 Pengfei Yi1,2†Tianqing Li1,2†Lianping Xu3Xin Li1,2,4Haiyan Wang4
Pengfei Yi1,2†Tianqing Li1,2†Lianping Xu3Xin Li1,2,4Haiyan Wang4 Yingcai Ma1,2Yunxiao Ma1,2Yawei Sun1,2Na Li1,2Qi Zhong5
Yingcai Ma1,2Yunxiao Ma1,2Yawei Sun1,2Na Li1,2Qi Zhong5 Xuelian Ma1,2
Xuelian Ma1,2 Gang Yao1,2*
Gang Yao1,2*- 1College of Veterinary Medicine, Xinjiang Agricultural University, Ürümqi, China
- 2Xinjiang Key Laboratory of New Drug Research and Development for Herbivores, Ürümqi, China
- 3Animal Disease Control and Diagnosis Center of Bayingolin Mongol Autonomous Prefecture, Korla, China
- 4Animal Disease Control and Diagnosis Center of Altay Prefecture, Altay, China
- 5Institute of Veterinary Research, Xinjiang Academy of Animal Science, Ürümqi, China
Infectious bovine rhinotracheitis virus (IBRV) is a globally prevalent pathogen that causes respiratory disease in cattle. Emerging evidence suggests that specific bacterial taxa and gut microbial community compositions are strongly associated with viral pathogenesis, by either enhancing or mitigating disease outcomes. This not only impacts the host’s gastrointestinal physiology but also affects distant organs, including the lungs, liver, and brain. However, the impact of IBRV infection on changes in gut microbiota composition and its association with MetaCyc metabolic pathways remains poorly understood. In this study, based on an epidemiological survey of one-month-old Angus calves in a large-scale Angus beef cattle breeding farm consists of four breeding areas located in Maigaiti County of Kashi Prefecture, China. Alterations in the gut microbiota of 10 IBRV-infected Angus calves (IBRV-positive group, P) compared with their 10 healthy counterparts (IBRV-negative group, N), as well as their correlations with MetaCyc metabolic pathways, were investigated using 16S rRNA sequencing. In comparison with N, both Simpson, Shannon and Pielou_e indices of alpha diversity were elevated in P, and the beta diversity showed a marked separation between N and P. The relative abundance of phylum Firmicutes_C was significantly increased, whereas that of phyla Bacteroidota, Cyanobacteria, and Firmicutes_D were reduced in P. The relative abundance of Genera Dialister and Klebsiella were enriched, while that of Lactobacillus and Blautia_A were depleted in P. Four distinct MetaCyc metabolic pathways were significantly altered, DENITRIFICATION-PWY, PWY-6906, and P101-PWY were significantly decreased in P, while PWY-7446 was significantly increased. Correlation analysis showed that in N, Faecalimonas was positively correlated with both P101-PWY and PWY-6906, and Limousia was positively correlated with P101-PWY. Faecalimonas was positively correlated with PWY-7446, and Klebsiella was positively correlated with DENITRIFICATION-PWY in P. Our results reveal that IBRV infection is associated with significant changes in the gut microbial community and its predicted metabolic functions, which may be linked to the host’s systemic response to the infection. This study provides preliminary data on the association between IBRV infection and gut microbiota profiles, laying a theoretical foundation for future investigations into IBRV pathogenesis and potential targeted prevention strategies.
1 Introduction
Infectious Bovine Rhinotracheitis Virus (IBRV), also known as Bovine Herpesvirus Type 1 (BHV-1), is a highly contagious virus that causes severe respiratory infections in cattle (Pardon et al., 2011; Wedgwood et al., 2020). Taxonomically, BHV-1 belongs to the subfamily Alphaherpesvirinae and the genus Varicellovirus (Nandi et al., 2009; Biswas et al., 2013). The respiratory symptoms is the most prevalent clinical manifestation of IBRV infection, primarily affecting the nasal cavity and trachea. Early-stage symptoms typically include fever, lethargy, anorexia, dyspnea, excessive nasal discharge, nasal mucosal congestion and ulceration, and swelling of the nasal planum (Nandi et al., 2009; Guo JunQing et al., 2019). In the absence of secondary infections, the disease is usually self-limiting and responds well to treatment. However, recovered cattle remain latent carriers of the virus and can continue to shed it into the environment (Schudel et al., 1986). In cases where symptoms are severe, recovery is challenging, and mortality rates are high (Epj, 1977). Additionally, immunosuppression is a significant characteristic of IBRV infection, which increases the risk of secondary infections and results in substantial indirect economic losses (Wang et al., 2024). Currently, there are no specific antiviral drugs for the treatment of this disease; vaccination remains the primary strategy for prevention and control (Ma et al., 2024).
The gut microbiota comprises a diverse and abundant community of microorganisms residing in the gastrointestinal tract (de Abreu Nascimento et al., 2023). The dynamic interactions between the gut microbiota and the host form a complex ecosystem that plays a vital role in maintaining host health (Barathan et al., 2024). In healthy individuals, the gut microbiota maintains homeostasis through a delicate balance with the host and its external environment (Obata and Pachnis, 2016). However, disruptions caused by factors such as weaning, dietary changes, and environmental alterations can lead to gut microbiota dysbiosis, which has been associated with a variety of diseases, including gastrointestinal disorders, metabolic syndromes, and cancers (Caserta et al., 2023; Tian et al., 2024). Research indicates that gut microbiota is involved in the pathogenesis of various pathogens, with dysbiosis potentially impairing immune responses and increasing the risk of pathogen infections (Pickard et al., 2017). Moreover, gut microbiota is also associated with the pathogenesis of non-gastrointestinal viruses. Mohammed et al. observed an association between gut microbiota dysbiosis and Hepatitis C virus infection, suggesting that microbial alterations may be involved in the disease process (El-Mowafy et al., 2021). Xing et al. (2022) demonstrated that gut microbiota plays a significant role in modulating immune responses to influenza virus infections. Recent reports suggest a close relationship between gut microbiota and the pathogenesis of human immunodeficiency virus and respiratory syncytial virus (Geng et al., 2020; Ji et al., 2021). Also, changes in the composition of certain microorganisms or changes in microbiota-host interactions may increase the susceptibility of mice to bovine viral diarrhea virus (Zhang et al., 2024). Infectious Bovine Rhinotracheitis Virus (IBRV) is a common respiratory virus in cattle, however the correlation between gut microbiota changes and IBRV infection has not been reported. Furthermore, studies have shown that various viruses, such as influenza virus, measles virus, and hepatitis virus, can significantly disrupt host cell metabolic responses by altering carbon, lipid, and amino acid metabolic pathways during infection (Cheng et al., 2020). The MetaCyc database is a resource providing extensive information on metabolic pathways from various organisms that may offer greater detail and specificity for certain metabolic pathways or networks than KEGG pathways do. (Karp et al., 2009; Caspi et al., 2017). Therefore, this study aims to investigate the alterations in gut microbiota in calves infected with IBRV and their association with MetaCyc metabolic pathways, in order to provide a foundation and theoretical basis for future research on the pathogenesis of respiratory viral diseases in calves.
2 Materials and methods
2.1 Epidemiological survey of respiratory diseases in calves
In 2021, a one-year epidemiological survey of respiratory diseases focusing on one-month-old calves was conducted in a large Angus beef cattle breeding farm consists of four breeding areas located in Maigaiti County of Kashi Prefecture, China. The calf clinical symptoms related to respiratory diseases included fever (rectal temperature above 39.5 °C), coughing (dry or wet cough, exacerbated by activity), and dyspnea (extended head and neck, labored breathing, pronounced abdominal breathing, and accompanying swallowing movements). The study protocol was approved by the Animal Ethics Committee of Xinjiang Agricultural University (2020036 and 2024023).
2.1.1 Sampling
Nasal and fecal samples were collected using sterile swabs inserted deeply into the nasal cavities and rectums from one-month-old calves showing clinical symptoms related to respiratory diseases, as well as randomly from healthy counterparts without the aforementioned respiratory symptoms. The samples were then sealed in cryovials and preserved in liquid nitrogen for future use.
2.1.2 PCR detection for IBRV
The UL27 gene sequence of IBRV deposited in GenBank (Accession No.: MK654723.1) was analyzed using DNAstar, DNAman, and online tools such as IEDB and Bepipred. Amino acid residues from positions 273 to 393, identified with high antigenicity, were selected as the target region. Primers were designed using Primer 5.0 and synthesized by Shanghai Bioengineering Co., Ltd. The primer sequences were F: 5′-CTCTACCGCACGGGCACCT-3′ and R: 5′-GCGGCTCTCGTCTCGCA-3′. The target gene (360 bp) was amplified using a total reaction volume of 15 μL. The reaction conditions were 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s followed by a final extension at 72 °C for 10 min.
