 Marcus Yee1
Marcus Yee1 Michael J. Trimble1,2
Michael J. Trimble1,2 Kazal Ghosh3
Kazal Ghosh3 Giselle Hughes3Daniel Knowles3Jun Duan4
Giselle Hughes3Daniel Knowles3Jun Duan4 Stephen Raverty3
Stephen Raverty3 Glenna McGregor3
Glenna McGregor3 William W. L. Hsiao1,2*
William W. L. Hsiao1,2*- 1Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, Canada
- 2Department of Health Sciences, Simon Fraser University, Burnaby, BC, Canada
- 3Animal Health Centre, Ministry of Agriculture and Food, Government of British Columbia, Abbotsford, BC, Canada
- 4Faculty of Health Sciences, Simon Fraser University, Burnaby, BC, Canada
Introduction: Bovine abortions result in significant economic losses to dairy producers, and bacteria are among the most common causes of these abortions. In 2021, Streptococcus pluranimalium was isolated from a dairy abortion case for the first time in British Columbia (BC), Canada. This bacterium has previously been recovered from the reproductive tracts of dairy cattle and various other species, including humans.
Methods: Between 2021 and 2023, S. pluranimalium was isolated from the placenta, fetal lung, and/or fetal abomasal contents of 10 aborted dairy fetuses submitted for routine abortion diagnostics. This study was conducted to better characterize the genotype of these 10 isolates. The histopathology of the bovine abortions was examined, and the BC strains were sequenced using Nanopore technology and underwent bioinformatic analysis.
Results: The BC strains had an average genome size of 2,313,582 base pairs and an average GC content of 38.59%. Based on whole genome phylogeny, the BC strains were clustered together and distinctly separated from other publicly available strains of this species from different regions and isolation sources. Through Clusters of Orthologous Groups analysis, the BC strains contained a larger proportion of genes associated with the mobilome. Additionally, although we identified only a few antibiotic resistance genes or virulence factors (VFs) in these strains, several of these genes were located within prophage sequences.
Discussion: Although the clinical and pathological significance of these bacteria in most abortion cases remains unclear, our findings underscore the importance of continued surveillance and research into uncommon pathogens to better understand their biology and potential impact on human and animal health.
1 Introduction
Bovine abortions result in significant economic losses for dairy producers worldwide. Reported bovine abortion rates range from 3 to 10% and can reach up to 30% in some herds. These abortions are attributed to a variety of factors, including genetic abnormalities, pathogen exposure, nutritional conditions, iatrogenic (medications) and teratogenic compounds, and hormonal fluctuations (Thurmond et al., 2005; Mee, 2023). Abortions are associated with various etiologic agents, including bacteria, fungi, protozoa, and viruses. Among these pathogens, infections caused by bacteria contribute significantly to morbidity, accounting for approximately 32–58% of all bovine abortion cases (Hecker et al., 2023). Pathogen exposure and invasion generally occur retrograde via the lower urogenital tract, or hematogenously through bacteremia, leading to localization in the placenta and developing fetus. These infections may be opportunistic, associated with normal commensal organisms, or result from exposure to contagious pathogens.
The dairy industry is the third largest agricultural sector in Canada (Pierre, 2017). In British Columbia (BC), particularly in the Fraser Valley, milk production plays a vital role in the regional economy. Due to the significant impact of fetal loss on dairy production, there has been an ongoing survey of bovine abortions at the Animal Health Centre (AHC) in Abbotsford, BC, to identify the causes of fetal loss. Between 2021 and 2023, a novel microbe, Streptococcus pluranimalium, was recovered from 10 aborted bovine fetuses. Prior to this period, this bacterium had not been identified in any bovine fetal samples submitted to the AHC. Given the limited knowledge about the natural history of this bacterium and the unknown risk of zoonotic transmission, a One Health approach was adopted to better characterize the molecular features of these isolates.
S. pluranimalium is a Gram-positive bacterium that is most closely related to Streptococcus hyovaginalis, Streptococcus halotolerans, and Streptococcus thoraltensis (Pan et al., 2018). The species has a broad tissue tropism and is capable of infecting a wide array of hosts, including canaries, chickens, cats, goats, and tilapia (Pan et al., 2018; Ghazvini et al., 2019). In bovine cases, S. pluranimalium has been linked to various diseases, such as vulvovaginitis, brain abscesses, tonsillitis, and mastitis, and reproductive issues, such as abortion (Foster et al., 2008; Twomey et al., 2012). Typically, this species transmits through blood, milk, and other infectious secretions from animals (Pan et al., 2018). There have been few reported cases of human infections, which manifested as suppurative meningitis, brain abscesses, endocarditis, and septicemia (Aryasinghe et al., 2014; Ananieva et al., 2023). Despite its wide range of potential hosts and zoonotic potential, there are few publicly available genomes of this species. As the genotype of this bacterium has not been well characterized, we performed whole-genome sequencing of 10 S. pluranimalium strains isolated from aborted bovine fetuses.
2 Materials and methods
2.1 Case material
The Animal Health Centre (AHC) is the provincial veterinary diagnostic laboratory for British Columbia and is accredited by the American Association of Veterinary Laboratory Diagnosticians (AAVLD). Case accessions included a range of tissue samples and whole carcasses obtained from local producers, veterinarians, and the general public. Bovine fetuses presented for necropsy underwent a standardized protocol, during which body weight and crown-to-rump length are recorded. All organ systems were examined internally and externally, and tissue samples were collected for routine bacteriology, histopathology, and other ancillary testing as deemed appropriate by a veterinary pathologist. Samples for bacteriology included the abomasal content, placenta, and lung tissues. These tissues were processed using conventional techniques, which included initial surface searing, followed by inoculation onto blood agar and MacConkey agar plates (Oxoid, ON). The samples were then incubated aerobically for up to 48 h. For any abortion cases, selective Salmonella culture was performed on Hektoen and XLT4 agar plates (Oxoid, ON), followed by selective enrichment in selenite broth. The colonies that grew on the agar plates were identified using biochemical tests and MALDI-TOF mass spectrometry (Bruker, ON), and the results were recorded in the proprietary veterinary information management system, VADDS&Vetstar developed by Advanced Technology Corp (Ramsey, NJ). For this study, S. pluranimalium isolates were archived at -20C for further analysis. The subsamples of the isolates were stored at -80C. Additional tissue samples were systematically collected for histopathological analysis, processed with an automated processor, embedded in paraffin, and sectioned at 5 um. These sections were stained with hematoxylin and eosin using an automated stainer, cover-slipped, and then reviewed by board-certified veterinary pathologists. Microscopic lesions were graded on a scale from 0 (no apparent lesions) to 4 (severe lesions). A summary case report was prepared. Based on the histopathology results, ancillary diagnostic studies were conducted to screen for Neospora caninum, bovine viral diarrhea, Chlamydophila abortus, and Ureoplasma diversum (Hewinson et al., 1997), along with radial immunodiffusion testing for bovine IgG and IgM in select cases.
