 Hui Cao
Hui Cao Yun Shen1
Yun Shen1- 1Department of Nutrition and Food Safety, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing, China
- 2Jiangsu Provincial Medical Key Laboratory of Pathogenic Microbiology in Emerging Major Infectious Diseases, Nanjing, China
Non-typhoidal Salmonella (NTS) poses a significant global health burden due to its association with gastroenteritis and rising antimicrobial resistance (AMR). This study conducted a genomic analysis of 62 Salmonella isolates from outpatient cases in Jiangsu, China, to monitor the epidemiological characteristics of NTS, including genetic diversity, AMR profiles, and resistance transmission mechanisms 18 serovars and 21 sequence types (STs) were identified by whole genome sequencing, with S. enteritidis (27.42%) and S. typhimurium (19.35%) predominating. 61 resistance genes from ten different antimicrobial categories were found by genotypic AMR screening. 90.32% of isolates had β-lactam resistance genes, indicating a high frequency of extended-spectrum β-lactamases (ESBL). Serovar-dependent resistance patterns were highlighted by the most varied AMR profile (40/61 genes) found in S. typhimurium. The co-occurrence of genes for aminoglycoside resistance, sul2, and blaTEM indicated clustering driven by mobile genetic elements. A plasmid in a S. Stanley isolate harbored 12 AMR genes, which showed structural changes suggestive of horizontal gene transfer and active recombination. These findings underscore the role of plasmids in disseminating MDR and the urgent need for enhanced antimicrobial stewardship, food safety protocols, and One Health interventions to mitigate the spread of resistant Salmonella clones.
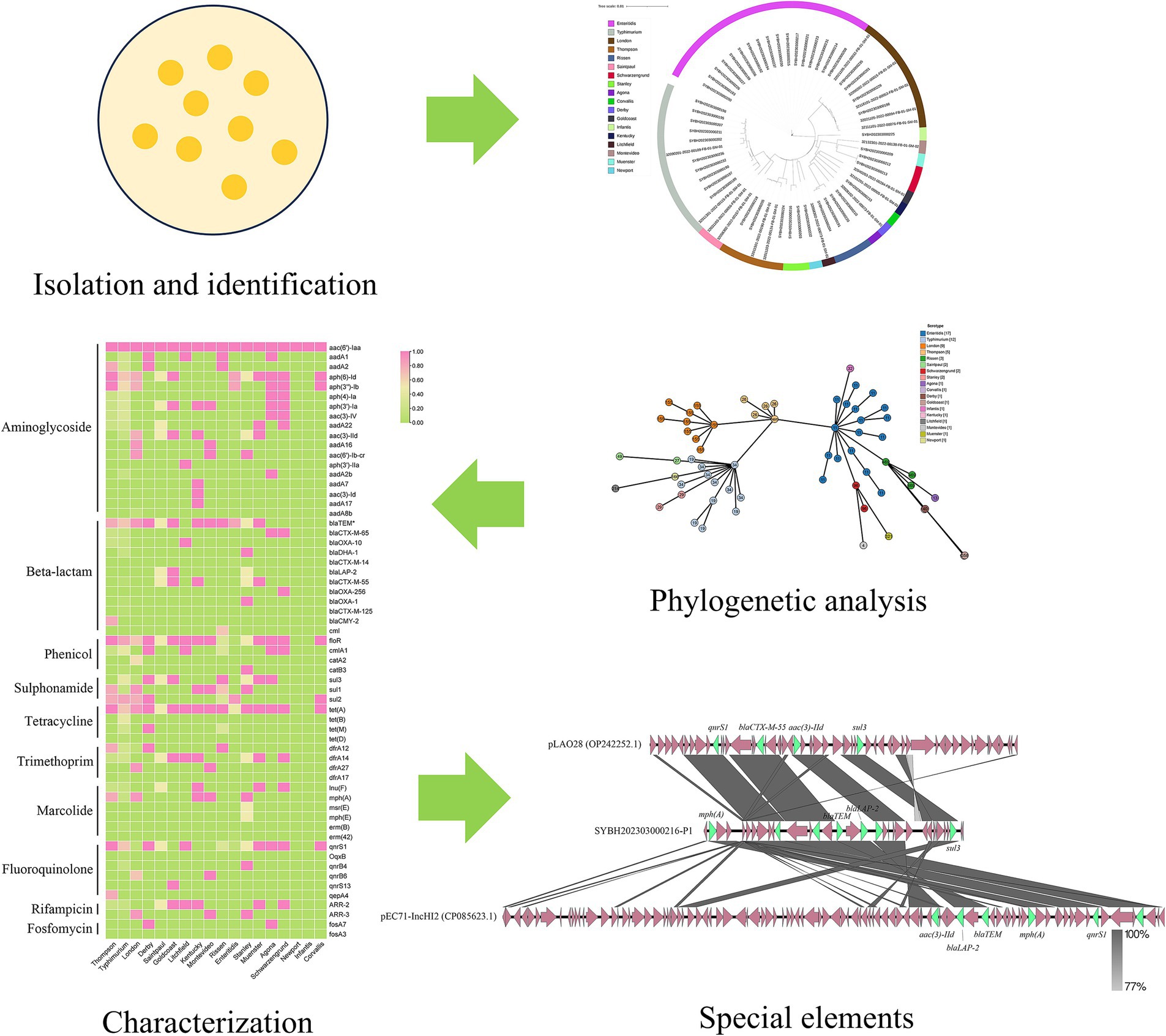
Graphical Abstract.
1 Introduction
Salmonella is a Gram-negative facultatively anaerobic bacterium, belonging to the Enterobacteriaceae family. Salmonella Genus is highly diversity, more than 2,600 Salmonella serovars have been described to date (Guibourdenche et al., 2010). Acute gastroenteritis is frequently caused by non-typhoidal Salmonella (NTS) all over the world (Alikhan et al., 2018). Even though the majority of human cases of salmonellosis are mild and self-limiting, dehydration brought on by severe vomiting and diarrhea can occasionally be fatal. Every year, NTS causes over 180 million instances of gastroenteritis and over 300,000 fatalities worldwide (Kirk et al., 2015; Besser, 2018). Salmonellosis incidence was estimated to be 626.5 cases per 100,000 people in China, where NTS is the second most frequent bacterium causing foodborne outbreaks (Chen J. et al., 2024). Salmonella might contaminate animal carcasses during transport, slaughtering, and then transmitted to humans via the farm-to-fork route, causing severe infections and threatening public health systems (Pan et al., 2018).
Treatment of bacterial infections is severely hampered by the misuse of antimicrobial drugs. Salmonella poses significant risks to public health and security because it is thought to be a significant reservoir of genes that cause antibiotic resistance (Jajere, 2019). Effective preventative and dietary control measures have previously reduced the general incidence of NTS; nevertheless, the emergence of multidrug-resistant clones in recent years has resulted in a large rise in the burden of human NTS infection (Xiang et al., 2020; Wang et al., 2020). Resistance to β-lactam antibiotics has increased over the past 20 years, and in some countries, contaminated food products have been shown to contribute to the spread of Salmonella isolates that produce extended-spectrum β-lactamase (ESBL) and the resulting health risks (Balta et al., 2024). The occurrence of multi-drug resistant bacteria lead to more complicate situation in Salmonella infection treatment and is regarded as a serious threat to public health because it limits the options for therapeutic treatment of patients (Collignon, 2013). Salmonella isolates recovered from animal farms, food chain processes, foods, and humans have been found to be resistant to various antibiotics (Elbediwi et al., 2019; Klemm Elizabeth et al., 2018).
Strategies for reducing the spread of disease will make more sense if the risk factors linked to bacterial infections are understood. In order to enhance our understanding of NTS infections, we used whole genome sequencing to examine the genetic diversity, MLST, antimicrobial resistance, virulence genes, and serotype of Salmonella isolates obtained from outpatients in Jiangsu province, China.
2 Materials and methods
2.1 Collection and identification of bacterial isolates
A total of 62 Salmonella isolates were isolated from the feces of patients with clinical diarrhea. These patients were from sentinel hospitals for foodborne diseases in Jiangsu Province. The suspected cause of the disease was the consumption of contaminated food. Diarrhea is defined as having three or more bowel movements within 24 h, accompanied by abnormal stool characteristics such as watery stool, loose stool, or mucus stool. MacConkey and Salmonella and Shigella (SS) plates were used to identify and purify those isolates, suspected Salmonella colonies on SS agar were 1–2 mm in size, circular, transparent, smooth and with a black centre, the colonies will be cultured in LB and the species of all isolates were confirmed as Salmonella by using mass spectrometry at first (Kim et al., 2022).