2.2 Gut microbiota analysis by 16S rRNA gene sequencing
2.2.1 Grouping
Based on the results of PCR testing, 10 fecal samples from calves exhibiting respiratory clinical symptoms and then identified as positive solely for IBRV were randomly selected as the IBRV-positive group (P). Similarly, 10 fecal samples from healthy calves, identified as negative for IBRV and other respiratory viruses, were assigned to the IBRV-negative group (N).
2.2.2 16S rRNA gene sequencing
Genomic DNA was extracted using the Omega Viral DNA Quick Extraction Kit (Rehner et al., 2022). DNA concentration and purity were measured using a spectrophotometer, and the quality of the extracted DNA was assessed by agarose gel electrophoresis. The target fragments were then amplified by PCR. The amplified products were analyzed using 1.2% agarose gel electrophoresis, and the target fragments were recovered using the Axygen Gel Recovery Kit. Based on preliminary quantification results from electrophoresis, the recovered PCR products were subjected to quantitative fluorescence analysis. The V3 and V4 variable regions were selected for paired-end sequencing of community DNA fragments using the Illumina platform. Sequencing services were provided by Shanghai Paisenno Biotechnology Co., Ltd., China.
The Divisive Amplicon Denoising Algorithm 2 (DADA2) method was used for primer removal, quality filtering, denoising, merging, and chimera removal steps (Callahan et al., 2016). Each library was analyzed separately for these steps. R language scripts were used to statistically analyze the length distribution of high-quality sequences across all samples. Each deduplicated sequence generated by DADA2 was referred to as an Amplicon Sequence Variant (ASV). ASV sequences were compared with the Greengenes database (DeSantis et al., 2006), and each ASV in the ASV table was annotated taxonomically at the phylum, class, order, family, and genus levels. After distinguishing the samples, ASV clustering and taxonomic analysis were performed using the Usearch software platform. Alpha-diversity and beta-diversity analyses were conducted using the mothur software based on ASV data, and community structure was statistically analyzed at various taxonomic levels based on taxonomic information. Anosim (Analysis of Similarities) analysis was performed using the vegan package in R programming language.
2.3 Data statistics and analysis
Data were presented as mean ± standard error (SE). Differences in the relative abundance of gut microbiota at the phylum and genus levels between the N and P groups were analyzed using the Mann–Whitney test in GraphPad Prism version 10. * Denotes p < 0.05, meaning significant difference, ** denotes p < 0.01, meaning extremely significant difference. Benjamini-Hochberg false discovery rate (FDR) correction was applied to control for multiple hypothesis testing. Functional predictions based on 16S rRNA gene sequences were performed using the PICRUSt2 pipeline and functionally annotated using the MetaCyc database as a reference (DeSantis et al., 2006). Correlation analysis between the differential genera and significantly changed MetaCyc pathways in the N and P groups was conducted using Spearman’s rank correlation. All correlation analyses and visualizations were performed in R (version 4.4.2) using the ggplot2 and corrplot packages.
3 Results
3.1 Epidemiological survey of respiratory diseases in calves and IBRV detection
A total of 11,215 calves were born from December 2020 to November 2021, of which 922 exhibited clinical symptoms such as fever, cough, and dyspnea, accounting for 8.2% of the total number of births (922/11,215). Of these, 98 calves died, resulting in a mortality rate of 10.6% (98/922).
A total of 197 calves manifesting clinical symptoms including fever, cough, and dyspnea were randomly sampled from the field. Additionally, 30 calves without clinical symptoms were randomly selected. Nasal and rectal swabs were collected, and PCR testing was performed on nasal swabs for IBRV detection. The results showed that the IBRV positive rate was 23.35% (46/197).
3.2 The composition and diversity of the gut microbiota in healthy and IBRV-infected Angus calves
3.2.1 The composition and the differences in the relative abundance of gut microbiota
The composition of top 20 phyla and the differentials in the gut microbiota of calves between N and P group were shown in Figures 1A,B, in which there were 4 phyla with significant differences, i.e., in comparison with N group, the relative abundance of Firmicutes_C increased extremely in P group (p < 0.01), whereas that of Bacteroidota, Firmicutes_D and Cyanobacteria decreased extremely in P group (p < 0.01).
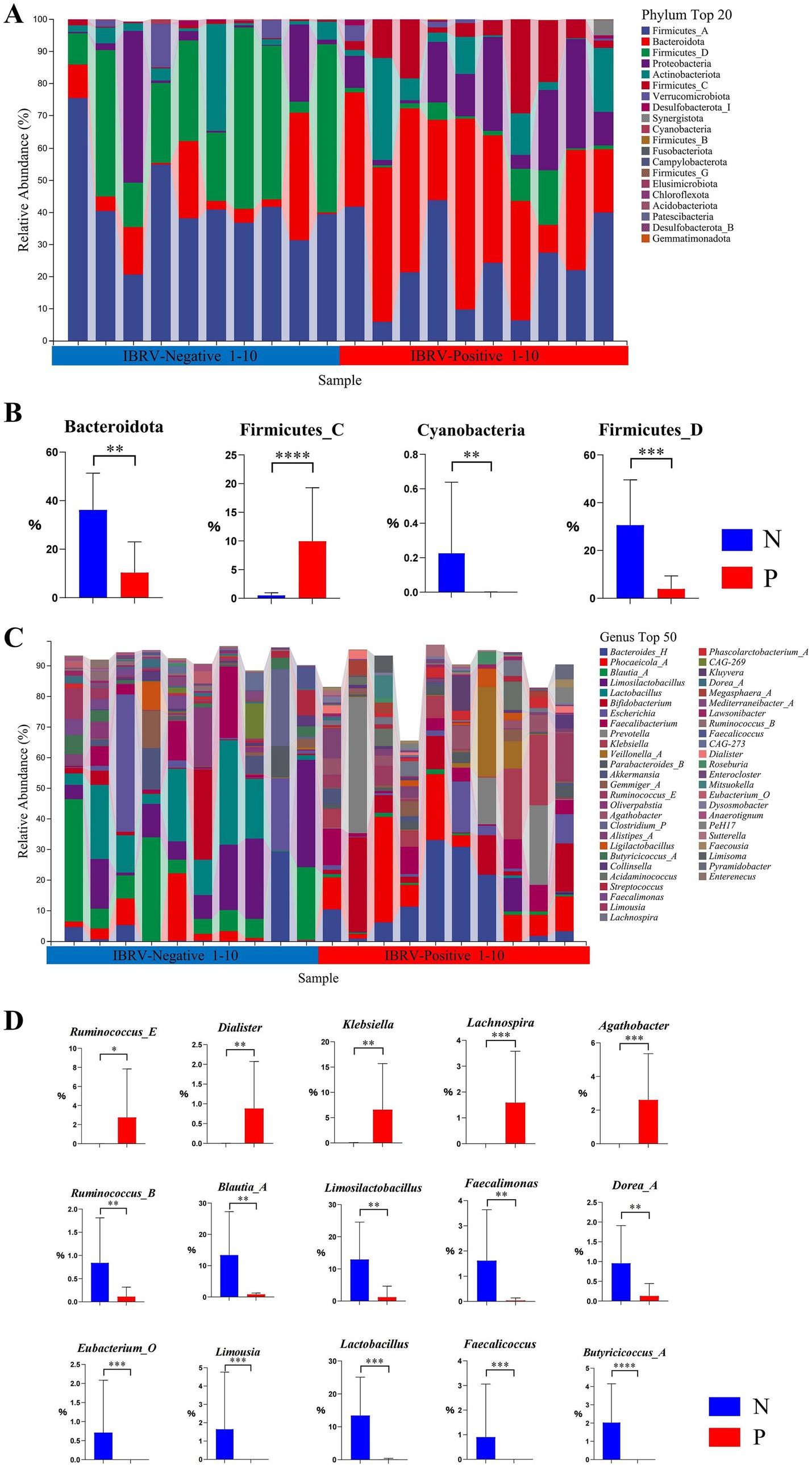
Figure 1. Composition and differential relative abundance of the top 20 phyla (A,B) and top 50 genera (C,D) between the N and P groups. (A) Top 20 phyla based on relative abundance in the N and P groups. (B) Differentially abundant phyla between the N and P groups. (C) Top 50 genera based on relative abundance in the N and P groups. (D) Differentially abundant genera between the N and P groups (N represents IBRV-Negative and P represents IBRV-Positive).
The composition of the genera top 50 and the differentials in the gut microbiota of calves between N and P group were shown in Figures 1C,D, in which there were 15 genera showed significant differences, i.e., in comparison with N group, the relative abundance of Ruminococcus_E increased significantly (p < 0.05), and that of Klebsiella, Lachnospira, Agathobacter and Dialister increased extremely in P group (p < 0.01), whereas that of Ruminococcus_B, Blautia_A, Limosilactobacillus, Faecalimonas, Dorea_A, Eubacterium_O, Limousia, Lactobacillus, Faecalicoccus, and Butyricicoccus_A decreased extremely in P group (p < 0.01).
