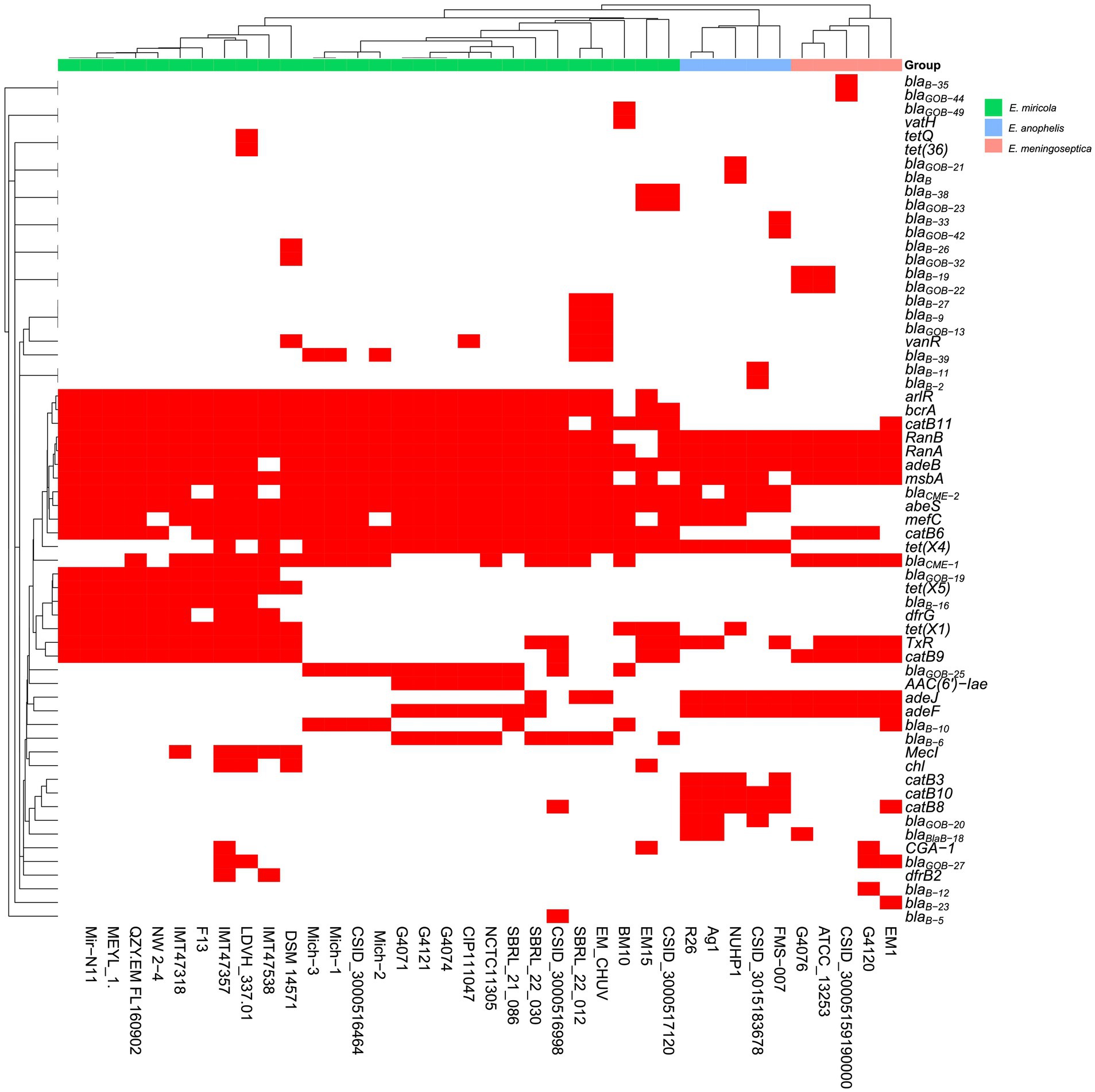
Figure 9. The presence and absence of predicted antibiotic resistance gene based on whole-genome sequence for each isolate. Red squares represented the presence of the related antibiotic resistance genes and white squares represented the absence of the related antibiotic resistance genes.
