 Qingshang Bi
Qingshang Bi Mengru Liu2,3†
Mengru Liu2,3† Jun Cheng
Jun Cheng Yuzhu Dai
Yuzhu Dai- 1Department of Clinical Laboratory, Third Sanatorium, Air Force Healthcare Center for Special Services, Hangzhou, China
- 2School of Laboratory Medicine, Bengbu Medical University, Bengbu, China
- 3Department of Clinical Laboratory, The 903rd Hospital of The People’s Liberation Army, Zhejiang, China
- 4Department of Health Economic, The 903rd Hospital of The People’s Liberation Army, Zhejiang, China
Rapid detection of infectious diseases is critical for global public health prevention and control. However, the use of traditional molecular diagnostic methods, including PCR, has been limited because of their cumbersome procedures, complex equipment requirements, operation at different temperatures, and the level of expertise required for operation. Isothermal amplification technology (IAT) provides a rapid, sensitive, specific, simple and less costly method for diagnosing infectious diseases, which has led to revolutionary breakthroughs in molecular diagnostics. This paper summarizes recent progress in IAT technology, which focuses on the principles and applications of core technologies such as NASBA, LAMP, RPA, and RAA. In addition, the combination of IATs with the CRISPR/Cas system, which further revolutionizes nucleic acid detection technology, is explored in this review.
1 Introduction
Infectious diseases, as defined as pathological disorders caused by invasive pathogenic microorganisms including bacteria, viruses, fungi, and parasites, continue to pose significant threats to global public health. These conditions are clinically distinguished from noncommunicable diseases by their transmissible nature and external pathogenic origin. Globally, infectious disease remains a leading cause of death and disability and a growing challenge to health security and human progress (Nii-Trebi, 2017). According to the World Health Organization (WHO), 13 million people died from communicable diseases in 2021, and people in low- and middle-income countries are far more likely to die from communicable diseases than from noncommunicable diseases (WHO, 2021). Rapid diagnosis of infectious diseases is critical to stop their spread. Although traditional molecular diagnostic techniques (such as PCR) are highly sensitive, their dependence on thermal cyclers, specialized laboratories, and skilled operators severely hinders their application in remote areas and during outbreaks (Obande and Banga Singh, 2020).
As a revolutionary breakthrough in molecular diagnostics, IAT overcomes the numerous limitations of traditional PCR. It enables rapid, portable detection of infectious diseases through an isothermal reaction process, delivers fast results, has high efficiency, and is compatible with simple equipment (Wang et al., 2023). Nucleic acid sequence-based amplification (NASBA) for rapid diagnostic detection of pathogenic viruses from RNA genomes was introduced in 1991 (Compton, 1991). In 2000, Notomi et al. developed loop-mediated isothermal amplification (LAMP), which has gained prominence because of its high specificity and rapidity (Notomi et al., 2000). Recombinase polymerase amplification (RPA), published in 2006 and commercialized by TwistDx, has emerged as a leading IAT (Piepenburg et al., 2006). A Chinese research team subsequently developed recombinase-aided amplification (RAA) based on its technical principles (Munawar, 2022). RAA has been used to optimize enzyme systems, control costs, and enhance local adaptability, offering unique advantages in emergency response scenarios such as SARS-CoV-2 screening and primary care. With the continuous improvement of IATs, combinations with other detection technologies, such as the CRISPR/Cas system, have been developed. Among the CRISPR/Cas associated proteins, the Cas12 and Cas13 proteins have been widely used for DNA and RNA detection (Guk et al., 2023). Mature and stable methods, such as SHERLOCK (Kellner et al., 2019) and DETECTR (Chen et al., 2018; Kellner et al., 2019), when combined with IAT, offer more versatile, portable and cost-effective solutions for infectious disease detection. This paper reviews recent advancements in IATs, focusing on NASBA, LAMP, RPA, RAA, and CRISPR/Cas-integrated IATs. Its core mechanism, technical characteristics, research progress, and current limitations are discussed, with the aim of providing theoretical support for accurate infectious disease diagnosis and public health prevention strategies.
2 Nucleic acid sequence-based amplification
NASBA is an isothermal RNA-specific amplification technique that was first proposed by Compton (1991). Its core principle involves achieving exponential amplification of target RNA by copying the natural replication process of viral RNA in host cells. The entire reaction system was maintained at a constant temperature (41°C) and relied on the synergistic action of three enzymes paired with a set of specific primers. Avian myeloblastosis virus (AMV) reverse transcriptase converts RNA templates to cDNA, and RNase H degrades RNA strands in hybridized strands to release single-stranded cDNA. T7 RNA polymerase then transcribes a large amount of single-stranded RNA using promoter-containing double-stranded DNA as a template. In the cyclic phase, the newly synthesized RNA serves as a template for repeating the reverse transcription-degradation-transcription step. Through a noncyclic phase for initial template activation and sustained self-amplification in the cyclic phase, template RNA can be exponentially amplified by a factor of 109 to 1012 within 2 h. This method achieves highly efficient target RNA amplification via a continuous, isothermal process (Figure 1). NASBA amplification products can be detected by agarose gel electrophoresis (Jean et al., 2004), microchip electrophoresis (MCE) (Liu et al., 2015), electrochemiluminescence (ECL) (Wacharapluesadee and Hemachudha, 2001) and real-time fluorescence (Aufdembrink et al., 2020).
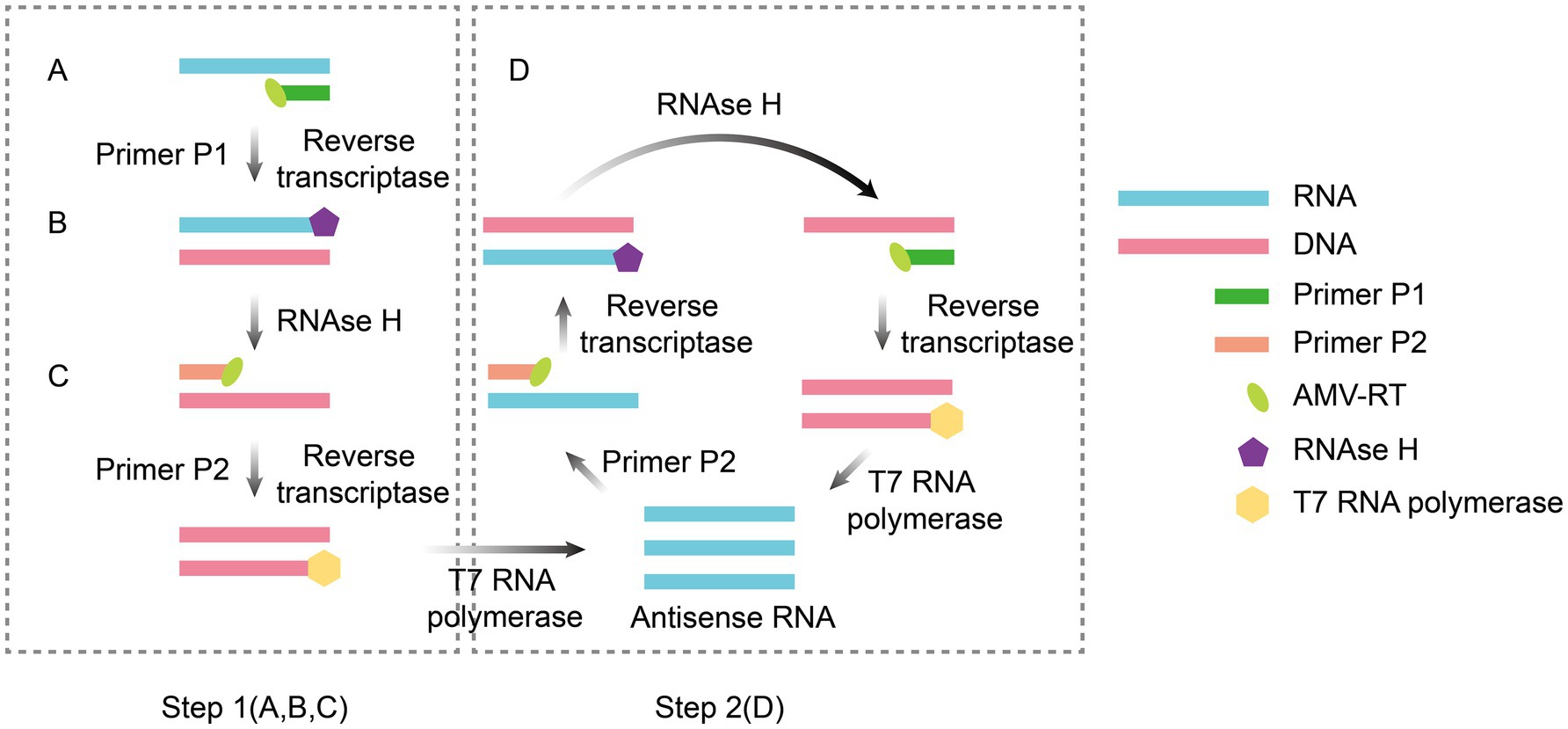
Figure 1. Principle of NASBA amplification. The reaction system contains two primers (F1 and F2) and three enzymes (AMV-RT, RNase H, and T7 RNA polymerase). AMV reverse transcriptase uses RNA as a template to synthesize cDNA (Step 1A), and after hydrolysis of the RNA strand by RNase H (Step 1B), F2 binds to the cDNA to produce double-stranded T7 promoter-containing DNA (Step 1C), which is transcribed by T7 RNA polymerase to produce large amounts of RNA. The new RNA is then used as a template to repeat reverse transcription, hydrolysis, and double-strand synthesis, triggering the T7 transcription loop for self-cycling amplification (Step 2D). The figure was drawn using Adobe Illustrator 2021 software.
The emergence of human immunodeficiency virus (HIV) in the 1990s accelerated the development of NASBA, paving the way for faster, more reliable diagnostic methods for HIV tests. The optimization of HIV-1 detection with NASBA Technology was developed by Kievits et al. (1991). Damen et al. quantified hepatitis C virus RNA (HCV-RNA) using nucleic acid sequence-based amplification with quantitative testing (NASBA-QT) and compared the results with those of two commonly used commercial assays (HCV branched DNA assay and HCV MONITOR assay) (Damen et al., 1999). NASBA-QT has demonstrated high sensitivity and reliability in the assay, with a quantitative detection limit of 103 copies/100 μL and a qualitative detection limit of 102.3 copies/100 μL. Compared with the bDNA method, this method has significant advantages (more than a 10-fold increase in sensitivity), and the quantitative results are highly consistent, with comparable sensitivity to that of the HCV MONITOR method, while the latter has systematically lower values (Damen et al., 1999). In addition, Mohammadi-Yeganeh et al. developed a molecular beacon-based multiplex NASBA for the simultaneous detection of HIV-1 and HCV in plasma samples (Mohammadi-Yeganeh et al., 2012). Using the multiplex NASBA technique, Lau et al. successfully detected a wide range of human respiratory viruses, including influenza A, influenza B, respiratory syncytial virus (RSV), and coxsackievirus (CSV) (Lau et al., 2010). Reed et al. developed an innovative label-free colorimetric nucleic acid assay for rapid visual detection of Zika virus (ZIKV) by integrating isothermal nucleic acid amplification with enzymatic signaling mechanisms (Reed et al., 2019). The method demonstrated a detection limit of 106 copies/ml with a clinical sensitivity of 97.64%. The method can be completed within 2 h and selectively differentiates between Dengue and West Nile viruses, which are closely related to the Zika virus (Reed et al., 2019). At the onset of the 2020 COVID-19 pandemic, real-time PCR-based molecular testing became critical for confirming diagnostic results, but its high cost has limited its implementation in resource-poor areas (Wang C. et al., 2020). A real-time NASBA for detecting SARS-CoV-2 was developed by Kia et al. (2023). Rapid amplification of viral RNA at a constant temperature was achieved by designing primers and probes specific to the RdRp and N genes. This method has a detection limit of 200 copies/ml with 97.64% clinical sensitivity, demonstrating performance comparable to that of real-time PCR. Crucially, it eliminates the need for expensive thermal cycling equipment and significantly reduces costs (Kia et al., 2023). Owing to its simple primer design and excellent reproducibility, this method is suitable for rapid screening in resource-limited settings, offering a cost-effective technological alternative to the existing global SARS-CoV-2 assay.
In general, NASBA can avoid DNA interference by directly amplifying RNA targets and is particularly well suited for detecting RNA viruses. However, the NASBA approach has several limitations. First, because the specificity of the reaction depends on thermally unstable enzymes, the reaction temperature should not exceed 42°C. This characteristic also makes the reaction prone to primer dimerization and nonspecific amplification, which increases the likelihood of false positives. Second, the target RNA sequence should be between 120 and 250 nucleotides in length since sequences shorter or longer than this range reduce amplification efficiency (Xue et al., 2022; Liu et al., 2023).
