 Lian Xu1,2
Lian Xu1,2 Ji-Quan Sun
Ji-Quan Sun- 1Jiangsu Key Laboratory for Organic Solid Waste Utilization, Educational Ministry Engineering Center of Resource-saving Fertilizers, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, Nanjing Agricultural University, Nanjing, China
- 2Ministry of Education Key Laboratory of Ecology and Resource Use of the Mongolian Plateau, School of Ecology and Environment, Inner Mongolia University, Hohhot, China
Phenolic acids (PAs), which can exert toxic effects on seed germination and plant growth, are the most common allelopathic substances found in soils. To better understand the degradation fates of PAs in the rhizosphere of halophytes, five haloalkaliphilic PA-degrading bacteria, which were identified as three novel species of Kocuria (namely, Kocuria rhizosphaerae sp. nov., Kocuria kalidii sp. nov., and Kocuria rhizosphaericola sp. nov.), were obtained from the rhizosphere and bulk soil of the halophyte Kalidium cuspidatum. All five Kocuria strains could efficiently degrade ferulic acid (FA) and cinnamic acid (CA) under saline-alkaline conditions. Genomic and transcriptomic analyses revealed that the acrylic groups of FA and CA were first converted to a carboxyl via the coenzyme A (CoA)-dependent non-β-oxidation pathway by the five Kocuria strains. However, the five Kocuria strains selected different aromatic ring-cleavage ways for the degradation of the benzoic derivatives intermediates of the two compounds. The protocatechuate result from FA was then thoroughly degraded through an aromatic ring-opening reaction catalyzed by protocatechuate 3,4-dioxygenase (PcaGH), and the β-ketoadipic acid pathway. At the same time, the yield of benzoate originated from CA was subsequently converted to catechol by the benzoate 1,2-dioxygenase system (BenABCD) or phenylacetyl-CoA epoxidase (PaaABCD) and further completed the ring-cleavage by catechol 1,2-dioxygenase or catechol 2,3-dioxygenase (two non-PcaGH dioxygenases). The comparative genomic analysis revealed that the genes for phenolic acids hydroxylation, protocatechuate 3,4-dioxygenation, and those involved in the β-ketoadipic acid pathways are universal in the Kocuria strains. It is also demonstrated that the Kocuria strains maintain their osmotic balance by accumulating potassium, rather than biosynthesizing organic osmoprotectants, under hypersaline conditions.
Highlights
• Five haloalkaliphilic phenolic acid (PA)-degrading bacteria isolated from the rhizosphere of halophyte.
• The five strains were identified as three novel species within the genus Kocuria.
• The Kocuria genus exhibits exceptional genetic versatility and adaptive capability.
• The five strains used different pathways to degrade ferulic acid (FA) and cinnamic acid (CA).
• Phenol acid-degrading abilities are universal in the genus Kocuria.
1 Introduction
Phenolic acids (PAs), as the most common allelochemicals in ecosystems, usually refer to the hydroxylated derivatives of benzoate and cinnamic acids (CA), that is, CA, ferulic acid (FA), p-hydroxybenzoate (PHA), protocatechuate (PCA), and vanillic acid (VA). These compounds are common intermediates during the decomposition of lignin and can also be secreted by the plant roots (Margesin et al., 2021). In addition, PAs are also common pollutants in industrial wastewater and sewage (González et al., 1990; Marchiosi et al., 2020). Majority of the PAs have been demonstrated to have antibiotic capacity. For example, FA not only irreversibly damaged the bacterial cell membrane but also significantly inhibited the adhesion of bacterial cells, the formation of biofilms, and the bacterial community structures (Borges et al., 2012; Kang et al., 2020; Matejczyk et al., 2024; Rao et al., 2023). A high concentration of PAs—a type of autotoxic substance in the soil—can inhibit the growth and development of plants, resulting in crop yield reduction (Ma et al., 2023; Xie et al., 2023).
Biodegradation by microbes is a crucial mechanism for regulating the concentration of PAs in the soil ecosystem. Many microbes from multiple habitats were found to be able to effectively metabolize various PAs in the soil environments (Margesin et al., 2021, 2022; Wang et al., 2022) and industrial process (Genethliou et al., 2020; Liu et al., 2022; Oshlag et al., 2020). However, fewer reports concerned the degradation fates of PAs in hypersaline-alkaline conditions, except two PAs-degrading bacteria from the rhizosphere soil of the halophyte Suaeda salsa, namely, Acinetobacter suaedae C16S1 and Devosia rhizosphaerae RR2S18 (Tian et al., 2024; Xu et al., 2024b).
CA and FA are the two most common PAs in rhizosphere soils. The degradation of the two PAs occurs mainly through two major strategies. The first strategy is to convert the acryl group to the carboxyl before the aromatic ring. For example, majority of bacteria initiate the degradation of the two compounds by converting them to central ring-fission intermediates [protocatechuic acid (PCA) or benzoic acids], catalyzed step by feruloyl-CoA synthase, hydroxycinnamoyl-CoA hydratase-lyase, and aldehyde dehydrogenase (Lubbers et al., 2019). The produced central intermediates are then consecutively degraded in a ring-cleavage step and via β-ketoadipic acid pathways to various low-molecular-weight organic acids, which finally enter the tricarboxylic acid (TCA) cycle (Harwood and Parales, 1996). Another strategy involved the two kinds of compounds undergoing aromatic ring fission without converting the acryl group to a carboxyl group. For example, the CA was directly oxidized into cinnamic acid-dihydrodiol and then to 2,3-dihydroxycinnamic acids through a set of enzymes encoded by the hca cluster. The produced 2,3-dihydroxycinnamic acids undergo extradiol ring cleavage and are ultimately degraded to Krebs cycle intermediates (Díaz et al., 1998).
Members of the genus Kocuria, belonging to the family Micrococcaceae within the phylum Actinomycetota, are usually characterized as coccoid, strictly aerobic, Gram-stain positive, mesophilic bacteria (Stackebrandt et al., 1995). Majority of them are recognized as halophilic and sourced from high-salt related environments (Figure 1a). Some of them have been exhibited to be versatile at degrading pollutants, such as polycyclic aromatic hydrocarbons, naphthalene (Ezima et al., 2024; Cui et al., 2024; Khandelwal et al., 2024), and pentachlorophenol (Karn et al., 2011), along with heavy oil (Huang et al., 2012) and poultry feathers (Coello et al., 2000). This suggests that the members of Kocuria may play an important role in recycling carbon and energy in saline environments. Although a substantial amount of research has shown that the genus is present in hypersaline habitats, there has been less research on the ecological roles of the genus in the environment.
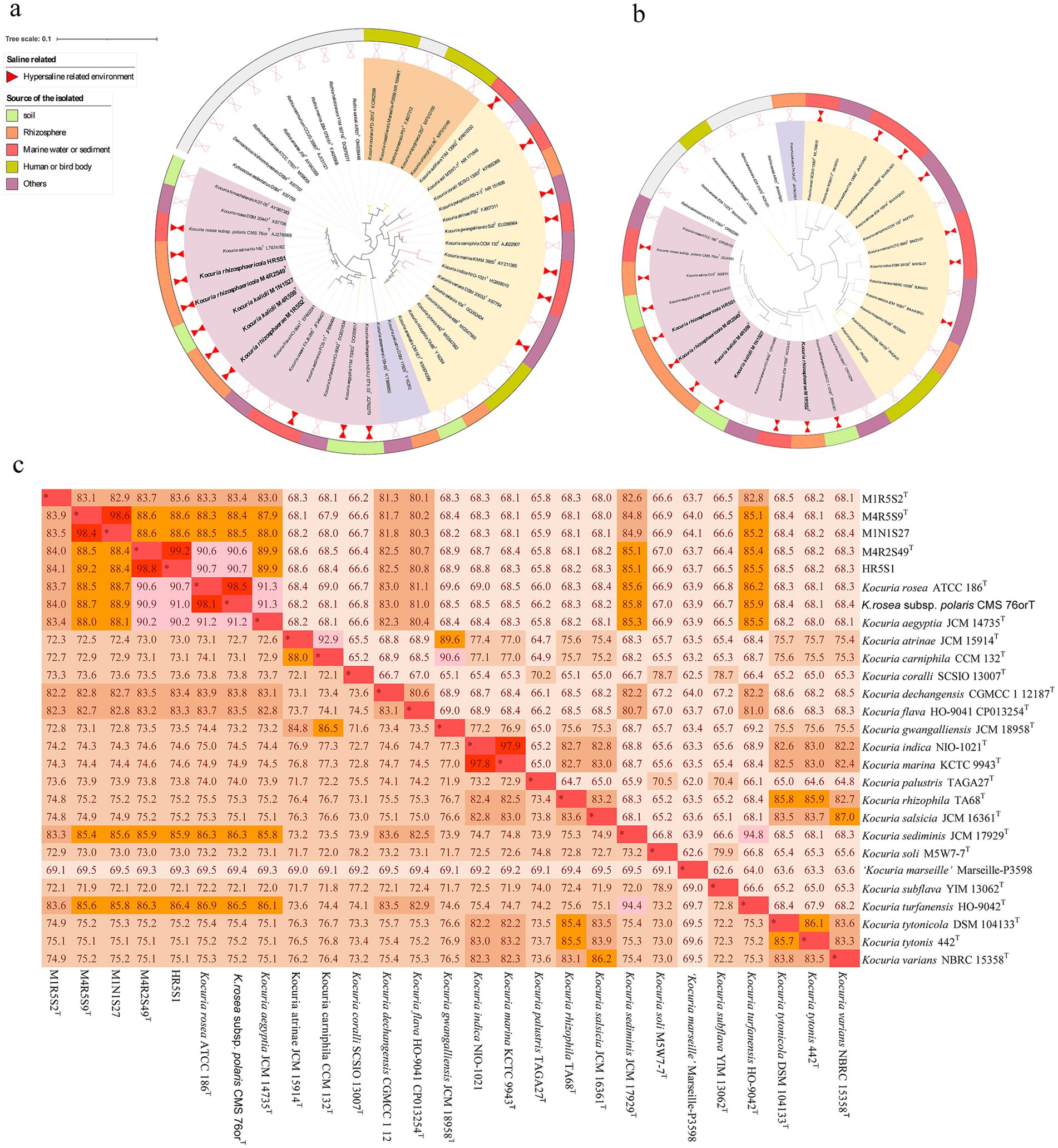
Figure 1. A phylogenetic tree based on the 16S rRNA gene sequences constructed using the neighbor-joining algorithm (a), and a phylogenomic tree based on 876 core genes using OrthoFinder (b) Both bars 0.1 substitutions per nucleotide (a)/amino acids (b) position. The isolation source and clusters for strains were marked with different colors. The ANI and AAI values between the Kocuria strains (c).