3.6 Virulence-associated genes of the various Elizabethkingia miricola strains
The virulence factors in the selected Elizabethkingia species were predicted using the VFDB (Chen et al., 2016). Consistent with previous studies on virulence factors in Elizabethkingia, genes involved in capsular polysaccharide biosynthesis, elongation factors, heat shock proteins, phospholipases, catalases, peroxidases, and various other factors were found in nearly all of the selected E. miricola genomes (Supplementary Table S5). These genes were reported to participate in stress survival, immune modulation, and environmental adherence (Kukutla et al., 2014; Teo et al., 2014; Breurec et al., 2016; Janda and Lopez, 2017; Perrin et al., 2017; Lin et al., 2019; Zajmi et al., 2022; Andriyanov et al., 2024). For example, htpB, fimH, rmlA and EF-Tu involved in capsule polysaccharide biosynthesis, adherence and invasion were found (Supplementary Table S5). They may facilitate evasion or neutralization of host immune responses and play a pivotal role in biofilm formation and surface colonization (Frees et al., 2004; Zarankiewicz et al., 2012; Hu et al., 2022a; Puah et al., 2022). Iron/heme utilization genes including iutA and hemL are conserved in most of E. miricola (Supplementary Table S5). E. miricola also carried many enzymes likely involved in pathogenesis. For example, the katG gene, conserved across Elizabethkingia species, encodes a catalase-peroxidase heme enzyme known to participate in iron metabolism and stress responses (Ratledge and Dover, 2000; Skaar, 2010; Chen et al., 2020). Urease genes (ureB and ureG) were found in E. miricola, which may play a pivotal role in the pathogenesis of gram-negative bacteria by facilitating survival and colonization in acidic environments (Supplementary Table S5).
4 Discussion
Elizabethkingia miricola is an emerging pathogen that poses a significant threat to human health (Zajmi et al., 2022; Soler-Iborte et al., 2024; Wu et al., 2024). The infection control of Elizabethkingia is challenging due to its intrinsic resistance to many antibiotics, difficult identification, unclear transmission pathways, and ability to persist in healthcare settings (Zajmi et al., 2022; Huang et al., 2024; Wu et al., 2024). Moreover, it can cause various diseases because it has been frequently isolated from the oral cavity, sputum, pulmonary abscesses, urine, CSF, and blood specimens (Choi et al., 2019; Huang et al., 2024). However, little is known about genetic compositions and features of these pathogens (Hu et al., 2020; Wu et al., 2024). To better understand its pathogenic potentials and multi-drug resistance mechanisms, we systematically conducted a comparative genomic analysis between human and frog isolates and its phylogenetic neighbors E. anophelis and E. meningoseptica (Lin et al., 2019; Puah et al., 2022).
The accurate identification of Elizabethkingia to the species level is challenging using morphological observations, routine biochemical tests, and MALDI-TOF MS in the clinical microbiology laboratory (Ransangan et al., 2013; Nicholson et al., 2018; Kadi et al., 2022; Puah et al., 2022). As demonstrated in this study and others, E. miricola was misidentified as E. anophelis or E. meningoseptica. Updates to the latest MALDI-TOF MS libraries are necessary for definitive species identification (McTaggart et al., 2019; Hem et al., 2022; Kadi et al., 2022). 16S rRNA sequence is helpful for molecular identification but it is limited in its taxonomic utility due to the sequence conservation in Elizabethkingia (Nicholson et al., 2018; Kadi et al., 2022). For example, Lee et al. reported that the homology of the two 16S rRNA sequences of E. miricola BM10 was more than 98% with E. anophelis R26, E. meningoseptica ATCC 13253, and E. miricola GTC 862 (Lee et al., 2019). However, strain E. miricola BM10 showed a low ANI value (93.36% identity) with that in three type strains in the genus Elizabethkingia, which agrees that it may need to be reclassified to a new genus within Elizabethkingia. Therefore, whole genomic sequence analysis, together with ANI or dDDH, may be used to correctly identify E. miricola in the future (Eriksen et al., 2017; McTaggart et al., 2019; Hem et al., 2022).