In addition, NASBA is expected to be deeply integrated with molecular beacons, microfluidic technology, etc., to achieve real-time quantitative detection and automated operation. For example, the combination of a G4-ThT fluorescence biosensor and NASBA can detect viral RNA at concentrations as low as 2 copies/μL with real-time detection capability and can be integrated into highly automated systems (such as microfluidic chips) (Guoshuai et al., 2022). The combination of NASBA and the CRISPR system can effectively improve detection sensitivity and specificity. Jung et al. developed and optimized a one-pot NASBA-Cas13a nucleic acid detection method for rapid and sensitive detection of SARS-CoV-2 RNA fragments with a sensitivity of 20–200 aM (Jung et al., 2025). NASBA does not require complex thermal cycling equipment. When combined with a CRISPR portable detection system, it enables the development of low-cost, rapidly deployable field detection tools suitable for remote areas or for responding to sudden epidemics.
3 Loop-mediated isothermal amplification
LAMP is a nucleic acid amplification method based on strand substitution developed by Notomi et al. (2000). The core mechanism involves the targeted identification of 6–8 regions of the target gene using 4–6 specific primers (F3/B3, FIP/BIP, LF/LB), followed by efficient amplification by the Bst DNA polymerase (which exhibits strand displacement activity) at a constant temperature of 60–65°C (Figure 2). Loop primers (LF/LB) bind to the stem–loop structure and significantly accelerate the amplification process (Nagamine et al., 2002). Within 30–60 min, exponential amplification of target sequences (up to 109–1010-fold) can be achieved. In the presence of reverse transcriptase, the target RNA can also be amplified after being reverse-transcribed into complementary cDNA (Talap et al., 2022). The reaction product comprises numerous alternating, inversely repeated DNA fragments. This result can be detected directly through turbidimetric methods (via magnesium pyrophosphate precipitation) (Mori et al., 2001), fluorescent dye labeling (Soliman and El-Matbouli, 2005), or colorimetric approaches (Tomita et al., 2008), all of which do not require complex instrumentation.
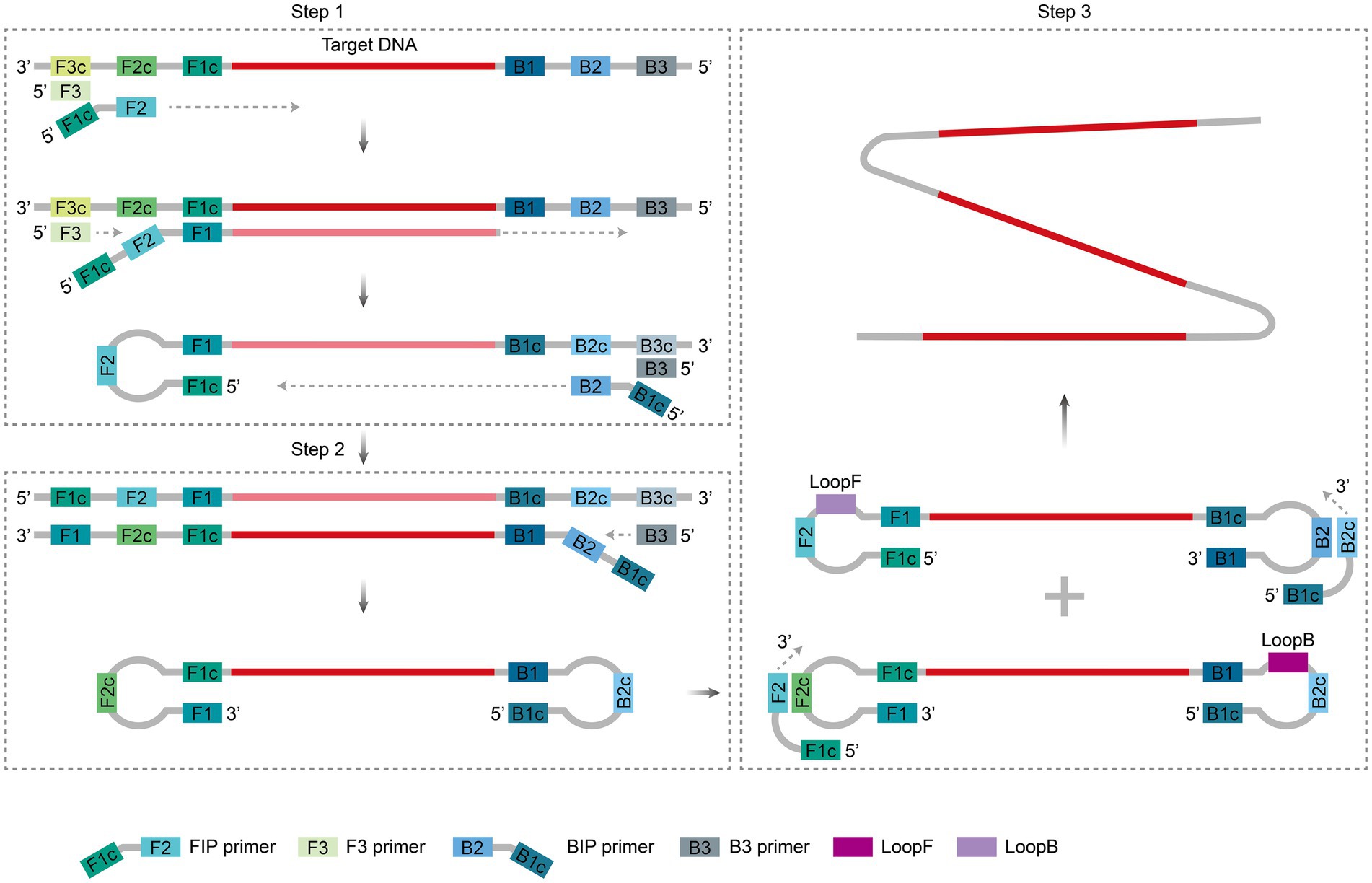
Figure 2. Principle of LAMP amplification. During the initiation phase, the FIP and F3 primers bind sequentially to the target sequence, which is extended by Bst polymerase, displacing the single strand to form a single-stranded loop structure (Step 1); subsequently, the BIP and B3 primers are extended in the reverse direction, generating dumbbell-shaped DNA with loops at both ends that self-fold into a stem–loop template (Step 2). During the cycling phase, the stem–loop template triggers multistep branching amplification through sustained binding, extension, and strand displacement by FIP and BIP primers, forming a polycyclic structure and generating many DNA products containing reverse repeat sequences (Step 3). Two ring primers, LooF and LooB, also bind to the stem–loop structure to accelerate its amplification process. The figure was drawn using Adobe Illustrator 2021 software.
At present, LAMP is widely used in the detection of infectious diseases. Lim et al. (2024) developed a point-of-care testing (POCT) system based on reverse transcription loop-mediated isothermal amplification (RT-LAMP) technology to detect SARS-CoV-2, influenza A and B, and RSV in a single reaction tube. The system combines a nucleic acid-free extraction process with an integrated microfluidic device through multiplex primer design, enabling visual fluorescence interpretation within 30 min. The detection limit was as low as 35–1,000 copies per sample, making it suitable for resource-limited scenarios. However, cost optimization of the device and successful clinical validation are needed to expand its applications. Wang et al. developed a multiplex detection technique combining loop-mediated isothermal amplification and a lateral flow biosensor (LAMP-LFB) for detecting the Mycobacterium tuberculosis complex (MTBC) (Wang et al., 2021). The method successfully completed the MTBC assay in less than 1 h, demonstrating a sensitivity of 10 fg. Clinical validation revealed 82% sensitivity and 97.7% specificity in samples, with significantly superior clinical performance compared with bacterial culture (47% sensitivity) and Xpert MTB/RIF (54% sensitivity). This technology has also been employed to detect Entamoeba histolytica (pathogenic amoeba), nonpathogenic Entamoeba species, pathogenic Leptospira spp., Listeria monocytogenes, SARS-CoV-2(2 genes), Enterococcus faecalis and Staphylococcus aureus (Nurul Najian et al., 2016; Foo et al., 2017; Wang et al., 2017; Ledlod et al., 2020; Zhu et al., 2020). Xie et al. integrated LAMP with a self-driven microfluidic chip, achieving automatic pump-less sample loading through the permeability of polydimethylsiloxane (PDMS) material combined with multiple primer design and fluorescence visualization (Xie et al., 2021). This system enables triplex nucleic acid detection of hepatitis B virus (HBV), HCV, and HIV to be completed within 50 min at a constant temperature of 63°C, demonstrating a sensitivity of up to 2 copies/μL. The clinical validation results were completely consistent with the qPCR findings. This method has also been used to detect Escherichia coli O157: H7 and Neisseria meningitidis (Dou et al., 2014; Guo et al., 2015). Microfluidic chips based on polymethyl methacrylate (PMMA) have been used for the detection of Escherichia coli and Enterococcus spp., achieving a limit of detection (LOD) of 4 copies per well within 35 min (Fu et al., 2021). This method has also been applied to detect Vibrio parahaemolyticus and Salmonella enterica (Wu et al., 2021b; Zhu et al., 2023).
With the continuous development of LAMP technology and the integration of various visualization techniques, instant nucleic acid detection has become more accessible and reliable, but the high complexity of LAMP primer design, intricate product structures, cross-interference in multiplex detection and the risk of aerosol contamination remain critical considerations in practical applications (Soroka et al., 2021).
Compared with conventional PCR technology, LAMP generates long DNA strands and cauliflower-like DNA structures, which limits its practicality in molecular biology by producing smeared or multiple bands during gel electrophoresis. In contrast, PCR typically yields single distinct bands. This characteristic makes specific product identification for LAMP more challenging in gel-based analysis. For multiplex detection, traditional multiplex LAMP (mLAMP) strategies, such as restriction enzyme site labeling, often suffer from incomplete enzymatic digestion, resulting in complex band patterns that are difficult to interpret (Crego-Vicente et al., 2024). While molecular barcoding or nanoparticle probe technologies can differentiate targets, they require sequencing or expensive reagents, and they involve time-consuming postprocessing steps and increased contamination risks (Oliveira et al., 2020; Ludwig et al., 2021). Furthermore, the complex product architecture of LAMP prevents target differentiation through melting curve analysis. Probe-based detection methods are also constrained by the inherent strand displacement activity of LAMP (Crego-Vicente et al., 2024). To address these challenges, microfluidic chip technology has demonstrated promise in physically separating primers for different targets into isolated reaction chambers. This spatial segregation effectively minimizes primer competition, streamlines detection workflows, and reduces costs (Liu et al., 2024). Such microfluidic platforms may overcome current limitations to enable efficient, cost-effective, and scalable mLAMP solutions. Additionally, the complexity of primer design introduces uncertainties into detection outcomes. Although Primer Explorer V5 remains the most widely used online tool for LAMP primer design, it occasionally fails to identify optimal targets automatically, necessitating labor-intensive manual optimization. The substantial length of key primers (FIP/BIP: 30–40 bases) and the requirement for multiple primer sets dramatically increase the risk of self-hybridization and nonspecific amplification, potentially causing false-positive results. Avoiding this risk often demands iterative experimental validation to optimize primer combinations (Soroka et al., 2021).
4 Recombinase polymerase amplification
RPA, an isothermal nucleic acid amplification technology first developed in 2006 by a team led by the British scientist Piepenburg et al. (2006), mimics the mechanism of viral DNA replication. It employs recombinant enzymes (e.g., the T4 UvsX protein) to guide primers toward double-stranded DNA binding with single-stranded binding proteins (e.g., T4 gp32) to stabilize the template, and strand-displacing polymerases (e.g., Bsu polymerase). The system enables rapid exponential amplification of target sequences at a constant temperature of 37–42°C, requiring only 20–30 min to amplify trace amounts of nucleic acids (as low as 1–10 copies) to detectable levels (Figure 3). The entire process requires no thermal cycling equipment and relies on the synergy of the enzyme system to achieve efficient and rapid nucleic acid replication. The amplification products generated from RPA can be detected via endpoint (post- amplification) or real-time (during amplification) assays, with RPA coupled with a lateral flow strip (RPA-LFS) and real-time RPA being two of the most widely used methods.