During an ongoing study on the plant-bacteria interaction, a total of 2,670 isolates were collected from the rhizosphere soil and non-rhizosphere soil of Kalidium cuspidatum, a common halophyte in the northwestern inner continent of China. Among them, five PAs-degrading strains of Kocuria shared low 16S ribosomal RNA (rRNA) gene similarities with current strains, and exhibited high PA-degrading abilities under saline-alkaline conditions. To better understand the roles of the genus in PAs degradation, we will elucidate their accurate taxonomic position and their PAs-degrading mechanism through multiple-omics analyses.
2 Materials and methods
2.1 Source of the strains
The soil samples used to isolate the strains were collected from the rhizosphere of K. cuspidatum and the bulk saline soil in Tumd Right Banner, Baotou, Inner Mongolia, China. Approximately 1.0 g rhizosphere soil was collected by hand-shaking roots to remove the adhering soil (Barillot et al., 2013), while the bulk soil was collected from soil without any plants growing next to the plants. Among them, two samples of rhizosphere soil were designated R2 and R5, while the bulk saline soil sample was designated N1. The values of the pH, concentration of salt, and organic matter were 9.1, 3.20 mS cm−1, and 2.94 g kg−1 for R2, 10.2, 6.63 mS cm−1, and 1.09 g kg−1 for R5, and 9.3, 7.77 mS cm−1, and 2.9 g kg−1 for N1 (Xu et al., 2024a), respectively. The strains were isolated using a protocol as previously described. Briefly, the 10-fold diluted soil suspensions were plated on M1 agar (g L−1: 2 sodium pyruvate, 1.0 L-asparagine, 0.1 (NH4)2SO4, 0.1 KCl, 30 MgSO4·7H2O, 0.05 FeSO4·7H2O, and 20 agar), M4 agar (g L−1: 2.5 cellulose, 2.0 sodium pyruvate, 0.25 KNO3, 1.0 proline, 0.2 MgSO4·7H2O, 0.2 K2HPO4, 0.5 CaCl2, 0.01 FeSO4·7H2O, 30 NaCl, and 20 agar), and artificial seawater agar (g L−1: 24.0 NaCl, 5.4 MgCl2·H2O, 5.0 tryptone, 4.0 Na2SO4, 1.5 CaCl2·H2O, 1.0 yeast extract, 0.68 KCl, 0.5 NH4Cl, 0.1 KBr, 0.2 NaHCO3, 0.2 Na2HPO4, 0.025 H3BO3, 0.024 SrCl2·6H2O, 0.002 NaF, and 20.0 agar, pH 8.0). The inoculated plates were then incubated at 30°C in the dark for a week. The colonies present on the agar plates with typical characteristics were picked and purified on new agar plates. The purified isolates were deposited in 15% glycerol solutions at −80°C. The isolates’ PAs-degrading capacities were primarily determined according to their growth in modified minimal salt medium (MMSM; g L−1: 3.5 K2HPO4, 0.2 KH2PO4, 30 NaCl, 1.0 (NH4)2SO4, and 0.2 MgSO4 7H2O; pH 8.0) which was added 100 mg L−1 PHA as sole organic carbon source using a protocol as previously described (Xu et al., 2024b).
Strain Kocuria rosea CGMCC 4.7049T (= ATCC 186T), which was used as a reference for the chemotaxonomic and phenotypic tests, was obtained from the China General Microbiological Culture Collection Center (CGMCC).
2.2 Phylogenetic analysis based on the 16S rRNA gene and whole genome
After 2 days of cultivation in high-salt LB broth (LB: g L−1: 5.0 yeast extract, 10.0 tryptone, and 30 NaCl; pH 8.0), the collected cell pellets were used as sources to for extract the total DNA using a DNA extraction kit (TianGen, Beijing, China) following the manufactures’ specification. The 16S rRNA genes of the five strains were amplified using universal primer sets (27F/1492R) and sequenced as previously described (Sun et al., 2017). The almost complete 16S rRNA gene sequences obtained were first used to determine their primary taxonomic positions using Basic Local Alignment Search Tool (BLAST) in NCBI.1 Their accurate taxonomic positions were determined by constructing a phylogenetic tree of the five strains along with the reference strains using the neighbor-joining (NJ) algorithm in MEGA software version 6.1 (Tamura et al., 2013). The topology of the tree was evaluated using the bootstrap methods.
To sequence the whole genome of the five strains, the short-insert (~350 bp) libraries were constructed using the Illumina library preparation kit (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions. All the libraries were then sequenced using an Illumina NovaSeq platform with the paired-end 150 (PE150) strategy. The raw reads were subsequently trimmed for quality using Trimmomatic version 0.35. The clean data were used for subsequent analyses. The draft genomes were assembled using SOAPdenovo2. Contig statistics, including N50 and the largest contig size, were calculated using QUality ASsessment Tool (QUAST) (Gurevich et al., 2013). The depth of coverage was determined using BEDTools (Quinlan and Hall, 2010). The sequencing generated 332–575-fold mean coverage for the genomes of the five strains. The authenticity of the genomes was checked using 16S rRNA genes. The completeness and contamination of the whole genome of the five strains were evaluated using CheckM (Parks et al., 2015). The completeness and contamination values of the strains and the reference strains are provided in Supplementary Table S1. The overall genome indices, including calculation of average nucleotide identity b (ANIb) and average amino-acid identity (AAI) values, were analyzed using fast alignment-free computation of whole-genome average nucleotide identity (FastANI) (Hernández-Salmerón and Moreno-Hagelsieb, 2022) and EzAAI (Kim et al., 2021), respectively. A phylogenomic tree was constructed based on 876 core genes using OrthoFinder (Emms and Kelly, 2015, 2019) with default parameters.
2.3 Polyphasic taxonomy
The growth range of temperature, pH, and concentration of NaCl of the five strains was determined in a modified LB broth as previously described (Xu et al., 2024a). The abilities to utilize carbon were determined in MMSM as previously described (Xu et al., 2022). The cell morphology was observed using a transmission electron microscope (HT7800; Hitachi, Tokyo, Japan).
The polar lipids of these five strains, along with their common reference strain K. rosea CGMCC 4.7049T, were extracted using a chloroform and methanol (1:2; v/v) mixture and detected using thin-layer chromatography as previously described (Kates, 1986). The respiratory quinones of these strains were extracted with chloroform/methanol (2:1; v/v), and determined using a high-pressure liquid chromatograph (HPLC; LC-16A; Shimadzu) equipped with a photodiode array detector (SPD-M40A) and a Kromasil 100−5 C18 separation column (internal diameter, 4.6 mm; length, 20 cm) as previously described (Komagata and Suzuki, 1987).
2.4 Degradation of the PAs
The PA-degrading abilities of the five strains were determined in MMSM with a protocol described in the literature (Tian et al., 2024). The inocula of the five strains were prepared in LB broth and harvested by centrifugation at 3,000g for 5 min. The cell pellets were rinsed and resuspended in MMSM at a final concentration of optical density (OD600) of 1.0. The cell suspensions were subsequently inoculated at 1% (v/v) into MMSM that contained 150 mg L−1 FA or 150 mg L−1 CA. The inoculated cultures were incubated at 150 r min−1 and 30°C in the dark. MMSM that contained PAs but were not inoculated with the strains were used as a control to evaluate the natural loss of the compounds. The cultures were sampled at 12, 24, 36, 48, 72, 96, 156, 204, 252, 300, 348, and 396 h.
The cell densities in the cultures were determined using an ultraviolet (UV) spectrophotometer (Shimadzu UV-1780, Shimadzu, Kyoto, Japan). The concentration of the residual PAs was determined using an HPLC equipped with a photodiode array detector (SPD-M40A) and a Kromasil 100−5 C18 separation column following the protocol ass previously described (Sun et al., 2012). Methanol:water (7:3, v/v) was used as the mobile phase at a flow rate of 0.7 mL min−1. FA and CA were recorded at 287 and 288 nm and 30°C with the detection limit being 0.01 mg L−1.
2.5 Transcriptomic analyses
Strains M4R2S49T, M1R5S2T, and M4R5S9T were selected for the transcriptomic analyses. Cells of the three strains in MMSM that contained CA or FA were harvested at 9 h of incubation for the total transcriptome analysis. The total RNA was extracted and evaluated as previously described (Xu et al., 2024b).
A total of 3 μg of RNA per sample was used to construct the sequencing libraries with an NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, United States) according to the manufacturer’s instructions. The rRNA removal, RNA fragmentation, first-strand cDNA synthesis, second-strand cDNA synthesis, conversion of the remaining overhangs to blunt ends, and adenylation of 3′ ends of DNA fragments were performed as previously described (Xu et al., 2024b). The library fragments were then purified using an AMPure XP system (Beckman Coulter, Brea, CA, United States) to select cDNA fragments of 150–200 bp in length. After cluster generation on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina), the library preparations were sequenced on an Illumina HiSeq platform, and paired-end reads were generated. The transcriptomic data were first normalized using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as the reference, and taking the sample cultivated with tryptone as the control.
2.6 Comparative genomic analyses
As of July 2024, 232 genomes of the Kocuria strain, including metagenome-assembled genomes (MAGs) deposited in GenBank. For the accuracy of the analysis, only 168 genomes with high quality (completeness >99% and contamination <1%) were selected for comparative genomic analysis. The pan- and core-genomes of the Kocuria strains were analyzed using the Bacterial Pan Genome Analysis tool (BPGA) pipeline version 1.3 (Chaudhari et al., 2016). The pan-genome genes were sorted out using Roray (Brynildsrud et al., 2016) with the default parameters.
2.7 Deposition of data
The 16S rRNA and whole genome sequence for these five strains were deposited in GenBank/EMBL/DDBJ under the accession numbers PQ498817-PQ498821, and JBISWJ000000000, JBISWK000000000, JBISWL000000000, JBISWM000000000, and JBISWN0000000000, respectively.