Unlike other Elizabethkingia species, E. miricola is well known to cause meningitis-like diseases in frogs and cause outbreaks from time to time. Zoonotic transmission from invertebrates to humans is possible but needs more investigation (Ransangan et al., 2013; Zajmi et al., 2022; Huang et al., 2024; Soler-Iborte et al., 2024). Most human isolates formed a different clade from those found in frogs; however, two strains, one from Minnesota and another from Brazil, were phylogenetically close to frog isolates. The genome size, total gene numbers, and average GC contents in isolates from human patients were not significantly different from those isolated from frogs (p > 0.05). Moreover, the pan-genomes of E. miricola showed that it is evolving through the loss or gain of various genes, which is similar to these observations in E. anophelis, E. meningoseptica, and other flavobacteria. E. miricola lives in diverse environments, including aquatic, terrestrial environments, vertebrates, and invertebrate animals (McTaggart et al., 2019; Hem et al., 2022). It seems that the bacterium adapts to the respective niche environments (Hu et al., 2020; Yang et al., 2021; Soler-Iborte et al., 2024). Thus, it is expected that E. miricola has an open pan-genome.
Resistance to β-lactams, tetracycline, and aminoglycosides is particularly concerning, as these drugs are commonly used for treating gram-negative bacterial infections (Hu et al., 2020; Comba et al., 2022; Huang et al., 2024). Comba et al. (2022) reported that 92% (11/12), 50% (6/12), and 83.3% (10/12) of E. miricola isolates from the United States were susceptible to piperacillin-tazobactam, ciprofloxacin, and TMP-SMX, respectively (Comba et al., 2022). However, our three isolates were resistant to both piperacillin and piperacillin-tazobactam but remained susceptible to ciprofloxacin and TMP-SMX. In contrast, Wu et al. (2024) found that 100% of 71 Elizabethkingia isolates from China were resistant to piperacillin, and 64% were resistant to piperacillin-tazobactam (Wu et al., 2024). These findings suggest that Elizabethkingia strains from diverse regions and environments may evolve distinct antibiotic resistance mechanisms (Hu et al., 2022b). However, the clinical significance of these variations remains uncertain due to the lack of standardized interpretive breakpoints for antimicrobial resistance in Elizabethkingia spp. (Comba et al., 2022).
A diverse array of drug-resistance genes has been reported in E. anophelis and E. meningoseptica (Breurec et al., 2016; McTaggart et al., 2019; Comba et al., 2022; Hem et al., 2022; Andriyanov et al., 2024; Wu et al., 2024). However, the profiles of MDR genes have not been comprehensively documented in E. miricola. Our results revealed that E. miricola harbors two distinct metallo-β-lactamase (MBL) genes (BlaB and GOB), as well as the CME gene. Remarkably, we found diverse subtypes of the GOB and BlaB genes in E. miricola. Chang et al. (2019) investigated the amino acid sequences of BlaB and GOB and divided them into 22 and 25 different types in Elizabethkingia, respectively (Chang et al., 2019). Their phylogenetic analysis showed BlaB and GOB are species-specific proteins. However, the simultaneous presence of both MBL and CME genes may explain its wide resistance to various β-lactams and combination with the β-lactam inhibitors in Elizabethkingia. Even novel β-lactamase inhibitors did not significantly enhance the activity of β-lactams (Yasmin et al., 2023). It is possible that those β-lactamase inhibitors may be better on certain β-lactamases while they had little effect on MBLs in Elizabethkingia (Kuo et al., 2021). Our discoveries showed that MBLs are intrinsically present in all E. miricola, which may confer resistance to Ampicillin/Sulbactam and Piperacillin/Tazobactam (Chang et al., 2019, 2021; Yasmin et al., 2023). Some of them carried several copies of MBL subtypes. Notably, BlaB-39 (encoding a class B β-lactamase, subclass B1) was unique to Michigan isolates and E. miricola EM_CHUV, suggesting distinct evolutionary pathways. In contrast, GOB-25 (subclass B3) was absent in frog isolates, while BlaB-16 was exclusively found in frog strains, highlighting unique gene distributions across environments. Two new chromosomal MBL (blaBlaB-16 and blaGOB-19) variants were found in the genome of E. miricola FL160902 (Hu et al., 2020, 2022b). Homologous expression of the two genes in E. coli resulted in increased MICs of most β-lactams, including imipenem, meropenem and ampicillin (Hu et al., 2020). blaGOB-13 and blaB-9 carbapenemase-encoding genes were reported in E. miricola EM_CHUV (Kuo et al., 2021). Chen et al. (2024) investigated the individual contributions of blaB, blaGOB and blaCME on MICs of β-lactams in Elizabethkingia, showing that the constitutively and highly expressed blaB gene significantly increased MICs of carbapenems, decreasing their efficacy in vivo (Berlin et al., 2015). Additionally, their studies demonstrated that CME raised MICs for ceftazidime and cefepime (Berlin et al., 2015). Collectively, regardless of allelic combinations, our findings underline the complexity of β-lactam resistance in E. miricola. Resistance genes detected in Michigan isolates included aadS, artR, tex(x4), bcrA, and catB6, which can contribute to the multidrug resistance of Elizabethkingia and complicate clinical treatment.