3.2.2 The diversity of gut microbiota
Alpha and beta diversity of gut microbiota between the N and P groups were shown in Figures 2A,B. The Simpson, Shannon, and Pielou_e indices were significantly higher in the P group than in the N group (p < 0.05). In contrast, the Goods_coverage index was significantly lower in the P group than in the N group (p = 0.013).
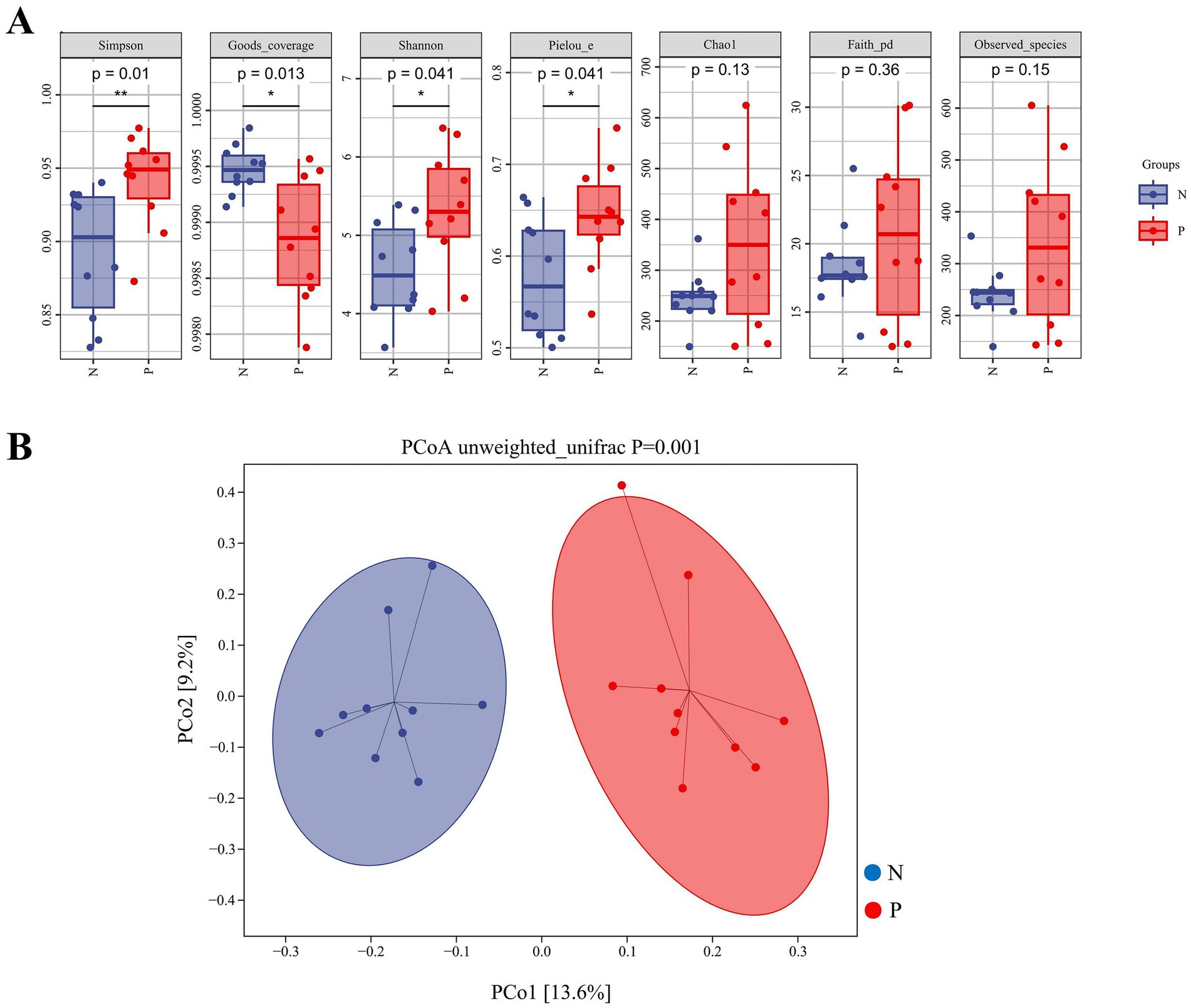
Figure 2. Indices of alpha-diversity (A) and beta-diversity (B) in the gut microbiota of calves between N and P group. (A) Box plots showing alpha diversity indices including Simpson, Shannon, Pielou_e, Chao1, Faith_pd, Observed_species, and Goods_coverage. The vertical axes represent the values of each alpha diversity index. p-values were calculated using the Wilcoxon rank-sum test; p < 0.05 was considered statistically significant. (B) Principal coordinate analysis (PCoA) based on unweighted UniFrac distances. The axes represent the first (PCo1) and second (PCo2) principal coordinates, explaining 13.6 and 9.2% of the variation among samples, respectively. Each dot represents a sample, and the ellipses denote 95% confidence intervals. The group difference was tested by PERMANOVA (p = 0.001) (N represents IBRV-Negative and P represents IBRV-Positive).
Beta diversity analysis by PCoA revealed that PCo1 was accounted for 13.6% of the variance, and PCo2 accounted for 9.2%. Further Unweighted_unifrac Adonis analysis indicated there was a highly significant independent distribution of gut microbiota between the P group and N group (p = 0.001) (Figure 2B).
3.3 Functional prediction of gut microbiota
3.3.1 Functional prediction in gut microbiota based on MetaCyc metabolic pathway
As shown in Figure 3, the MetaCyc database was used to predict the primary metabolic functions of the gut microbiota in groups N and P. The major functional pathways were identified, i.e., biosynthesis, degradation/utilization/assimilation, detoxification, generation of precursor metabolite and energy, glycan pathways, macromolecular modifications, and metabolic clusters. At level 2 in above 7 categories, a total of 59 metabolic pathways were detected, in which pathways with the higher relative abundance were primarily enriched in the biosynthesis, degradation/utilization/assimilation, and the generation of precursor metabolites and energy.
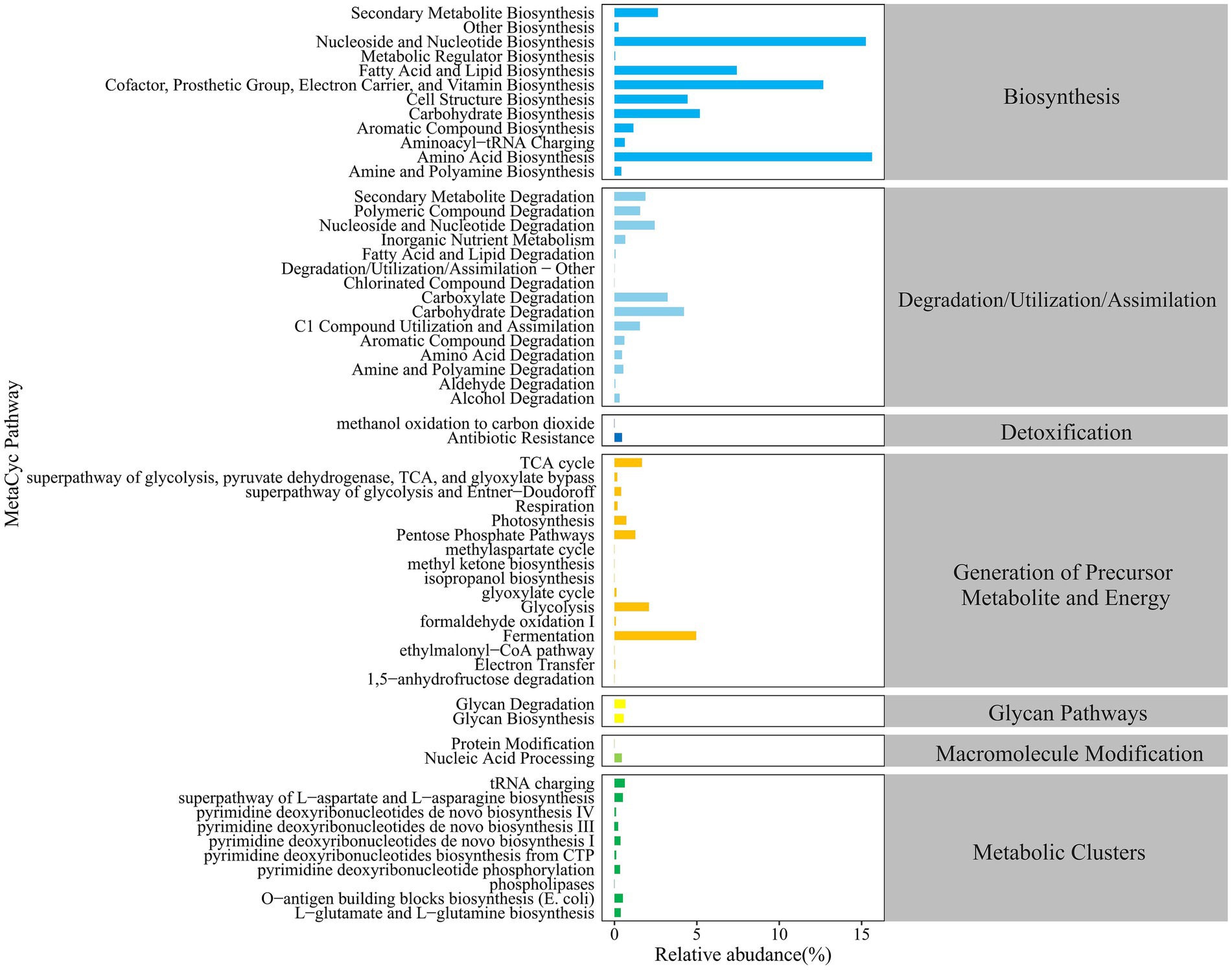
Figure 3. Statistics of MetaCyc metabolic pathways in the Gut Microbiota of calves.