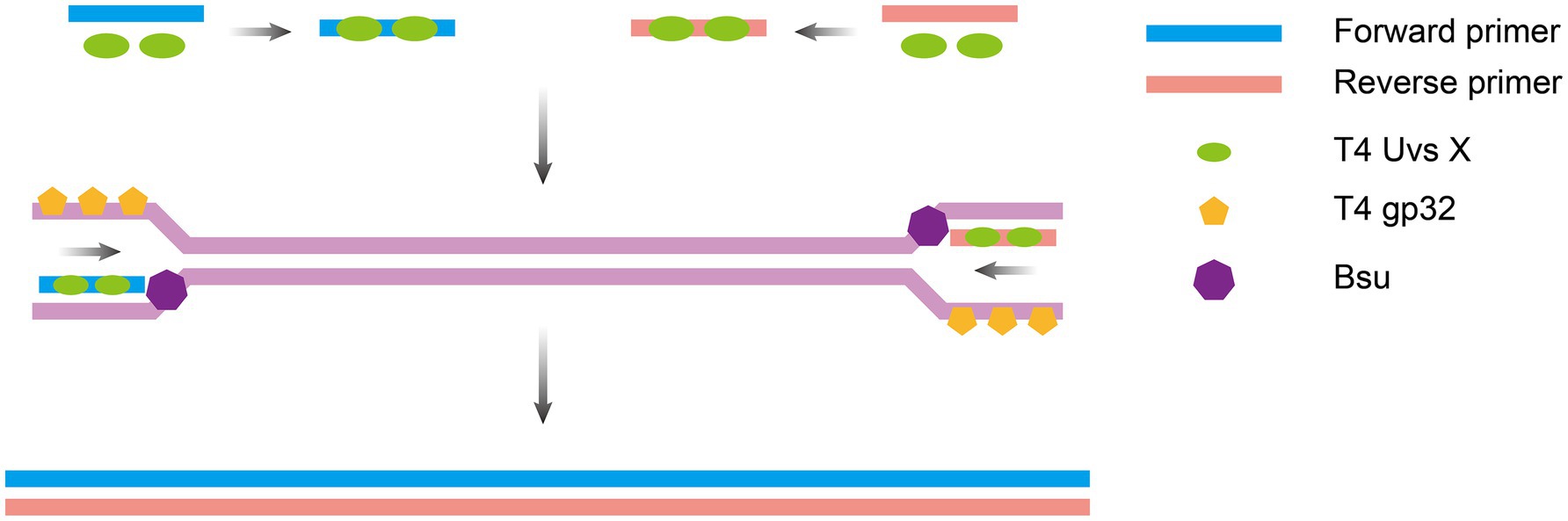
Figure 3. Principle of RPA amplification. Recombinase-primer complexes anneal to double-stranded DNA, inducing localized strand separation to generate single-stranded DNA regions; single-strand binding proteins stabilize the template, and strand-displacing polymerase extends the primer and displaces the complementary strand, creating a new double strand. The displaced strand serves as a template for subsequent amplification cycles, driving recombinase complex binding and strand synthesis. The figure was drawn using Adobe Illustrator 2021 software.
RPA-LFS has been widely adopted for infectious disease testing because it allows for rapid visual interpretation of results with the naked eye. Ji et al. developed a rapid RPA-LFS-based assay for Staphylococcus haemolyticus that achieved nucleic acid amplification in 8 min and 1 min of visual interpretation. The assay demonstrated a sensitivity of 0.147 CFU/μL and showed 100 and 98.73% consistency with qPCR and conventional culture, respectively, in validation with clinical samples (Ji et al., 2023). RPA-LFS is also applicable for detecting Streptococcus pneumoniae with high sensitivity (3.32 CFU/μL) and specificity within 15 min, and the clinical sample validation results were highly concordant with those of PCR (98.18% concordance rate). In addition, RPA-LFS shows no cross-reactivity with other hepatitis viruses, with the lowest detection limits of 10 copies/ml for HBV and 100 copies/ml for hepatitis E viruses (HEVs) (Zhang et al., 2021; Li M. et al., 2023). In real-time RPA, fluorescent probes are combined to enable real-time monitoring of RPA amplification of target sequences. Zhang et al. developed a real-time RPA assay for Yersinia enterocolitica detection in intestinal samples, achieving detection within 20 min with a sensitivity of 104 ng/μL (Zhang et al., 2023). Ying et al. applied real-time RPA for subtyping human papillomavirus (HPV) 16 and 18, achieving 1,000 copies/μL sensitivity (Ying et al., 2023).
RPA technology offers several unique advantages during the detection of infectious diseases, including simple primer design, streamlined amplification protocols, rapid reaction kinetics, and high sensitivity and specificity. It supports the detection of both DNA and RNA targets (when combined with reverse transcriptase) (Lobato and O'Sullivan, 2018) and is compatible with multiple detection methods, such as fluorescent probes, lateral flow dipsticks, and integration with CRISPR-based systems (Kellner et al., 2019). However, as a relatively new method with a short development history, RPA also has certain limitations. For example, its amplification fragment length is typically restricted to 100–500 bp, which hinders long-sequence analysis. Primer design requires rigorous structural optimization to avoid nonspecific amplification. Low and isothermal amplification of the RPA can easily lead to false positives and must be combined with other technologies to improve specificity. Zhang et al. developed a method by combining the CRISPR/Cas12a/13a systems, leveraging the cleavage specificity of Cas12a and Cas13a to detect HPV16 and HPV18 simultaneously with high specificity and sensitivity (10 copies/μL) (Zhang et al., 2024). Additionally, the current limitations in multichannel parallel processing capability impede its efficient application in large-scale screening scenarios (Lobato and O'Sullivan, 2018; Wu et al., 2024). Despite these limitations, further exploration and enhancement of its strengths could position RPA as a mainstream nucleic acid amplification technology in the future.
5 Recombinase-aided amplification
RAA technology is an isothermal nucleic acid amplification technology proposed and optimized by a Chinese research team in 2010 (Shen et al., 2019). RAA employs recombinant enzymes to recognize target sequences and unwind double-stranded DNA (typically derived from fungi or bacteria, such as bacterial RecA or engineered enzymes). This process enables single-stranded binding proteins to protect the template and facilitates DNA polymerase-mediated strand exchange synthesis of the new strand. This synthesis occurs in conjunction with bispecific primers and fluorescent probes for real-time monitoring (Nie et al., 2022). The entire amplification process shares key characteristics with RPA, including operation at a constant temperature (37–42°C) and rapid completion (within approximately 30 min) (Figure 4). The final product, a mixture of double-stranded DNA and single-stranded DNA, can be directly detected using methods such as fluorescent probes, lateral flow strips, or electrophoresis (Wu K. et al., 2021; Lin et al., 2022; Wang K. et al., 2022).
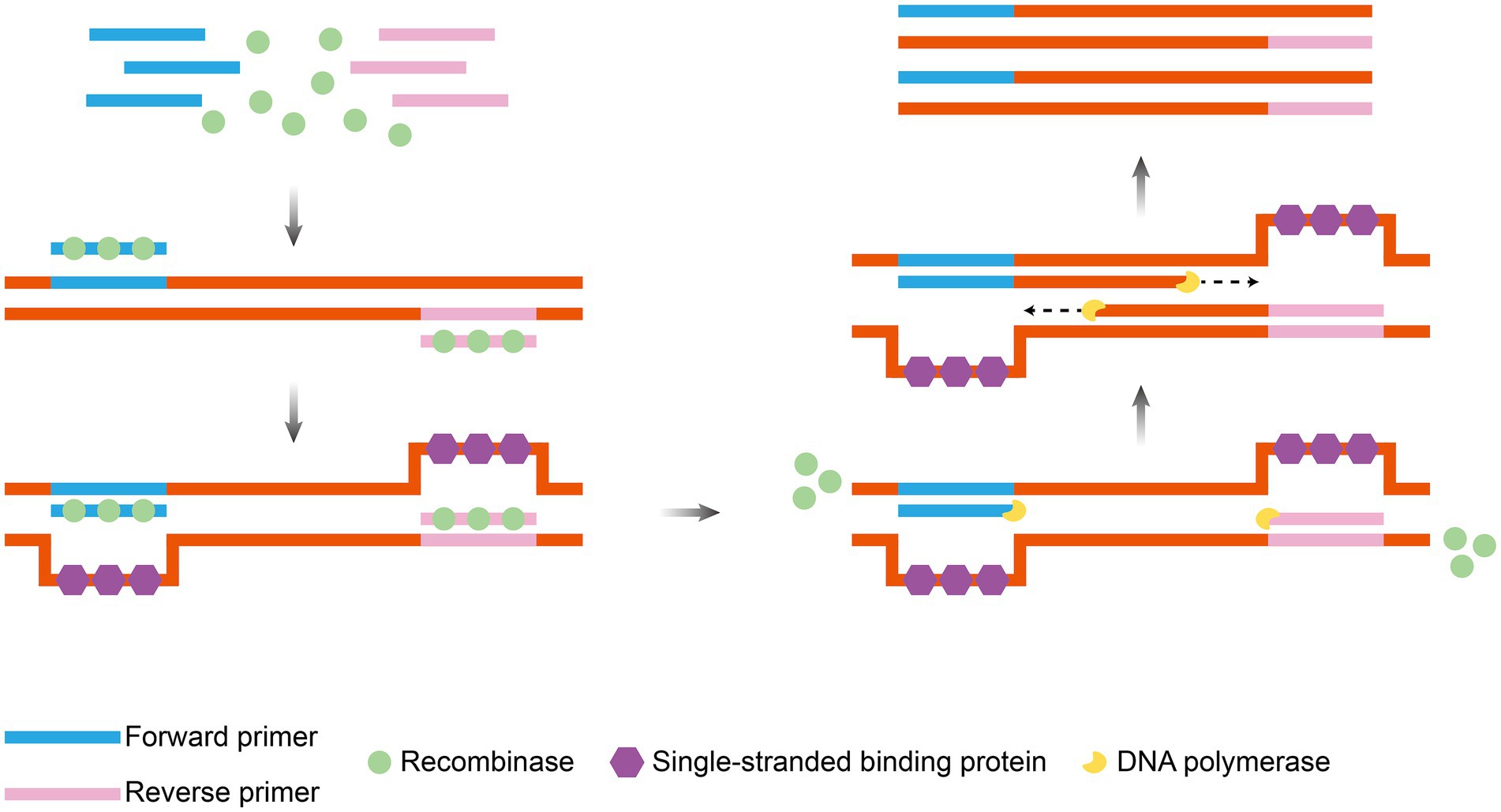
Figure 4. Principle of RAA amplification. Under isothermal conditions, recombinase, single-strand binding protein and DNA polymerase operate synergistically: recombinase targets and binds to specific DNA sequences, induces strand unwinding, single-strand binding protein stabilizes the single-stranded template, and DNA polymerase catalyzes strand extension to drive exponential amplification of target DNA. The figure was drawn using Adobe Illustrator 2021 software.
RAA represents an advanced isothermal amplification technique. With technological advancements in RAA, the development of RAA-derived variants has enabled more convenient and rapid detection approaches for infectious diseases. For example, Bai et al. developed a real-time fluorescent RAA-based assay for HBV that can be performed in a single tube at 39°C for 40 min, with a minimum detection limit of 100 IU/mL, a sensitivity of 97.18%, and a specificity of 100% for clinical samples (Bai et al., 2020). To detect methicillin-resistant Staphylococcus aureus (MRSA), real-time RAA fluorescence technology has a detection limit of 10 copies/μL, with 97.01% consistency with traditional qPCR results, and high specificity without cross-reactivity with other clinically relevant bacteria (Ding et al., 2022). Li et al. developed a reverse transcription-recombinase-assisted amplification (RT-RAA)-based POCT assay for SARS-CoV-2 nucleic acids in combination with a lateral flow strip (LFS) (Zheng et al., 2021). Through specific targeting of the viral nucleocapsid (N) gene, the assay was performed at 39°C for 30 min, with a limit of detection of 1 copy/μL. Method validation revealed no cross-reactivity with human coronavirus, influenza A/B virus, RSV, or HBV. The clinical evaluation of 100 samples (13 positive, 87 negative) demonstrated 100% sensitivity and specificity compared with RT-qPCR, with advantages such as rapidity, high precision, and room-temperature operation, highlighting its suitability for POCT applications. It has been validated for detecting avian influenza virus subtype H5 (Li Y. et al., 2023), senecavirus A (Wang W. et al., 2022) and HCV (Wang H. et al., 2022). Integration with the CRISPR/Cas system enhances nucleic acid detection specificity for low-copy-number targets. For example, Qian et al. combined the CRISPR/Cas12a system with RT-RAA isothermal amplification to achieve highly sensitive (0.1 copies/μL), rapid (30–40 min) detection of norovirus subtypes GII.4 and GII.17 without complex equipment, demonstrating over 95% clinical concordance with clinical samples and showing no cross-reactivity with related viruses (Qian et al., 2022).
The RAA assay, a novel isothermal amplification technique, has been widely used for detecting various pathogens. Unlike RPA assays, RAA technology has been optimized in the formulation to adapt to China’s pathogen detection. The RAA employs three core enzymes: recombinase UvsX, DNA polymerase, and single-stranded DNA-binding protein (Xue et al., 2020). A key advantage of RAA technology lies in its ability to perform amplification under optimized conditions at 37°C or even room temperature, enabling the acquisition of target amplification products within 30 min. This process achieves exponential amplification of target DNA without requiring auxiliary heating equipment, making it particularly suitable for on-site pathogen detection. Furthermore, the RAA reaction system mandates strict complementarity between primers and templates, with primer lengths restricted to 30–35 nucleotides. This stringent requirement ensures both the precision and specificity of the RAA method, distinguishing it from other amplification approaches.