2.2 Genomic DNA extraction and sequencing
Genomic DNA of all Salmonella isolates was extracted by using FastPure Bacterial DNA Isolation Mini Kit (Vazyme, China) following the manufacturer’s instructions. Whole genome sequencing was performed on the Illumina NovaSeq PE150 platform, and the quality of the raw data was controlled by using fastQC,1 and filtered by using Trim-galore,2 followed by de novo assembly using SOAP denovo (version 2.04) (Li et al., 2010; Li et al., 2008). The quality of these genome files was assessed by checkM2 (Chklovski et al., 2023).
2.3 Serotyping and MLST
The serovar, serogroup and O antigen of the Salmonella isolates was predicted by using SISTR (Salmonella In Silico Typing Resource) (Yoshida et al., 2016). Multilocus serotyping was performed by using mlst.3
2.4 Genome wide phylogenic analysis
Whole genome phylogenetic tree was constructed by using Mashtree (Katz et al., 2019), and visualized by iTOL (Letunic and Bork, 2019). cgMLST was performed to further analyze the phylogenetic difference between these Salmonella isolates, all genomes were annotated by Prokka (Seemann, 2014), and the core genome was collected from the result of Roary (Page et al., 2015), then Bac_cgMLST4 was used to perform the cgMLST. A minimum spanning tree was generated to analyze the relationship between genomic characteristics and the typing results by using GrapeTree (Zhou et al., 2018).
2.5 Antimicrobial resistance genes analysis
Antimicrobial susceptibility of these Salmonella isolates was tested following Clinical & Laboratory Standards Institute (CLSI) 2021 using VITEK® 2 COMPACT (bioMérieux). The following antibiotics were used in this study: ampicillin (AMP), Ampicillin-Sulbactam (AMS), imipenem (IPM), tetracycline (TET), gentamicin (GEN), chloramphenicol (CHL), Colistin (CT), azithromycin (AZM), cefotaxime (CTX), ceftazidime (CAZ), cefazolin (CFZ), cefoxitin (CFX), ciprofloxacin (CIP), and nalidixic acid (NAL).
The present of antimicrobial resistance genes (AMRGs) in every genome of Salmonella isolates was predicted by using ResFinder (Bortolaia et al., 2020), the threshold was identity > 90%, coverage > 60%. The result was organized and a headmap was built by using TBtools (Chen et al., 2020). Further, the frequency of the combinations of AMRGs were calculated.
2.6 Scanning the plasmid replicons
The assembled contigs were then used to predict plasmid replicons and by using PlasmidFinder 2.1,5 the threshold was identity > 95%, coverage > 60%.
2.7 Comparison of the antimicrobial resistance gene carrying elements
To further discuss the hazard of highly burden of AMRGs, the elements carrying the greatest number of AMRGs were analyzed. The genome was scaffolded by RagTag (Alonge et al., 2022), the comparison between closest elements were performed using Easyfig (Sullivan et al., 2011).
2.8 Statistical analysis
Basic Python calculation method was used in this study to count the number of groups. No comparison of groups was performed in this study. If not stated, all tools we used in this study were used according to default Settings.
3 Results
3.1 Population structure of Salmonella
62 Salmonella isolates have been isolated and identified from outpatients from Jiangsu province in this study (Table 1). A diverse serogroup distribution was discovered, including B, C1, C2, C3, D1, E1, F. Further, a total number of 18 serovars were identified, among them, S. enteritidis is predominant (17/62; 27.42%), followed by S. typhimurium (12/62; 19.35%), S. London (9/62; 14.52%) and S. Thompson (5/62; 8.06%) (Table 2).
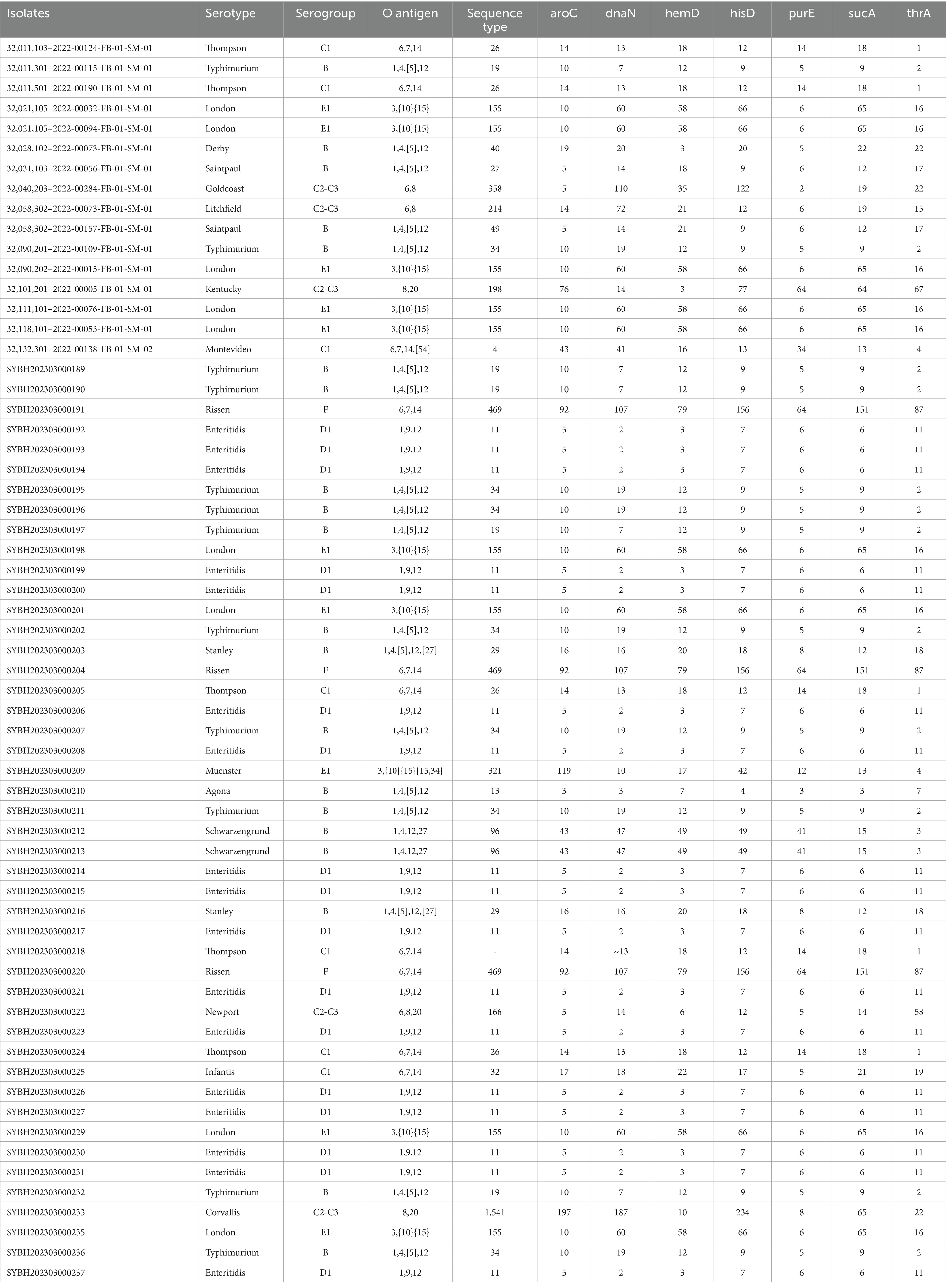
Table 1. Information of 62 Salmonella isolates isolated in this study.