3 Results
3.1 Isolation and taxonomy of the strains
A total of five haloalkaliphilic strains, which were isolated from the rhizosphere soil of K. cuspidatum (namely strains HR5S1, M1R5S2T, M4R5S9T, and M4R2S49T) and bulk saline soils (strain M1N1S27), were selected for this study. A phylogenetic tree based on the 16S rRNA genes showed that the five strains clustered with K. rosea ATCC 186T, K. rosea subsp. polaris CMS 76orT, and Kocuria himachalensis K07-05T, and formed three independent clades: M4R5S9T with M1N1S27, M4R2S49T with HR5S1, and M1R5S2T alone (Figure 1a). The five strains shared the highest 16S rRNA gene similarities with their partner within the clades and then with K. rosea ATCC 186T. However, the phylogenomic tree based on the core genome showed a somewhat different phylogeny relationship. For example, on the phylogenomic tree, only the clade of strains M4R2S49T and HR5S1 tightly clustered with strains of K. rosea ATCC 186T and K. rosea subsp. polaris CMS 76orT, and K. himachalensis K07-05T, while the two other clades were located outside of the branch (Figure 1b). The ANIb and AAI values between the strains within the two clades of M4R5S9T and M1N1S27, and M4R2S49T and HR5S1 were all >95.0%, indicating that the strains within each clade belonged to the same species (Chun et al., 2018). Meanwhile, all the ANIb and AAI values of the five strains from the other type strains, including their closest relative K. rosea ATCC 186T, were <95.0% (Figure 1c), demonstrating they can be differentiated from the current species. Many characteristics can be used to distinguish the five strains of three clades from their closest related strain, K. rosea ATCC 186T (=CGMCC 4.7049T) (Supplementary Table S2). Based on the phylogenetic and phenotypic results, three clades formed by five new isolates should be identified as three novel species within the genus Kocuria. Therefore, K. rhizosphaerae sp. nov. (type strain: M1R5S2T = CGMCC 1.64778T = JCM 37379T), Kocuria kalidii sp. nov. (type strain: M4R5S9T = CGMCC 1.64776T = JCM 37381T; non-type strain: M1N1S27), and Kocuria rhizosphaericola sp. nov. (type strain: M4R2S49T = CGMCC 1.64777T = JCM 37380T; non-type strain: HR5S1) were proposed. The descriptions of the three novel species are shown in Table 1.
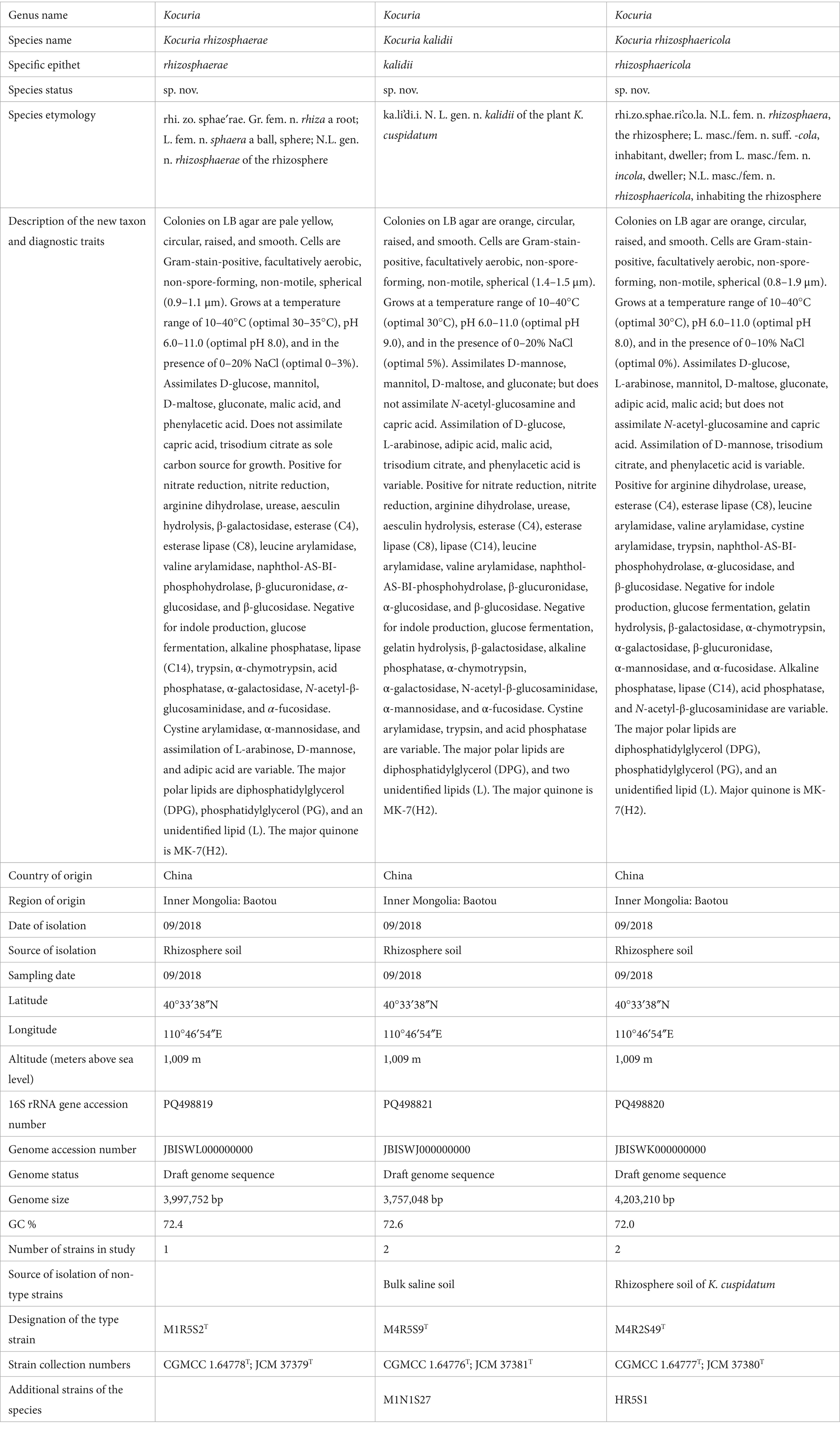
Table 1. Description of the three novel species within the genus Kocuria.
3.2 Characteristics of the degradation of PAs by the five Kocuria strains
All five Kocuria strains efficiently degraded FA and CA in the presence of 3% NaCl at pH 8.0 (Figure 2). The strains belong to a species that exhibited similar degradation characteristics. Overall, the strains degraded CA quickly compared to FA, which may be attributed to the different molecular structures of these compounds. The presence of a methoxy group in FA made it difficult to degrade. In detail, strain M1R5S2T degraded PAs the quickest: it completely degraded 150 mg L−1 CA within 24 h, and 150 mg L−1 FA within 60 h. While strains M4R5S9T and M1N1S27 degraded the same amount of FA within 160 h, and CA within 210 h. Compared to strains M4R5S9T and M1N1S27, strains M4R2S49T and HR5S1 took much longer time (>300 h) to degrade the same amount of FA, but less time for the degradation of CA. Additionally, the degradation of PAs consistently increased the cell density. In detail, the cell densities (OD600) of the five strains increased from 0.05 to 0.1–0.45 in the samples that contained 150 mg L−1 FA as the sole carbon source, while the cell densities increased from 0.05 to 0.20–0.30 in the samples that contained 150 mg L−1 CA, indicating that these five strains could utilize these two kinds of PAs as the sole carbon source for strains’ growth (Figure 2). It is notable that the same amount of CA resulted in much less biomass from M4R5S9T and M1N1S27 (OD600 < 0.2) compared to the three other strains, which corresponded to the lower degradation rate of CA by these two strains.
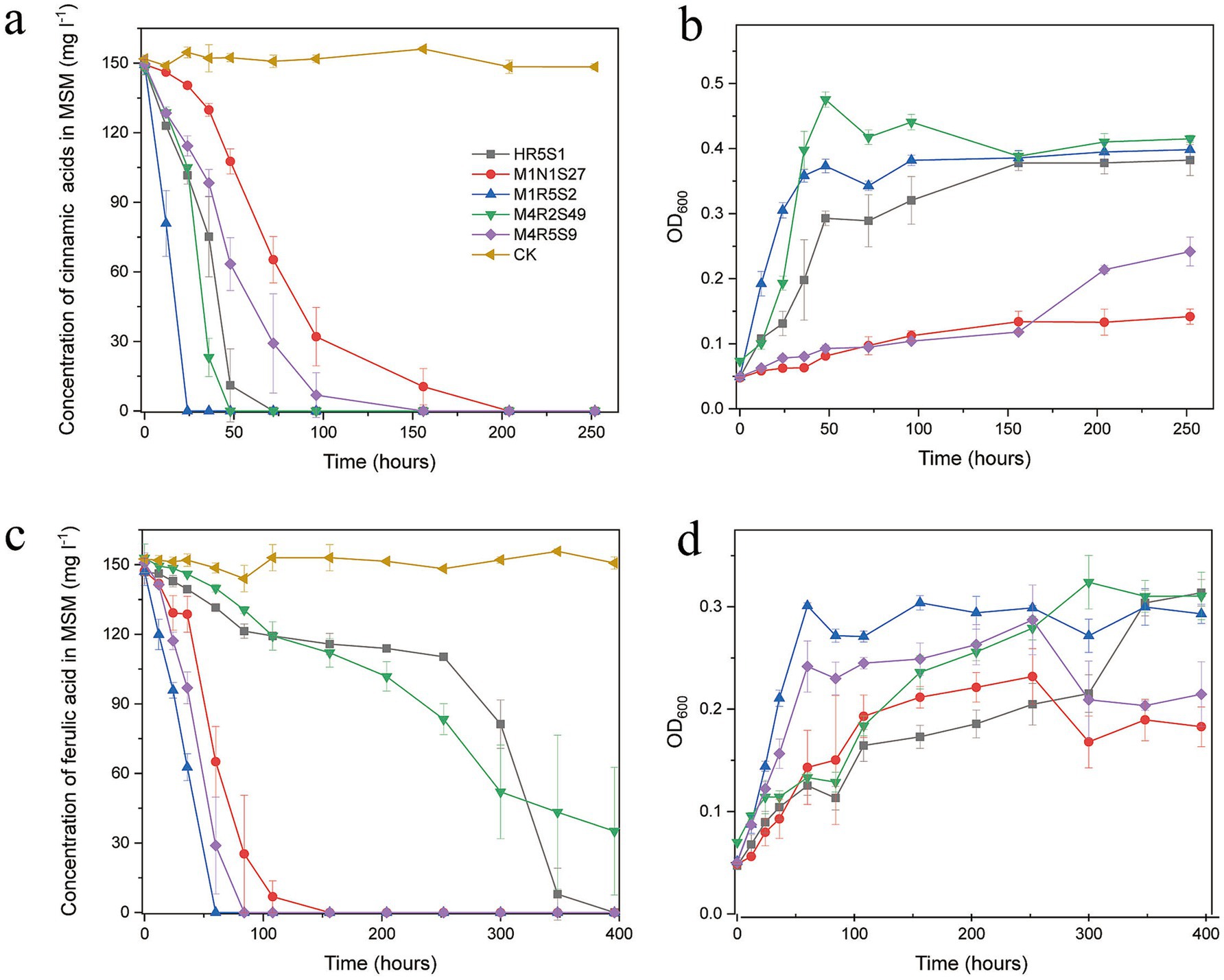
Figure 2. (A,C) The five Kocuria strains degraded FA and CA in MMSM (pH 8.0 and 3.0% NaCl) as the sole carbon and energy source for the strains’ growth. The initial concentration of both PAs is 150 mg L−1. (B,D) The biomass of the strains was determined by measuring the optical density at 600 nm (OD600). The values in the figures are the average of the 3 times, while the bars are error bars. MMSM, which contained FA or CA but without inoculating any strains, was used as a control (CK) to evaluate the natural loss of compounds. All five strains did not obviously grow in MMSMs without any PAs (data not shown).