However, owing to the very high sensitivity of RAA, which can easily cause false-positive results, cross-contamination should be avoided throughout the process. This prevention requires vigilant control throughout sample collection, reagent preparation, amplification reactions, and result analysis. Key recommendations include maintaining separate laboratory zones (e.g., sample handling, reagent preparation, and amplification areas) with proper ventilation; using disposable, nuclease-free consumables and filtered pipette tips; practicing aseptic techniques (e.g., gentle pipetting, frequent glove changes); aliquoting reagents to avoid repeated thawing; incorporating negative and positive controls; optimizing primer/probe specificity; and implementing regular training for personnel. Stringent adherence to these protocols, coupled with standardized operating procedures, can effectively mitigate contamination risks and ensure accurate, reliable RAA results. In addition, nonspecific products may be generated during the reaction because of the short length of the target genes, and primer dimer formation may be caused by excess primers and specific interactions between primer molecule (Kellner et al., 2019; Wang et al., 2019). As with other IATs, the practical application of RAA technology also faces the same challenges. First, achieving on-site sample preprocessing (e.g., nucleic acid extraction) in environments requiring immediate sample processing remains difficult; however, this step is critical for obtaining high-quality nucleic acid templates essential for amplification. Second, achieving highly sensitive and specific multitarget isothermal amplification detection under single closed-tube conditions presents a significant technical hurdle. The ability to detect multiple targets in a single reaction simultaneously is vital for efficient and comprehensive pathogen identification. To broaden the applicability of RAA and fully unlock its potential, current research and development efforts are focused on addressing these issues to increase the practicality and effectiveness of RAA in real-world settings.
6 Isothermal nucleic acid amplification combined with the CRISPR/Cas system
Isothermal nucleic acid amplification combined with the CRISPR/Cas system is a molecular diagnostic tool adapted from the bacterial immune system. The clustered regularly interspaced short palindromic repeats sequence (CRISPR) system, which evolved as a bacterial defense mechanism against viral invasion, consists of the enzyme Cas nuclease and a guide RNA (gRNA). The gRNA recognizes a specific DNA or RNA sequence, and the Cas enzyme precisely cuts the target (Makarova and Koonin, 2015). The research team combined the system’s targeted cleavage capability with isothermal amplification technology. Once the Cas protein recognizes the target sequence in a sample, it triggers its cleavage activity, which in turn enables visual detection using downstream techniques such as fluorescent labeling, lateral flow test strips, or electrochemical signaling (Figure 5) (Gootenberg et al., 2017; Kaminski et al., 2021).
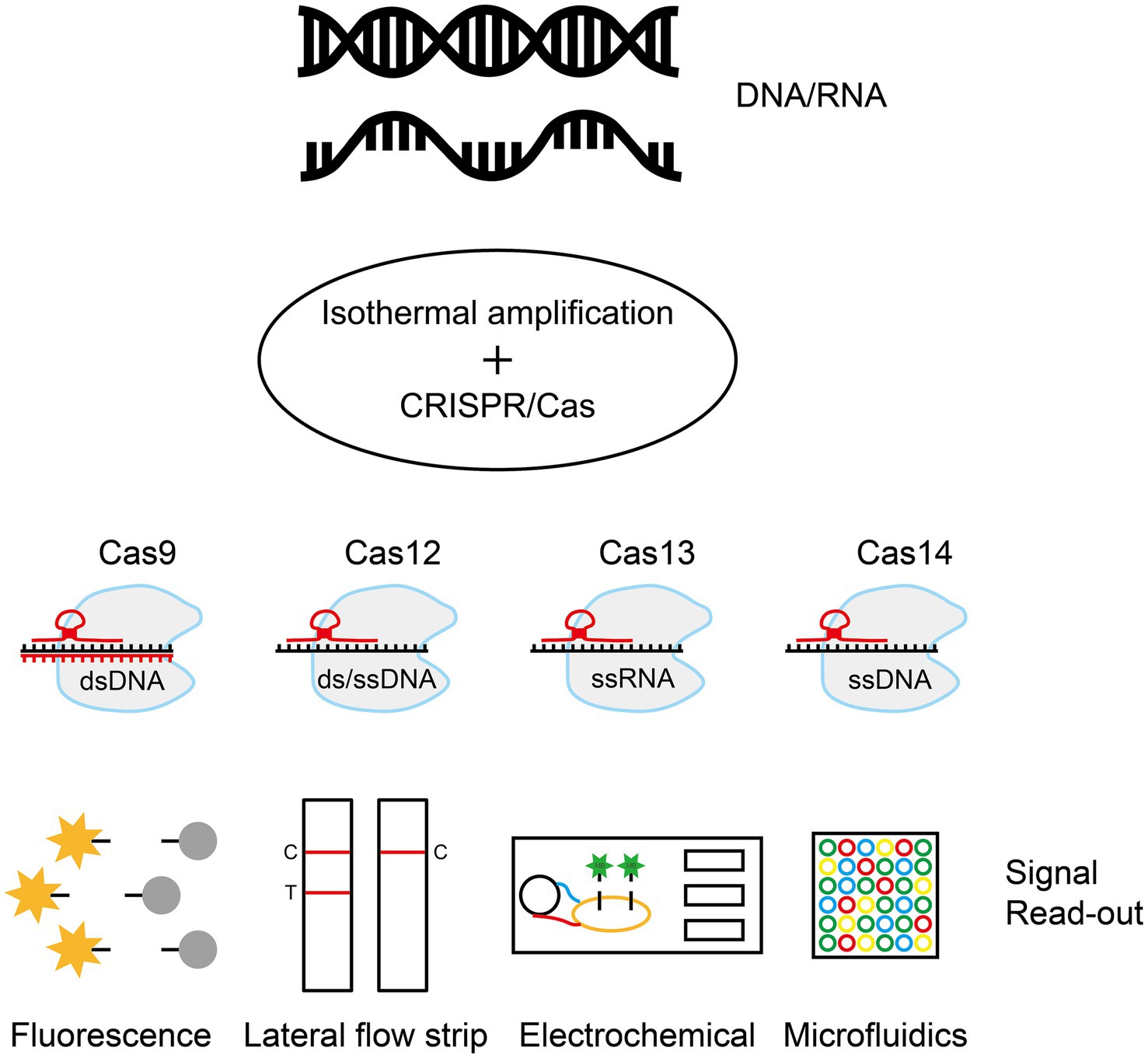
Figure 5. Isothermal amplification combined with the CRISPR/Cas system. Through rapid isothermal amplification of DNA/RNA, the CRISPR/Cas system performs targeted cleavage of the target sequence and signal recognition using downstream techniques such as fluorescent labeling, lateral chromatography test strips, or electrochemical signaling. The figure was drawn with Adobe Illustrator 2021 software.
6.1 Combined CRISPR/Cas9 and isothermal amplification of nucleic acids
Pardee et al. combined NASBA with the CRISPR/Cas9 system to develop a molecular diagnostic assay called NASBA-CRISPR cut (NASBACC), which discriminates ZIKV genotypes (e.g., American vs. African strains) with single-base resolution (Pardee et al., 2016). In this assay, researchers used NASBA to amplify viral RNA and incorporated trigger sequences that activate a toehold switch sensor into the amplification products. gRNAs were designed to target single-nucleotide polymorphism (SNP) sites specific to the American strain, which contains NGG protospacer adjacent motif (PAM) sequences. Cas9 recognized the PAM site of the American strain and cleaved the amplified DNA, resulting in the truncation of the RNA product, a lack of a trigger sequence to activate the sensor, and the maintenance of the original color (yellow). In the African strain, the absence of the PAM sequence prevented Cas9-mediated cleavage, leaving the RNA product intact and harboring a trigger sequence that was bound to the toehold switch, activating LacZ reporter gene expression and inducing a color change from yellow to purple (Pardee et al., 2016). This method was used to identify Zika virus in the Americas and Zika virus in Africa.
The CRISPR/Cas9-mediated lateral flow nucleic acid assay (CASLFA) integrates the CRISPR/Cas9 system, lateral flow assay (LFA), and RPA/PCR-based DNA/RNA detection methods (Wang et al., 2020b). It uses a Cas9/sgRNA complex to bind to target double-stranded DNA and releases nontarget single-stranded regions. These regions hybridize with a gold nanoparticle (AuNP) probe, generating a visual signal on a lateral flow strip. CASLFA can detect Listeria monocytogenes, transgenic rice (35S promoter), and African swine fever virus (ASFV) with limits as low as hundreds of genome copies, and when combined with RPA, sample-to-result analysis can be completed in less than 40 min. In validation, CASLFA accurately identified 27 ASFV-positive samples among 110 swine serum suspects, showing 100% concordance with real-time PCR results in terms of sensitivity and specificity (Wang et al., 2020b). Additionally, the CRISPR/Cas9-mediated triple-line lateral flow assay (TL-LFA), combined with multiplex reverse transcription RPA (RT-RPA), enables rapid simultaneous detection of the SARS-CoV-2 E gene and Orf1ab gene on a single test strip. This method achieves a sensitivity of 100 RNA copies per 25 μL reaction system. Clinical validation using 64 nasopharyngeal samples demonstrated 100% negative predictive agreement and 97.14% positive predictive agreement (Xiong et al., 2021). This approach offers an accurate and convenient diagnostic solution for detecting COVID-19 in resource-limited settings.
6.2 Combined CRISPR/Cas12 and isothermal amplification of nucleic acids
The DNA endonuclease-targeted CRISPR trans reporter (DETECTR), developed by Chen et al. in 2018, is a molecular diagnostic technology that integrates isothermal RPA with Cas12a enzymatic activity (Chen et al., 2018). The target sequence is amplified by RPA, and the gRNA directs the Cas12a protein to recognize and bind to the target sequence. Upon target engagement, Cas12a cleaves the double-stranded DNA (cis cleavage) and simultaneously activates trans cleavage activity, releasing fluorescent signals through nonspecific cleavage of the free single-stranded DNA (ssDNA) probe. Xu et al. developed a rapid detection method for Bacillus anthracis based on DETECTR, achieving fast (<40 min), highly sensitive (nearly two-copy level), and specific detection of the pathogen’s nucleic acid (Xu et al., 2023). This method also enabled the discriminatory detection of HPV16/18 (Chen et al., 2018), Japanese encephalitis virus genotypes I, III, and V (Kwak et al., 2023) and African swine fever virus (Wang et al., 2020a), demonstrating its versatility in differentiating infections caused by these pathogens. OR-DETECTR is an assay based on RT-RPA and DETECTR technology. Researchers have optimized the reaction components such that the assay can be performed in a single test tube, requires no special equipment, and can be completed in less than 1 h. It has been used to detect the H1N1 and SARS-CoV-2 viruses (Sun et al., 2021).
Lee et al. established a rapid, sensitive and visual detection method for the Escherichia coli O157: H7 gene using the LAMP-CRISPR/Cas12a system (Lee and Oh, 2022). The target gene sequence is amplified by LAMP. Cas12a binds to the amplified sequence and then cleaves the fluorescently labeled ssDNA probe, releasing a fluorescent signal. This approach has also been reported for detecting Salmonella spp., Neisseria meningitidis and Pseudomonas aeruginosa (Mukama et al., 2020; Wu et al., 2021a).
6.3 Combined CRISPR/Cas13 and isothermal amplification of nucleic acids
SHERLOCK (Specific High-sensitivity Enzymatic Reporter UnLOCKing) is a molecular diagnostic method based on the CRISPR-LWA-Cas13a system developed by Feng Zhang’s team in 2017 (Gootenberg et al., 2017). This method uses the targeted RNA recognition ability and cleavage activity of the Cas13a protein to achieve highly sensitive and specific detection of DNA or RNA target sequences. These sequences are amplified using RPA (for DNA) or RT-RPA (for RNA), with DNA amplicons transcribed to ssRNA by T7 RNA polymerase. gRNA guides the LwaCas13a protein to recognize target sequences, activating nonspecific cleavage of fluorescent ssRNA reporter probes to generate detectable signals. SHERLOCK technology has been employed in the detection of the Zika and Dengue viruses, demonstrating high sensitivity and specificity (Gootenberg et al., 2017). Allan-Blitz et al. developed a dual-molecule diagnostic system based on the CRISPR/Cas13a-powered SHERLOCK platform. This system enables pathogen detection by targeting the porA gene (with a sensitivity of 14 strains of Neisseria gonorrhoeae and no cross-reactivity with 3 non-gonococcal Neisseria strains) while simultaneously predicting ciprofloxacin resistance through the identification of mutations in the gyrase A (gyrA) gene (91/91 mutation sites validated using 20 resistant strains and 3 susceptible strains, with DNA sequencing confirming 100% concordance) (Allan-Blitz et al., 2023).
However, SHERLOCK technology still faces limitations, including the lack of quantitative capability and reliance on fluorescence detection equipment for readout. Therefore, the team further optimized this approach in 2018 by developing multiple detection capabilities based on the first generation, with higher sensitivity and more flexible application scenarios (Gootenberg et al., 2018). SHERLOCKv2 allows for the simultaneous detection of four different target nucleic acids (DNA or RNA) by employing four different Cas proteins (LwaCas13a, PsmCas13b, CcaCas13b and AsCas12a). Each Cas protein is guided by a specific crRNA. Upon target recognition and activation, each Cas protein cleaves various types of reporter probes, such as fluorescently labeled RNA or DNA, enabling parallel multitarget detection. Furthermore, by incorporating the Csm6 protein (a CRISPR-associated nuclease), the system achieves signal amplification. When Cas13 cleaves its target RNA, it releases fragments that activate Csm6. Activated Csm6 then cleaves additional reporter probes, amplifying the signal through a cascade reaction and increasing the sensitivity to 2 aM. SHERLOCKv2 combines fluorescence detection with lateral flow strips (colorimetric readouts), offering quantification, visual interpretability, and flexible multiformat reporting, thereby expanding its utility in nucleic acid diagnostics (Gootenberg et al., 2018).