3.3.2 Differential MetaCyc metabolic pathways in the gut microbiota
As shown in Figure 4, there were 4 differential pathways identified at level 3 in MetaCyc database for function predictions of gut microbiota between the P and N groups. In comparison with group N, the relative abundance of the nitrate reduction I (DENITRIFICATION-PWY) in inorganic nutrient metabolism and respiration, the chitin derivatives degradation (PWY-6906) in carbohydrate degradation, and the ectoine biosynthesis (P101-PWY) in amide, amidine, amine, and polyamine biosynthesis in group P was significantly or extremely decreased (p < 0.05 or p < 0.01), while that of sulfoquinovose degradation I (PWY-7446) in carbohydrate degradation was extremely increased.
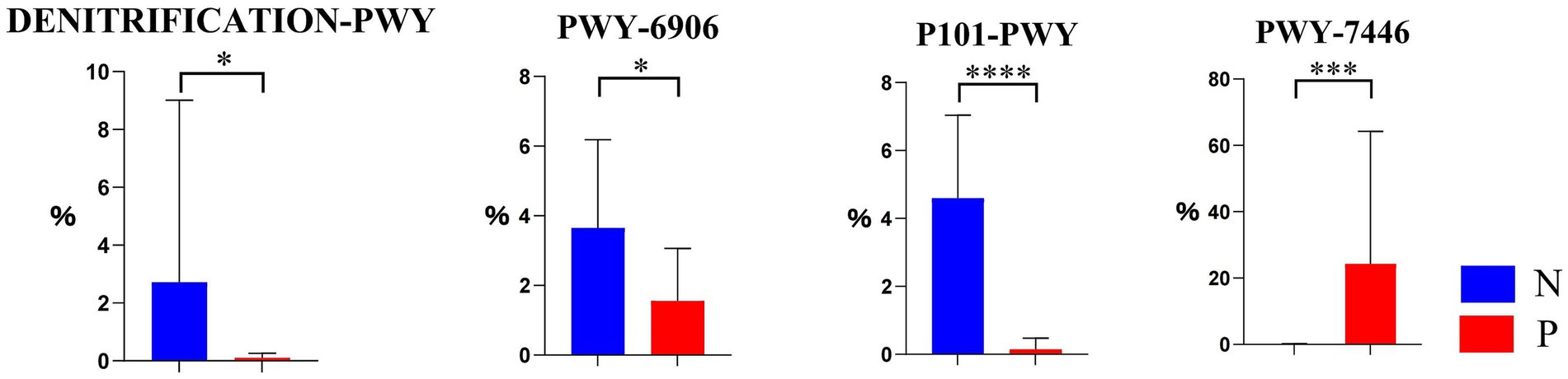
Figure 4. The differential MetaCyc metabolic pathways in the gut microbiota of calves between N and P group. The y-axis indicates the relative functional abundance (%), predicted by PICRUSt2 and normalized by 16S rRNA gene copy number (N represents IBRV-Negative and P represents IBRV-Positive).
3.3.3 Differential genera in contribution to differential MetaCyc metabolic pathway
A Mann–Whitney test was conducted to compare 15 distinct genera identified in Section 3.2.1 that contribute to the differential MetaCyc metabolic pathways of the gut microbiota in groups N and P. The results revealed significant differences in genera within the ectoine biosynthesis (P101-PWY) and the chitin derivative degradation pathway (PWY-6906) between the two groups. However, no significant genera differences were observed in the nitrate reduction I pathway (DENITRIFICATION-PWY) and the sugar degradation pathway (PWY-7446, sulfoglycolysis).
3.3.3.1 Differential genera in the ectoine biosynthesis pathway
As illustrated in the ectoine biosynthesis (P101-PWY) in Figure 5, the relative abundances of Dorea_A, Bacteroides_H, Limousia, Faecalimonas, and CAG-273 were significantly lower (p < 0.05), and that of Faecalicoccus, Lactobacillus, and Butyricicoccus_A was extremely lower (p < 0.01) in group P, if compared to group N.
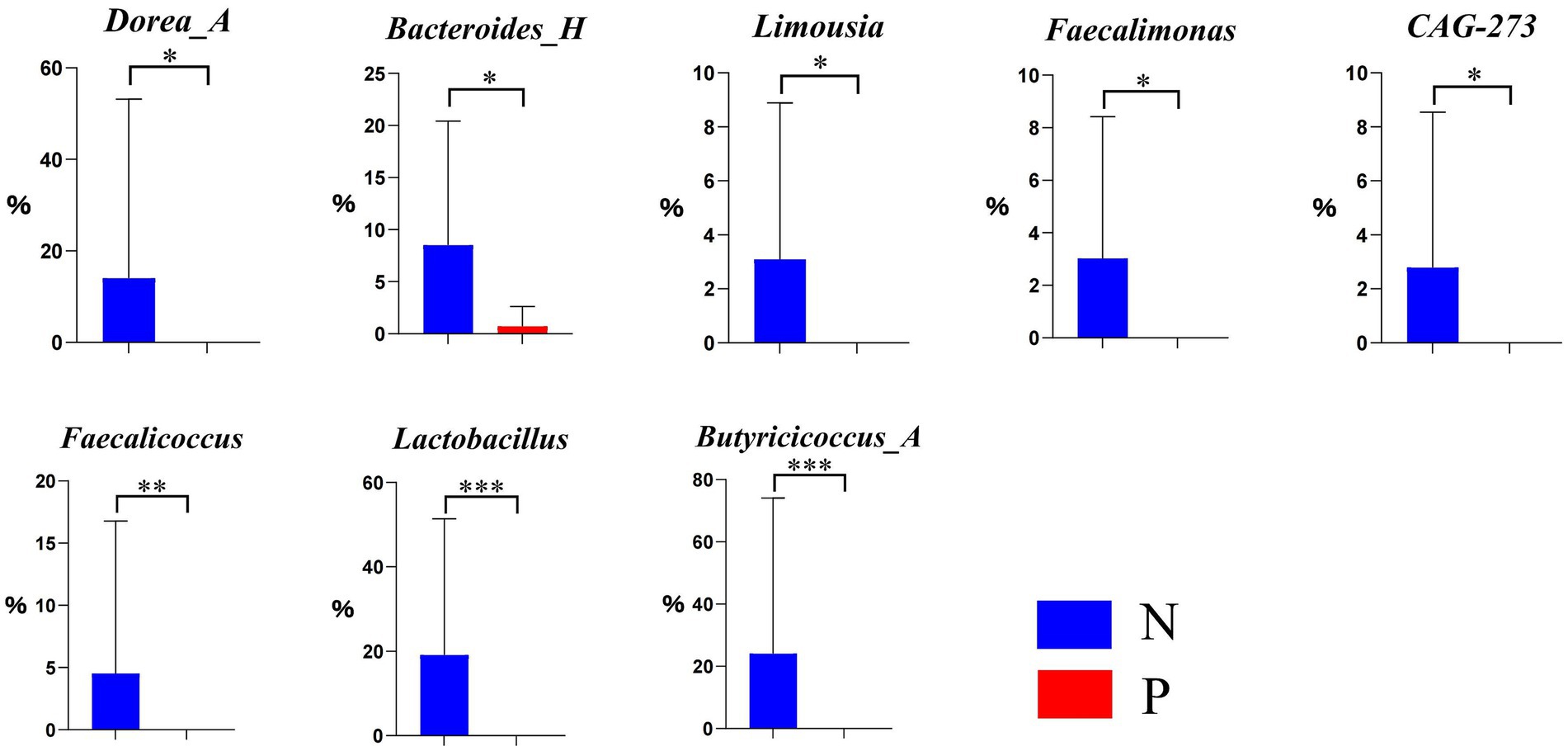
Figure 5. Differential genera in the ectoine biosynthesis pathway P101-PWY between N and P group (N represents IBRV-Negative and P represents IBRV-Positive).
3.3.3.2 Differential genera in the chitin derivative degradation pathway
As illustrated in the chitin derivative degradation pathway (PWY-6906) in Figure 6, the relative abundance of Agathobacter was significantly higher (p < 0.05), and that of Klebsiella was extremely higher (p < 0.01) in group P, if compared to group N.
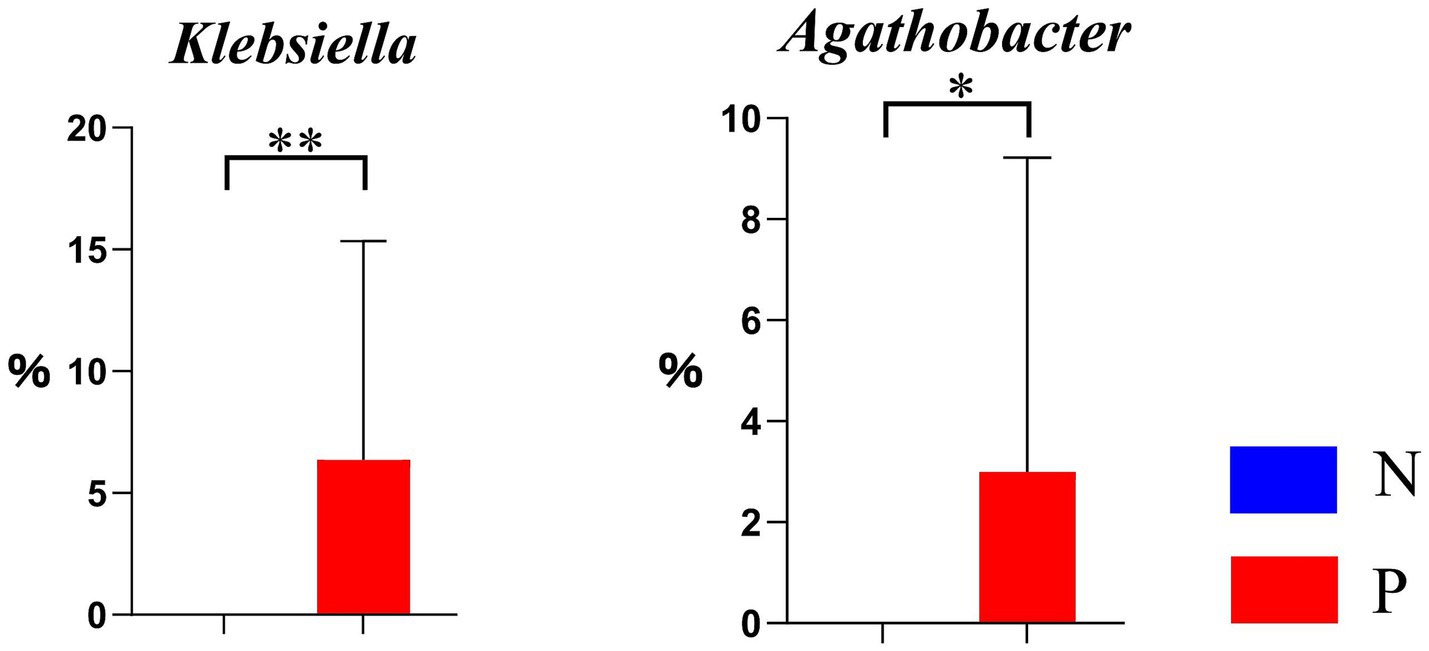
Figure 6. Differential genera in the chitin derivative degradation pathway PWY-6906 between N and P groups (N represents IBRV-Negative and P represents IBRV-Positive).
3.3.4 Correlation analysis between differential genera of gut microbiota and differential MetaCyc metabolic pathways
A correlation analysis was conducted between the 15 differentially abundant genera and the 4 differential MetaCyc metabolic pathways. In N group, two differential genera were significantly correlated with two metabolic pathways (Figure 7A). Specifically, Faecalimonas was highly significantly positively correlated with the ectoine biosynthesis pathway (P101-PWY) (p < 0.05, r = 0.736). Limousia was significantly positively correlated with the ectoine biosynthesis pathway (p < 0.05, r = 0.648). Additionally, Faecalimonas showed a highly significant positive correlation with the chitin derivatives degradation pathway (PWY-6906) (p < 0.01, r = 0.815).
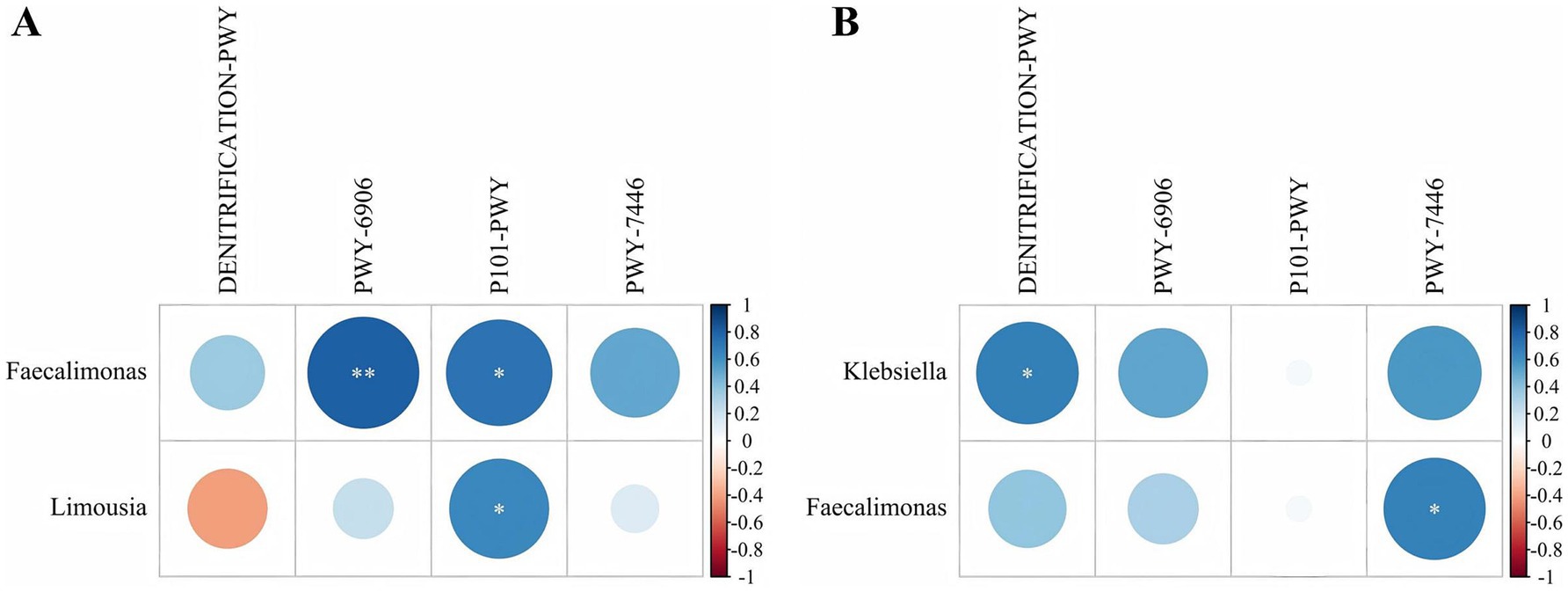
Figure 7. Correlation between the differential genera and differential MetaCyc metabolic pathways in group N and P. (A) Group N correlation. (B) Group P correlation (N represents IBRV-Negative and P represents IBRV-Positive).
In P group, two differential genera were significantly correlated with two metabolic pathways (Figure 7B). Faecalimonas showed a significant positively correlation with the same pathway (p < 0.05, r = 0.679). Additionally, Klebsiella was significantly positively correlated with the nitrate reduction I pathway (DENITRIFICATION-PWY) (p < 0.05, r = 0.683).
4 Discussion
Respiratory viral infections are known to influence gut microbiota composition, as reported for influenza and respiratory syncytial virus (RSV) (Haak et al., 2018; Groves et al., 2020). Similarly, SARS-CoV-2 has been shown to disrupt gut microbial ecology and induce intestinal inflammation (Zuo et al., 2020; Li et al., 2021). IBRV infection led to notable changes in the relative abundance of specific phyla and genera, with increases in Bacteroidota, and decreases in Firmicutes_D and Verrucomicrobia. Bacteroidota, an essential component of the gut microbiota, has been linked to inflammation and respiratory diseases; its dysregulation may exacerbate lung inflammation via the gut-lung axis, as seen in asthma patients (Chiu et al., 2021; Zhao et al., 2023). Firmicutes is known to regulate immune function, supporting immune balance under normal conditions, and its reduced abundance may weaken the immune system’s defense against pathogens (Duan et al., 2022). Normally, Verrucomicrobia can modulate host immune responses to a certain extent (Zhang et al., 2022b); its abundance may be altered when viral infections disrupt gut microbiota balance (Zhang et al., 2022a). During rotavirus infections, for example, the gut microbiota ecosystem is disrupted, affecting the growth and metabolism of Verrucomicrobia (Jang et al., 2018). At the genus level, the relative abundance of Klebsiella increased, while Lactobacillus and Blautia_A decreased. Klebsiella, which is highly pathogenic, includes Klebsiella pneumoniae, the most common species (Conde-Pérez et al., 2024), and some studies suggest that Klebsiella may act synergistically with viruses to worsen disease progression (Yasir et al., 2023). Lactobacillus enhances immune function by modulating immune cell activity and protects mucosal barriers in the gut and respiratory tract by strengthening tight junction protein expression, thus resisting viral invasion of cells (Peng et al., 2022; Shi et al., 2022). Additionally, Lactobacillus can reduce viral infection at colonization sites, such as the gut, through competitive exclusion (Deng et al., 2021; Bu et al., 2023). Blautia_A also plays a role in immune regulation, supporting immune homeostasis and enhancing gut barrier function (Mukherjee et al., 2020; Zhao et al., 2022). The results suggest that these changes in the abundance of specific phyla and genera are closely associated with IBRV infection.
Viral infections have been shown to significantly alter the composition and diversity of the gut microbiota. For example, in mouse models of influenza virus infection, the abundance of beneficial gut bacteria decreases while certain opportunistic pathogens increase (Sun et al., 2020). Similarly, changes in gut microbiota diversity and the proportions of specific genera have been observed following SARS-CoV-2 infection (Nguyen et al., 2023). In the present study, alpha diversity analysis revealed that gut microbiota diversity was higher in IBRV-infected calves compared to healthy controls. Beta diversity analysis further demonstrated significant compositional differences between the two groups. Previous studies indicate a dynamic and complex interaction between viral infections and gut microbiota alterations (Groves et al., 2018; Vujkovic-Cvijin and Somsouk, 2019). Viral infections can elicit immune responses that not only target the pathogen but also influence the intestinal environment, thereby reshaping the gut microbial community (Kloepfer and Kennedy, 2023). Such changes may damage the intestinal mucosa, increasing susceptibility to secondary infections (Preveden et al., 2017). Moreover, IBRV infection significantly affected the functional potential of the gut microbiota. Multiple differential MetaCyc metabolic pathways were identified between IBRV-infected and healthy calves, particularly those related to biosynthesis, degradation/utilization/assimilation, precursor metabolite generation, and energy production. These alterations suggest that IBRV infection may influence host physiological processes such as nutrient absorption, metabolic regulation, and immune function (Li et al., 2022; Singh et al., 2024). Notably, nitrate reduction I (DENITRIFICATION-PWY) and chitin derivatives degradation (PWY-6906) pathways were significantly downregulated in the infected group. Nitrate is involved in nitrification and reduction processes critical for nitrogen metabolism in animals (Rizzatti et al., 2017), while chitin metabolism contributes to nitrogen cycling and elemental balance (Deng et al., 2022). Both are essential for maintaining nitrogen homeostasis and energy metabolism. Additionally, the ectoine biosynthesis pathway (P101-PWY) was markedly downregulated in IBRV-infected calves. Ectoine is a protective molecule that helps microorganisms regulate intracellular osmotic pressure and maintain membrane and protein integrity under osmotic stress (Ojeda et al., 2015; Damacena-Angelis et al., 2017). Conversely, the sulfoglycolysis pathway (PWY-7446) was significantly upregulated following IBRV infection. The metabolic products of this pathway, such as butyrate, are beneficial to the host; for instance, butyrate serves as a key energy source for intestinal epithelial cells (Xiao et al., 2023). For instance, butyrate, produced during carbohydrate degradation, serves as a primary energy source for intestinal epithelial cells (Sharma et al., 2021). Furthermore, correlation analysis between the 15 differentially abundant genera and the four differential MetaCyc pathways revealed that Faecalimonas showed a highly significant positively correlation with the chitin derivatives degradation pathway (PWY-6906), potentially reflecting its role in maintaining host metabolism (Tlaskalová-Hogenová et al., 2004). Whereas Faecalimonas and Limousia in the gut microbiota of healthy calves were significantly positively correlated with ectoine biosynthesis (P101-PWY). These genera, commonly found in the gut microbiota, are reported to play roles in immune regulation by promoting mucus secretion from intestinal epithelial cells, which helps maintain the integrity of the gut mucosal barrier (Viggiano et al., 2015; Rastogi and Singh, 2022). These functional alterations suggest that IBRV infection not only changes the structural composition of gut microbiota but also impairs essential microbial functions that help maintain host health. The downregulation of pathways such as ectoine biosynthesis pathway, nitrate reduction I, and chitin degradation indicates a reduced microbial capacity for osmotic protection, nitrogen balance, and barrier support. This may weaken the host’s mucosal defense and immune homeostasis, thereby increasing susceptibility to IBRV infection and exacerbating its progression. Conversely, the upregulation of the sulfoquinovose degradation I pathway may reflect a microbial adaptation to inflammation or dysbiosis. Collectively, these results support a potential mechanistic link between gut microbiota metabolic function and IBRV pathogenesis. In IBRV-infected calves, the ectoine biosynthesis pathway (P101-PWY) was significantly downregulated, with the relative abundances of Faecalimonas and Limousia in this pathway markedly lower than those in healthy calves. In the gut microbiota of IBRV-infected calves, Klebsiella was significantly positively correlated with the nitrate reduction I pathway (DENITRIFICATION-PWY), whereas Faecalimonas was significantly positively correlated with the sulfoglycolysis pathway (PWY-7446), which was not consistent with the increase in relative abundance of Klebsiella and decrease in relative abundance of Faecalimonas after IBRV infection. This suggests that there may be a discrepancy between changes in abundance of some genera and changes in abundance of metabolic pathways during alterations in the gut microbiota (Li et al., 2023). The exact reasons for this situation are expected to be explored in future study.
5 Conclusion
This study demonstrated significant alterations in the gut microbiota of IBRV-infected calves and their association with MetaCyc metabolic pathways. The gut microbiota may play a crucial role in regulating the host’s response to IBRV infection through gut microbial metabolic activities linked to the onset and progression of the disease. Further investigations are warranted to clarify the causal relationship between gut microbiota alterations and IBRV pathogenesis, as well as the mechanisms by which specific bacterial taxa influence the IBRV infection process. It would facilitate the development of strategies for preventing and treating IBRV infection via gut microbiota intervention.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/genbank/, accession number MK654723.1.
Ethics statement
The animal study was approved by I would be glad to explain that, the calves used in the study “The technology of fecal bacteria transplantation to prevent and control calf diarrhea has been popularized” by Dr. Xuelian Ma et al. were commercial livestock raised from some of the large-scale farms in the Southern Xinjiang region and owned by the large-scale farms. Before the study, the calves used were raised by local large-scale farms (Southern Xinjiang region). At the end of the study, all calves were reared normally in each scale farm. In such case, the informed consent from any client was exempted, and the whole research procedures were reviewed and permitted by The Animal Welfare and Ethics Committee of Xinjiang Agricultural University (Approval Number: 2024023). I would be glad to explain that, the calves used in the study “The differential gut microbiota and their MetaCyc pathways in IBRV infected Angus calves” by Dr. Pengfei Yi et al. were commercial livestock raised from some of the large-scale farms in the Southern Xinjiang region and owned by the large-scale farms. Before the study, the calves used were raised by local large-scale farms (Southern Xinjiang region). At the end of the study, all calves were reared normally in each scale farm. In such case, the informed consent from any client was exempted, and the whole research procedures were reviewed and permitted by The Animal Welfare and Ethics Committee of Xinjiang Agricultural University (Approval Number: 2020036). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
PY: Conceptualization, Validation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft. TL: Formal analysis, Software, Validation, Visualization, Writing – original draft. LX: Investigation, Resources, Writing – review & editing. XL: Investigation, Resources, Writing – review & editing. HW: Investigation, Writing – review & editing, Methodology. YcM: Methodology, Writing – review & editing, Data curation. YxM: Methodology, Writing – review & editing, Investigation. YS: Writing – review & editing, Data curation, Supervision. NL: Data curation, Supervision, Writing – review & editing. QZ: Supervision, Writing – review & editing, Resources, Software. XM: Funding acquisition, Project administration, Resources, Writing – review & editing. GY: Resources, Supervision, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant No. 32460870), the Major Science and Technology Projects of Xinjiang Uygur Autonomous Region (Grant No. 2023A02007-2) and the Science and Technology Projects for Rural Revitalization in Xinjiang (Grant No. 2024NC025).
