 Gaopeng Liu
Gaopeng Liu Chengzhi Mao
Chengzhi Mao Qi Li
Qi Li Da Huo
Da Huo Tao Li
Tao Li- 1State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 2College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing, China
Ralstonia spp. are highly adaptable bacteria that are widely distributed across diverse environments. Here, we isolated four Ralstonia pickettii (R. pickettii) genomes from cultures of Dolichospermum spp., and using a comparative genomic framework of 228 Ralstonia genomes. We performed phylogenetic analyses that grouped them into water, soil, plant, and human-associated clades based on their predominant isolation habitats. Fluorescence in situ hybridization revealed minimal physical interactions between R. pickettii and cyanobacterial cells, indicating a commensal or independent ecological relationship. Distinct differences in carbohydrate-active enzymes (CAZymes) and secondary metabolite profiles were observed between water and human-associated dominant groups compared to plant-associated dominant groups, highlighting potential niche-specific adaptations. The water-associated dominant groups harbored antibiotic resistance genes, including CeoB and OXA-type β-lactamase genes. These genes are typically linked to human-associated strains, suggesting potential horizontal gene transfer or shared selective pressures, and the gene content of T3SS is reduced. Notably, water-associated dominant groups exhibited a unique pyrimidine degradation pathway, potentially enabling the utilization of exogenous pyrimidines to support survival in nutrient-limited aquatic environments. We propose that the gene content loss of T3SS and the acquisition of specialized metabolic pathways reflect adaptive strategies of Ralstonia spp. for thriving in aquatic free-living niches.
1 Introduction
Ralstonia spp., Gram-negative bacteria in the family Burkholderiaceae, are widely distributed across diverse environments, including water, plants, hospitals, and soil (Ryan and Adley, 2014). While their adaptability to various habitats has been extensively studied, the ecological roles and adaptation mechanisms of Ralstonia spp. in natural water bodies remain largely unexplored (Yuan et al., 2024; Silva et al., 2024). Previous studies have suggested that genomic islands (GIs) and horizontal gene transfer (HGT) may contribute to the survival of R. pickettii in drinking water systems (Yuan et al., 2024), yet little is known about their strategies for adapting to freshwater ecosystems. Several species of Ralstonia are opportunistic pathogens with significant ecological and clinical impacts (Fluit et al., 2021; Ryan et al., 2006). R. pickettii, in particular, is known to contaminate hospital water systems, increasing the risk of secondary infections. It can also pass through 0.22 μm filter membranes, potentially causing contamination in sterile drug formulations (Demirdag et al., 2022). Meanwhile, factors such as the type III secretion system (T3SS), exopolysaccharides (EPS), and flagella have been identified as key pathogenic determinants in R. solanacearum (Peeters et al., 2013). Natural water bodies are rich in microorganisms, which play a vital role in element cycling and energy flow processes. For instance, marine microalgae, which play a crucial role in the global carbon cycle as primary producers (Liu et al., 2020). However, the ecological adaptation mechanisms of R. pickettii in non-clinical aquatic environments remain unclear.
However, the ecological significance of Ralstonia extends beyond pathogenicity. Whole-genome studies have revealed that R. eutropha possess complete pathways for bioplastic synthesis, while deeper insights have shown that R. pickettii harbors a complete pathway for microplastic degradation (Ryan et al., 2007; Pohlmann et al., 2006). Comparative genomics has revealed key metabolic adaptations in prokaryotes. A comparison between pathogenic and non-pathogenic R. solanacearum genomes identified horizontal gene transfer and gene loss events (Ailloud et al., 2015). In addition, non-synonymous polymorphisms in type III effectors were found to contribute to differences in host range (Ailloud et al., 2015). These virulence genes evolve at a faster rate than the whole of the genome (Remenant et al., 2010). However, a comprehensive phylogenetic and ecological analysis of the genus Ralstonia remains lacking. With over 600 genomes from different habitats now available in public databases, there is an unprecedented opportunity to explore the evolutionary relationships and ecological adaptations of Ralstonia species across diverse environments. The ecological functions of microorganisms vary across different habitats. Their adaptability is facilitated by mechanisms like HGT and GIs such as antibiotic resistance genes (ARGs), CAZymes, and the type secretion system (TSS) (Zhao et al., 2023; Bernabeu et al., 2024). Getting more understanding of these variations offers a genomic perspective for elucidating the adaptive traits and ecological roles of microorganisms.
In this study, we investigated a Dolichospermum spp. bloom event in a freshwater lake in Wuhan, China, and we performed isolation and cultivation, during which R. pickettii was found in significant abundance alongside cyanobacterial cells. We obtained four complete genomes from metagenomic data and retrieved genomic data of Ralstonia spp. from various habitats via public databases. These genomes were used for phylogenetic and comparative genomic analyses to uncover the ecological and metabolic adaptations of R. pickettii to aquatic environments. Additionally, we explored its potential interactions with Dolichospermum and examined how pathogenicity-related traits vary across different habitats.
2 Materials and methods
2.1 Sampling and identification
On May 17th, 2022, a water sample was collected from a natural lake (N: 30.546166, E: 114.353721) in Wuhan, Hubei Province, China. Algae filaments were separated, washed multiple times with distilled water droplets, and then cultured in CT medium. Ralstonia isolates were cultured on R2A medium, and after three successive generations of purification, single colonies were selected. The full-length 16S rRNA gene was then directly amplified from these colonies using 2 × EasyTaq® PCR SuperMix (TransGen Biotech, China) with primers 27F (5’-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5’-TACGGCTACCTTGTTACGACTT-3′). Bacterial identification was conducted using the online BLAST program.1 The ncbi-genome-download (v 0.3.3)2 tool was employed to download all genome sequences of the Ralstonia genus from the RefSeq section, and pyani (v 0.2.12) (Pritchard et al., 2016) software was used to calculate the average nucleotide identity (ANI) for further species identification under -m ANIm -g parameter. The culture used for sequencing was maintained at 28 ± 1°C with a speed of 100 rpm in the incubator. The genomic collection information was compiled based on submission data from NCBI, combined with statistical information from published articles.
2.2 Genome sequencing, assembly, and annotation
Metagenomic DNA samples obtained from culture were extracted using the DNA Gel Extraction kit according to the manufacturer’s instructions (Axygen, United States). DNA concentration was measured using a Nanodrop 2000 spectrophotometer (Thermo Scientific, United States). Library construction and sequencing were conducted at BENAGEN Biotechnology Co., Ltd. Sequencing was performed using the Illumina MI seq PE150 platform for next-generation sequencing and the Nanopore PromethION platform for third-generation sequencing, generating 5 Gb of data, respectively. Offline data processing was performed using the fast Guppy basecaller (v 6.3.8) software for accurate base calling with a high-accuracy model and subsequent quality control. FastQC (v 0.11.9) (Schmieder and Edwards, 2011) was used for quality control, and raw data with a quality score below Q20 was discarded. Filtering sequences shorter than 5,000 bp, de novo assembly was conducted using the Flye (v 2.9.1)—Racon (v 1.5.0) model (Vaser et al., 2017; Kolmogorov et al., 2019) with nanopore sequences under default parameter, followed by three rounds of correction and polishing. Next, Illumina sequencing data was used for two rounds of polishing of the de novo genome using NextPolish (v 1.4.1) (Hu et al., 2020) software to eliminate sequencing errors under default parameter. R. pickettii genomes data were extracted from the complete metagenomic dataset and annotated using Prokka (v 1.14.6) software under default parameter (Seemann, 2014). Full-length 16S rRNA sequences were extracted from the genome using barrnap (v 0.9)3 software and identified using the online BLAST program.4
2.3 Phylogenetic analysis
For comparative analysis, we retrieved 428 Ralstonia spp. genome sequences and their annotations from the NCBI GenBank database. We selected 228 genomes with completeness above 99% (evaluated using CheckM (v 1.2.1) (Parks et al., 2015) software under lineage_wf mode) and fewer than 100 contigs for further analysis. Accession numbers of the genomes included in this study and their genomic features are shown in Supplementary Table S1. Protein sequences were extracted from GenBank files using a custom python script, and single-copy orthologous gene families were identified for the 231 genomes (including three species selected as outgroups) using OrthoFinder (v 2.5.5) (Emms and Kelly, 2019) software under -a 50 -M msa parameters. We identified 421 single-copy orthologous proteins with an average length of more than 100 bp for further analysis. After alignment and trimming using MAFFT (v 7.520) (Katoh et al., 2002) software under --auto parameters and TrimAl (v 1.4) software under -automated1 parameters (Capella-Gutiérrez et al., 2009), respectively, conserved proteins for each species were concatenated using SeqKit (v 2.5.1) (Shen et al., 2016) software. The concatenated amino acid sequence of the 421 gene families was used to construct a maximum likelihood (ML) phylogenomic tree with ultrafast 1,000 bootstrap replicates using iqtree2 (v 2.2.5) software under -m MFP -B 1000 -bnni parameters (Nguyen et al., 2015). The amino acid substitution model was automatically selected the best-fit model suggest by iqtree2 software. Phylogenetic trees were visualized and beautified using iTOL (v 5.0) (Letunic and Bork, 2021).