An intermediate metabolite, which shared a similar ultraviolet spectrum and retention time (5.6 min) to VA, was detected during the degradation of FA by strain M4R5S9T. The same metabolites were also detected during the degradation of FA by strains M1R5S2T and M1N1S27 (data not shown).
3.3 Predicted degradation pathways in the five strains of Kocuria
The CoA-dependent β-oxidation pathway, by which FA is converted to PCA, is a common way to degrade FA. This pathway is usually catalyzed by FCS, FadB, Vdh, and VanAB (Figure 3a). Although all five strains were proven to be capable of degrading CA and FA, only strain M1N1S27 was found to harbor a specialized feruloyl CoA synthesis gene (fcs) and hydroxycinnamoyl-CoA hydratase-lyase gene (fadB) (Figure 3a). However, there are many predicted enzymes with similar catalytic functions in the five strains. For example, the 2-succinylbenzoate-CoA ligase gene menE functions similarly to fcs, and an enoyl-CoA hydratase gene (ech) is similar in function to fadB. The transcriptomic analysis showed that menE and at least one of the enoyl-CoA hydratase genes were significantly upregulated by both PAs (Figure 3c). The annotated vanillin dehydrogenase was not upregulated. Still, a phenylacetaldehyde dehydrogenase (named feaB) was remarkably upregulated by both PAs. All five strains harbored the vanillate O-demethylase genes vanAB, which were responsible for the conversion of a methoxy group on the aromatic ring of vanillate (or its derivative) to a hydroxyl group. Because CA lacks a methoxy group, the vanillate o-demethylase genes vanAB should be upregulated by FA, and not upregulated by CA. The transcriptome result confirmed this hypothesis. There are three other O-demethylase genes (two vanillate O-demethylase genes and a syringate O-demethylase gene desA) in the genome of strain M1N1S27 (Figure 3a). However, all these demethylase genes were not upregulated by FA or CA, which suggests that the other O-demethylase genes may not be involved in the degradation of FA. The transcriptomic results indicated that all five strains could convert FA to PCA, and convert CA to benzoate via CoA-dependent β-oxidation pathways.
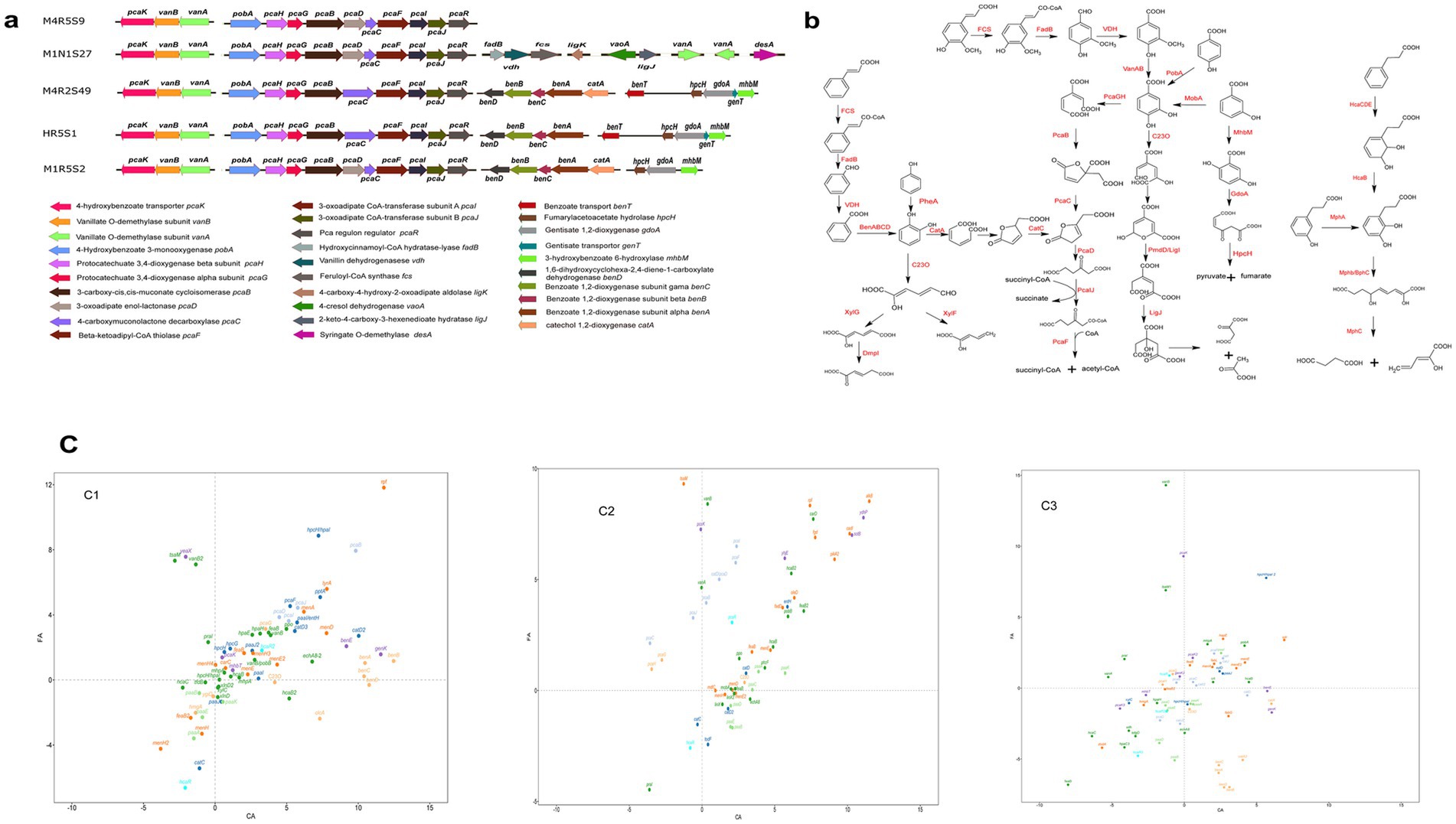
Figure 3. Genes related to aromatic compounds degradation in the genomes of the five strains (a), and the predicted degradation pathways according to the annotated genes (b), and the regulation of aromatic compounds-degrading genes in strains M1R5S2T (C1), M4R5S9T (C2) and M4R2S49T (C3) by FA and CA. The values of the genes were first normalized using the GDPAH gene as a reference and using cells cultivated in LB as the control. The normalized values were then used for logarithmic calculation using 1.5 as the base number. The X-axis represents the expression of the genes induced by CA, while the Y-axis reflects the expression of the related genes induced by FA. The values are positive for the expression of the genes. A value >0 means gene upregulation, while a value <0 means gene downregulation (c). More detailed gene names are listed in Supplementary Table S3.
After the conversion of an acryl group to a carboxyl group on the aromatic ring of CA/FA by the CoA-dependent non-β-oxidation pathway, the yield PCA from FA can directly undergo an enzymatic aromatic ring-opening reaction, but benzoate from CA cannot. The benzoate, which resulted from CA, should be converted into a derivative of catechol or other diphenol compounds before it can be ring-opened for further degradation. Strains M1N1S27, M4R2S49T, HR5S1, and M1R5S2T harbored a benABCD operon, which was responsible for the direct conversion of benzoate to catechol. The transcriptomic analyses showed that benABCD was significantly upregulated by CA and downregulated by FA in strains M4R2S49T and M1R5S2T. In addition, a phenylacetyl-CoA epoxidase gene operon (paaABCDE), which was responsible for the reduction of phenylacetyl-CoA (PA-CoA) to form 1,2-epoxyphenylacetyl-CoA, was found in all five strains. However, this operon was upregulated by CA in M4R5S9T and downregulated by CA or FA in strains M4R2S49T and M1R5S2T. This may be an alternative pathway for the conversion of benzoate to catechol in strain M4R5S9T. In addition, there are many other hydroxylase genes in these five strains. A p-hydroxybenzoate 3-hydroxylase gene (pobA or praI) located in the pca operon was found in all five strains, indicating that all five strains could convert PHA to PCA. Furthermore, strains M4R2S49T, M1N1S27, M4R5S9T, and M1R5S2T harbored the m-hydroxybenzoate 4-hydroxylase gene (mobA), which enables the host to degrade m-hydroxybenzoate to PCA. In addition, strains M4R5S9T, HR5S1, and M1R5S2T could convert m-hydroxybenzoate to gentisic acid catalyzed by 3-hydroxybenzoate 6-hydroxylase (XlnD) (Figure 3). However, all these hydroxybenzoate hydroxylase genes were not upregulated by FA or CA, that because these genes were not directly involved in the degradation of FA and CA.
Aromatic ring fission is another key step during the degradation of aromatic compounds. The genome analyses revealed that all five strains harbored several gene clusters involved in the ring-cleavage of aromatic compounds. A pca gene cluster, which includes pcaRJIFCDBGH and is involved in the degradation of PCA, was annotated in all five strains. Specifically, the ring of PCA, which was obtained from FA or the hydroxylation of PBA, was then opened due to the catalysis of protocatechuate 3,4-dioxygenase (PcaGH). The produced straight chain dicarboxylic acid was subsequently degraded to succinyl-CoA and acetyl-CoA after consecutive reactions, which were catalyzed by the enzymes encoded by the pca cluster (Figure 3b). Although all five strains harbored the pca gene cluster, there were some minor differences among them. Strains M4R5S9T, M1N1S27, and M1R5S2T completely converted 4-carboxymuconolactone to 3-oxoadipate using two independent enzymes (PcaC and PcaD), while that in strains M4R2S49T and HR5S1 was completed by a fusion enzyme PcaC (Figure 3a). In addition, all five strains still harbored other ring-fission genes, such as that for catechol 1,2-dioxygenase (C12O, including catA and benA), and the gene for catechol 2,3-dioxygenase (C23O). It is notable that two catechol 1,2-dioxygenae genes (namely, catA and catA2) and a catechol 2,3-dioxygenase gene were significantly upregulated by CA, and downregulated by FA in the three strains. In contrast, pcaGH was upregulated by FA, but downregulated by CA. This suggests that these strains employ two different pathways to degrade CA and FA after the removal of the acryl group from the aromatic ring via CoA-dependent non-β-oxidation. pcaGH was responsible for the ring-opening of the PCA produced from FA, while catA or C23O genes, which were not located in the pca-operon, were responsible for the ring fission of catechol that resulted from CA.