While the mechanisms underlying Elizabethkingia pathogenesis remain largely unknown, we found some virulence factors (VFs) involved in adherence, antiphagocytosis and immune evasion commonly conserved in E. miricola (Perrin et al., 2017; Hu et al., 2024). For example, the capD gene in E. miricola strain FL160902 is located in the conserved region of the Wzy-dependent capsule synthesis gene cluster (Hu et al., 2024). Deletion of capD results in an impaired capsule structure, notably reducing cell wall thickness. The mutant strain exhibits a significantly lower survival rate in complement-mediated killing assays and an increased capacity to evade macrophage phagocytosis. In frog infection models, the absence of the polysaccharide capsule attenuates virulence. Additionally, capD deletion increases bacterial surface hydrophobicity while reducing desiccation resistance and biofilm formation (Hu et al., 2024).
Elizabethkingia miricola is well known to cause bloodstream infections and meningitis (Howard et al., 2020; Gao et al., 2021). Therefore, it is important to understand its iron/heme utilization mechanisms in the disease courses (Chen et al., 2015, 2020). iutA is part of the iucABCD-iutA operon responsible for aerobactin biosynthesis and uptake in E. miricola genomes (Chen et al., 2020). Under the iron-limited condition, gene expression of iutA was significantly upregulated, indicating its important roles in iron metabolism (Chen et al., 2020). Chen et al. (2020) further demonstrated that deleting the aerobactin biosynthesis gene cluster in E. anophelis impairs iron uptake, increases oxidative damage from H2 O2, and reduces biofilm formation (Chen et al., 2020). Interestingly, typical alpha or beta hemolysis activity was not observed on sheep blood agar after culturing E. miricola strains for 24 h, despite the prediction of a hemolysin gene (hlyB). Andriyanov et al. (2024) similarly reported that E. anophelis exhibited no hemolysis on 5% bovine blood agar and showed delayed hemolysis on 5% O-human blood agar (Andriyanov et al., 2024). Clear lysis zones appeared only beneath dense colonies with higher biomass (Andriyanov et al., 2024). This suggests that Elizabethkingia may employ a slower or more regulated mechanism to break down red blood cells and access iron/heme, possibly preventing excessive host immune activation (Choby and Skaar, 2016; Donegan et al., 2019). If delayed hemolysis enables sustained iron acquisition in vivo, it might help explain the bacterium’s persistence during infections (Choi et al., 2019; Huang et al., 2024). Ureases catalyze the hydrolysis of urea into ammonia and carbon dioxide, which increases the local pH, countering acidic conditions such as those found in the stomach or urinary tract (Konieczna et al., 2012). This process can lead to chronic infections and complications. As in Helicobacter (Elbehiry et al., 2025), it is possible that the ammonia produced by urease in E. miricola can be toxic to host cells, disrupt epithelial integrity, and elicit an inflammatory response, further aiding in tissue colonization and immune evasion. However, further studies are warranted to elucidate the mechanisms underlying these putative virulence factors.
In conclusion, our study provides the first detailed report on the isolation and characterization of three pathogenic E. miricola strains in the United States. All three isolates demonstrated high levels of resistance to most major antimicrobial agent families. Notably, discrepancies between antibiotic susceptibility testing methods underscore the urgent need for standardized interpretative guidelines for Elizabethkingia spp., particularly to enhance clinical decision-making. Through comprehensive whole-genome sequencing analysis of E. miricola strains isolated from humans, frogs, condensation water, and termites, we identified a diverse repertoire of resistance genes, including blaB, blaGOB, and blaCME, which confer resistance to a broad range of β-lactams. Of particular concern, we identified frog-derived isolates closely related to clinical strains, emphasizing the potential for cross-habitat transmission. Key genes involved in stress regulation, adherence, and immune modulation were conserved across both frog and clinical isolates, further supporting the likelihood of transmission from aquatic environments to clinical settings. This adaptability not only enhances their resistance and virulence but also underscores the significant threats they pose to both human and animal health.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, JBEUGN000000000, JBEUGO000000000, and JBEUGP000000000.