2.4 Fluorescence in situ hybridization and microscopy
The cultures in logarithmic growth phase were centrifuged at 8000 rpm for 10 min, washed three times with sterile PBS (phosphate buffered saline), and fixed overnight at 4°C in 4% paraformaldehyde. Glass slides were pretreated with 0.1% polylysine (Sangon Biotech, China), onto which the fixed cultures were added and dried at 45°C for more than 20 min to allow adsorption onto the slides. After fixation, pre-hybridization was performed using a hybridization solution and incubated at 40°C for 1 h. Subsequently, a 30 μL solution of probe (5‘-GCAAGGCCTCATGCTATAG-3’, diluted 1,5 v:v in 30% formamide) was added and incubated overnight at 40°C in a moist chamber. Washing steps included 15 min washes with 2 × SSC (Saline sodium citrate), followed by two 7 min washes with 1 × SSC, 15 min wash with 0.5 × SSC, and 45 min incubation with diluted branch probe at 40°C in a moist chamber, followed by 7 min washes with 2 × SSC, 1 × SSC, 0.5 × SSC, and 0.1 × SSC. Incubation at 42°C for 3 h with a fluorescent signal probe in a moist chamber was followed by the same washing process. DAPI solution (2 μg/mL) was incubated for 20 min away from light, and the samples were gently rinsed three times with sterile PBS before sealing the slides with an anti-fluorescence quenching agent. The dilution ratio of SSC solution is expressed as volume ratio (v/v). All reagents were obtained from Wuhan Servicebio Biotechnology Co., Ltd. The design and synthesis of R. pickettii’s fluorescent probe were completed by Wuhan Servicebio Biotechnology Co., Ltd. Observations and panoramic scanning were conducted using a Leica microscope Aperio VERSA 8 (v 1.4.0.125). DAPI staining was observed using ultraviolet excitation luminescence, Dolichospermum sp. self-luminescence was observed using an excitation wavelength of 510–560 nm (G-2A), and R. pickettii was observed emitting green fluorescence in the wavelength range of 460–500 nm.
2.5 Comparative genomics analysis
Gene function and secretion system prediction were conducted using KofamScan (v 1.3.0) (Aramaki et al., 2020) software with database version 20,231,127 under default parameter, and the software recommended annotation was selected as the final result. CAZymes annotation was performed using dbCAN2 (v 4.0.0) (Zhang et al., 2018) against the dbCAN2 database (v12) with hmmer-based comparison (coverage >0.35 and e-value <1e-15), dbCAN-sub (coverage >0.35 and e-value <1e-15), and diamond (e-value <1e-10) methods. The annotation predicted by hmmer was used as the statistical standard after integrating the results from three methods. Antibiotic resistance genes (ARGs) and virulence factors were predicted using abricate (v 1.0.1) software5 with the Card (Zankari et al., 2012) and VFDB (Chen et al., 2005) databases, respectively (identity >80%). Bacterial secretion systems prediction was extracted from KofamScan software annotated results, and gene names and classification information can be queried through KO numbers. Annotate secondary metabolites using online6 and detection strictness used strict, and other parameters using default methods (Blin et al., 2023).
A python script was employed to screen OGs (orthogroups genes) present in all strains, based on the OrthoFinder results excluding outgroups, shared metabolism representing extremely conservative sequences that present in all genomes. One transporter was classified as incomplete because one of its constituent proteins was missing. Nevertheless, this protein showed over 50% identity to the corresponding Escherichia coli protein in BLASTp analysis, indicating it may have been overlooked during the prediction process. Such cases are defined as “dismissed” transporter proteins. To investigate the unique metabolic content of each group, an exact Fisher’s test (p < 0.05) was performed. This analysis identified whether certain metabolic pathways were specifically lost in individual groups, thereby revealing their unique metabolic profiles. The longest aligned sequence representing each selected OG was chosen, and all sequences were merged for annotation using KofamScan (v 1.3.0) (Aramaki et al., 2020) software with database version 20,231,127. The annotation table was reconstructed using KEGG-reconstruct7 to visualize metabolic pathways. A simplified metabolic diagram was created using Adobe Illustrator CC 2019 based on the metabolic pathway results. Gene family contraction and expansion in the Ralstonia genus were analyzed using Badirate (v 1.35) (Librado et al., 2012) software based on the BDI-CSP-FR model, providing important insights into species evolution. A standard binary phylogenetic tree, with duplicate values removed, was used for the analysis of gene family contraction and expansion.
2.6 Statistical analysis and data visualization
Data were analyzed and visualized using R (v 4.3.1) in RStudio (v 2023.6.0.421) with the R packages pheatmap (v 1.0.12) and ggplot2 (v 2.3.4). Results for CAZymes, resistance genes, and virulence factors were generated using pheatmap (v 1.0.12) and gplots (v 4.3.1). Boxplots for genome-related information were created using ggplot2. The vegan package (v 2.6.4) was utilized for analysis of similarities (anosim) to distinguish among different clades based on CAZymes and secondary metabolites.
3 Results
3.1 Localize Ralstonia pickettii in Dolichospermum culture
Fluorescence in situ hybridization (FISH) was conducted to investigate the spatial relationship between Dolichospermum sp. and R. pickettii in culture. Individual filaments were isolated under a microscope and washed with sterile water before being transferred to CT medium, a commonly used culture medium for cyanobacteria. During the exponential growth phase of Dolichospermum sp., appropriate amounts of culture were collected under sterile conditions for further analysis. The morphology of Dolichospermum filaments is distinct, emitting red excitation light R. pickettii rarely attaches on the Dolichospermum cells (Figures 1A–C). However, there is a small amount of aggregation of R. pickettii surrounding the filaments. The abundance of green fluorescent signals from R. pickettii in the field of view indicates its high presence in the culture. However, the absence of a stable parasitic relationship suggests that R. pickettii is likely to exist in a free-living association with Dolichospermum, either in culture or in the natural environment.
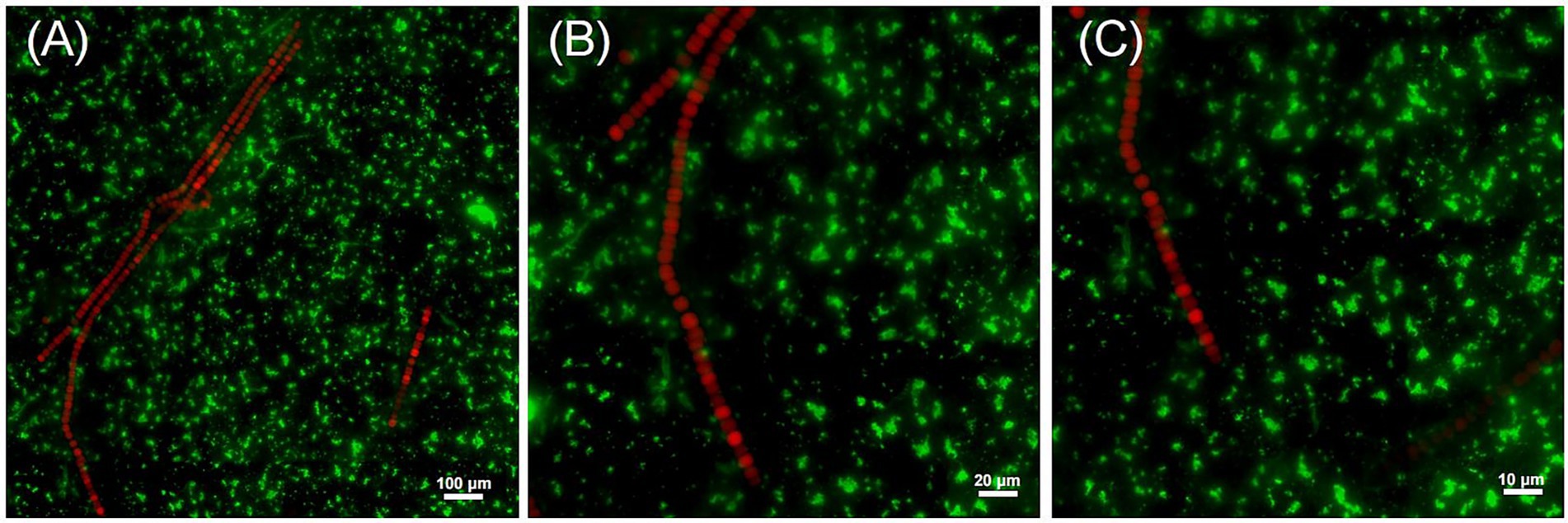
Figure 1. The relative positional relationship between R. pickettii and Dolichospermum sp. in the culture medium. Using a probe designed based on the 16 s rRNA sequence of R. pickettii, light is activated within the 460–500 nm wavelength range (green), whereas the self-luminescence of blue-green algae (red) is activated within the 510–560 nm (G-2A) wavelength range. Maximum intensity projection of stack fluorescence images acquired at magnifications of ×10 (A), ×20 (B), and ×100 (C) is shown.