To determine the number of S. pluranimalium isolates recovered at the AHC, a database search covering the period from 2008 to 2024 was performed using the parameters of bovine, fetus, and abortion.
2.2 Sample extraction, library preparation, and sequencing
Bacterial isolates were recovered from freezer stocks by initial culturing on tryptic soy agar plates supplemented with 5% defibrinated sheep’s blood and incubating overnight at 37°C. A single colony was picked and inoculated into tryptic soy broth supplemented with 5% defibrinated sheep’s blood, and the culture was shaken overnight at 37°C. DNA extractions were performed using Qiagen’s DNeasy Blood & Tissue kit (Toronto, ON), following the supplemental steps for the pretreatment of Gram-positive bacteria. The extracted DNA was then assessed for purity and concentration using gel electrophoresis, Nanodrop, and Qubit.
The samples were then prepared for sequencing on MinION, Oxford Nanopore Technologies (ONT) (Oxford, UK), using their Native Barcoding Kit (EXP-NBD114). This process involved repairing the DNA and performing end-preparation using the NEBNext FFPE DNA Repair Mix and the NEBNext Ultra II End Repair/dA-Tailing Module reagents (New England Biolabs, NEB; Whitby, ON). The samples were purified using AMPure XP beads, and the native barcodes were then ligated onto the fragments using Blunt/TA Ligase Master Mix (NEB). The samples were purified using AMPure XP beads, quantified, and then pooled in equimolar amounts. The ONT adapters were ligated to the barcoded pool using T4 Ligase (NEB) before being loaded onto an R10.3 flow cell, following ONT specifications. The samples were sequenced for 72 h and base-called using the MinKNOW (v22.03.6) high-accuracy base-calling model.
2.3 Sequence analysis
Raw reads had adaptors trimmed using Porechop v0.2.4 (Wick, 2018) and were quality-filtered using NanoFilt v2.8.0 (using the parameters -q 10, −l 300, −-headcrop 40) (De Coster et al., 2018). The filtered reads were assembled using Flye v2.9.1 (Kolmogorov et al., 2019) and polished using Medaka v1.11.1 (ONT, 2024). The genomes were annotated using Bakta v1.7 (Schwengers et al., 2021), and a Maximum likelihood phylogenetic tree was created using the tool autoMLST (accessed August 23, 2022) (Alanjary et al., 2019). Genome completeness was assessed using BUSCO v5.7.1 (Simão et al., 2015). The Virulence Factor Database (accessed May 10, 2024) (Liu et al., 2022) and Abricate v1.0.0 (Seemann, 2020) were used to search for the presence of virulence factors. The Comprehensive Antibiotic Resistance Database (CARD) v3.2.9 (McArthur et al., 2013), coupled with RGI v6.0.2, was used to examine antibiotic resistance genes (ARGs). Clusters of Orthologous Groups analysis was conducted using the EggNOG-mapper v2.1.9 (Cantalapiedra et al., 2021) and EggNOG Database v5.0.2 (Huerta-Cepas et al., 2018). Plasmids were identified using Mobsuite v3.0.3 (Robertson and Nash, 2018), and phage sequences were identified using Virsorter v2.2.3 (Guo et al., 2021).
3 Results
3.1 Identification of microorganisms in aborted fetuses
Between 2008 and 2024, 382 fetuses were submitted to the AHC for diagnostic evaluation. Each case was reviewed individually, and the signalment, gross pathology, histopathology, bacteriology, and molecular results were tabulated and scored. Using MALDI-TOF mass spectrometry, S. pluranimalium was identified in 10 fetal samples, which then underwent further characterization. In these 10 samples, additional bacterial isolates were taxonomically identified at the species level or, if the species level could not be determined, at the genus level. Overall, 13 bacterial species and genera were isolated from the lung, stomach contents, and placenta (Supplementary Table 1). These included S. pluranimalium (n = 11), Streptococcus uberis (n = 2), the Glutamicibacter genus (n = 2), the Staphylococcus genus (n = 3), Enterococcus saccharolyticus (n = 1), the Psychrobacter genus (n = 1), the Arthrobacter genus (n = 3), Aerococcus viridans (n = 1), the Vibrio genus (n = 1), the Acinetobacter genus (n = 5), the Corynebacterium genus (n = 2), Enterococcus faecium (n = 1), and Escherichia coli (n = 5). In addition, sample BC-AHC-08 contained the fungi Aspergillus fumigatus and a species of Candida. The presence of three other pathogens—Neospora caninum (n = 8), Bovine Viral Diarrhea Virus (n = 1), and Ureoplasma diversum (n = 1)—was confirmed either through polymerase chain reaction or serology.
3.2 Lesion distribution
The histopathology results indicated the presence of several lesions in the aborted fetuses (Table 1), affecting various tissues including the brain, lungs, heart, muscles, kidneys, and eyes. Lesions in the eyes (n = 6) and placenta (n = 7) were the most common. Sample BC-AHC-03 exhibited the most extensive lesions, affecting six different tissues. No histopathological abnormalities were apparent in sample BC-AHC-05.
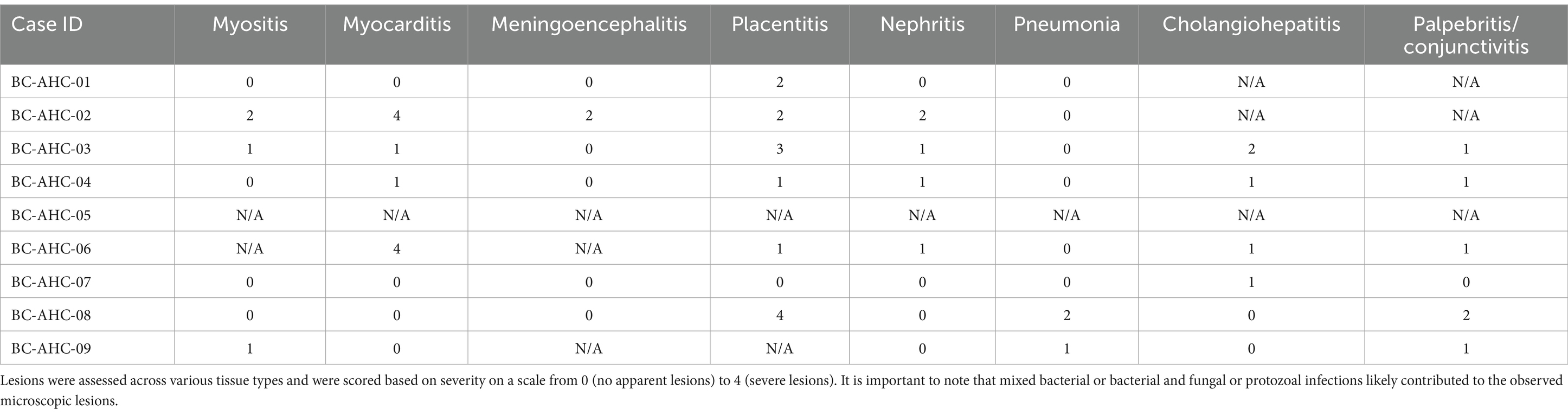
Table 1. Histopathological findings of aborted bovine fetuses.