Author contributions
SC: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. GA: Data curation, Investigation, Methodology, Writing – review & editing. JB: Data curation, Formal analysis, Visualization, Writing – review & editing. EW: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project was funded by Startup Grant, the Seed Grant of College of Health and Human Sciences at Northern Illinois University (awarded to SC) and NIH grant R37AI21884 (awarded to EW).
Acknowledgments
The authors expressed their gratitude to the Department of Microbiology at Henry Ford Health System (Jackson, Michigan) and for their assistance in antimicrobial susceptibility determination and identification by MALDI-ToF and Dr. Marty Soehnlen at Michigan Department of Health and Human Services to provide the E. miricola strains.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1582121/full#supplementary-material
Footnotes
References
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–D699. doi: 10.1093/nar/gkac920
Andriyanov, P., Zhurilov, P., Menshikova, A., Tutrina, A., Yashin, I., and Kashina, D. (2024). Large-scale genomic analysis of Elizabethkingia anophelis. BMC Genomics 25:1015. doi: 10.1186/s12864-024-10921-y
Barakat, M., Ortet, P., and Whitworth, D. E. (2013). P2RP: a web-based framework for the identification and analysis of regulatory proteins in prokaryotic genomes. BMC Genomics 14:269. doi: 10.1186/1471-2164-14-269
Berlin, K., Koren, S., Chin, C.-S., Drake, J. P., Landolin, J. M., and Phillippy, A. M. (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630. doi: 10.1038/nbt.3238
Breurec, S., Criscuolo, A., Diancourt, L., Rendueles, O., Vandenbogaert, M., Passet, V., et al. (2016). Genomic epidemiology and global diversity of the emerging bacterial pathogen Elizabethkingia anophelis. Sci. Rep. 6:30379. doi: 10.1038/srep30379
Chang, T.-Y., Chen, H.-Y., Chou, Y.-C., Cheng, Y.-H., and Sun, J.-R. (2019). In vitro activities of imipenem, vancomycin, and rifampicin against clinical Elizabethkingia species producing BlaB and GOB metallo-beta-lactamases. Eur. J. Clin. Microbiol. Infect. Dis. 38, 2045–2052. doi: 10.1007/s10096-019-03639-3
Chang, Y., Zhang, D., Niu, S., Chen, Q., Lin, Q., and Zhang, X. (2021). MBLs, rather than efflux pumps, led to Carbapenem resistance in Fosfomycin and Aztreonam/avibactam resistant Elizabethkingia anophelis. Infect. Drug Resist. 14, 315–327. doi: 10.2147/IDR.S294149
Chen, S., Bagdasarian, M., and Walker, E. D. (2015). Elizabethkingia anophelis: molecular manipulation and interactions with mosquito hosts. Appl. Environ. Microbiol. 81, 2233–2243. doi: 10.1128/AEM.03733-14
Chen, S., Johnson, B. K., Yu, T., Nelson, B. N., and Walker, E. D. (2020). Elizabethkingia anophelis: physiologic and transcriptomic responses to iron stress. Front. Microbiol. 11:804. doi: 10.3389/fmicb.2020.00804
Chen, S., Soehnlen, M., Blom, J., Terrapon, N., Henrissat, B., and Walker, E. D. (2019). Comparative genomic analyses reveal diverse virulence factors and antimicrobial resistance mechanisms in clinical Elizabethkingia meningoseptica strains. PLoS One 14:e0222648. doi: 10.1371/journal.pone.0222648
Chen, S., Soehnlen, M., Downes, F. P., and Walker, E. D. (2017). Insights from the draft genome into the pathogenicity of a clinical isolate of Elizabethkingia meningoseptica Em3. Stand. Genomic Sci. 12:56. doi: 10.1186/s40793-017-0269-8