To enable large-scale multiplexed nucleic acid detection, Ackerman et al. developed a high-throughput platform using CARMEN-Cas13 (Ackerman et al., 2020). The core mechanism enables simultaneous differentiation of 169 human-associated viruses by PCR or RPA amplification of target sequences followed by the use of microfluidic arrays with the trans-cleaving activity of Cas13. Simultaneously, comprehensive subtyping of influenza A strains and multiple identification of dozens of HIV drug-resistant mutations have been achieved (Ackerman et al., 2020).
6.4 Combined CRISPR/Cas14 and isothermal amplification of nucleic acids
Cas14-DETECTR, a Cas14-based DNA detection platform developed by Aquino-Jarquin et al. in 2019, integrates Cas14’s highly specific ssDNA recognition with trans-cleavage activity for sensitive and specific nucleic acid detection (Harrington et al., 2018; Aquino-Jarquin, 2019). The workflow involves RPA amplification of target sequences, crRNA-guided specific binding of the Cas14 protein to ssDNA targets, and activation-driven nonspecific cleavage of the fluorescent ssDNA reporter probes. The Cas14-DETECTR method has been shown to detect the human E3 ubiquitin protein ligase (HERC2) gene accurately (Harrington et al., 2018). The Cas14 protein exhibits high-fidelity target sequence recognition, making Cas14-DETECTR a high-fidelity detection tool for identifying medically important pathogens and SNPs (Aquino-Jarquin, 2019). Chen et al. developed a naked-eye colorimetric assay using multiplexed isothermal amplification with CRISPR/Cas14a for aflatoxin B1 (AFB1), employing magnetic/gold nanocomposite probes (MAPs) (Chen et al., 2023). This technology uses an aptamer competition mechanism to capture the triggered toxin, triggers a DNA signal amplification reaction and produces a visual color change by CRISPR/Cas14a-specific cleavage of MAPs, with a detection limit of 31.90 pg./mL. It demonstrates excellent specificity and accuracy in real-world samples. To address the challenges of Helicobacter pylori antibiotic resistance detection, Lai et al. developed the Cas14VIDet visual detection platform, which integrates ultrarapid PCR with the CRISPR/Cas14 system in a single-tube reaction format (Lai et al., 2025). This innovative approach overcomes the limitations of conventional methods by enabling the precise identification of SNPs without requiring a PAM sequence. The platform achieves exceptional sensitivity at 100 CFU/mL (single-colony level) and allows for visual interpretation of resistance genes within 10 min. When validated with 50 clinical samples, it demonstrated 100% sensitivity, specificity, and accuracy in detecting levofloxacin resistance genes, showing complete consistency with the Sanger sequencing results. This breakthrough provides an efficient solution for guiding precise antibiotic selection in H. pylori infection management.
In summary, the IAT-CRISPR/Cas system leverages its highly specific nucleic acid recognition ability by combining isothermal amplification and signal conversion mechanisms to achieve efficient detection of various pathogens. This approach offers high sensitivity, low cost, and simple operation, making it suitable for POCT applications (Shao et al., 2019; Mao et al., 2023). However, CRISPR/Cas systems still face several challenges in nucleic acid detection. First, owing to the off-target effects of the CRISPR/Cas system, gRNAs can recognize and induce cleavage at nontarget DNA or RNA sequences, leading to false-positive test results. To mitigate this problem, strategies include optimizing the Cas protein and CRISPR/Cas base sequences to obtain more stable mutants and adjusting the gRNA sequence length, mismatch tolerance, GC content, and reaction conditions (Hajiahmadi et al., 2019). Second, the recognition of target sequences by Cas effector proteins is limited by specific PAM sequences, restricting their widespread application. Solutions involve introducing PAM sequences into PCR and LAMP amplification products, using PAM-containing primers, or developing CRISPR/Cas systems with broader PAM recognition (Harrington et al., 2018; Li et al., 2018). Lastly, the activity of CRISPR/Cas systems directly affects detection sensitivity, requiring continuous optimization of Cas proteins and corresponding gRNAs or crRNAs to increase system activity (Hajiahmadi et al., 2019; Arizti-Sanz et al., 2020).
In addition, when nucleic acid amplification is coupled with CRISPR/Cas detection systems, CRISPR/Cas-based diagnostics face challenges in commercialization, contamination risks, and insufficient sensitivity. The key issue lies in effectively amplifying nucleic acid signals to levels detectable by Cas systems. To address this need, CRISPR/Cas12-based diagnostic technologies (e.g., SHERLOCK) can be optimized into independent steps of nucleic acid amplification and CRISPR/Cas detection to increase sensitivity, but this optimization increases procedural complexity and cross-contamination risks. Alternatively, single-step nucleic acid detection strategies exhibit lower sensitivity, whereas two-step approaches suffer from operational complexity and commercialization hurdles. Future advancements aim to integrate CRISPR/Cas systems into single-reaction formats for improved efficiency. For example, Hu et al. developed a light-controlled single-tube RPA-CRISPR/Cas12a DNA detection system that demonstrated promising sensitivity (Hu et al., 2022). One pot of SHINE (Streamlined Highlighting of Infections to Navigate Epidemics) enables visualized fluorescence readouts, reducing contamination rates and facilitating interpretation through a smartphone application, but it still lacks robust clinical evidence, comprehensive functionality, and scalability for widespread clinical applications (Arizti-Sanz et al., 2020). Future advancements should focus on device miniaturization, optimizing multiplex detection technologies, expanding clinical applications across diverse scenarios, and establishing standardized protocols (Gootenberg et al., 2018; Kaminski et al., 2021).
7 Conclusion
IAT is a method for rapid amplification of nucleic acid fragments at a constant temperature, which enables efficient and sensitive amplification of DNA/RNA through a specific enzyme-catalyzed reaction without thermal cycling. Based on their operational simplicity and rapid reaction kinetics, IATs are suitable for immediate detection and resource-limited environments, including rapid on-site diagnostic and pathogen detection. The comparative technical specifications of different IATs (Table 1) and conventional PCRs (Table 2) visually demonstrate the advantages of the IAT in terms of convenience, efficiency and sensitivity for pathogen detection. Therefore, IAT is considered a highly efficient approach for rapid POCT. In addition, combining IATs with the CRISPR/Cas system enables rapid and sensitive molecular detection by integrating the precise recognition capability of the CRISPR gene editing tool with the high efficiency of isothermal nucleic acid amplification technology. Table 3 shows the application of different Cas systems combined with IATs for the detection of pathogens and related technical indicators (Table 3).
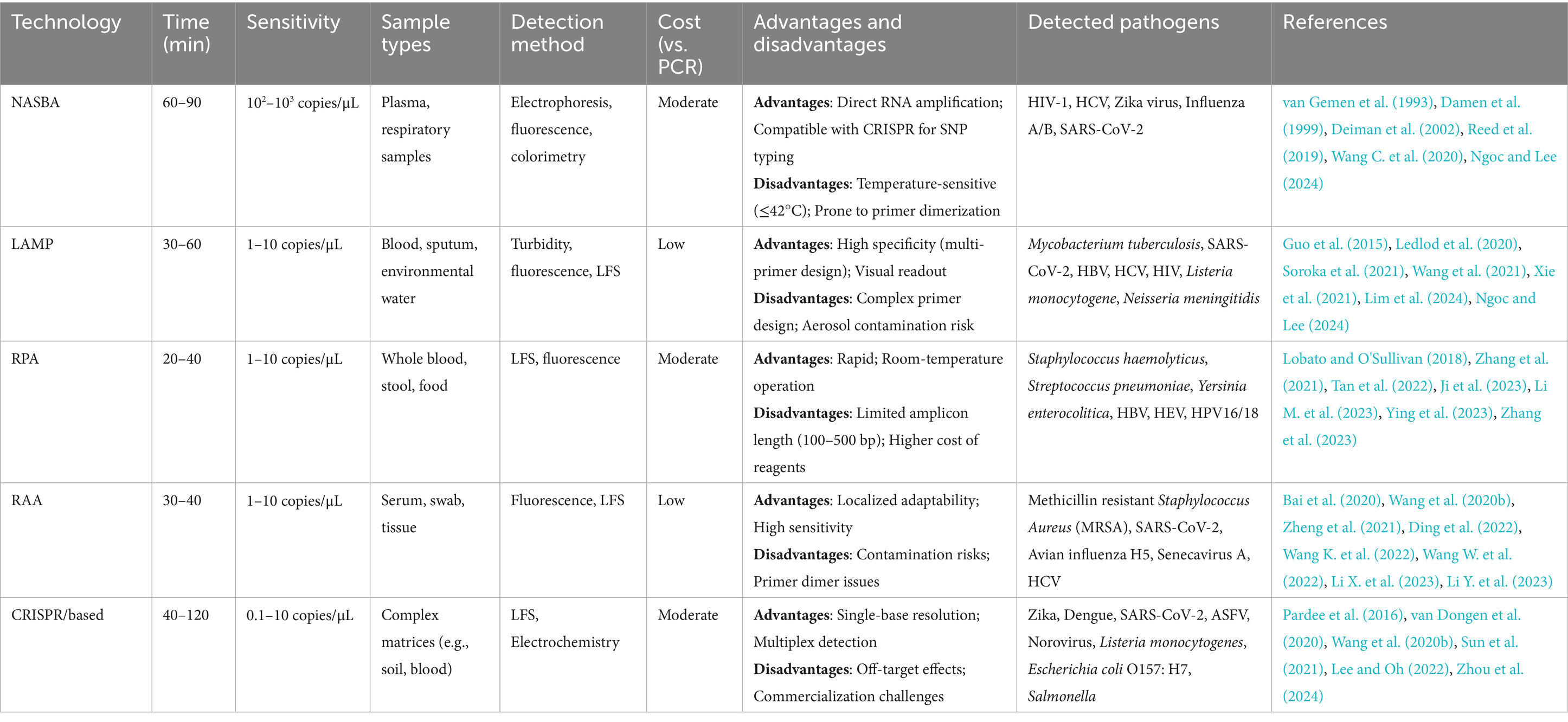
Table 1. Key metrics comparison of isothermal amplification technologies.
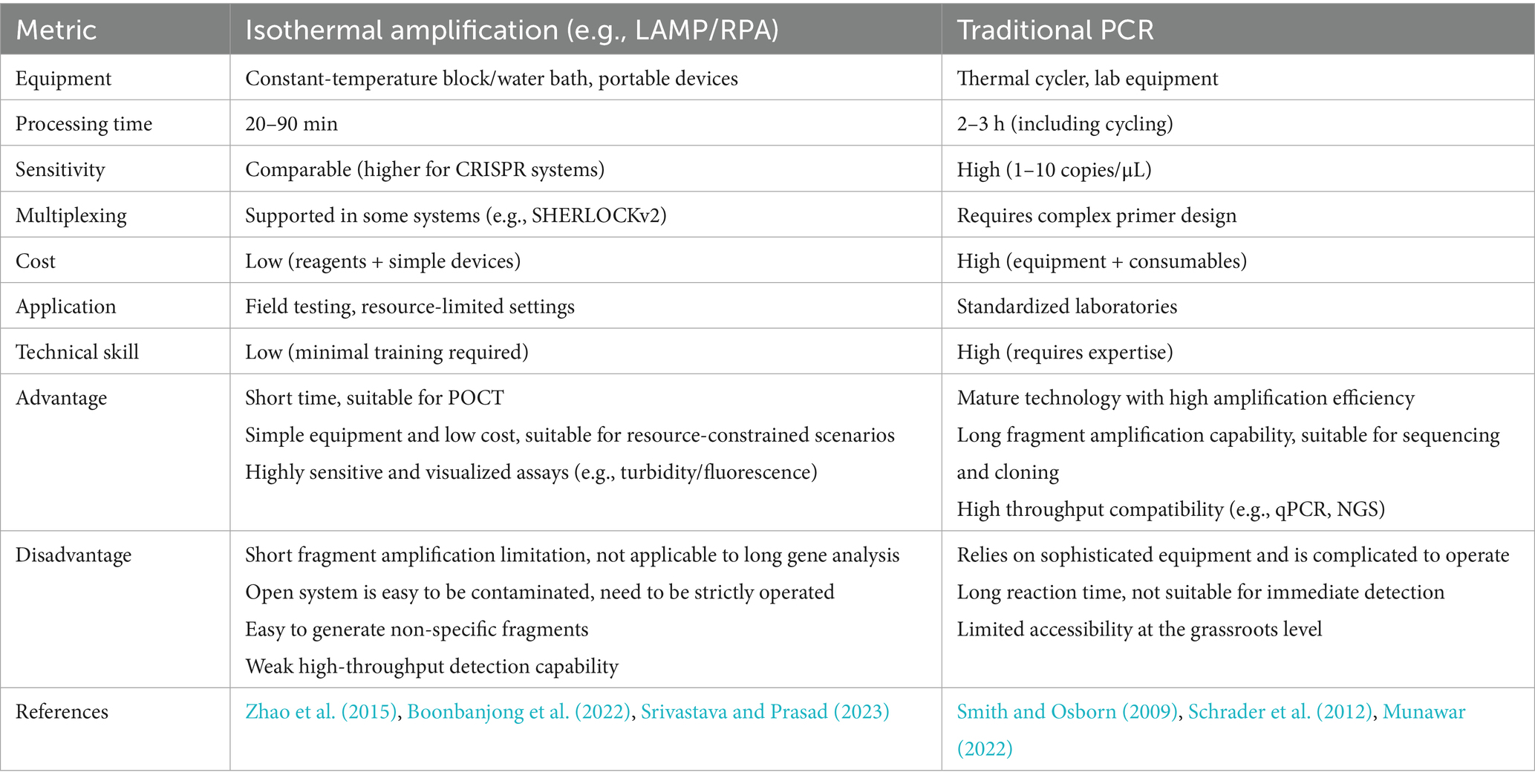
Table 2. Isothermal amplification technologies vs. traditional PCR.