3.3 Whole-genome sequencing of Streptococcus pluranimalium
Among the 10 fetal samples with MALDI-TOF-confirmed S. pluranimalium, isolates from 9 were further characterized using molecular methods. Whole-genome sequencing was performed on one isolate from each sample, except for sample BC-AHC-09, from which two isolates were sequenced: BC-AHC-09_a isolated from the lung and BC-AHC-09_b isolated from the stomach. The BC strains had an average genome size of 2,313,582 base pairs (bp) and an average GC content of 38.59%, similar to the reference strain TH11417 (Pan et al., 2018), which has a genome size of 2,065,522 and a GC content of 38.65 (Table 2). The BC strains had an average of 15.2 contigs per assembly and an average genome completeness of 96.63%, as assigned by BUSCO.
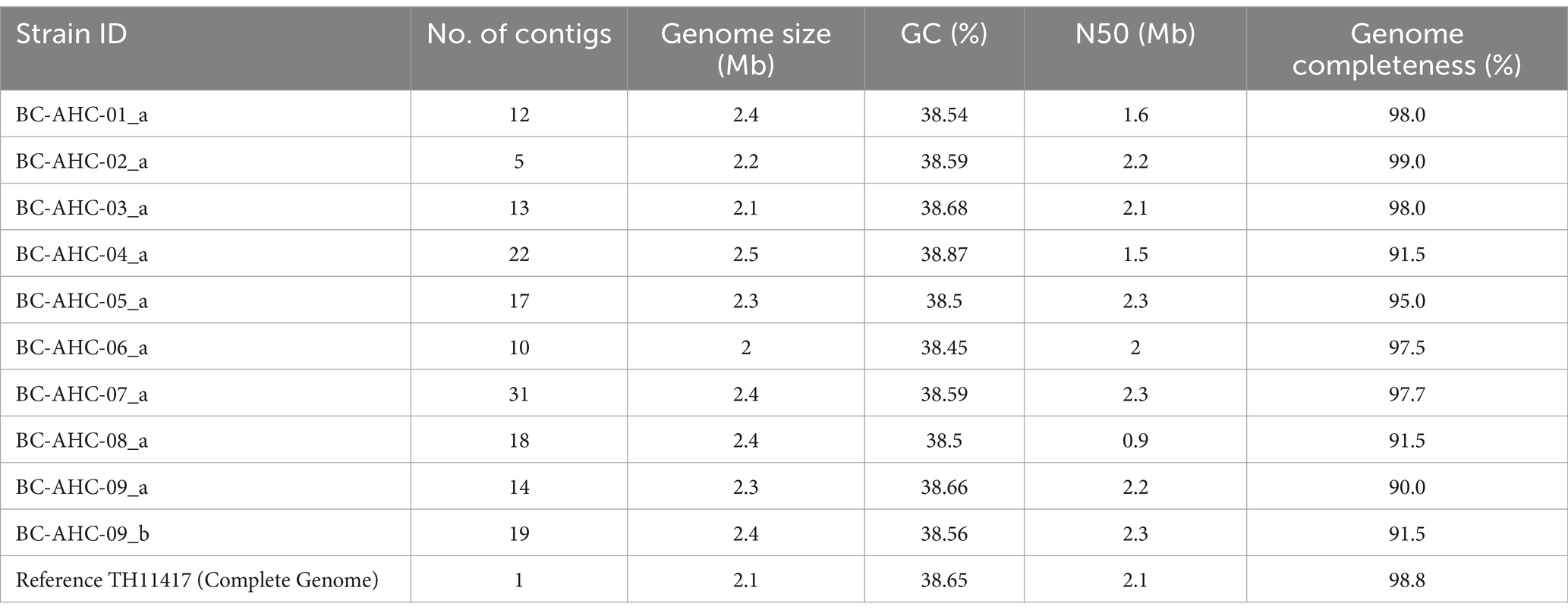
Table 2. Whole-genome sequence assembly statistics of the BC S. pluranimalium strains.
3.4 Phylogeny
A maximum likelihood phylogenetic tree was constructed based on the analysis of core, single-copy genes present in all strains (Figure 1). The phylogenetic tree was constructed using the 10 BC genomes and 6 other publicly available genomes from the NCBI as of July 2024. The six genomes from the NCBI include TH11417 (Pan et al., 2018), 14A0014 (Rodriguez Campos et al., 2018), Colony612 (Buathong et al., 2021), SP21-2 (Xu et al., 2023), SP28 (Zhu, 2023), and SUG2384 (Holman et al., 2024). The BC strains formed a distinct clade, with strain BC-AHC-06_a being the most divergent. Strains BC-AHC-09_a and BC-AHC-09_b were isolated from the same sample but were separated on the phylogenetic tree. The publicly available strain, 14A0014, which was also recovered from a bovine abortion sample, was the most genetically distinct compared to the BC strains.