Chen, P.-J., Tan, M.-C., Huang, W.-C., Hsu, S.-Y., Chen, T.-L., Yang, C.-Y., et al. (2024). The individual contributions of Bla B, Bla GOB and Bla CME on MICs of β-lactams in Elizabethkingia anophelis. J. Antimicrob. Chemother. 79, 1577–1580. doi: 10.1093/jac/dkae137
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Choby, J. E., and Skaar, E. P. (2016). Heme synthesis and acquisition in bacterial pathogens. J. Mol. Biol. 428, 3408–3428. doi: 10.1016/j.jmb.2016.03.018
Choi, M. H., Kim, M., Jeong, S. J., Choi, J. Y., Lee, I.-Y., Yong, T.-S., et al. (2019). Risk factors for Elizabethkingia acquisition and clinical characteristics of patients, South Korea. Emerg. Infect. Dis. 25, 42–51. doi: 10.3201/eid2501.171985
Comba, I. Y., Schuetz, A. N., Misra, A., Friedman, D. Z. P., Stevens, R., Patel, R., et al. (2022). Antimicrobial susceptibility of Elizabethkingia species: report from a reference laboratory. J. Clin. Microbiol. 60:e0254121. doi: 10.1128/jcm.02541-21
Coyle, A. L. (2017). Elizabethkingia anophelis. Nursing (Brux) 47, 61–63. doi: 10.1097/01.NURSE.0000512887.67622.84
Dieckmann, M. A., Beyvers, S., Nkouamedjo-Fankep, R. C., Hanel, P. H. G., Jelonek, L., Blom, J., et al. (2021). EDGAR3.0: comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 49, W185–W192. doi: 10.1093/nar/gkab341
Donegan, R. K., Moore, C. M., Hanna, D. A., and Reddi, A. R. (2019). Handling heme: the mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 133, 88–100. doi: 10.1016/j.freeradbiomed.2018.08.005
Elbehiry, A., Marzouk, E., Abalkhail, A., Sindi, W., Alzahrani, Y., Alhifani, S., et al. (2025). Pivotal role of Helicobacter pylori virulence genes in pathogenicity and vaccine development. Front. Med. (Lausanne) 11:1523991. doi: 10.3389/fmed.2024.1523991
Eriksen, H. B., Gumpert, H., Faurholt, C. H., and Westh, H. (2017). Determination of Elizabethkingia diversity by MALDI-TOF mass spectrometry and whole-genome sequencing. Emerg. Infect. Dis. 23, 320–323. doi: 10.3201/eid2302.161321
Frees, D., Chastanet, A., Qazi, S., Sørensen, K., Hill, P., Msadek, T., et al. (2004). Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 54, 1445–1462. doi: 10.1111/j.1365-2958.2004.04368.x
Gao, H., Li, T., Feng, L., and Zhang, S. (2021). Elizabethkingia miricola causes intracranial infection: a case study. Front. Med. (Lausanne) 8:761924. doi: 10.3389/fmed.2021.761924
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Hem, S., Jarocki, V. M., Baker, D. J., Charles, I. G., Drigo, B., Aucote, S., et al. (2022). Genomic analysis of Elizabethkingia species from aquatic environments: evidence for potential clinical transmission. Curr. Res. Microb. Sci. 3:100083. doi: 10.1016/j.crmicr.2021.100083
Howard, J. C., Chen, K., Anderson, T., and Dalton, S. C. (2020). Elizabethkingia miricola bacteraemia in a haemodialysis patient. Access Microbiol. 2:acmi000098. doi: 10.1099/acmi.0.000098
Hu, R., Liu, F., Yu, F., Hou, J., Chen, D., and Gu, Z. (2024). capD deletion in the Elizabethkingia miricola capsular locus leads to capsule production deficiency and reduced virulence. Vet. Res. 55:148. doi: 10.1186/s13567-024-01394-8
Hu, S., Lv, Y., Xu, H., Zheng, B., and Xiao, Y. (2022a). Biofilm formation and antibiotic sensitivity in Elizabethkingia anophelis. Front. Cell. Infect. Microbiol. 12:953780. doi: 10.3389/fcimb.2022.953780
Hu, S., Xu, H., Meng, X., Bai, X., Xu, J., Ji, J., et al. (2022b). Population genomics of emerging Elizabethkingia anophelis pathogens reveals potential outbreak and rapid global dissemination. Emerg. Microbes Infect. 11, 2590–2599. doi: 10.1080/22221751.2022.2132880