3.2 Phylogenetic reconstruction
We selected 228 genomes with completeness above 99% and fewer than 100 contigs for further analysis. The genomic collection information was compiled based on submission data from NCBI, combined with statistical information from published articles. The ANI results of the 228 genomes (excluding 3 outgroups) (Supplementary Figure S1) indicate that the similarity between the four Ralstonia strains which we isolate from Dolichospermum sp. culture and R. pickettii is over 98%. Based on the primary habitat of the strains within each clade, the 228 species can be categorized into four groups: plant, human, soil and water (Figure 2A). In the plant group, there are R. solanacearum, R. pseudosolanacearum and R. syzygii. There are R. insidiosa and R. mannitolilytica in the human group, novel species are found in the soil group and R. pickettii in the water group. The average genome length and GC content of these 228 Ralstonia spp. genomes were 5.49 Mb and 64.99%, ranging from 4.38–7.02 Mb and 62.95–67.1%, respectively (Figure 2B). The greatest variation is the GC content across different habitats, clades associated with plant-hosts comprising the highest proportion, averaging 66.61%, while soil group exhibits the lowest average content at 63.60%. However, the number of CDSs (coding sequences) showed a significant increase in soil and water habitats compared with the plant group (Figure 2B). In this study, it was observed that the number of gene families in the common ancestor of the Ralstonia genus was relatively modest, with an increase in gene family count occurring concomitant with adaptation to diverse environments. The R. insidiosa gene family, which is associated with human, boasts the highest number of genes and may be implicated in its adaptation to various hosts or environment (Supplementary Figure S2).
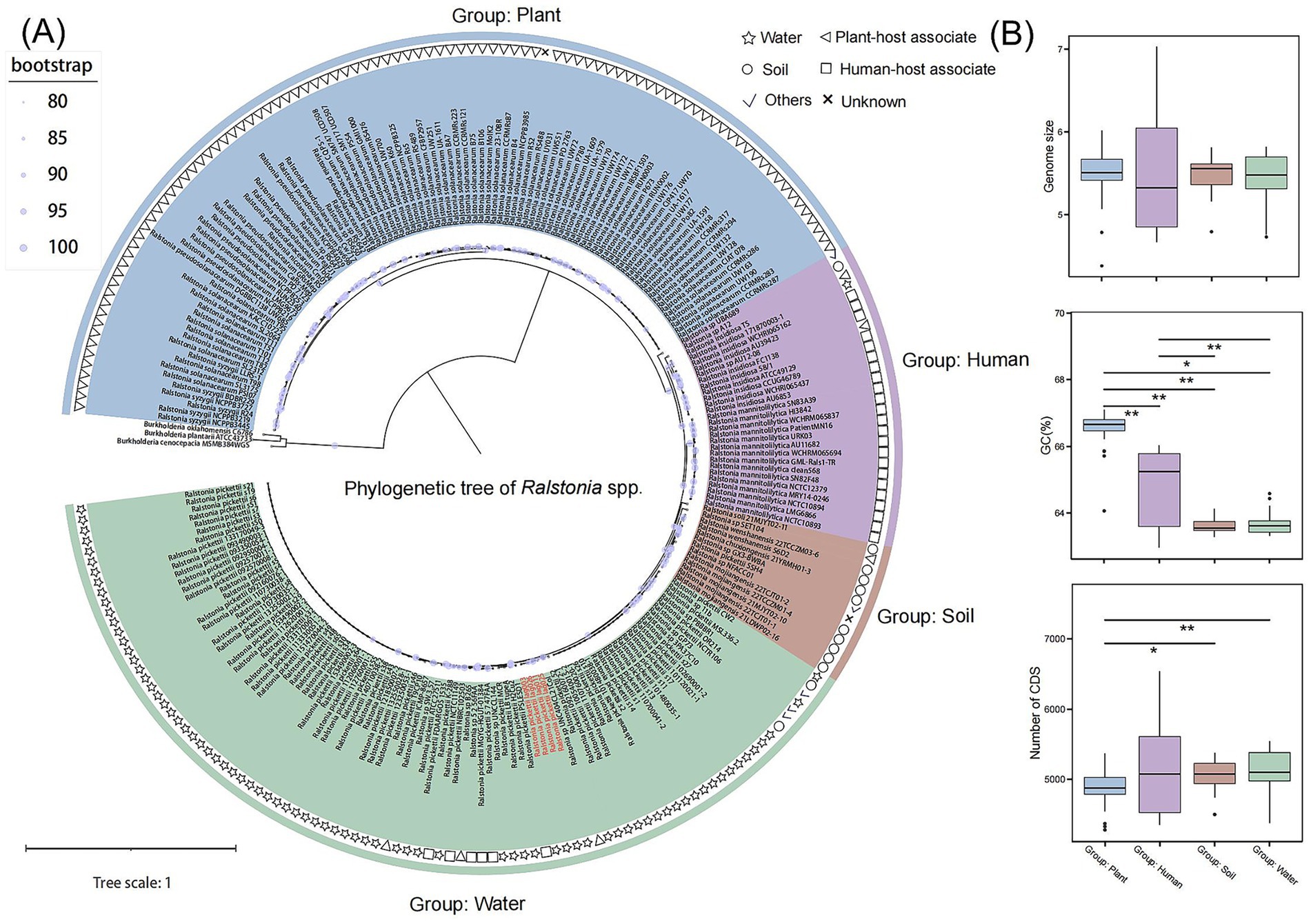
Figure 2. Genomic features and phylogenetic reconstruction of Ralstonia spp. genomes. (A) Maximum likelihood phylogenetic tree constructed from a concatenated alignment of 421 single-copy orthologous proteins. Nodes with bootstrap values ≥50 are indicated by solid circles. (B) Distributions of genome size, GC content (%), and number of coding sequences (CDSs) across different clades. Statistical significance was determined using the Wilcoxon rank-sum test: p < 0.05 (*), p < 0.01 (**).
3.3 Type of toxicity and infectivity in Ralstonia spp.
ARGs are present in nearly all genomes and can be divided into three types: OXA, ceoB, and sul2 (Figure 3). OXA refers to a class of β-lactamases that confer resistance to oxacillin and other β-lactam antibiotics. CeoB encodes an efflux pump component involved in resistance to chloramphenicol and other antimicrobials. Sul2 encodes a variant of dihydropteroate synthase, which mediates resistance to sulfonamide antibiotics. OXA is present in all environments except those associated with plant groups. CeoB is found in nearly all genomes except those from soil environments, with a notable absence observed in one human-associated lineage. The virulence factor bopC is exclusively distributed in soil group, inducing host cell necrosis. FlgG, a common virulence factor activating the host’s innate immunity, is found in some groups of plant and human genomes. In water-associated clades, all strains were found to harbor OXA- and ceoB-type ARGs, indicating a potentially greater threat to host organisms. These findings highlight the importance of monitoring ARGs in R. pickettii to minimize the risk of infection associated with environmental exposure or water usage. These findings indicate that Ralstonia spp. exhibit distinct patterns of ARGs and virulence factor accumulation across different environments, with aquatic groups showing greater abundance, potentially reflecting the complexity of their ecological niches. Notably, two specific ARGs were identified in aquatic-associated groups and were present in nearly every genome within these clades.