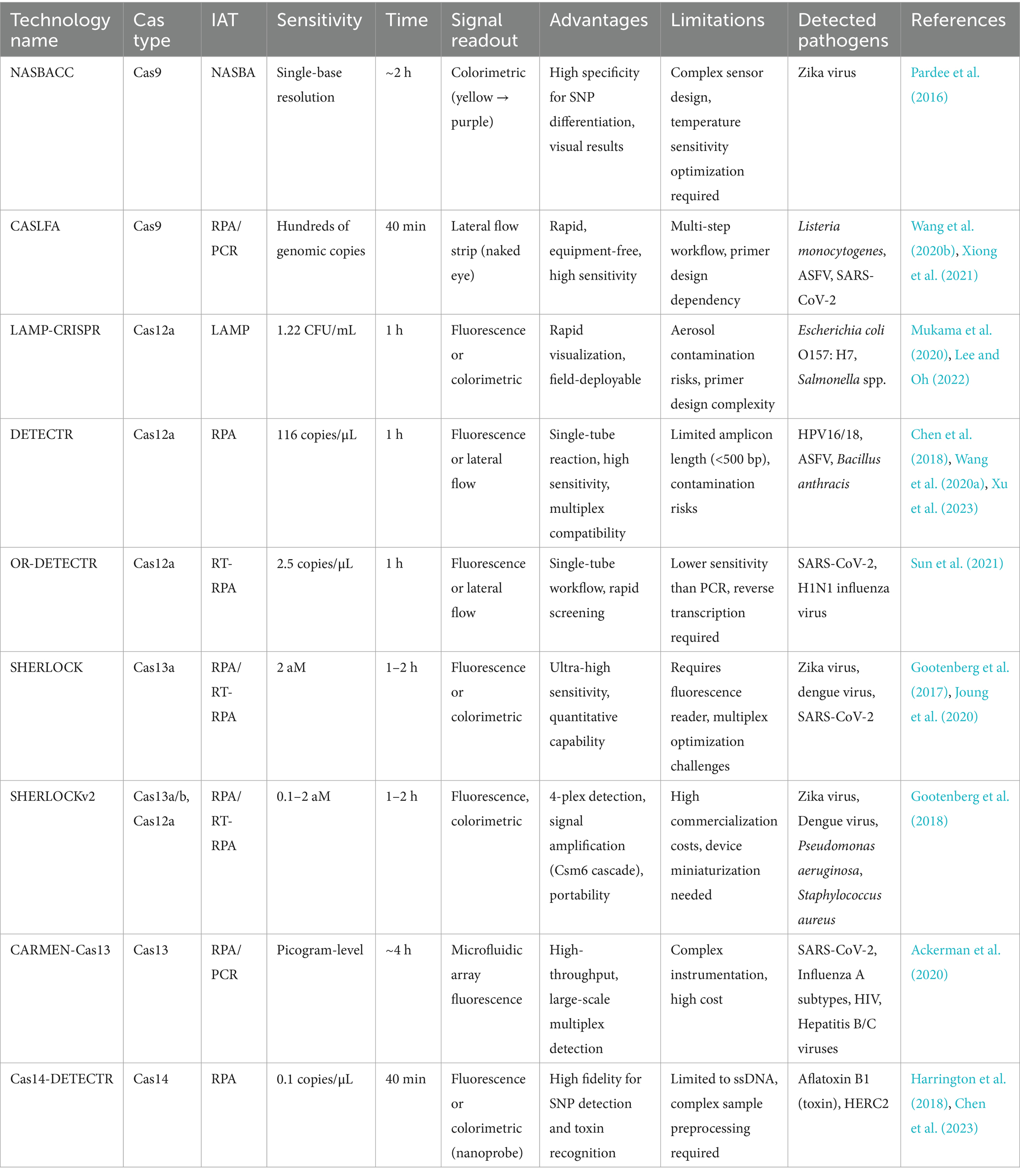
Table 3. CRISPR/Cas systems combined with isothermal amplification technologies.
8 Summary and outlook
Traditional molecular diagnostic methods, such as PCR, rely on thermal cyclers and specialized laboratories. This requirement makes meeting the demand for POCT in remote areas and during outbreaks difficult. In contrast, IAT represents a revolutionary breakthrough, offering an isothermal reaction, minimal equipment requirements, and exceptional speed and efficiency. We focus on the principles and applications of core technologies such as NASBA, LAMP, RPA, and RAA. NASBA enables isothermal amplification of RNA targets through reverse transcriptase and RNA polymerase but is limited by temperature sensitivity and nonspecific amplification. LAMP uses a chain-substitution polymerase and multi-primer design to achieve rapid detection but has complex primer design and risks aerosol contamination. RPA and RAA simplify the process by combining recombinase and polymerase, which significantly increases assay speed, but they are limited by fragment length. In addition, IAT technologies combined with the CRISPR/Cas system, such as SHERLOCK and DETECTR, have achieved ultrahigh sensitivity and multiplex detection through highly specific detection and signal amplification by nucleases, demonstrating significant value in outbreaks of diseases such as Zika virus and COVID-19.
The concretization and expansion of future technological trajectories should prioritize the synergistic integration of innovation and practical implementation. First, the development of machine learning-driven intelligent platforms (e.g., AlphaFold-assisted enzyme engineering or CRISPR scan-optimized gRNA design platforms) is aimed at mitigating challenges related to nonspecific amplification in NASBA and off-target effects in CRISPR/Cas systematically (Moreno-Mateos et al., 2015; Cui et al., 2018; Peccati et al., 2023). By predicting enzymatic thermal stability and target binding affinities, these tools enable precise modulation of molecular interactions, thereby increasing reaction specificity. Additionally, machine learning algorithms can optimize primer design protocols, quantitatively assess primer ensemble performance, and minimize primer dimer formation and nonspecific amplification events, streamlining nucleic acid synthesis workflows (Dwivedi-Yu et al., 2023).
Second, advancements in miniaturized, automated pathogen detection systems leverage integrated technologies spanning isothermal amplification techniques, CRISPR-based signal transduction, and microfluidic chip architectures to create “sample-in, result-out” diagnostic devices (Qin et al., 2019; Didarian and Azar, 2025). Miniaturization enables seamless integration into mobile terminals or POCT kits, overcoming geographical barriers and democratizing access to diagnostics. End-to-end automation of sample preparation, reaction orchestration, and signal detection not only reduces operator-dependent errors but also expedites assay turnaround times, ensuring robust analytical sensitivity and specificity. The synergy of these technologies has accelerated the implementation of POCT, supporting public health emergency responses and equitable access to medical resources. In remote areas and during sudden outbreaks, such advancements will emerge as pivotal technological forces for safeguarding health and lives.
In the future, IAT will further advance molecular diagnostics, particularly in POCT scenarios in resource-limited areas. These technologies are uniquely fast, specific, and sensitive, enabling rapid and accurate pathogen detection. As they evolve, they hold the potential to transform diagnostics, personalized medicine, and environmental monitoring, making high-quality analytical tools accessible across diverse settings. IAT is poised to become a cornerstone of next-generation precision medicine, delivering efficient, portable, and cost-effective solutions for global infectious disease prevention and control.
Author contributions
QB: Writing – review & editing, Writing – original draft. ML: Writing – review & editing, Writing – original draft. LY: Writing – original draft, Writing – review & editing. JC: Resources, Conceptualization, Supervision, Writing – review & editing. QS: Conceptualization, Writing – review & editing, Resources, Supervision. YD: Writing – review & editing, Methodology, Resources, Supervision, Funding acquisition. LZ: Writing – review & editing, Funding acquisition, Supervision, Methodology, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Zhejiang Province (No. LGF20H200009) and the Independent Research Project of the 903rd Hospital of PLA (No. ZJ202301).
Acknowledgments
We thank all authors for their contribution to the writing of this review on progress in the application of isothermal amplification technology in the diagnostic of infectious diseases.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ackerman, C. M., Myhrvold, C., Thakku, S. G., Freije, C. A., Metsky, H. C., Yang, D. K., et al. (2020). Massively multiplexed nucleic acid detection with Cas13. Nature 582, 277–282. doi: 10.1038/s41586-020-2279-8
Allan-Blitz, L. T., Shah, P., Adams, G., Branda, J. A., Klausner, J. D., Goldstein, R., et al. (2023). Development of Cas13a-based assays for Neisseria gonorrhoeae detection and gyrase a determination. mSphere 8:e0041623. doi: 10.1128/msphere.00416-23
Aquino-Jarquin, G. (2019). CRISPR-Cas14 is now part of the artillery for gene editing and molecular diagnostic. Nanomedicine 18, 428–431. doi: 10.1016/j.nano.2019.03.006
Arizti-Sanz, J., Freije, C. A., Stanton, A. C., Petros, B. A., Boehm, C. K., Siddiqui, S., et al. (2020). Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 11:5921. doi: 10.1038/s41467-020-19097-x
Aufdembrink, L. M., Khan, P., Gaut, N. J., Adamala, K. P., and Engelhart, A. E. (2020). Highly specific, multiplexed isothermal pathogen detection with fluorescent aptamer readout. RNA 26, 1283–1290. doi: 10.1261/rna.075192.120
Bai, X., Ma, X., Li, M., Li, X., Fan, G., Zhang, R., et al. (2020). Field applicable detection of hepatitis B virus using internal controlled duplex recombinase-aided amplification assay and lateral flow dipstick assay. J. Med. Virol. 92, 3344–3353. doi: 10.1002/jmv.25778
Boonbanjong, P., Treerattrakoon, K., Waiwinya, W., Pitikultham, P., and Japrung, D. (2022). Isothermal amplification Technology for Disease Diagnosis. Biosensors (Basel) 12:677. doi: 10.3390/bios12090677
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. doi: 10.1126/science.aar6245
Chen, J., Ren, B., Wang, Z., Wang, Q., Bi, J., and Sun, X. (2023). Multiple isothermal amplification coupled with CRISPR-Cas14a for the naked-eye and colorimetric detection of aflatoxin B1. ACS Appl. Mater. Interfaces 15, 55423–55432. doi: 10.1021/acsami.3c13331
Compton, J. (1991). Nucleic acid sequence-based amplification. Nature 350, 91–92. doi: 10.1038/350091a0
Crego-Vicente, B., Del Olmo, M. D., Muro, A., and Fernández-Soto, P. (2024). Multiplexing LAMP assays: a methodological review and diagnostic application. Int. J. Mol. Sci. 25:6374. doi: 10.3390/ijms25126374
Cui, Y., Xu, J., Cheng, M., Liao, X., and Peng, S. (2018). Review of CRISPR/Cas9 sgRNA design tools. Interdiscip. Sci. 10, 455–465. doi: 10.1007/s12539-018-0298-z
Damen, M., Sillekens, P., Cuypers, H. T., Frantzen, I., and Melsert, R. (1999). Characterization of the quantitative HCV NASBA assay. J. Virol. Methods 82, 45–54. doi: 10.1016/s0166-0934(99)00079-8
Deiman, B., van Aarle, P., and Sillekens, P. (2002). Characteristics and applications of nucleic acid sequence-based amplification (NASBA). Mol. Biotechnol. 20, 163–180. doi: 10.1385/mb:20:2:163
Didarian, R., and Azar, M. T. (2025). Microfluidic biosensors: revolutionizing detection in DNA analysis, cellular analysis, and pathogen detection. Biomed. Microdevices 27:10. doi: 10.1007/s10544-025-00741-6
Ding, X., Wang, H., Cui, M., Cheng, M., Zhao, Q., Bai, Y., et al. (2022). Development of a real-time recombinase-aided amplification method to rapidly detect methicillin-resistant Staphylococcus aureus. Microorganisms 10:2351. doi: 10.3390/microorganisms10122351
Dou, M., Dominguez, D. C., Li, X., Sanchez, J., and Scott, G. (2014). A versatile PDMS/paper hybrid microfluidic platform for sensitive infectious disease diagnosis. Anal. Chem. 86, 7978–7986. doi: 10.1021/ac5021694
Dwivedi-Yu, J. A., Oppler, Z. J., Mitchell, M. W., Song, Y. S., and Brisson, D. (2023). A fast machine-learning-guided primer design pipeline for selective whole genome amplification. PLoS Comput. Biol. 19:e1010137. doi: 10.1371/journal.pcbi.1010137
