 Mankgopo Kgatle1,2,3*
Mankgopo Kgatle1,2,3* Saidon Mbambara1,2,4
Saidon Mbambara1,2,4 Leon Khoza1,2
Leon Khoza1,2 Olalekan Fadebi1,2
Olalekan Fadebi1,2 Tivani Mashamba-Thompson5
Tivani Mashamba-Thompson5 Mike Sathekge1,2
Mike Sathekge1,2- 1Department of Nuclear Medicine, University of Pretoria and Steve Biko Academic Hospital, Pretoria, South Africa
- 2Nuclear Medicine Research Infrastructure (NuMeRI), Steve Biko Academic Hospital, Pretoria, South Africa
- 3Department of Medicine, University of Cape Town and Groote Schuur Hospital, Cape Town, South Africa
- 4Department of Biomedical Sciences, Tropical Diseases Research Center, Ndola, Zambia
- 5Faculty of Health Sciences, School of Health System and Public Health, University of Pretoria, Pretoria, South Africa
Oncogenic viruses typically manipulate host cellular mechanisms to drive tumorigenesis. They exploit pioneering transcription factors to modify gene expression, enabling uncontrolled proliferation. These viruses alter chromatin accessibility and induce chromatin remodelling, disrupting DNA repair and promoting viral genome integration. Additionally, epigenetic reprogramming through mechanisms like DNA methylation and histone modifications silences tumor suppressor genes and activates oncogenes. Understanding these mechanisms is critical for identifying more improved therapeutic targets, improving diagnostics, and predicting disease progression. Advances in this field can guide the development of innovative treatments and early detection tools. This comprehensive review synthesizes existing knowledge on the contributions of oncogenic viruses such as hepatitis B virus (HBV), hepatitis C virus (HCV), human papillomavirus (HPV), and human T-cell leukaemia virus type 1 (HTLV-1), Epstein–Barr virus (EBV), human herpesvirus 8 (HHV-8), and Merkel cell polyomavirus (MCV) to cancer development, highlighting their therapeutic relevance and driving forward research in viral oncogenesis.
Introduction
Oncogenic viruses such as hepatitis B virus (HBV), hepatitis C virus (HCV), human papillomavirus (HPV), human T-cell leukaemia virus type 1 (HTLV-1), Epstein–Barr virus (EBV), human herpesvirus 8 (HHV-8), and Merkel cell polyomavirus (MCV) significantly influence host cellular mechanisms to drive oncogenesis (Mohanty and Harhaj, 2023; Krump and You, 2018). These viruses significantly influence host cellular mechanisms by disrupting transcription factor activity, altering chromatin accessibility, and reprogramming epigenetic processes (Krump and You, 2018; Cirillo et al., 2002). Such disruptions lie at the heart of their role in tumorigenesis (McLaughlin-Drubin and Münger, 2009; Seeger and Mason, 2000; Young and Rickinson, 2004; Mesri et al., 2014).
Many oncogenic viruses integrate into accessible regions of the host genome, activating oncogenes while silencing tumor suppressor genes (McLaughlin-Drubin and Münger, 2009; Seeger and Mason, 2000; Young and Rickinson, 2004; Mesri et al., 2014). This process disrupts normal transcriptional activities to establish persistent infections, promote cell survival, and fuel oncogenesis (Young and Rickinson, 2004; Mesri et al., 2014). Particularly, pioneer transcription factors (PTFs) are often co-opted by these viruses to remodel condensed chromatin and recruit other proteins, thereby enhancing access to the transcriptional machinery essential for gene expression (Cirillo et al., 2002). The reprogramming facilitated by these PTFs involves key epigenetic modifications such as chromatin remodelling, histone alterations, and DNA methylation (Iwafuchi-Doi and Zaret, 2014). Oncogenic viruses, including HBV, HCV, HPV, HTLV-1, EBV, HHV-8, and MCV utilize these mechanisms to manipulate host chromatin, thereby advancing disease progression (Krump and You, 2018).
Chromatin accessibility further determines which genomic regions are available for transcription (Clapier et al., 2017). Oncogenic viruses like HBV and HPV, which integrate into the host genome, can exploit local chromatin states to modulate host gene expression. Although EBV typically persists as an episome, it can still impact host chromatin structure and gene regulation by interacting with epigenetic modifiers, even without integration. However, in certain cases, such as certain lymphomas and EBV-associated gastric carcinomas, EBV may integrate into the host genome (Péneau et al., 2022; Dai et al., 2021). This chromatin remodelling activity interferes with DNA repair mechanisms and apoptosis, ultimately fostering genomic instability and supporting viral genome integration into the host DNA—processes that collectively drive tumorigenesis (Dai et al., 2021).
Epigenetic reprogramming is another hallmark of viral oncogenesis (Krump and You, 2018). Oncogenic viruses deploy mechanisms like DNA methylation and histone modification to deactivate tumor suppressor genes and activate oncogenes (Warburton et al., 2021). For example, EBV and HTLV-1 leverage viral proteins to manipulate the host’s epigenetic machinery, thereby creating a cellular environment conducive to cancer development (Dai et al., 2021; Banerjee et al., 2024; Soliman et al., 2021). These changes are essential for maintaining viral survival while promoting tumor progression (Soliman et al., 2021).
A deeper understanding of these processes is critical for advancing cancer diagnostics, identifying therapeutic targets, and developing strategies to impede disease progression. This review consolidates current knowledge on the roles of transcription factors, chromatin remodelling, and epigenetic reprogramming in viral-induced malignancies, emphasizing their importance in therapeutic research and clinical applications. The focus of our study on HBV, HCV, HPV, HTLV-1, EBV, HHV8, and MCV stems from their classification as oncogenic viruses due to their direct role in causing cancers. By disrupting cellular mechanisms through pathways such as transcription factor activity, chromatin accessibility, and epigenetic reprogramming, these viruses remain central to the scope of our study.
A “peek” into PTFS and epigenetics
Pioneer transcription factors and chromatin remodelling
PTFs are a unique class of transcription factors that can bind to closed, heterochromatic regions and initiate chromatin remodelling (King and Klose, 2017; Soufi et al., 2015; Sinha et al., 2023). They are termed “pioneer factors” because they can “open” or remodel chromatin at specific genomic sites, making these regions transcriptionally active. Unlike conventional transcription factors, PTFs interact with nucleosome-bound DNA, thus unlocking previously inaccessible DNA sequences and make them transcriptionally active (Cirillo et al., 2002; Soufi et al., 2015). By recruiting chromatin remodelers, histone modifiers, and DNA methylation machinery, PTFs establish active or poised transcriptional states that drive gene transcription (Soufi et al., 2015; Sinha et al., 2023). These factors are critical in oncogenic virus-mediated transformation, as they reprogram host chromatin landscapes to activate both viral and host oncogenes while silencing tumor suppressor genes (Neugebauer et al., 2023).
Key mechanisms of epigenetic regulation
Epigenetic regulation controls gene expression without altering the underlying DNA sequence (Cavalli and Heard, 2019). This process involves mechanisms such as chromatin remodelling, histone modifications, and DNA methylation, which collectively influence chromatin structure and gene accessibility (Jaenisch and Bird, 2003). Chromatin, composed of DNA and histone proteins, exists in two main states: euchromatin, which is loosely packed and transcriptionally active, and heterochromatin, which is tightly packed and repressive (Lippman et al., 2004; Morrison and Thakur, 2021; Teperino et al., 2010; Zhang et al., 2015). ATP-dependent complexes such as chromodomain helicase DNA-binding (CHD), switch/sucrose non-fermentable (SWI/SNF), imitation switch (ISWI), and INO80, dynamically reorganize chromatin to regulate gene accessibility (Clapier et al., 2017). Proper chromatin is essential for biological processes like stem cell differentiation, stress responses, and cancer progression, while disruptions in these processes can lead to abnormal gene expression and tumorigenesis (Clapier et al., 2017; Mansisidor and Risca, 2022).
Histone proteins, which form the core of nucleosomes, undergo post-translational modifications (PTMs) that regulate chromatin structure and gene expression (Pietropaolo et al., 2021). These modifications include acetylation, methylation, phosphorylation, ubiquitination, and SUMOylation (Jones and Baylin, 2007). For instance, acetylation by histone acetyltransferases (HATs) loosens histone-DNA interactions to promote gene transcription, while histone methylation can either activate or repress transcription, depending on the specific marker (Mac and Moody, 2020). Aberrant histone modifications are often associated with cancer, as they disrupt genes vital for tumor suppression and immune responses (Figure 1).
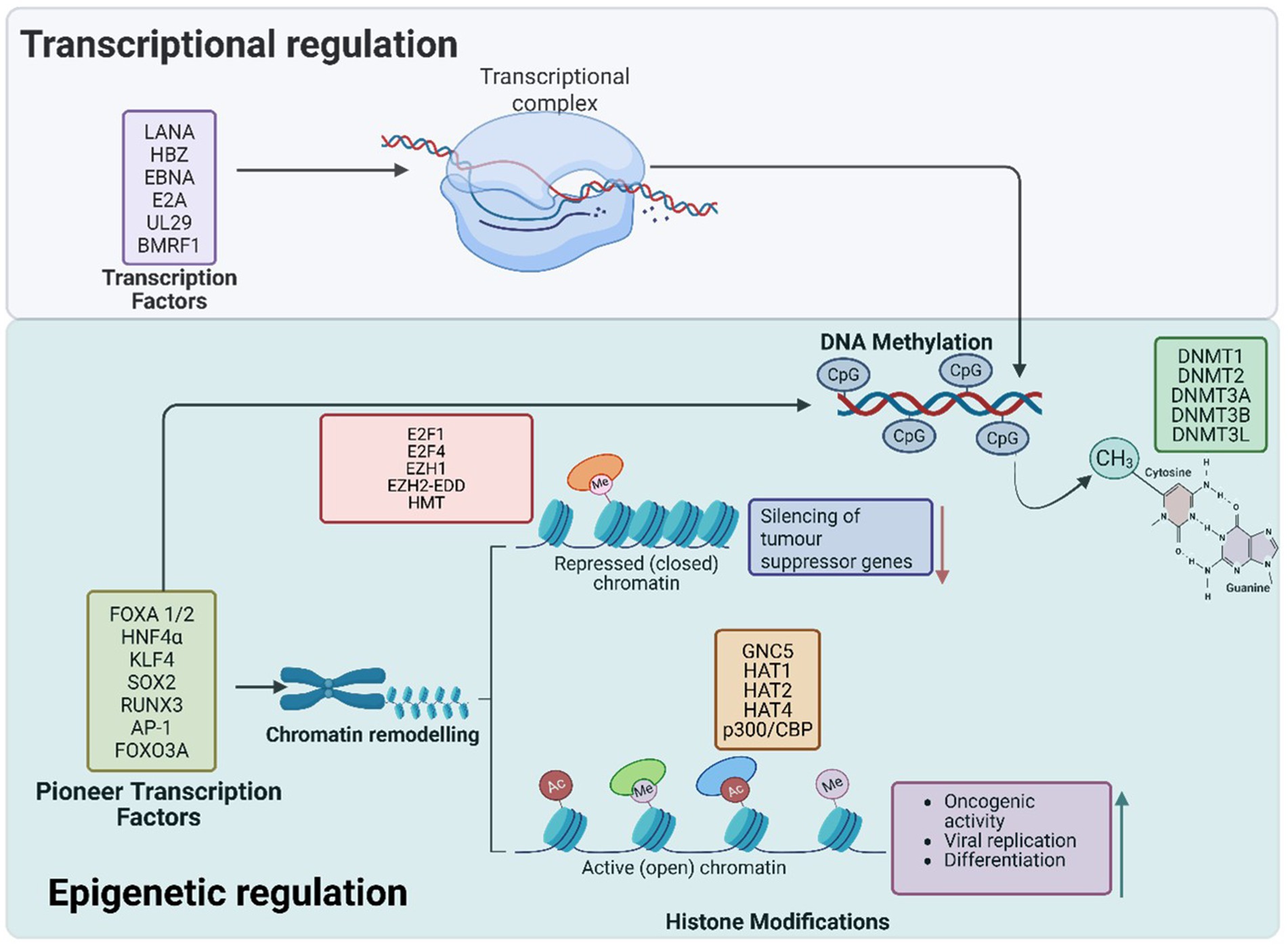
Figure 1. Pioneer transcription factors play a critical role in chromatin remodelling and epigenetic reprogramming during oncogenic transformation. They bind to closed or opened chromatin regions, facilitating the recruitment of chromatin remodelers and histone-modifying enzymes to establish active or repressive transcriptional states. Through histone acetylation and methylation, PTFs activate oncogenes such as TERT and CCNE1 while repressing tumor suppressor genes like p16, pRB, and PTEN. Additionally, DNA methylation at CpG sites further reinforces the silencing of tumor suppressor genes, promoting immune evasion and cellular transformation. Collectively, these epigenetic modifications disrupt normal cellular function, driving cancer progression. The transcription factors in the upper purple box are viral in origin.
DNA methylation and demethylation
DNA methylation, the addition of methyl groups to cytosine residues at CpG dinucleotides, is typically associated with transcriptional repression (Moore et al., 2013). This process is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT2, and DNMT3. Hypermethylation in promoter regions silences tumor suppressor genes, whereas hypomethylation activates oncogenes, promoting uncontrolled cell proliferation (Andreescu, 2024; Varley et al., 2013). Global DNA hypomethylation, common in aging and cancer, can also lead to genomic instability and activation of transposable elements (Varley et al., 2013). Therapeutically, DNMT inhibitors like azacitidine and decitabine are used therapeutically to reactivate silenced tumor suppressor genes.
Conversely, DNA demethylation is mediated by ten-eleven translocation (TET) enzymes—TET1, TET2, and TET3—which convert 5-methylcytosine to intermediate products that restore cytosine to its unmethylated state. TET enzymes are essential for embryonic development and stem cell differentiation (Ito et al., 2010; Ono et al., 2021; Tahiliani et al., 2009). Reduced TET activity can result in hypermethylation of tumor suppressor genes, contributing to cancer development (Thienpont et al., 2016; Rasmussen and Helin, 2016).
Histone methylation and polycomb repressive complexes
Histone methylation, orchestrated by histone methyltransferases (HMTs) and demethylases (HDMs), plays a key role in transcriptional regulation (Klose and Zhang, 2007). Certain methylation marks, such as H3K4me3 and H3K36me3, are linked to transcriptional activation, while others, such as H3K27me3 and H4K20me3, are associated with transcriptional repression (Ratner, 2021). The repressive mark H3K27me3, catalyzed by the polycomb repressive complex 2 (PRC2) via its Enhancer of Zeste Homolog 1 / 2 (EZH2/EZH1) subunits, is a critical signal for PRC activity. This mark recruits PRC1, which ubiquitinates histone H2A at lysine 119 (H2AK119Ub), leading to chromatin compaction and gene silencing (Martin and Moorehead, 2020). H3K27me3 is vital for repressing developmental genes, such as HOX genes, and for maintaining cell cycle regulation. Loss of this mark can result in differentiation defects, loss of stem cell identity, and cancer progression (Jones and Baylin, 2007).
Additionally, certain histone marks can counteract PRC-mediated repression. For example, genes with both H3K4me3 (an active mark) and H3K27me3 exist in a “bivalent” state in stem cells, allowing them to remain poised for either activation or repression. Other marks, such as H3K9me3, generally do not overlap with PRC activity, illustrating distinct silencing pathways. Additional histone modifications, such as phosphorylation, ubiquitination, and SUMOylation, influence chromatin dynamics, DNA repair, and gene expression (Ryu and Hochstrasser, 2021).
Hepatitis B virus and hepatocellular carcinoma
HBV virology
HBV is a partially double-stranded DNA virus belonging to the Hepadnaviridae family that primarily targets hepatocytes, causing hepatitis B (Krump and You, 2018; Karayiannis, 2017) It is recognized as a major oncogenic virus, contributing to hepatocellular carcinoma (HCC) through persistent infection, chronic inflammation, and epigenetic reprogramming (Karayiannis, 2017; Kgatle et al., 2017). HBV’s small, circular DNA genome (~3.2 kb) contains four overlapping open reading frames (ORFs) encoding essential viral proteins: the S gene (surface antigen, HBsAg), the C gene (core antigen, HBcAg, and HBeAg), the P gene (DNA polymerase), and the X gene (encoding HBx protein, which plays a key role in viral replication and oncogenesis) (Karayiannis, 2017; Robinson et al., 1974).
Unlike most DNA viruses, HBV replicates through an RNA intermediate, utilizing reverse transcription. Upon infection, the viral genome is converted into covalently closed circular DNA (cccDNA) in the nucleus, which serves as a template for transcription (Karayiannis, 2017). HBV is transmitted through blood, sexual contact, and perinatal routes from mother to child (di Filippo Villa and Navas, 2023).
Clinically, HBV infection can range from asymptomatic to acute hepatitis, presenting with symptoms such as jaundice, fatigue, and liver inflammation (Mistry and Yeoman, 2023). Chronic HBV infection significantly increases the risk of HCC development due to its persistent impact on host cellular and molecular mechanisms.
HBV integrates into the host genome, leading to long-term disruptions in chromatin accessibility and gene regulation (Péneau et al., 2022). HBV DNA integrates into active chromatin regions, where it modifies histone and DNA methylation marks to alter host gene expression (Péneau et al., 2022). These changes activate oncogenes and silence tumor suppressor genes, contributing to oncogenesis (Karayiannis, 2017; Kgatle et al., 2017). This capacity to regulate host epigenetics is central to HBV’s role in promoting hepatocyte transformation and the development of HCC (Péneau et al., 2022; Kgatle et al., 2017).
HBV-mediated regulation of PTFS
HBV utilizes PTFs to remodel chromatin, enhancing oncogenic pathway activation, sustaining viral replication, and altering hepatocyte identity as described in Figure 2. These changes contribute to chronic liver disease and the progression to hepatocellular carcinoma (HCC) (Levrero and Zucman-Rossi, 2016).
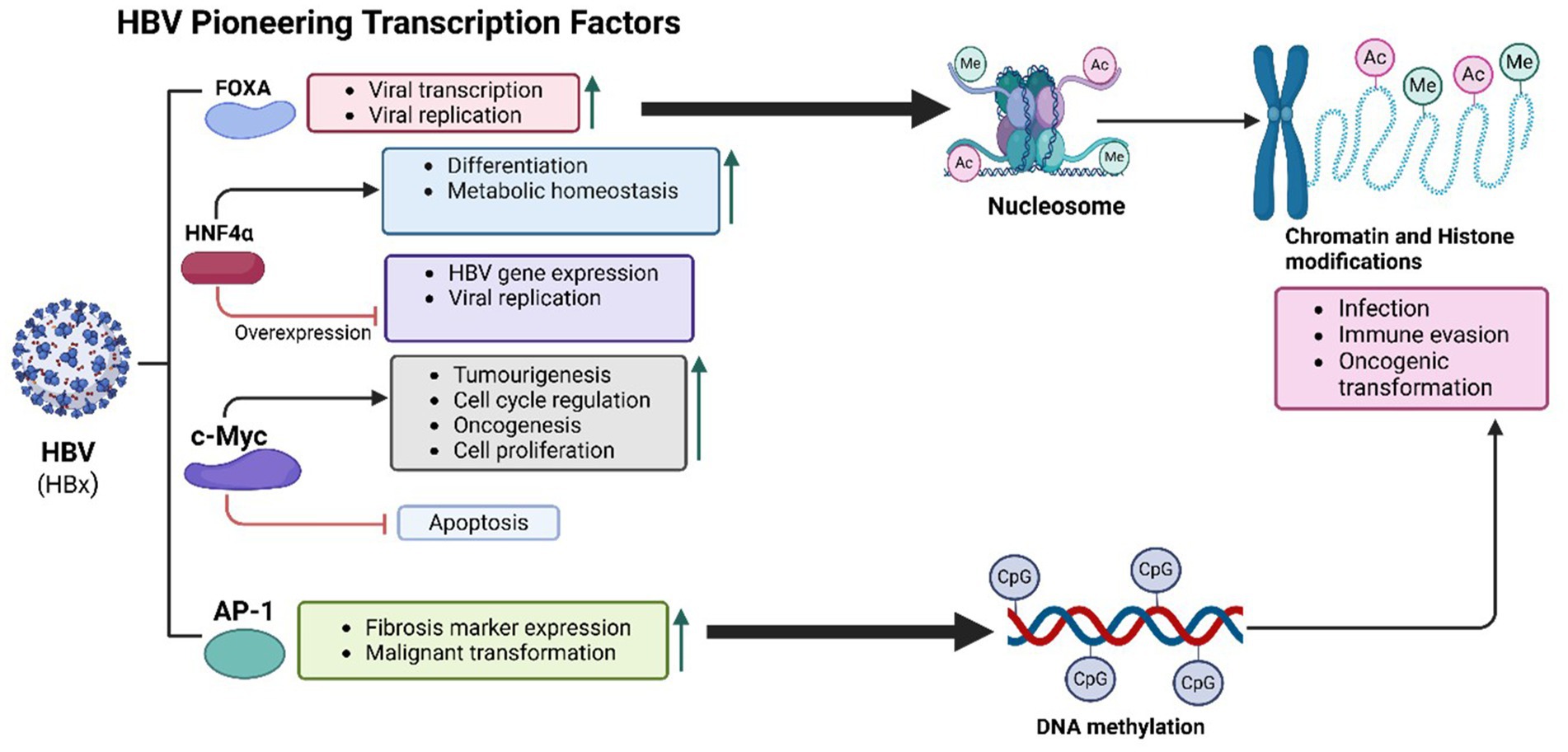
Figure 2. HBV-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. The HBx protein of HBV plays a central role in oncogenesis by recruiting histone-modifying enzymes to alter chromatin structure, enhancing histone acetylation and methylation. These modifications activate oncogenes and silence tumor suppressor genes. HBV integrates into actively transcribed chromatin regions, such as TERT and CCNE1 loci, disrupting cell cycle regulation and promoting tumor development. Interactions between HBx and transcription factors like FOXA1 and FOXA2 further increase chromatin accessibility and transcriptional activity, activating pro-oncogenic pathways and enabling immune evasion. Additionally, DNA methylation at CpG islands silences tumor suppressor genes, driving chronic liver disease and hepatocyte transformation.
Key liver-specific PTFs involved in HBV-mediated oncogenesis include the Forkhead box A (FoxA) family, particularly FoxA1 and FoxA2 (Chen et al., 2024). These transcription factors are essential for hepatic development and function, facilitating transcription by opening chromatin at liver-specific gene loci (Lee et al., 2005). HBV’s regulatory protein HBx interacts directly with FOXA1 and FOXA2, enhancing their chromatin-binding ability and transcriptional activity (Liu et al., 2023). This interaction activates pro-oncogenic pathways such as TGF-β, Wnt/β-catenin, and MYC, promoting liver fibrosis, immune evasion, and cell proliferation, which collectively advance HCC progression (Yan et al., 2024). Additionally, FOXA factors facilitate HBV transcription, sustaining viral replication and chronic infection. FOXA1 also targets PIK3R1, inhibiting the PI3K/Akt signalling pathway and thereby reducing HCC cell proliferation, migration, and invasion (He et al., 2017). The enrichment of FOXA1/2 binding sites in HBV-infected hepatocytes underscores their role in viral persistence and oncogenesis (Nevola et al., 2023).
Hepatocyte nuclear factor 4 alpha (HNF4α), another liver-specific PTF, is disrupted by HBV and implicated in liver cancer progression (Chen et al., 2024; Kotulkar et al., 2023). HNF4α regulates hepatocyte differentiation, metabolic homeostasis, and liver-specific gene expression, controlling genes involved in lipid metabolism, glucose regulation, and detoxification (Huck et al., 2021). Normally, HNF4α prevents oncogenic transformation by maintaining a differentiated hepatocyte state (Teeli et al., 2021). In HBV-infected hepatocytes, HNF4α competes with SOX9 to bind the EnhII/Cp region of the HBV genome, forming a feedback loop in viral replication (Yang et al., 2020). While overexpression of HNF4α reduces HBV replication by activating the NF-κB pathway and decreasing viral protein production, HBV disrupts HNF4α function, driving hepatocyte dedifferentiation and increased proliferation associated with early hepatocarcinogenesis (Teeli et al., 2021; Zhao et al., 2012).
Activator Protein-1 (AP-1), a transcription factor complex composed of c-Fos and c-Jun, is another target of HBV that enhances chromatin accessibility (Song et al., 2023). HBV infection sustains AP-1 activation, which is associated with increased expression of fibrosis markers and liver disease progression, ultimately heightening cancer risk. The interaction of HBx with AP-1 upregulates inflammatory and survival genes, exacerbating chronic liver inflammation and malignant transformation (Yang et al., 2019). Persistent AP-1 activity, mediated by signalling pathways involving SIRT1, Jab1, and JNK/c-Yun, amplifies HBV replication and fosters an environment conducive to HCC development (Park et al., 2020; Tanaka et al., 2025).
Beyond these transcription factors, HBV exploits inflammatory regulators such as STAT3 and NF-κB to sustain chronic immune activation and oncogenesis (Zakeri et al., 2024). Persistent activation of STAT3 and NF-κB, which are essential for cytokine signalling and immune responses, drives inflammation, fibrosis, and cirrhosis—major precursors to HCC (Zhao et al., 2020). HBV proteins, particularly HBc antigen, enhance chromatin binding of STAT3 and NF-κB, supporting the transcription of genes involved in cell survival, proliferation, and immune evasion (Jiang et al., 2021). Additionally, HBx-mediated activation of NF-κB increases inflammatory cytokines like IL-6 and TNF-α, perpetuating a pro-inflammatory environment conducive to hepatocarcinogenesis (Sivasudhan et al., 2022). Chronic NF-κB activation in HBV-related HCC is linked to poorer prognoses and increased tumor burden, underscoring the role of inflammation in HBV-driven oncogenesis (Sivasudhan et al., 2022).
Chromatin accessibility and epigenetic reprogramming in HBV-related HCC
HBV utilizes its ability to manipulate host chromatin structure as a pivotal strategy for viral replication and carcinogenesis (Levrero and Zucman-Rossi, 2016). Through alterations in chromatin accessibility and epigenetic regulation, HBV promotes persistent infection, immune evasion, and oncogenic transformation (Levrero and Zucman-Rossi, 2016).
HBV integrates its DNA into the host genome, predominantly targeting actively transcribed, open chromatin regions (Figure 2). These regions, associated with actively expressed genes, create a favourable environment for viral replication (Péneau et al., 2022). HBV DNA integration can profoundly affect host gene expression, leading to the activation or silencing of genes critical for cell proliferation, survival, and differentiation (Péneau et al., 2022). Certain genomic loci are preferentially targeted by HBV, particularly those linked to cancer-related genes, and are observed more frequently in tumours than in non-tumor liver tissues (Péneau et al., 2022).