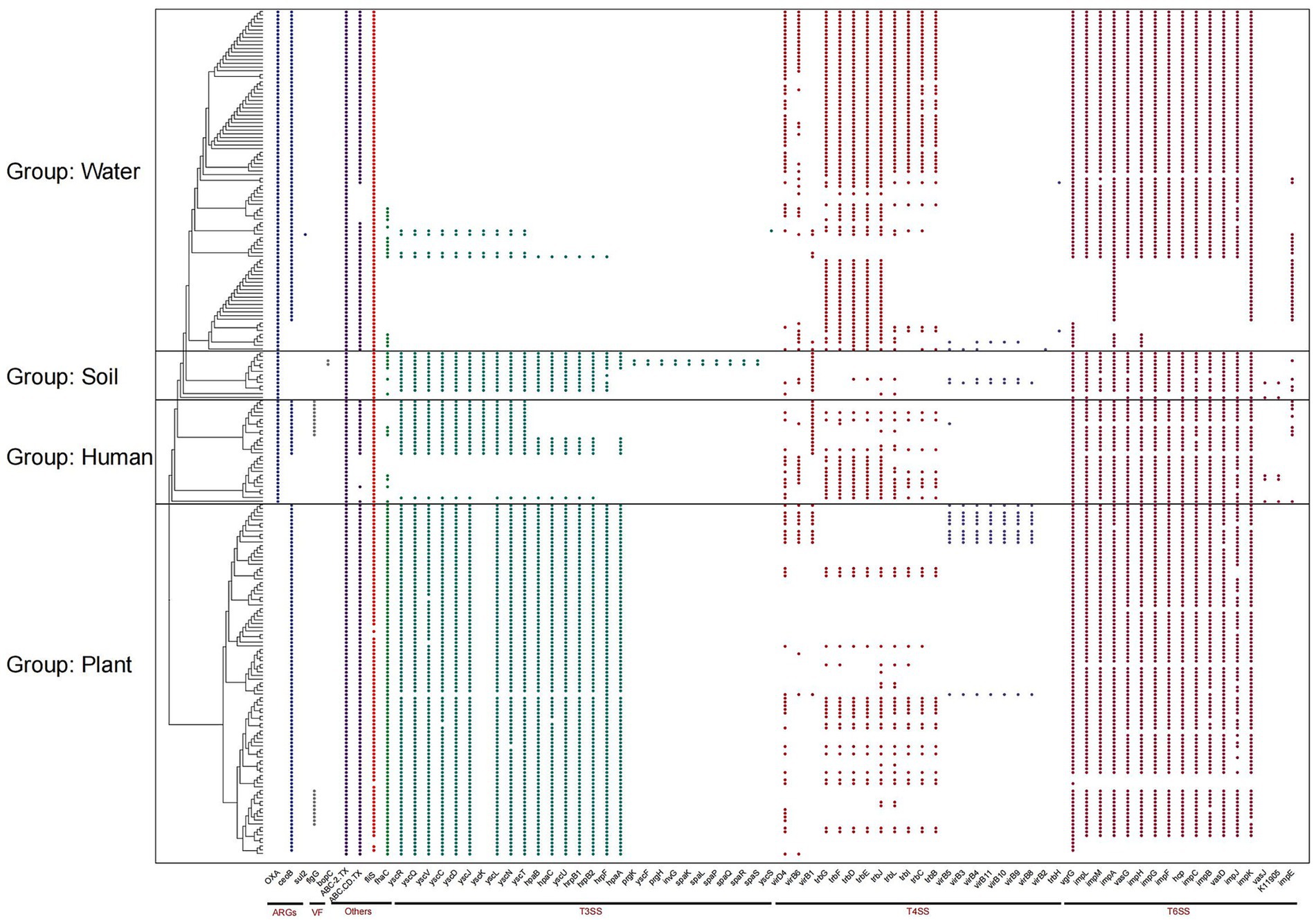
Figure 3. Composition of antibiotic resistance genes (ARGs) and bacterial secretion systems in Ralstonia spp. genomes. The figure shows the distribution of ARGs, virulence factors (VFs), type III secretion systems (T3SS), type IV secretion systems (T4SS), type VI secretion systems (T6SS), and other secretion systems in Ralstonia spp. genomes.
The bacterial secretion system plays a crucial role in the invasion of pathogenic microorganisms into hosts and serves as an indicator of toxicity. All genomes in this study contain complete T2SS, the typical T2SS is a secretion system that transports substances from the periplasmic space to the outer membrane of bacteria, encoded by approximately 12 to 15 gsp (general secretion pathway) genes, indicating a potential infection ability in Ralstonia spp. T3Es secreted by T3SS play a pivotal role in pathogenesis. Interestingly, the number of T3SS proteins is significantly lower in water group compared to other groups. While T4SS proteins is present in all clades, but with a higher presence observed in water group. Conversely, T6SS exhibits a certain degree of deficiency in water group.
3.4 CAZymes in Ralstonia genus
CAZymes are involved in biological processes related to carbohydrate synthesis and metabolism. They play roles in synthesis (GTs), degradation (GHs, PLs, CEs, AAs), and recognition (CBMs). The proportion of GHs, involved in carbohydrate degradation, was more abundant in the plant group compared to other groups (Figure 4A). Conversely, there were more GTs in the human group, which may be related to host invasion. There were no significant differences observed among PLs, CEs, CBMs, and AAs across the groups. The group of water species contains a different abundance GTs, indicating that different water habitat can also affect the composition of CAZymes. NMDS results revealed differences between the plant group and those groups with other habitats (R = 0.776, p = 0.001) based on bray-curtis distance (Figure 4B).
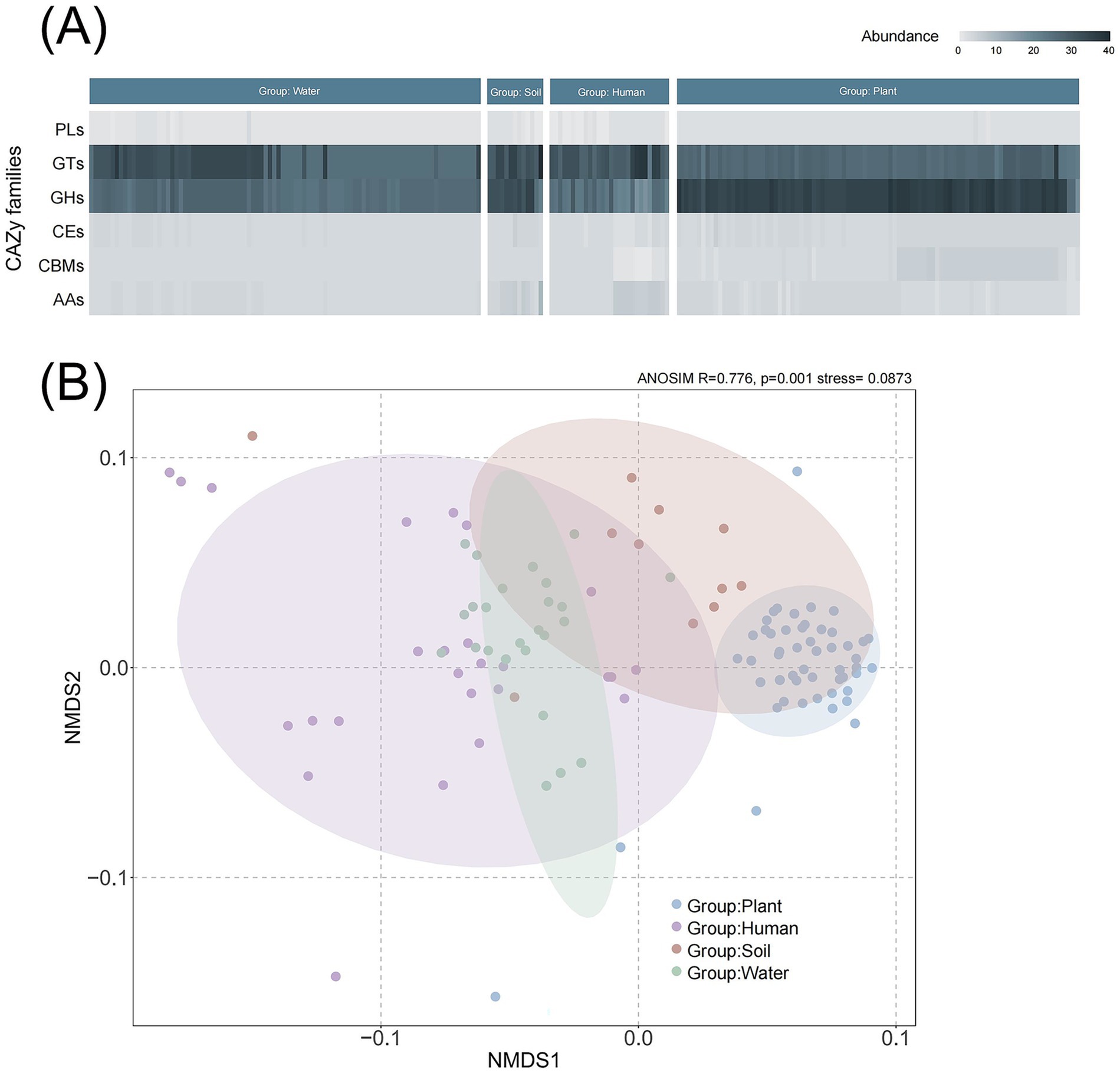
Figure 4. Composition and comparative analysis of carbohydrate-active enzymes (CAZymes) in Ralstonia spp. genomes. (A) Diversity and abundance of CAZymes across different clades. CAZyme genes are classified into six categories: PL (polysaccharide lyases), GT (glycosyltransferases), GH (glycoside hydrolases), CE (carbohydrate esterases), CBM (carbohydrate-binding modules), and AA (auxiliary activities). (B) NMDS (non-metric multidimensional scaling) analysis of enzyme composition among clades based on Bray–Curtis distance.