In addition to the ring-fission pathway that utilized catechol or its derivatives as an intermediate, the usage of gentisic acid as the substrate was another ring-fission pathway. Three of the five novel isolates harbored gentisate 1,2-dioxygenase (GdoA), by which the host could convert gentisic acid to 3-maleylpyruvate. The aromatic ring-opened product 3-maleylpyruvate was subsequently degraded to pyruvate and fumarate, which can be thoroughly degraded or transformed into other chemicals as a carbon skeleton for growth through the TCA cycle (Figure 3b). However, not all these genes were significantly upregulated by both PAs, indicating that the pathways represented by these genes were not involved in the degradation of either compound.
In addition to the CoA-dependent non-β-oxidation, CA can also be directly dioxygenated at the 2- and 3-sites of the aromatic ring, which was consecutively catalyzed by HcaA1A2CD and HcaB. The production of 2,3-dihydroxy-CA was then ring-opened by MphB. In the three strains whose transcriptomes were analyzed, the hcaB genes were upregulated by CA and downregulated by FA, suggesting that the CA-2,3-dioxygenation may also be a branch degradation pathway of CA. However, no hcaA1A2CD genes or mphB gene were found in all these genomes (Figures 3, 4).
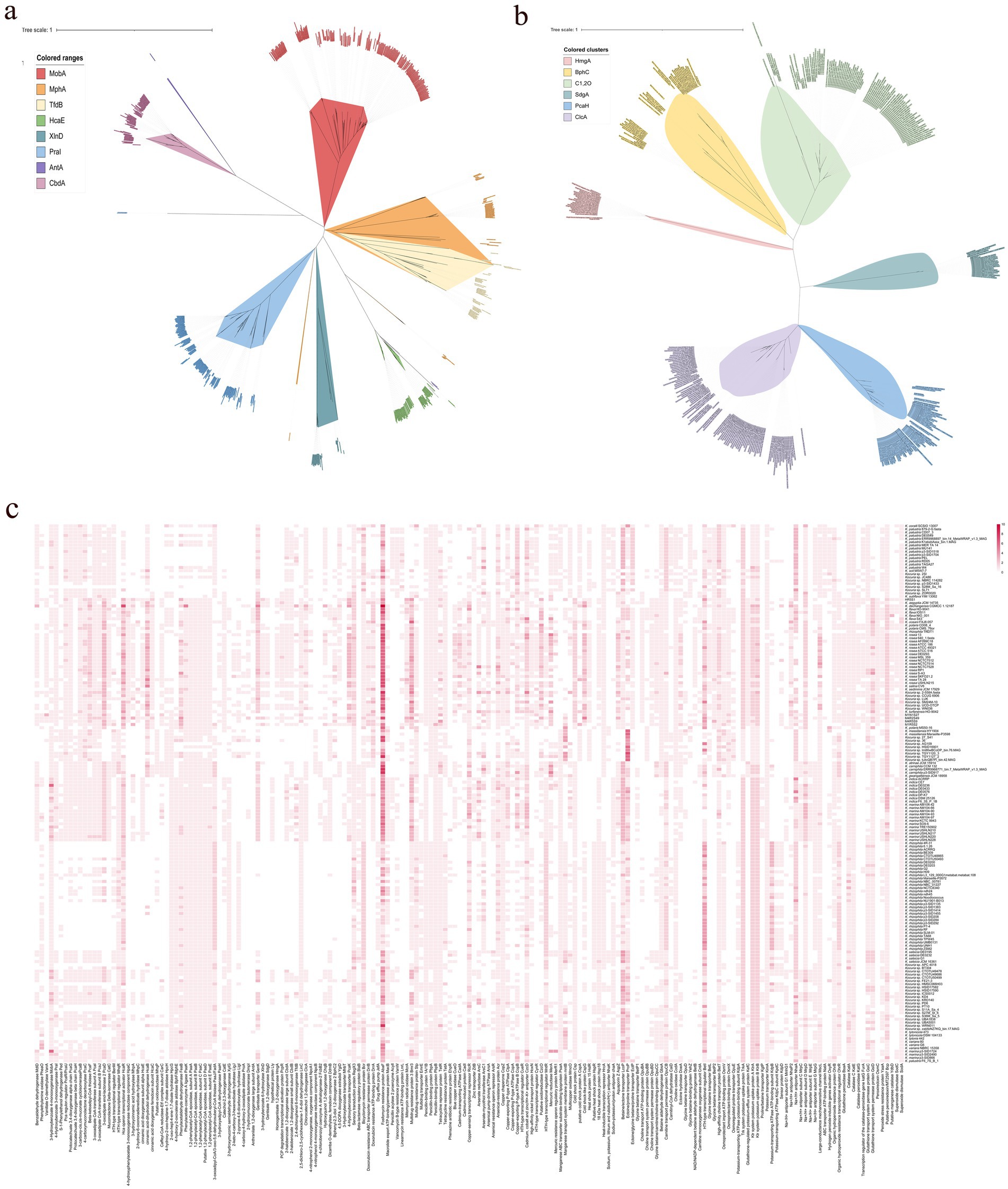
Figure 4. The phylogenetic trees based on aromatic compounds hydroxylase (a) and aromatic ring-opening dioxygenase (b), and the distribution of enzymes relating to aromatic compounds degradation, antibiotic resistance, osmotic balance maintenance, and oxidative stress defense (c).
As the transportation of the PAs, a 4-hydroxybenzoate transporter gene (named pcaK) was consistently upregulated by FA but downregulated (in strains M4R5S9T and M4R2S49T) or upregulated at a low level (in strains M1R5S2T) by CA. This suggests that this gene may play a key role in the uptake of FA by these strains. In contrast, two genes, genK encoding the gentisate transporter and benE (benzoate/H + symporter), were upregulated by CA but downregulated by FA in strains M4R2S49T and M1R5S2T, suggesting that they may play vital roles during the transmembrane absorption of CA. In strain M1R5S2T, three transporter genes (namely, sotB, responsible for sugar efflux transporter, and two inner membrane transporter genes: ydhP and yhjE) were all significantly upregulated by both PAs.
3.4 Comparative genome feature of the genus Kocuria
A comparative genomic analysis showed that the 168 selected Kocuria genomes contained 42,983 gene families, and the numbers of core, accessory, and unique genes were 231, 29,888, and 12,864, respectively. According to Heaps’ law, the pan-genome of Kocuria remains open (b = 0.44), indicating that with each newly added genome, the number of new genes increases the genetic repertoire of the species (Supplementary Figure S1).
All strains of Kocuria harbored the glycolysis/gluconeogenesis pathway, the pentose phosphate pathway, and the citrate cycle pathways, indicating that all strains of Kocuria could utilize glucose, fructose, and even starch as their carbon source for growth. In terms of the biosynthesis of amino acids, the genus strains can synthesize all types of amino acids except tryptophan with the simple substrates from carbohydrate metabolism. Majority of the Kocuria strains harbored various genes involved in the reduction of nitrate, nitrite, and sulfate (Supplementary Figure S2). These metabolic abilities facilitate the case in which the Kocuria strains can obtain carbon, nitrogen, sulfur, and energy from the environment, thereby enhancing their adaptability to diverse environments.
It was also revealed that all the strains of Kocuria harbored the betaine aldehyde dehydrogenase gene gbsA, carnitinyl-CoA dehydratase caiD, ectoine hydrolase doeA, and choline oxidase gene coda (Figure 4c). However, only seven ectB genes and two ectC genes, which are two key genes involved in the biosynthesis of ectoine, were found in these strains. No ectA gene was found, indicating that the Kocuria strains cannot synthesize ectoine as an osmoprotectant by themselves under hypersaline conditions. However, many osmoprotectant-binding proteins and transporter genes were found in these 168 genomes. For example, a total of 319 betaine/ectoine transporter gene lcoP, and 265 betaine/proline transporter gene proP were annotated in 155 and 140 Kocuria strains, respectively. In addition, majority of them could bind and transport the choline via a high-affinity choline transporter BetT (223 genes). Some of the Kocuria strains could absorb proline through the osmoprotectant proline transporter OsmX. The accumulation of potassium in the cytoplasm is another crucial mechanism for maintaining osmotic balance. Many ktrAB (296 and 303 genes from all 168 strains, respectively), trkA (309 genes from 163 strains), and kimA (164 genes from 164 strains), were annotated in the genomes of these strains. Furthermore, the Kocuria strains also harbored Na+/H+ antiporter genes, by which the cells could export sodium. These results indicate that the Kocuria strains could maintain the osmotic balance via several mechanisms.
Many PAs-degrading genes are ubiquitous in the Kocuria genomes. For example, a total of 185 mobA genes were annotated in 142 Kocuria genomes, indicating that majority of the Kocuria strains possess the ability to degrade m-hydroxybenzoate. A total of 116 genomes contained the praI (or named pobA) gene. Furthermore, 136 Kocuria strains harbored at least one gene of the pca cluster, indicating that the biodegradation of PCA via the β-ketoadipic acid pathway was quietly present in the genus Kocuria. In addition to the β-ketoadipic acid degradation pathways, 91 protocatechuate-4,5-dioxygenase genes (or catechol 2,3-dioxygenase genes) were found in 90 Kocuria strains. A total of 31 xlnD and 27 sdgD genes were identified in 28 and 26 Kocuria strains, respectively, indicating that these strains could degrade m-hydroxybenzoate via the gentisate-degrading pathway. Majority of the Kocuria strains harbored paa clusters, indicating that these strains could degrade phenylacetate via the Paa-degradation pathways. It is notable that 59 genomes of Kocuria harbored hcaE genes, a key enzyme that catalyzes the dioxygenation reaction of cinnamic acid to 2,3-diohydro-2,3-dihydroxycinnamic acid. A total of 167 hcaB genes, which were involved in the reaction of 2,3-dihydro-2,3-dihydroxycinnamic acid to 2,3-dihydroxycinnamic acid, were found in 68 strains of Kocuria. No mohB gene, which catalyzes the aromatic ring-cleavage reaction, was annotated in the genomes (Figure 4). However, several potential enzymes could substitute for MohB. For example, bphC, a gene involved in the cleavage of aromatic rings, was found in Majority of the Kocuria strains. The ring-opened product could be subsequently degraded to the small molecule dicarboxylic acid, which was catalyzed by MhpC or the other isoenzymes (Figure 4c). The results of comparative genomic analysis revealed that strains of the genus Kocuria generally have multiple pathways to degrade PAs and other aromatic compounds, indicating the great application potential of the genus Kocuria in bioremediation.
4 Discussion
In this study, five haloalkaliphilic Kocuria strains were isolated from the rhizosphere and bulk saline soils of the halophyte K. cuspidatum. All five strains were able to degrade several PAs at pH 8.0 in the presence of a high concentration of salt. It is generally believed that the Kocuria strains have a strong ability to degrade pollutants and tolerate abiotic stress. Not only they degrade organic pollutants, such as fuel oil or heavy oil (Huang et al., 2012; Promsing et al., 2021), and Aflatoxin B1 (Wang and Nan, 2025), they can also tolerate heavy metals and high concentration of salt. However, only Kocuria sp. TIBETAN4, a strain sourced from a saline lake located in northwestern China, could degrade phenol via the ortho-pathway under saline-alkaline conditions (Wu et al., 2018). There is less information on the degradation of PAs by the Kocuria strains. To the best of our knowledge, this is the first report about the degradation of PAs by Kocuria strains at saline-alkaline conditions. This should facilitate their use as agents for the bioremediation of saline-alkaline environments contaminated with PAs.
Although all five strains could degrade CA, FA, and some other PAs, they exhibited significant differences in the degradation rates of CA and FA. These differences may be attributed to the diversity of PA-degrading pathways. The genomic annotation and transcriptomic analyses revealed that all five strains utilized the CoA-dependent non-β-oxidation, protocatechuate 3,4-dioxygenation ring-open, and the β-ketoadipic acid pathway to degrade FA. After the conversion of CA to benzoate, some of them utilized the BenABCD enzyme system to convert benzoate to catechol; however, strain M4R5S9T did not harbor the ben-operon. In strain M4R5S9T, the paa-operon genes were upregulated by CA and downregulated by FA. No other pathway genes were significantly upregulated by CA. In strains M4R2S49T and M1R5S2T, the genes in the paa operon were not upregulated by both PAs. These findings suggest that strain M4R5S9T may select the paa pathway as its alternative way to degrade CA, owning to its inability to convert benzoate to catechol. These may explain why CA was slowly degraded by strain M4R5S9T. Notably, all five strains may select PcaGH dioxygenases for the aromatic-ring cleavage of PCA from the degradation of FA and non-PcaGH dioxygenases (CatA and C23O) for the ring-cleavage of benzoate from the degradation of CA. These findings indicated that CatA was primarily responsible for the ring-opening of catechol or its derivatives, which lack a carboxyl group on the aromatic ring. At the same time, the PcaGH was primarily responsible for the degradation of PCA or its derivatives. Naturally, the genes in the pca operon were also responsible for the degradation of the ring-opening products of PCA. While the β-ketoadipic acid pathway, which was not encoded by a gene within the pca operon was utilized for the degradation of catechol originating from CA. The presence of these multiple enzyme systems may improve the efficiency of bacterial strain metabolism of PAs in various environments.
The comparative genomic analysis revealed that the majority of Kocuria strains universally harbored PAs-degrading genes, such as the aromatic ring-opening genes, pcaGH, sdgD, the C23O gene, and bphC. As the monomers of lignin, the PAs are abundant in soils inhabited by these bacteria (Xie et al., 2023). The powerful PA-degrading abilities endow the Kocuria strains with higher competitiveness, which widens the territory in which they can survive. Alternatively, the PAs are also the most common allelochemicals in the soils, particularly in the rhizosphere (Peracchi et al., 2024). Low concentration of PAs could promote the growth and development of the host plant, while high concentrations may inhibit their growth and decrease the rate of seed germination (Li et al., 2015). Therefore, the Kocuria strains in the rhizosphere may play an important role in regulating the concentration of PAs in the rhizosphere environment, resulting in beneficial effects on the host plant. The correlation analysis between the genes and environments revealed that many of the genes were highly related to the isolation source of the host. Many of the aromatic compounds-degrading genes were positively related to the rhizosphere habitats, including phenylacetaldehyde dehydrogenase gene feaB, 3-dydroxyadipyl-CoA dehydrogenase paaH, 2-hydroxymuconate tautomerase dmpI, 3-hydroxybenzoate transporter mhbT, dicamba o-demethylase ddmA, bphC, 4-hydroxyacetophenone monooxygenase hapE, and several genes related to the degradation of phenylacetate (Figure 5). In addition, hipO, npcA, and vanA, 4-hydroxybenzoate transporter gene pcaK, 3-hydroxycinamic acid hydroxylase mhpA, and hydroxybenzoate 6-hydroxylase gene xlnD were positively related to the plant rhizosphere and soil, and negatively associated with seawater and the other sources, indicating the higher abundance in terrestrial ecological environments (Figure 5). These findings imply that the existence of these genes may be the result of coevolution between bacterial strains and specific environments.
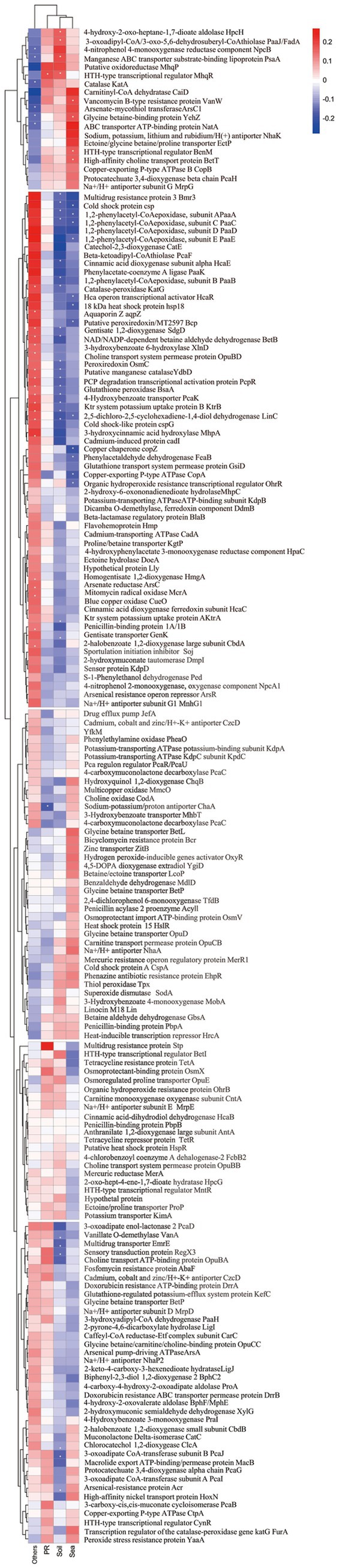
Figure 5. Correlation of the gene distribution and the strain isolation source.
The calculation for the analysis of the habitats, from which the strains were isolated, showed that many of Kocuria strains originated from the hypersaline habitats, such as seawater, and saline-alkaline soil (Figure 1a). However, less research has been conducted on the mechanism of tolerance to salt stress tolerance in this genus. The five Kocuria strains were able to degrade the PAs under a moderate saline condition, which once again confirms the extremely high environmental adaptability of the genus. Bacteria usually adopt two strategies to respond to osmotic stress. The first is to accumulate organic osmoprotectants and potassium, while the second is to export the sodium (Sunita et al., 2020; Tsujii et al., 2020). Glycine-betaine and ectoine are the two most common organic osmoprotectants in halophilic bacteria. This study revealed that Kocuria strains primarily depend on the absorption of osmoprotectants from their environments to maintain osmotic balance, rather than producing them through biosynthesis. However, majority of the genus strains harbored multiple potassium ion absorption channel genes, including ktrAB, trkA, and kimA. In addition, the strains harbored Na+/H+ antiporter genes, which enabled the cells to export sodium. During the degradation test, the medium (MMSM) does not contain organic osmoprotectants, but it contains sufficient potassium ions. These results suggest that absorbing potassium ions and expelling sodium ions is a major way for the Kocuria strains to maintain their osmotic balance. The comparative genomic analysis also revealed that majority of the Kocuria strains harbored the bicyclomycin resistance gene bcr (232 genes distributed across 163 genomes) and a total of 671 of the fosfomycin resistance gene abaF. The penicillin-binding protein genes ponA (146 genes in 146 genomes), pbpA (136 genes in 136 genomes), and pbpB (168 genes in 166 genomes), and the tetracycline resistance gene tetA (110 genes in 116 genomes) and its regulator gene tetR (170 genes in 144 genomes) were also found in the majority of Kocuria strains (Figure 4c). A total of 281 multidrug resistance protein 3 genes (bmr3) were found in 132 Kocuria strains. These genes endow the genus with the universal ability to resist antibiotics, facilitating their colonization in the rhizosphere. In terms of heavy metal resistance, many genes that encoded resistance to heavy metals were also annotated in the Kocuria strains. For example, 214 acr genes (in 161 genomes), which were responsible for arsenical resistance, were annotated in these strains. In addition, 118 arsC genes encoding arsenate reductase were found in 94 genomes, indicating that Kocuria strains universally harbor arsenate resistance. A total of 150 mercuric reductase genes, merA, were annotated in these 90 Kocuria genomes (Figure 4c). In addition, oxidative stress within the cells is the direct reason why cells die in various extreme environments. All of the Kocuria strains harbored at least one way to remove the oxidative molecules in time, including organic hydroperoxide resistance gene (ohrB, 151 genes in 148 strains) and its regulator gene (ohrR, 172 genes in 134 strains), katA (187 genes in 164 strains) and katE (160 genes in 159 strains), and sodA (169 genes from 166 strains). Furthermore, the Kocuria strains also harbored other genes for removing oxidative agents. In all these manners, the Kocuria strains could adapt better to various stresses in the environment, undoubtedly increasing the abundance of bacterial strains in extreme habitats and the remediation efficiency of the PA-contaminating environment, and the efficiency of colonization in the rhizosphere of plants. These findings of comparative genomic analysis can provide important guidance for the stimulation of the genus Kocuria in the environment.
The analysis described above naturally leads to a question: how and when did these Kocuria strains acquire their high PA-degrading abilities? Bacteria primarily acquire genetic resources through two pathways. The first pathway is to acquire them from their ancestry cells, which is known as vertical gene transfer (VGT), while another way is to obtain the genes from the other cells or other organisms by transformation or transfection, which is called horizontal gene transfer (HGT) (Coleman et al., 2021). The phylogenomic analysis showed that these strains of Kocuria could be subdivided into five clusters. Cluster I includes Kocuria subflava, Kocuria coralli, Kocuria soli, Kocuria palustris, and some unidentified species of Kocuria. Cluster II is composed of Kocuria flava, Kocuria dechangensis, Kocuria sediminis, Kocuria turfanensis, Kocuria oceani, Kocuria segyptia, and Kocuria salina, the three novel species identified in this study, and the type species K. rosea, as well as a misidentified Kocuria rhizophilia strain. Cluster III only contains the misidentified strain K. rosea subsp. polaris MS50-16. Cluster IV consists of “K. massiliensis” and eight unidentified Kocuria strains, while Cluster V primarily consists of Kocuria atrinae, Kocuria gwangalliensis, Kocuria carniphila, Kocuria marina, Kocuria indica, Kocuria Marina, Kocuria varians, Kocuria salsicia, Kocuria Tytonicola, and some unidentified Kocuria strains (Figure 6). The topology of the phylogenetic tree based on a single amino acid sequence of PcaG, CatE, BphC, PraI, TfdB, CbdA, and MobA was primarily similar to the phylogenomic tree, suggesting that these genes were mostly obtained from VGT. However, those based on the amino acid sequence of Acr and HcaB were quite different from that of the phylogenomic tree, suggesting these genes may have been obtained via HGT. In addition, some genes, such as hgmA and sdgA, were only present in the Cluster II strains (Figure 4), indicating that the Kocuria strains in Cluster II possess additional pathways to degrade aromatic compounds compared to the other four clusters. It is also revealed that the members of Cluster II have a high proportion that harbor genes to degrade aromatic compounds (Figure 6). This may be because the common ancestor of these clusters accidentally obtained the genes from other organisms by HGT.
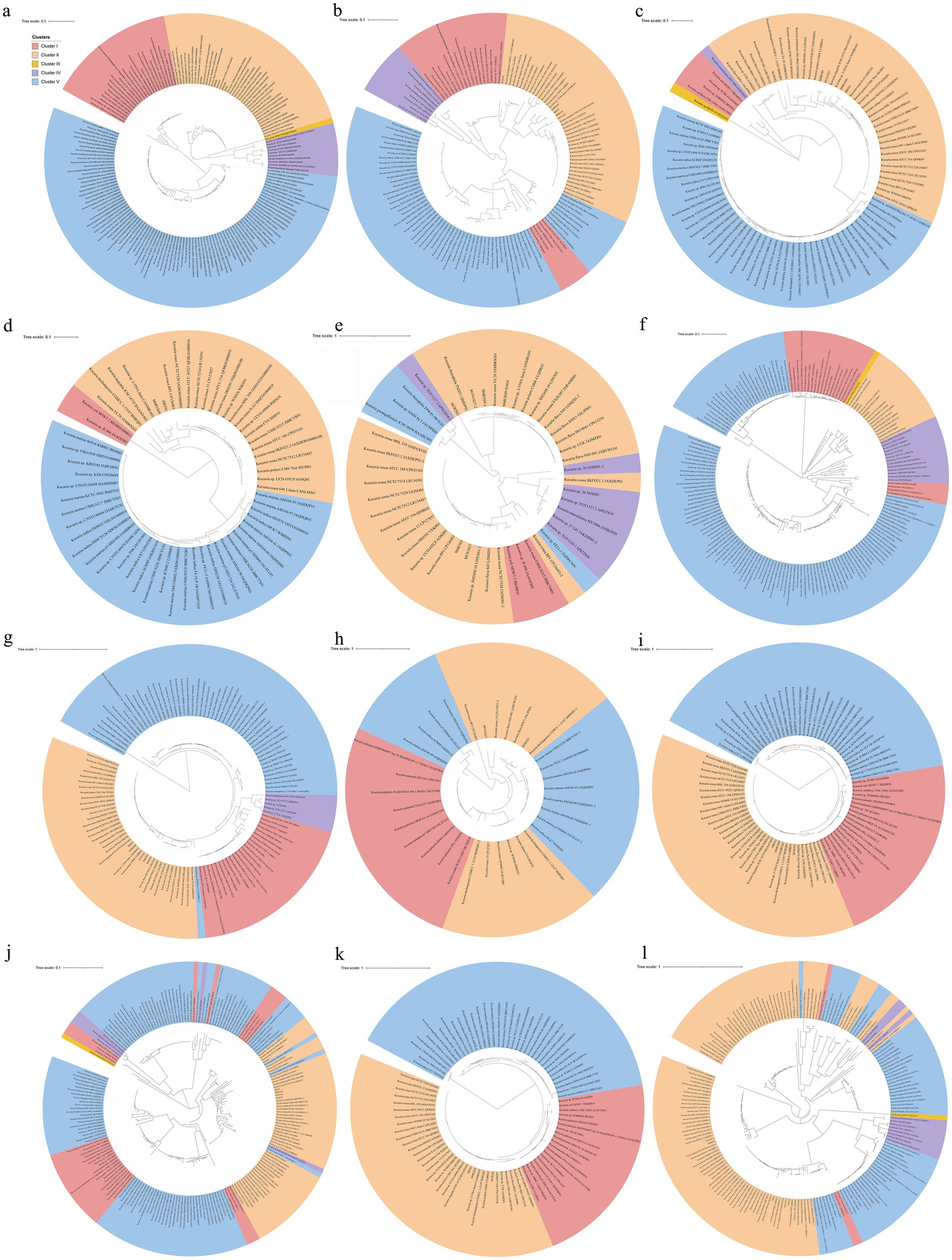
Figure 6. Phylogenomic tree of Kocuria strains (including non-type strains) based on core genome using GTDB (a), and the phylogenetic trees based on the single amino sequence of PcaG (b), CatE (c), HcaE (d), BphC (e), MobA (f), PraI (g), TfdB (h), CbdA (i), Acr (j), CzcD (k), and HcaB (l). The five clusters were subdivided according to the topological relation on the phylogenomic tree.
5 Conclusion
A total of five halotolerant and halophilic Kocuria strains were isolated from the rhizosphere soil and bulk saline soil of the euhalophyte K. cuspidatum. The five strains were identified as three novel species of Kocuria using a polyphasic taxonomy analysis, namely, K. rhizosphaerae sp. nov. (type strain M1R5S2T), K. kalidii sp. nov. (type strain M4R5S9T; non-type strain M1N1S27), and K. rhizosphaericola sp. nov. (type strain M4R2S49T; non-type strain HR5S1). All five strains could degrade FA and CA as their sole carbon and energy source for their growth under saline-alkaline conditions. The genomic and transcriptomic analyses revealed that all five strains degraded FA via the CoA-dependent non-β-oxidation pathway, and protocatechuate 3,4-dioxygenase and the β-ketoadipic acid pathway, while they degraded CA via the CoA-dependent non-β-oxidation pathway, benzoate 1,2-dioxygenase for conversion of benzoate to catechol, catechol 1,2-dioxygenase or catechol 2,3-dioxygenase for ring fission, and the β-ketoadipic acid pathway. A comparative genomic analysis revealed that PAs-degrading genes, including PAs hydroxylase or dioxygenase genes, and catechol and its derivative ring-opening genes are universal in the Kocuria strains. In addition, the Kocuria strains could maintain their osmotic balance by absorbing organic osmoprotectants from the environment, taking up potassium through multiple pathways, and effluxing sodium through Na+/H+ antiporter channel. This study illustrates the mechanisms that are utilized by the Kocuria strains to adapt to extreme environmental condition and rhizosphere of halophyte, and provide important guidance for the stimulation of the genus of Kocuria in the environment, and demonstrates the enormous potential for the application of these strains in the remediation of PA pollution in saline-alkaline environments and their pivotal roles of Kocuria strains on PA degradation in ecosystem.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
LX: Visualization, Writing – original draft, Investigation. R-QS: Investigation, Writing – review & editing. J-HZ: Investigation, Writing – review & editing. H-MW: Investigation, Writing – review & editing. BS: Conceptualization, Writing – review & editing. J-QS: Funding acquisition, Conceptualization, Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported in part by the National Natural Science Foundation of China (grant numbers: 32260022 and 32460025), and the High-Level Talent Start-Up Research Project of Inner Mongolia University (No. 21800–5185133).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1626161/full#supplementary-material
Footnotes
References
Barillot, C. D. C., Sarde, C. -O., Bert, V., Tarnaud, E., and Cochet, N. (2013). A standardized method for the sampling of rhizosphere and rhizoplan soil bacteria associated to a herbaceous root system. Ann. Microbiol. 63, 471–476. doi: 10.1007/s13213-012-0491-y
Borges, A., Saavedra, M. J., and Simoes, M. (2012). The activity of ferulic and gallic acids in biofilm prevention and control of pathogenic bacteria. Biofouling 28, 755–767. doi: 10.1080/08927014.2012.706751
Brynildsrud, O., Bohlin, J., Scheffer, L., and Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with scoary. Genome Biol. 17:238. doi: 10.1186/s13059-016-1108-8
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Coello, N., Vidal, L., and Bretaña, A. (2000). Isolation of a poultry feather-degrading Kocuria rosea strain. Rev. Cient-Fac. Cien. V. 10, 107–113.
Coleman, G. A., Davín, A. A., Mahendrarajah, T. A., Szánthó, L. L., Spang, A., Hugenholtz, P., et al. (2021). A rooted phylogeny resolves early bacterial evolution. Science 372:eabe0511. doi: 10.1126/science.abe0511
Cui, J. Q., He, Z. Q., Chen, K., Ntakirutimana, S., Liu, Z. H., Bai, H., et al. (2024). Lignin-derived compounds assisted with Kocuria marina H-2 and Pseudomonas putida B6-2 co-culture enhanced naphthalene biodegradation. Bioresour. Technol. 413:131512. doi: 10.1016/j.biortech.2024.131512
Díaz, E., Ferrández, A., and García, J. L. (1998). Characterization of the hca cluster encoding the dioxygenolytic pathway for initial catabolism of 3-phenylpropionic acid in Escherichia coli K-12. J. Bacteriol. 180, 2915–2923. doi: 10.1128/JB.180.11.2915-2923.1998
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Ezima, E. N., Awotula, A. O., Adegbesan, B. O., Osonuga, I. O., Agu, G. C., Adesanya, E. O., et al. (2024). Biodegradation of naphthalene using Kocuria rosea isolated from a sawmill in Ikenne, Southwestern Nigeria. Pure Appl. Chem. 96, 1375–1381. doi: 10.1515/pac-2023-1136
Genethliou, C., Kornaros, M., and Dailianis, S. (2020). Biodegradation of olive mill wastewater phenolic compounds in a thermophilic anaerobic upflow packed bed reactor and assessment of their toxicity in digester effluents. J. Environ. Manag. 255:109882. doi: 10.1016/j.jenvman.2019.109882
González, M. D., Moreno, E., Quevedo-Sarmiento, J., and Ramos-Cormenzana, A. (1990). Studies on antibacterial activity of waste waters from olive oil mills (alpechin): inhibitory activity of phenolic and fatty acids. Chemosphere 20, 423–432. doi: 10.1016/0045-6535(90)90073-3
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harwood, C. S., and Parales, R. E. (1996). The beta-ketoadipate pathway and the biology of self-identity. Ann. Rev. Microbiol. 50, 553–590. doi: 10.1146/annurev.micro.50.1.553
Hernández-Salmerón, J. E., and Moreno-Hagelsieb, G. (2022). FastANI, mash and dashing equally differentiate between Klebsiella species. PeerJ 10:e13784. doi: 10.7717/peerj.13784
Huang, T. P., Chen, J., Yang, Q. Y., Zhang, Y. K., and Ding, R. F. (2012). Biodegradation of heavy oil with high asphaltene content by Kocuria sp. Asian J. Chem. 24, 2717–2720.