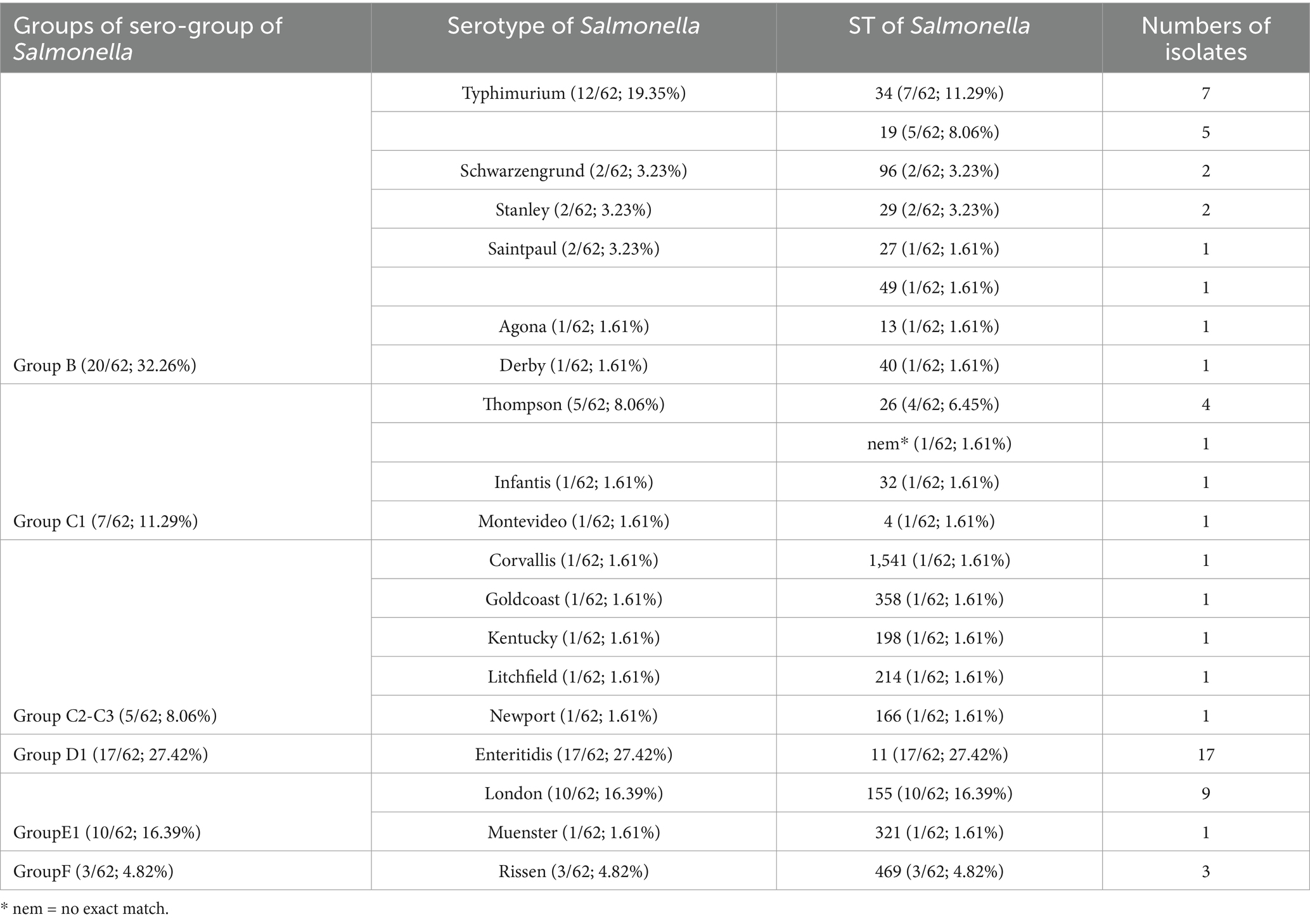
Table 2. Typing details of the Salmonella isolates.
Multi-locus sequence typing shows that 21 sequence types (STs) were identified with the dominance of ST11 (17/62; 27.42%), ST155 (10/62; 16.39%) and ST34 (7/62; 11.29%). There are 3 serovars presented multiple STs, which is S. typhimurium (ST34, ST19), S. Saintpaul (ST27, ST49), and S. Thompson (ST26 and 1 no exact match), and the rest of them presented only one ST (Table 2).
3.2 Phylogenomic analysis
Through sequencing quality assessment, the average genomes size of those genome files is 4,905,831 bp, with a N50 between 143, 671 bp and 411,631 bp, the average N50 is 233,405 bp (Supplementary Table 1). A phylogenetic tree based on the whole genome sequence was constructed for these Salmonella isolates to determine their epidemiological relatedness, and for all identified serovars, the isolates belong to the same serovar is clustered in the same clade. This finding suggested the serovar of Salmonella was closely related to genome evolution (Figure 1).
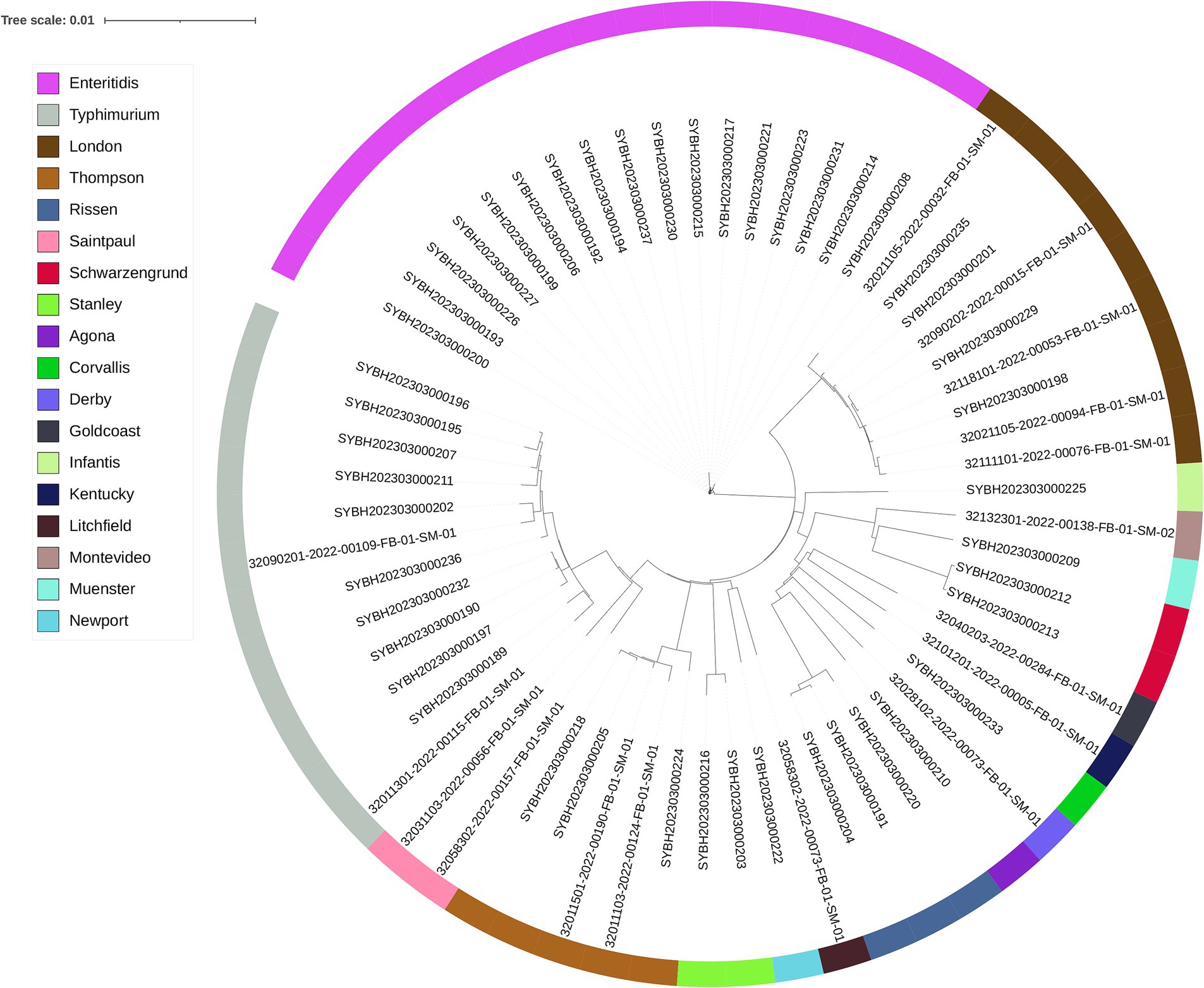
Figure 1. The phylogenomic tree of 62 Salmonella isolates constructed by Mashtree. Isolates are annotated with the serotype predicted by SISTR.
A cgMLST analysis was performed to further analyze the correlationship between genome evolution and typing data, S. Thompson isolates is the key node of these isolates, it composed the backbone of cgMLST diagram with S. enteritidis, S. typhimurium ST34 and S. London. Among them, S. enteritidis and S. typhimurium presented a more important role, because all the other isolates with different serovars or STs were branched from them, 13 from S. enteritidis, and 6 from S. typhimurium (Figure 2).