3.5 Metabolism analysis of Ralstonia spp.
The metabolism of microorganisms is highly diverse. The core genome of the Ralstonia spp. consists of only 12 pathways or biochemical processes under strict conditions (present in all genomes), including amino acid metabolism (leucine biosynthesis, proline biosynthesis and degradation), energy metabolism (cytochrome-c oxidase and oxygen oxidoreductase), metabolism of cofactors and vitamins (lipoic acid biosynthesis), carbohydrate metabolism (glycolysis of three-carbon compounds, nucleotide sugar biosynthesis, ribose-phosphate pyrophosphokinase), and nucleotide metabolism (adenine ribonucleotide biosynthesis, guanine ribonucleotide biosynthesis, ATP phosphohydrolase). There are three shared ABC transporters, including absorption of branched-chain amino acids, D-methionine, and efflux of LPS (lipopolysaccharide). Dismiss transporters, defined as lacking one protein component, four inward transport systems as Fe3+, phosphate, phosphonate, and glutamate/aspartate, which were identified through whole-genome BLASTp searches (identity >50%) using E. coli sequences as reference indicated by the yellow dashed arrows in Figure 5.
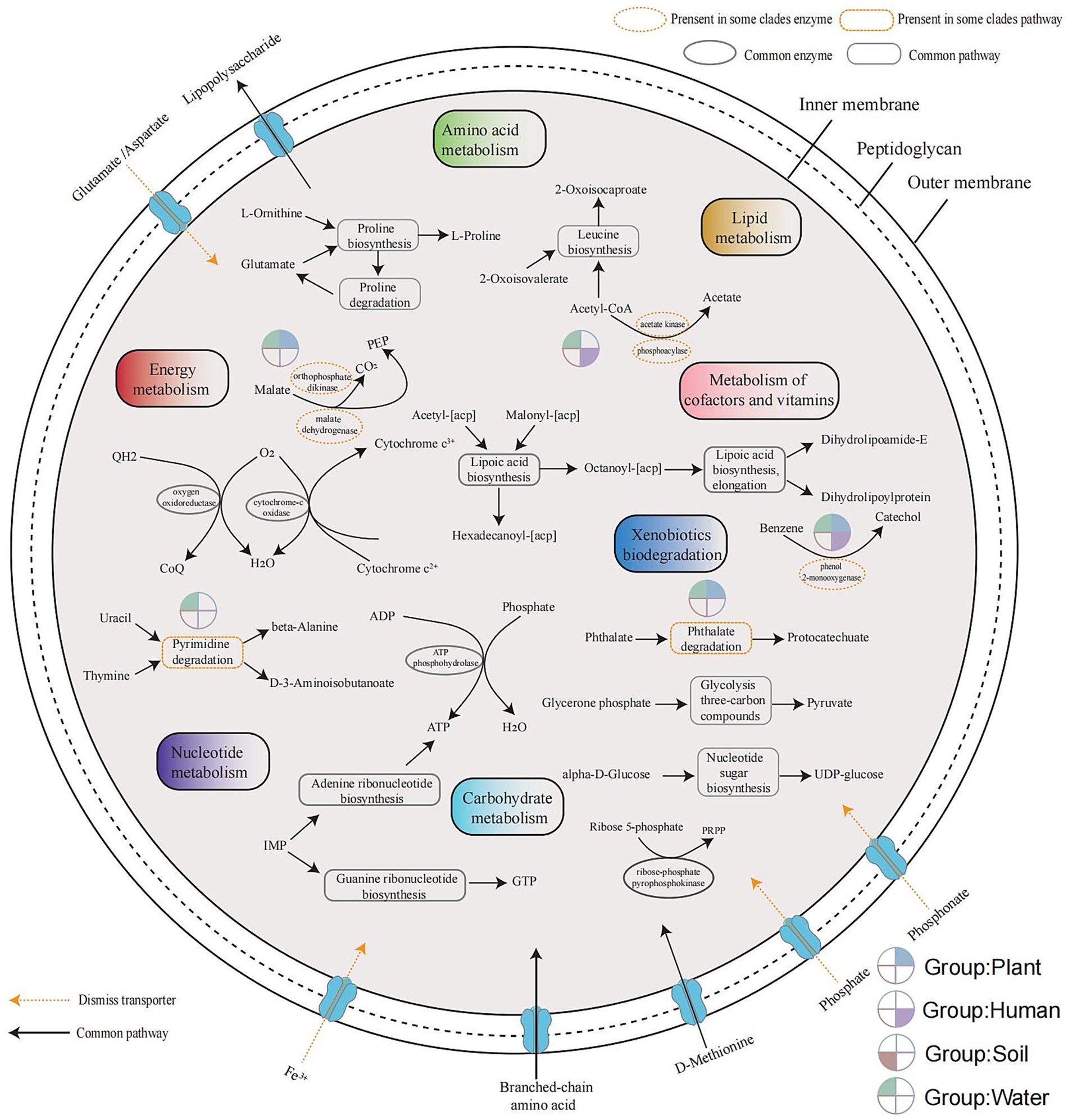
Figure 5. Overview of the metabolic potential across different habitats. Metabolic pathways were inferred from the orthogroup gene count matrix (excluding outgroups) obtained from OrthoFinder software results. Shared genomic metabolic pathways represent highly conserved sequences present in all genomes. Metabolic differences among clades were assessed using Fisher’s exact test (p < 0.05). Incomplete transporters, identified by sequence loss, were aligned to the corresponding E. coli protein sequences using BLASTp, with identity thresholds above 50%.
The phthalate degradation and acetate synthesis pathways are observed in plant and water groups, while phenol 2-monooxygenase is absent in soil group. The pyrimidine degradation pathway is only present in the group of water, while PEP (phosphoenolpyruvate) synthesis is present in plant and water groups. There are four types of ABC transporters observed with dismiss based on BLASTp result in all genomes, including Fe3+, phosphate, phosphonate and glutamate/aspartate.
3.6 Secondary metabolite prediction from Ralstonia spp. genomes
The prediction of secondary metabolites in Ralstonia spp. using antiSMASH software revealed a total of 16 secondary metabolites, with varying proportions observed across different habitats (Figure 6A). The average secondary metabolite content of each genome in different habitat is highest in the plant group, with NRPS (Nonribosomal peptides) content being the highest. However, in the human and soil groups, the average content of arylpolyene is the highest, while in water group, the content of redox-cofactor is the highest. Interestingly, the content of redox-cofactor in the plant group is extremely low, while the content of T1PKS is higher than the others. At the same time, the results of NMDS analysis also indicate differences in secondary metabolites between plant group and other groups (stress value <0.2, R = 0.545, p = 0.001) (Figure 6B).