Kang, J. M., Liu, L., Liu, Y. F., and Wang, X. Y. (2020). Ferulic acid inactivates Shigella flexneri through cell membrane destruction, biofilm retardation, and altered gene expression. J. Agric. Food Chem. 68, 7121–7131. doi: 10.1021/acs.jafc.0c01901
Karn, S. K., Chakrabarti, S. K., and Reddy, M. S. (2011). Degradation of pentachlorophenol by Kocuria sp. CL2 isolated from secondary sludge of pulp and paper mill. Biodegradation 22, 63–69. doi: 10.1007/s10532-010-9376-6
Khandelwal, A., Nain, L., Singh, S. B., Varghese, E., Sharma, A., Gupta, S., et al. (2024). Bacteria and fungi mediated degradation of poly aromatic hydrocarbons and effect of surfactant Tween-80. Int. J. Environ. Anal. Chem. 104, 27–42. doi: 10.1080/03067319.2021.2015584
Kim, D., Park, S., and Chun, J. (2021). Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 59, 476–480. doi: 10.1007/s12275-021-1154-0
Komagata, K., and Suzuki, K. (1987). Lipid and cell wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Li, H. Q., Zhang, L. L., Jiang, X. W., and Liu, Q. Z. (2015). Allelopathic effects of phenolic acids on the growth and physiological characteristics of strawberry plants. Allelopath. J. 35, 61–75.
Liu, Y., Zhao, Z. H., Yang, H. W., Fu, L., and Zhou, D. D. (2022). Trace phenolic acids simultaneously enhance degradation of chlorophenol and biofuel production by Chlorella regularis. Water Res. 218:118524. doi: 10.1016/j.watres.2022.118524
Lubbers, R. J. M., Dilokpimol, A., Visser, J., Mäkelä, M. R., Hildén, K. S., and de Vries, R. P. (2019). A comparison between the homocyclic aromatic metabolic pathways from plant-derived compounds by bacteria and fungi. Biotechnol. Adv. 37:107396. doi: 10.1016/j.biotechadv.2019.05.002
Ma, H. Y., Li, J. J., Luo, A. H., Lv, H. P., Ren, Z. T., Yang, H. K., et al. (2023). Vanillin, a newly discovered autotoxic substance in long-term potato continuous cropping soil, inhibits plant growth by decreasing the root auxin content and reducing adventitious root numbers. J. Agric. Food Chem. 71, 16993–17004. doi: 10.1021/acs.jafc.3c05027
Marchiosi, R., dos Santos, W. D., Constantin, R. P., de Lima, R. B., Soares, A. R., Finger-Teixeira, A., et al. (2020). Biosynthesis and metabolic actions of simple phenolic acids in plants. Phytochem. Rev. 19, 865–906. doi: 10.1007/s11101-020-09689-2
Margesin, R., Ludwikowski, T. M., Kutzner, A., and Wagner, A. O. (2022). Low-temperature biodegradation of lignin-derived aromatic model monomers by the cold-adapted yeast Rhodosporidiobolus colostri isolated from alpine forest soil. Microorganisms 10:515. doi: 10.3390/microorganisms10030515
Margesin, R., Volgger, G., Wagner, A. O., Zhang, D., and Poyntner, C. (2021). Biodegradation of lignin monomers and bioconversion of ferulic acid to vanillic acid by Paraburkholderia aromaticivorans AR20-38 isolated from alpine forest soil. Appl. Micrbiol. Biotechnol. 105, 2967–2977. doi: 10.1007/s00253-021-11215-z
Matejczyk, M., Ofman, P., Juszczuk-Kubiak, E., Swislocka, R., Shing, W. L., Kesari, K. K., et al. (2024). Biological effects of vanillic acid, iso-vanillic acid, and orto-vanillic acid as environmental pollutants. Ecotoxicol. Environ. Saf. 277:116383. doi: 10.1016/j.ecoenv.2024.116383
Oshlag, J. Z., Ma, Y. J., Morse, K., Burger, B. T., Lemke, R. A., Karlen, S. D., et al. (2020). Anaerobic degradation of syringic acid by an adapted strain of Rhodopseudomonas palustris. Appl. Environ. Microbiol. 86:e01888-19. doi: 10.1128/AEM.01888-19
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peracchi, L. M., Panahabadi, R., Barros-Rios, J., Bartley, L. E., and Sanguinet, K. A. (2024). Grass lignin: biosynthesis, biological roles, and industrial applications. Front. Plant Sci. 15:26. doi: 10.3389/fpls.2024.1343097
Promsing, S., Pokethitiyook, P., Kruatrachue, M., Ounjai, P., Meetam, M., Onparn, N., et al. (2021). Rhizoremediation of fuel oil by Vetiveria zizanioides in association with Kocuria sp. no. MU1 and Micrococcus luteus WN01. ScienceAsia 47, 96–105. doi: 10.2306/scienceasia1513-1874.2021.015
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rao, L. Z., Gu, D. C., Xiang, X. J., Zeng, J., Wu, Y. C., Lin, X. G., et al. (2023). Impact of lignin constituents on the bacterial community and polycyclic aromatic hydrocarbon co-metabolism in an agricultural soil. Environ. Pollut. 333:122105. doi: 10.1016/j.envpol.2023.122105
Stackebrandt, E., Koch, C., Gvozdiak, O., and Schumann, P. (1995). Taxonomic dissection of the genus Micrococcus: Kocuria gen. nov., Nesterenkonia gen. nov., Kytococcus gen. nov., Dermacoccus gen. nov., and Micrococcus Cohn 1872 gen. emend. Int. J. Syst. Bacteriol. 45, 682–692. doi: 10.1099/00207713-45-4-682
Sun, J. -Q., Xu, L., Guo, Y., Li, W. L., Shao, Z. -Q., Yang, Y. -L., et al. (2017). Kribbella deserti sp nov., isolated from rhizosphere soil of Ammopiptanthus mongolicus. Int. J. Syst. Evol. Microbiol. 67, 692–696. doi: 10.1099/ijsem.0.001697
Sun, J. Q., Xu, L., Tang, Y. Q., Chen, F. M., and Wu, X. L. (2012). Simultaneous degradation of phenol and n-hexadecane by Acinetobacter strains. Bioresour. Technol. 123, 664–668. doi: 10.1016/j.biortech.2012.06.072
Sunita, K., Mishra, I., Mishra, J., Prakash, J., and Arora, N. K. (2020). Secondary metabolites from halotolerant plant growth promoting rhizobacteria for ameliorating salinity stress in plants. Front. Microbiol. 11:567768. doi: 10.3389/fmicb.2020.567768
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tian, J., Xu, L., and Sun, J. Q. (2024). Taxonomic characterization and comparative genomic analysis of a novel Devosia species revealed that phenolic acid-degrading traits are ubiquitous in the Devosia genus. Environ. Res. 261:119724. doi: 10.1016/j.envres.2024.119724
Tsujii, M., Tanudjaja, E., and Uozumi, N. (2020). Diverse physiological functions of cation proton antiporters across bacteria and plant cells. Int. J. Mol. Sci. 21:4566. doi: 10.3390/ijms21124566
Wang, J. Y., and Nan, J. (2025). Comparative genomic and transcriptomic analysis provides new insights into the aflatoxin B1 biodegradability by Kocuria rosea from deep sea. Microorganisms 13:875. doi: 10.3390/microorganisms13040875
Wang, D. D., Sun, L., Yin, Z. Q., Cui, S. Y., Huang, W. W., and Xie, Z. H. (2022). Insights into genomic evolution and the potential genetic basis of Klebsiella variicola subsp. variicola ZH07 reveal its potential for plant growth promotion and autotoxin degradation. Microbiol. Spectr. 10:e00846-22. doi: 10.1128/spectrum.00846-22
Wu, L. Y., Ali, D. C., Liu, P., Peng, C., Zhai, J. X., Wang, Y., et al. (2018). Degradation of phenol via ortho-pathway by Kocuria sp. strain TIBETAN4 isolated from the soils around Qinghai Lake in China. PLoS One 13:e0199572. doi: 10.1371/journal.pone.0199572
Xie, Z., Zhao, S., Li, Y., Deng, Y., Shi, Y., Chen, X., et al. (2023). Phenolic acid-induced phase separation and translation inhibition mediate plant interspecific competition. Nat. Plants 9, 1481–1499. doi: 10.1038/s41477-023-01499-6
Xu, L., Huang, X.-X., Wang, H.-T., Tang, S.-K., Shen, B., and Sun, J.-Q. (2022). Description and characterization of three endophytic Bacillaceae from the halophyte Suaeda salsa: Paenalkalicoccus suaedae gen. nov., sp. nov., Cytobacillus suaedae sp. nov., and Bacillus suaedae sp. nov. Int. J. Syst. Evol. Microbiol. 72:005337. doi: 10.1099/ijsem.0.005337
Xu, L., Wei, H. M., Sun, Y. N., Wu, Q., Gao, X. Y., Shen, B., et al. (2024a). Halomonas rhizosphaerae sp. nov. and Halomonas kalidii sp. nov., two novel moderate halophilic phenolic acid-degrading species isolated from saline soil. Syst. Appl. Microbiol. 47:126488. doi: 10.1016/j.syapm.2024.126488
Keywords: phenolic acids, biodegradation, Kocuria , comparative genomic analysis, osmotic stress, polyphasic taxonomy
Citation: Xu L, Sun R-Q, Zeng J-H, Wei H-M, Shen B and Sun J-Q (2025) Multiple-omics analysis of three novel haloalkaliphilic species of Kocuria revealed that the phenolic acid-degrading abilities are ubiquitous in the genus. Front. Microbiol. 16:1626161. doi: 10.3389/fmicb.2025.1626161
Edited by:
Muhammad Zahid Mumtaz, Gansu Agricultural University, ChinaReviewed by:
Kusum Dhakar, Academy of Sciences of the Czech Republic (ASCR), CzechiaNavneet Joshi, Mody University of Science and Technology, India
Copyright © 2025 Xu, Sun, Zeng, Wei, Shen and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Quan Sun, c3VuanE4MUAxNjMuY29t