Foo, P. C., Chan, Y. Y., Mohamed, M., Wong, W. K., Nurul Najian, A. B., and Lim, B. H. (2017). Development of a thermostabilised triplex LAMP assay with dry-reagent four target lateral flow dipstick for detection of Entamoeba histolytica and non-pathogenic Entamoeba spp. Anal. Chim. Acta 966, 71–80. doi: 10.1016/j.aca.2017.02.019
Fu, J., Chiang, E. L. C., Medriano, C. A. D., Li, L., and Bae, S. (2021). Rapid quantification of fecal indicator bacteria in water using the most probable number - loop-mediated isothermal amplification (MPN-LAMP) approach on a polymethyl methacrylate (PMMA) microchip. Water Res. 199:117172. doi: 10.1016/j.watres.2021.117172
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., and Zhang, F. (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. doi: 10.1126/science.aaq0179
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi: 10.1126/science.aam9321
Guk, K., Yi, S., Kim, H., Bae, Y., Yong, D., Kim, S., et al. (2023). Hybrid CRISPR/Cas protein for one-pot detection of DNA and RNA. Biosens. Bioelectron. 219:114819. doi: 10.1016/j.bios.2022.114819
Guo, Z., Yu, T., He, J., Liu, F., Hao, H., Zhao, Y., et al. (2015). An integrated microfluidic chip for the detection of bacteria - a proof of concept. Mol. Cell. Probes 29, 223–227. doi: 10.1016/j.mcp.2015.05.005
Guoshuai, J., Xiaomeng, X., Zengdan, G., Xingxing, H., Qi, P., Hanbing, Z., et al. (2022). A rapid and high sensitivity RNA detection based on NASBA and G4-ThT fluorescent biosensor. Sci. Rep. 12:10076. doi: 10.1038/s41598-022-14107-y
Hajiahmadi, Z., Movahedi, A., Wei, H., Li, D., Orooji, Y., Ruan, H., et al. (2019). Strategies to increase on-target and reduce off-target effects of the CRISPR/Cas9 system in plants. Int. J. Mol. Sci. 20:3719. doi: 10.3390/ijms20153719
Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., et al. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842. doi: 10.1126/science.aav4294
Hu, M., Qiu, Z., Bi, Z., Tian, T., Jiang, Y., and Zhou, X. (2022). Photocontrolled crRNA activation enables robust CRISPR-Cas12a diagnostics. Proc. Natl. Acad. Sci. USA 119:e2202034119. doi: 10.1073/pnas.2202034119
Jean, J., D'Souza, D. H., and Jaykus, L. A. (2004). Multiplex nucleic acid sequence-based amplification for simultaneous detection of several enteric viruses in model ready-to-eat foods. Appl. Environ. Microbiol. 70, 6603–6610. doi: 10.1128/aem.70.11.6603-6610.2004
Ji, T., Zhang, J., Gao, Y., Zhao, C., and Gao, X. (2023). A rapid and visual detection of Staphylococcus haemolyticus in clinical specimens with RPA-LFS. Anal. Chim. Acta 1273:341534. doi: 10.1016/j.aca.2023.341534
Joung, J., Ladha, A., Saito, M., Kim, N. G., Woolley, A. E., Segel, M., et al. (2020). Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 383, 1492–1494. doi: 10.1056/NEJMc2026172
Jung, J. K., Dreyer, K. S., Dray, K. E., Muldoon, J. J., George, J., Shirman, S., et al. (2025). Developing, characterizing, and modeling CRISPR-based point-of-use pathogen diagnostics. ACS Synth. Biol. 14, 129–147. doi: 10.1021/acssynbio.4c00469
Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F., and Collins, J. J. (2021). CRISPR-based diagnostics. Nat. Biomed. Eng. 5, 643–656. doi: 10.1038/s41551-021-00760-7
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O., and Zhang, F. (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. doi: 10.1038/s41596-019-0210-2
Kia, V., Tafti, A., Paryan, M., and Mohammadi-Yeganeh, S. (2023). Evaluation of real-time NASBA assay for the detection of SARS-CoV-2 compared with real-time PCR. Ir. J. Med. Sci. 192, 723–729. doi: 10.1007/s11845-022-03046-2
Kievits, T., van Gemen, B., van Strijp, D., Schukkink, R., Dircks, M., Adriaanse, H., et al. (1991). NASBA isothermal enzymatic in vitro nucleic acid amplification optimized for the diagnosis of HIV-1 infection. J. Virol. Methods 35, 273–286. doi: 10.1016/0166-0934(91)90069-c
Kwak, N., Park, B. J., and Song, Y. J. (2023). A CRISPR-Cas12a-based diagnostic method for Japanese encephalitis virus genotypes I, III, and V. Biosensors (Basel) 13:769. doi: 10.3390/bios13080769
Lai, Y., Guo, K., Zhu, C., Lv, Y., Wu, H., Zhang, L., et al. (2025). Cas14VIDet: a visual instant method free from PAM restriction for antibiotic resistance bacteria detection. Biosens. Bioelectron. 268:116884. doi: 10.1016/j.bios.2024.116884
Lau, L. T., Feng, X. Y., Lam, T. Y., Hui, H. K., and Yu, A. C. (2010). Development of multiplex nucleic acid sequence-based amplification for detection of human respiratory tract viruses. J. Virol. Methods 168, 251–254. doi: 10.1016/j.jviromet.2010.04.027
Ledlod, S., Bunroddith, K., Areekit, S., Santiwatanakul, S., and Chansiri, K. (2020). Development of a duplex lateral flow dipstick test for the detection and differentiation of Listeria spp. and Listeria monocytogenes in meat products based on loop-mediated isothermal amplification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1139:121834. doi: 10.1016/j.jchromb.2019.121834
Lee, S. Y., and Oh, S. W. (2022). Filtration-based LAMP-CRISPR/Cas12a system for the rapid, sensitive and visualized detection of Escherichia coli O157:H7. Talanta 241:123186. doi: 10.1016/j.talanta.2021.123186
Li, S. Y., Cheng, Q. X., Wang, J. M., Li, X. Y., Zhang, Z. L., Gao, S., et al. (2018). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4:20. doi: 10.1038/s41421-018-0028-z
Li, M., Li, T., Hao, X., Liu, Y., Lan, H., and Zhou, C. (2023). Preliminary investigation of hepatitis E virus detection by a recombinase polymerase amplification assay combined with a lateral flow strip. J. Vet. Diagn. Invest. 35, 396–399. doi: 10.1177/10406387231167119
Li, Y., Shang, J., Wang, Y., Luo, J., Jiang, W., Yin, X., et al. (2023). Establishment of two assays based on reverse transcription recombinase-aided amplification technology for rapid detection of H5 subtype avian influenza virus. Microbiol. Spectr. 11:e0218623. doi: 10.1128/spectrum.02186-23
Li, X., Zhu, S., Zhang, X., Ren, Y., He, J., Zhou, J., et al. (2023). Advances in the application of recombinase-aided amplification combined with CRISPR-Cas technology in quick detection of pathogenic microbes. Front. Bioeng. Biotechnol. 11:1215466. doi: 10.3389/fbioe.2023.1215466
Lim, J., Koprowski, K., Stavins, R., Xuan, N., Hoang, T. H., Baek, J., et al. (2024). Point-of-care multiplex detection of respiratory viruses. ACS Sens. 9, 4058–4068. doi: 10.1021/acssensors.4c00992
Lin, H., Zhao, S., Ye, Y., Shao, L., Jiang, N., and Yang, K. (2022). A fluorescent recombinase aided amplification assay for detection of Babesia microti. Korean J. Parasitol. 60, 201–205. doi: 10.3347/kjp.2022.60.3.201
Liu, Q., Jin, X., Cheng, J., Zhou, H., Zhang, Y., and Dai, Y. (2023). Advances in the application of molecular diagnostic techniques for the detection of infectious disease pathogens (review). Mol. Med. Rep. 27:104. doi: 10.3892/mmr.2023.12991
Liu, Q., Lin, X., Lin, L., Yi, L., Li, H., and Lin, J. M. (2015). A comparative study of three different nucleic acid amplification techniques combined with microchip electrophoresis for HPV16 E6/E7 mRNA detection. Analyst 140, 6736–6741. doi: 10.1039/c5an00944h
Liu, J., Zeng, Z., Li, F., Jiang, B., Nie, Y., Zhang, G., et al. (2024). Portable and simultaneous detection of four respiratory pathogens through a microfluidic LAMP and real-time fluorescence assay. Analyst 149, 5091–5100. doi: 10.1039/d4an00748d
Lobato, I. M., and O'Sullivan, C. K. (2018). Recombinase polymerase amplification: basics, applications and recent advances. Trends Analyt. Chem. 98, 19–35. doi: 10.1016/j.trac.2017.10.015
Ludwig, K. U., Schmithausen, R. M., Li, D., Jacobs, M. L., Hollstein, R., Blumenstock, K., et al. (2021). LAMP-Seq enables sensitive, multiplexed COVID-19 diagnostics using molecular barcoding. Nat. Biotechnol. 39, 1556–1562. doi: 10.1038/s41587-021-00966-9
Makarova, K. S., and Koonin, E. V. (2015). Annotation and classification of CRISPR-Cas systems. Methods Mol. Biol. 1311, 47–75. doi: 10.1007/978-1-4939-2687-9_4
Mao, X., Xu, M., Luo, S., Yang, Y., Zhong, J., Zhou, J., et al. (2023). Advancements in the synergy of isothermal amplification and CRISPR-cas technologies for pathogen detection. Front. Bioeng. Biotechnol. 11:1273988. doi: 10.3389/fbioe.2023.1273988
Mohammadi-Yeganeh, S., Paryan, M., Mirab Samiee, S., Kia, V., and Rezvan, H. (2012). Molecular beacon probes-base multiplex NASBA real-time for detection of HIV-1 and HCV. Iran. J. Microbiol. 4, 47–54.