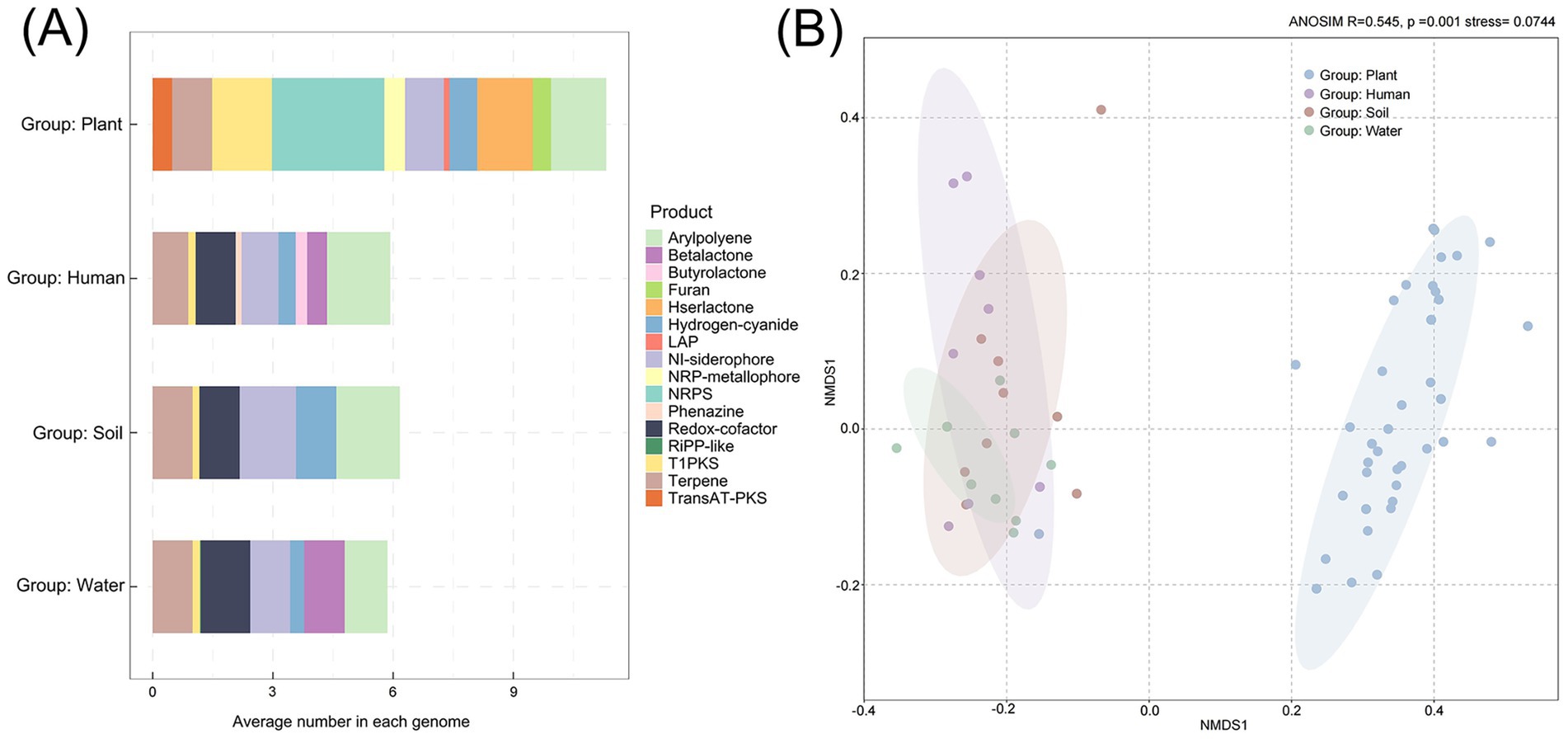
Figure 6. Prediction of secondary metabolites in Ralstonia spp. from different habitats based on antiSMASH software result. (A) Average number of secondary metabolites predicted per genome across different habitats. (B) NMDS analysis of clades based on secondary metabolite profiles using Bray–Curtis distances.
4 Discussion
Our comparative genomic analysis sheds light on the ecological and evolutionary adaptations of Ralstonia spp., with a particular focus on the aquatic habitat lineage. By reconstructing the phylogenomic relationships of 228 genomes, we identified four clades based on dominate habitat (plant, human, soil, and water). Notably, the water-associated R. pickettii lineage exhibited unique genomic features, including reduced T3SS genes content, diverse ARGs, and a distinctive pyrimidine degradation pathway, all of which suggest adaptations to oligotrophic aquatic environments. These findings contribute to a deeper understanding of the ecological roles of Ralstonia spp., particularly in aquatic microbial communities.
4.1 The relationship between Ralstonia pickettii and Dolichospermum sp.
Algae bloom is an important manifestation of microecological disorder in the water environment, manifested by an abnormal increase in the abundance of algae, extremely serious in China (Huo et al., 2021). Dolichospermum as one of bloom-forming cyanobacterium can produce toxins, including microcystin, which inhibit protein phosphatases, enhance cell membrane permeability, and cause DNA damage (Ji et al., 2024; Hitzfeld et al., 2000; Jong et al., 2020; Winter et al., 2021). These effects heighten the selection pressure for ARGs and facilitate HGT, potentially leading to a greater diversity of ARGs in aquatic environments of R. pickettii. We isolate R. pickettii from cyanobacterium cultures, enabling to preliminarily explore its possible relationship with algae blooms. The connection appears to be initially related to EPS include proteins, glyoxylates and lipids, which is critical for microbial aggregation (Liu et al., 2023; Naveed et al., 2019; Guan et al., 2024) and the microbial community can produce VB12 to promote the growth of cyanobacteria (Xie et al., 2016). The FISH results (Figure 1) indicate that R. pickettii may free living in lakes and achieve coexistence with cyanobacteria in a free state in the culture. When Dolichospermum is under a low-nitrogen environment, R. pickettii can promote pyrimidine degradation, supplement nitrogen sources, and prevent Dolichospermum dying for nitrogen deficiency.
4.2 Inconsistencies between the phylogeny of certain species and ecological habitats
We performed a preliminary screening of genomes based on completeness and retrieved associated habitat information from the NCBI database at the time of genome submission. For genomes with incomplete or missing habitat annotations, additional information was obtained through manual curation from published literature and relevant databases. Phylogenetic analysis revealed four distinct clades, which we classified into four habitat-based groups: Plant, Human, Soil, and Water. These classifications were derived objectively from the available data, without any prior assumptions.
Notably, discrepancies were observed between the phylogenetic placement and the recorded habitats of certain species. This may be attributed to the high environmental adaptability of Ralstonia species. Previous studies have shown that strains of the R. solanacearum species complex (plant pathogens) can persist in water and soil for extended periods (Alvarez et al., 2008; de Pedro-Jové et al., 2023). Moreover, human activities such as water consumption and excretion may facilitate the introduction of human-associated strains into aquatic or soil environments, while surface runoff following rainfall may contribute to the transfer of soil-associated strains into water bodies.
4.3 T3SS deficiency and ARGs diversity increased in water group
Many Ralstonia species are opportunistic pathogens that infect humans and plants, causing respiratory failure and leading to significant economic losses, respectively (Fluit et al., 2021; Ryan et al., 2006). The reasons for the diversity of ARGs are closely related to the microenvironment, where ARGs can be transferred among various microorganisms through mobile genetic elements (MGEs), including transposons, plasmids, and insertion sequences, thereby facilitating the adaptive evolution of resistant bacteria in this environment (Bellanger et al., 2014; Gootz, 2010). ARGs may potentially interact with native microbes in freshwater and estuarine ecosystems, leading to modifications in bacterial ecology and subsequent changes in microbial community structure, which influence ecosystem sustainability and function (Ohore et al., 2022). Therefore, the increased diversity of ARGs (OXA, sul2 and ceoB) in the aquatic community observed in this study may linked to the water ecological environment.
Gram-negative bacteria possess six types of secretion systems, labeled from I to VI. R. solanacearum relies on the TSS for the production of EPS, cell appendages, and protein secretions (Costa et al., 2015). In certain microbial communities, Bacteroidales utilize the T6SS system to shape the formation and evolution of these communities (García-Bayona et al., 2021). Simultaneously, the triggered of T4SS also facilitates the dissemination of resistance genes within microbial communities (Zhao et al., 2021). The absence of T3SS in certain species found in the human host group may be attributable to their origin in aquatic environments preceding human colonization. The absence of T3SS-associated genes in aquatic Ralstonia spp., in contrast to their terrestrial counterparts, suggests a distinct adaptive strategy to life in aquatic environments.
4.4 CAZymes and secondary metabolites different between plant group and others
Additionally, we explored the survival patterns of different groups in the habitats through CAZymes differences. CAZymes exhibit a higher prevalence in the genomes of pathogens, potentially associated with the degradation of complex carbohydrate structures within hosts, such as plant cell walls (Huang et al., 2018). Currently, GTs have been implicated in various pathogenic bacteria, including enterotoxigenic E. coli, Photorhabdus asymbiotica, Pseudomonas aeruginosa (Szymanski and Wren, 2005; Lu et al., 2015). In this study, the abundance of GTs showed variation even in the water group, potentially due to differences in aquatic environments. The increased synthesis of polysaccharides might contribute to the enhanced adaptability of bacteria to complex aquatic environment.
Secondary metabolites are substances that use primary metabolites as precursors and do not have specific biological functions. Due to differences in hosts or environments, there are significant distinctions between plant-associated groups and other groups. For example, NRPS, a diverse class of peptide compounds, plays an important role as an antibiotic in clinical applications, including telomycin, griselimycin, and lugdunin (Dang and Süssmuth, 2017). These findings suggest that plant-associated Ralstonia spp. possess distinct adaptive traits that may contribute to their successful colonization and survival within specific ecological niches. The proportion of redox-cofactor and betalactone are the highest in aquatic environments than other habitats. Redox cofactor are molecules that contains multiple types and can enhance the ability of microorganisms to adapt to various extreme environments (Somayaji et al., 2022). A redox cofactor enables microorganisms to adapt to shifts in redox states across diverse and complex environments, potentially associate its function with the variability of aquatic ecosystems (Guedes da Silva et al., 2020). Betalactone compounds contain multiple antibiotics to enhance microbial resistance to bacterial and fungal invasions and are an important way to obtain dominant community ecological niches (Tymiak et al., 1985; Robinson et al., 2019). The results indicate that Ralstonia spp. has adapted to a variable environment and produced different types of secondary metabolites.