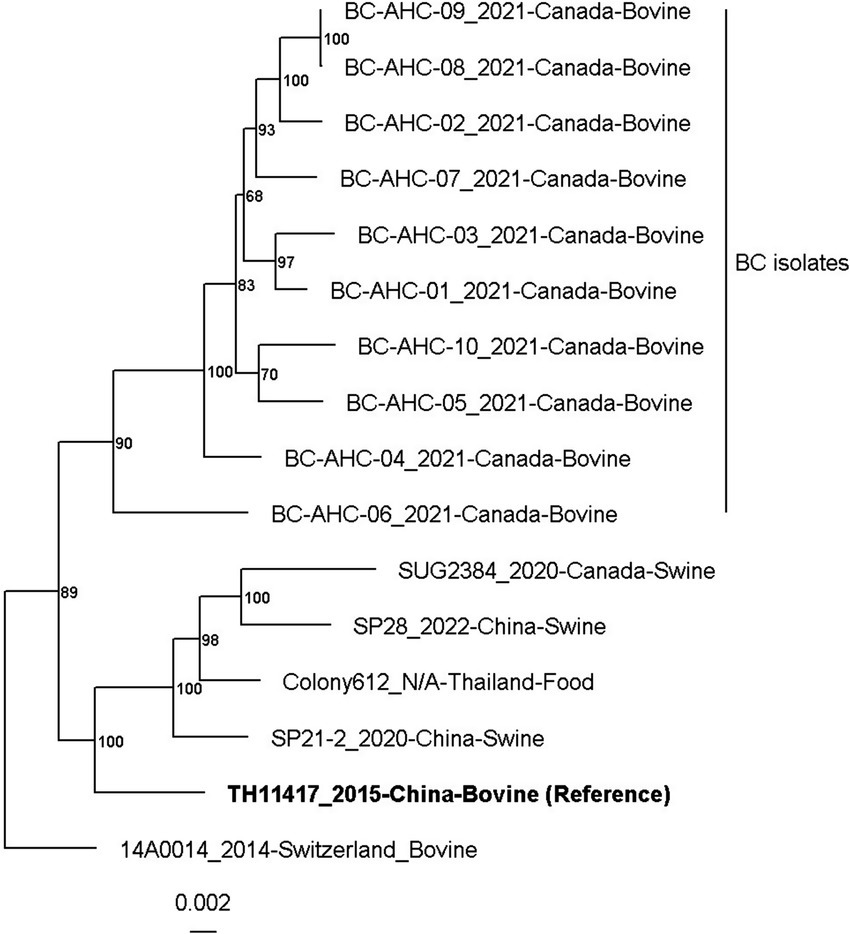
Figure 1. Maximum likelihood phylogenetic tree of S. pluranimalium strains with multilocus sequence typing analysis using the tool autoMLST.
3.5 Clusters of orthologous groups
Clusters of orthologous groups (COGs) is a method of phylogenetic classification that clusters each gene into a COG ID and assigns each COG ID to a functional category. The COG analysis, supported by Fisher’s exact test, revealed that the BC strains, except for strain BC-AHC-06_a, contained a significantly higher proportion of COG IDs associated with category X compared to other S. pluranimalium strains (p < 0.05) (Figure 2A). The BC strains, excluding BC-AHC-06_a, had an average of 12.5% of all COG IDs associated with category X. In comparison, strain BC-AHC-06_a had only 2.7% of its COG IDs associated with category X, which is similar to the non-BC strains that had an average of 3.2% of their COG IDs. With the exception of strain BC-AHC-06_a, many COG IDs associated with category X that were in high abundance were shared among all of the BC strains (Figure 2B).
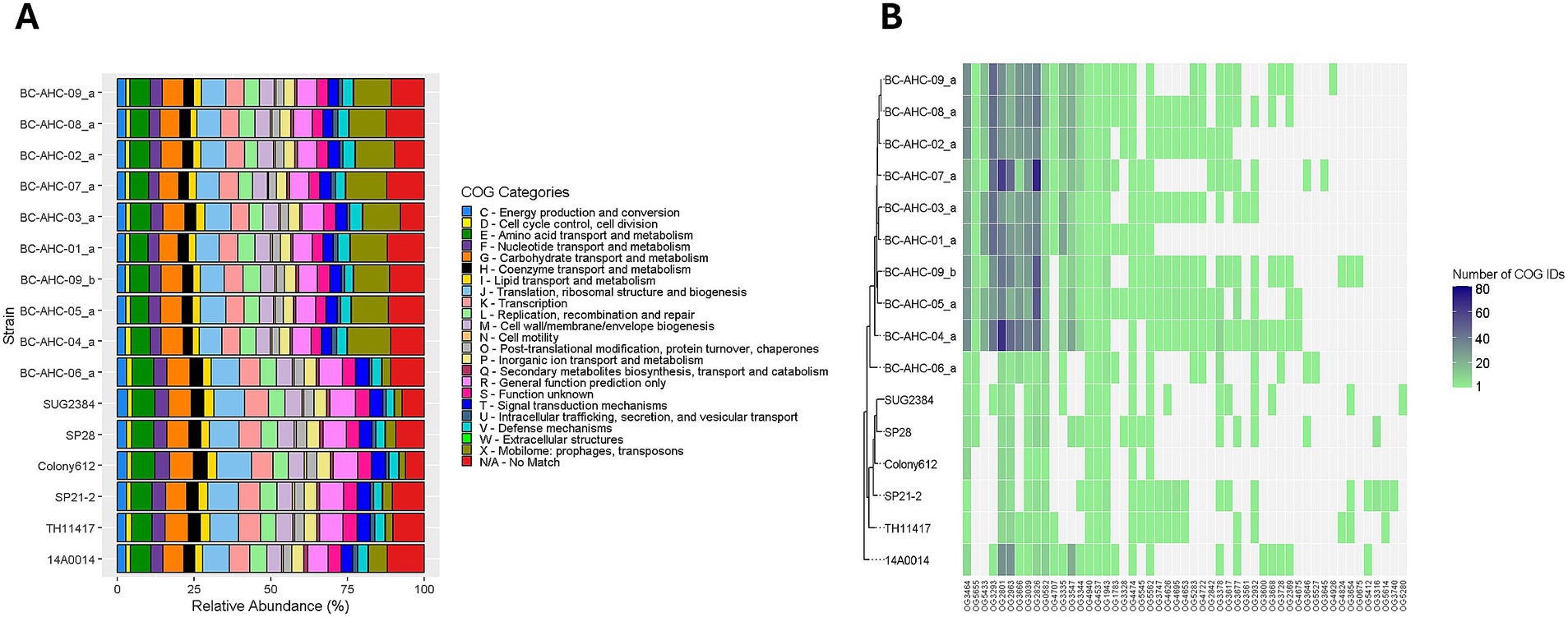
Figure 2. (A) Distribution of COG functional categories of each gene in S. pluranimalium genomes. (B) Heatmap showing the number of COG IDs associated with Category X in the S. pluranimalium genomes. COG IDs not present in a strain are colored light gray, while COG IDs present in a strain are represented by a gradient from light green to navy blue.
3.6 Virulence factors
Utilizing the tool Abricate and the Virulence Factor Database, seven different virulence factors (VFs) were identified in the BC strains, encompassing four virulence classes: immune modulation, stress survival, exotoxin, and adherence (Table 3). All BC strains contained the gene cluster rfbA, rfbB, and rfbC, with an 80–82% nucleotide identity. The VFs tig/ropA, plr/gapA, and eno were also found in all BC strains, with identities of approximately 82, 89, and 90%, respectively. Strain BC-AHC-06_a contained the SSU98_0978 virulence factor, with a nucleotide identity of 95%.