Key hotspots for integration include telomerase reverse transcriptase (TERT), mixed lineage leukaemia 4 (MLL4), cyclin E1 (CCNE1), CCNA2, aryl-hydrocarbon receptor repressor (AHRR), and tumor protein p53 binding protein 1 (TP53BP1) (Péneau et al., 2022). Integration at the TERT locus, influenced by viral enhancers, drives overexpression of telomerase, contributing to cellular immortality, a hallmark of cancer cells (Péneau et al., 2022). Integration at the MLL4 locus disrupts transcription factors involved in cell differentiation, promoting oncogenesis (Dong et al., 2024). At the CCNE1 locus, integration induces cyclin E1 overexpression, enabling uncontrolled cell cycle progression—a critical step in carcinogenesis (Caldon and Musgrove, 2010). Integration at TP53BP1 impairs DNA damage response pathways, disrupting p53 tumor suppressor functions and driving genomic instability (Péneau et al., 2022). Clinically, HCC with a high number of HBV insertions is associated with younger patients and poorer prognoses (Péneau et al., 2022). These integration events often coincide with structural chromatin changes, including disrupted topology, gene fusions, enhancer hijacking, and abnormal oncogene activation.
HBV integration alters the three-dimensional chromatin architecture, leading to chimeric gene formation and enhancer hijacking, both of which contribute to aberrant transcription of oncogenes (Rosenkranz, 2023). Gene fusions arise from the combination of viral and host genes at integration sites, producing fusion proteins with oncogenic properties (Rosenkranz, 2023). HBV also displaces host enhancers, promoting inappropriate activation of genes associated with tumorigenesis (Levrero and Zucman-Rossi, 2016).
Epigenetic modifications, such as histone changes, are central to HBV’s manipulation of chromatin structure and gene expression (Bannister and Kouzarides, 2011). HBV infection induces specific histone acetylation and methylation changes that alter chromatin accessibility, driving transcription of both viral and host oncogenic genes (Hensel et al., 2017). The HBx protein plays a crucial role in this process, recruiting HATs like p300/CBP, which acetylate histones at lysine residues (H3K9ac and H3K27ac) to facilitate active transcription (Wang et al., 2013). Increased histone acetylation at promoter regions enhances transcription of genes critical for viral replication and oncogenesis.
HBV also activates the oncogene c-Myc, a key regulator of cell proliferation, through chromatin remodelling mechanisms (Jiang et al., 2021). The HBx protein activates c-Myc via the Ras/Raf/ERK1/2 pathway, upregulating the HSP90α promoter to enhance tumor cell invasion (Jiang et al., 2021). Additionally, HBx recruits HATs to oncogene promoters, increasing chromatin accessibility and driving the expression of cell cycle regulators necessary for uncontrolled proliferation (Haery et al., 2015). The widespread histone acetylation induced by HBx at oncogenic loci fosters hepatocyte proliferation and resistance to apoptosis (Rajan et al., 2020).
HBx-driven epigenetic reprogramming, particularly through activation of oncogenes like c-Myc, is integral to early liver tumor development (Rajan et al., 2020). HBx enhances c-Myc expression by promoting histone acetylation, increasing chromatin accessibility at oncogenic loci. Enrichment of H3K27ac, a marker of active enhancers, in HBV-infected liver cells drives transcription of genes involved in cell cycle regulation and survival (Andrisani, 2021). Conversely, HBx recruits histone methyltransferases like EZH2 and SUV39H1 to introduce repressive marks (H3K27me3 and H3K9me3), silencing key tumor suppressor genes (Yang et al., 2020; Chen et al., 2018). For example, EZH2-mediated repression of p16INK4A, Rb, and PTEN contributes to unchecked cell cycle progression and HBV-driven oncogenesis (Mui et al., 2017).
DNA methylation at CpG islands represents another epigenetic alteration linked to HBV infection. Elevated methylation levels at tumor suppressor gene promoters, including CDKN2A encoding p16, contribute to transcriptional silencing and hepatocarcinogenesis (Song et al., 2014; Wong et al., 2019). Hypermethylation of CDKN2A suppresses its role in regulating the G1-to-S phase cell cycle transition, enhancing cellular survival and tumor progression (Casciano et al., 2012). Similarly, methylation of SOCS1, which negatively regulates JAK–STAT signalling, heightens STAT3 activation, promoting cell proliferation and survival (Boosani and Agrawal, 2015). Other genes, such as APC, GSTP1, and RASSF1A, also undergo methylation-induced silencing, further driving HBV-associated HCC (Niller et al., 2012; Rongrui et al., 2014).
Hepatitis c virus and HCC
HCV virology
HCV is a hepatotropic, positive-sense, single-stranded RNA oncogenic virus classified within the Flaviviridae family and the Hepacivirus genus (Krump and You, 2018). It is a key etiological agent responsible for HCC, as its progressive infection, chronic liver inflammation, and cirrhosis, especially when untreated drive disease advancement (Krump and You, 2018; Virzì et al., 2018; Fiehn et al., 2024). The progression to HCC is attributed to persistent inflammation, viral protein expression, oxidative stress, and dysregulated signalling pathways, all contributing to genomic instability (Virzì et al., 2018). Approximately 80% of HCV infections become chronic, with 15–30% progressing to cirrhosis within two decades(Fiehn et al., 2024; Zhao et al., 2021). For patients with cirrhosis, the annual risk of developing HCC ranges between 1–4% (Fiehn et al., 2024; Pan et al., 2024; Khullar and Firpi, 2015). Globally, about 71 million people are infected with HCV, and 3–4 million new cases occur annually, highlighting its significant impact on public health (Pan et al., 2024).
HCV is a small, spherical virus approximately 50 nm in diameter. It is enveloped by a lipid membrane embedded with glycoproteins E1 and E2, which are critical for viral entry into host cells (Dearborn and Marcotrigiano, 2020). The HCV genome spans 9.6 kb and comprises a single open reading frame (ORF) flanked by untranslated regions (UTRs) at both ends (Romero-López and Berzal-Herranz, 2020). Upon infection, the ORF is translated into a large polyprotein (~3,000 amino acids), which is cleaved into structural proteins (Core, E1, E2, p7) and non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B) (Ashfaq et al., 2011; Dubuisson, 2007). These proteins play vital roles in viral replication, immune evasion, and pathogenesis (Ashfaq et al., 2011; Dubuisson, 2007).
The virus enters hepatocytes by binding to host cell receptors such as CD81, SR-B1, CLDN1, and OCLN, followed by internalization through clathrin-mediated endocytosis. Once inside the host cell, the viral genome is translated into a polyprotein, which is subsequently cleaved by viral and host proteases. Viral replication occurs on modified endoplasmic reticulum (ER) membranes, forming a specialized structure known as the membranous web (Sasvari and Nagy, 2010; Wolff et al., 2020). The RNA polymerase NS5B synthesizes a complementary negative-strand RNA, which serves as a template for producing new positive-strand RNAs (Sasvari and Nagy, 2010; Wolff et al., 2020). These RNAs are assembled into virions with core proteins and enveloped glycoproteins (E1 and E2) before being released through the host’s secretory pathway, often associated with very low-density lipoproteins (VLDL) (Vieyres et al., 2014).
HCV does not integrate its genome into the host DNA (Schinzari et al., 2015). However, it induces oncogenesis by altering chromatin accessibility and modulating epigenetic mechanisms via interactions with host PTFs and epigenetic modifiers (Morselli and Dieci, 2022). HCV proteins such as NS5A and Core are known to interact with histone-modifying enzymes, leading to changes in histone acetylation, DNA methylation, and gene expression (Żychowska et al., 2024).
HCV-mediated regulation of PTFS
HCV significantly contributes to oncogenesis by exploiting PTFs such as FoxA1, FoxA2, HNF4α, STAT3, NF-κB, and c-Myc (Żychowska et al., 2024). These PTFs play a crucial role in maintaining an open chromatin structure, enabling liver-specific gene expression. Through this epigenetic regulation, HCV promotes hepatic gene reprogramming, viral persistence, immune evasion, chronic inflammation, and oncogenesis. Furthermore, HCV disrupts the expression of regulatory RNA molecules such as microRNAs (e.g., miR-122) and long non-coding RNAs (e.g., MALAT1), modifying host transcriptional networks without the need for direct genome integration (Liu et al., 2024; Okushin et al., 2021).
HCV’s Core and NS5A proteins interact with FOXA1 and FOXA2, altering their normal functions and driving aberrant gene expression (Figure 3). This disruption leads to the upregulation of pro-oncogenic genes such as AFP and components of the TGF-β signalling pathway, driving processes like fibrosis, epithelial-mesenchymal transition (EMT), and tumor progression. By reshaping chromatin, HCV establishes a landscape favourable to viral replication while priming hepatocytes for malignant transformation, accelerating the progression of liver disease and promoting a tumor-supportive microenvironment (Żychowska et al., 2024).
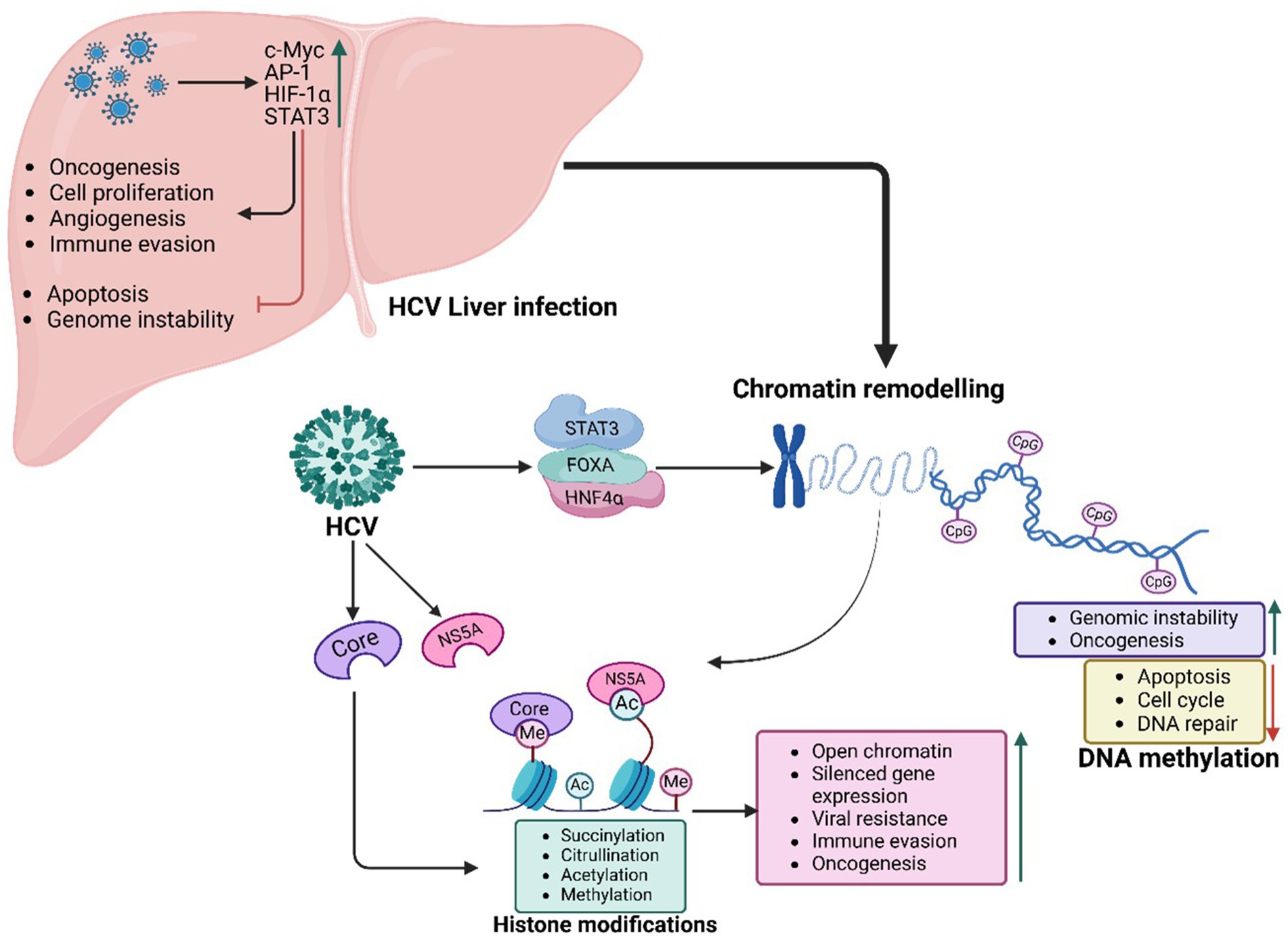
Figure 3. HCV-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. HCV proteins Core and NS5A drive HCC by exploiting PTFs like FOXA1, FOXA2, HNF4α, STAT3, NF-κB, and c-Myc and altering epigenetic mechanisms (e.g., H3K27ac marks), including histone modifications, DNA methylation, and chromatin remodelling. These changes, coupled with circadian disruptions, activate oncogenes, silence tumor suppressors, and sustain tumorigenic potential, even after viral clearance.
As observed with HBV, HNF4α acts as a central regulator of hepatocyte identity, differentiation, and metabolic homeostasis (Trauner and Halilbasic, 2011). In HCV-infected cells, the virus suppresses HNF4α expression and activity, reducing chromatin accessibility and driving hepatocyte dedifferentiation (Amicone and Marchetti, 2018). This reprogramming enhances vulnerability to oncogenic transformation while fostering metabolic disruptions, inflammation, and genomic instability, hallmarks of HCV-induced oncogenesis (Amicone and Marchetti, 2018). Continuous suppression of HNF4α activity during chronic HCV infection is associated with more aggressive tumor phenotypes and poorer clinical outcomes in HCC patients (Vallianou et al., 2016).
In response to HCV infection, oncogenic transcription factors such as c-Myc, AP-1, HIF-1α, and STAT3 become activated, promoting uncontrolled cell proliferation, angiogenesis, and immune evasion (Vishnoi et al., 2020). The viral protein NS5A plays a key role in activating c-Myc, recruiting HATs to specific gene loci. This epigenetic remodelling upregulates genes involved in cell cycle progression, ribosome biogenesis, and nucleotide metabolism, driving unchecked hepatocyte proliferation. NS5A also interacts with the AP-1 transcription factor complex, inducing chromatin remodelling at genes linked to inflammation and oncogenesis. Sustained activation of c-Myc and AP-1 disrupts regulatory networks, enhancing oncogenic transformation, apoptosis resistance, and genomic instability, thus accelerating HCC progression (Vishnoi et al., 2020).
Chronic inflammation is a hallmark of HCV pathogenesis and serves as a driving factor for liver fibrosis, cirrhosis, and tumorigenesis (Hoshida et al., 2014). Transcriptional regulators such as STAT3 and NF-κB play critical roles in shaping the chromatin landscape of immune response and survival genes (Kaszycki and Kim, 2025). Persistent activation of STAT3 and NF-κB during HCV infection promotes the expression of genes involved in cytokine signalling, anti-apoptotic pathways, and immune evasion (Virzì et al., 2018). These factors remodel chromatin at loci associated with fibrosis and survival, creating an epigenetic environment that expedites the progression from chronic liver disease to HCC (El Taghdouini et al., 2015).
Chromatin accessibility and epigenetic reprogramming in HCV-related HCC
HCV disrupts host pTF regulation, significantly altering the epigenetic landscape to enhance viral replication and drive hepatocyte transformation (Figure 3). These changes contribute to chronic liver disease, fibrosis, and HCC (Okushin et al., 2021; Yuan et al., 2025). Epigenetic reprogramming in HCV infection is largely mediated by histone modifications, which influence chromatin accessibility and gene expression (Hlady et al., 2022).
HCV proteins, particularly NS5A and Core, interfere with chromatin remodelling factors, causing widespread epigenetic reprogramming and abnormal gene expression (Çevik et al., 2017). These viral proteins recruit HATs like p300 and CBP, leading to increased acetylation of histone H3 and H4, which enhances chromatin accessibility at oncogenic loci (Gruber et al., 2019; Chen et al., 2022). Concurrently, HCV induces HDACs, resulting in hypoacetylation that silences tumor suppressor genes, fostering malignant transformation (Domovitz and Gal-Tanamy, 2021). Dysregulated histone methylation further exacerbates oncogenesis, as HMTs and HDMs are either recruited or inhibited by HCV, leading to silencing of tumor suppressor genes or activation of oncogenic pathways (Zeisel et al., 2021). Additionally, the virus alters nucleosome remodelling and histone variant deposition, reshaping chromatin to sustain viral replication while promoting genomic instability, inflammation, and uncontrolled cell proliferation, creating a carcinogenic microenvironment (Lieberman, 2016).
HCV-induced disruptions extend to chromatin remodelling complexes, essential for nucleosome positioning and gene regulation (Pietropaolo et al., 2021). The virus targets ATP-dependent chromatin remodelers, including the SWI/SNF and ISWI complexes, to manipulate host transcriptional programs, ensuring its persistence and oncogenesis (Pietropaolo et al., 2021). Disruption of these complexes silences tumor suppressor genes and activates pro-oncogenic pathways (Hu et al., 2021). For example, HCV’s interference with BRG1 and BRM components shifts gene expression toward pro-inflammatory and oncogenic states, amplifying NF-κB and STAT3 signalling, which promotes fibrosis, inflammation, and genomic instability, accelerating HCC progression (Tonon, 2016; Pfefferlé and Vallelian, 2024). Several analyses identified genes like MORF4L1, HDAC1, VPS72, and RUVBL2 as key ATP-dependent chromatin remodelling-related genes (ACRRGs) influencing HCC prognosis (Xu et al., 2021). Functional studies confirmed that MORF4L1 enhances cancer stemness through Hedgehog signalling, highlighting its role in tumor growth and metastasis (Xu et al., 2021).
Emerging research emphasizes the critical role of histone post-translational modifications including succinylation, citrullination, and acetylation in regulating chromatin architecture and influencing tumor growth, metastasis, and metabolic reprogramming (Wang et al., 2025). Genome-wide analyses reveal that HCV infection induces changes in histone marks, such as H3K4me3 and H3K9ac, which are linked to oncogenic pathways (Perez et al., 2019). Even after achieving a sustained virologic response (SVR) with direct-acting antivirals (DAAs), HCV leaves behind persistent epigenetic changes. These alterations, particularly in H3K27ac, remain beyond viral clearance and continue to drive oncogenic processes, contributing to the elevated risk of HCC post-SVR (Perez et al., 2019).
Studies also explore how HCV reshapes the 3D structure of the host genome through chromatin-organizing factors like CTCF and cohesin, which influence both viral and cellular genome configurations. These structural changes can promote a persistent pro-oncogenic epigenetic landscape even after viral clearance (Kim and Lieberman, 2024). Additionally, HCV infection disrupts the circadian regulation of gene expression in the liver, disturbing chromatin remodelling pathways and promoting a pro-tumorigenic environment (Mukherji et al., 2024). Such disruptions reveal how HCV exploits circadian mechanisms for chronic infection and oncogenic transformation.
Aberrant DNA methylation is another hallmark of HCV-induced epigenetic reprogramming. HCV proteins, particularly Core and NS5A, modulate DNMTs, resulting in hypermethylation of tumor suppressor gene promoters and hypomethylation of oncogenes. These changes promote genomic instability, silencing genes involved in cell cycle regulation, apoptosis, and DNA repair, while activating proto-oncogenes to enhance tumor progression (Cheng et al., 2021). Methylation patterns associated with HCV-induced cirrhosis and HCC serve as biomarkers for early detection and risk stratification. Distinct methylation profiles in HCV-driven HCC compared to HBV-driven HCC highlight virus-specific mechanisms underlying oncogenesis, offering opportunities for precision medicine approaches (Kim and Lieberman, 2024).
Human papillomavirus and cervical cancer
HPV virology
HPV, a circular, double-stranded DNA (dsDNA) virus from the Papillomaviridae family, is a small, non-enveloped pathogen primarily infecting epithelial tissues (Liu et al., 2021). Comprising around 52 genera, HPV is highly oncogenic and epitheliotropic, with a genome of approximately 8 kb. It is a major cause of cervical cancer, anogenital malignancies, specific head and neck squamous cell carcinomas (HNSCCs), and nasopharyngeal cancer (NPC), accounting for approximately 70% of cervical cancer cases. Its genome is divided into three regions: the early (E genes, including the oncogenes E6 and E7 essential for replication and oncogenesis), the late (L genes responsible for structural proteins), and the upstream regulatory region (URR) that governs transcription and replication. The long control region (LCR) harbours regulatory sequences essential for controlling viral gene expression (Kurvinen et al., 2000).
HPV is a sexually transmitted virus categorized into low, medium, and high-risk strains, with the International Agency for Research on Cancer (IARC) identifying 12 high-risk genotypes. Among its over 200 recognized types, low-risk strains like HPV-6 and HPV-11 are associated with benign conditions such as genital warts, whereas high-risk strains, including HPV-16 and HPV-18, are strongly linked to cervical, anogenital, and oropharyngeal cancers. High-risk strains significantly contribute to various malignancies, such as cervical, penile, and head and neck cancers (Kidd et al., 2017).
HPV infects basal epithelial cells through microabrasions in the skin or mucosa. Infections with high-risk HPV strains involve integration of the viral genome into the host DNA, which disrupts normal cell cycle regulation. Key viral proteins, E6 and E7, drive oncogenesis by deactivating tumor suppressors like p53 and pRb, resulting in unchecked cell proliferation and immune evasion (Yim and Park, 2005). Persistent high-risk HPV infections significantly increase the risk of cancer, causing nearly all cervical cancers, 90% of anal cancers, and substantial proportions of vaginal and oropharyngeal cancers (Egawa, 2023). The virus evades immune detection by downregulating antigen presentation and interfering with interferon signalling, thereby prolonging infections (Yim and Park, 2005).
Preventive strategies have been highly effective in reducing HPV-associated diseases. Vaccines such as Gardasil and Cervarix target high-risk HPV types, significantly decreasing cervical cancer rates (Cheng et al., 2020). Early detection methods, including Pap smears and HPV DNA testing, are essential for identifying precancerous lesions. Treatment options such as surgical removal, cryotherapy, and immune-modulating therapies help manage HPV-related conditions. However, the absence of specific antiviral therapies for HPV remains a limitation in combating the virus (Pathak et al., 2022).
HPV-mediated regulation of PTFS
HPV utilizes various host PTFs to regulate its oncogenic processes, transitioning from latent infection to active viral replication and contributing to malignancy (Figure 4). Among these, AP-1 plays a pivotal role by binding to LCR enhancer elements and activating early HPV genes, including E6 and E7 (Liu et al., 2002; Yee, 2013). AP-1 remodels chromatin by recruiting p300/CBP to induce H3K27 acetylation (H3K27ac), enhancing chromatin accessibility and enabling transcription of the E6/E7 promoters. This process sustains viral oncogene expression and promotes malignancy (Wang et al., 2025; Yee, 2013). Furthermore, AP-1 activates host oncogenes such as MYC and cyclin E/cyclin-dependent kinase (CDK), facilitating cell cycle progression and proliferation. Its persistent activity in HPV-driven cancers reinforces oncogenesis (McLaughlin-Drubin and Münger, 2009).
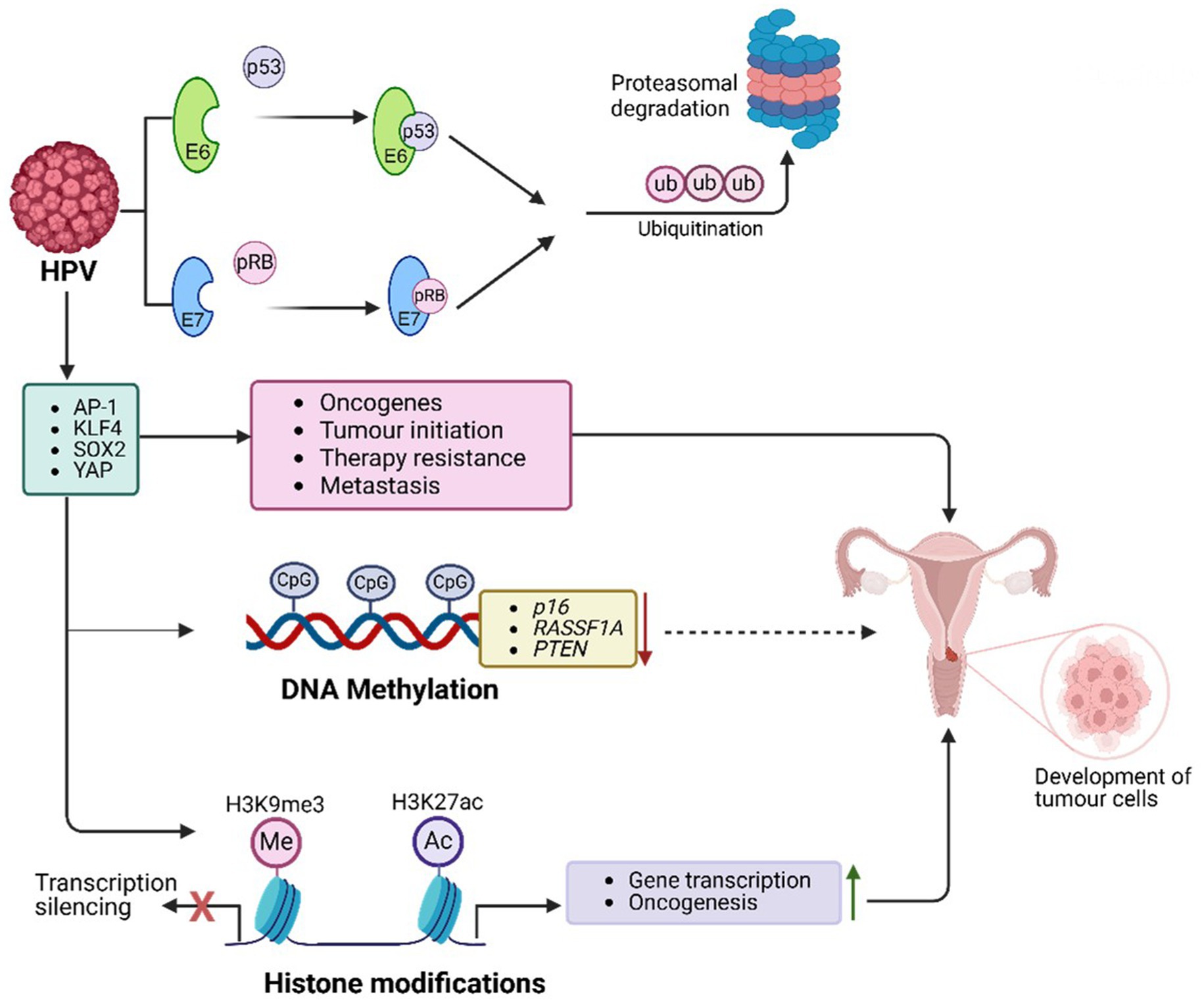
Figure 4. HPV-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. HPV drives cancer through epigenetic alterations. Viral proteins E6 and E7 modify host chromatin to activate oncogenes and silence tumor suppressor genes (e.g., p16, RASSF1A), disrupting cell cycle regulation. E7 degrades pRB, releasing E2F transcription factors, enhancing survival and proliferation genes (e.g., MYC, CDK2). HPV manipulates PTFs like AP-1, KLF4, YAP/TEAD, and SOX2 to sustain viral oncogene transcription, remodel chromatin, and drive malignancy. These changes, combined with chromatin remodelling, histone modifications (e.g., H3K27ac), and DNA methylation, lead to genomic instability and promote cervical, anogenital, and oropharyngeal cancers.