4.5 A distinctive pyrimidine degradation pathway in the water group
The metabolism of microorganisms is diverse, consists of only 12 pathways or biochemical processes represents an extremely conservative function under strict conditions (present in all genomes). Although many metabolic pathways in the phylum Proteobacteria are known to be conserved, our predictions were based on protein annotations derived from genome data using computational tools. Due to inherent limitations in reference databases and algorithmic approaches, these predictions may be incomplete. However, the probability of incomplete annotation is expected to be consistent across all genomes. Therefore, instead of focusing solely on conserved core metabolic functions, our primary interest lies in identifying adaptive metabolic differences associated with distinct environmental niches. In the water group, the metabolic types are the most abundant, the group has acetate metabolism, phenol 2-monooxygenase, phthalates, malate dehydrogenase and PEP production. The pyrimidine degradation pathway enables assimilation of nitrogen and carbon for growth (Yin et al., 2019). R. pickettii in the water group employs this pyrimidine degradation strategy, which may relate to its ability to survive in relatively oligotrophic media (Matache et al., 2024). There is an abundance of pyrimidine in water, and its degradation produces urea. This urea serves as a primary nitrogen source for microorganisms, enabling them to adapt to low-nitrogen environments such as the space environment (Berg and Jørgensen, 2006; Maruyama et al., 2009; Santiago and West, 1999). Moreover, increased urea production may enhance nitrogen fixation by Dolichospermum sp., suggesting a potential interaction between R. pickettii and Dolichospermum sp.
In conclusion, this study highlights the ecological and evolutionary adaptations of Ralstonia spp., revealing four clades associated with soil, water, plant, and human groups based on dominate habitats. Water-associated R. pickettii strains show unique adaptations to aquatic environments, including reduced T3SS gene content, and a pyrimidine degradation pathway that supports survival in oligotrophic conditions. ARGs such as OXA and ceoB, enriched in water-associated strains, underscore the dynamic nature of microbial communities in aquatic ecosystems. Secondary metabolite profiles further reveal habitat-specific metabolic strategies. In water-associated strains, redox cofactors are likely to enhance resilience to oxidative stress, while β-lactones may confer competitive advantages. During algal bloom events, R. pickettii appears to coexist with cyanobacteria, potentially utilizing extracellular polymeric substances as a nutrient source. These findings provide new insights into the adaptive evolution of Ralstonia spp., particularly in aquatic ecosystems, and raise important questions about the ecological roles of R. pickettii during cyanobacterial blooms.
5 Limitations of the study
Due to the fact that this study is based on genome prediction results, additional direct experimental evidence is needed to support this interaction. Establish an indoor simulation group to observe the growth of Dolichospermum sp. and R. pickettii in coexistence, as well as the response to adding R. pickettii at different EPS concentrations. However, due to the difficulty in purifying Dolichospermum, indoor simulation experiments continue to encounter significant challenges. Additionally, we sought to verify whether the ability to degrade pyrimidine is exclusive to these groups. However, this idea has proven difficult to pursue due to the lack of available strains of different habitats.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA1111332.
Author contributions
GL: Writing – original draft, Writing – review & editing, Data curation. CM: Writing – review & editing, Writing – original draft. QL: Project administration, Writing – review & editing, Data curation, Supervision. DH: Funding acquisition, Writing – review & editing, Data curation, Writing – original draft, Supervision, Project administration. TL: Project administration, Writing – review & editing, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Key Research and Development Program of China (No. 2020YFA0907402), China Postdoctoral Science Foundation (No. 2021M703430), and the National Natural Science Foundation of China (No. 92251304).
Acknowledgments
We thank Guangxin Wang (Analysis and Testing Center, IHB, CAS) for assistance with fluorescence microscopy scanning and analysis. Additionally, we would like to express our appreciation to Shengjie Sun (Tianjin Agricultural University) for his valuable contributions to the discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1625651/full#supplementary-material
Footnotes
1. ^http://blast.ncbi.nlm.nih.gov/Blast.cgi
2. ^https://github.com/kblin/ncbi-genome-download
3. ^https://github.com/tseemann/barrnap
4. ^http://blast.ncbi.nlm.nih.gov/Blast.cgi
5. ^https://github.com/tseemann/abricate
References
Ailloud, F., Lowe, T., Cellier, G., Roche, D., Allen, C., and Prior, P. (2015). Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genomics 16, 1–11. doi: 10.1186/s12864-015-1474-8
Alvarez, B., López, M. M., and Biosca, E. G. J. M. (2008). Survival strategies and pathogenicity of Ralstonia solanacearum phylotype II subjected to prolonged starvation in environmental water microcosms. Microbiology 154, 3590–3598. doi: 10.1099/mic.0.2008/019448-0
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., et al. (2020). Kofam KOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Bellanger, X., Guilloteau, H., Bonot, S., and Merlin, C. (2014). Demonstrating plasmid-based horizontal gene transfer in complex environmental matrices: a practical approach for a critical review. Sci. Total Environ. 493, 872–882. doi: 10.1016/j.scitotenv.2014.06.070
Berg, G. M., and Jørgensen, N. O. (2006). Purine and pyrimidine metabolism by estuarine bacteria. Aquat. Microb. Ecol. 42, 215–226. doi: 10.3354/ame042215
Bernabeu, M., Cabello-Yeves, E., Flores, E., Samarra, A., Kimberley Summers, J., Marina, A., et al. (2024). Role of vertical and horizontal microbial transmission of antimicrobial resistance genes in early life: insights from maternal-infant dyads. Curr. Opin. Microbiol. 77:102424. doi: 10.1016/j.mib.2023.102424
Blin, K., Shaw, S., Augustijn, H. E., Reitz, Z. L., Biermann, F., Alanjary, M., et al. (2023). antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50. doi: 10.1093/nar/gkad344
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. J. B. (2009). Trim Al: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Dang, T., and Süssmuth, R. D. (2017). Bioactive peptide natural products as lead structures for medicinal use. Acc. Chem. Res. 50, 1566–1576. doi: 10.1021/acs.accounts.7b00159
de Pedro-Jové, R., Corral, J., Rocafort, M., Puigvert, M., Azam, F. L., Vandecaveye, A., et al. (2023). Gene expression changes throughout the life cycle allow a bacterial plant pathogen to persist in diverse environmental habitats. PLoS Pathog. 19:e1011888. doi: 10.1371/journal.ppat.1011888
Demirdag, T. B., Ozkaya-Parlakay, A., Bayrakdar, F., Gulhan, B., Yuksek, S. K., Yildiz, S. S., et al. (2022). An outbreak of Ralstonia pickettii bloodstream infection among pediatric leukemia patients. J. Microbiol. Immunol. Infect. 55, 80–85. doi: 10.1016/j.jmii.2020.12.004
Emms, D. M., and Kelly, S. J. G. (2019). Ortho finder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Fluit, A. C., Bayjanov, J. R., Aguilar, M. D., Cantón, R., Tunney, M. M., Elborn, J. S., et al. (2021). Characterization of clinical Ralstonia strains and their taxonomic position. Antonie Van Leeuwenhoek 114, 1721–1733. doi: 10.1007/s10482-021-01637-0
García-Bayona, L., Coyne, M. J., and Comstock, L. E. (2021). Mobile type VI secretion system loci of the gut Bacteroidales display extensive intra-ecosystem transfer, multi-species spread and geographical clustering. PLoS Genet. 17:e1009541. doi: 10.1371/journal.pgen.1009541
Gootz, T. D. (2010). The global problem of antibiotic resistance. Crit. Rev. Immunol. 30, 93–79. doi: 10.1615/critrevimmunol.v30.i1.60
Guan, Y., Yu, G., Jia, N., Han, R., and Huo, D. (2024). Spectral characteristics of dissolved organic matter in Plateau Lakes: identifying eutrophication indicators in Southwest China. Eco. Inform. 82:102703. doi: 10.1016/j.ecoinf.2024.102703