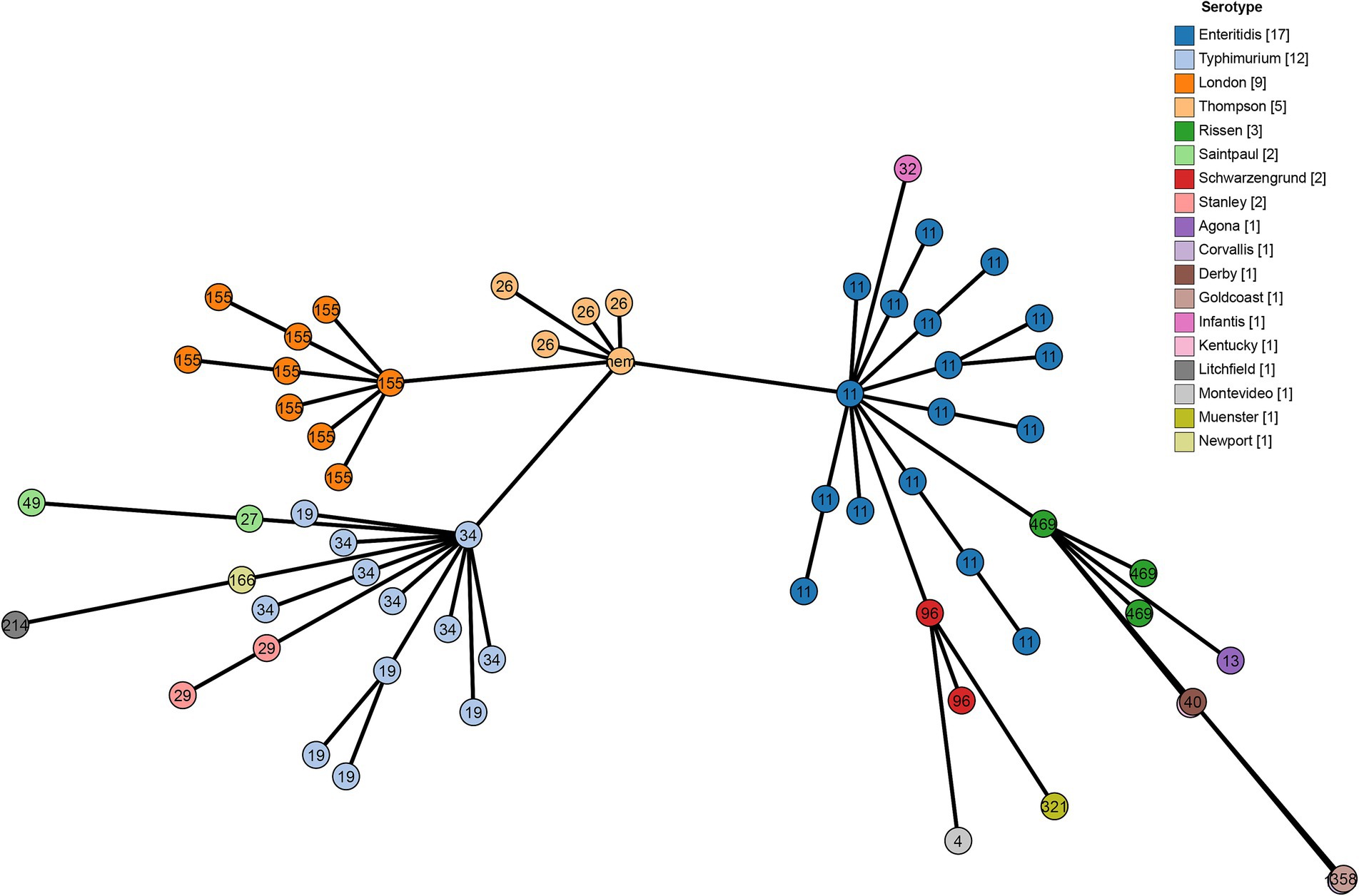
Figure 2. Minimum spanning tree using cgMLST profiles of 62 Salmonella isolates. The colors are marked by the serotype predicted by SISTR and the sequence types are marked by numbers.
3.3 Genotypic and phenotypic of antimicrobial resistance
Now it is easy to screen the encoded antimicrobial resistance genes (AMRGs) using whole genome sequencing. A total number of 61 different AMRGs belong to 10 different antimicrobial categories were detected. Except the aac (6′)-Iaa gene presented in all Salmonella isolates, the other most prevalent AMRG is tet(A) (45/62, 72.58%), blaTEM (44/62, 70.97%), aph(6)-Id (42/62, 67.74%) and aph(3″)-Ib (37/62, 59.68%) (Figure 3), and for the antimicrobial categories is Aminoglycoside (62/62, 100%), Beta-Lactam (56/62, 90.32%), Sulphonamide (53/62, 85.48%) and Tetracycline (49/62, 79.03%) (Table 3). The antimicrobial phenotype was also tested, and most of the phenotype could correspond to the genotype (Table 4).

Figure 3. The heatmap of antimicrobial resistance genes (ARGs) in the studied Salmonella isolates.
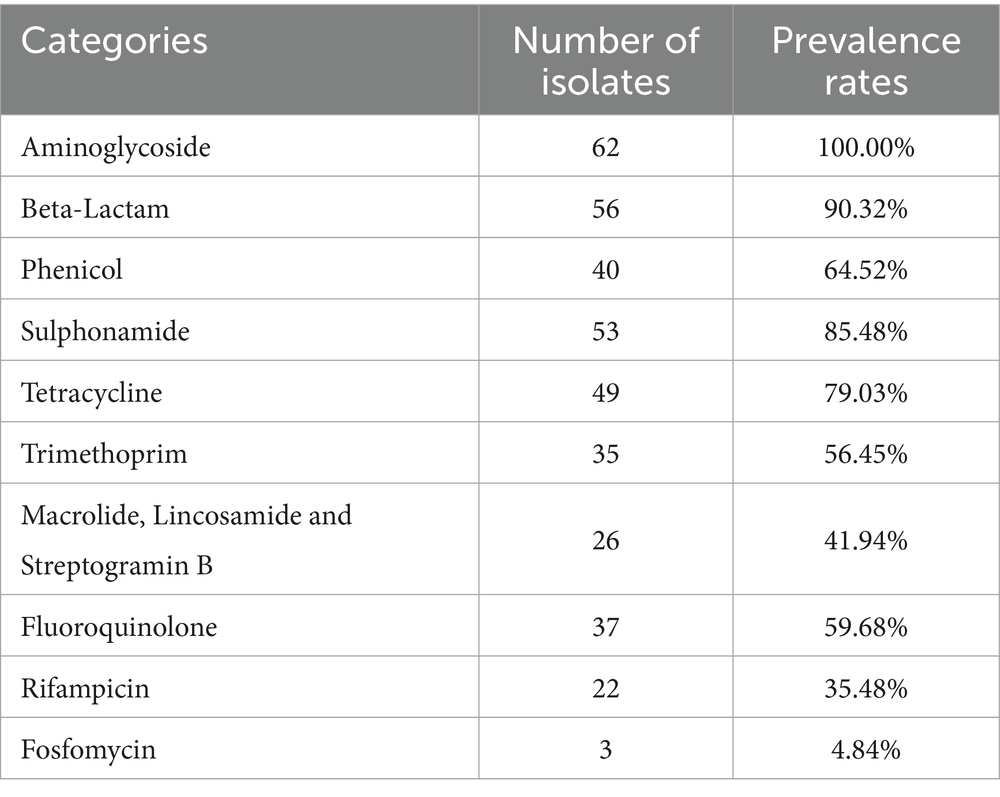
Table 3. Prevalence of antimicrobial resistance genes in 62 Salmonella isolates.

Table 4. Antimicrobial resistance phenotype of 62 Salmonella isolates isolated in this study.
3.4 Relationship between antimicrobial resistance and serovar
In order to understand the prevalence of AMRGs among different serovars, after screened the AMRG in all those Salmonella isolates. Among those serovars, S. typhimurium harbors most diversified AMRGs (40/61, 65.57%), followed by S. Thompson (24/61, 39.34%) and S. Stanley (21/61, 34.42%). Meanwhile, there are two serovars contain only one AMRG, aac(6′)-Iaa, which is located on chromosome (Figure 4). This data elucidated that the prevalence of AMRGs is associated with serovars and the distribution biased.