Krϋppel-like factor 4 (KLF4) also acts as an HPV PTF, enabling transcription by decondensing compacted chromatin (Tiwari et al., 2013; Yang and Zheng, 2012). In HPV-positive cancers, KLF4 promotes viral oncogene transcription by targeting silenced genomic regions, generating accessible chromatin structures (Gunasekharan et al., 2016). Upregulated through post-transcriptional and post-translational modifications, KLF4 affects specific target genes, including TCHHL1, VIM, ACTN1, and POT1, driving tumor growth by sustaining cell cycle progression, a proliferative state, and resistance to differentiation signals in HPV-infected epithelial cells (Gunasekharan et al., 2016). Overexpression of KLF4, mediated by E6 and E7, is crucial for HPV genome amplification and late gene expression. Silencing KLF4 using shRNAs disrupts these processes (Gunasekharan et al., 2016). Additionally, KLF4 protein collaborates with chromatin remodelers like SWI/SNF to enhance accessibility, reprogram endothelial enhancers, and establish transcriptional networks supporting oncogenic gene expression (Moonen et al., 2022). Enhancer-promoter loops involving KLF4 regulate genes like BMPR2, SMAD5, and DUSP5, stabilizing chromatin and sustaining E6/E7 oncogene expression (Gunasekharan et al., 2016).
The HPV E6/E7 oncoproteins also target Yes-associated protein 1 (YAP1) and large tumor suppressor kinases 1 and 2 (LATS1/2), key regulators of the Hippo signalling pathway (Blakely et al., 2024). By degrading YAP1 and LATS1/2, E6/E7 block YAP phosphorylation, promoting its nuclear translocation and interaction with TEAD transcription factors. The activated YAP/TEAD complexes increase chromatin accessibility, facilitating transcription of genes essential for cell survival, proliferation, and apoptosis resistance (Blakely et al., 2024; Zhao et al., 2023). This results in the upregulation of oncogenic targets such as CCND1, which drives the G1/S cell cycle transition, and BIRC5 (Survivin), an anti-apoptotic protein. Persistent YAP/TEAD activation in HPV-associated cancers underscores the Hippo pathway as a therapeutic target (Chiou et al., 2003; Montalto and De Amicis, 2020).
Another key factor, SOX2, is upregulated by HPV infection to maintain stemness and self-renewal. Elevated SOX2 expression in HPV-positive cancers contributes to tumor initiation, therapy resistance, and metastasis by promoting cancer stem-like phenotypes (Mamun et al., 2020). SOX2-positive cells show increased tumorigenic potential and resistance to differentiation cues, driving disease progression (Mamun et al., 2020). SOX2 interacts with chromatin modifiers like HATs and chromatin remodelers, facilitating epigenetic reprogramming in HPV-infected epithelial cells (Soto et al., 2017). Moreover, it collaborates with transcription factors like AP-1, KLF4, and TEAD/YAP to enhance chromatin accessibility at oncogene loci, further sustaining the malignant transformation (Mamun et al., 2020).
Overall, the above evidence underscores the interconnected roles of AP-1, KLF4, YAP1, and SOX2 in HPV-driven malignancies as displayed in Figure 4, demonstrating their contributions to chromatin remodelling, viral gene transcription, and oncogenic progression.
Chromatin accessibility and epigenetic reprogramming in HPV-related cervical cancer
HPV demonstrates its oncogenic potential by interacting with host chromatin, reprogramming chromatin architecture, and regulating gene transcription (Figure 4). These activities lead to cell cycle disruption and immune evasion (McLaughlin-Drubin and Münger, 2009; Doorbar et al., 2012). The virus relies on transcriptional regulators like P97 and P670 to manipulate host chromatin for viral replication and oncogenesis (Castro-Oropeza and Piña-Sánchez, 2022). Additionally, HPV may play a role in the metastasis of aggressive breast cancers by activating specific transcription factors (Ghoreshi et al., 2023). Despite lacking intrinsic chromatin-modifying enzymes, HPV reprograms host epigenetic machinery through episomal maintenance and genome integration, ultimately enhancing viral transcription and oncogenesis.
During infection, HPV targets the basal layer of stratified epithelia, maintaining its genome as a low-copy episome in basal cell nuclei (reviewed in 146). Episomal maintenance is essential for viral persistence and stable replication alongside host DNA during cell division. Viral proteins E1 and E2 facilitate replication and partitioning of the viral genome to daughter cells, enabling HPV to persist without immediate genome integration (McBride, 2017). However, the integration of viral DNA into the host genome represents a pivotal step in malignancy progression. This process disrupts normal chromatin architecture and gene regulation, potentially inactivating or aberrantly expressing host genes (Zhang et al., 2016). For instance, insertional mutagenesis may disable tumor suppressor genes, driving uncontrolled proliferation (Zhang et al., 2016). Furthermore, integration introduces binding sites for host transcription factors, reorganizing local chromatin structure and forming new topologically associating domains (TADs), which aberrantly regulate gene expression. These changes contribute to oncogenesis by activating oncogenes or repressing tumor suppressor genes (Zhang et al., 2016).
HPV preferentially integrates into common fragile sites (CFSs), genomic regions prone to breakage and instability under replication stress (Warburton et al., 2021). Such integration disrupts chromatin folding, promotes genomic instability, and fosters chromosomal rearrangements and mutations—hallmarks of cancer progression. While episomal HPV genomes maintain controlled viral gene expression (Durzynska et al., 2017), integration results in the loss of regulatory elements, leading to unchecked expression of E6 and E7 oncoproteins (Yeo-Teh et al., 2018). This dysregulation bypasses cellular checks, sustaining oncogene expression, promoting proliferation, and inhibiting tumor suppressors. Integration also induces DNA methylation at tumor suppressor gene promoters, silencing them while opening chromatin at oncogene loci, enhancing expression of genes involved in cell cycle progression and survival (Soto et al., 2017). These epigenetic changes create a permissive environment for cancer progression (Mac and Moody, 2020).
HPV oncoproteins E6 and E7 play central roles in epigenetic reprogramming by influencing chromatin remodelling, histone modifications, and DNA methylation (Sen et al., 2018). E6 promotes p53 degradation via ubiquitination mediated by E6-associated protein (E6AP), impairing p53-dependent chromatin repression and DNA repair pathways. This drives unchecked proliferation and survival. The viral genome forms a chromatin-like structure with host histones, subject to histone modifications. While H3K27ac enhances early gene transcription, promoting oncogenesis, H3K9me3 is linked to transcriptional silencing and latency (Mac and Moody, 2020). Chromatin remodelers like HATs and HDACs dynamically regulate these marks, controlling viral DNA accessibility (Mac and Moody, 2020). E6 of high-risk HPV further activates oncogenes like EGFR and c-MET by destabilizing histone demethylase KDM5C (Chen et al., 2018). Inhibition of Sp1 impacts active histone marks and HPV-host chromatin interactions, reducing oncogene expression and enhancing immune checkpoint gene expression (Cao et al., 2024).
E7 targets retinoblastoma protein (pRB) for proteasomal degradation, releasing E2F transcription factors and driving chromatin opening at cell cycle-related genes like CDK2, Cyclin A, and MYC (Yeo-Teh et al., 2018). This disrupts critical cell cycle checkpoints, enabling uncontrolled proliferation. E7 recruits p300/CBP histone acetyltransferases, catalysing H3K27ac, a hallmark of active enhancers, to drive the transcription of proliferation and survival genes (Wang et al., 2022). Additionally, E7 inhibits HDAC1, maintaining an open chromatin state and ensuring sustained oncogene expression (Wang et al., 2022).
E6 also promotes global DNA hypermethylation through DNMT1, silencing tumor suppressor genes such as p16, RASSF1A, and PTEN, while downregulating TET enzymes responsible for DNA demethylation. This leads to hypermethylated DNA, suppressing tumor suppressor genes and advancing carcinogenesis (Sen et al., 2018).
Human t-cell leukaemia virus type 1 and ATLL
HTLV-1 virology
HTLV-1, a member of the Retroviridae family, is a well-established causative agent of oncogenic and inflammatory diseases, most notably adult T-cell leukaemia/lymphoma (ATLL), a malignancy of CD4 + T cells (Eusebio-Ponce et al., 2019). ATLL is characterized by uncontrolled proliferation of infected CD4 + T cells, affecting organs such as the lymph nodes, liver, or spleen (Graham et al., 2014). It is categorized into four subtypes: acute, chronic, smouldering, and lymphoma, with the acute subtype being the most prevalent (Letafati et al., 2023). HTLV-1 has a 9 kb genome consisting of +ssRNA and encodes structural and enzymatic genes such as gag, pro, pol, and env (Azodi et al., 2017). HTLV-1 also expresses accessory genes, including the transactivator protein Tax, transcribed from the sense strand, which is critical for cellular transformation and transcriptional activation of the 5′ LTR promoter region (Enose-Akahata et al., 2017). In contrast, HBZ, encoded from the antisense strand, produces the HBZ protein, which counteracts Tax activities (Carcone et al., 2022).
Upon infecting host cells, HTLV-1 undergoes reverse transcription, converting its +ssRNA genome into dsDNA, which integrates into the host genome. The viral genome is flanked by long terminal repeats (LTR) at the 5′ and 3′ ends (Matsuoka and Mesnard, 2020; Martinez et al., 2019). These LTR regions, comprising U3, R, and U5 domains, regulate key processes like viral transcription, polyadenylation, and integration, ensuring efficient viral gene expression (Ernzen and Panfil, 2022).
HTLV-1-mediated T-cell malignancies, including lymphomas and leukaemia’s, result from dysregulated T-cell development and poor clinical outcomes (Andreescu, 2024). Epigenetic modifications, such as chromatin remodelling and alterations in miRNA activity, play a crucial role in transcriptional regulation of viral and host genes, including Tax and HBZ (Pietropaolo et al., 2021). These changes exploit host transcription factors such as NF-κB, AP-1, and STAT3, driving increased chromatin accessibility, tumorigenesis, immune evasion, and altered gene expression (Dong et al., 2024). The Tax protein activates oncogenes and inflammatory pathways, while HBZ represses tumor suppressor genes, ensuring continued immune evasion (Dong et al., 2024).
By reprogramming the host’s genetic and epigenetic landscapes, HTLV-1 creates conditions favourable for malignancy (Yamagishi et al., 2018). Emerging therapeutic approaches, including epigenetic regulators, NF-κB inhibitors, and immunotherapy, hold promise for managing ATLL and other HTLV-1-associated disorders (Dong et al., 2024).
HTLV-1-mediated regulation of PTFS
HTLV-1 employs PTFs to evade immune responses, promote T-cell transformation, and drive leukaemia progression (Figure 5). By manipulating host gene expression, HTLV-1 regulates its replication while avoiding immune detection (Mohanty and Harhaj, 2023). The viral protein HBZ facilitates immune evasion by leveraging the NF-κB signalling pathway. Concurrently, Tax, another key viral protein, contains nuclear localization (NLS) and nuclear export (NES) signals, enabling its movement between the nucleus and cytoplasm, where it activates NF-κB and drives viral gene expression (Mohanty and Harhaj, 2023). Mutations in Tax often render it undetectable in peripheral blood mononuclear cells (PBMCs) of ATLL-infected individuals, as Tax-expressing cells are highly immunogenic and targeted by cytotoxic T cells. Mutations in the HTLV-1 Tax protein can reduce its detectability in peripheral blood mononuclear cells (PBMCs) of individuals with ATLL. Tax-expressing cells are highly immunogenic and are actively targeted by cytotoxic T cells (Mohanty and Harhaj, 2023). Consequently, certain mutations may alter Tax’s structure or expression, enabling infected cells to evade immune surveillance and diminishing detectable Tax levels in PBMCs. However, wild-type Tax is generally considered highly immunogenic, while specific mutations can either maintain or reduce this immunogenicity depending on their impact on Tax function and recognition by the immune system (Mohanty and Harhaj, 2023). Additionally, Tax suppresses IRF3 activity by interacting with TBK1, which reduces antiviral IFN-1 responses. It also activates SOCS1 through NF-κB, leading to IRF3 degradation, dysregulated IFN-β secretion, and suppression of TLR4 signalling, further aiding viral immune evasion (Mohanty and Harhaj, 2023).
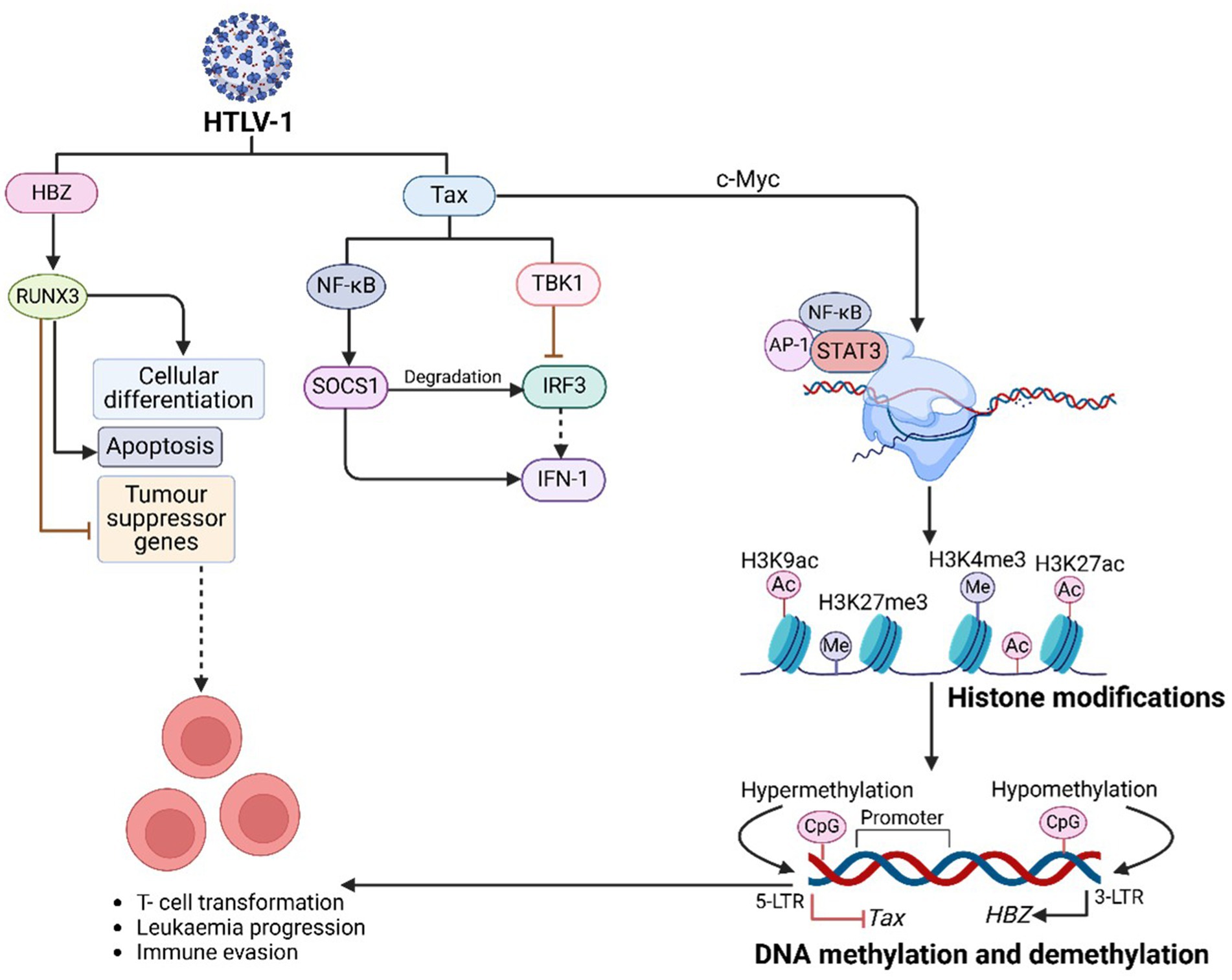
Figure 5. HTLV-1-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. HTLV-1 + ssRNA genome integrates into active euchromatin regions, leveraging LTRs for viral gene expression. HTLV-1 promotes oncogenesis through chromatin remodelling (through, e.g., SWI/SNF, CHD, INO80), histone modifications, and DNA methylation, driven by its Tax and HBZ proteins. Tax enhances inflammation and transcription, while HBZ silences tumor suppressor genes, enabling malignant progression. Targeting these pathways with epigenetic therapies offers potential treatment for ATLL.
HBZ binds to RUNX3, a runt-related transcription factor that regulates apoptosis and differentiation. By displacing RUNX3 from tumor suppressor gene promoters, HBZ ensures tumor suppressor gene inactivation. Mutations or abnormal expression of RUNX3 are strongly associated with cancer development, highlighting the oncogenic potential of HBZ (Kulkarni et al., 2018).
The Tax protein activates c-Myc, a proto-oncogene that regulates transcription of cancer-related genes. This activation results in histone acetylation at oncogenic promoters such as CCND1 and E2F1 (Wallbillich and Lu, 2023). c-Myc interacts with key signalling pathways, including Wnt/β-catenin, JAK/STAT, MAPK, and NF-κB. By recruiting HATs like p300/CBP and TIP60, c-Myc enhances chromatin accessibility at super-enhancers, promoting aggressive malignancies such as ATLL (Sundeep et al., n.d.). Dysregulated bromodomain and extraterminal domain (BET) activity, involving BRD4, further amplifies oncogenic transcription through H3K122 acetylation. BET and HAT inhibitors have shown potential in targeting these pathways to combat c-Myc-driven leukemogenesis (Table 1) (Verbeke et al., 2025).
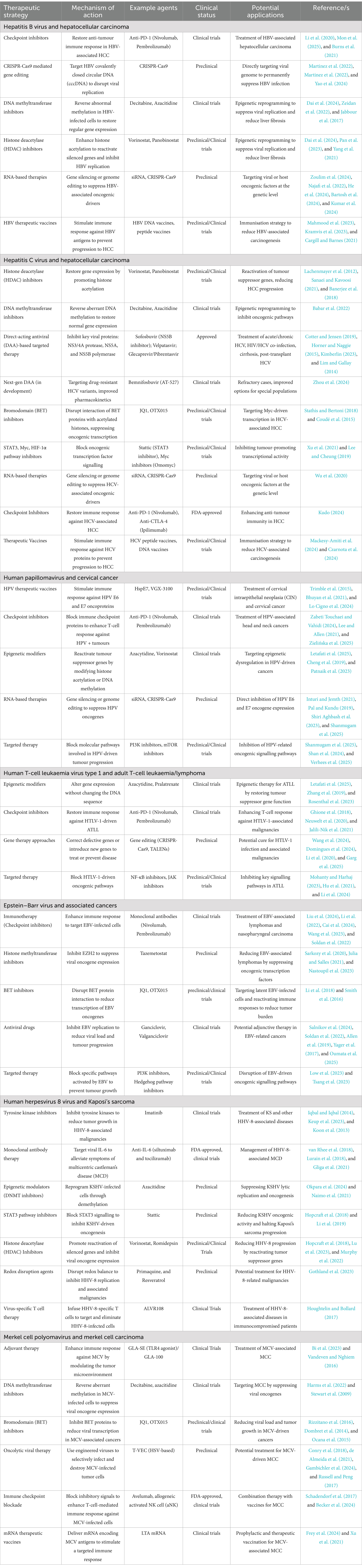
Table 1. Emerging therapeutic strategies targeting epigenetic and transcriptional dysregulation in oncogenic viruses.
Tax induces the AP-1 transcription factor complex (c-Fos and c-Jun), remodelling chromatin at genes involved in T-cell activation. This activation promotes the expression of oncogenic genes such as IL-2Rα (CD25), a marker of ATLL proliferation (Ahmadi Ghezeldasht et al., 2023). Tax-dependent AP-1 activation drives the expression of cytokines like IL-2, IL-8, and TNF-α, contributing to deregulated phenotypes in HTLV-1-infected T cells (Gazon et al., 2017). Tax also interacts with c-Jun NH2-terminal kinases (JNKs), modulating cell proliferation and apoptosis, thereby influencing viral replication and transformation. JNK inhibitors have demonstrated potential in reducing HTLV-1-driven T-cell activation, highlighting the therapeutic significance of this pathway (Bangham and Ratner, 2015).
Tax recruits STAT3 to super-enhancers, increasing chromatin accessibility at loci such as BCL-xL, Myc, and IL-21, driving tumor progression (Jhan and Andrechek, 2016). Super-enhancers play a pivotal role in activating oncogenic gene expression. Meanwhile, HBZ inhibits STAT5, blocking apoptosis and promoting immune escape. It also represses NF-κB activity and innate immune responses (Mohanty and Harhaj, 2023; Enose-Akahata et al., 2017). By suppressing IFN-β production through the inhibition of IRF3 activation, HBZ prevents apoptosis by downregulating pro-apoptotic genes (Chattopadhyay et al., 2011).
Super-enhancers are critical in regulating HTLV-1 transcription. Transcription factors such as CREB/ATF and AP-1 interact with Tax-responsive elements (TREs), driving viral gene expression (Pluta et al., 2020; Giam and Semmes, 2016) These mechanisms enhance cell survival, proliferation, and transformation (Giam and Semmes, 2016; Bellon et al., 2024). Activated STAT3, stimulated by cytokines or growth factors, promotes cell differentiation, proliferation, and resistance to apoptosis, further contributing to malignancy (Wang et al., 2022). Targeting STAT3 with inhibitors like tofacitinib offers a promising therapeutic strategy for suppressing oncogenic pathways driving ATLL (Adesoye et al., 2024).
Chromatin accessibility and epigenetic reprogramming in HTLV-1 and ATLL
HTLV-1 leverages epigenetic mechanisms to alter host chromatin structure, ensuring persistent infection and promoting oncogenesis (Figure 5). These changes influence gene expression, contributing to the development of ATLL (Mizuike et al., 2025). The virus integrates into transcriptionally active euchromatin regions, often targeting genes associated with cellular growth and survival. This integration facilitates viral persistence and cellular transformation. In infected CD4 + T cells, HTLV-1 replicates through either proviral DNA duplication or transcription into mRNA, enabling the production of viral proteins. The viral transactivator protein Tax plays a critical role in chromatin remodelling, affecting gene expression and RNA splicing (Kim and Lieberman, 2024). HTLV-1 integration frequently occurs near transcription start sites and interacts with key transcription factors like STAT1 and p53, as well as enzymes such as PP2A and BRG1, which regulate transcription at these loci (Ahuja et al., 2014). The viral proteins Tax and HBZ are central to the progression of ATLL by regulating cell signalling and gene expression (Akbarin et al., 2024). Tax activates transcription from methylated HTLV-1 LTRs by interacting with MBD2. Histone modifications associated with transcriptional activation, such as H3K4me3, H3K9ac, and H3K27ac, are frequently observed, while the reprogramming of H3K27me3 by PCR3 disrupts gene regulation and drives ATLL progression. Epigenetic co-activators like EP300 and CBP enhance the expression of genes involved in DNA repair, apoptosis, and proliferation. Concurrently, these epigenetic changes silence tumor suppressor genes, undermining genomic stability (Letafati et al., 2025).
Tax constitutively activates NF-κB, driving chromatin remodelling at inflammatory and survival gene loci. By degrading IκBα, Tax enables NF-κB to regulate genes such as IL-2, IL-9, and BCL-xL, promoting cell proliferation and apoptosis resistance (Bellon et al., 2024). Additionally, Tax recruits BRD4 to sustain NF-κB transcriptional activity, facilitating viral replication and malignancy. This process operates through both canonical pathways, involving phosphorylation of the IKK complex, and non-canonical pathways, mediated by IKKα-dependent processing of p100 to p52 (Su et al., 2024). Targeting these pathways with NF-κB inhibitors shows potential for reversing Tax-mediated malignancies (Hleihel et al., 2023).
HTLV-1 recruits chromatin remodelers such as SWI/SNF, CHD, and INO80 to restructure chromatin. The Tax protein interacts with histone-modifying enzymes like HATs and HDACs, recruiting p300/CBP to enhance histone acetylation at H3K9ac and H3K27ac marks, driving transcriptional activation. Conversely, Tax associates with transcriptional repressors like SIRT1 and methyltransferases such as SUV39H1 and SMYD3, influencing transcription through methylation at H3K4 and H3K27 sites (Ratner, 2021; Mizuike et al., 2025). Loss of RUNX3 function also induces chromatin repression through marks like H3K27me3, silencing key apoptosis-related genes (Kulkarni et al., 2018).
Distinct methylation patterns are observed in HTLV-1-infected cells. The 5′ LTR of the proviral genome is hypermethylated, silencing the Tax gene, while the 3′ LTR is hypomethylated, sustaining HBZ gene expression. These modifications help HTLV-1 evade immune responses and establish latency. Treatments with hypomethylating agents, such as 5-azacytidine, can reactivate viral gene transcription in latently infected cells, emphasizing the regulatory role of DNA methylation in HTLV-1 persistence (Pietropaolo et al., 2021; Mizuike et al., 2025).
Aberrant DNA methylation in ATLL cells affects several host genes critical for cellular function (Geissler et al., 2024). Hypermethylation silences tumor suppressor genes like CDKN2A and BMP6, leading to unchecked proliferation and altered signalling (Geissler et al., 2024; Wajed et al., 2001). Similarly, genes such as KLF4 and EGR3, crucial for cell cycle regulation and apoptosis, are silenced, enabling resistance to activation-induced cell death (Pietropaolo et al., 2021). Hypermethylation also impacts zinc finger transcription factors and MHC class I proteins, allowing ATLL cells to evade immune detection(Letafati et al., 2025; Paixão et al., 2006). Clinically, the extent of promoter-associated CpG island hypermethylation is correlated with poor prognosis in ATLL patients (Paixão et al., 2006). Preclinical studies suggest that hypomethylating agents could target these epigenetic changes, offering therapeutic potential (Letafati et al., 2025).
Epstein–BARR virus and cancer
EBV virology
EBV, also known as human herpesvirus 4 (HHV-4), is a widespread lymphotropic gamma-herpesvirus with a biphasic lifecycle comprising both lytic and latent phases (Cao et al., 2024). This double-stranded linear DNA virus is primarily transmitted through saliva, where it establishes replicative infections in the oropharynx and a lifelong latent presence in oral epithelial cells and B cells (Zhang et al., 2024; Dasari et al., 2023; Parija, 2023). EBV’s persistent infection, often asymptomatic, has oncogenic potential and is associated with several malignancies, including nasopharyngeal carcinoma, EBV-associated gastric carcinoma, Burkitt lymphoma, and Hodgkin lymphoma, particularly in immunosuppressed individuals (Zhou et al., 2025; Looi et al., 2021; Su et al., 2023).
The oncogenic capacity of EBV arises from its large genome, which encodes numerous viral genes capable of manipulating the cellular environment. These genes co-opt PTFs, alter host epigenetic machinery, and influence key oncogenic pathways, producing medically significant viral strains that drive cancer development (Wen et al., 2022; Das and Kundu, 2025).