Acknowledgments
Acknowledge goes to Xinjiang Daolang Sunshine Animal Husbandry Science & Technology Co. for providing experimental animal and management assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barathan, M., Ng, S. L., Lokanathan, Y., Ng, M. H., and Law, J. X. (2024). The profound influence of gut microbiome and extracellular vesicles on animal health and disease. Int. J. Mol. Sci. 25:4024. doi: 10.3390/ijms25074024
Biswas, S., Bandyopadhyay, S., Dimri, U., and H Patra, P. (2013). Bovine herpesvirus-1 (BHV-1)–a re-emerging concern in livestock: a revisit to its biology, epidemiology, diagnosis, and prophylaxis. Vet. Q. 33, 68–81. doi: 10.1080/01652176.2013.799301
Bu, Y., Liu, Y., Liu, Y., Cao, J., Zhang, Z., and Yi, H. (2023). Protective effects of bacteriocin-producing Lactiplantibacillus plantarum on intestinal barrier of mice. Nutrients 15:3518. doi: 10.3390/nu15163518
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caserta, S., Genovese, C., Cicero, N., Toscano, V., Gangemi, S., and Allegra, A. (2023). The interplay between medical plants and gut microbiota in cancer. Nutrients 15:3327. doi: 10.3390/nu15153327
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2017). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639. doi: 10.1093/nar/gkx935
Cheng, M.-L., Chien, K.-Y., Lai, C.-H., Li, G.-J., Lin, J.-F., and Ho, H.-Y. (2020). Metabolic reprogramming of host cells in response to enteroviral infection. Cells 9:473. doi: 10.3390/cells9020473
Chiu, Y.-C., Lee, S.-W., Liu, C.-W., Lin, R. C.-J., Huang, Y.-C., Lan, T.-Y., et al. (2021). Comprehensive profiling of the gut microbiota in patients with chronic obstructive pulmonary disease of varying severity. PLoS One 16:e0249944. doi: 10.1371/journal.pone.0249944
Conde-Pérez, K., Aja-Macaya, P., Buetas, E., Trigo-Tasende, N., Nasser-Ali, M., Rumbo-Feal, S., et al. (2024). The multispecies microbial cluster of Fusobacterium, Parvimonas, Bacteroides and Faecalibacterium as a precision biomarker for colorectal cancer diagnosis. Mol. Oncol. 18, 1093–1122. doi: 10.1002/1878-0261.13604
Damacena-Angelis, C., Oliveira-Paula, G. H., Pinheiro, L. C., Crevelin, E. J., Portella, R. L., Moraes, L. A. B., et al. (2017). Nitrate decreases xanthine oxidoreductase-mediated nitrite reductase activity and attenuates vascular and blood pressure responses to nitrite. Redox Biol. 12, 291–299. doi: 10.1016/j.redox.2017.03.003
De Abreu Nascimento, M., Pimenta, N. D. M. A., Polastri, V. A. D. M. P., Chamon, R. C., and Figueiredo, M. S. (2023). Immunonutrients and intestinal microbiota: a gap in the literature. Crit. Rev. Food Sci. Nutr. 64, 13058–13071. doi: 10.1080/10408398.2023.2260468
Deng, Z., Han, D., Wang, Y., Wang, Q., Yan, X., Wang, S., et al. (2021). Lactobacillus casei protects intestinal mucosa from damage in chicks caused by Salmonella pullorum via regulating immunity and the Wnt signaling pathway and maintaining the abundance of gut microbiota. Poult. Sci. 100:101283. doi: 10.1016/j.psj.2021.101283
Deng, F., Lin, Z.-B., Sun, Q.-S., Min, Y., Zhang, Y., Chen, Y., et al. (2022). The role of intestinal microbiota and its metabolites in intestinal and extraintestinal organ injury induced by intestinal ischemia reperfusion injury. Int. J. Biol. Sci. 18, 3981–3992. doi: 10.7150/ijbs.71491
Desantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/aem.03006-05
Duan, X., Chen, P., Xu, X., Han, M., and Li, J. (2022). Role of gastric microorganisms other than Helicobacter pylori in the development and treatment of gastric diseases. Biomed. Res. Int. 2022:6263423. doi: 10.1155/2022/6263423
El-Mowafy, M., Elgaml, A., El-Mesery, M., Sultan, S., Ahmed, T. A. E., Gomaa, A. I., et al. (2021). Changes of gut-microbiota-liver axis in hepatitis C virus infection. Biology 10:55. doi: 10.3390/biology10010055
Geng, S.-T., Zhang, Z.-Y., Wang, Y.-X., Lu, D., Yu, J., Zhang, J.-B., et al. (2020). Regulation of gut microbiota on immune reconstitution in patients with acquired immunodeficiency syndrome. Front. Microbiol. 11:594820. doi: 10.3389/fmicb.2020.594820
Groves, H. T., Cuthbertson, L., James, P., Moffatt, M. F., Cox, M. J., and Tregoning, J. S. (2018). Respiratory disease following viral lung infection alters the murine gut microbiota. Front. Immunol. 9:182. doi: 10.3389/fimmu.2018.00182
Groves, H. T., Higham, S. L., Moffatt, M. F., Cox, M. J., and Tregoning, J. S. (2020). Respiratory viral infection alters the gut microbiota by inducing inappetence. MBio 11:e03236–19. doi: 10.1128/mbio.03236-19
Guo Junqing, G. J., Li Qingmei, L. Q., and Jones, C. (2019). The bovine herpesvirus 1 regulatory proteins, bICP4 and bICP22, are expressed during the escape from latency. J. Neurovirol. 25, 42–49. doi: 10.1007/s13365-018-0684-7
Haak, B. W., Littmann, E. R., Chaubard, J.-L., Pickard, A. J., Fontana, E., Adhi, F., et al. (2018). Impact of gut colonization with butyrate producing microbiota on respiratory viral infection following allo-HCT. Blood, 2978–2986. doi: 10.1182/blood-2018-01-828996
Jang, J.-Y., Kim, S., Kwon, M.-S., Lee, J., Yu, D.-H., Song, R.-H., et al. (2018). Rotavirus-mediated alteration of gut microbiota and its correlation with physiological characteristics in neonatal calves. J. Microbiol. 57, 113–121. doi: 10.1007/s12275-019-8549-1
Ji, J.-J., Sun, Q.-M., Nie, D.-Y., Wang, Q., Zhang, H., Qin, F.-F., et al. (2021). Probiotics protect against RSV infection by modulating the microbiota-alveolar-macrophage axis. Acta Pharmacol. Sin. 42, 1630–1641. doi: 10.1038/s41401-020-00573-5
Karp, P. D., Paley, S. M., Krummenacker, M., Latendresse, M., Dale, J. M., Lee, T. J., et al. (2009). Pathway tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79. doi: 10.1093/bib/bbp043
Kloepfer, K. M., and Kennedy, J. L. (2023). Childhood respiratory viral infections and the microbiome. J. Allergy Clin. Immunol. 152, 827–834. doi: 10.1016/j.jaci.2023.08.008
Li, Y., Lan, Y., Zhang, S., and Wang, X. (2022). Comparative analysis of gut microbiota between healthy and diarrheic horses. Front. Vet. Sci. 9:882423. doi: 10.3389/fvets.2022.882423
Li, Y., Mao, K., Zang, Y., Lu, G., Qiu, Q., Ouyang, K., et al. (2023). Revealing the developmental characterization of rumen microbiome and its host in newly received cattle during receiving period contributes to formulating precise nutritional strategies. Microbiome 11:238. doi: 10.1186/s40168-023-01682-z
Li, S., Yang, S., Zhou, Y., Disoma, C., Dong, Z., Du, A., et al. (2021). Microbiome profiling using shotgun metagenomic sequencing identified unique microorganisms in COVID-19 patients with altered gut microbiota. Front. Microbiol. 12:712081. doi: 10.3389/fmicb.2021.712081
Ma, Y., Guo, X., He, Q., Liu, L., Li, Z., Zhao, X., et al. (2024). Integrated analysis of microRNA and messenger RNA expression profiles reveals functional microRNA in infectious bovine rhinotracheitis virus-induced mitochondrial damage in Madin-Darby bovine kidney cells. BMC Genomics 25:158. doi: 10.1186/s12864-024-10042-6
Mukherjee, A., Lordan, C., Ross, R. P., and Cotter, P. D. (2020). Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 12:1802866. doi: 10.1080/19490976.2020.1802866
Nandi, S., Kumar, M., Manohar, M., and Chauhan, R. S. (2009). Bovine herpes virus infections in cattle. Anim. Health Res. Rev. 10, 85–98. doi: 10.1017/S1466252309990028
Nguyen, L. H., Okin, D., Drew, D. A., Battista, V. M., Jesudasen, S. J., Kuntz, T. M., et al. (2023). Metagenomic assessment of gut microbial communities and risk of severe COVID-19. Genome Med. 15:49. doi: 10.1186/s13073-023-01202-6
Obata, Y., and Pachnis, V. (2016). The effect of microbiota and the immune system on the development and organization of the enteric nervous system. Gastroenterology 151, 836–844. doi: 10.1053/j.gastro.2016.07.044
Ojeda, P., Bobe, A., Dolan, K., Leone, V., and Martinez, K. (2015). Nutritional modulation of gut microbiota — the impact on metabolic disease pathophysiology. J. Nutr. Biochem. 28, 191–200. doi: 10.1016/j.jnutbio.2015.08.013
Pardon, B., De Bleecker, K., Dewulf, J., Callens, J., Boyen, F., Catry, B., et al. (2011). Prevalence of respiratory pathogens in diseased, non-vaccinated, routinely medicated veal calves. Vet. Rec. 169:278. doi: 10.1136/vr.d4406
Peng, X., Ed-Dra, A., Song, Y., Elbediwi, M., Nambiar, R. B., Zhou, X., et al. (2022). Lacticaseibacillus rhamnosus alleviates intestinal inflammation and promotes microbiota-mediated protection against Salmonella fatal infections. Front. Immunol. 13:973224. doi: 10.3389/fimmu.2022.973224
Pickard, J. M., Zeng, M. Y., Caruso, R., and Núñez, G. (2017). Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89. doi: 10.1111/imr.12567
Preveden, T., Scarpellini, E., Milić, N., Luzza, F., and Abenavoli, L. (2017). Gut microbiota changes and chronic hepatitis C virus infection. Expert Rev. Gastroenterol. Hepatol. 11, 813–819. doi: 10.1080/17474124.2017.1343663
Rastogi, S., and Singh, A. (2022). Gut microbiome and human health: exploring how the probiotic genus Lactobacillus modulate immune responses. Front. Pharmacol. 13:1042189. doi: 10.3389/fphar.2022.1042189
Rehner, J., Schmartz, G. P., Groeger, L., Dastbaz, J., Ludwig, N., Hannig, M., et al. (2022). Systematic cross-biospecimen evaluation of DNA extraction kits for long- and short-read multi-metagenomic sequencing studies. Genomics Proteomics Bioinformatics. doi: 10.1016/j.gpb.2022.05.006
Rizzatti, G., Lopetuso, L. R., Gibiino, G., Binda, C., and Gasbarrini, A. (2017). Proteobacteria: a common factor in human diseases. Biomed. Res. Int. 2017, 1–7. doi: 10.1155/2017/9351507
Schudel, A. A., Carrillo, B. J., Wyler, R., and Metzler, A. E. (1986). Infections of calves with antigenic variants of bovine herpesvirus 1 (BHV-1) and neurological disease. J. Veterinary Med. Ser. B 33, 303–310. doi: 10.1111/j.1439-0450.1986.tb00036.x
Sharma, M., Abayakoon, P., Epa, R., Jin, Y., Lingford, J. P., Shimada, T., et al. (2021). Molecular basis of Sulfosugar selectivity in Sulfoglycolysis. ACS Cent. Sci. 7, 476–487. doi: 10.1021/acscentsci.0c01285
Shi, Z., Guan, N., Sun, W., Sun, T., Niu, L., Li, J., et al. (2022). Protective effect of Levilactobacillus brevis against Yersinia enterocolitica infection in mouse model via regulating MAPK and NF-κB pathway. Probiotics Antimicrob. Proteins 14, 830–844. doi: 10.1007/s12602-022-09957-x
Singh, A. S., Pathak, D., Devi, M. S., Anifowoshe, A. T., and Nongthomba, U. (2024). Antibiotic alters host’s gut microbiota, fertility, and antimicrobial peptide gene expression Vis-à-Vis ampicillin treatment on model organism Drosophila melanogaster. Int. Microbiol. 27, 1665–1676. doi: 10.1007/s10123-024-00507-9
Sun, Y., He, Z., Li, J., Gong, S., Yuan, S., Li, T., et al. (2020). Gentamicin induced microbiome adaptations associate with increased BCAA levels and enhance severity of influenza infection. Front. Immunol. 11:608895. doi: 10.3389/fimmu.2020.608895
Tian, Q. B., Chen, S. J., Xiao, L. J., Xie, J. Q., Zhao, H. B., and Zhang, X. (2024). Potential effects of nutrition-induced alteration of gut microbiota on inflammatory bowel disease: a review. J. Dig. Dis.. doi: 10.1111/1751-2980.13256
Tlaskalová-Hogenová, H., Stepánková, R., Hudcovic, T., Tucková, L., Cukrowska, B., Lodinová-Zádníková, R., et al. (2004). Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol. Lett. 93, 97–108. doi: 10.1016/j.imlet.2004.02.005
Viggiano, D., Ianiro, G., Vanella, G., Bibbò, S., Bruno, G., Simeone, G., et al. (2015). Kaledo, a board game for nutrition education of children and adolescents at school: cluster randomized controlled trial of healthy lifestyle promotion. Eur J Pediatr, 174:217–228. doi: 10.1007/s00431-014-2381-8
Vujkovic-Cvijin, I., and Somsouk, M. (2019). HIV and the gut microbiota: composition, consequences, and avenues for amelioration. Curr. HIV/AIDS Rep. 16, 204–213. doi: 10.1007/s11904-019-00441-w
Wang, Y., Shang, J., Li, Z., Zhang, A., and Cheng, Y. (2024). Establishment and application of a rapid diagnostic method for BVDV and IBRV using recombinase polymerase amplification-lateral flow device. Front. Vet. Sci. 11:1360504. doi: 10.3389/fvets.2024.1360504
Wedgwood, S., Gerard, K., Halloran, K., Hanhauser, A., Monacelli, S., Warford, C., et al. (2020). Intestinal dysbiosis and the developing lung: the role of toll-like receptor 4 in the gut-lung Axis. Front. Immunol. 11:357. doi: 10.3389/fimmu.2020.00357
Xiao, J., Wei, Z., Yang, C., Dai, S., Wang, X., and Shang, Y. (2023). The gut microbiota in experimental abdominal aortic aneurysm. Front. Cardiovasc. Med. 10:1051648. doi: 10.3389/fcvm.2023.1051648
Xing, J.-H., Shi, C.-W., Sun, M.-J., Gu, W., Zhang, R.-R., Chen, H.-L., et al. (2022). Lactiplantibacillus plantarum 0111 protects against influenza virus by modulating intestinal microbial-mediated immune responses. Front. Microbiol. 13:820484. doi: 10.3389/fmicb.2022.820484
Yasir, M., Al-Sharif, H. A., Al-Subhi, T., Sindi, A. A., Bokhary, D. H., El-Daly, M. M., et al. (2023). Analysis of the nasopharyngeal microbiome and respiratory pathogens in COVID-19 patients from Saudi Arabia. J. Infect. Public Health 16, 680–688. doi: 10.1016/j.jiph.2023.03.001
Zhang, Y., Hu, J., Tan, H., Zhong, Y., and Nie, S. (2022a). Akkermansia muciniphila, an important link between dietary fiber and host health. Curr. Opin. Food Sci. 47:100905. doi: 10.1016/j.cofs.2022.100905
Zhang, Z., Huang, J., Li, C., Zhao, Z., Cui, Y., Yuan, X., et al. (2024). The gut microbiota contributes to the infection of bovine viral diarrhea virus in mice. J. Virol. 98:e0203523. doi: 10.1128/jvi.02035-23
Zhang, H., Zuo, Y., Zhao, H., Zhao, H., Wang, Y., Zhang, X., et al. (2022b). Folic acid ameliorates alcohol-induced liver injury via gut–liver axis homeostasis. Front. Nutr. 9:989311. doi: 10.3389/fnut.2022.989311
Zhao, X., Hu, M., Zhou, H., Yang, Y., Shen, S., You, Y., et al. (2023). The role of gut microbiome in the complex relationship between respiratory tract infection and asthma. Front. Microbiol. 14:1219942. doi: 10.3389/fmicb.2023.1219942
Zhao, X., Jiang, L., Fang, X., Guo, Z., Wang, X., Shi, B., et al. (2022). Host-microbiota interaction-mediated resistance to inflammatory bowel disease in pigs. Microbiome 10:115. doi: 10.1186/s40168-022-01303-1
Keywords: infectious bovine rhinotracheitis virus, Angus calves, 16S rRNA, gut microbiota, metabolic pathways
Citation: Yi P, Li T, Xu L, Li X, Wang H, Ma Y, Ma Y, Sun Y, Li N, Zhong Q, Ma X and Yao G (2025) The differential gut microbiota and their MetaCyc pathways in IBRV infected Angus calves. Front. Microbiol. 16:1588341. doi: 10.3389/fmicb.2025.1588341
Edited by:
Yu Bai, Tianjin University of Science and Technology, ChinaReviewed by:
Yangyi Hao, China Agricultural University, ChinaYuanyuan Li, Shihezi University, China
Copyright © 2025 Yi, Li, Xu, Li, Wang, Ma, Ma, Sun, Li, Zhong, Ma and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Yao, eWdAeGphdS5lZHUuY24=
†These authors have contributed equally to this work