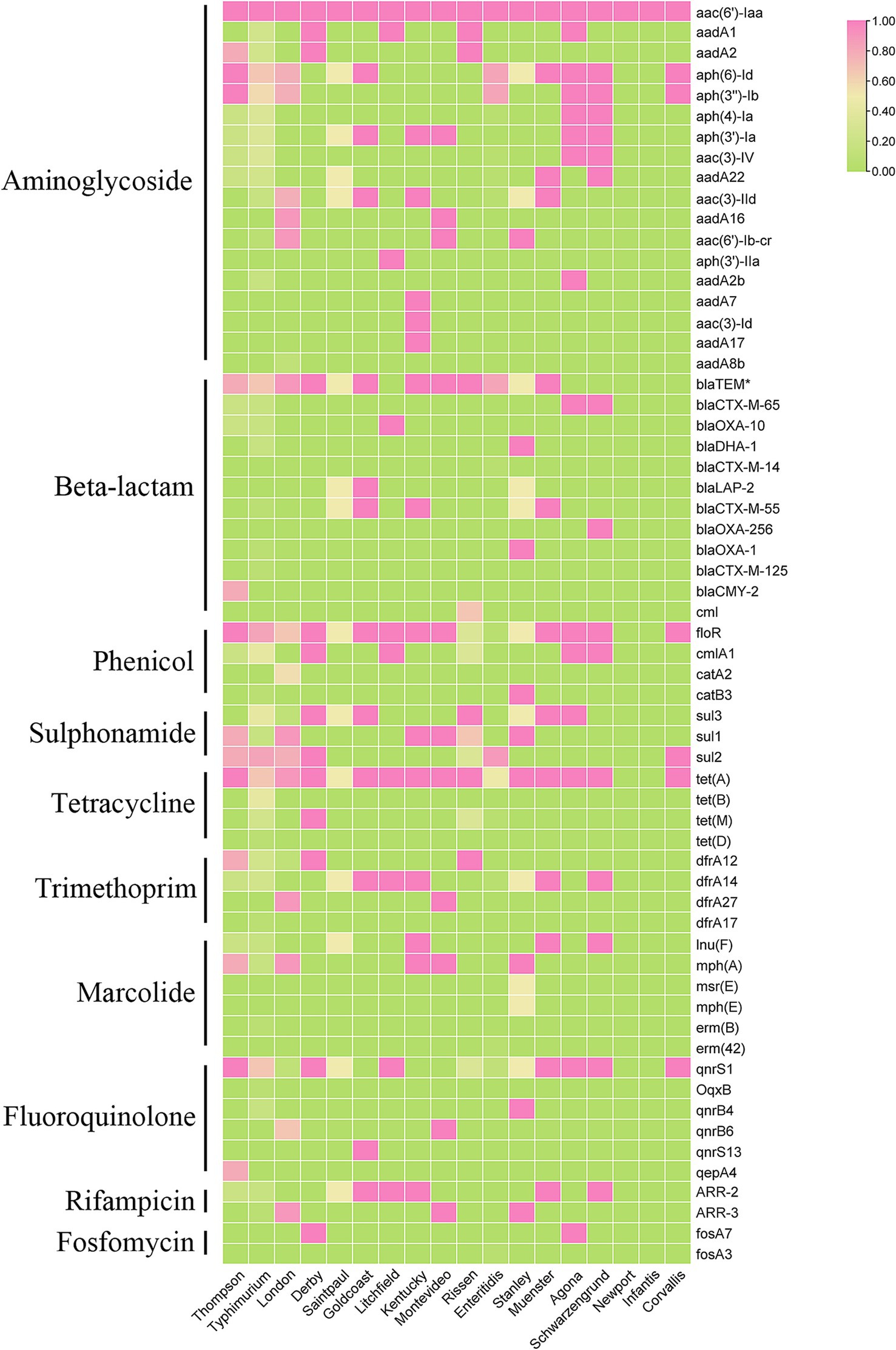
Figure 4. The heatmap of represent the relationship between antimicrobial resistance genes (ARGs) and serotypes of 62 Salmonella isolates.
3.5 Co-existence of antimicrobial resistance genes
We further analyzed that which AMRGs are prefer to exist together, considering the presence frequency of AMRGs against Aminoglycoside class drug is extremely high and the resistance gene aac(6′)-Iaa is located in chromosome and presence in every genome, we calculated the combinations twice, excluded aac(6′)-Iaa and excluded all AMRGs against Aminoglycoside class drug. Aph(6)-Id+aph(3″)-Ib+sul2 (31 times) is most frequent when only exclude aac(6′)-Iaa, followed by aph(6)-Id+aph(3″)-Ib+blaTEM (28 times) and aph(6)-Id+blaTEM+sul2 (27 times), we can see that these combinations are all composed by Aminoglycoside AMRGs with a Beta-Lactam AMRG blaTEM and/or a Sulphonamide AMRG sul2. When exclude the AMRGs against Aminoglycoside class drug, the most frequent combination is blaTEM+floR+tet(A) (24 times), equal with floR+tet(A) + qnrS1 (24 times) and blaTEM+sul2 + tet(A) (24 times), Tetracycline resistance gene tet(A) exist in every combination, and we can also found Phenicol resistance gene floR and Fluoroquinolone resistance gene qnrS1 (Table 5).
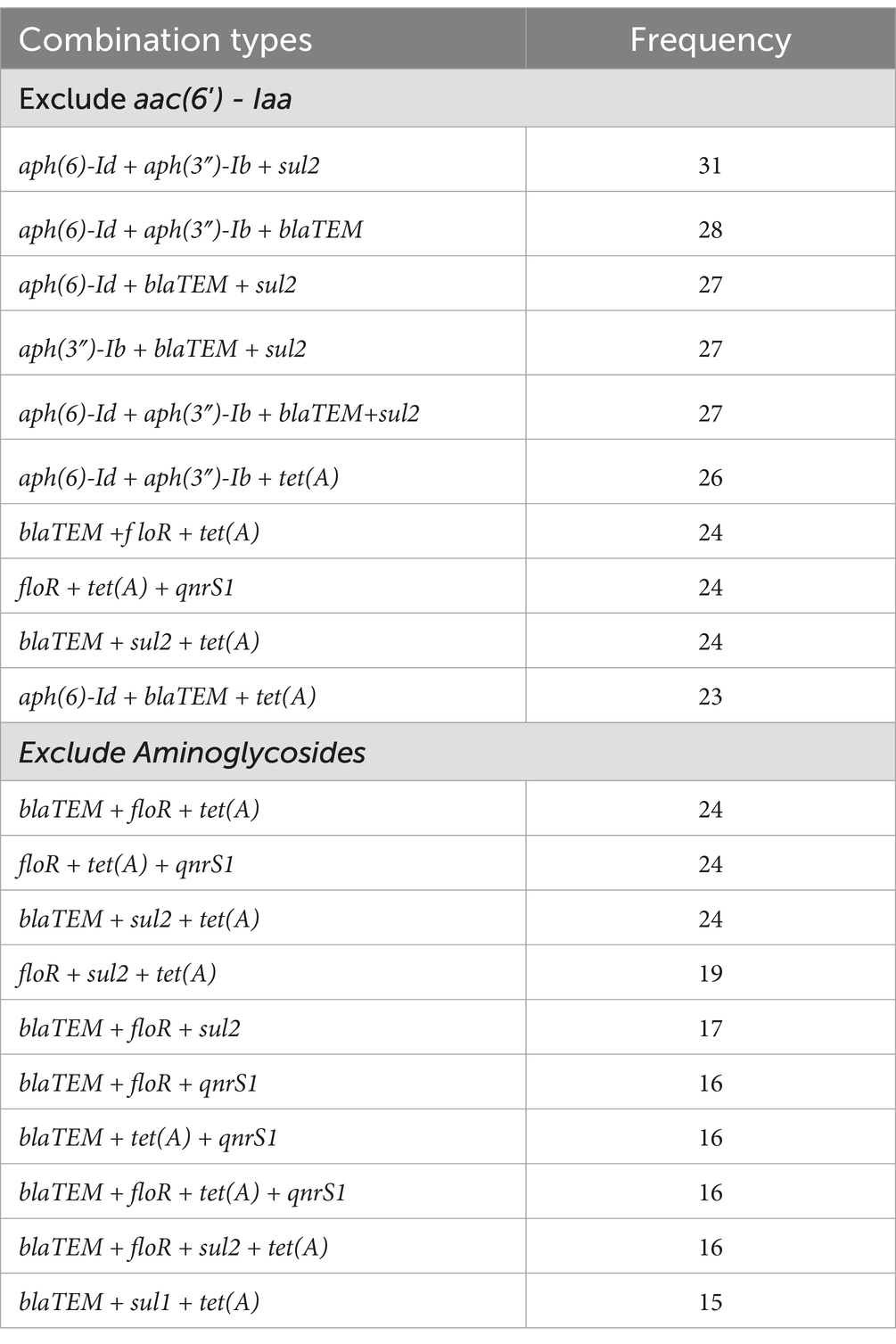
Table 5. Top 10 most frequent antimicrobial resistance gene combinations of 62 Salmonella isolates (The elements in the combination > = 3).
3.6 Plasmid replicons
Plasmid carrying AMRGs is one of the reasons that the spread of antimicrobial resistance, among 62 Salmonella isolates, 58 of them were detected carrying at least one plasmid (58/62, 93.55%), 5 of them even carrying 6 different plasmids. 28 different plasmids were detected, where the plasmid Col(pHAD28)_1 was the most prevalent (22/62, 35.48%), followed by IncFIB(S)_1, and IncFII(S)_1, IncFII(pAR0022)_1 (17/62, 27.42%) and IncX1_4 (16/62, 25.81%). Further, we found that S. typhimurium harbors more diversified plasmid (n = 19) compared with other serovars, followed by S. enteritidis (n = 13). However, only 8 of 18 serovars were identified harboring plasmids. Notably, the co-occurrence between plasmid replicons and antimicrobial resistance genes was mainly observed in S. typhimurium (Figure 5) 0.3.7 Characterization of plasmids carrying multiple AMRGs.

Figure 5. The heatmap of plasmids distribution in different Salmonella isolates. The strength of the colors corresponds to the numerical value of the prevalence of the plasmids.
Among these Salmonella isolates, a S. Stanley isolate contain 21 AMRGs, which is the most of them. We tried to discuss how this bacterial isolate contains that much AMRGs, through further analysis, more than half (12 of 21) AMRGs are detected located in one plasmid. The distribution of these AMRGs is wide, but still 7 of them are very close, and we compared this part of this plasmid with other plasmids with high homology, we can see that even for these closest sequences, there are still a lot of difference, rearrangement, translocation, inversion could be observed (Figure 6), The number of AMRGs in these sequences are different, the accumulation of AMRGs could be the result of gene exchange.
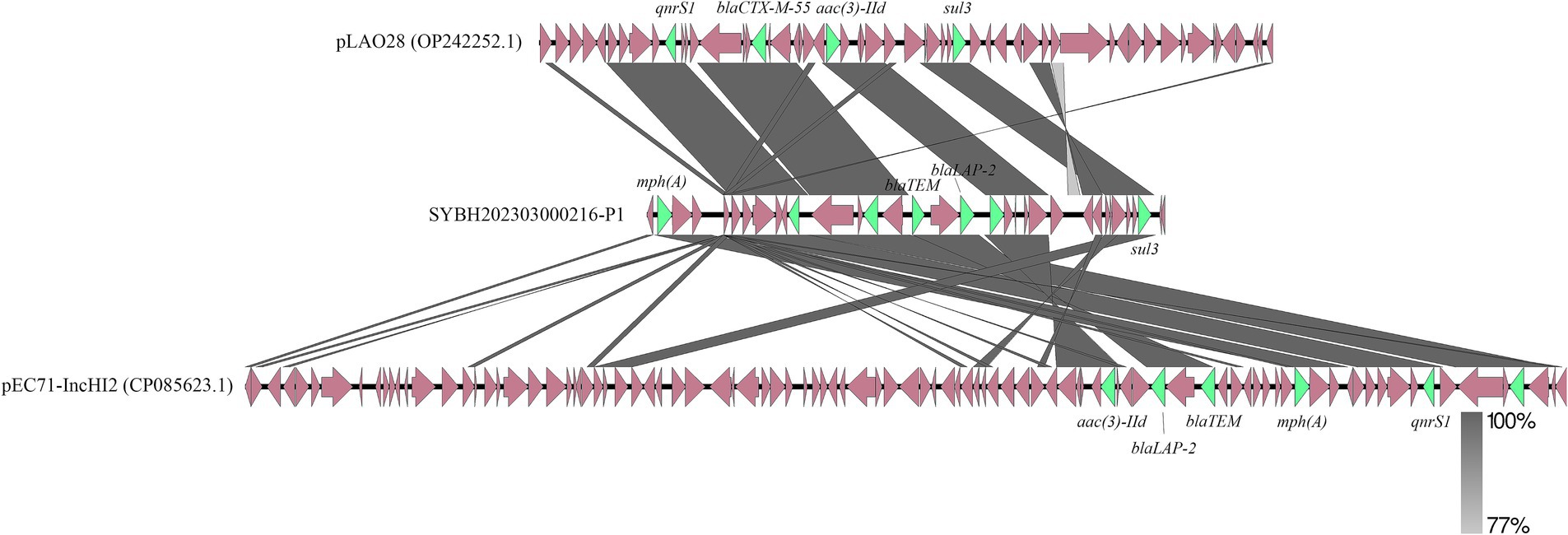
Figure 6. Genetic comparison of the structure of SYBH202303000216-P1 with pLAO28 (Accession No. OP242252.1) and pEC71-IncHI2 (Accession No. CP085623.1). The antimicrobial resistance genes are shown in green and the rest of the genes are shown in reddish brown.
4 Discussion
A major global public health concern, non-typhoidal salmonellosis (NTS) has a huge financial and medical impact on a global scale (Paudyal et al., 2018). The present study provides a comprehensive genomic analysis of Salmonella isolates from outpatients in Jiangsu, China, emphasizing the genetic diversity, antimicrobial resistance (AMR) profiles, and possible processes behind the spread of resistance. Our results highlight the serious public health issues that NTS presents, especially in light of the growing worldwide burden of foodborne diseases and multidrug resistance (MDR).
The dominance of S. enteritidis (27.42%) and S. typhimurium (19.35%) among the 62 isolates aligns with global epidemiological trends, where these serovars are frequently associated with foodborne transmission and human salmonellosis (Majowicz et al., 2010). The detection of 18 serovars and 21 sequence types (STs) reflects substantial genetic diversity, consistent with the high adaptability of Salmonella (Cheng et al., 2019). Notably, the prevalence of ST11 (related with S. enteritidis) and ST34 (associated with S. typhimurium) was high, which is consistent with findings from other areas where these STs are connected to MDR outbreaks and phenotypes (Balasubramanian et al., 2019; Woh et al., 2025), further corroborates findings from the Chinese Local Salmonella Genome Database and other studies, which highlights ST34 as a pandemic MDR clone responsible for pediatric infections and livestock-associated outbreaks (Wang et al., 2023a,b). The serotype-specific genomic evolution impacts epidemiological trends comes from the phylogenetic grouping of isolates by serovar (Yoshida et al., 2016).
When the antimicrobial resistance genes in those NTS isolates were screened using the WGS technique, 61 genes encoding resistance to 10 categories were found. Except of the aac(6′)-Iaa gene, which confers intrinsic aminoglycoside resistance, is 100% harboring the chromosome of NTS isolates, high prevalence of tet(A) (72.58%), blaTEM (70.97%), and sulfonamide resistance genes (85.48%) was detected, underscores the widespread use of β-lactams, tetracyclines, and sulfonamides in clinical and agricultural settings (Alekshun and Levy, 2007; Ramirez and Tolmasky, 2010; Nazir et al., 2025). One of the most important public health concerns is the increase of Salmonella isolates that are resistant to antibiotics (Eng et al., 2015). Alarmingly, 90.32% of isolates had β-lactam resistance genes, which is indicative of the increase in Salmonella that produce extended-spectrum β-lactamase (ESBL) worldwide, reflect a national crisis in antibiotic misuse, as reported in studies analyzing >35,000 Salmonella isolates across China (Wang et al., 2023a). Therapeutic choices are severely limited by such developments, especially in areas where antibiotic abuse is prevalent (Okeke et al., 2024).
The serovar-dependent AMR patterns shown here underscore the relevance of clonal proliferation in resistance diffusion. S. typhimurium isolates had the most varied AMRGs (40/61), consistent with its status as an MDR “high-risk” clone (Pitti et al., 2023; Medalla et al., 2016). Conversely, some serotypes, like S. infantis and S. Newport, showed fewer resistance genes. This discrepancy might be due to the limited number of isolates, but S. enteritidis, the other dominant serotype, also only has barely half of the S. typhimurium resistance genes (17/61). This data implies that horizontal transmission of the AMRG containing genetic components may change depending on the serovar.
The co-occurrence of blaTEM, sul2, aph(6)-Id, and aph(3″)-Ib in more than 50% of isolates suggests that resistance genes may located closely in the genome, and these combinatorial patterns are frequently associated with mobile genetic elements (MGEs), which promote the clustering of resistance genes (Yan et al., 2024). The combination of floR+tet(A) + qnrS1, which was detected in 24 isolates, suggests co-selection of tetracycline, phenicol, and fluoroquinolone resistance, possibly driven by agricultural use of these antibiotics, which emphasized the importance of One Health in disease control. Given that fluoroquinolones, such as ciprofloxacin, are first-line treatments for severe salmonellosis, this occurrence makes therapy more difficult (Randall Luke et al., 2006; Chen Y. et al., 2024). The risk levels associated with antimicrobial resistance (AMR) in Salmonella isolates from Jiangsu are stratified by the clinical urgency of the compromised antibiotics, transmission potential of resistance genes, and the possibility of treatment failures. Resistance to β-lactams, particularly extended-spectrum β-lactamases (ESBLs) like blaTEM (detected in 70.97% of isolates), poses a critical risk (Okeke et al., 2024). Since ciprofloxacin is the first-line treatment for invasive NTS, fluoroquinolone resistance (qnrS1 in 24 isolates) represents a high risk. Resistance compromises empirical treatment, the agricultural origin threat was highlighted by a previous study that showed a correlation between human treatment failures and qnrS1-positive Salmonella in poultry farms (Chen Y. et al., 2024). We found that the plasmid is widely distributed among those Salmonella isolates, and that the co-occurrence of MDR genes on mobile genetic elements (MGEs) increases risks by facilitating rapid horizontal gene transfer. Plasmids frequently carry integrons and transposons, which allow “resistance gene cassettes” to accumulate (Li et al., 2023).
Concern over β-lactam antibiotics is growing due to a combination of variables in both human and veterinary antibiotic treatments, as evidenced by the high frequency of blaTEM (70.97%) and ESBL-associated resistance genes. Overuse of antibiotics on farms and the release of active antimicrobials into the environment favor resistant bacterial lineages in those settings, putting humans and animals at risk of a future pandemic from an incurable bacterial infection. The misuse and overuse of β-lactam antibiotics in human medicine, such as prescribing them inappropriately for viral infections or not finishing treatment courses, have created sustained selective pressure that has allowed the proliferation of resistance mechanisms like β-lactamase enzymes and efflux pumps (Okeke et al., 2024).
In veterinary medicine, β-lactams are also often used to prevent illness and promote growth, frequently at subtherapeutic dosages that encourage the survival of resistant bacteria. blaCTX-M genes were significantly enriched in Salmonella by the agricultural usage of ceftiofur, a third-generation cephalosporin, in chicken farms, which then spread to humans via contaminated meat (Yang et al., 2022). Additionally, resistance is increased by lax rules on the disposal of antibiotics and environmental contamination. Residual β-lactams in agricultural and hospital wastewater generate resistance gene reservoirs, which facilitate horizontal movement between various bacterial populations (Yan et al., 2024). One Health strategy is needed to address this, which includes investing in wastewater treatment technology to stop the spread of resistance genes, prohibiting the non-therapeutic use of antibiotics in cattle, and enforcing stronger antibiotic stewardship in clinics.
We examined one isolate that had the highest amount of AMRGs among these isolates in order to better understand the horizontal transfer of antimicrobial resistance genes in Salmonella. The fact that over half of the AMRGs were found in a plasmid highlights the critical role that plasmids play in the spread of resistance. Plasmids containing AMRGs showed structural differences (rearrangements, inversions) that were suggestive of active recombination, according to comparative analysis. Similar results have been shown for Salmonella from food goods and cattle, where plasmids serve as conduits for the transmission of resistance throughout ecosystems (Li et al., 2023; Huang et al., 2025; Wang et al., 2019). The mosaic nature of these plasmids suggests ongoing evolution under antibiotic selection pressure, with implications for One Health frameworks.
The high rate of MDR Salmonella in Jiangsu outpatients highlights the urgent need for improved monitoring and antibiotic management. The prevalence of fluoroquinolone resistance (qnrS1) and ESBL-associated blaTEM is similar to patterns in clinical Enterobacteriaceae, where these genes jeopardize empirical treatment (Nüesch-Inderbinen et al., 2023; Aworh et al., 2023). Salmonella infection management in China, where NTS is the second most common cause of foodborne outbreaks, requires stronger laws governing the use of antibiotics in agriculture as well as better food safety procedures to break the chain of transmission from farm to fork (Chen J. et al., 2024).
As a result of clonal growth and plasmid-mediated horizontal gene transfer, our genomic study indicates a significant burden of MDR Salmonella in Jiangsu. The need for intersectoral interventions spanning healthcare, agriculture, and policy to slow the spread of resistant strains is highlighted by the convergence of diverse AMRGs in clinically relevant serovars such as S. typhimurium. To stop the spread of NTS, it is essential to improve food safety systems, invest in innovative treatments, and strengthen antimicrobial stewardship.
Data availability statement
The sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA025182) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Author contributions
HC: Conceptualization, Data curation, Visualization, Writing – original draft. YS: Formal analysis, Investigation, Writing – original draft. KM: Investigation, Resources, Writing – original draft. DZ: Methodology, Resources, Writing – review & editing. YX: Writing – review & editing. XQ: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1587421/full#supplementary-material
Footnotes
1. ^https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
2. ^https://github.com/FelixKrueger/TrimGalore
3. ^https://github.com/tseemann/mlst
References
Alekshun, M. N., and Levy, S. B. (2007). Molecular mechanisms of antibacterial multidrug resistance. Cell 128, 1037–1050. doi: 10.1016/j.cell.2007.03.004
Alikhan, N.-F., Zhou, Z., Sergeant, M. J., and Achtman, M. (2018). A genomic overview of the population structure of Salmonella. PLoS Genet. 14:e1007261. doi: 10.1371/journal.pgen.1007261
Alonge, M., Lebeigle, L., Kirsche, M., Jenike, K., Ou, S., Aganezov, S., et al. (2022). Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23:258. doi: 10.1186/s13059-022-02823-7
Aworh, M. K., Kwaga, J. K. P., Hendriksen, R. S., Okolocha, E. C., Harrell, E., and Thakur, S. (2023). Quinolone-resistant Escherichia coli at the interface between humans, poultry and their shared environment- a potential public health risk. One Health Outlook 5:2. doi: 10.1186/s42522-023-00079-0
Balasubramanian, R., Im, J., Lee, J.-S., Jeon, H. J., Mogeni, O. D., Kim, J. H., et al. (2019). The global burden and epidemiology of invasive non-typhoidal Salmonella infections. Hum. Vaccin. Immunother. 15, 1421–1426. doi: 10.1080/21645515.2018.1504717
Balta, I., Lemon, J., Murnane, C., Pet, I., Vintila, T., McCleery, D., et al. (2024). The one health aspect of climate events with impact on foodborne pathogens transmission. One Health 19:100926. doi: 10.1016/j.onehlt.2024.100926
Besser, J. M. (2018). Salmonella epidemiology: a whirlwind of change. Food Microbiol. 71, 55–59. doi: 10.1016/j.fm.2017.08.018
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, J., Huang, L., An, H., Wang, Z., Kang, X., Yin, R., et al. (2024). One health approach probes zoonotic non-typhoidal Salmonella infections in China: a systematic review and meta-analysis. J. Glob. Health 14:04256. doi: 10.7189/jogh.14.04256
Chen, Y., Liu, L., Guo, Y., Chu, J., Wang, B., Sui, Y., et al. (2024). Distribution and genetic characterization of fluoroquinolone resistance gene qnr among Salmonella strains from chicken in China. Microbiol. Spectr. 12, e03000–e03023. doi: 10.1128/spectrum.03000-23
Cheng, R. A., Eade, C. R., and Wiedmann, M. (2019). Embracing diversity: differences in virulence mechanisms, disease severity, and host adaptations contribute to the success of Nontyphoidal Salmonella as a foodborne pathogen. Front. Microbiol. 10:10. doi: 10.3389/fmicb.2019.01368
Chklovski, A., Parks, D. H., Woodcroft, B. J., and Tyson, G. W. (2023). CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212. doi: 10.1038/s41592-023-01940-w
Collignon, P. (2013). Superbugs in food: a severe public health concern. Lancet Infect. Dis. 13, 641–643. doi: 10.1016/S1473-3099(13)70141-5
Elbediwi, M., Li, Y., Paudyal, N., Pan, H., Li, X., Xie, S., et al. (2019). Global burden of Colistin-resistant Bacteria: mobilized Colistin resistance genes study (1980–2018). Microorganisms 7:461. doi: 10.3390/microorganisms7100461
Eng, S.-K., Pusparajah, P., Ab Mutalib, N.-S., Ser, H.-L., Chan, K.-G., and Lee, L.-H. (2015). Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front. Life Sci. 8, 284–293. doi: 10.1080/21553769.2015.1051243
Guibourdenche, M., Roggentin, P., Mikoleit, M., Fields, P. I., Bockemühl, J., Grimont, P. A. D., et al. (2010). Supplement 2003–2007 (no. 47) to the white-Kauffmann-Le minor scheme. Res. Microbiol. 161, 26–29. doi: 10.1016/j.resmic.2009.10.002
Huang, J., Alzahrani, K. O., Zhou, G., Alsalman, S. A., Alsufyani, A. T., Alotaibi, N. M., et al. (2025). Genomic survey of multidrug resistant Salmonella enterica serovar Minnesota clones in chicken products. NPJ Antimicrob. Resist. 3:10. doi: 10.1038/s44259-025-00077-4
Jajere, S. M. (2019). A review of Salmonella enterica with particular focus on the pathogenicity and virulence factors, host specificity and antimicrobial resistance including multidrug resistance. Vet. World 12, 504–521. doi: 10.14202/vetworld.2019.504-521
Katz, L. S., Griswold, T., Morrison, S. S., Caravas, J. A., Zhang, S., den Bakker, H. C., et al. (2019). Mashtree: a rapid comparison of whole genome sequence files. J. Open Source Softw. 4:1762. doi: 10.21105/joss.01762
Kim, G. R., Kim, S. H., Kim, E.-Y., Park, E. H., Hwang, I. Y., Jeong, S. H., et al. (2022). Performance of MALDI-TOF mass spectrometry (VITEK MS) in the identification of Salmonella species. Microorganisms 10:1974. doi: 10.3390/microorganisms10101974
Kirk, M. D., Pires, S. M., Black, R. E., Caipo, M., Crump, J. A., Devleesschauwer, B., et al. (2015). World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med. 12:e1001921. doi: 10.1371/journal.pmed.1001921
Klemm Elizabeth, J., Shakoor, S., Page Andrew, J., Qamar Farah, N., Judge, K., Saeed Dania, K., et al. (2018). Emergence of an extensively drug-resistant Salmonella enterica Serovar Typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation Cephalosporins. MBio 9:e00105-18. doi: 10.1128/mBio.00105-18
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Li, Y., Zhang, P., Du, P., Mu, Y., Cui, S., Fanning, S., et al. (2023). Insertion sequences mediate clinical ST34 monophasic Salmonella enterica serovar typhimurium plasmid polymorphism. Microbiol. Res. 272:127387. doi: 10.1016/j.micres.2023.127387
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Majowicz, S. E., Musto, J., Scallan, E., Angulo, F. J., Kirk, M., O'Brien, S. J., et al. (2010). For the international collaboration on enteric disease “burden of illness” S: the global burden of Nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 50, 882–889. doi: 10.1086/650733
Medalla, F., Gu, W., Mahon, B. E., Judd, M., Folster, J., Griffin, P. M., et al. (2016). Estimated incidence of antimicrobial drug-resistant Nontyphoidal Salmonella infections, United States, 2004-2012. Emerg. Infect. Dis. 23, 29–37. doi: 10.3201/eid2301.160771
Nazir, J., Manzoor, T., Saleem, A., Gani, U., Bhat, S. S., Khan, S., et al. (2025). Combatting Salmonella: a focus on antimicrobial resistance and the need for effective vaccination. BMC Infect. Dis. 25:84. doi: 10.1186/s12879-025-10478-5
Nüesch-Inderbinen, M., Heyvaert, L., Cernela, N., Zurfluh, K., Biggel, M., and Stephan, R. (2023). Emergence of blaSHV-12 and qnrS1 encoded on IncX3 plasmids: changing epidemiology of extended-spectrum ß-lactamases among Enterobacterales isolated from broilers. J. Glob. Antimicrob. Resist. 33, 194–200. doi: 10.1016/j.jgar.2023.03.008
Okeke, I. N., de Kraker, M. E. A., Van Boeckel, T. P., Kumar, C. K., Schmitt, H., Gales, A. C., et al. (2024). The scope of the antimicrobial resistance challenge. Lancet 403, 2426–2438. doi: 10.1016/S0140-6736(24)00876-6
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pan, H., Paudyal, N., Li, X., Fang, W., and Yue, M. (2018). Multiple food-animal-borne route in transmission of antibiotic-resistant Salmonella Newport to humans. Front. Microbiol. 9:9. doi: 10.3389/fmicb.2018.00023
Paudyal, N., Pan, H., Liao, X., Zhang, X., Li, X., Fang, W., et al. (2018). A Meta-analysis of major foodborne pathogens in Chinese food commodities between 2006 and 2016. Foodborne Pathog. Dis. 15, 187–197. doi: 10.1089/fpd.2017.2417
Pitti, M., Garcia-Vozmediano, A., Tramuta, C., Ce, R. C. L. G., Maurella, C., and Decastelli, L. (2023). Monitoring of antimicrobial resistance of Salmonella serotypes isolated from humans in Northwest Italy, 2012–2021. Pathogens 12:89. doi: 10.3390/pathogens12010089
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Randall Luke, P., Cooles Sue, W., Coldham Nick, C., Stapleton Ken, S., Piddock Laura, J. V., and Woodward Martin, J. (2006). Modification of Enrofloxacin treatment regimens for poultry experimentally infected with Salmonella enterica Serovar typhimurium DT104 to minimize selection of resistance. Antimicrob. Agents Chemother. 50, 4030–4037. doi: 10.1128/AAC.00525-06
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Wang, X.-C., Lei, C.-W., Kang, Z.-Z., Zhang, Y., and Wang, H.-N. (2019). IS26-mediated genetic rearrangements in Salmonella Genomic Island 1 of Proteus mirabilis. Front. Microbiol. 10:10. doi: 10.3389/fmicb.2019.02245
Wang, Y., Liu, Y., Lyu, N., Li, Z., Ma, S., Cao, D., et al. (2023a). The temporal dynamics of antimicrobial-resistant Salmonella enterica and predominant serovars in China. Natl. Sci. Rev. 10:nwac269. doi: 10.1093/nsr/nwac269
Wang, M., Qazi, I. H., Wang, L., Zhou, G., and Han, H. (2020). Salmonella Virulence and Immune Escape. Microorganisms 8:407. doi: 10.3390/microorganisms8030407
Wang, Y., Xu, X., Zhu, B., Lyu, N., Liu, Y., Ma, S., et al. (2023b). Genomic analysis of almost 8,000 Salmonella genomes reveals drivers and landscape of antimicrobial resistance in China. Microbiol. Spectr. 11, e02080–e02023. doi: 10.1128/spectrum.02080-23
Woh, P. Y., Chen, Y., Kwok, K. W. H., and Quiroga, J. (2025). Bayesian phylogeographic analysis infers cross-border transmission dynamics of drug-resistant Salmonella enteritidis. Microbiol. Spectr. 13, e02292–e02224. doi: 10.1128/spectrum.02292-24
Xiang, Y., Li, F., Dong, N., Tian, S., Zhang, H., Du, X., et al. (2020). Investigation of a salmonellosis outbreak caused by multidrug resistant Salmonella Typhimurium in China. Front. Microbiol. 11:11. doi: 10.3389/fmicb.2020.00801
Yan, J., Doublet, B., and Wiedemann, A. (2024). Trends in horizontal gene transfer research in Salmonella antimicrobial resistance: a bibliometric analysis. Front. Microbiol. 15:15. doi: 10.3389/fmicb.2024.1439664
Yang, L., Shen, Y., Jiang, J., Wang, X., Shao, D., Lam, M. M. C., et al. (2022). Distinct increase in antimicrobial resistance genes among Escherichia coli during 50 years of antimicrobial use in livestock production in China. Nat. Food 3, 197–205. doi: 10.1038/s43016-022-00470-6
Yoshida, C. E., Kruczkiewicz, P., Laing, C. R., Lingohr, E. J., Gannon, V. P. J., Nash, J. H. E., et al. (2016). The Salmonella in silico typing resource (SISTR): An open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One 11:e0147101. doi: 10.1371/journal.pone.0147101
Keywords: non-typhoidal Salmonella, antimicrobial resistance, whole genome sequencing, serotyping, plasmid-mediated resistance
Citation: Cao H, Shen Y, Ma K, Zheng D, Xu Y and Qiao X (2025) Molecular characterization of clinical non-typhoidal Salmonella isolates shows high antimicrobial resistance burden in Jiangsu, China. Front. Microbiol. 16:1587421. doi: 10.3389/fmicb.2025.1587421
Edited by:
Marc Jean Struelens, Université libre de Bruxelles, BelgiumReviewed by:
Chiranjit Chowdhury, National Chemical Laboratory (CSIR), IndiaYanan Wang, Henan Agricultural University, China
Manoj Kumawat, ICMR-National Institute for Research in Environmental Health, India
Copyright © 2025 Cao, Shen, Ma, Zheng, Xu and Qiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Qiao, anNjZGNxeEAxMjYuY29t; Yan Xu, Y2RjeHlAdmlwLnNpbmEuY29t