Hu, R., Zhang, Q., and Gu, Z. (2020). Whole-genome analysis of the potentially zoonotic Elizabethkingia miricola FL160902 with two new chromosomal MBL gene variants. J. Antimicrob. Chemother. 75, 526–530. doi: 10.1093/jac/dkz480
Huang, C., Kuo, S., and Lin, L. (2024). Mortality risk and antibiotic therapy for patients with infections caused by Elizabethkingia species—a meta-analysis. Medicina (B Aires) 60:1529. doi: 10.3390/medicina60091529
Janda, J. M., and Lopez, D. L. (2017). Mini review: new pathogen profiles: Elizabethkingia anophelis. Diagn. Microbiol. Infect. Dis. 88, 201–205. doi: 10.1016/j.diagmicrobio.2017.03.007
Kadi, H., Tanriverdi Cayci, Y., Yener, N., Gur Vural, D., Bilgin, K., and Birinci, A. (2022). 16s rRNA-based phylogenetic analyses of Elizabethkingia anophelis: detection of Elizabethkingia anophelis, a rare infectious agent from blood and determination of antibiotic susceptibility in Turkey. Indian J. Med. Microbiol. 40, 557–559. doi: 10.1016/j.ijmmb.2022.07.004
Konieczna, I., Zarnowiec, P., Kwinkowski, M., Kolesinska, B., Fraczyk, J., Kaminski, Z., et al. (2012). Bacterial urease and its role in long-lasting human diseases. Curr. Protein Pept. Sci. 13, 789–806. doi: 10.2174/138920312804871094
Kukutla, P., Lindberg, B. G., Pei, D., Rayl, M., Yu, W., Steritz, M., et al. (2014). Insights from the genome annotation of Elizabethkingia anophelis from the malaria vector Anopheles gambiae. PLoS One 9:e97715. doi: 10.1371/journal.pone.0097715
Kuo, S.-C., Tan, M.-C., Huang, W.-C., Wu, H.-C., Chen, F.-J., Liao, Y.-C., et al. (2021). Susceptibility of Elizabethkingia spp. to commonly tested and novel antibiotics and concordance between broth microdilution and automated testing methods. J. Antimicrob. Chemother. 76, 653–658. doi: 10.1093/jac/dkaa499
Lee, Y.-L., and Hsueh, P.-R. (2023). Emerging infections in vulnerable hosts: Stenotrophomonas maltophilia and Elizabethkingia anophelis. Curr. Opin. Infect. Dis. 36, 481–494. doi: 10.1097/QCO.0000000000000953
Lee, D., Kim, Y.-K., Kim, Y.-S., and Kim, T.-J. (2019). Elizabethkingia miricola BM10, a new symbiotic bacterium isolated from the hindgut of the termite Reticulitermes speratus KMT001. J. Korean Wood Sci. Technol. 47, 692–699. doi: 10.5658/WOOD.2019.47.6.692
Lin, J.-N., Lai, C.-H., Yang, C.-H., and Huang, Y.-H. (2019). Elizabethkingia infections in humans: from genomics to clinics. Microorganisms 7:295. doi: 10.3390/microorganisms7090295
Mallinckrodt, L., Huis Int Veld, R., Rosema, S., Voss, A., and Bathoorn, E. (2023). Review on infection control strategies to minimize outbreaks of the emerging pathogen Elizabethkingia anophelis. Antimicrob. Resist. Infect. Control 12:97. doi: 10.1186/s13756-023-01304-1
McTaggart, L. R., Stapleton, P. J., Eshaghi, A., Soares, D., Brisse, S., Patel, S. N., et al. (2019). Application of whole genome sequencing to query a potential outbreak of Elizabethkingia anophelis in Ontario, Canada. Access Microbiol. 1:e000017. doi: 10.1099/acmi.0.000017
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Nicholson, A. C., Gulvik, C. A., Whitney, A. M., Humrighouse, B. W., Graziano, J., Emery, B., et al. (2018). Revisiting the taxonomy of the genus Elizabethkingia using whole-genome sequencing, optical mapping, and MALDI-TOF, along with proposal of three novel Elizabethkingia species: Elizabethkingia bruuniana sp. nov., Elizabethkingia ursingii sp. nov., and Elizabethkingia occulta sp. nov. Antonie Van Leeuwenhoek 111, 55–72. doi: 10.1007/s10482-017-0926-3
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Perrin, A., Larsonneur, E., Nicholson, A. C., Edwards, D. J., Gundlach, K. M., Whitney, A. M., et al. (2017). Evolutionary dynamics and genomic features of the Elizabethkingia anophelis 2015 to 2016 Wisconsin outbreak strain. Nat. Commun. 8:15483. doi: 10.1038/ncomms15483
Puah, S. M., Fong, S. P., Kee, B. P., Puthucheary, S. D., and Chua, K. H. (2022). Molecular identification and biofilm-forming ability of Elizabethkingia species. Microb. Pathog. 162:105345. doi: 10.1016/j.micpath.2021.105345
Ransangan, J., Zainuri, N., Lal, T. M., Jintoni, B., and Chung, V. S. (2013). Identification of Elizabethkingia meningoseptica from American bullfrog (Rana catesbeiana) farmed in Sabah, Malaysia using PCR method and future management of outbreak. Malays. J. Microbiol. doi: 10.21161/mjm.44012
Ratledge, C., and Dover, L. G. (2000). Iron metabolism in pathogenic bacteria. Ann. Rev. Microbiol. 54, 881–941.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Skaar, E. P. (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Soler-Iborte, E., Rivera-Izquierdo, M., and Valero-Ubierna, C. (2024). Opportunistic Elizabethkingia miricola infections in intensive care unit, Spain. Emerg. Infect. Dis. 30, 834–837. doi: 10.3201/eid3004.231491
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Teo, J., Tan, S. Y.-Y., Liu, Y., Tay, M., Ding, Y., Li, Y., et al. (2014). Comparative genomic analysis of malaria mosquito vector-associated novel pathogen Elizabethkingia anophelis. Genome Biol. Evol. 6, 1158–1165. doi: 10.1093/gbe/evu094
Wu, C., Xiong, L., Liao, Q., Zhang, W., Xiao, Y., and Xie, Y. (2024). Clinical manifestations, antimicrobial resistance and genomic feature analysis of multidrug-resistant Elizabethkingia strains. Ann. Clin. Microbiol. Antimicrob. 23:32. doi: 10.1186/s12941-024-00691-6
Yang, C., Liu, Z., Yu, S., Ye, K., Li, X., and Shen, D. (2021). Comparison of three species of Elizabethkingia genus by whole-genome sequence analysis. FEMS Microbiol. Lett. 368:fnab018. doi: 10.1093/femsle/fnab018
Yasmin, M., Rojas, L. J., Marshall, S. H., Hujer, A. M., Cmolik, A., Marshall, E., et al. (2023). Characterization of a novel pathogen in immunocompromised patients: Elizabethkingia anophelis —exploring the scope of resistance to contemporary antimicrobial agents and β-lactamase inhibitors. Open Forum Infect. Dis. 10:ofad014. doi: 10.1093/ofid/ofad014
Yoon, S.-H., Ha, S., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zajmi, A., Teo, J., and Yeo, C. C. (2022). Epidemiology and characteristics of Elizabethkingia spp. infections in Southeast Asia. Microorganisms 10:882. doi: 10.3390/microorganisms10050882
Keywords: Elizabethkingia miricola , genome analysis, antimicrobial resistance, molecular identification, pangenomes
Citation: Chen S, Agah G, Blom J and Walker ED (2025) Molecular characterization, comparative genome analysis and resistance determinants of three clinical Elizabethkingia miricola strains isolated from Michigan. Front. Microbiol. 16:1582121. doi: 10.3389/fmicb.2025.1582121
Edited by:
Arryn Craney, Petrified Bugs LLC, United StatesReviewed by:
Jozsef Soki, University of Szeged, HungaryAnna Kopf, Technical University Dresden, Germany
Shaowu Li, Chinese Academy of Fishery Sciences, China
Copyright © 2025 Chen, Agah, Blom and Walker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Chen, c2NoZW4xQG5pdS5lZHU=