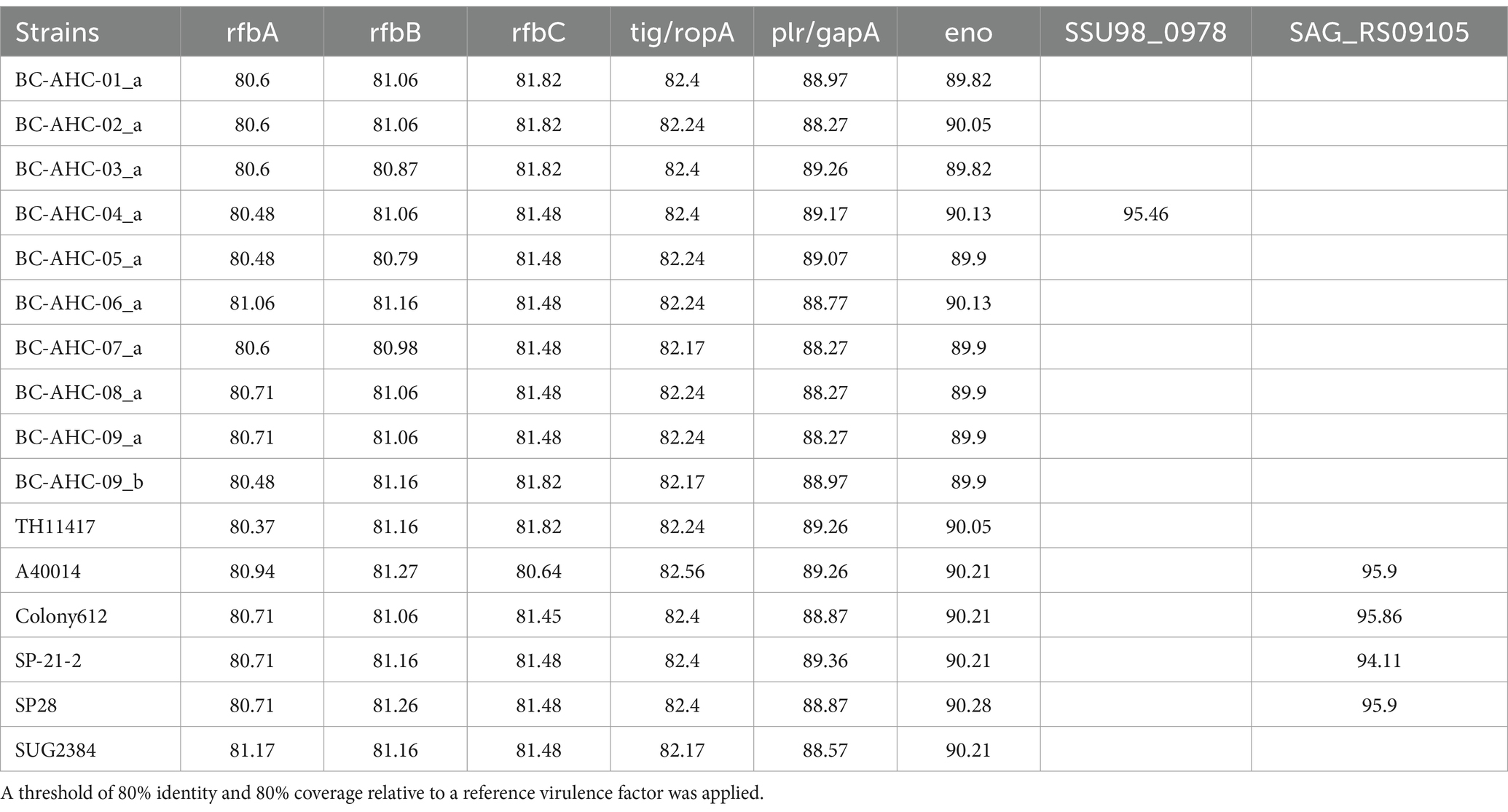
Table 3. Percent identity of virulence factors in S. pluranimalium strains, identified using the Virulence Factor Database.
3.7 Antimicrobial resistance genes
Utilizing the Resistance Gene Identifier tool and the Comprehensive Antibiotic Resistance Database, several genes conferring resistance to multiple antibiotics were identified using strict and perfect criteria (Figure 3). Antibiotic resistance genes (ARGs) conferring resistance to a number of antibiotics such as glycopeptides (vanY genes), lincosamides (lnuC), tetracycline (tet(M)), and macrolides (mreA) were detected (Table 2). Strains BC-AHC-08_a and BC-AHC-09_a contained the largest number of ARGs, with four each.
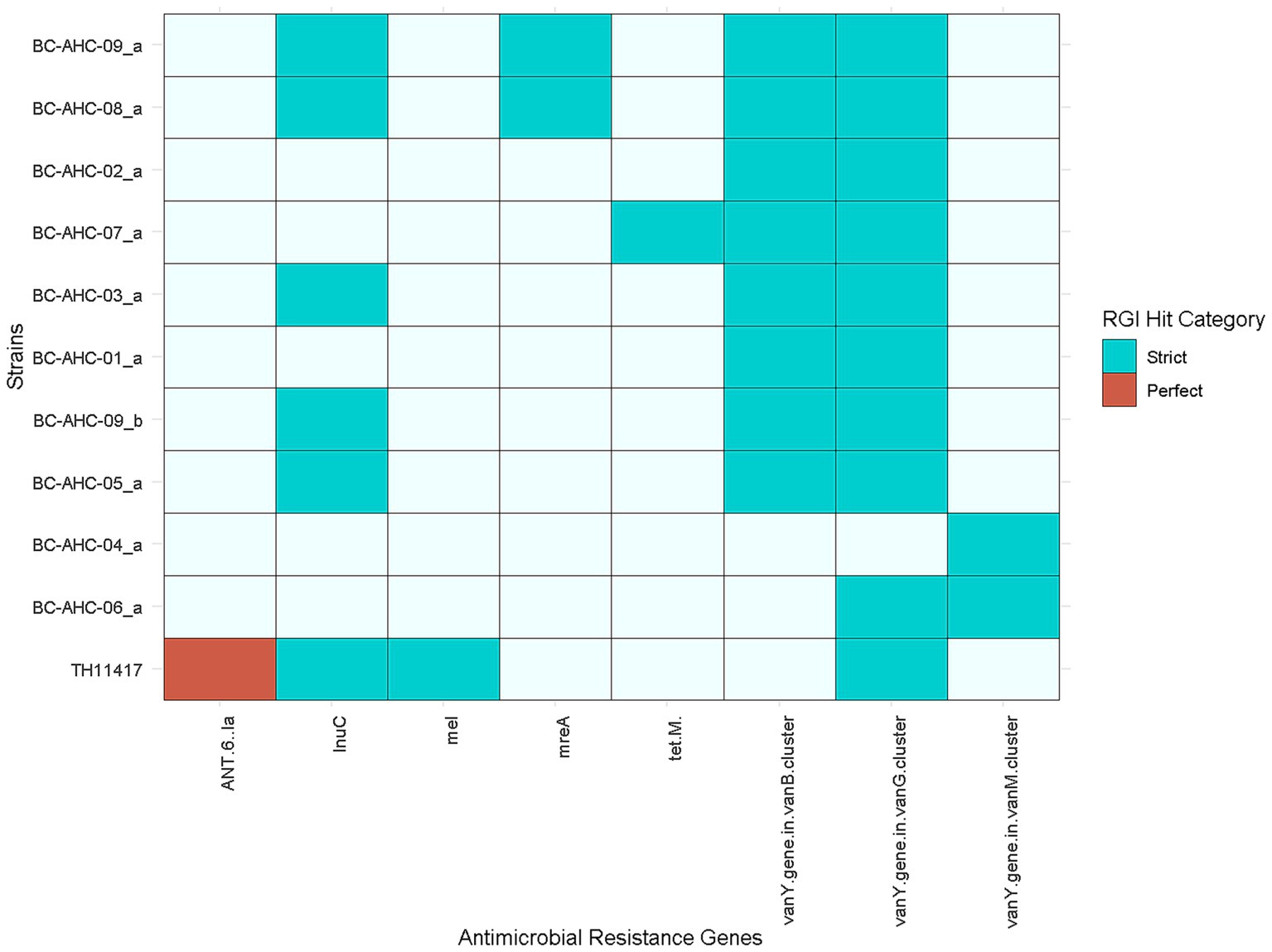
Figure 3. Presence of ARGs identified using the Resistance Gene identifier tool and the Comprehensive Antibiotic Resistance Gene Database. Perfect and strict hits were used.
3.8 Plasmids
Using the tool MOB-suite, two novel plasmids were identified. The first was a 10.8 kb plasmid identified in strains BC-AHC-01_a and BC-AHC-02_a, and the second was a 5.4 kb plasmid identified in strains BC-AHC-03_a, BC-AHC-04_a, BC-AHC-08_a, and BC-AHC-09_b. Both plasmids showed the closest similarity to NSUI060 (Athey et al., 2016), a 5.6 kb Streptococcus suis plasmid. These S. pluranimalium plasmids did not contain any known virulence factors or antimicrobial resistance genes, had the MOBV relaxase type, and were classified as mobilizable.
3.9 Prophages
Using VirSorter2, a machine learning tool that predicts phage sequences based on features such as the number of hallmark viral genes, the percentage of viral genes, and the mean GC content, we identified 41 putative prophages in the BC strains (Supplementary Table 2). Their lengths ranged from 1,024 bp to 295,435 bp. Each BC strain had an average of 291,212 bp of phage sequences. In comparison, the non-BC strains Colony612 and SP28 did not contain any phage sequences. The four other non-BC strains had an average of 147,501 bp of phage sequences per strain. Strains BC-AHC-03_a and BC-AHC-09_b contained two virulence factors, tig/ropA and plr/gapA, located on a phage sequence. The eno virulence factor was also located on a phage sequence in strain BC-AHC-06_a. Strain BC-AHC-08_a contained a phage sequence with three ARGs: the vanY gene in the vanB cluster, mreA, and lnuC. In addition, strain BC-AHC-09_a contained one phage sequence with one ARG, lnuC.
4 Discussion
Bovine abortions cause significant economic losses and are frequently associated with bacterial infections. Since 2021, S. pluranimalium, an emerging pathogen, has been identified in a subset of aborted fetuses in BC. Notably, MALDI-TOF analysis was only implemented at the AHC in 2018; prior to this period, the biochemical tests used for bacterial identification were not capable of distinguishing this pathogen. In addition to S. pluranimalium, 12 other bacterial isolates were recovered from these aborted fetuses. The most common bacterial species identified were Escherichia coli (n = 5) and the Acinetobacter genus (n = 5). Evidence of exposure to the protozoan parasite, Neospora caninum, was also found in eight fetuses. Given the non-sterile conditions at the farm site, it is not unusual to observe a diversity of microbial isolates in the submitted case materials (Hecker et al., 2023). This situation complicates the determination of the cause of fetal death in cases of mixed infection bovine abortion (Barkallah et al., 2014). Although we cannot definitively state that S. pluranimalium was the cause of abortion, the discovery of this bacterium from multiple tissues with occasional active inflammation warrants further investigation.
In this study, we conducted whole-genome sequencing on 10 S. pluranimalium isolates, greatly increasing the number of publicly available genomes for this species (Table 2 and Supplementary Table 1). The phylogenetic analysis revealed a distinct BC clade, with strain BC-AHC-06_a being the most divergent. Strains BC-AHC-09_a and BC-AHC-09_b were both isolated from the same case, with one isolate recovered from the lung and the other from the stomach (Figure 1). These strains appeared separately on the phylogenetic tree, indicating a mixed infection of two S. pluranimalium strains. Interestingly, the public strain 14AA0014, which was also isolated in Canada and associated with bovine abortions, was the most distinct compared to BC strains.
In BC strains, we found a larger number of COG IDs linked to COG category X compared to the other S. pluranimalium strains (Figure 2B). This category is listed as the mobilome and encompasses genes associated with transposons, prophages, and plasmid replication (Galperin et al., 2014). The exception was strain BC-AHC-06_a, which was the most divergent strain on the phylogenetic tree. Analysis of the location of these COG X IDs showed that they were distributed evenly across the genomes. Although the BC strains contained more prophage sequences, as predicted by VirSorter2, and these phage sequences contained many COG category X genes, removing the phage-located COG IDs did not alter the overall trend. The BC strains, except for BC-AHC-06_a, still exhibited a significantly higher percentage of COG category X genes. Although these findings further substantiate the BC clade, differentiating the BC strains from other S. pluranimalium strains, further research is needed to understand the higher prevalence of genes associated with mobilizable elements in the BC strains.
Similar to other S. pluranimalium strains, the BC strains contained limited antimicrobial resistance genes. Several of the BC strains shared the vanY gene from the vanG cluster and the lnuC gene with the reference S. pluranimalium strain TH11417 (Figure 3) (Pan et al., 2018). The vanY gene confers resistance to glycopeptides, while the lnuC gene confers resistance to lincosamides. The BC-AHC-07_a strain contained the tet(M) gene, which confers resistance to tetracycline. This gene was not present in any other S. pluranimalium genome as of July 2024. Both BC and non-BC S. pluranimalium strains contained six virulence factors, indicating that these factors are likely part of the core genome for the species. These virulence genes include the gene cluster rfbA, rfbB, and rfbC. These genes are involved in capsule synthesis and are traditionally associated with O-antigen production in Gram-negative bacteria; however, they have been reported in Streptococcus thermophilus (Bank et al., 2022). The three other virulence factors present in all S. pluranimalium strains are tig/ropA, a trigger factor linked to stress tolerance, plr/gapA, a GAPDH homolog involved in host cell adherence, and eno, an enzyme linked to the binding of human plasminogen (Antikainen et al., 2007; Purves et al., 2010; Wu et al., 2011). BC-AHC-04_a was the only BC strain to have the virulence factor SSU98_0978, an agglutinin receptor involved in adhesion (Forsgren et al., 2010).
We also identified two novel plasmids, both showing the closest similarity to a Streptococcus suis plasmid from Canada (Athey et al., 2016). The larger plasmid, measuring 10.8 kb, was found in strains BC-AHC-01_a and BC-AHC-02_a. The smaller plasmid, measuring 5.4 kb, was found in strains BC-AHC-03_a, BC-AHC-04_a, BC-AHC-09_a, and BC-AHC-09_b. Interestingly, further analysis revealed that the larger plasmid in BC-AHC-01_a and BC-AHC-02_a appeared to be an expanded version of the smaller plasmid, representing a duplication of its genetic content. No known virulence factors or antimicrobial resistance genes were found on these plasmids.
The BC strains contained a higher number of longer phage sequences compared to the other S. pluranimalium strains. Notably, two virulence factors, tig/ropA and plr/gapA, were identified within a phage sequence in two BC strains. In addition, another BC strain contained a phage sequence with the eno virulence factor. One BC strain contained a phage sequence with three ARGs: vanY in the vanB cluster, mreA, and lnuC. In contrast, another strain contained a phage sequence with lnuC. As these virulence factors and ARGs present on the phage sequences are potentially mobilizable, these findings highlight the need for vigilant monitoring to prevent the potential transmission of these genes from S. pluranimalium to other species, given its broad host tropism.
5 Conclusion
We performed whole-genome sequencing of 10 BC S. pluranimalium isolates, a previously unreported species in this region. The BC strains clustered separately from other S. pluranimalium strains but shared similar attributes, including a low number of detected VFs and ARGs. Overall, our findings underscore the importance of continued surveillance and research on rare and novel bacterial isolates to enhance our understanding of their biology and potential impact on human and animal health.
Data availability statement
All genome sequences generated in this study have been deposited in the GenBank database under the BioProject accession number PRJNA1251123.
Ethics statement
Ethical approval was not required for the studies involving animals in accordance with the local legislation and institutional requirements because the fetuses were submitted to the diagnostic service provided by the Animal Health Centre, Abbotsford, BC. On presenting the fetus to the Animal Health Centre, each producer or veterinarian signed a waiver included in the submission form for permission to (1) undertake and complete the requested testing and diagnostics and (2) inform the submitter that data from these investigations may be used for ongoing statistical surveillance of production animal health in British Columbia. As the fetuses comprised spontaneous abortions and were dead at the time of submission, no IACUC review was warranted. In this case series, no fetuses were actively solicited from producers and no fetuses were obtained by induced abortion of pregnant dams. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
MY: Formal analysis, Software, Visualization, Writing – original draft, Writing – review & editing, Investigation. MT: Methodology, Supervision, Writing – review & editing, Investigation. KG: Conceptualization, Investigation, Methodology, Writing – review & editing, Supervision. GH: Data curation, Writing – review & editing. DK: Data curation, Writing – review & editing. JD: Data curation, Writing – review & editing. SR: Conceptualization, Investigation, Methodology, Writing – review & editing, Supervision. GM: Methodology, Writing – review & editing, Conceptualization, Investigation, Supervision. WH: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to extend our sincere thanks to the bacteriology laboratory scientists at the Animal Health Centre for conducting the bacterial cultures. We particularly thank Giselle Hughes and Daniel Knowles for their contributions to bacterial sequencing. We would also like to acknowledge Jun Duan for his role in sequence assembly. We also extend our appreciation to all submitters involved in these cases. We would like to thank the Michael Smith Health research funding as well as the Canadian Institutes of Health Research and Canada Graduate Scholarships — Master’s program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1603770/full#supplementary-material
SUPPLEMENTARY TABLE 1 | Sample metadata and Pathology.
SUPPLEMENTARY TABLE 2 | Bacteriophages identified in Streptococcus pluranimalium strains.
References
Alanjary, M., Steinke, K., and Ziemert, N. (2019). AutoMLST: an automated web server for generating multi-locus species trees highlighting natural product potential. Nucleic Acids Res. 47, W276–W282. doi: 10.1093/nar/gkz282
Ananieva, M., Faustova, M., Loban, G., Avetikov, D., Tkachenko, P., Bobyr, V., et al. (2023). Biological properties of Streptococcus pluranimalium as the new human pathogen. Open access Macedonian. J. Med. Sci. 11, 53–57. doi: 10.3889/oamjms.2023.10990
Antikainen, J., Kuparinen, V., Lähteenmäki, K., and Korhonen, T. K. (2007). Enolases from gram-positive bacterial pathogens and commensal lactobacilli share functional similarity in virulence-associated traits. FEMS Immunol. Med. Microbiol. 51, 526–534. doi: 10.1111/j.1574-695X.2007.00330.x
Aryasinghe, L., Sabbar, S., Kazim, Y., Awan, L. M., and Khan, H. K. (2014). Streptococcus pluranimalium: a novel human pathogen? Int. J. Surg. Case Rep. 5, 1242–1246. doi: 10.1016/j.ijscr.2014.11.029
Athey, T. B., Teatero, S., Takamatsu, D., Wasserscheid, J., Dewar, K., Gottschalk, M., et al. (2016). Population structure and antimicrobial resistance profiles of Streptococcus suis serotype 2 sequence type 25 strains. PLoS One 11:e0150908. doi: 10.1371/journal.pone.0150908
Bank, N. C., Singh, V., and Rodriguez-Palacios, A. (2022). Classification of Parabacteroides distasonis and other Bacteroidetes using O-antigen virulence gene: Rfb A-typing and hypothesis for pathogenic vs. probiotic strain differentiation. Gut Microbes 14:1997293. doi: 10.1080/19490976.2021.1997293
Barkallah, M., Gharbi, Y., Hassena, A. B., Slima, A. B., Mallek, Z., Gautier, M., et al. (2014). Survey of infectious etiologies of bovine abortion during mid-to late gestation in dairy herds. PLoS One 9:e91549. doi: 10.1371/journal.pone.0091549
Buathong, R., Joyjinda, Y., Rodpan, A., Yomrat, S., Ponpinit, T., Ampoot, W., et al. (2021). "Streptococcus pluranimalium strain Colony 612 chromosome". (NCBI Accession No. NZ_CP078544). Available at: https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP078544
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). Nano pack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Forsgren, N., Lamont, R. J., and Persson, K. (2010). Two intramolecular isopeptide bonds are identified in the crystal structure of the Streptococcus gordonii SspB C-terminal domain. J. Mol. Biol. 397, 740–751. doi: 10.1016/j.jmb.2010.01.065
Foster, G., Barley, J., Howie, F., Falsen, E., Moore, E., Twomey, D. F., et al. (2008). Streptococcus pluranimalium in bovine reproductive disease. Vet. Rec. 163:638. doi: 10.1136/vr.163.21.638
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2014). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Ghazvini, K., Karbalaei, M., Kianifar, H., and Keikha, M. (2019). The first report of Streptococcus pluranimalium infection from Iran: a case report and literature review. Clin Case Rep 7, 1858–1862. doi: 10.1002/ccr3.2374
Guo, J., Bolduc, B., Zayed, A. A., Varsani, A., Dominguez-Huerta, G., Delmont, T. O., et al. (2021). VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9:37. doi: 10.1186/s40168-020-00990-y
Hecker, Y. P., González-Ortega, S., Cano, S., Ortega-Mora, L. M., and Horcajo, P. (2023). Bovine infectious abortion: a systematic review and meta-analysis. Front Vet Sci 10:1249410. doi: 10.3389/fvets.2023.1249410
Hewinson, R. G., Griffiths, P. C., Bevan, B. J., Kirwan, S. E., Field, M. E., Woodward, M. J., et al. (1997). Detection of Chlamydia psittaci DNA in avian clinical samples by polymerase chain reaction. Vet. Microbiol. 54, 155–166. doi: 10.1016/s0378-1135(96)01268-0
Holman, D. B., Gzyl, K. E., and Kommadath, A. (2024). Florfenicol administration in piglets co-selects for multiple antimicrobial resistance genes. mSystems 9, e0125024–e0101224. doi: 10.1128/msystems.01250-24
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2018). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–d917. doi: 10.1093/nar/gkab1107
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/aac.00419-13
Mee, J. F. (2023). Invited review: bovine abortion-incidence, risk factors and causes. Reprod. Domest. Anim. 58, 23–33. doi: 10.1111/rda.14366
ONT, (2024). Medaka [Online]. Github. Available online at: https://github.com/nanoporetech/medaka (Accessed June, 2022).
Pan, Y., An, H., Fu, T., Zhao, S., Zhang, C., Xiao, G., et al. (2018). Characterization of Streptococcus pluranimalium from a cattle with mastitis by whole genome sequencing and functional validation. BMC Microbiol. 18:182. doi: 10.1186/s12866-018-1327-0
Pierre, M. S. (2017). VISTA on the Agri-food industry and the farm community Changes in Canadians’ preferences for milk and dairy products. Oxford University Press: Nucleic Acids Research.
Purves, J., Cockayne, A., Moody, P. C., and Morrissey, J. A. (2010). Comparison of the regulation, metabolic functions, and roles in virulence of the glyceraldehyde-3-phosphate dehydrogenase homologues gapA and gapB in Staphylococcus aureus. Infect. Immun. 78, 5223–5232. doi: 10.1128/iai.00762-10
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom 4:e000206. doi: 10.1099/mgen.0.000206
Rodriguez Campos, S., Gobeli Brawand, S., Brodard, I., Rychener, L., and Perreten, V. (2018). "Streptococcus pluranimalium strain 14A0014 chromosome, complete genome". (NCBI Accession No. CP022601). Available at: https://www.ncbi.nlm.nih.gov/nuccore/CP022601
Schwengers, O., Jelonek, L., Dieckmann, M. A., Beyvers, S., Blom, J., and Goesmann, A. (2021). Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 7:000685. doi: 10.1099/mgen.0.000685
Seemann, T. (2020). ABRicate [Online]. Github. Available online at: https://github.com/tseemann/abricate (Accessed September, 2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Thurmond, M. C., Branscum, A. J., Johnson, W. O., Bedrick, E. J., and Hanson, T. E. (2005). Predicting the probability of abortion in dairy cows: a hierarchical Bayesian logistic-survival model using sequential pregnancy data. Prev. Vet. Med. 68, 223–239. doi: 10.1016/j.prevetmed.2005.01.008
Twomey, D. F., Carson, T., Foster, G., Koylass, M. S., and Whatmore, A. M. (2012). Phenotypic characterisation and 16S rRNA sequence analysis of veterinary isolates of Streptococcus pluranimalium. Vet. J. 192, 236–238. doi: 10.1016/j.tvjl.2011.05.007
Wick, R. (2018). Porechop [Online]. Github. Available online at: https://github.com/rrwick/Porechop (Accessed June, 2022).
Wu, T., Zhao, Z., Zhang, L., Ma, H., Lu, K., Ren, W., et al. (2011). Trigger factor of Streptococcus suis is involved in stress tolerance and virulence. Microb. Pathog. 51, 69–76. doi: 10.1016/j.micpath.2010.10.001
Xu, C. W., Zhou, X., Zhang, X. L., Zhou, Q., Qi, H. X., Li, Y. X., et al. (2023). Whole genome sequence of Streptococcus pluranimalium SP21-2, a porcine strain harbouring optr a and lsa (E) with chromosomal location. J. Glob. Antimicrob. Resist. 35, 101–103. doi: 10.1016/j.jgar.2023.09.007
Zhu, Y. (2023). Streptococcus pluranimalium strain SP28 chromosome, complete genome. (NCBI Accession No. CP121201). Available at: https://www.ncbi.nlm.nih.gov/nuccore/CP121201
Keywords: Streptococcus pluranimalium, bovine abortion, whole genome sequencing, antimicrobial resistance (AMR), animal health
Citation: Yee M, Trimble MJ, Ghosh K, Hughes G, Knowles D, Duan J, Raverty S, McGregor G and Hsiao WWL (2025) The genotypic characterization of Streptococcus pluranimalium from aborted bovine fetuses in British Columbia, Canada. Front. Microbiol. 16:1603770. doi: 10.3389/fmicb.2025.1603770
Edited by:
Hong Yin, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Hasan Faisal Hussein Kahya, University of Mosul, IraqMiroslav Benić, Croatian Veterinary Institute, Croatia
Copyright © 2025 Yee, Trimble, Ghosh, Hughes, Knowles, Duan, Raverty, McGregor and Hsiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William W. L. Hsiao, d3doc2lhb0BzZnUuY2E=