EBV infects more than 95% of the global adult population, establishing a lifelong latent infection characterized by periodic reactivation and viral shedding (Murata et al., 2021; Damania et al., 2022). The virus demonstrates remarkable adaptability in its gene expression programs, transitioning between lytic replication and multiple latency types. Each latency form is defined by the expression of specific viral genes including Epstein–Barr nuclear antigens (EBNAs, EBNA1-2, 3A-3C) and latent membrane proteins (LMPs), which are crucial for maintaining the viral genome and modulating host immune responses. These programs are tightly regulated by epigenetic modifications and the activity of recruited transcription factors, enabling EBV to adapt to diverse cellular environments while promoting oncogenesis (Kong and Giulino-Roth, 2024).
Epigenetic changes, such as chromatin remodelling and transcriptional reprogramming, play pivotal roles in the development of EBV-associated malignancies. These modifications are key to understanding the mechanisms by which EBV contributes to cancer progression, illustrating how the virus shapes host gene expression to support its lifecycle and oncogenic strategies.
EBV-mediated regulation of PTFS
EBV manipulates host PTFs to remodel chromatin and sustain oncogenic programs (Figure 6). A central player in this process is the EBV immediate-early protein BZLF1 (also known as Zta or ZEBRA), which functions as a viral pioneer factor facilitating the transition from latency to the lytic cycle (Bernaudat et al., 2022). BZLF1 interacts with chromatin remodelling complexes such as BRG1-containing SWI/SNF and HATs, enabling chromatin opening at viral promoters and initiating lytic reactivation (Asha and Sharma-Walia, 2021). Additionally, BZLF1 counteracts repression mediated by PRC2, activating the transcription of viral genes essential for the lytic phase of infection (Asha and Sharma-Walia, 2021).
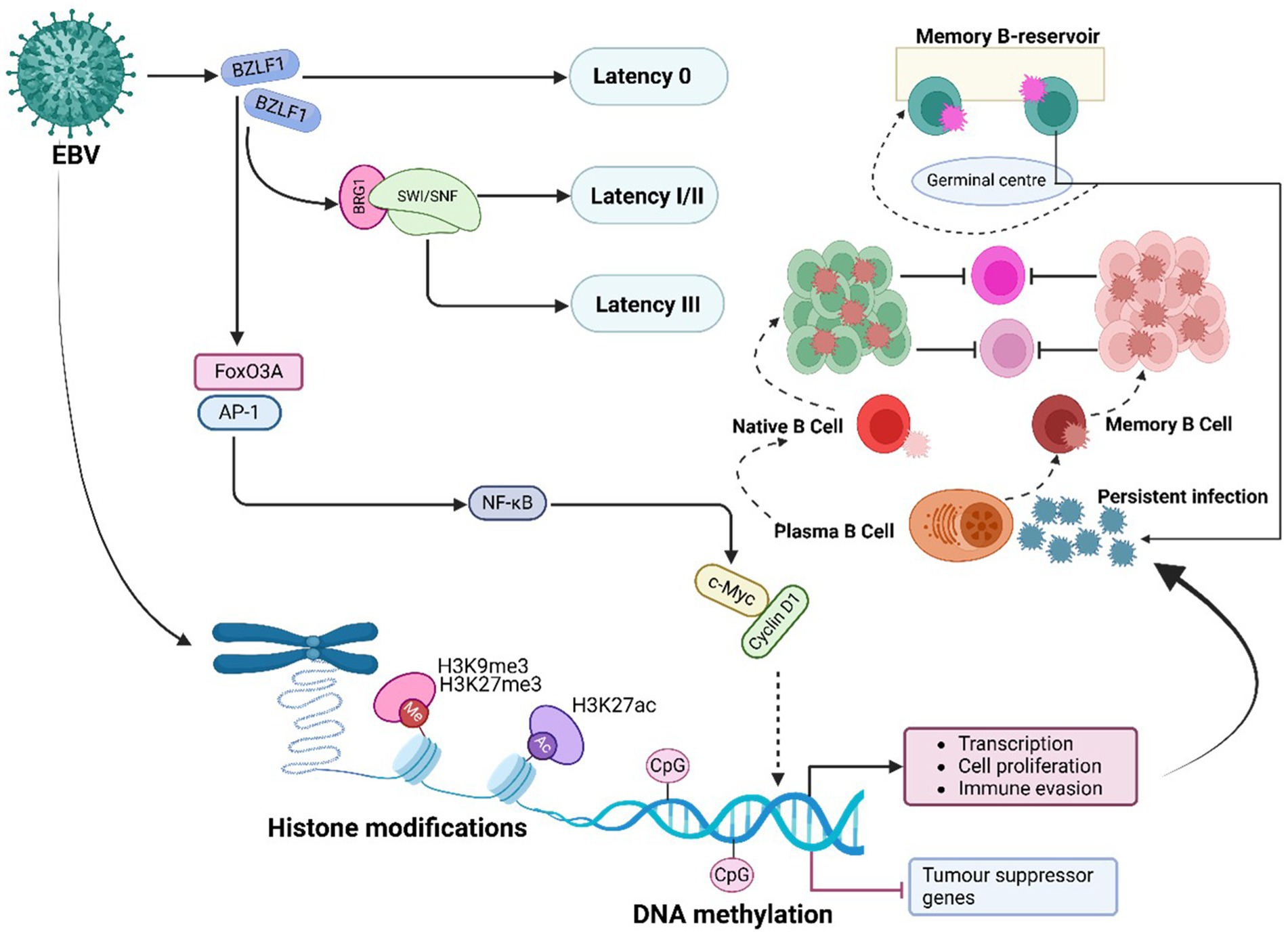
Figure 6. EBV-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. EBV alters host and viral chromatin through histone (e.g., H3K9me3, H3K27me3) and DNA modifications, silencing tumor suppressor genes (e.g., p16, RASSF1A, CDH1, PTEN) and activating oncogenic pathways. These changes enhance immune evasion, cell proliferation, and malignant transformation, contributing to cancer development.
Another crucial pTF affected by EBV is FoxO3A, a tumor suppressor that is downregulated in EBV-infected cells. This downregulation disrupts chromatin accessibility at pro-apoptotic gene loci, reducing apoptosis and enhancing B cell survival (Neugebauer et al., 2023; Huang et al., 2021). Loss of FoxO3A function significantly contributes to the development of lymphomas. Similarly, the viral protein LMP1 persistently activates NF-κB signalling in EBV-associated Hodgkin lymphoma, promoting chromatin opening at loci encoding inflammatory cytokines like IL-6 and anti-apoptotic proteins like BCL-2. This activation supports the survival and proliferation of EBV-infected cells (Banerjee et al., 2024; Chakravorty et al., 2022).
In addition to targeting FoxO3A and NF-κB, EBV exploits the AP-1 transcription factor complex (FOS/JUN dimer) to drive oncogenic transcriptional programs (Song et al., 2023). The viral protein LMP1 activates AP-1, which recruits BRD4, a bromodomain protein that amplifies the transcription of oncogenic factors such as c-Myc and CCND1. In Burkitt lymphoma, EBNA2 works in conjunction with p300, a histone acetyltransferase, to acetylate chromatin at the c-Myc locus, enhancing cellular proliferation and contributing to oncogenesis (Kasprzyk et al., 2021).
Chromatin accessibility in EBV infection and cancer development
EBV undergoes chromatinization upon entering the nucleus (Figure 6). This process involves extensive histone modifications and DNA methylation, which regulate viral gene expression and establish distinct latency programs: Latency I, II, and III (Scott, 2017). Each latency phase is defined by specific chromatin landscapes and is associated with malignancies (Ho et al., 2023). In Latency I, which is typically observed in Burkitt lymphoma, most viral genes are silenced through repressive histone modifications such as H3K9me3 and H3K27me3, alongside DNA methylation (Chen et al., 2024; Shareena and Kumar, 2023). By contrast, Latency II and III, seen in Hodgkin lymphoma and NPC, involve the reactivation of oncogenic viral genes like LMP1 and LMP2A via chromatin-opening mechanisms (Mesri et al., 2014; Zhou et al., 2025). Transitions between these latency programs are tightly regulated by epigenetic factors (Kong and Giulino-Roth, 2024).
Epigenetic modifications are central to EBV’s capacity to manipulate host chromatin, ensuring viral persistence and promoting oncogenic transformation (Dai et al., 2021; Soliman et al., 2021; Zhou et al., 2025). By dynamically altering chromatin accessibility, EBV maintains latency, evades immune responses, drives uncontrolled cellular proliferation, and suppresses tumor suppressor gene transcription (Zhou et al., 2025). For instance, the viral protein LMP1 recruits HATs, such as p300/CBP, to enhance H3K27ac, activating genes involved in cell proliferation and immune evasion (Dai et al., 2021; Soliman et al., 2021; Zhou et al., 2025). Additionally, EBNA3C interacts with histone methyltransferases like EZH2, a component of PRC2, depositing H3K27me3 at tumor suppressor loci. This modification silences genes such as p16 and RASSF1A, contributing to cancer development (Banerjee et al., 2024).
In B cells, EBV infection removes repressive histone marks, including H3K9me3, H3K27me3, and H4K20me3, thereby enhancing chromatin accessibility and regulating genes linked to the cell cycle and apoptosis (Zhou et al., 2025). Viral protein-induced histone modifications destabilize normal cellular processes, facilitating malignant transformation. These changes, affecting both the viral and host genomes, are essential for establishing and maintaining latency while driving cellular transformation (Zhou et al., 2025; Niller et al., 2016).
Both viral and host DNA methylation play crucial roles in the development of EBV-associated cancers. During latent infection, the EBV genome undergoes extensive methylation to regulate viral gene expression. This widespread CpG island methylation, known as the CpG island methylator phenotype (CIMP), is frequently observed in EBV-associated cancers, such as gastric carcinoma and NPC (Salnikov et al., 2024; Leong and Lung, 2021). The viral protein BZLF1 preferentially binds methylated CpG motifs in key viral promoters, facilitating the transition from latency to lytic infection (Zhang et al., 2022). This binding bypasses repressed chromatin states without requiring active DNA demethylation, promoting efficient lytic viral gene expression and ensuring persistence and oncogenic potential (Zhang et al., 2022).
EBV also induces widespread aberrant DNA methylation in the host genome, silencing tumor suppressor genes and advancing tumor progression. Latent membrane proteins (LMP1 and LMP2A) upregulate DNMT1, DNMT3A, and DNMT3B, resulting in global DNA hypermethylation and repression of tumor suppressor genes (Kuss-Duerkop et al., 2018). Key tumor suppressor genes silenced through EBV-induced hypermethylation include CDH1, which disrupts cell adhesion and promotes invasion and metastasis, and RASSF10, which suppresses apoptosis and cell proliferation (Pietropaolo et al., 2021). Moreover, LMP2A activates the STAT3 pathway to enhance DNMT1 activity, leading to promoter hypermethylation of PTEN, thereby driving uncontrolled cell proliferation and survival (Pietropaolo et al., 2021). Hypermethylation of genes such as CDH1 and RASSF10 may serve as biomarkers for early detection of EBV-associated cancers. Detecting these methylation changes in tissue biopsies or circulating tumor DNA can aid early diagnosis and improve prognosis assessments.
Human herpesvirus 8
HHV-8 virology
Human herpesvirus 8 (HHV-8), also referred to as Kaposi’s sarcoma-associated herpesvirus (KSHV), belongs to the Rhadinovirus genus and Gammaherpesvirinae subfamily within the herpesvirus family (Gessain, 2008; Sarid et al., 2020). It is a recognized causative agent of Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (MCD), conditions that are particularly prevalent among immunocompromised individuals, such as those living with HIV/AIDS (Sarid et al., 2020; Chan et al., 2000; Pei et al., 2020). HHV-8 drives cancer development through intricate interactions with the host’s cellular machinery, involving the regulation of transcription factors, chromatin remodelling, and epigenetic reprogramming (Pei et al., 2020).
HHV-8 is predominantly transmitted through saliva, though other routes include sexual contact, blood transfusion, organ transplantation, and, less commonly, mother-to-child transmission (Tan and Pinsky, 2017; Jenkins et al., 2002). The virus infects various cell types, such as endothelial cells, B-cells, monocytes, and epithelial cells, enabling angiogenesis and immune evasion (Knowlton et al., 2013). Individuals with weakened immune systems, including those with HIV/AIDS or transplant recipients, are especially vulnerable to cancers associated with HHV-8 (Sunil et al., 2010).
Structurally, HHV-8 features an enveloped, icosahedral capsid containing a double-stranded DNA genome measuring 140–170 kilobases (Bai et al., 2024). Tegument proteins within the capsid play crucial roles in the virus’s functionality; Its genome is composed of conserved genes responsible for replication, latency, and structural integrity, alongside unique genes that facilitate immune evasion, angiogenesis, and cell proliferation (Bai et al., 2024). Key genes include ORF50 (transcription activator, RTA), which drives reactivation from latency; LANA (ORF73), which aids in latency maintenance and apoptosis inhibition; vIL-6, a cytokine linked to inflammation; and vGPCR, which promotes angiogenesis and cell growth (Losay and Damania, 2025).
The replication cycle of HHV-8 occurs in two phases. During the latent phase, the viral genome exists in the nucleus as circular DNA, with limited gene expression (e.g., LANA, vCyclin) aimed at suppressing apoptosis and evading immune detection (Losay and Damania, 2025). This phase is vital for the virus’s long-term persistence and potential to cause tumours (Losay and Damania, 2025). The lytic phase, triggered by environmental stressors like hypoxia or immune suppression, is characterized by the reactivation of the virus mediated by ORF50 (RTA) (Sarid et al., 2020; Aneja and Yuan, 2017). This leads to the production of new virus particles, cell destruction, and further spread of the infection (Aneja and Yuan, 2017).
HHV-mediated regulation of PTFS
HHV-8-induced oncogenesis involves a complex network of molecular pathways that drive cell survival, proliferation, and immune evasion (Figure 7). A central player in this process is the NF-κB signalling pathway, which is activated by viral FLICE inhibitory protein (vFLIP). An et al., (An et al., 2003) demonstrated that vFLIP stimulates NF-κB signalling, leading to increased cell proliferation and suppression of apoptosis. This promotes KS tumorigenesis through unchecked cell growth and resistance to programmed cell death (An et al., 2003). Kang et al., (Kang et al., 2008) further revealed that NF-κB activation supports inflammatory responses and angiogenesis, critical processes for the progression of KS and primary effusion lymphoma (PEL) (Kang et al., 2008).
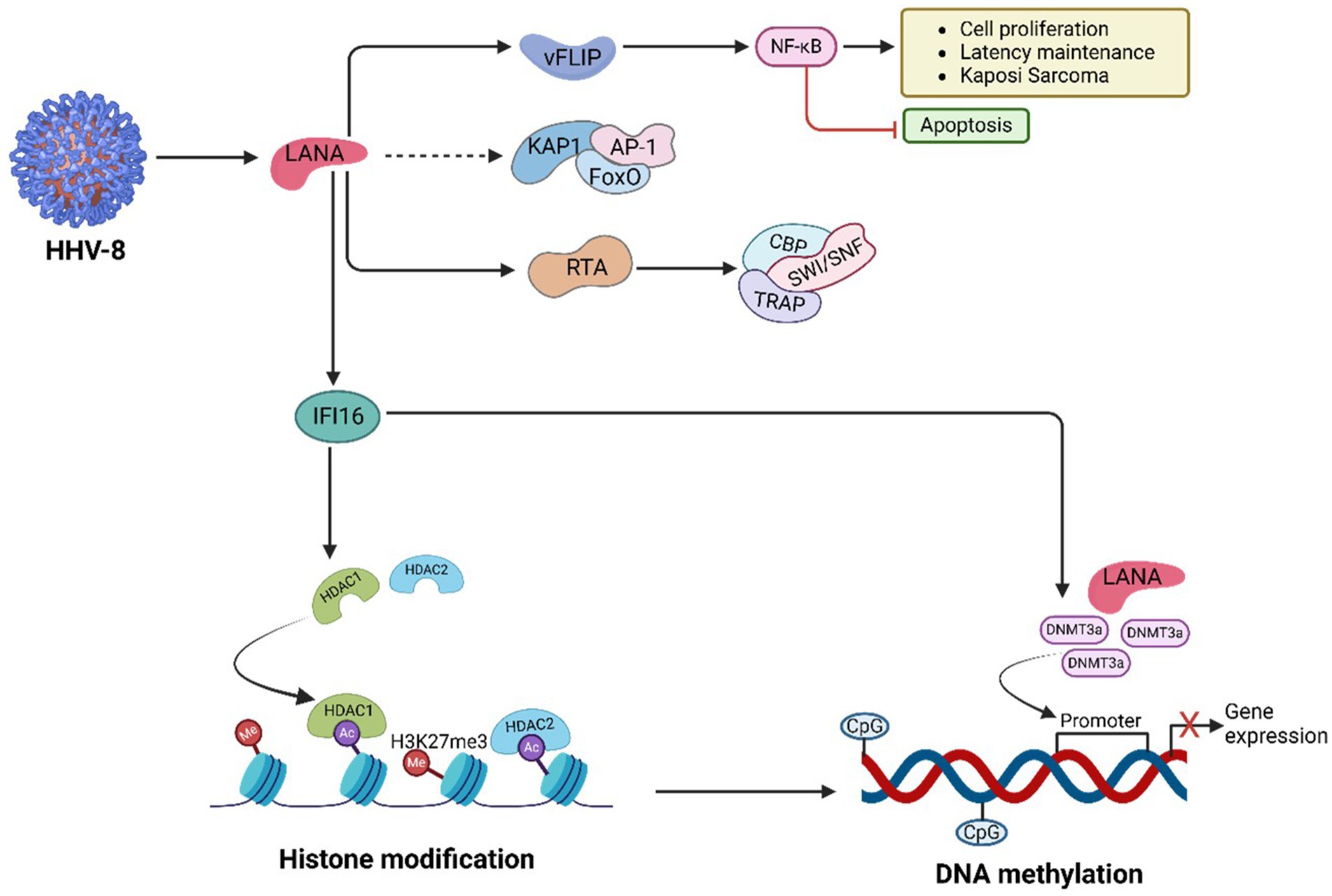
Figure 7. HHV-8-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. HHV-8 proteins such as LANA, vIL-6, and vGPCR, which facilitate immune evasion and angiogenesis. HHV-8 promotes histone modifications and DNA methylation, silencing tumor suppressor genes like p16INK4a and TGF-β receptor. LANA restructures host chromatin, recruits HDACs and DNMTs, and suppresses apoptosis pathways, enabling viral persistence and oncogenic progression. These processes collectively reshape the chromatin landscape and sustain HHV-8’s oncogenic potential.
The replication and transcription activator (RTA) protein of HHV-8 plays a significant role in viral gene expression by interacting with a short acidic sequence in its carboxyl region (Gwack et al., 2003). RTA recruits key factors such as CBP, the SWI/SNF chromatin remodelling complex, and the thyroid hormone receptor-associated protein (TRAP)/Mediator coactivator to viral promoters (Gwack et al., 2003). This recruitment is essential for loosening the nucleosome structure at replication sites, facilitating RTA-driven gene expression. Notably, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1), a tumor suppressor protein involved in hedgehog signalling, is recruited during this process and contributes to regulating tumorigenic pathways (Asha et al., 2020). Additionally, direct interactions between RTA and the Brg1 subunit of SWI/SNF and the TRAP230 subunit of TRAP/Mediator have been identified, emphasizing their involvement in HHV-8 gene regulation (Gwack et al., 2003).
The c-Myc proto-oncogene is another key factor in HHV-8-mediated cancers. Liu et al. (2007) showed that HHV-8’s LANA binds to c-Myc promoter regions, increasing its transcription. This upregulation drives cellular transformation and tumor proliferation in KS, highlighting c-Myc’s pivotal role in HHV-8 oncogenesis (Liu et al., 2007).
HHV-8 also exploits Sp1, a transcription factor, to regulate gene expression supporting cell growth and survival. Verma et al., (Verma et al., 2004) demonstrated that LANA interacts with Sp1 to control genes involved in the cell cycle, leading to the upregulation of pro-survival and proliferative pathways, which further promote KS progression (Verma et al., 2004).
In addition, HHV-8 manipulates AP-1 activity to foster a pro-oncogenic environment by upregulating IL-6 and c-Jun, an AP-1 component, through the MAPK pathway (Yang et al., 2023). This upregulation enhances the transcription of pro-inflammatory and pro-survival genes, sustaining the inflammatory and proliferative conditions characteristic of KS (Yang et al., 2023).
FoxO transcription factors, critical regulators of apoptosis and stress response, are inactivated during HHV-8 infection. Gao et al., (Lan et al., 2023) demonstrated that HHV-8 inhibits FoxO1 activity, suppressing apoptosis and promoting uncontrolled cell proliferation. They also showed that knocking down FoxO1 increases intracellular reactive oxygen species (ROS) levels, which disrupt HHV-8 latency and induce viral lytic reactivation. Similarly, Lan et al. (2023) found that FoxO3 is phosphorylated and inactivated during HHV-8 infection, further enhancing the survival of infected cells and driving tumor progression.
Together, these mechanisms demonstrate how HHV-8 reprograms host signalling and transcriptional machinery to sustain its persistence and promote oncogenesis. By hijacking pathways such as NF-κB, c-Myc, Sp1, AP-1, and FoxO PTFs, HHV-8 creates a favourable environment for cell proliferation, survival, and immune evasion, contributing to the development and progression of KS and other associated cancers.
Chromatin accessibility and epigenetic reprogramming HHV-8-related KS
HHV-8 drives tumorigenesis by altering histones and chromatin marks, creating conditions that favour viral persistence and oncogenic potential (Figure 7). These modifications involve processes such as histone acetylation, methylation, and deacetylation, which silence tumor suppressor genes while promoting the expression of viral genes. Collectively, these changes support the survival of the virus within host cells and enable cancer development (Han et al., 2024).
LANA is a key protein expressed during HHV-8 latency that plays a central role in maintaining viral latency while restructuring host chromatin. LANA interacts with the host protein IFI16, which facilitates the recruitment of HDAC1 and HDAC2 to specific promoter regions. This recruitment leads to the deacetylation of the RTA promoter, silencing its activity and reinforcing viral latency (Ghosh et al., 2025). By preventing the activation of tumor suppressor pathways, including p53-mediated apoptosis, LANA promotes the survival of infected cells and drives cancer progression (Ghosh et al., 2025). Additionally, LANA recruits the SWI/SNF chromatin remodelling complex to the viral genome, ensuring its stable maintenance within host cells. This mechanism supports viral gene expression during reactivation and underscores the multifaceted role of LANA in HHV-8-mediated oncogenesis (Gwack et al., 2003; Han et al., 2024; Ghosh et al., 2025).
Another host factor manipulated by HHV-8 is KAP1 (KRAB-associated protein 1), a transcriptional co-repressor that contributes to viral latency and oncogenesis (Sun et al., 2014). HHV-8 viral proteins recruit KAP1 to induce chromatin compaction and transcriptional silencing of host genes involved in critical processes such as cell cycle regulation, apoptosis, and immune evasion. These actions enable HHV-8 to establish latency and evade host immune surveillance, significantly promoting cancer progression (Sun et al., 2014).
DNA methylation also plays a pivotal role in HHV-8-mediated tumorigenesis. The viral protein LANA recruits DNMT3a to CpG regions on specific promoters, facilitating DNA methylation and repressing gene expression (Shamay et al., 2006). Additionally, HHV-8 microRNA miR-K12-4-5p increases DNMT1, DNMT3a, and DNMT3b levels by targeting their negative regulator, Rbl2, further driving methylation of both viral and host genes (Lu et al., 2010). This alteration silences tumor suppressor genes such as p16INK4a, TGF-β type II receptor (TbetaRII/TGFBR2), PDZ-LIM domain-containing protein 2 (PDLIM2), and CDH13, fostering unchecked cellular proliferation characteristic of Kaposi’s sarcoma (Shamay et al., 2006; Sun et al., 2015; Platt et al., 2002; Di Bartolo et al., 2008).
The PRC introduces the H3K27me3 histone mark, further facilitating CpG methylation and contributing to HHV-8 pathogenesis (Schlesinger et al., 2007; Günther and Grundhoff, 2010). Latent viral proteins such as vFLIP and LANA upregulate EZH2, the catalytic subunit of PRC, and assist in recruiting the complex to chromatin, reinforcing the oncogenic potential of the virus (Schlesinger et al., 2007; Günther and Grundhoff, 2010; He et al., 2012; Toth et al., 2016).
Together, these findings emphasize HHV-8’s ability to manipulate host epigenetic and chromatin regulatory mechanisms, including histone acetylation, methylation, and DNA methylation. By hijacking these pathways, the virus silences tumor suppressor genes, reshapes the chromatin landscape, and sustains its oncogenic potential. This highlights the intricate strategies employed by HHV-8 to drive tumorigenesis and persist within host cells.
Merkel cell polyomavirus
MCV virology
Merkel cell polyomavirus (MCV) is a human virus that plays a central role in the development of Merkel cell carcinoma (MCC), a rare and aggressive neuroendocrine skin cancer (Houben et al., 2023). MCV is present in over 80% of MCC cases, and its oncogenesis is linked to the viral integration into the host genome and the persistent expression of viral proteins (Feng et al., 2008; Pedersen et al., 2024).
MCV is a non-enveloped virus with a 45–50 nm icosahedral capsid composed primarily of the major structural protein VP1, which enables binding to host cell receptors (Houben et al., 2023; Feng et al., 2008; Pedersen et al., 2024). Minor capsid proteins VP2 and VP3 assist in capsid assembly and facilitate viral entry and genome release during infection (Houben et al., 2023).
MCV’s circular, double-stranded DNA genome is about 5,400 base pairs long and divided into early and late regions (Houben et al., 2023). The early region encodes regulatory proteins, including Large T antigen (LT) and Small T antigen (sT), which drive viral replication and manipulate host cell machinery. LT is critical for initiating viral DNA replication and inactivating tumor suppressor proteins like p53 and pRB, enabling cell survival and proliferation (Houben et al., 2023). Meanwhile, sT stabilizes LT, disrupts cell cycle regulation, and controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7 (Kwun et al., 2013). The late region encodes structural proteins (VP1, VP2, VP3) essential for forming new virus particles (Houben et al., 2023).
Key regulatory elements in the MCV genome include the origin of replication (Ori), promoters, enhancers, and a polyadenylation signal, are critical for MCV clonal integration in the host (Feng et al., 2008; Tolstov et al., 2009; Stakaitytė et al., 2014). These elements ensure precise viral DNA replication and gene expression, enabling the successful production of viral proteins and replication of the virus (Houben et al., 2023; Tolstov et al., 2009). The oncogenic process driven by MCV involves a complex interplay between pioneering transcription factors, chromatin remodelling, and epigenetic reprogramming, which collectively influence the cellular environment to promote malignant transformation.
MCV-mediated regulation of PTFS
MCV LT plays a central role in oncogenesis by targeting and inactivating the tumor suppressor proteins p53 and pRB (Figure 8). This disruption prevents cell cycle arrest and apoptosis, enabling uncontrolled cellular proliferation and survival. Shuda et al., (Shuda et al., 2008) demonstrated that LT binds to and inactivates these tumor suppressor pathways, allowing cells to bypass critical checkpoints and divide uncontrollably, a hallmark of tumorigenesis (Shuda et al., 2008). Moreover, LT, in conjunction with the ST antigen, further disrupts cellular checkpoints by targeting both pRB and p53 genes, leading to chromosomal instability and mutation accumulation, which drive MCC progression (Pedersen et al., 2024; Kellogg et al., 2022; Helmbold et al., 2009).
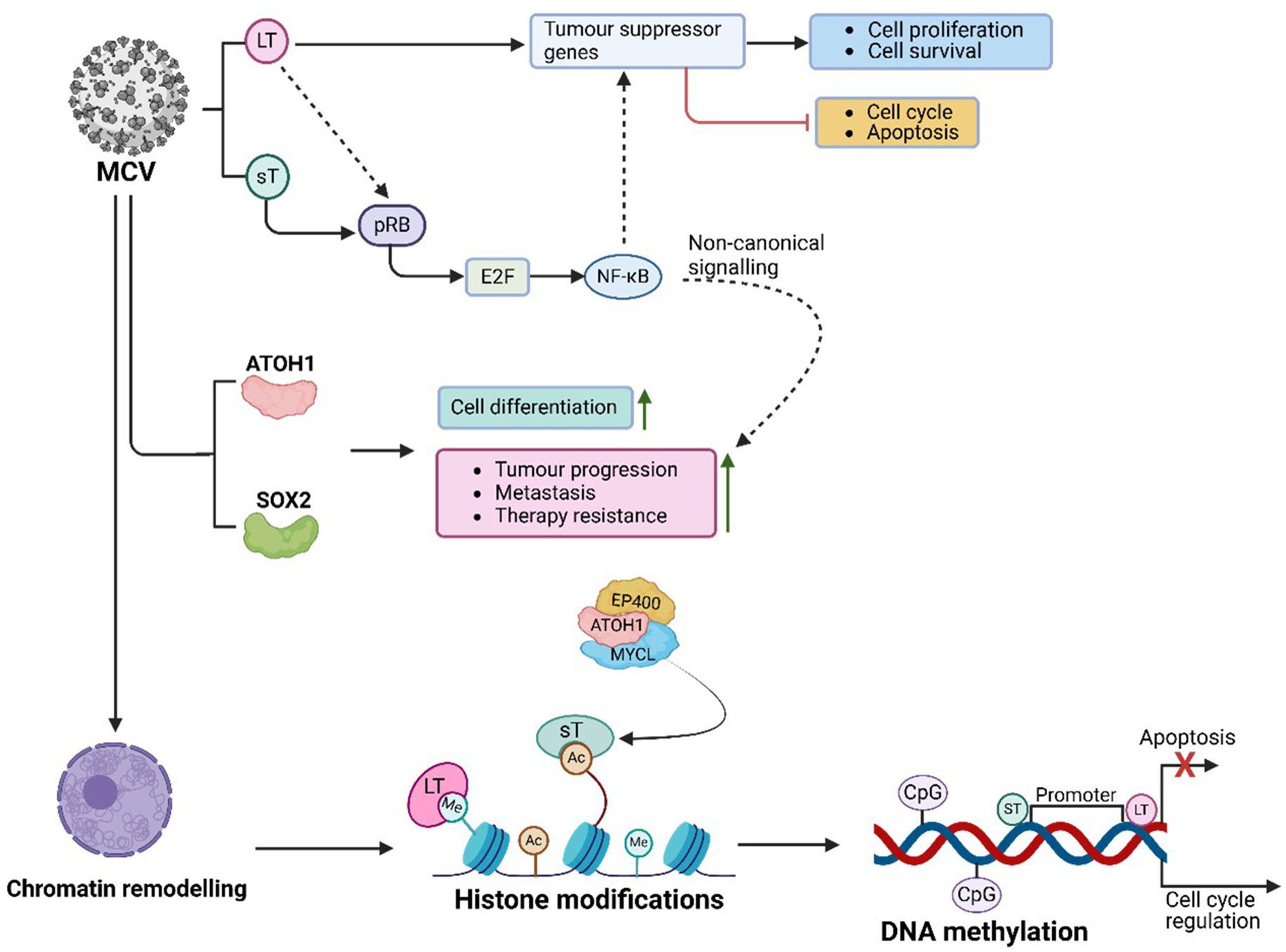
Figure 8. MCV-mediated PTFs regulation, chromatin remodelling and epigenetic reprogramming, and role in oncogenesis. MCV LT inactivates tumor suppressors (e.g., p53, pRB), recruits chromatin modifiers, and releases E2F transcription factors to promote cell cycle progression. Meanwhile, sT regulates complexes like MYCL and EP400, contributing to chromosomal instability and oncogene activation. MCV induces epigenetic reprogramming, including chromatin remodelling, histone modifications, and methylation of genes like CDKN2A and RASSF1A, silencing tumor suppressor genes and enhancing immune evasion. This reshaping of the chromatin landscape activates oncogenes, disrupts cellular processes, and drives MCC progression.
The inactivation of pRB1 by LT also results in the release of E2F transcription factors, which are crucial regulators of the cell cycle (Pedersen et al., 2024; Hesbacher et al., 2016). These E2F transcription factors are upregulated in MCV-positive MCC cells following pRB1 inactivation by LT. This upregulation drives the transcription of genes necessary for DNA replication and cell division, facilitating unchecked proliferation and contributing to the oncogenic phenotype of MCC (Pedersen et al., 2024; Hesbacher et al., 2016).
Atonal Homolog 1 (ATOH1) and SOX2 are PTFs that play vital roles in Merkel cell differentiation, and tumor-suppressing pathways (Harold et al., 2019). ATOH1, a basic helix–loop–helix (bHLH) transcription factor, is essential for the differentiation of neuroendocrine cells such as Merkel cells, which are involved in touch perception. Its expression in MCC correlates with advanced disease progression, underscoring its prognostic significance (Gambichler et al., 2017). When ATOH1 function is disrupted, tumor cells may lose their Merkel cell identity and acquire stem-like characteristics, leading to malignancy.
SOX2, known for its role in maintaining stem cell properties and pluripotency, is another key factor in sustaining the MCC phenotype. Frequently upregulated in MCC, particularly in MCV-positive tumours, SOX2 fosters a stem-like state that promotes tumor progression, metastasis, and therapy resistance (Harold et al., 2019). MCV LT drives the expression of both SOX2 and ATOH1 by inhibiting pRB, thereby maintaining the MCC phenotype. Notably, knocking down LT in MCV-positive MCC cells results in the loss of SOX2 and ATOH1 expression and induces a phenotypic conversion into a differentiated neuronal state. This transformation can also be triggered by inhibiting SOX2 alone, highlighting its pivotal role in maintaining MCC characteristics (Harold et al., 2019).
The viral oncoproteins, including LT and sT, also activate the non-canonical NF-κB signalling pathway, which supports MCC progression (Zhao et al., 2020; Gerer et al., 2017). NF-κB promotes tumor growth by driving the transcription of genes that inhibit apoptosis, advance the cell cycle, and enable immune evasion. Additionally, it contributes to inflammation, a key feature of the tumor microenvironment, and enhances the expression of pro-survival proteins such as BCL-2 and inhibitors of apoptosis (IAPs), thereby fostering MCC cell survival, proliferation, and therapeutic resistance (Zhao et al., 2020; Gerer et al., 2017).
In MCV-related MCC, the YAP/TEAD signalling axis influences gene expression related to cell proliferation, survival, and stem-like characteristics (Frost et al., 2023). Notably, there is an inverse correlation between neuroendocrine gene expression and the activity of YAP1 and WW domain-containing transcriptional regulator 1 (WWTR1) at both the transcript and protein levels in MCC samples (Frost et al., 2023). In MCV-positive MCC, YAP1 and WWTR1 induce cell cycle arrest by repressing LT expression through TEAD-dependent transcriptional mechanisms. This underscores the importance of YAP1/WWTR1 silencing in the development of MCV-related MCC and highlights the heterogeneity of neuroendocrine gene expression in MCC (Frost et al., 2023). Acting as PTFs, the YAP/TEAD complex can influence chromatin remodelling at critical regions involved in cell growth and survival, further establishing its role in MCC progression.
Chromatin accessibility and epigenetic reprogramming in MCV-related MCC
The integration of MCV into the host genome is a defining feature of MCC (Shuda et al., 2008). This integration often results in the persistent expression of viral proteins, including LT and ST antigens, which significantly alter chromatin structure and regulate gene expression (Rotondo et al., 2021). Feng et al. (2008) demonstrated that MCV integration disrupts tumor suppressor genes such as p53 and pRB, triggering chromatin rearrangements that activate oncogenes and promote cellular transformation (Feng et al., 2008). LT further contributes to tumorigenesis by binding to host chromatin, remodelling it to upregulate pro-proliferative genes (Feng et al., 2008; Lu et al., 2012). Sheng et al. (2018) also found that MCV integration modifies host chromatin methylation patterns, activating genes involved in cell cycle progression and suppressing apoptosis, ultimately facilitating both viral replication and oncogenesis.
Histone modifications play a pivotal role in regulating gene expression in MCV-associated MCC (Park et al., 2020; Sheng et al., 2018; Cheng et al., 2017). LT and ST drive histone modifications at key promoter regions to suppress apoptosis-related genes and activate those essential for cell cycle progression (Park et al., 2020; Sheng et al., 2018; Cheng et al., 2017). These viral proteins also interact with chromatin-modifying enzymes, with LT recruiting HDACs and chromatin remodelers to suppress tumor suppressor genes while activating genes that promote cell survival and proliferation (Sheng et al., 2018).
ATOH1, plays a crucial role in chromatin remodelling, regulating cell cycle progression, differentiation, and apoptosis (Park et al., 2020; Cheng et al., 2017). In MCC, ATOH1 promotes oncogenesis by recruiting MYCL to the EP400 histone acetyltransferase and chromatin remodelling complex (Cheng et al., 2017). However, Park et al., (Park et al., 2020) demonstrated that ST represses ATOH1 expression by forming a complex with MYCL and EP400 to activate lysine-specific demethylase 1A (LSD1A), REST corepressor 2 (RCOR2), and insulinoma-associated protein 1 (INSM1). LSD1 inhibition reduces MCC growth, emphasizing the antagonistic relationship between LSD1 and the non-canonical BAF chromatin remodelling complex in gene regulation and tumorigenesis (Park et al., 2020; Sheng et al., 2018; DeCaprio, 2021). Similarly, SOX2 aids chromatin remodelling, regulating cancer progression genes and preserving cancer stem cell characteristics, further solidifying the combined role of ATOH1 and SOX2 in MCC oncogenesis (Harold et al., 2019; DeCaprio, 2021).
MCV MT recruits Src family kinases via its SH3 motif, leading to its phosphorylation at the SH2 domain (Peng et al., 2023). Phosphorylated MT then recruits and activates phospholipase C gamma 1 (PLCγ1), which triggers inflammatory signalling via NF-κB. NF-κB activity promotes chromatin remodelling and gene expression changes, further linking these processes to histone modifications and tumorigenesis (Peng et al., 2023).
DNA methylation also plays a central role in MCV-driven oncogenesis. This epigenetic reprogramming silences tumor suppressor genes, contributing to tumor progression. Sato et al., (Helmbold et al., 2009) observed the frequent silencing of genes such as CDKN2A (encoding p16INK4a) and RASSF1A in MCV-positive MCC tumours, facilitating immune evasion and unchecked proliferation (Helmbold et al., 2009). LT and sT mediate this silencing by reshaping the chromatin landscape to favour tumorigenesis.
Additionally, PCR2-mediated reduction of the suppressive H3K27me3 histone mark has been observed in MCV-positive MCC tumours, suggesting its involvement in MCC pathogenesis (Busam et al., 2017). Wang et al., (Wang et al., 2012) further demonstrated that LT and sT interact with bromodomain protein 4 (Brd4), reshaping the host chromatin landscape to drive uncontrolled cell growth. Brd4, which regulates transcriptional activity by binding to acetylated lysines on histones, interacts with LT and colocalizes with the viral replication origin complex in the nucleus, enabling viral DNA replication by recruiting replication factor C (RFC) (Wang et al., 2012). Disrupting the Brd4-LT interaction halts replication, while blocking Brd4’s chromatin role enhances viral replication, revealing distinct functions in viral replication and transcription regulation (Wang et al., 2012).
Together, these studies reveal how MCV manipulates chromatin and epigenetic machinery, including histone acetylation and methylation, to integrate into the host genome, alter gene expression, and drive MCC progression. This hijacking of the host’s regulatory pathways highlights MCV’s oncogenic potential.
Therapeutic implications
Oncogenic viruses such as HBV, HCV, HPV, HTLV-1, HHV-8, and MCV significantly impact cancer progression by altering epigenetic programming and chromatin accessibility, leading to disrupted gene expression and tumorigenesis. Addressing these challenges, innovative therapies target epigenetic mechanisms and transcriptional dysregulation to counteract viral effects as described in Table 1.
For HBV and HCV, DNMT inhibitors (e.g., decitabine) and HDAC inhibitors (e.g., vorinostat) reverse epigenetic silencing of tumor suppressor genes, while BET inhibitors and CRISPR technologies provide new avenues for managing viral integration and oncogenesis (Zhang et al., 2019; Wu et al., 2020). When combined with direct-acting antivirals (DAAs), HDAC and DNMT inhibitors aim to reduce the long-term risk of HCC associated with HCV infection (Dai et al., 2024). HPV-induced cancers are also managed with HDAC and DNMT inhibitors, YAP/TEAD pathway inhibitors, and immunotherapies like pembrolizumab, offering comprehensive treatment options (Bertagnin et al., 2023; Martinez et al., 2022; Martinez et al., 2022; Yao et al., 2024; Zeidan et al., 2022; Jabbour et al., 2017; Pan et al., 2023; Yang et al., 2021).
Epigenetic therapies also target EBV-driven malignancies, leveraging DNMT and HDAC inhibitors to reverse viral-induced changes, while BET inhibitors and immune checkpoint therapies enhance the host immune response (Leong and Lung, 2021; Sarkozy et al., 2020; Julia and Salles, 2021; Nastoupil et al., 2023). Similarly, in HTLV-1-associated cancers, DNMT and HDAC inhibitors, BET inhibitors, and CRISPR-based techniques mitigate oncogenic processes by disrupting viral transcription and chromatin modifications (Letafati et al., 2025; Wang et al., 2024; Sarkozy et al., 2020; Julia and Salles, 2021; Nastoupil et al., 2023; Li et al., 2018; Smith et al., 2016). BET inhibitors, which target BRD4, inhibit the progression of Tax-infected cells, while emerging CRISPR technologies show potential for targeting key oncogenic genes, such as Tax and HBZ, to mitigate the virus’s impact (Letafati et al., 2025; Wang et al., 2024; Li et al., 2018; Smith et al., 2016).
For HHV-8-associated malignancies, therapeutic strategies include inhibiting NF-κB signalling, reversing viral latency through HDAC, DNMT, and EZH2 inhibitors, and suppressing IL-6/AP-1 signalling (Okpara et al., 2024; Naimo et al., 2021; Hopcraft et al., 2018; Li et al., 2019; Lu et al., 2023; Murphy et al., 2022). Chromatin remodelling inhibitors, FoxO activators, and immune therapies enhance tumor suppression, while antiviral agents like ganciclovir and RTA activators target viral replication and latency.
MCV-related MCC therapies focus on restoring tumor suppressors (e.g., p53, pRB), targeting transcription factors (ATOH1, SOX2), and modulating pathways like NF-κB and YAP/TEAD. Additionally, epigenetic interventions and checkpoint inhibitors offer a multifaceted approach to managing MCC (Harms et al., 2022; Stewart et al., 2009; Rizzitano et al., 2016; Dombret et al., 2014; Ocana et al., 2015).
These diverse, virus-specific approaches highlight the advancement of therapeutic options targeting the intricate molecular mechanisms underlying oncogenic viruses, offering promising directions for combating virus-associated cancers.
Conclusion and future research perspectives
Oncogenic viruses, including HBV, HCV, HPV, HTLV-1, EBV, HHV-8, and MCV, employ sophisticated strategies to manipulate host chromatin architecture and epigenetic mechanisms, driving persistent infection and tumorigenesis. These viruses exploit pathways to silence tumor suppressor genes, promote uncontrolled proliferation, and evade immune responses. For example, HBV and HCV reprogram chromatin and epigenetic landscapes to sustain infection and foster the inflammatory microenvironment that leads to hepatocellular carcinoma. HPV utilizes chromatin remodelling and transcriptional reprogramming to dysregulate key pathways like YAP-mediated signalling, silencing tumor suppressor genes through DNA hypermethylation and histone modifications. Similarly, EBV and HTLV-1 alter host gene expression through viral proteins interacting with chromatin remodelling complexes, while HHV-8 activates NF-κB signalling and disrupts transcription factor activity to sustain its oncogenic state. MCV impacts chromatin structure through epigenetic enzyme recruitment and transcription factor regulation, promoting proliferation, chromosomal instability, and immune evasion in MCC. Collectively, the ability of these viruses to hijack host regulatory processes underscores their oncogenic potential and highlights critical areas for therapeutic intervention.
Future research on oncogenic viruses should prioritize exploring how these pathogens manipulate epigenetic mechanisms, including histone modifications, DNA methylation, and chromatin remodelling complexes, to drive tumorigenesis. This knowledge could inform the development of epigenetic therapies, such as HDAC and DNMT inhibitors, tailored to virus-driven malignancies. Additionally, detailed investigations into the interactions between viral proteins and host transcription factors, such as YAP, c-Myc, SOX2, and NF-κB, may reveal novel therapeutic targets to disrupt the oncogenic reprogramming of transcriptional networks. Understanding immune evasion mechanisms employed by these viruses to bypass immune responses is another vital area of study, with potential to enhance immune checkpoint inhibitor efficacy when combined with epigenetic therapies. The identification of virus-specific biomarkers reflecting chromatin and epigenetic alterations could enable earlier detection and personalized treatment strategies for virus-associated cancers. Furthermore, research into resistance mechanisms against therapies, including checkpoint inhibitors and epigenetic drugs, could guide the development of more durable treatment options. Lastly, a deeper examination of the interplay between chromatin remodelling complexes and viral oncoproteins could uncover innovative approaches for interrupting the progression of virus-driven malignancies. These perspectives offer promising opportunities for advancing the understanding of oncogenic viruses and designing effective, targeted interventions.
Author contributions
MK: Methodology, Supervision, Writing – review & editing, Resources, Writing – original draft, Investigation. SM: Writing – review & editing, Writing – original draft. LK: Writing – review & editing, Writing – original draft. OF: Writing – review & editing, Writing – original draft. TM-T: Writing – review & editing, Writing – original draft. MS: Funding acquisition, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Figures were created with BioRender.com (Agreement reference numbers: VF282V31I8; CP282V3ZCG, RP282V9N13, AO282V5V7F, GO282VAM98, CQ282V7LZA, XC282V8621, and AD282VA4L9).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Gen AI was used in the creation of this manuscript. Grammatical errors were reviewed and corrected using AI.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adesoye, T., Tripathy, D., Hunt, K. K., and Keyomarsi, K. (2024). Exploring novel Frontiers: leveraging STAT3 signaling for advanced Cancer therapeutics. Cancers 16:492. doi: 10.3390/cancers16030492
Ahmadi Ghezeldasht, S., Blackbourn, D. J., Mosavat, A., and Rezaee, S. A. (2023). Pathogenicity and virulence of human T lymphotropic virus type-1 (HTLV-1) in oncogenesis: adult T-cell leukemia/lymphoma (ATLL). Crit. Rev. Clin. Lab. Sci. 60, 189–211. doi: 10.1080/10408363.2022.2157791
Ahuja, R., Jamal, A., Nosrati, N., Pandey, V., Rajput, P., Saxena, N., et al. (2014). Human oncogenic viruses and cancer. Curr. Sci. 107, 768–785.
Akbarin, M. M., Rezaee, S. A., Farjami, Z., Rahimi, H., and Rafatpanah, H. (2024). The role of CREB and MAPK signaling pathways in ATLL patients. AIDS Res. Ther. 21:81. doi: 10.1186/s12981-024-00664-x
Allen, U. D., and Preiksaitis, J. K.Practice tAIDCo (2019). Post-transplant lymphoproliferative disorders, Epstein-Barr virus infection, and disease in solid organ transplantation: guidelines from the American Society of Transplantation infectious diseases Community of Practice. Clin. Transpl. 33:e13652. doi: 10.1111/ctr.13652
Amicone, L., and Marchetti, A. (2018). Microenvironment and tumor cells: two targets for new molecular therapies of hepatocellular carcinoma. Transl. Gastroenterol. Hepatol. 3:24. doi: 10.21037/tgh.2018.04.05
An, J., Sun, Y., Sun, R., and Rettig, M. B. (2003). Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene 22, 3371–3385. doi: 10.1038/sj.onc.1206407
Andreescu, M. (2024). Epigenetic alterations that are the backbone of immune evasion in T-cell malignancies. Cureus. 16:e51662. doi: 10.7759/cureus.51662
Andrisani, O. (2021). Epigenetic mechanisms in hepatitis B virus-associated hepatocellular carcinoma. Hepatoma Research 7:12. doi: 10.20517/2394-5079.2020.83
Aneja, K. K., and Yuan, Y. (2017). Reactivation and lytic replication of Kaposi's sarcoma-associated herpesvirus: An update. Front. Microbiol. 8:613. doi: 10.3389/fmicb.2017.00613
Asha, K., Balfe, N., and Sharma-Walia, N. (2020). Concurrent control of the Kaposi's sarcoma-associated herpesvirus life cycle through chromatin modulation and host hedgehog signaling: a new Prospect for the therapeutic potential of Lipoxin A4. J. Virol. 94, 10–128. doi: 10.1128/JVI.02177-19
Asha, K., and Sharma-Walia, N. (2021). Targeting host cellular factors as a strategy of therapeutic intervention for herpesvirus infections. Front. Cell. Infect. Microbiol. 11:603309. doi: 10.3389/fcimb.2021.603309
Ashfaq, U. A., Javed, T., Rehman, S., Nawaz, Z., and Riazuddin, S. (2011). An overview of HCV molecular biology, replication and immune responses. Virol. J. 8:161. doi: 10.1186/1743-422X-8-161
Azodi, S., Nair, G., Enose-Akahata, Y., Charlip, E., Vellucci, A., Cortese, I., et al. (2017). Imaging spinal cord atrophy in progressive myelopathies: HTLV-I-associated neurological disease (HAM/TSP) and multiple sclerosis (MS). Ann. Neurol. 82, 719–728. doi: 10.1002/ana.25072
Babar, Q., Saeed, A., Tabish, T. A., Pricl, S., Townley, H., and Thorat, N. (2022). Novel epigenetic therapeutic strategies and targets in cancer. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 1868:166552. doi: 10.1016/j.bbadis.2022.166552
Bai, L., Xu, J., Zeng, L., Zhang, L., and Zhou, F. (2024). A review of HSV pathogenesis, vaccine development, and advanced applications. Mol. Biomed. 5:35. doi: 10.1186/s43556-024-00199-7
Banerjee, A., Dass, D., Mukherjee, S., Kaul, M., Harshithkumar, R., Bagchi, P., et al. (2024). The ‘Oma’s of the gammas—Cancerogenesis by γ-herpesviruses. Viruses 16:1928. doi: 10.3390/v16121928
Banerjee, N. S., Moore, D. W., Broker, T. R., and Chow, L. T. (2018). Vorinostat, a pan-HDAC inhibitor, abrogates productive HPV-18 DNA amplification. Proc. Natl. Acad. Sci. 115, E11138–E11147. doi: 10.1073/pnas.1801156115
Bangham, C. R., and Ratner, L. (2015). How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr. Opin. Virol. 14, 93–100. doi: 10.1016/j.coviro.2015.09.004
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Bartosh, U. I., Dome, A. S., Zhukova, N. V., Karitskaya, P. E., and Stepanov, G. A. (2024). CRISPR/Cas9 as a new antiviral strategy for treating hepatitis viral infections. Int. J. Mol. Sci. 25:334. doi: 10.3390/ijms25010334
Becker, J. C., Stang, A., Schrama, D., and Ugurel, S. (2024). Merkel cell carcinoma: integrating epidemiology, immunology, and therapeutic updates. Am. J. Clin. Dermatol. 25, 541–557. doi: 10.1007/s40257-024-00858-z
Bellon, M., Yeh, C.-h., Bai Xue, T., and Nicot, C. (2024). The HTLV-I oncoprotein tax inactivates the tumor suppressor FBXW7. J. Virol. 98:e00405-24. doi: 10.1128/jvi.00405-24
Bernaudat, F., Gustems, M., Günther, J., Oliva, M. F., Buschle, A., Göbel, C., et al. (2022). Structural basis of DNA methylation-dependent site selectivity of the Epstein-Barr virus lytic switch protein ZEBRA/Zta/BZLF1. Nucleic Acids Res. 50, 490–511. doi: 10.1093/nar/gkab1183
Bertagnin, C., Messa, L., Pavan, M., Celegato, M., Sturlese, M., Mercorelli, B., et al. (2023). A small molecule targeting the interaction between human papillomavirus E7 oncoprotein and cellular phosphatase PTPN14 exerts antitumoral activity in cervical cancer cells. Cancer Lett. 571:216331. doi: 10.1016/j.canlet.2023.216331
Bhuyan, P. K., Dallas, M., Kraynyak, K., Herring, T., Morrow, M., Boyer, J., et al. (2021). Durability of response to VGX-3100 treatment of HPV16/18 positive cervical HSIL. Hum. Vaccin. Immunother. 17, 1288–1293. doi: 10.1080/21645515.2020.1823778
Bi, Y., Xia, C., Zhang, X., and Liu, H. (2023). Targeted treatments after chemoradiotherapy failure in a patient with relapsed, advanced non-small cell lung cancer with on-therapy circulating tumor biomarker monitoring: a case report. Oncol. Lett. 26:407. doi: 10.3892/ol.2023.13993
Blakely, W. J., Hatterschide, J., and White, E. A. (2024). HPV18 E7 inhibits LATS1 kinase and activates YAP1 by degrading PTPN14. MBio 15:e0181124. doi: 10.1128/mbio.01811-24
Boosani, C. S., and Agrawal, D. K. (2015). Methylation and microRNA-mediated epigenetic regulation of SOCS3. Mol. Biol. Rep. 42, 853–872. doi: 10.1007/s11033-015-3860-3
Burns, E. A., Muhsen, I. N., Anand, K., Xu, J., Umoru, G., Arain, A. N., et al. (2021). Hepatitis B virus reactivation in Cancer patients treated with immune checkpoint inhibitors. J. Immunother. 44, 132–139. doi: 10.1097/CJI.0000000000000358
Busam, K. J., Pulitzer, M. P., Coit, D. C., Arcila, M., Leng, D., Jungbluth, A. A., et al. (2017). Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Modern Pathol 30, 877–883. doi: 10.1038/modpathol.2017.8
Cai, M., Wang, Y., Ma, H., Yang, L., and Xu, Z. (2024). Advances and challenges in immunotherapy for locally advanced nasopharyngeal carcinoma. Cancer Treat. Rev. 131:102840. doi: 10.1016/j.ctrv.2024.102840
Caldon, C. E., and Musgrove, E. A. (2010). Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell Div. 5:2. doi: 10.1186/1747-1028-5-2
Cao, C., Xu, Q., Zhu, Z., Xu, M., Wei, Y., Lin, S., et al. (2024). Three-dimensional chromatin analysis reveals Sp1 as a mediator to program and reprogram HPV-host epigenetic architecture in cervical cancer. Cancer Lett. 588:216809. doi: 10.1016/j.canlet.2024.216809
Carcone, A., Journo, C., and Dutartre, H. (2022). Is the HTLV-1 retrovirus targeted by host restriction factors? Viruses 14:1611. doi: 10.3390/v14081611
Cargill, T., and Barnes, E. (2021). Therapeutic vaccination for treatment of chronic hepatitis B. Clin. Exp. Immunol. 205, 106–118. doi: 10.1111/cei.13614
Casciano, J. C., Bagga, S., Yang, B., and Bouchard, M. J. (2012). Modulation of cell proliferation pathways by the hepatitis B virus X protein: A potential contributor to the development of hepatocellular carcinoma. Hepatocellular carcinoma-basic research InTech: Rijeka, Croatia, 103–152.
Castro-Oropeza, R., and Piña-Sánchez, P. (2022). Epigenetic and transcriptomic regulation landscape in HPV+ cancers: biological and clinical implications. Front. Genet. 13:886613. doi: 10.3389/fgene.2022.886613
Cavalli, G., and Heard, E. (2019). Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499. doi: 10.1038/s41586-019-1411-0
Çevik, R. E., Cesarec, M., Da Silva Filipe, A., Licastro, D., McLauchlan, J., and Marcello, A. (2017). Hepatitis C virus NS5A targets nucleosome assembly protein NAP1L1 to control the innate cellular response. J. Virol. 91:18. doi: 10.1128/JVI.00880-17
Chakravorty, S., Afzali, B., and Kazemian, M. (2022). EBV-associated diseases: current therapeutics and emerging technologies. Front. Immunol. 13:1059133. doi: 10.3389/fimmu.2022.1059133
Chan, P. K., Ng, H. K., Cheung, J. L., and Cheng, A. F. (2000). Survey for the presence and distribution of human herpesvirus 8 in healthy brain. J. Clin. Microbiol. 38, 2772–2773. doi: 10.1128/JCM.38.7.2772-2773.2000
Chattopadhyay, S., Yamashita, M., Zhang, Y., and Sen, G. C. (2011). The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 85, 3708–3716. doi: 10.1128/JVI.02133-10
Chen, Y., Liang, R., Li, Y., Jiang, L., Ma, D., Luo, Q., et al. (2024). Chromatin accessibility: biological functions, molecular mechanisms and therapeutic application. Signal Transduct. Target. Ther. 9:340. doi: 10.1038/s41392-024-02030-9
Chen, X., Loo, J. X., Shi, X., Xiong, W., Guo, Y., Ke, H., et al. (2018). E6 protein expressed by high-risk HPV activates super-enhancers of the EGFR and c-MET oncogenes by destabilizing the histone demethylase KDM5C. Cancer Res. 78, 1418–1430. doi: 10.1158/0008-5472.CAN-17-2118
Chen, C., Wang, Z., Ding, Y., Wang, L., Wang, S., Wang, H., et al. (2022). DNA methylation: from Cancer biology to clinical perspectives. FBL. 27:27(12). doi: 10.31083/j.fbl2712326
Chen, G., Zhang, L., Wang, R., and Xie, Z. (2024). Histone methylation in Epstein-Barr virus-associated diseases. Epigenomics. 16, 865–877. doi: 10.1080/17501911.2024.2345040
Cheng, Y., He, C., Wang, M., Ma, X., Mo, F., Yang, S., et al. (2019). Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4:62. doi: 10.1038/s41392-019-0095-0
Cheng, Q. J., Ohta, S., Sheu, K. M., Spreafico, R., Adelaja, A., Taylor, B., et al. (2021). NF-κB dynamics determine the stimulus specificity of epigenomic reprogramming in macrophages. Science 372, 1349–1353. doi: 10.1126/science.abc0269
Cheng, J., Park, D. E., Berrios, C., White, E. A., Arora, R., Yoon, R., et al. (2017). Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 13:e1006668. doi: 10.1371/journal.ppat.1006668
Cheng, L., Wang, Y., and Du, J. (2020). Human papillomavirus vaccines: An updated review. Vaccine 8:391. doi: 10.3390/vaccines8030391
Chiou, S. K., Jones, M. K., and Tarnawski, A. S. (2003). Survivin - an anti-apoptosis protein: its biological roles and implications for cancer and beyond. Med. Sci. Monit. 9, Pi25–Pi29.
Cirillo, L. A., Lin, F. R., Cuesta, I., Friedman, D., Jarnik, M., and Zaret, K. S. (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. doi: 10.1016/S1097-2765(02)00459-8
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Conry, R. M., Westbrook, B., McKee, S., and Norwood, T. G. (2018). Talimogene laherparepvec: first in class oncolytic virotherapy. Hum. Vaccin. Immunother. 14, 839–846. doi: 10.1080/21645515.2017.1412896
Cotter, T. G., and Jensen, D. M. (2019). Glecaprevir/pibrentasvir for the treatment of chronic hepatitis C: design, development, and place in therapy. Drug Des. Devel. Ther. 13, 2565–2577. doi: 10.2147/DDDT.S172512
Coudé, M.-M., Braun, T., Berrou, J., Dupont, M., Bertrand, S., Masse, A., et al. (2015). BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 6, 17698–17712. doi: 10.18632/oncotarget.4131
Czarnota, A., Raszplewicz, A., Sławińska, A., Bieńkowska-Szewczyk, K., and Grzyb, K. (2024). Minicircle-based vaccine induces potent T-cell and antibody responses against hepatitis C virus. Sci. Rep. 14:26698. doi: 10.1038/s41598-024-78049-3
Dai, W., Qiao, X., Fang, Y., Guo, R., Bai, P., Liu, S., et al. (2024). Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct. Target. Ther. 9:332. doi: 10.1038/s41392-024-02039-0
Dai, E., Zhu, Z., Wahed, S., Qu, Z., Storkus, W. J., and Guo, Z. S. (2021). Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 20:171. doi: 10.1186/s12943-021-01464-x
Damania, B., Kenney, S. C., and Raab-Traub, N. (2022). Epstein-Barr virus: biology and clinical disease. Cell 185, 3652–3670. doi: 10.1016/j.cell.2022.08.026
Das, C., and Kundu, C. N. (2025). Decoding the molecular complexity of viruses in human cancer: insights into host cell infection, oncogenesis, and therapeutic prospects. Crit. Rev. Microbiol. 1–24. doi: 10.1080/1040841X.2025.2461045
Dasari, V., McNeil, L. K., Beckett, K., Solomon, M., Ambalathingal, G., Thuy, T. L., et al. (2023). Lymph node targeted multi-epitope subunit vaccine promotes effective immunity to EBV in HLA-expressing mice. Nat. Commun. 14:4371. doi: 10.1038/s41467-023-39770-1
de Almeida, N. A. A., Ribeiro, C. R. A., Raposo, J. V., and de Paula, V. S. (2021). Immunotherapy and gene therapy for oncoviruses infections: a review. Viruses 13:822. doi: 10.3390/v13050822
Dearborn, A. D., and Marcotrigiano, J. (2020). Hepatitis C virus structure: defined by what it is not. Cold Spring Harb. Perspect. Med. 10, 1–10. doi: 10.1101/cshperspect.a036822
DeCaprio, J. A. (2021). Molecular pathogenesis of Merkel cell carcinoma. Annu. Rev. Pathol. 16, 69–91. doi: 10.1146/annurev-pathmechdis-012419-032817
Di Bartolo, D. L., Cannon, M., Liu, Y. F., Renne, R., Chadburn, A., Boshoff, C., et al. (2008). KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111, 4731–4740. doi: 10.1182/blood-2007-09-110544
di Filippo Villa, D., and Navas, M. C. (2023). Vertical transmission of hepatitis B virus-an update. Microorganisms 11:1140. doi: 10.3390/microorganisms11051140
Dombret, H., Preudhomme, C., Berthon, C., Raffoux, E., Thomas, X., Vey, N., et al. (2014). A phase 1 study of the BET-Bromodomain inhibitor OTX015 in patients with advanced acute leukemia. Blood 124:117. doi: 10.1182/blood.V124.21.117.117
Domingues, W., Folgosi, V., Sanabani, S. S., Leite Junior, P. D., Assone, T., and Casseb, J. (2024). Novel approaches for HTLV-1 therapy: innovative applications of CRISPR-Cas9. Rev. Inst. Med. Trop. Sao Paulo 66:e48. doi: 10.1590/S1678-9946202466048
Domovitz, T., and Gal-Tanamy, M. (2021). Tracking down the epigenetic footprint of HCV-induced Hepatocarcinogenesis. J. Clin. Med. 10:551. doi: 10.3390/jcm10030551
Dong, W., Wang, H., Li, M., Li, P., and Ji, S. (2024). Virus-induced host genomic remodeling dysregulates gene expression, triggering tumorigenesis. Front. Cell. Infect. Microbiol. 14:1359766. doi: 10.3389/fcimb.2024.1359766
Doorbar, J., Quint, W., Banks, L., Bravo, I. G., Stoler, M., Broker, T. R., et al. (2012). The biology and life-cycle of human papillomaviruses. Vaccine 30, F55–F70. doi: 10.1016/j.vaccine.2012.06.083
Dubuisson, J. (2007). Hepatitis C virus proteins. World J. Gastroenterol. 13, 2406–2415. doi: 10.3748/wjg.v13.i17.2406
Durzynska, J., Lesniewicz, K., and Poreba, E. (2017). Human papillomaviruses in epigenetic regulations. Mutat. Res. Rev. Mutat. Res. 772, 36–50. doi: 10.1016/j.mrrev.2016.09.006
Egawa, N. (2023). Papillomaviruses and cancer: commonalities and differences in HPV carcinogenesis at different sites of the body. Int. J. Clin. Oncol. 28, 956–964. doi: 10.1007/s10147-023-02340-y
El Taghdouini, A., Sørensen, A. L., Reiner, A. H., Coll, M., Verhulst, S., Mannaerts, I., et al. (2015). Genome-wide analysis of DNA methylation and gene expression patterns in purified, uncultured human liver cells and activated hepatic stellate cells. Oncotarget 6, 26729–26745. doi: 10.18632/oncotarget.4925
Enose-Akahata, Y., Vellucci, A., and Jacobson, S. (2017). Role of HTLV-1 tax and HBZ in the pathogenesis of HAM/TSP. Front. Microbiol. 8:2563. doi: 10.3389/fmicb.2017.02563
Ernzen, K. J., and Panfil, A. R. (2022). Regulation of HTLV-1 transformation. Biosci. Rep. 42, 1–10. doi: 10.1042/BSR20211921
Eusebio-Ponce, E., Anguita, E., Paulino-Ramirez, R., and Candel, F. J. (2019). HTLV-1 infection: an emerging risk. Pathogenesis, epidemiology, diagnosis and associated diseases. Rev. Esp. Quimioter 32, 485–496. doi: 10.1073/pnas.77.12.7415
Feng, H., Shuda, M., Chang, Y., and Moore, P. S. (2008). Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100. doi: 10.1126/science.1152586
Fiehn, F., Beisel, C., and Binder, M. (2024). Hepatitis C virus and hepatocellular carcinoma: carcinogenesis in the era of direct-acting antivirals. Curr. Opin. Virol. 67:101423. doi: 10.1016/j.coviro.2024.101423
Frey, A., Perry, C., Olino, K., and Ishizuka, J. J. (2024). Development of an mRNA therapeutic vaccine for virally driven Merkel cell carcinoma. J. Clin. Oncol. 42:2637. doi: 10.1200/JCO.2024.42.16_suppl.2637
Frost, T. C., Gartin, A. K., Liu, M., Cheng, J., Dharaneeswaran, H., Keskin, D. B., et al. (2023). YAP1 and WWTR1 expression inversely correlates with neuroendocrine markers in Merkel cell carcinoma. J. Clin. Invest. 133:e157171. doi: 10.1172/JCI157171
Gambichler, T., Mohtezebsade, S., Wieland, U., Silling, S., Höh, A. K., Dreißigacker, M., et al. (2017). Prognostic relevance of high atonal homolog-1 expression in Merkel cell carcinoma. J. Cancer Res. Clin. Oncol. 143, 43–49. doi: 10.1007/s00432-016-2257-6
Gambichler, T., Schrama, D., Käpynen, R., Weyer-Fahlbusch, S. S., Becker, J. C., Susok, L., et al. (2024). Current Progress in vaccines against Merkel cell carcinoma: a narrative review and update. Vaccine 12:533. doi: 10.3390/vaccines12050533
Garg, P., Singhal, G., Pareek, S., Kulkarni, P., Horne, D., Nath, A., et al. (2025). Unveiling the potential of gene editing techniques in revolutionizing cancer treatment: a comprehensive overview. Biochimica et Biophysica Acta (BBA) Reviews Cancer 1880:189233. doi: 10.1016/j.bbcan.2024.189233
Gazon, H., Barbeau, B., Mesnard, J. M., and Peloponese, J. M. Jr. (2017). Hijacking of the AP-1 signaling pathway during development of ATL. Front. Microbiol. 8:2686. doi: 10.3389/fmicb.2017.02686
Geissler, F., Nesic, K., Kondrashova, O., Dobrovic, A., Swisher, E. M., Scott, C. L., et al. (2024). The role of aberrant DNA methylation in cancer initiation and clinical impacts. Therap. Adv. Med. Oncol. 16:17588359231220511. doi: 10.1177/17588359231220511
Gerer, K. F., Erdmann, M., Hadrup, S. R., Lyngaa, R., Martin, L. M., Voll, R. E., et al. (2017). Preclinical evaluation of NF-κB-triggered dendritic cells expressing the viral oncogenic driver of Merkel cell carcinoma for therapeutic vaccination. Therap. Adv. Med. Oncol. 9, 451–464. doi: 10.1177/1758834017712630
Gessain, A. (2008). L’herpèsvirus humain 8 (HHV-8): aspects cliniques, épidémiologiques et clonalité des maladies tumorales associées. Bull. Acad. Natl Med. 192, 1189–1206. doi: 10.1016/S0001-4079(19)32717-7
Ghione, P., Moskowitz, A. J., De Paola, N. E. K., Horwitz, S. M., and Ruella, M. (2018). Novel immunotherapies for T cell lymphoma and leukemia. Curr. Hematol. Malig. Rep. 13, 494–506. doi: 10.1007/s11899-018-0480-8
Ghoreshi, Z. A., Molaei, H. R., and Arefinia, N. (2023). The role of DNA viruses in human cancer. Cancer Inform 22:11769351231154186. doi: 10.1177/11769351231154186
Ghosh, A., Britto, J., Chandran, B., and Roy, A. (2025). IFI16 recruits HDAC1 and HDAC2 to deacetylate the Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA), facilitating latency. J. Virol. 99, e01549–e01524. doi: 10.1128/jvi.01549-24
Giam, C. Z., and Semmes, O. J. (2016). HTLV-1 infection and adult T-cell leukemia/lymphoma-a tale of two proteins: tax and HBZ. Viruses 8:161. doi: 10.3390/v8060161
Gliga, S., Orth, H. M., Lübke, N., Timm, J., Luedde, T., and Jensen, B.-E. O. (2021). Multicentric Castleman’s disease in HIV patients: a single-center cohort diagnosed from 2008 to 2018. Infection 49, 945–951. doi: 10.1007/s15010-021-01618-5
Gothland, A., Jary, A., Grange, P., Leducq, V., Beauvais-Remigereau, L., Dupin, N., et al. (2023). Harnessing redox disruption to treat human herpesvirus 8 (HHV-8) related malignancies. Antioxidants. 12:84. doi: 10.3390/antiox12010084
Graham, R. L., Burch, M., and Krause, J. R. (2014). Adult T-cell leukemia/lymphoma. Proc. (Baylor Univ. Med. Cent.) 27, 235–238. doi: 10.1080/08998280.2014.11929123
Gruber, J. J., Geller, B., Lipchik, A. M., Chen, J., Salahudeen, A. A., Ram, A. N., et al. (2019). HAT1 coordinates histone production and acetylation via H4 promoter binding. Mol. Cell 75, 711–24.e5. doi: 10.1016/j.molcel.2019.05.034
Gunasekharan, V. K., Li, Y., Andrade, J., and Laimins, L. A. (2016). Post-transcriptional regulation of KLF4 by high-risk human papillomaviruses is necessary for the differentiation-dependent viral life cycle. PLoS Pathog. 12:e1005747. doi: 10.1371/journal.ppat.1005747
Günther, T., and Grundhoff, A. (2010). The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6:e1000935. doi: 10.1371/journal.ppat.1000935
Gwack, Y., Baek, H. J., Nakamura, H., Lee, S. H., Meisterernst, M., Roeder, R. G., et al. (2003). Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23, 2055–2067. doi: 10.1128/MCB.23.6.2055-2067.2003
Haery, L., Thompson, R. C., and Gilmore, T. D. (2015). Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 6, 184–213. doi: 10.18632/genesandcancer.65
Han, C., Niu, D., and Lan, K. (2024). Rewriting viral fate: epigenetic and transcriptional dynamics in KSHV infection. Viruses 16:1870. doi: 10.3390/v16121870
Harms, P. W., Verhaegen, M. E., Vo, J. N., Tien, J. C., Pratt, D., Su, F., et al. (2022). Viral status predicts the patterns of genome methylation and Decitabine response in Merkel cell carcinoma. J. Invest. Dermatol. 142, 641–652. doi: 10.1016/j.jid.2021.07.173
Harold, A., Amako, Y., Hachisuka, J., Bai, Y., Li, M. Y., Kubat, L., et al. (2019). Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 116, 20104–20114. doi: 10.1073/pnas.1907154116
He, M., Zhang, W., Bakken, T., Schutten, M., Toth, Z., Jung, J. U., et al. (2012). Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 72, 3582–3592. doi: 10.1158/0008-5472.CAN-11-2876
He, S., Zhang, J., Zhang, W., Chen, F., and Luo, R. (2017). FOXA1 inhibits hepatocellular carcinoma progression by suppressing PIK3R1 expression in male patients. J. Exp. Clin. Cancer Res. 36:175. doi: 10.1186/s13046-017-0646-6
He, W., Zheng, Z., Zhao, Q., Zhang, R., and Zheng, H. (2024). Targeting HBV cccDNA levels: key to achieving complete cure of chronic hepatitis B. Pathogens 13:1100.
Helmbold, P., Lahtz, C., Enk, A., Herrmann-Trost, P., Marsch, W., Kutzner, H., et al. (2009). Frequent occurrence of RASSF1A promoter hypermethylation and Merkel cell polyomavirus in Merkel cell carcinoma. Mol. Carcinog. 48, 903–909. doi: 10.1002/mc.20540
Hensel, K. O., Rendon, J. C., Navas, M. C., Rots, M. G., and Postberg, J. (2017). Virus-host interplay in hepatitis B virus infection and epigenetic treatment strategies. FEBS J. 284, 3550–3572. doi: 10.1111/febs.14094
Hesbacher, S., Pfitzer, L., Wiedorfer, K., Angermeyer, S., Borst, A., Haferkamp, S., et al. (2016). RB1 is the crucial target of the Merkel cell polyomavirus large T antigen in Merkel cell carcinoma cells. Oncotarget 7, 32956–32968. doi: 10.18632/oncotarget.8793
Hlady, R. A., Zhao, X., El Khoury, L. Y., Luna, A., Pham, K., Wu, Q., et al. (2022). Interferon drives HCV scarring of the epigenome and creates targetable vulnerabilities following viral clearance. Hepatology 75, 983–996. doi: 10.1002/hep.32111
Hleihel, R., Skayneh, H., de Thé, H., Hermine, O., and Bazarbachi, A. (2023). Primary cells from patients with adult T cell leukemia/lymphoma depend on HTLV-1 tax expression for NF-κB activation and survival. Blood Cancer J. 13:67. doi: 10.1038/s41408-023-00841-7
Ho, J. W. Y., Li, L., Wong, K. Y., Srivastava, G., and Tao, Q. (2023). Comprehensive profiling of EBV gene expression and promoter methylation reveals latency II viral infection and sporadic abortive lytic activation in peripheral T-cell lymphomas. Viruses 15:423. doi: 10.3390/v15020423
Hopcraft, S. E., Pattenden, S. G., James, L. I., Frye, S., Dittmer, D. P., and Damania, B. (2018). Chromatin remodeling controls Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 14:e1007267. doi: 10.1371/journal.ppat.1007267
Horner, S. M., and Naggie, S. (2015). Successes and challenges on the road to cure hepatitis C. PLoS Pathog. 11:e1004854. doi: 10.1371/journal.ppat.1004854
Hoshida, Y., Fuchs, B. C., Bardeesy, N., Baumert, T. F., and Chung, R. T. (2014). Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 61, S79–S90. doi: 10.1016/j.jhep.2014.07.010
Houben, R., Celikdemir, B., Kervarrec, T., and Schrama, D. (2023). Merkel cell polyomavirus: infection, genome, transcripts and its role in development of Merkel cell carcinoma. Cancers 15:444. doi: 10.3390/cancers15020444
Houghtelin, A., and Bollard, C. M. (2017). Virus-specific T cells for the immunocompromised patient. Front. Immunol. 8, 1–10. doi: 10.3389/fimmu.2017.01272
Hu, X., li, J., Fu, M., Zhao, X., and Wang, W. (2021). The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target. Ther. 6:402. doi: 10.1038/s41392-021-00791-1
Huang, D. N., Wang, S., Sooranna, S. R., and Miao, J. H. (2021). The efficacy of natural bioactive compounds for the treatment of nasopharyngeal carcinoma. Mini Rev. Med. Chem. 21, 1679–1691. doi: 10.2174/1389557521666210105113831
Huck, I., Morris, E. M., Thyfault, J., and Apte, U. (2021). Hepatocyte-specific hepatocyte nuclear factor 4 alpha (HNF4) deletion decreases resting energy expenditure by disrupting lipid and carbohydrate homeostasis. Gene Expr. 20, 157–168. doi: 10.3727/105221621X16153933463538
Inturi, R., and Jemth, P. (2021). CRISPR/Cas9-based inactivation of human papillomavirus oncogenes E6 or E7 induces senescence in cervical cancer cells. Virology 562, 92–102. doi: 10.1016/j.virol.2021.07.005
Iqbal, N., and Iqbal, N. (2014). Imatinib: a breakthrough of targeted therapy in cancer. Chemotherapy research and practice. 2014:357027, 1–9. doi: 10.1155/2014/357027
Ito, S., D’Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. doi: 10.1038/nature09303
Iwafuchi-Doi, M., and Zaret, K. S. (2014). Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692. doi: 10.1101/gad.253443.114
Jabbour, E., Short, N. J., Montalban-Bravo, G., Huang, X., Bueso-Ramos, C., Qiao, W., et al. (2017). Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 130, 1514–1522. doi: 10.1182/blood-2017-06-788497
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33, 245–254. doi: 10.1038/ng1089
Jalili-Nik, M., Soltani, A., Mashkani, B., Rafatpanah, H., and Hashemy, S. I. (2021). PD-1 and PD-L1 inhibitors foster the progression of adult T-cell leukemia/lymphoma. Int. Immunopharmacol. 98:107870. doi: 10.1016/j.intimp.2021.107870
Jenkins, F. J., Hoffman, L. J., and Liegey-Dougall, A. (2002). Reactivation of and primary infection with human herpesvirus 8 among solid-organ transplant recipients. J. Infect. Dis. 185, 1238–1243. doi: 10.1086/340237
Jhan, J. R., and Andrechek, E. R. (2016). Stat3 accelerates Myc induced tumor formation while reducing growth rate in a mouse model of breast cancer. Oncotarget 7, 65797–65807. doi: 10.18632/oncotarget.11667
Jiang, Y., Han, Q., Zhao, H., and Zhang, J. (2021). The mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatocellular Carcinoma. 8, 435–450. doi: 10.2147/JHC.S307962
Jones, P. A., and Baylin, S. B. (2007). The epigenomics of cancer. Cell 128, 683–692. doi: 10.1016/j.cell.2007.01.029
Julia, E., and Salles, G. (2021). EZH2 inhibition by tazemetostat: mechanisms of action, safety and efficacy in relapsed/refractory follicular lymphoma. Future Oncol. 17, 2127–2140. doi: 10.2217/fon-2020-1244
Kang, T., Ye, F. C., Gao, S. J., and Wang, L. D. (2008). Angiogenesis, Kaposi's sarcoma and Kaposi's sarcoma-associated herpesvirus. Virol. Sin. 23, 449–458. doi: 10.1007/s12250-008-2998-8
Karayiannis, P. (2017). Hepatitis B virus: virology, molecular biology, life cycle and intrahepatic spread. Hepatol. Int. 11, 500–508. doi: 10.1007/s12072-017-9829-7
Kasprzyk, M. E., Sura, W., and Dzikiewicz-Krawczyk, A. (2021). Enhancing B-cell malignancies-on repurposing enhancer activity towards Cancer. Cancers 13:3270. doi: 10.3390/cancers13133270
Kaszycki, J., and Kim, M. (2025). Epigenetic regulation of transcription factors involved in NLRP3 inflammasome and NF-kB signaling pathways. Front. Immunol. 16:1529756. doi: 10.3389/fimmu.2025.1529756
Kellogg, C., KV, L., and Tsigelny, I. F. (2022). Interactions of large T-antigen (LT) protein of polyomaviruses with p53 unfold their cancerogenic potential. J. Biomol. Struct. Dyn. 40, 5243–5252. doi: 10.1080/07391102.2020.1869097
Keup, C., Kimmig, R., and Kasimir-Bauer, S. (2023). The diversity of liquid biopsies and their potential in breast Cancer management. Cancers 15:5463. doi: 10.3390/cancers15225463
Kgatle, M. M., Spearman, C. W., Kalla, A. A., and Hairwadzi, H. N. (2017). DNA oncogenic virus-induced oxidative stress, genomic damage, and aberrant epigenetic alterations. Oxidative Med. Cell. Longev. 2017:3179421. doi: 10.1155/2017/3179421
Khullar, V., and Firpi, R. J. (2015). Hepatitis C cirrhosis: new perspectives for diagnosis and treatment. World J. Hepatol. 7, 1843–1855. doi: 10.4254/wjh.v7.i14.1843
Kidd, L. C., Chaing, S., Chipollini, J., Giuliano, A. R., Spiess, P. E., and Sharma, P. (2017). Relationship between human papillomavirus and penile cancer-implications for prevention and treatment. Transl. Androl. Urol. 6, 791–802. doi: 10.21037/tau.2017.06.27
Kim, K.-D., and Lieberman, P. M. (2024). Viral remodeling of the 4D nucleome. Exp. Mol. Med. 56, 799–808. doi: 10.1038/s12276-024-01207-0
Kimberlin, D. W. (2023). “295 - Antiviral Agents” in Principles and practice of pediatric infectious diseases. ed. S. S. Long. 6th ed (Philadelphia: Elsevier), 1583–1598.
King, H. W., and Klose, R. J. (2017). The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 6:6. doi: 10.7554/eLife.22631
Klose, R. J., and Zhang, Y. (2007). Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318. doi: 10.1038/nrm2143
Knowlton, E. R., Lepone, L. M., Li, J., Rappocciolo, G., Jenkins, F. J., and Rinaldo, C. R. (2013). Professional antigen presenting cells in human herpesvirus 8 infection. Front. Immunol. 3:427. doi: 10.3389/fimmu.2012.00427
Kong, I. Y., and Giulino-Roth, L. (2024). Targeting latent viral infection in EBV-associated lymphomas. Front. Immunol. 15, 1–10. doi: 10.3389/fimmu.2024.1342455
Koon, H. B., Krown, S. E., Lee, J. Y., Honda, K., Rapisuwon, S., Wang, Z., et al. (2013). Phase II trial of imatinib in AIDS-associated Kaposi's sarcoma: AIDS malignancy consortium protocol 042. J. Clin. Oncol. 32, 402–408. doi: 10.1200/JCO.2012.48.6365
Kotulkar, M., Robarts, D. R., and Apte, U. (2023). HNF4α in hepatocyte health and disease. Semin. Liver Dis. 43, 234–244. doi: 10.1055/a-2097-0660
Kramvis, A., Mammas, I. N., and Spandidos, D. A. (2023). Exploring the optimal vaccination strategy against hepatitis B virus in childhood (review). Biomed Rep. 19:48. doi: 10.3892/br.2023.1631
Krump, N. A., and You, J. (2018). Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 16, 684–698. doi: 10.1038/s41579-018-0064-6
Kudo, M. (2024). Nivolumab plus Ipilimumab: a novel first-line combination immunotherapy for Unresectable hepatocellular carcinoma. Liver Cancer 13, 459–467. doi: 10.1159/000540801
Kulkarni, M., Tan, T. Z., Syed Sulaiman, N. B., Lamar, J. M., Bansal, P., Cui, J., et al. (2018). RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget 9, 14175–14192. doi: 10.18632/oncotarget.24419
Kumar, A., Combe, E., Mougené, L., Zoulim, F., and Testoni, B. (2024). Applications of CRISPR/Cas as a toolbox for hepatitis B virus detection and therapeutics. Viruses 16:1565. doi: 10.3390/v16101565
Kurvinen, K., Yliskoski, M., Saarikoski, S., Syrjänen, K., and Syrjänen, S. (2000). Variants of the long control region of human papillomavirus type 16. Eur. J. Cancer 36, 1402–1410. doi: 10.1016/s0959-8049(00)00121-0
Kuss-Duerkop, S. K., Westrich, J. A., and Pyeon, D. (2018). DNA tumor virus regulation of host DNA methylation and its implications for immune evasion and oncogenesis. Viruses 10:82. doi: 10.3390/v10020082
Kwun, H. J., Shuda, M., Feng, H., Camacho, C. J., Moore, P. S., and Chang, Y. (2013). Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 14, 125–135. doi: 10.1016/j.chom.2013.06.008
Lachenmayer, A., Toffanin, S., Cabellos, L., Alsinet, C., Hoshida, Y., Villanueva, A., et al. (2012). Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 56, 1343–1350. doi: 10.1016/j.jhep.2012.01.009
Lan, J., Wang, Y., Yue, S., Xu, D., Li, Y., Peng, X., et al. (2023). Targeting FoxO proteins induces lytic reactivation of KSHV for treating herpesviral primary effusion lymphoma. PLoS Pathog. 19:e1011581. doi: 10.1371/journal.ppat.1011581
Lee, M. Y., and Allen, C. T. (2021). Immunotherapy for HPV malignancies. Semin. Radiat. Oncol. 31, 361–370. doi: 10.1016/j.semradonc.2021.02.008
Lee, C., and Cheung, S. T. (2019). STAT3: An emerging therapeutic target for hepatocellular carcinoma. Cancers 11, 1–20. doi: 10.3390/cancers11111646
Lee, C. S., Friedman, J. R., Fulmer, J. T., and Kaestner, K. H. (2005). The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–947. doi: 10.1038/nature03649
Leong, M. M. L., and Lung, M. L. (2021). The impact of Epstein-Barr virus infection on epigenetic regulation of host cell gene expression in epithelial and lymphocytic malignancies. Front. Oncol. 11:629780. doi: 10.3389/fonc.2021.629780
Letafati, A., Mehdigholian Chaijani, R., Edalat, F., Eslami, N., Askari, H., Askari, F., et al. (2025). Advances in epigenetic treatment of adult T-cell leukemia/lymphoma: a comprehensive review. Clin. Epigenetics 17:39. doi: 10.1186/s13148-025-01841-z
Letafati, A., Mozhgani, S. H., Marjani, A., Amiri, A., Siami, Z., Mohammaditabar, M., et al. (2023). Decoding dysregulated angiogenesis in HTLV-1 asymptomatic carriers compared to healthy individuals. Medical Oncol. 40:317. doi: 10.1007/s12032-023-02177-5
Levrero, M., and Zucman-Rossi, J. (2016). Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 64, S84–S101. doi: 10.1016/j.jhep.2016.02.021
Li, W., Duan, X., Chen, X., Zhan, M., Peng, H., Meng, Y., et al. (2022). Immunotherapeutic approaches in EBV-associated nasopharyngeal carcinoma. Front. Immunol. 13:1079515. doi: 10.3389/fimmu.2022.1079515
Li, B., Yan, C., Zhu, J., Chen, X., Fu, Q., Zhang, H., et al. (2020). Anti-PD-1/PD-L1 blockade immunotherapy employed in treating hepatitis B virus infection-related advanced hepatocellular carcinoma: a literature review. Front. Immunol. 11:1037. doi: 10.3389/fimmu.2020.01037
Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., and Zhao, X. (2020). Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 5:1. doi: 10.1038/s41392-019-0089-y
Li, N., Yang, L., Qi, X. K., Lin, Y. X., Xie, X., He, G. P., et al. (2018). BET bromodomain inhibitor JQ1 preferentially suppresses EBV-positive nasopharyngeal carcinoma cells partially through repressing c-Myc. Cell Death Dis. 9:761. doi: 10.1038/s41419-018-0789-1
Li, Y., Zhao, B., Peng, J., Tang, H., Wang, S., Peng, S., et al. (2024). Inhibition of NF-κB signaling unveils novel strategies to overcome drug resistance in cancers. Drug Resist. Updat. 73:101042. doi: 10.1016/j.drup.2023.101042
Li, T., Zhu, Y., Cheng, F., Lu, C., Jung, J. U., and Gao, S. J. (2019). Oncogenic Kaposi's sarcoma-associated herpesvirus upregulates Argininosuccinate synthase 1, a rate-limiting enzyme of the Citrulline-nitric oxide cycle, to activate the STAT3 pathway and promote growth transformation. J. Virol. 93, 1–17. doi: 10.1128/JVI.01599-18
Lieberman, P. M. (2016). Epigenetics and genetics of viral latency. Cell Host Microbe 19, 619–628. doi: 10.1016/j.chom.2016.04.008
Lim, P. J., and Gallay, P. A. (2014). Hepatitis C NS5A protein: two drug targets within the same protein with different mechanisms of resistance. Curr. Opin. Virol. 8, 30–37. doi: 10.1016/j.coviro.2014.04.012
Lippman, Z., Gendrel, A. V., Black, M., Vaughn, M. W., Dedhia, N., McCombie, W. R., et al. (2004). Role of transposable elements in heterochromatin and epigenetic control. Nature 430, 471–476. doi: 10.1038/nature02651
Liu, Y., Li, J. Z., Yuan, X. H., Adler-Storthz, K., and Che, Z. (2002). An AP-1 binding site mutation in HPV-16 LCR enhances E6/E7 promoter activity in human oral epithelial cells. Virus Genes 24, 29–37. doi: 10.1023/a:1014081803232
Liu, F., Liao, Z., and Zhang, Z. (2023). MYC in liver cancer: mechanisms and targeted therapy opportunities. Oncogene 42, 3303–3318. doi: 10.1038/s41388-023-02861-w
Liu, J., Martin, H. J., Liao, G., and Hayward, S. D. (2007). The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 81, 10451–10459. doi: 10.1128/JVI.00804-07
Liu, X., Shen, H., Zhang, L., Huang, W., Zhang, S., and Zhang, B. (2024). Immunotherapy for recurrent or metastatic nasopharyngeal carcinoma. NPJ Precis. Oncol. 8:101. doi: 10.1038/s41698-024-00601-1
Liu, H., Xu, J., Yang, Y., Wang, X., Wu, E., Majerciak, V., et al. (2021). Oncogenic HPV promotes the expression of the long noncoding RNA lnc-FANCI-2 through E7 and YY1. Proc. Natl. Acad. Sci. USA 118:e2014195118. doi: 10.1073/pnas.2014195118
Lo Cigno, I., Calati, F., Girone, C., Catozzo, M., and Gariglio, M. (2024). High-risk HPV oncoproteins E6 and E7 and their interplay with the innate immune response: uncovering mechanisms of immune evasion and therapeutic prospects. J. Med. Virol. 96:e29685. doi: 10.1002/jmv.29685
Looi, C. K., Hii, L. W., Chung, F. F., Mai, C. W., Lim, W. M., and Leong, C. O. (2021). Roles of inflammasomes in Epstein-Barr virus-associated nasopharyngeal cancer. Cancers 13, 1–28. doi: 10.3390/cancers13081786
Losay, V. A., and Damania, B. (2025). Unraveling the Kaposi sarcoma-associated herpesvirus (KSHV) lifecycle: An overview of latency, lytic replication, and KSHV-associated diseases. Viruses 17:177. doi: 10.3390/v17020177
Low, Y. H., Loh, C. J. L., Peh, D. Y. Y., Chu, A. J. M., Han, S., and Toh, H. C. (2023). Pathogenesis and therapeutic implications of EBV-associated epithelial cancers. Front. Oncol. 13:1202117. doi: 10.3389/fonc.2023.1202117
Lu, G., Jin, S., Lin, S., Gong, Y., Zhang, L., Yang, J., et al. (2023). Update on histone deacetylase inhibitors in peripheral T-cell lymphoma (PTCL). Clin. Epigenetics 15:124. doi: 10.1186/s13148-023-01531-8
Lu, F., Stedman, W., Yousef, M., Renne, R., and Lieberman, P. M. (2010). Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 84, 2697–2706. doi: 10.1128/JVI.01997-09
Lu, F., Tsai, K., Chen, H. S., Wikramasinghe, P., Davuluri, R. V., Showe, L., et al. (2012). Identification of host-chromosome binding sites and candidate gene targets for Kaposi's sarcoma-associated herpesvirus LANA. J. Virol. 86, 5752–5762. doi: 10.1128/JVI.07216-11
Lurain, K., Yarchoan, R., and Uldrick, T. S. (2018). Treatment of Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Hematol. Oncol. Clin. North Am. 32, 75–88. doi: 10.1016/j.hoc.2017.09.007
Mac, M., and Moody, C. A. (2020). Epigenetic regulation of the human papillomavirus life cycle. Pathogens 9, 1–18. doi: 10.3390/pathogens9060483
Mackesy-Amiti, M.-E., Gutfraind, A., Tatara, E., Collier, N. T., Cotler, S. J., Page, K., et al. (2024). Modeling of randomized hepatitis C vaccine trials: bridging the gap between controlled human infection models and real-word testing. PNAS Nexus 4, 1–8. doi: 10.1093/pnasnexus/pgae564
Mahmood, F., Xu, R., Awan, M. U. N., Song, Y., Han, Q., Xia, X., et al. (2023). HBV vaccines: advances and development. Vaccine 11, 1–26. doi: 10.3390/vaccines11121862
Mamun, M. A., Mannoor, K., Cao, J., Qadri, F., and Song, X. (2020). SOX2 in cancer stemness: tumor malignancy and therapeutic potentials. J. Mol. Cell Biol. 12, 85–98. doi: 10.1093/jmcb/mjy080
Mansisidor, A. R., and Risca, V. I. (2022). Chromatin accessibility: methods, mechanisms, and biological insights. Nucleus 13, 236–276. doi: 10.1080/19491034.2022.2143106
Martin, C. J., and Moorehead, R. A. (2020). Polycomb repressor complex 2 function in breast cancer (review). Int. J. Oncol. 57, 1085–1094. doi: 10.3892/ijo.2020.5122
Martinez, M. P., Al-Saleem, J., and Green, P. L. (2019). Comparative virology of HTLV-1 and HTLV-2. Retrovirology 16:21. doi: 10.1186/s12977-019-0483-0
Martinez, M. G., Combe, E., Inchauspe, A., Mangeot, P. E., Delberghe, E., Chapus, F., et al. (2022). CRISPR-Cas9 targeting of hepatitis B virus covalently closed circular DNA generates transcriptionally active Episomal variants. MBio 13:e0288821. doi: 10.1128/mbio.02888-21
Martinez, M. G., Smekalova, E., Combe, E., Gregoire, F., Zoulim, F., and Testoni, B. (2022). Gene editing technologies to target HBV cccDNA. Viruses 14, 1–22. doi: 10.3390/v14122654
Matsuoka, M., and Mesnard, J.-M. (2020). HTLV-1 bZIP factor: the key viral gene for pathogenesis. Retrovirology 17:2. doi: 10.1186/s12977-020-0511-0
McBride, A. A. (2017). Mechanisms and strategies of papillomavirus replication. Biol. Chem. 398, 919–927. doi: 10.1515/hsz-2017-0113
McLaughlin-Drubin, M. E., and Münger, K. (2009). Oncogenic activities of human papillomaviruses. Virus Res. 143, 195–208. doi: 10.1016/j.virusres.2009.06.008
Mesri, E. A., Feitelson, M. A., and Munger, K. (2014). Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe 15, 266–282. doi: 10.1016/j.chom.2014.02.011
Mistry, N., and Yeoman, A. (2023). Clinical and laboratory assessment of symptomatic and asymptomatic liver disease. Medicine 51, 305–310. doi: 10.1016/j.mpmed.2023.02.009
Mizuike, J., Suzuki, K., Tosaka, S., Kuze, Y., Kobayashi, S., Nakashima, M., et al. (2025). Rewired chromatin structure and epigenetic gene dysregulation during HTLV-1 infection to leukemogenesis. Cancer Sci. 116, 513–523. doi: 10.1111/cas.16388
Mohanty, S., and Harhaj, E. W. (2023). Mechanisms of innate immune sensing of HTLV-1 and viral immune evasion. Pathogens 12, 1–22. doi: 10.3390/pathogens12050735
Mon, H.-C., Lee, P.-C., Hung, Y.-P., Hung, Y.-W., Wu, C.-J., Lee, C.-J., et al. (2025). Functional cure of hepatitis B in patients with cancer undergoing immune checkpoint inhibitor therapy. J. Hepatol. 82, 51–61. doi: 10.1016/j.jhep.2024.07.018
Montalto, F. I., and De Amicis, F. (2020). Cyclin D1 in Cancer: a molecular connection for cell cycle control, adhesion and invasion in tumor and stroma. Cells 9, 1–15. doi: 10.3390/cells9122648
Moonen, J. R., Chappell, J., Shi, M., Shinohara, T., Li, D., Mumbach, M. R., et al. (2022). KLF4 recruits SWI/SNF to increase chromatin accessibility and reprogram the endothelial enhancer landscape under laminar shear stress. Nat. Commun. 13:4941. doi: 10.1038/s41467-022-32566-9
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. doi: 10.1038/npp.2012.112
Morrison, O., and Thakur, J. (2021). Molecular complexes at Euchromatin, heterochromatin and Centromeric chromatin. Int. J. Mol. Sci. 22, 1–28. doi: 10.3390/ijms22136922
Morselli, M., and Dieci, G. (2022). Epigenetic regulation of human non-coding RNA gene transcription. Biochem. Soc. Trans. 50, 723–736. doi: 10.1042/BST20210860
Mui, U. N., Haley, C. T., and Tyring, S. K. (2017). Viral oncology: molecular biology and pathogenesis. J. Clin. Med. 6, 1–58. doi: 10.3390/jcm6120111
Mukherji, A., Jühling, F., Simanjuntak, Y., Crouchet, E., Del Zompo, F., Teraoka, Y., et al. (2024). An atlas of the human liver diurnal transcriptome and its perturbation by hepatitis C virus infection. Nat. Commun. 15:7486. doi: 10.1038/s41467-024-51698-8
Murata, T., Sugimoto, A., Inagaki, T., Yanagi, Y., Watanabe, T., Sato, Y., et al. (2021). Molecular basis of Epstein-Barr virus latency establishment and lytic reactivation. Viruses 13, 1–20. doi: 10.3390/v13122344
Murphy, S. A., Mapes, N. J., Dua, D., and Kaur, B. (2022). Histone modifiers at the crossroads of oncolytic and oncogenic viruses. Mol. Ther. 30, 2153–2162. doi: 10.1016/j.ymthe.2022.02.006
Naimo, E., Zischke, J., and Schulz, T. F. (2021). Recent advances in developing treatments of Kaposi's sarcoma herpesvirus-related diseases. Viruses 13, 1–16. doi: 10.3390/v13091797
Najafi, S., Tan, S. C., Aghamiri, S., Raee, P., Ebrahimi, Z., Jahromi, Z. K., et al. (2022). Therapeutic potentials of CRISPR-Cas genome editing technology in human viral infections. Biomed. Pharmacother. 148:112743. doi: 10.1016/j.biopha.2022.112743
Nastoupil, L. J., Salles, G., Leonard, J. P., Morschhauser, F., Veazey, K. J., Perrot, V., et al. (2023). An externally controlled trial to indirectly compare tazemetostat plus lenalidomide and rituximab versus lenalidomide and rituximab after at least one prior systemic therapy in patients with refractory and relapsed follicular lymphoma. Blood 142:4410. doi: 10.1182/blood-2023-181807
Neugebauer, E., Bastidas-Quintero, A. M., Weidl, D., and Full, F. (2023). Pioneer factors in viral infection. Front. Immunol. 14, 1–9. doi: 10.3389/fimmu.2023.1286617
Neuwelt, A., Al-Juhaishi, T., Davila, E., and Haverkos, B. (2020). Enhancing antitumor immunity through checkpoint blockade as a therapeutic strategy in T-cell lymphomas. Blood Adv. 4, 4256–4266. doi: 10.1182/bloodadvances.2020001966
Nevola, R., Beccia, D., Rosato, V., Ruocco, R., Mastrocinque, D., Villani, A., et al. (2023). HBV infection and host interactions: the role in viral persistence and oncogenesis. Int. J. Mol. Sci. 24:7651. doi: 10.3390/ijms24087651
Niller, H. H., Banati, F., Ay, E., and Minarovits, J. (2012). Epigenetic changes in virus-associated neoplasms In Patho-Epigenetics of Disease. Eds. J. Minarovits and H. H. Niller (New York: Springer) pp. 179–225.
Niller, H. H., Banati, F., Salamon, D., and Minarovits, J. (2016). Epigenetic alterations in Epstein-Barr virus-associated diseases. In Patho-Epigenetics of Infectious Disease. Eds. J. Minarovits and H. H. Niller (Springer International Publishing) pp. 39–69.
Ocana, A., Corrales-Sanchez, V., Peña, J. P., Serrano-Heras, G., Gascon-Escribano, M. J., Montero, J. C., et al. (2015). Anti-proliferative activity of bromodomain inhibitors JQ1 and OTX015 in triple negative breast cancer. J. Clin. Oncol. 33:e12078-e. doi: 10.1200/jco.2015.33.15_suppl.e12078
Okpara, M. O., Vaaltyn, M. C., Watson, J. L., Alhassan, M., Albericio, F., de la Torre, B. G., et al. (2024). Modulators of the hop-HSP90 protein–protein interaction disrupt KSHV lytic replication. ACS Infect. Dis. 10, 3853–3867. doi: 10.1021/acsinfecdis.4c00429
Okushin, K., Suzuki, R., Tsutsumi, T., Okamoto, K., Ikeuchi, K., Kado, A., et al. (2021). Change in hepatitis C virus positivity among needle-stick injury source patients: a 10-year experience in a Japanese tertiary hospital. BMC Infect. Dis. 21:399. doi: 10.1186/s12879-021-06117-4
Ono, R., Masuya, M., Inoue, N., Shinmei, M., Ishii, S., Maegawa, Y., et al. (2021). Tet1 is not required for myeloid leukemogenesis by MLL-ENL in novel mouse models. PLoS One 16:e0248425. doi: 10.1371/journal.pone.0248425
Oumata, N., Zhong, Q., Zhang, Y., Galons, H., Andrei, G., and Zeng, M. (2025). Emerging drugs for Epstein-Barr virus associated-diseases. Eur. J. Med. Chem. 287:117386. doi: 10.1016/j.ejmech.2025.117386
Paixão, V. A., Vidal, D. O., Caballero, O. L., Vettore, A. L., Tone, L. G., Ribeiro, K. B., et al. (2006). Hypermethylation of CpG island in the promoter region of CALCA in acute lymphoblastic leukemia with central nervous system (CNS) infiltration correlates with poorer prognosis. Leuk. Res. 30, 891–894. doi: 10.1016/j.leukres.2005.11.016
Pal, A., and Kundu, R. (2019). Human papillomavirus E6 and E7: the cervical cancer hallmarks and targets for therapy. Front. Microbiol. 10:3116. doi: 10.3389/fmicb.2019.03116
Pan, Z., Seto, W.-K., Liu, C.-J., Mao, Y., Alqahtani, S. A., and Eslam, M. (2024). A literature review of genetics and epigenetics of HCV-related hepatocellular carcinoma: translational impact. Hepatobiliary Surgery Nutr. 13, 650–661. doi: 10.21037/hbsn-23-562
Pan, Y., Xia, H., He, Y., Zeng, S., Shen, Z., and Huang, W. (2023). The progress of molecules and strategies for the treatment of HBV infection. Front. Cell. Infect. Microbiol. 13:1128807. doi: 10.3389/fcimb.2023.1128807
Parija, S. C. (2023). Textbook of microbiology and immunology. Vol. 1579. Berlin, Heidelberg, Germany: Springer.
Park, D. E., Cheng, J., McGrath, J. P., Lim, M. Y., Cushman, C., Swanson, S. K., et al. (2020). Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 22, 603–615. doi: 10.1038/s41556-020-0503-2
Pathak, P., Pajai, S., and Kesharwani, H. (2022). A review on the use of the HPV vaccine in the prevention of cervical Cancer. Cureus 14:e28710. doi: 10.7759/cureus.28710
Patnaik, E., Madu, C., and Lu, Y. (2023). Epigenetic modulators as therapeutic agents in Cancer. Int. J. Mol. Sci. 24, 1–13. doi: 10.3390/ijms241914964
Pedersen, E. A., Verhaegen, M. E., Joseph, M. K., Harms, K. L., and Harms, P. W. (2024). Merkel cell carcinoma: updates in tumor biology, emerging therapies, and preclinical models. Front. Oncol. 14:1413793. doi: 10.3389/fonc.2024.1413793
Pei, Y., Wong, J. H.-Y., and Robertson, E. S. (2020). Herpesvirus epigenetic reprogramming and oncogenesis. Annual Review Virol. 7, 309–331. doi: 10.1146/annurev-virology-020420-014025
Péneau, C., Imbeaud, S., La Bella, T., Hirsch, T. Z., Caruso, S., Calderaro, J., et al. (2022). Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 71, 616–626. doi: 10.1136/gutjnl-2020-323153
Peng, W. Y., Abere, B., Shi, H., Toland, S., Smithgall, T. E., Moore, P. S., et al. (2023). Membrane-bound Merkel cell polyomavirus middle T protein constitutively activates PLCγ1 signaling through Src-family kinases. Proc. Natl. Acad. Sci. USA 120:e2316467120. doi: 10.1073/pnas.2316467120
Perez, S., Kaspi, A., Domovitz, T., Davidovich, A., Lavi-Itzkovitz, A., Meirson, T., et al. (2019). Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 15:e1008181. doi: 10.1371/journal.pgen.1008181
Pfefferlé, M., and Vallelian, F. (2024). Transcription factor NRF2 in shaping myeloid cell differentiation and function. Adv. Exp. Med. Biol. 1459, 159–195. doi: 10.1007/978-3-031-62731-6_8
Pietropaolo, V., Prezioso, C., and Moens, U. (2021). Role of virus-induced host cell epigenetic changes in Cancer. Int. J. Mol. Sci. 22, 1–51. doi: 10.3390/ijms22158346
Platt, G., Carbone, A., and Mittnacht, S. (2002). p16INK4a loss and sensitivity in KSHV associated primary effusion lymphoma. Oncogene 21, 1823–1831. doi: 10.1038/sj.onc.1205360
Pluta, A., Jaworski, J. P., and Douville, R. N. (2020). Regulation of expression and latency in BLV and HTLV. Viruses 12, 1–31. doi: 10.3390/v12101079
Rajan, P. K., Udoh, U. A., Sanabria, J. D., Banerjee, M., Smith, G., Schade, M. S., et al. (2020). The role of histone acetylation−/methylation-mediated apoptotic gene regulation in hepatocellular carcinoma. Int. J. Mol. Sci. 21, 1–21. doi: 10.3390/ijms21238894
Rasmussen, K. D., and Helin, K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750. doi: 10.1101/gad.276568.115
Ratner, L. (2021). Epigenetic regulation of human T-cell leukemia virus gene expression. Microorganisms 10, 1–12. doi: 10.3390/microorganisms10010084
Rizzitano, S., Cavanè, A., Piazzoni, M., Vendramin, A., Gimondi, S., Biancon, G., et al. (2016). Synergistic anti-tumor efficacy of BET inhibitors JQ1 and Otx-015 in combination with Dasatinib in preclinical models of T-cell lymphomas. Blood 128:3967. doi: 10.1182/blood.V128.22.3967.3967
Robinson, W. S., Clayton, D. A., and Greenman, R. L. (1974). DNA of a human hepatitis B virus candidate. J. Virol. 14, 384–391. doi: 10.1128/jvi.14.2.384-391.1974
Romero-López, C., and Berzal-Herranz, A. (2020). The role of the RNA-RNA Interactome in the hepatitis C virus life cycle. Int. J. Mol. Sci. 21:1479. doi: 10.3390/ijms21041479
Rongrui, L., Na, H., Zongfang, L., Fanpu, J., and Shiwen, J. (2014). Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 20, 1715–1725. doi: 10.2174/13816128113199990533
Rosenkranz, AL (2023). Determining secondary structural elements of hepatitis B virus pregenomic RNA and preliminary analysis of the DDB1-HBx interaction as a therapeutic target. (Doctoral dissertation, Drexel University).
Rosenthal, A. C., Munoz, J. L., and Villasboas, J. C. (2023). Clinical advances in epigenetic therapies for lymphoma. Clin. Epigenetics 15:39. doi: 10.1186/s13148-023-01452-6
Rotondo, J. C., Mazziotta, C., Lanzillotti, C., Tognon, M., and Martini, F. (2021). Epigenetic dysregulations in Merkel cell polyomavirus-driven Merkel cell carcinoma. Int. J. Mol. Sci. 22:11464. doi: 10.3390/ijms222111464
Russell, S. J., and Peng, K.-W. (2017). Oncolytic Virotherapy: a contest between apples and oranges. Mol. Ther. 25, 1107–1116. doi: 10.1016/j.ymthe.2017.03.026
Ryu, H. Y., and Hochstrasser, M. (2021). Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 49, 6043–6052. doi: 10.1093/nar/gkab280
Salnikov, M. Y., MacNeil, K. M., and Mymryk, J. S. (2024). The viral etiology of EBV-associated gastric cancers contributes to their unique pathology, clinical outcomes, treatment responses and immune landscape. Front. Immunol. 15, 1–17. doi: 10.3389/fimmu.2024.1358511
Sanaei, M., and Kavoosi, F. (2021). Effect of vorinostat on INK4 family and HDACs 1, 2, and 3 in pancreatic cancer and hepatocellular carcinoma. Res. Pharmaceutical Sci. 16, 260–268. doi: 10.4103/1735-5362.314824
Sarid, R., Dünn-Kittenplon, D., and Calabrò, M. L. (2020). “Human herpesviruses: Kaposi’s sarcoma and other malignancies” in Viral infections of humans: Epidemiology and control. eds. R. A. Kaslow, L. R. Stanberry, and A. M. Powers (New York, NY: Springer US), 1–72.
Sarkozy, C., Morschhauser, F., Dubois, S., Molina, T., Michot, J. M., Cullières-Dartigues, P., et al. (2020). A LYSA phase Ib study of Tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin. Cancer Res. 26, 3145–3153. doi: 10.1158/1078-0432.CCR-19-3741
Sasvari, Z., and Nagy, P. D. (2010). Making of viral replication organelles by remodeling interior membranes. Viruses 2, 2436–2442. doi: 10.3390/v2112436
Schadendorf, D., Paul, N., Shailender, B., Axel, H., Philippe, S., Lisa, M., et al. (2017). Immune evasion mechanisms and immune checkpoint inhibition in advanced Merkel cell carcinoma. Onco Targets Ther 6:e1338237. doi: 10.1080/2162402X.2017.1338237
Schinzari, V., Barnaba, V., and Piconese, S. (2015). Chronic hepatitis B virus and hepatitis C virus infections and cancer: synergy between viral and host factors. Clin. Microbiol. Infect. 21, 969–974. doi: 10.1016/j.cmi.2015.06.026
Schlesinger, Y., Straussman, R., Keshet, I., Farkash, S., Hecht, M., Zimmerman, J., et al. (2007). Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 39, 232–236. doi: 10.1038/ng1950
Scott, R. S. (2017). Epstein-Barr virus: a master epigenetic manipulator. Curr. Opin. Virol. 26, 74–80. doi: 10.1016/j.coviro.2017.07.017
Seeger, C., and Mason, W. S. (2000). Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64, 51–68. doi: 10.1128/MMBR.64.1.51-68.2000
Sen, P., Ganguly, P., and Ganguly, N. (2018). Modulation of DNA methylation by human papillomavirus E6 and E7 oncoproteins in cervical cancer. Oncol. Lett. 15, 11–22. doi: 10.3892/ol.2017.7292
Shamay, M., Krithivas, A., Zhang, J., and Hayward, S. D. (2006). Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 103, 14554–14559. doi: 10.1073/pnas.0604469103
Shan, K. S., Bonano-Rios, A., Theik, N. W. Y., Hussein, A., and Blaya, M. (2024). Molecular targeting of the Phosphoinositide-3-protein kinase (PI3K) pathway across various cancers. Int. J. Mol. Sci. 25:1973. doi: 10.3390/ijms25041973
Shanmugam, G., Jeyaraj, G., and Sarkar, K. (2025). Molecular mechanisms and diagnostic innovations in HPV-associated head and neck squamous cell carcinomas: insights into integration, epigenetic modifications, and biomarker applications. Oral Oncol. Rep. 13:100710. doi: 10.1016/j.oor.2024.100710
Shareena, G., and Kumar, D. (2023). Epigenetics of Epstein Barr virus - a review. Biochim. Biophys. Acta Mol. basis Dis. 1869:166838. doi: 10.1016/j.bbadis.2023.166838
Sheng, W., LaFleur, M. W., Nguyen, T. H., Chen, S., Chakravarthy, A., Conway, J. R., et al. (2018). LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–63.e19. doi: 10.1016/j.cell.2018.05.052
Shiri Aghbash, P., Hemmat, N., Baradaran, B., and Bannazadeh Baghi, H. (2023). SiRNA-E6 sensitizes HPV-16-related cervical cancer through oxaliplatin: an in vitro study on anti-cancer combination therapy. Eur. J. Med. Res. 28:42. doi: 10.1186/s40001-023-01014-9
Shuda, M., Feng, H., Kwun, H. J., Rosen, S. T., Gjoerup, O., Moore, P. S., et al. (2008). T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 105, 16272–16277. doi: 10.1073/pnas.0806526105
Sinha, K. K., Bilokapic, S., Du, Y., Malik, D., and Halic, M. (2023). Histone modifications regulate pioneer transcription factor cooperativity. Nature 619, 378–384. doi: 10.1038/s41586-023-06112-6
Sivasudhan, E., Blake, N., Lu, Z., Meng, J., and Rong, R. (2022). Hepatitis B viral protein HBx and the molecular mechanisms modulating the hallmarks of hepatocellular carcinoma: a comprehensive review. Cells 11:741. doi: 10.3390/cells11040741
Smith, E., Stiff, A., Patton, J. T., Carson, W. III, Lapalombella, R., and Baiocchi, R. A. (2016). BET Bromodomain inhibition selectively targets the Epstein-Barr virus oncogene LMP1 while promoting virus-specific, adaptive anti-tumor activity. Blood 128:46. doi: 10.1182/blood.V128.22.46.46
Soldan, S. S., Messick, T. E., and Lieberman, P. M. (2022). Therapeutic approaches to Epstein-Barr virus cancers. Curr. Opin. Virol. 56:101260. doi: 10.1016/j.coviro.2022.101260
Soliman, S. H. A., Orlacchio, A., and Verginelli, F. (2021). Viral manipulation of the host epigenome as a driver of virus-induced oncogenesis. Microorganisms 9, 1–40. doi: 10.3390/microorganisms9061179
Song, D., Lian, Y., and Zhang, L. (2023). The potential of activator protein 1 (AP-1) in cancer targeted therapy. Front. Immunol. 14:1224892. doi: 10.3389/fimmu.2023.1224892
Song, W., Liu, Y., Liu, Y., Zhang, C., Yuan, B., Zhang, L., et al. (2014). Increased p16 DNA methylation in mouse thymic lymphoma induced by irradiation. PLoS One 9:e93850. doi: 10.1371/journal.pone.0093850
Soto, D., Song, C., and McLaughlin-Drubin, M. E. (2017). Epigenetic alterations in human papillomavirus-associated cancers. Viruses 9, 1–18. doi: 10.3390/v9090248
Soufi, A., Garcia, M. F., Jaroszewicz, A., Osman, N., Pellegrini, M., and Zaret, K. S. (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. doi: 10.1016/j.cell.2015.03.017
Stakaitytė, G., Wood, J. J., Knight, L. M., Abdul-Sada, H., Adzahar, N. S., Nwogu, N., et al. (2014). Merkel cell polyomavirus: molecular insights into the most recently discovered human tumour virus. Cancers 6, 1267–1297. doi: 10.3390/cancers6031267
Stathis, A., and Bertoni, F. (2018). BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24–36. doi: 10.1158/2159-8290.CD-17-0605
Stewart, D. J., Issa, J. P., Kurzrock, R., Nunez, M. I., Jelinek, J., Hong, D., et al. (2009). Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin. Cancer Res. 15, 3881–3888. doi: 10.1158/1078-0432.CCR-08-2196
Su, L., Bu, J., Yu, J., Jin, M., Meng, G., and Zhu, X. (2024). Comprehensive review and updated analysis of DNA methylation in hepatocellular carcinoma: from basic research to clinical application. Clin. Transl. Med. 14:e70066. doi: 10.1002/ctm2.70066
Su, Z. Y., Siak, P. Y., Leong, C. O., and Cheah, S. C. (2023). The role of Epstein-Barr virus in nasopharyngeal carcinoma. Front. Microbiol. 14:1116143. doi: 10.3389/fmicb.2023.1116143
Sun, R., Liang, D., Gao, Y., and Lan, K. (2014). Kaposi's sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 88, 7331–7344. doi: 10.1128/JVI.00596-14
Sun, F., Xiao, Y., and Qu, Z. (2015). Oncovirus Kaposi sarcoma herpesvirus (KSHV) represses tumor suppressor PDLIM2 to persistently activate nuclear factor κB (NF-κB) and STAT3 transcription factors for tumorigenesis and tumor maintenance. J. Biol. Chem. 290, 7362–7368. doi: 10.1074/jbc.C115.637918
Sundeep, D., Umadevi, K., Masood, J., Deshmane, V. L., Raj, D. P., Varadharaj, E. K., et al. (n.d.). Human T-cell leukemia virus (HTLV)-induced carcinogenesis and its molecular detection. In Viral Oncology. Boca Raton, Taylor and Francis online, UK: CRC Press, 197–236.
Sunil, M., Reid, E., and Lechowicz, M. J. (2010). Update on HHV-8-associated malignancies. Curr. Infect. Dis. Rep. 12, 147–154. doi: 10.1007/s11908-010-0092-5
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Tan, S. K., and Pinsky, B. A. (2017). “Chapter 8 - molecular testing for herpes viruses” in Diagnostic molecular pathology. eds. W. B. Coleman and G. J. Tsongalis (Cambridge, Massachusetts, USA: Academic Press), 89–101.
Tanaka, A., Ishitsuka, Y., Ohta, H., Takenouchi, N., Nakagawa, M., Koh, K. R., et al. (2025). Integrative analysis of ATAC-seq and RNA-seq for cells infected by human T-cell leukemia virus type 1. PLoS Comput. Biol. 21:e1012690. doi: 10.1371/journal.pcbi.1012690
Teeli, A. S., Łuczyńska, K., Haque, E., Gayas, M. A., Winiarczyk, D., and Taniguchi, H. (2021). Disruption of tumor suppressors HNF4α/HNF1α causes tumorigenesis in liver. Cancers 13, 1–29. doi: 10.3390/cancers13215357
Teperino, R., Schoonjans, K., and Auwerx, J. (2010). Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 12, 321–327. doi: 10.1016/j.cmet.2010.09.004
Thienpont, B., Steinbacher, J., Zhao, H., D'Anna, F., Kuchnio, A., Ploumakis, A., et al. (2016). Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68. doi: 10.1038/nature19081
Tiwari, N., Meyer-Schaller, N., Arnold, P., Antoniadis, H., Pachkov, M., van Nimwegen, E., et al. (2013). Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS One 8:e57329. doi: 10.1371/journal.pone.0057329
Tolstov, Y. L., Pastrana, D. V., Feng, H., Becker, J. C., Jenkins, F. J., Moschos, S., et al. (2009). Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 125, 1250–1256. doi: 10.1002/ijc.24509
Toth, Z., Papp, B., Brulois, K., Choi, Y. J., Gao, S. J., and Jung, J. U. (2016). LANA-mediated recruitment of host Polycomb repressive complexes onto the KSHV genome during De novo infection. PLoS Pathog. 12:e1005878. doi: 10.1371/journal.ppat.1005878
Trauner, M., and Halilbasic, E. (2011). Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology 140:e1-12, 1120–1125. doi: 10.1053/j.gastro.2011.02.044
Trimble, C. L., Morrow, M. P., Kraynyak, K. A., Shen, X., Dallas, M., Yan, J., et al. (2015). Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet 386, 2078–2088. doi: 10.1016/S0140-6736(15)00239-1
Tsang, C. M., Wu, M., and Lo, K. W. (2023). Molecular signaling pathways in nasopharyngeal carcinoma. In: Medical Radiology. Berlin, Heidelberg: Springer. p. 1–23.
Vallianou, I., Dafou, D., Vassilaki, N., Mavromara, P., and Hadzopoulou-Cladaras, M. (2016). Hepatitis C virus suppresses hepatocyte nuclear factor 4 alpha, a key regulator of hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 78, 315–326. doi: 10.1016/j.biocel.2016.07.027
van Rhee, F., Voorhees, P., Dispenzieri, A., Fosså, A., Srkalovic, G., Ide, M., et al. (2018). International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 132, 2115–2124. doi: 10.1182/blood-2018-07-862334
Vandeven, N., and Nghiem, P. (2016). Rationale for immune-based therapies in Merkel polyomavirus-positive and -negative Merkel cell carcinomas. Immunotherapy 8, 907–921. doi: 10.2217/imt-2016-0009
Varley, K. E., Gertz, J., Bowling, K. M., Parker, S. L., Reddy, T. E., Pauli-Behn, F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567. doi: 10.1101/gr.147942.112
Verbeke, S., Bourdon, A., Lafon, M., Chaire, V., Frederic, B., Naït Eldjoudi, A., et al. (2025). Dual inhibition of BET and EP300 has antitumor activity in undifferentiated pleomorphic sarcomas and synergizes with ferroptosis induction. Transl. Oncol. 52:102236. doi: 10.1016/j.tranon.2024.102236
Verhees, F., Demers, I., Legemaate, D., Jacobs, R., Hoeben, A., Kremer, B., et al. (2025). Exploring the antiproliferative effect of PI3K/Akt/mTOR pathway and CDK4/6 inhibitors in human papillomavirus-positive and -negative head and neck squamous cell carcinoma cell lines. Int. J. Oncol. 66:13. doi: 10.3892/ijo.2025.5719
Verma, S. C., Borah, S., and Robertson, E. S. (2004). Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78, 10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004
Vieyres, G., Dubuisson, J., and Pietschmann, T. (2014). Incorporation of hepatitis C virus E1 and E2 glycoproteins: the keystones on a peculiar virion. Viruses 6, 1149–1187. doi: 10.3390/v6031149
Virzì, A., Roca Suarez, A. A., Baumert, T. F., and Lupberger, J. (2018). Oncogenic signaling induced by HCV infection. Viruses 10, 1–23. doi: 10.3390/v10100538
Vishnoi, K., Viswakarma, N., Rana, A., and Rana, B. (2020). Transcription factors in Cancer development and therapy. Cancers 12, 1–32. doi: 10.3390/cancers12082296
Wajed, S. A., Laird, P. W., and DeMeester, T. R. (2001). DNA methylation: an alternative pathway to cancer. Ann. Surg. 234, 10–20. doi: 10.1097/00000658-200107000-00003
Wallbillich, N. J., and Lu, H. (2023). Role of c-Myc in lung cancer: Progress, challenges, and prospects. Chin. Med. J. Pulmonary Critical Care Med. 1, 129–138. doi: 10.1016/j.pccm.2023.07.001
Wang, S., Chen, S., Zhong, Q., and Liu, Y. (2023). Immunotherapy for the treatment of advanced nasopharyngeal carcinoma: a promising new era. J. Cancer Res. Clin. Oncol. 149, 2071–2079. doi: 10.1007/s00432-022-04214-8
Wang, T. T., Hirons, A., Doerflinger, M., Morris, K. V., Ledger, S., Purcell, D. F. J., et al. (2024). Current state of therapeutics for HTLV-1. Viruses 16:1616. doi: 10.3390/v16101616
Wang, X., Li, J., Schowalter, R. M., Jiao, J., Buck, C. B., and You, J. (2012). Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 8:e1003021. doi: 10.1371/journal.ppat.1003021
Wang, Z., Liu, Z., Lv, M., Luan, Z., Li, T., and Hu, J. (2025). Novel histone modifications and liver cancer: emerging frontiers in epigenetic regulation. Clin. Epigenetics 17:30. doi: 10.1186/s13148-025-01838-8
Wang, H. Q., Man, Q. W., Huo, F. Y., Gao, X., Lin, H., Li, S. R., et al. (2022). STAT3 pathway in cancers: past, present, and future. MedComm 3:e124. doi: 10.1002/mco2.124
Wang, F., Marshall, C. B., and Ikura, M. (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cellular Molecular Life Sciences 70, 3989–4008. doi: 10.1007/s00018-012-1254-4
Warburton, A., Markowitz, T. E., Katz, J. P., Pipas, J. M., and McBride, A. A. (2021). Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. NPJ Genom. Med. 6:101. doi: 10.1038/s41525-021-00264-y
Wen, K. W., Wang, L., Menke, J. R., and Damania, B. (2022). Cancers associated with human gammaherpesviruses. FEBS J. 289, 7631–7669. doi: 10.1111/febs.16206
Wolff, G., Melia, C. E., Snijder, E. J., and Bárcena, M. (2020). Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 28, 1022–1033. doi: 10.1016/j.tim.2020.05.009
Wong, C. C., Li, W., Chan, B., and Yu, J. (2019). Epigenomic biomarkers for prognostication and diagnosis of gastrointestinal cancers. Semin. Cancer Biol. 55, 90–105. doi: 10.1016/j.semcancer.2018.04.002
Wu, X., Ma, W., Mei, C., Chen, X., Yao, Y., Liu, Y., et al. (2020). Description of CRISPR/Cas9 development and its prospect in hepatocellular carcinoma treatment. J. Exp. Clin. Cancer Res. 39:97. doi: 10.1186/s13046-020-01603-0
Xu, D., Jiang, S., He, Y., Jin, X., Zhao, G., and Wang, B. (2021). Development of a therapeutic vaccine targeting Merkel cell polyomavirus capsid protein VP1 against Merkel cell carcinoma. NPJ vaccines. 6:119. doi: 10.1038/s41541-021-00382-9
Xu, J., Lin, H., Wu, G., Zhu, M., and Li, M. (2021). IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front. Oncol. 11:760971. doi: 10.3389/fonc.2021.760971
Yager, J. E., Magaret, A. S., Kuntz, S. R., Selke, S., Huang, M. L., Corey, L., et al. (2017). Valganciclovir for the suppression of Epstein-Barr virus replication. J. Infect. Dis. 216, 198–202. doi: 10.1093/infdis/jix263
Yamagishi, M., Fujikawa, D., Watanabe, T., and Uchimaru, K. (2018). HTLV-1-mediated epigenetic pathway to adult T-cell leukemia-lymphoma. Front. Microbiol. 9:1686. doi: 10.3389/fmicb.2018.01686
Yan, W., Rao, D., Fan, F., Liang, H., Zhang, Z., and Dong, H. (2024). Hepatitis B virus X protein and TGF-β: partners in the carcinogenic journey of hepatocellular carcinoma. Front. Oncol. 14:1407434. doi: 10.3389/fonc.2024.1407434
Yang, Y. M., Kim, S. Y., and Seki, E. (2019). Inflammation and liver cancer: molecular mechanisms and therapeutic targets. Semin. Liver Dis. 39, 26–42. doi: 10.1055/s-0038-1676806
Yang, Z., Xiang, Q., and Nicholas, J. (2023). Direct and biologically significant interactions of human herpesvirus 8 interferon regulatory factor 1 with STAT3 and Janus kinase TYK2. PLoS Pathog. 19:e1011806. doi: 10.1371/journal.ppat.1011806
Yang, Y., Yan, Y., Chen, Z., Hu, J., Wang, K., Tang, N., et al. (2021). Histone deacetylase inhibitors Romidepsin and Vorinostat promote hepatitis B virus replication by inducing cell cycle arrest. J. Clin. Transl. Hepatol. 9, 160–168. doi: 10.14218/JCTH.2020.00105
Yang, W. T., and Zheng, P. S. (2012). Krüppel-like factor 4 functions as a tumor suppressor in cervical carcinoma. Cancer 118, 3691–3702. doi: 10.1002/cncr.26698
Yang, H., Zhou, Y., Mo, J., Xiang, Q., Qin, M., Liu, W., et al. (2020). SOX9 represses hepatitis B virus replication through binding to HBV EnhII/Cp and inhibiting the promoter activity. Antivir. Res. 177:104761. doi: 10.1016/j.antiviral.2020.104761
Yao, Z. Q., Schank, M. B., Zhao, J., El Gazzar, M., Wang, L., Zhang, Y., et al. (2024). The potential of HBV cure: an overview of CRISPR-mediated HBV gene disruption. Front Genome Editing 6:6. doi: 10.3389/fgeed.2024.1467449
Yee, GPC (2013). Targeting the transcription factor c-Jun in cervical cancer cells. Doctoral dissertation: UNSW Sydney Yeo.
Yeo-Teh, N. S. L., Ito, Y., and Jha, S. (2018). High-risk human Papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int. J. Mol. Sci. 19, 1–27. doi: 10.3390/ijms19061706
Yim, E. K., and Park, J. S. (2005). The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 37, 319–324. doi: 10.4143/crt.2005.37.6.319
Young, L. S., and Rickinson, A. B. (2004). Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768. doi: 10.1038/nrc1452
Yuan, H., Xu, R., Li, S., Zheng, M., Tong, Q., Xiang, M., et al. (2025). The malignant transformation of viral hepatitis to hepatocellular carcinoma: mechanisms and interventions. MedComm. 6:e70121. doi: 10.1002/mco2.70121
Zabeti Touchaei, A., and Vahidi, S. (2024). MicroRNAs as regulators of immune checkpoints in cancer immunotherapy: targeting PD-1/PD-L1 and CTLA-4 pathways. Cancer Cell Int. 24:102. doi: 10.1186/s12935-024-03293-6
Zakeri, M., Atefe, P. J., Ahmad, T., Alireza, Z., and Razizadeh, M. H. (2024). The role of NF-κB signaling pathway in viral hepatitis and gastroenteritis: pathogenesis and therapeutic applications. Future Virol. 19, 435–452. doi: 10.1080/17460794.2024.2415216
Zeidan, A. M., Fenaux, P., Gobbi, M., Mayer, J., Roboz, G. J., Krauter, J., et al. (2022). Prospective comparison of outcomes with azacitidine and decitabine in patients with AML ineligible for intensive chemotherapy. Blood 140, 285–289. doi: 10.1182/blood.2022015832
Zeisel, M. B., Guerrieri, F., and Levrero, M. (2021). Host epigenetic alterations and hepatitis B virus-associated hepatocellular carcinoma. J. Clin. Med. 10, 1–16. doi: 10.3390/jcm10081715
Zhang, T., Cooper, S., and Brockdorff, N. (2015). The interplay of histone modifications - writers that read. EMBO Rep. 16, 1467–1481. doi: 10.15252/embr.201540945
Zhang, F., Li, W., Zheng, X., Ren, Y., Li, L., and Yin, H. (2024). The novel immune landscape of immune-checkpoint blockade in EBV-associated malignancies. FASEB J. 38:e70139. doi: 10.1096/fj.202301980RR
Zhang, R., Shen, C., Zhao, L., Wang, J., McCrae, M., Chen, X., et al. (2016). Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int. J. Cancer 138, 1163–1174. doi: 10.1002/ijc.29872
Zhang, Q., Wang, S., Chen, J., and Yu, Z. (2019). Histone deacetylases (HDACs) guided novel therapies for T-cell lymphomas. Int. J. Med. Sci. 16, 424–442. doi: 10.7150/ijms.30154
Zhang, L., Wang, R., and Xie, Z. (2022). The roles of DNA methylation on the promotor of the Epstein-Barr virus (EBV) gene and the genome in patients with EBV-associated diseases. Appl. Microbiol. Biotechnol. 106, 4413–4426. doi: 10.1007/s00253-022-12029-3
Zhao, Z., Hong, W., Zeng, Z., Wu, Y., Hu, K., Tian, X., et al. (2012). Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4α in vitro and in vivo. J. Biol. Chem. 287, 30181–30190. doi: 10.1074/jbc.M112.370312
Zhao, J., Jia, Y., Shen, S., Kim, J., Wang, X., Lee, E., et al. (2020). Merkel cell polyomavirus small T antigen activates noncanonical NF-κB signaling to promote tumorigenesis. Molecular Cancer Res. 18, 1623–1637. doi: 10.1158/1541-7786.MCR-20-0587
Zhao, P., Malik, S., and Xing, S. (2021). Epigenetic mechanisms involved in HCV-induced hepatocellular carcinoma (HCC). Front. Oncol. 11:677926. doi: 10.3389/fonc.2021.677926
Zhao, Y., Sheldon, M., Sun, Y., and Ma, L. (2023). New insights into YAP/TAZ-TEAD-mediated gene regulation and biological processes in Cancer. Cancers 15, 1–32. doi: 10.3390/cancers15235497
Zhou, X. J., Good, S. S., Pietropaolo, K., Huang, Q., Moussa, A., Hammond, J. M., et al. (2024). Bemnifosbuvir (BEM, AT-527), a novel nucleotide analogue inhibitor of the hepatitis C virus NS5B polymerase. Expert Opin. Investig. Drugs 33, 9–17. doi: 10.1080/13543784.2024.2305137
Zhou, Y.-Q., Jiang, J.-X., He, S., Li, Y.-Q., Cheng, X.-X., Liu, S.-Q., et al. (2025). Epstein-Barr virus hijacks histone demethylase machinery to drive epithelial malignancy progression through KDM5B upregulation. Signal Transduct. Target. Ther. 10:83. doi: 10.1038/s41392-025-02163-5
Zielińska, M. K., Ciążyńska, M., Sulejczak, D., Rutkowski, P., and Czarnecka, A. M. (2025). Mechanisms of resistance to anti-PD-1 immunotherapy in melanoma and strategies to overcome it. Biomol. Ther. 15:269. doi: 10.3390/biom15020269
Zoulim, F., Chen, P.-J., Dandri, M., Kennedy, P. T., and Seeger, C. (2024). Hepatitis B virus DNA integration: implications for diagnostics, therapy, and outcome. J. Hepatol. 81, 1087–1099. doi: 10.1016/j.jhep.2024.06.037
Keywords: chromatin accessibility, DNA methylation, EBV, epigenetics, epigenetics reprogramming, oncogenic viruses, pioneer transcription factors
Citation: Kgatle M, Mbambara S, Khoza L, Fadebi O, Mashamba-Thompson T and Sathekge M (2025) The role of pioneering transcription factors, chromatin accessibility and epigenetic reprogramming in oncogenic viruses. Front. Microbiol. 16:1602497. doi: 10.3389/fmicb.2025.1602497
Edited by:
Eleftherios Paschalis Ilios, Harvard University, United StatesReviewed by:
Haifei Jiang, Mayo Clinic, United StatesCollin Anthony O’Leary, Cornell College, United States
Copyright © 2025 Kgatle, Mbambara, Khoza, Fadebi, Mashamba-Thompson and Sathekge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mankgopo Kgatle, a2dhdGxlLm1hbmtnb3BvQGdtYWlsLmNvbQ==