Guedes da Silva, L., Olavarria Gamez, K., Castro Gomes, J., Akkermans, K., Welles, L., Abbas, B., et al. (2020). Revealing the metabolic flexibility of “Candidatus Accumulibacter phosphatis” through redox cofactor analysis and metabolic network modeling. Appl. Environ. Microbiol. 86, e00808–e00820. doi: 10.1128/AEM.00808-20
Hitzfeld, B. C., Höger, S. J., and Dietrich, D. R. (2000). Cyanobacterial toxins: removal during drinking water treatment, and human risk assessment. Environ. Health Perspect. 108, 113–122. doi: 10.1289/ehp.00108s1113
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). Next polish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Huang, L., Zhang, H., Wu, P., Entwistle, S., Li, X., Yohe, T., et al. (2018). dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 46, D516–D521. doi: 10.1093/nar/gkx894
Huo, D., Gan, N., Geng, R., Cao, Q., Song, L., Yu, G., et al. (2021). Cyanobacterial blooms in China: diversity, distribution, and cyanotoxins. Harmful Algae 109:102106. doi: 10.1016/j.hal.2021.102106
Ji, W., Ma, J., Zheng, Z., Al-Herrawy, A. Z., Xie, B., and Wu, D. (2024). Algae blooms with resistance in fresh water: potential interplay between Microcystis and antibiotic resistance genes. Sci. Total Environ. 940:173528. doi: 10.1016/j.scitotenv.2024.173528
Jong, M.-C., Harwood, C. R., Blackburn, A., Snape, J. R., and Graham, D. W. (2020). Impact of redox conditions on antibiotic resistance conjugative gene transfer frequency and plasmid fate in wastewater ecosystems. Environ. Sci. Technol. 54, 14984–14993. doi: 10.1021/acs.est.0c03714
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Librado, P., Vieira, F. G., and Rozas, J. J. B. (2012). Badirate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28, 279–281. doi: 10.1093/bioinformatics/btr623
Liu, Y., Yang, F., Liu, S., Zhang, X., and Li, M. (2023). Molecular characteristics of microalgal extracellular polymeric substances were different among phyla and correlated with the extracellular persistent free radicals. Sci. Total Environ. 857:159704. doi: 10.1016/j.scitotenv.2022.159704
Liu, D., Zhang, J., Lü, C., Xia, Y., Liu, H., Jiao, N., et al. (2020). Synechococcus sp. strain PCC7002 uses sulfide: quinone oxidoreductase to detoxify exogenous sulfide and to convert endogenous sulfide to cellular sulfane sulfur. MBio 11:10-1128. doi: 10.1128/mbio.03420-19
Lu, Q., Li, S., and Shao, F. (2015). Sweet talk: protein glycosylation in bacterial interaction with the host. Trends Microbiol. 23, 630–641. doi: 10.1016/j.tim.2015.07.003
Maruyama, J., Fukui, N., Kawaguchi, M., and Abe, I. (2009). Use of purine and pyrimidine bases as nitrogen sources of active site in oxygen reduction catalyst. J. Power Sources 194, 655–661. doi: 10.1016/j.jpowsour.2009.06.048
Matache, R., Deak, G., Jawdhari, A., Sadîca, I., Pop, C. E., Fendrihan, S., et al. (2024). First insights of the Danube sturgeon (Acipenser gueldenstaedtii) skin adherent microbiota. bioRxiv :2024.03.13.584882. [preprint]. doi: 10.1101/2024.03.13.584882
Naveed, S., Li, C., Lu, X., Chen, S., Yin, B., Zhang, C., et al. (2019). Microalgal extracellular polymeric substances and their interactions with metal (loid) s: a review. Crit. Rev. Environ. Sci. Technol. 49, 1769–1802. doi: 10.1080/10643389.2019.1583052
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ohore, O. E., Wei, Y., Wang, Y., Nwankwegu, A. S., and Wang, Z. (2022). Tracking the influence of antibiotics, antibiotic resistomes, and salinity gradient in modulating microbial community assemblage of surface water and the ecological consequences. Chemosphere 305:135428. doi: 10.1016/j.chemosphere.2022.135428
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peeters, N., Guidot, A., Vailleau, F., and Valls, M. (2013). Ralstonia solanacearum, a widespread bacterial plant pathogen in the post-genomic era. Mol. Plant Pathol. 14, 651–662. doi: 10.1111/mpp.12038
Pohlmann, A., Fricke, W. F., Reinecke, F., Kusian, B., Liesegang, H., Cramm, R., et al. (2006). Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24, 1257–1262. doi: 10.1038/nbt1244
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Remenant, B., Coupat-Goutaland, B., Guidot, A., Cellier, G., Wicker, E., Allen, C., et al. (2010). Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics 11, 1–16. doi: 10.1186/1471-2164-11-379
Robinson, S. L., Christenson, J. K., and Wackett, L. P. (2019). Biosynthesis and chemical diversity of β-lactone natural products. Nat. Prod. Rep. 36, 458–475. doi: 10.1039/c8np00052b
Ryan, M. P., and Adley, C. C. (2014). Ralstonia spp.: emerging global opportunistic pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 33, 291–304. doi: 10.1007/s10096-013-1975-9
Ryan, M. P., Pembroke, J. T., and Adley, C. C. (2006). Ralstonia pickettii: a persistent gram-negative nosocomial infectious organism. J. Hosp. Infect. 62, 278–284. doi: 10.1016/j.jhin.2005.08.015
Ryan, M. P., Pembroke, J. T., and Adley, C. C. (2007). Ralstonia pickettii in environmental biotechnology: potential and applications. J. Appl. Microbiol. 103, 754–764. doi: 10.1111/j.1365-2672.2007.03361.x
Santiago, M. F., and West, T. P. (1999). Metabolism of pyrimidine bases and nucleosides by Pseudomonas fluorescens biotype F. Microbios 154, 221–224.
Schmieder, R., and Edwards, R. J. B. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Seemann, T. J. B. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shen, W., Le, S., Li, Y., and Hu, F. (2016). SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. doi: 10.1371/journal.pone.0163962
Silva, P. H., Johnson-Silva, W., Albuquerque, G. M., Oliveira, V. L., Santos, L. V., Gonçalves, M. H., et al. (2024). Exploring the competitive potential of Ralstonia pseudosolanacearum and Ralstonia solanacearum: insights from a comparative adaptability study. Plant Pathol. 73, 898–914. doi: 10.1111/ppa.13848
Somayaji, A., Dhanjal, C. R., Lingamsetty, R., Vinayagam, R., Selvaraj, R., Varadavenkatesan, T., et al. (2022). An insight into the mechanisms of homeostasis in extremophiles. Microbiol. Res. 263:127115. doi: 10.1016/j.micres.2022.127115
Szymanski, C. M., and Wren, B. W. (2005). Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3, 225–237. doi: 10.1038/nrmicro1100
Tymiak, A. A., Culver, C. A., Malley, M. F., and Gougoutas, J. Z. (1985). Structure of obafluorin: an antibacterial. Beta.-lactone from Pseudomonas fluorescens. J. Org. Chem. 50, 5491–5495. doi: 10.1021/jo00350a010
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Winter, M., Buckling, A., Harms, K., Johnsen, P. J., and Vos, M. (2021). Antimicrobial resistance acquisition via natural transformation: context is everything. Curr. Opin. Microbiol. 64, 133–138. doi: 10.1016/j.mib.2021.09.009
Xie, M., Ren, M., Yang, C., Yi, H., Li, Z., Li, T., et al. (2016). Metagenomic analysis reveals symbiotic relationship among bacteria in Microcystis-dominated community. Front. Microbiol. 7:173284. doi: 10.3389/fmicb.2016.00056
Yin, J., Wei, Y., Liu, D., Hu, Y., Lu, Q., Ang, E. L., et al. (2019). An extended bacterial reductive pyrimidine degradation pathway that enables nitrogen release from β-alanine. J. Biol. Chem. 294, 15662–15671. doi: 10.1074/jbc.RA119.010406
Yuan, C., An, T., Li, X., Zou, J., Lin, Z., Gu, J., et al. (2024). Genomic analysis of Ralstonia pickettii reveals the genetic features for potential pathogenicity and adaptive evolution in drinking water. Front. Microbiol. 14:1272636. doi: 10.3389/fmicb.2023.1272636
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhao, Q., Guo, W., Luo, H., Xing, C., Wang, H., Liu, B., et al. (2021). Deciphering the transfers of antibiotic resistance genes under antibiotic exposure conditions: driven by functional modules and bacterial community. Water Res. 205:117672. doi: 10.1016/j.watres.2021.117672
Keywords: Ralstonia , comparative genomics, antibiotic resistance, pathogenic microorganisms, microbial evolution
Citation: Liu G, Mao C, Li Q, Huo D and Li T (2025) Comparative genomic analysis reveals the adaptive traits of Ralstonia spp. in aquatic environments. Front. Microbiol. 16:1625651. doi: 10.3389/fmicb.2025.1625651
Edited by:
Yizhi Sheng, China University of Geosciences, ChinaReviewed by:
Cecilia Susana Demergasso Semenzato, Universidad Católica del Norte, ChileLuminita Marutescu, University of Bucharest, Romania
Copyright © 2025 Liu, Mao, Li, Huo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Li, bGlxaUBpaGIuYWMuY24=; Da Huo, aHVvZGFAaWhiLmFjLmNu