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J. D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988. doi: 10.1038/nmeth.3543
Mori, Y., Nagamine, K., Tomita, N., and Notomi, T. (2001). Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 289, 150–154. doi: 10.1006/bbrc.2001.5921
Mukama, O., Wu, J., Li, Z., Liang, Q., Yi, Z., Lu, X., et al. (2020). An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosens. Bioelectron. 159:112143. doi: 10.1016/j.bios.2020.112143
Munawar, M. A. (2022). Critical insight into recombinase polymerase amplification technology. Expert. Rev. Mol. Diagn. 22, 725–737. doi: 10.1080/14737159.2022.2109964
Nagamine, K., Hase, T., and Notomi, T. (2002). Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 16, 223–229. doi: 10.1006/mcpr.2002.0415
Ngoc, L. T. N., and Lee, Y. C. (2024). Current trends in RNA virus detection via nucleic acid isothermal amplification-based platforms. Biosensors (Basel) 14:97. doi: 10.3390/bios14020097
Nie, M., Zhou, Y., Li, F., Deng, H., Zhao, M., Huang, Y., et al. (2022). Epidemiological investigation of swine Japanese encephalitis virus based on RT-RAA detection method. Sci. Rep. 12:9392. doi: 10.1038/s41598-022-13604-4
Nii-Trebi, N. I. (2017). Emerging and neglected infectious diseases: insights, advances, and challenges. Biomed. Res. Int. 2017, 1–15. doi: 10.1155/2017/5245021
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, 63e–663e. doi: 10.1093/nar/28.12.e63
Nurul Najian, A. B., Engku Nur Syafirah, E. A., Ismail, N., Mohamed, M., and Yean, C. Y. (2016). Development of multiplex loop mediated isothermal amplification (m-LAMP) label-based gold nanoparticles lateral flow dipstick biosensor for detection of pathogenic Leptospira. Anal. Chim. Acta 903, 142–148. doi: 10.1016/j.aca.2015.11.015
Obande, G. A., and Banga Singh, K. K. (2020). Current and future perspectives on isothermal nucleic acid amplification Technologies for Diagnosing Infections. Infect. Drug Resist. 13, 455–483. doi: 10.2147/idr.S217571
Oliveira, B. B., Veigas, B., Carlos, F. F., Sánchez-Melsió, A., Balcázar, J. L., Borrego, C. M., et al. (2020). Water safety screening via multiplex LAMP-au-nanoprobe integrated approach. Sci. Total Environ. 741:140447. doi: 10.1016/j.scitotenv.2020.140447
Pardee, K., Green, A. A., Takahashi, M. K., Braff, D., Lambert, G., Lee, J. W., et al. (2016). Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell 165, 1255–1266. doi: 10.1016/j.cell.2016.04.059
Peccati, F., Alunno-Rufini, S., and Jiménez-Osés, G. (2023). Accurate prediction of enzyme Thermostabilization with Rosetta using AlphaFold ensembles. J. Chem. Inf. Model. 63, 898–909. doi: 10.1021/acs.jcim.2c01083
Piepenburg, O., Williams, C. H., Stemple, D. L., and Armes, N. A. (2006). DNA detection using recombination proteins. PLoS Biol. 4:e204. doi: 10.1371/journal.pbio.0040204
Qian, W., Huang, J., Wang, T., Fan, C., Kang, J., Zhang, Q., et al. (2022). Ultrasensitive and visual detection of human norovirus genotype GII.4 or GII.17 using CRISPR-Cas12a assay. Virol. J. 19:150. doi: 10.1186/s12985-022-01878-z
Qin, P., Park, M., Alfson, K. J., Tamhankar, M., Carrion, R., Patterson, J. L., et al. (2019). Rapid and fully microfluidic Ebola virus detection with CRISPR-Cas13a. ACS Sens. 4, 1048–1054. doi: 10.1021/acssensors.9b00239
Reed, A. J., Connelly, R. P., Williams, A., Tran, M., Shim, B. S., Choe, H., et al. (2019). Label-free pathogen detection by a Deoxyribozyme Cascade with visual signal readout. Sens. Actuat. B Chem. 282, 945–951. doi: 10.1016/j.snb.2018.11.147
Schrader, C., Schielke, A., Ellerbroek, L., and Johne, R. (2012). PCR inhibitors - occurrence, properties and removal. J. Appl. Microbiol. 113, 1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x
Shao, N., Han, X., Song, Y., Zhang, P., and Qin, L. (2019). CRISPR-Cas12a coupled with platinum Nanoreporter for visual quantification of SNVs on a volumetric Bar-chart Chip. Anal. Chem. 91, 12384–12391. doi: 10.1021/acs.analchem.9b02925
Shen, X. X., Qiu, F. Z., Shen, L. P., Yan, T. F., Zhao, M. C., Qi, J. J., et al. (2019). A rapid and sensitive recombinase aided amplification assay to detect hepatitis B virus without DNA extraction. BMC Infect. Dis. 19:229. doi: 10.1186/s12879-019-3814-9
Smith, C. J., and Osborn, A. M. (2009). Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67, 6–20. doi: 10.1111/j.1574-6941.2008.00629.x
Soliman, H., and El-Matbouli, M. (2005). An inexpensive and rapid diagnostic method of koi herpesvirus (KHV) infection by loop-mediated isothermal amplification. Virol. J. 2:83. doi: 10.1186/1743-422x-2-83
Soroka, M., Wasowicz, B., and Rymaszewska, A. (2021). Loop-mediated isothermal amplification (LAMP): the better sibling of PCR? Cells 10:1931. doi: 10.3390/cells10081931
Srivastava, P., and Prasad, D. (2023). Isothermal nucleic acid amplification and its uses in modern diagnostic technologies. 3 Biotech 13:200. doi: 10.1007/s13205-023-03628-6
Sun, Y., Yu, L., Liu, C., Ye, S., Chen, W., Li, D., et al. (2021). One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J. Transl. Med. 19:74. doi: 10.1186/s12967-021-02741-5
Talap, J., Shen, M., Yu, L., Zeng, S., and Cai, S. (2022). RT-LAMP assay combining multi-fluorescent probes for SARS-CoV-2 RNA detection and variant differentiation. Talanta 248:123644. doi: 10.1016/j.talanta.2022.123644
Tan, M., Liao, C., Liang, L., Yi, X., Zhou, Z., and Wei, G. (2022). Recent advances in recombinase polymerase amplification: principle, advantages, disadvantages and applications. Front. Cell. Infect. Microbiol. 12:1019071. doi: 10.3389/fcimb.2022.1019071
Tomita, N., Mori, Y., Kanda, H., and Notomi, T. (2008). Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3, 877–882. doi: 10.1038/nprot.2008.57
van Dongen, J. E., Berendsen, J. T. W., Steenbergen, R. D. M., Wolthuis, R. M. F., Eijkel, J. C. T., and Segerink, L. I. (2020). Point-of-care CRISPR/Cas nucleic acid detection: recent advances, challenges and opportunities. Biosens. Bioelectron. 166:112445. doi: 10.1016/j.bios.2020.112445
van Gemen, B., Kievits, T., Schukkink, R., van Strijp, D., Malek, L. T., Sooknanan, R., et al. (1993). Quantification of HIV-1 RNA in plasma using NASBA during HIV-1 primary infection. J. Virol. Methods 43, 177–187. doi: 10.1016/0166-0934(93)90075-3
Wacharapluesadee, S., and Hemachudha, T. (2001). Nucleic-acid sequence based amplification in the rapid diagnosis of rabies. Lancet 358, 892–893. doi: 10.1016/s0140-6736(01)06041-x
Wang, X., He, S., Zhao, N., Liu, X., Cao, Y., Zhang, G., et al. (2020a). Development and clinical application of a novel CRISPR-Cas12a based assay for the detection of African swine fever virus. BMC Microbiol. 20:282. doi: 10.1186/s12866-020-01966-6
Wang, C., Horby, P. W., Hayden, F. G., and Gao, G. F. (2020). A novel coronavirus outbreak of global health concern. Lancet 395, 470–473. doi: 10.1016/s0140-6736(20)30185-9
Wang, K., Huang, R., Zhang, L., Liu, D., and Diao, Y. (2022). Recombinase-aided amplification combined with lateral flow (LF-RAA) assay for rapid AAV genome detection. ACS Omega 7, 47832–47839. doi: 10.1021/acsomega.2c05660
Wang, Y., Li, H., Wang, Y., Zhang, L., Xu, J., and Ye, C. (2017). Loop-mediated isothermal amplification label-based gold nanoparticles lateral flow biosensor for detection of Enterococcus faecalis and Staphylococcus aureus. Front. Microbiol. 8:192. doi: 10.3389/fmicb.2017.00192
Wang, M., Liu, H., Ren, J., Huang, Y., Deng, Y., Liu, Y., et al. (2023). Enzyme-assisted nucleic acid amplification in molecular diagnosis: a review. Biosensors (Basel) 13:160. doi: 10.3390/bios13020160
Wang, X., Wang, G., Wang, Y., Quan, S., Qi, H., Sun, L., et al. (2021). Development and preliminary application of multiplex loop-mediated isothermal amplification coupled with lateral flow biosensor for detection of Mycobacterium tuberculosis complex. Front. Cell. Infect. Microbiol. 11:666492. doi: 10.3389/fcimb.2021.666492
Wang, X., Xiong, E., Tian, T., Cheng, M., Lin, W., Wang, H., et al. (2020b). Clustered regularly interspaced short palindromic repeats/Cas9-mediated lateral flow nucleic acid assay. ACS Nano 14, 2497–2508. doi: 10.1021/acsnano.0c00022
Wang, R. H., Zhang, H., Zhang, Y., Li, X. N., Shen, X. X., Qi, J. J., et al. (2019). Development and evaluation of recombinase-aided amplification assays incorporating competitive internal controls for detection of human adenovirus serotypes 3 and 7. Virol. J. 16:86. doi: 10.1186/s12985-019-1178-9
Wang, H., Zhang, Y., Zhou, J., Li, M., Chen, Y., Liu, Y., et al. (2022). Rapid visual detection of hepatitis C virus using reverse transcription recombinase-aided amplification-lateral flow dipstick. Front. Cell. Infect. Microbiol. 12:816238. doi: 10.3389/fcimb.2022.816238
Wang, W., Zhou, L., Ge, X., Han, J., Guo, X., Chen, Y., et al. (2022). Development of a VP2-based real-time fluorescent reverse transcription recombinase-aided amplification assay to rapidly detect Senecavirus A. Transbound. Emerg. Dis. 69, 2828–2839. doi: 10.1111/tbed.14435
WHO (2021). World Health Organization the top 10 causes of death. Available online at: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
Wu, H., Chen, Y., Shi, Y., Wang, L., Zhang, M., Wu, J., et al. (2021a). Carrying out pseudo dual nucleic acid detection from sample to visual result in a polypropylene bag with CRISPR/Cas12a. Biosens. Bioelectron. 178:113001. doi: 10.1016/j.bios.2021.113001
Wu, H., Qian, S., Peng, C., Wang, X., Wang, T., Zhong, X., et al. (2021b). Rotary valve-assisted fluidic system coupling with CRISPR/Cas12a for fully integrated nucleic acid detection. ACS Sens. 6, 4048–4056. doi: 10.1021/acssensors.1c01468
Wu, S., Yu, W., Fu, X., Yu, X., Ye, Z., Zhang, M., et al. (2024). Advances in virus detection techniques based on recombinant polymerase amplification. Molecules 29:4972. doi: 10.3390/molecules29204972
Wu, K., Zhang, Y., Zeng, S., Liu, X., Li, Y., Li, X., et al. (2021). Development and application of RAA nucleic acid test strip assay and double RAA gel electrophoresis detection methods for ASFV and CSFV. Front. Mol. Biosci. 8:811824. doi: 10.3389/fmolb.2021.811824
Xie, C., Chen, S., Zhang, L., He, X., Ma, Y., Wu, H., et al. (2021). Multiplex detection of blood-borne pathogens on a self-driven microfluidic chip using loop-mediated isothermal amplification. Anal. Bioanal. Chem. 413, 2923–2931. doi: 10.1007/s00216-021-03224-8
Xiong, E., Jiang, L., Tian, T., Hu, M., Yue, H., Huang, M., et al. (2021). Simultaneous dual-gene diagnosis of SARS-CoV-2 based on CRISPR/Cas9-mediated lateral flow assay. Angew. Chem. Int. Ed. Engl. 60, 5307–5315. doi: 10.1002/anie.202014506
Xu, J., Bai, X., Zhang, X., Yuan, B., Lin, L., Guo, Y., et al. (2023). Development and application of DETECTR-based rapid detection for pathogenic Bacillusanthracis. Anal. Chim. Acta 1247:340891. doi: 10.1016/j.aca.2023.340891
Xue, G., Li, S., Zhang, W., Du, B., Cui, J., Yan, C., et al. (2020). Reverse-transcription recombinase-aided amplification assay for rapid detection of the 2019 novel coronavirus (SARS-CoV-2). Anal. Chem. 92, 9699–9705. doi: 10.1021/acs.analchem.0c01032
Xue, T., Lu, Y., Yang, H., Hu, X., Zhang, K., Ren, Y., et al. (2022). Isothermal RNA amplification for the detection of viable pathogenic Bacteria to estimate the Salmonella virulence for causing enteritis. J. Agric. Food Chem. 70, 1670–1678. doi: 10.1021/acs.jafc.1c07182
Ying, J., Mao, L., Tang, Y., Fassatoui, M., Song, W., Xu, X., et al. (2023). Development and validation of real-time recombinase polymerase amplification-based assays for detecting HPV16 and HPV18 DNA. Microbiol. Spectr. 11:e0120723. doi: 10.1128/spectrum.01207-23
Zhang, K., Li, Q., Wang, K., Zhang, Q., Ma, C., Yang, G., et al. (2024). RPA-CRISPR-Cas-mediated dual lateral flow assay for the point-of-care testing of HPV16 and HPV18. Bioconjug. Chem. 35, 1797–1804. doi: 10.1021/acs.bioconjchem.4c00375
Zhang, H., Zhao, M., Hu, S., Ma, K., Li, J., Zhao, J., et al. (2023). Establishment of a real-time recombinase polymerase amplification for rapid detection of pathogenic Yersinia enterocolitica. Pathogens 12:226. doi: 10.3390/pathogens12020226
Zhang, B., Zhu, Z., Li, F., Xie, X., and Ding, A. (2021). Rapid and sensitive detection of hepatitis B virus by lateral flow recombinase polymerase amplification assay. J. Virol. Methods 291:114094. doi: 10.1016/j.jviromet.2021.114094
Zhao, Y., Chen, F., Li, Q., Wang, L., and Fan, C. (2015). Isothermal amplification of nucleic acids. Chem. Rev. 115, 12491–12545. doi: 10.1021/acs.chemrev.5b00428
Zheng, Y. Z., Chen, J. T., Li, J., Wu, X. J., Wen, J. Z., Liu, X. Z., et al. (2021). Reverse transcription recombinase-aided amplification assay with lateral flow dipstick assay for rapid detection of 2019 novel coronavirus. Front. Cell. Infect. Microbiol. 11:613304. doi: 10.3389/fcimb.2021.613304
Zhou, J., Li, Z., Seun Olajide, J., and Wang, G. (2024). CRISPR/Cas-based nucleic acid detection strategies: trends and challenges. Heliyon 10:e26179. doi: 10.1016/j.heliyon.2024.e26179
Zhu, Y., Liu, J., Liu, S., Zhu, X., Wu, J., Zhou, Q., et al. (2023). CRISPR/Cas12a-assisted visible fluorescence for pseudo dual nucleic acid detection based on an integrated chip. Anal. Chim. Acta 1280:341860. doi: 10.1016/j.aca.2023.341860
Keywords: isothermal amplification technology, infectious diseases, CRISPR/Cas system, molecular diagnostics, nucleic acid amplification
Citation: Bi Q, Liu M, Yan L, Cheng J, Sun Q, Dai Y and Zou L (2025) Progress in the application of isothermal amplification technology in the diagnosis of infectious diseases. Front. Microbiol. 16:1601644. doi: 10.3389/fmicb.2025.1601644
Edited by:
Wenn-Chyau Lee, University of Malaya, MalaysiaReviewed by:
Shelley C. Rankin, University of Pennsylvania, United StatesInsaf Bel Hadj Ali, Pasteur Institute of Tunis, Tunisia
Copyright © 2025 Bi, Liu, Yan, Cheng, Sun, Dai and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuzhu Dai, ZHl6NTg5NUBxcS5jb20=; Lingli Zou, NTc1NzIzNDA3QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship