 Chunguang Ren
Chunguang Ren Yu Liu1
Yu Liu1- 1Guizhou Institute of Mountain Resources, Guiyang, China
- 2Guizhou Botanical Garden, Guiyang, China
Botryosphaeria dothidea is the main cause of soft rot in kiwifruit, significantly reducing both yield and quality. While chemical treatments are commonly used, their effectiveness is limited and they may pose environmental risks. As a result, biological control using Bacillus species has emerged as a promising alternative. In this study, we explored the antifungal mechanism of the biocontrol strain Bacillus velezensis LMY3-5 against kiwifruit soft rot through integrated genomic and transcriptomic analyses. In terms of antagonistic activity: B. velezensis LMY3-5 exhibits strong antagonism against B. dothidea, the causal agent of kiwifruit soft rot, showing a 70.44% biocontrol efficacy in inoculation tests. In terms of genomic features: genome 4.03 Mb circular chromosome with 46.5% GC content. Eight antimicrobial BGCs were identified, including those for surfactin, fengycin, bacillaene, bacillibactin, and others, explaining its biocontrol potential. In terms of transcriptomic insights: during coculture with B. dothidea, 114 DEGs (31 upregulated, 93 downregulated) were detected. Downregulated: flagellar assembly and chemotaxis genes (suggesting reduced motility during antagonism). Upregulated: genes linked to fengycin, siderophores, and lysozyme production, critical for antifungal activity. In terms of mechanism and implications lipopeptides (e.g., fengycin) and siderophores are key in inhibiting fungal growth. Findings support LMY3-5’s potential as a biocontrol agent for plant protection against B. dothidea. The main conclusion of this study is LMY3-5 combats B. dothidea via antimicrobial metabolites, with genomics and transcriptomics revealing its molecular basis for biocontrol. This strain holds promise for sustainable plant disease management. This may provide a theoretical basis for the potential application of B. velezensis LMY3-5 in the field of plant protection in the future.
1 Introduction
The kiwifruit (Actinidia spp.), also known as the Chinese gooseberry, is native to China. Currently, more than 30 countries cultivate kiwifruit globally, with China being the largest producer in terms of both cultivation area and yield. As the world’s leading kiwifruit-producing region, China’s kiwifruit industry is widely distributed across Shaanxi, Sichuan, Anhui, Hunan, Zhejiang, and Guizhou provinces. According to 2021 statistics, Guizhou Province has a kiwifruit cultivation area of 673,000 mu (approximately 44,867 hectares) with an annual output of 220,000 metric tons, ranking third and fourth in the nation for cultivation area and yield, respectively. The kiwifruit industry has become a pillar of rural revitalization in Guizhou and plays an indispensable role in the province’s national economy (Source: Guizhou Provincial Department of Agriculture and Rural Affairs, https://nynct.guizhou.gov.cn/).
However, with the continuous expansion of kiwifruit cultivation, disease incidence has also increased significantly. In particular, postharvest soft rot disease leads to substantial fruit decay during storage and marketing, with an average incidence rate of 40–50% (Li et al., 2016; Li et al., 2017; Su et al., 2021). This disease severely compromises fruit quality and has caused substantial economic losses to both Guizhou’s and China’s kiwifruit industries.
The disease can be caused by individual or combined infections with fungi such as Botryosphaeria dothidea (Li et al., 2017), Diaporthe eres (Liu et al., 2021), Alternaria alternata (Li L. et al., 2023; Li W. et al., 2023), and Phomopsis phragmitis (Wang X. J. et al., 2021). These fungi infect fruits during the young fruit stage, with symptoms manifesting during storage. Among such infectious fungi, B. dothidea, which is a primary pathogen, can also cause fruit decay, stem or branch withering, and necrosis in various crops, such as cherry, blueberry, apple, yam, and peach (Hilario and Artur, 2020; Li et al., 2022; Tang et al., 2012; Wang F. et al., 2021; Yanli et al., 2018; Zhang et al., 2019). Presently, the prevention and control of kiwifruit soft rot mainly involves spraying chemical fungicides such as thiophanate-methyl and tebuconazole after fruit setting (Kim et al., 2013; Shin et al., 2021). However, long-term and large-scale spraying of chemical fungicides can lead to secondary environmental pollution and food safety issues, further increasing the drug resistance of pathogens (Bakirci et al., 2014; Yin et al., 2023). Therefore, a green, safe, and stable biological control method for controlling kiwifruit soft rot disease is required.
Bacillus species are gram-positive, endospore-forming bacteria widely distributed in nature. They are of high commercial value in agriculture due to their biocontrol potential, producing lipopeptides and polyketides that effectively suppress plant pathogens. Their spores are acid-, alkali-, heat-, and radiation-resistant, while their microbial agents are convenient for storage and transport and harmless to humans (Rabbee et al., 2019; Saxena et al., 2020). Bacillus spp. can be employed for the storage and preservation of fruits after picking. For example, Bacillus spp. have been used to control postharvest diseases of various fruits, such as blueberry gray mold (Lu et al., 2021), litchi downy mildew (Zheng et al., 2021), and strawberry gray mold (Wang F. et al., 2021). However, studies on the biological strains used for managing kiwifruit soft rot are limited. These biocontrol strains mainly include Fusicolla violacea (Li et al., 2021) and Bacillus subtilis (Fan et al., 2023). Therefore, other potential biocontrol strains need to be identified. In the early stages of this study, a Bacillus strain was isolated from kiwifruit that exerted an inhibitory effect on the growth of B. dothidea. Determining its biocontrol mechanism is necessary for addressing the current issue of soft rot disease in the kiwifruit industry. Developing efficient, safe, and effective microbial fungicides to address the excessive reliance on chemical pesticides for controlling kiwifruit soft rot is crucial.
In recent years, the use of whole-genome and transcriptome sequencing to investigate the types, structures, and synthesis pathways of microbial metabolites at the molecular level has emerged as a pioneering approach to mine antifungal functional genes, identify antifungal active substances, and study antifungal mechanisms and important gene metabolic regulatory networks (Lu et al., 2023; Mu et al., 2023; Zhi et al., 2017; Zhou et al., 2019a; Zhou et al., 2019b). Since 2012, the complete genome sequences of 856 strains of B. velezensis have been published, enabling the identification of various antimicrobial substances such as lipopeptides, surfactin, fengyuansu, and plantazolicin (data source: https://www.ncbi.nlm.nih.gov/). With sequencing technology developments and sequencing cost reduction, comparative genomics, transcriptomics, and other omics methods are increasingly used for analyzing the differences in the composition of interspecies gene clusters and types of metabolites, facilitating the discovery of new antimicrobial peptides, antagonistic genes (clusters), and synthesis pathways of antagonistic substances. Approaches based on these techniques have important reference values for studying the antifungal mechanisms of biocontrol bacteria.
In this study, we isolated a Bacillus strain (LMY3-5) from kiwifruits in Xiuwen County, Guizhou Province, China. This strain exerted an inhibitory effect on the growth of the kiwifruit soft rot pathogen, B. dothidea. This study aimed to comprehensively characterize the biocontrol potential of Bacillus strain LMY3-5 through multiple approaches: evaluating its efficacy in suppressing kiwifruit soft rot pathogenesis, performing whole-genome sequencing and annotation to identify potential biocontrol-related genes, elucidating the metabolic pathways and signal transduction mechanisms underlying its antifungal activity through transcriptomic analysis of differentially expressed genes (DEGs), and functionally screening for specific genes contributing to antifungal activity.
2 Materials and methods
2.1 Test strains and culture conditions
Bacillus velezensis LMY3-5 (CGMCC No: 29700) and B. dothidea were provided by the Molecular Laboratory of Guizhou Provincial Institute of Mountain Resources (city, country), and stored at −80°C. B. dothidea was isolated from diseased Xiuwen kiwifruits in Guizhou, whereas the biocontrol B. velezensis LMY3-5 strain was isolated from healthy Xiuwen kiwifruits in Guizhou. B. dothidea was cultured in potato dextrose agar (PDA) medium at 27°C for 3 d, whereas strain LMY3-5 was grown in 100 mL Luria-Bertani (LB) medium at 30°C and 200 rpm for 12 h to prepare the inoculum.
2.2 Antagonism assays of pathogenic and biocontrol bacteria
A mycelial block of B. dothidea (0.5 cm in diameter) was inoculated at the center of the PDA dish. For the experimental group, antagonistic LMY3-5 bacteria were inoculated equidistantly in the four directions (2.5 cm) from the center of the plate. For the control group, no bacteria were inoculated, and only the B. dothidea amycelial block was present. The experimental and control groups were cultured at 28°C for 5 d. Three biological replicates were used for all experiments.
2.3 Mycelial morphology and hyphal ultrastructure
Hyphae at the edge of the antifungal zone were selected for evaluation of the antagonistic effects of the LMY3-5 strain. Morphological changes were observed using a DM500 binocular microscope (Leica, Wetzlar, Germany) at a magnification of 400 × and an SU8010 SEM (Hitachi, Tokyo, Japan) (Mo et al., 2021).
2.4 In vivo inhibition of B. dothidea infection of kiwifruit
A cell-free supernatant (CFS) was prepared using the protocols described in a previous study (Liu et al., 2023). Strain LMY3-5 was cultured in LB medium at 30°C and 200 rpm for 1 d to obtain a seed culture. Next, 5 mL of the seed culture was further cultured in LB medium (100 mL) at 30°C and 200 rpm for 3 d. The supernatant was centrifuged (12,000 rpm for 15 min) and filtrated (0.22 μm sterile filter) to obtain the CFS.
Fresh and healthy kiwifruits of the same size were selected. The surface was disinfected with 75% alcohol for 30 s and washed with sterile water thrice. Three wounds were created on the surface of each kiwifruit using sterile toothpicks. Each wound was inoculated with a 5-mm mycelial plug of actively growing B. dothidea (cultured on PDA at 28°C for 5 d). Experimental groups: Immersed in 1, 2, 4, 8%, or 16% LMY3-5 CFS for 60 min. Negative control: Immersed in sterile water. After treatment, all fruits were placed in an incubator at 28°C and 90% RH. Disease progression was assessed after 7 d (photographed), with 30 fruits per group.
2.5 Whole genome sequencing and functional annotation of biocontrol strain LMY3-5
The genome of B. velezensis LMY3-5 was sequenced by Shanghai Meiji Biotechnology Co., Ltd. (city, country) using a combination of third-generation PacBio RS and second-generation Illumina sequencing platforms. The raw sequencing data were subjected to quality control, and low-quality reads and adaptors were removed. The reads were assembled using the SMRT Link v5.1.0 software (Pacific Biosciences, Menlo Park, CA, United States), and the sequencing data were submitted to the NCBI database. The coding sequence (CDS) regions in the genome were predicted using the Glimmer1, GeneMarkS, and Prodigal software, and the obtained genome sequences were submitted to six major databases (NR, Swiss-Prot, Pfam, EggNOG, GO, and KEGG) for functional annotation.
2.6 Comparative genomic analysis of LMY3-5
The gyrA gene sequence was extracted from the whole genome, and BLAST sequence similarity analysis was performed using GenBank. Sequences with high homology were downloaded, and a phylogenetic tree was constructed using the ML method with the MEGA 7.0 software. For accurate identification of the strain, the whole genome sequence of a closely related Bacillus strain was downloaded from the NCBI and EzBioCloud2 servers, and the genome-to-genome distance calculator (GGDC)3 was used to calculate the average nucleotide identity (ANI) and intergenome DNA–DNA molecular hybridization (DDH) genetic distance. ANI ≥ 95% and GGDC ≥ 70% were used as criteria for determining species status (Meier-Kolthoff et al., 2013; Yoon et al., 2017). The genome structure and collinearity of the CCBC3-3-1 and reference strains were analyzed using the Mauve v2.3.1 software (Darling et al., 2004). The antiSMASH 4.0 database (Medema et al., 2011) was used to identify gene clusters involved in secondary metabolite synthesis in strain LMY3-5.
2.7 Transcriptome extraction and sequencing analysis of LMY3-5
Transcriptome samples were prepared using the plate confrontation method, as described in Section 2.2. A PDA plate inoculated with LMY3-5 was used as control. LMY3-5 cells grown at 28°C for 3 d were collected, frozen in liquid nitrogen, and stored at −80°C. Each biological replicate comprised cells pooled from 10 plates, with three independent replicates per group.
Library construction was performed using the NEBNext® Ultra™ mRNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, United States). Sequencing was conducted by Shanghai Meiji Biomedical Technology Co., Ltd. (Shanghai, China) on an Illumina platform. Raw reads were quality-assessed with FastQC and filtered using Trimmomatic (Bolger et al., 2014) to remove low-quality sequences.
2.8 Differential gene identification and functional annotation
The reads were counted using the feature Counts (v2.0.3; from the Subread package). The DESeq2 package (v1.38.3; Bioconductor) was used to analyze the transcript levels of DEGs in treated samples, using FPKM ≥ 1 as the threshold of expression (Yu et al., 2012). Genes with values of p < 0.05 and | log2 (fold change) | ≥ 1 were categorized as DEGs. The clusterProfiler package (v4.6.2; Bioconductor) was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis on DEGs. Pathways with a p-value < 0.05 were considered significantly enriched pathways (Aramaki et al., 2020; Kanehisa, 2019; Kanehisa et al., 2023; Kanehisa and Goto, 2000).
2.9 Real-time PCR (qRT-PCR) validation
qRT-PCR validation maintained identical transcriptome conditions: cDNA was synthesized from the same LMY3-5 RNA sources described in Section 2.7—control (monoculture on PDA, 28°C, 3 d) and treatment (co-culture with B. dothidea)—using the PrimeScript cDNA Synthesis Kit (TransScript). Reactions with TB Green Premix Ex Taq (TaKaRa) employed primers designed via Primer 5.0 under thermal cycling: 95°C 30 s; 40 cycles of 95°C 10 s/60°C 30 s. The 16S rDNA gene served as the sole reference since transcriptome sequencing exclusively targeted LMY3-5 in co-culture systems. Gene expression was calculated by 2−ΔΔCQ (Adnan et al., 2011) with three technical replicates.
2.10 Statistical analyses
Validation of transcriptome data maintained identical co-culture conditions: RNA was extracted from B. velezensis LMY3-5 monoculture (PDA, 28°C, 48 h) as the control and LMY3-5/B. dothidea co-culture as the treatment. qRT-PCR reactions used SYBR Green Master Mix (Vazyme Q711-02) with gene-specific primers, amplifying targets over 40 cycles (95°C 30 s; 95°C 10 s/60°C 30 s per cycle) on a QuantStudio 6 Pro system. The rpoB (LMY3-5) and *EF-1α* (B. dothidea) genes served as references for 2 < sup > -ΔΔCt</sup > analysis.
All experimental data were analyzed using Microsoft Excel (v2010) and SPSS Statistics 25 (IBM Corp., Armonk, NY, USA). Significant differences between treatment groups were assessed by one-way analysis of variance (ANOVA), followed by Duncan’s multiple range test as a post hoc comparison. Statistical significance was defined at p < 0.05. Data visualization was performed using OriginPro 2021 (OriginLab Corp., Northampton, MA, United States).
3 Results
3.1 The inhibitory activity of LMY3-5 against B. dothidea
Strain LMY3-5 significantly inhibited B. dothidea-mediated kiwifruit soft rot, reducing lesion diameter by70.44% compared to untreated controls (p < 0.01). Morphological assessments revealed progressive mycelial degradation: colony assays demonstrated complete growth suppression with distinct inhibition halos (Figure 1A); light microscopy (400×) exhibited hyphal collapse and cytoplasmic shrinkage (Figure 1B); and scanning electron microscopy confirmed severe surface invaginations and structural rupture in pathogen hyphae (Figure 1C).
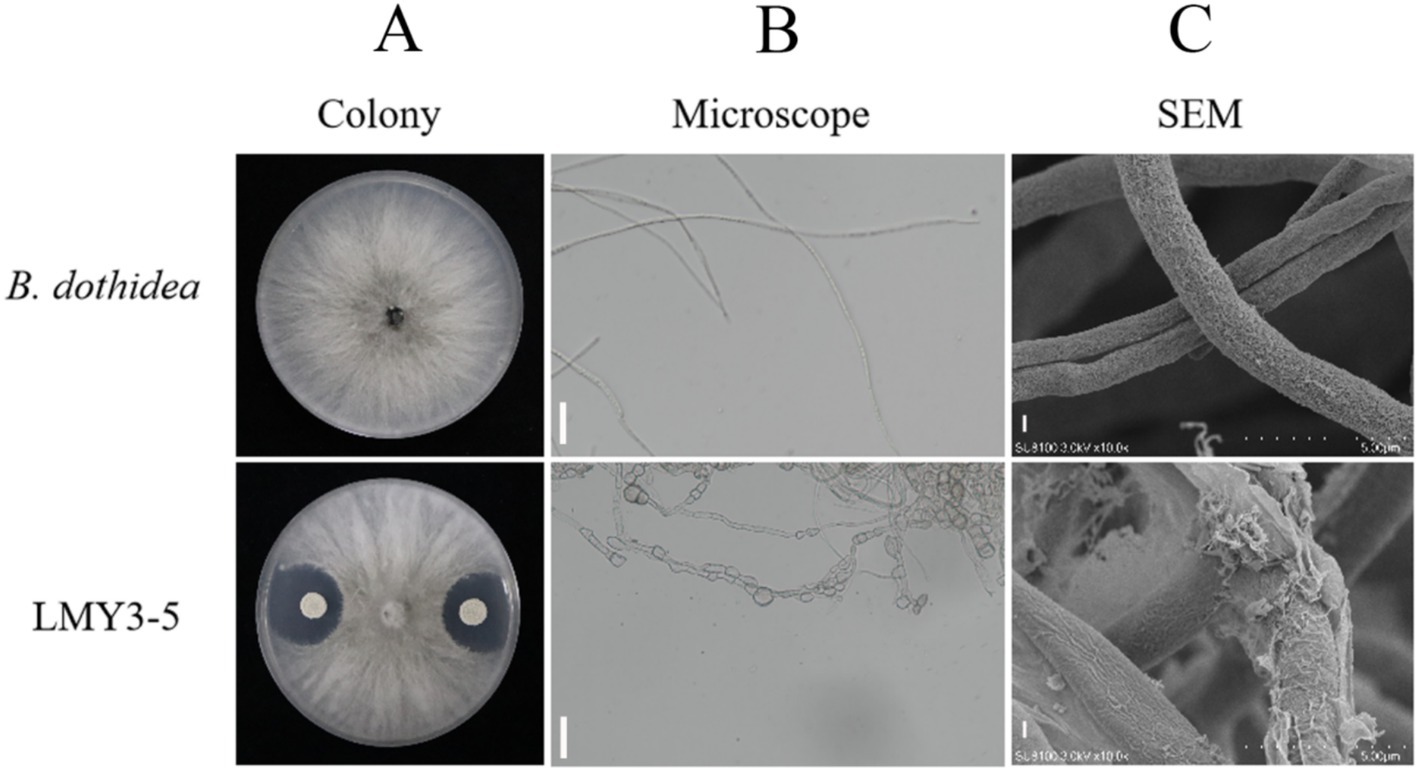
Figure 1. Antifungal activity of B. velezensis LMY3-5 against B. dothidea. (A) Dual-culture assay: top panel (B. dothidea only); bottom panel (co-culture with LMY3-5). (B) Light microscopy (400×) of hyphae (scale bar: 20 μm). (C) Scanning electron micrograph (scale bar: 5 μm).
3.2 LMY3-5-induced inhibition of soft rot development in kiwifruit
Kiwifruit treated with sterile fermentation filtrate began to soften around the inoculation site on the third day after inoculation, followed by the appearance of a large surface rot on the seventh day, at which point, the kiwifruit skin was torn open (Figure 2). The flesh became brown, and the fruit smelled sour. However, the fruits treated with LMY3-5 CFS exhibited only a small area of slight mechanical contusion around the inoculation site on the seventh day, which did not spread further, while the color and texture were consistent with those before inoculation. The inhibition rates of soft rot development in kiwifruit inoculated with B. dothidea and treated with 4, 8, and 16% LMY3-5 CFS were 43.2, 59.58, and 70.44%, respectively.
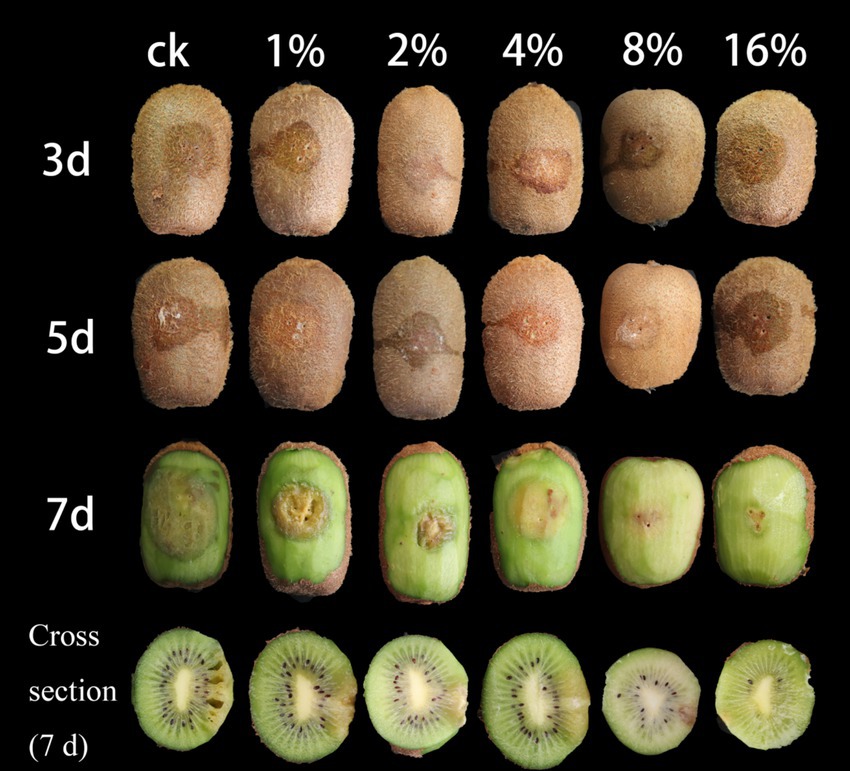
Figure 2. Suppression of B. dothidea-induced kiwifruit soft rot by LMY3-5 CFS treatment. Control (ck) denotes fruits treated with sterile water; numerical labels (1, 2, 4, 8, and 16%) represent concentration gradients of LMY3-5 cell-free supernatant. Images show disease progression at 3, 5, and 7 days post-inoculation.
3.3 lMY3-5 genome analysis
Whole genome sequencing revealed that the LMY3-5 genome was composed of a single circular chromosome with a length of 4,032,433 bp and GC content of 46.5%. No plasmid was detected. The chromosome contained 3,914 CDSs, with an average length of 910.21 bp, which accounted for 88.35% of the whole genome sequence. Notably, the LMY3-5 genome contained 27 rRNAs, 87 tRNAs, 82 sRNAs, and 87 tandem repeats but zero insertion sequences, comprising five gene islands (Figure 3).
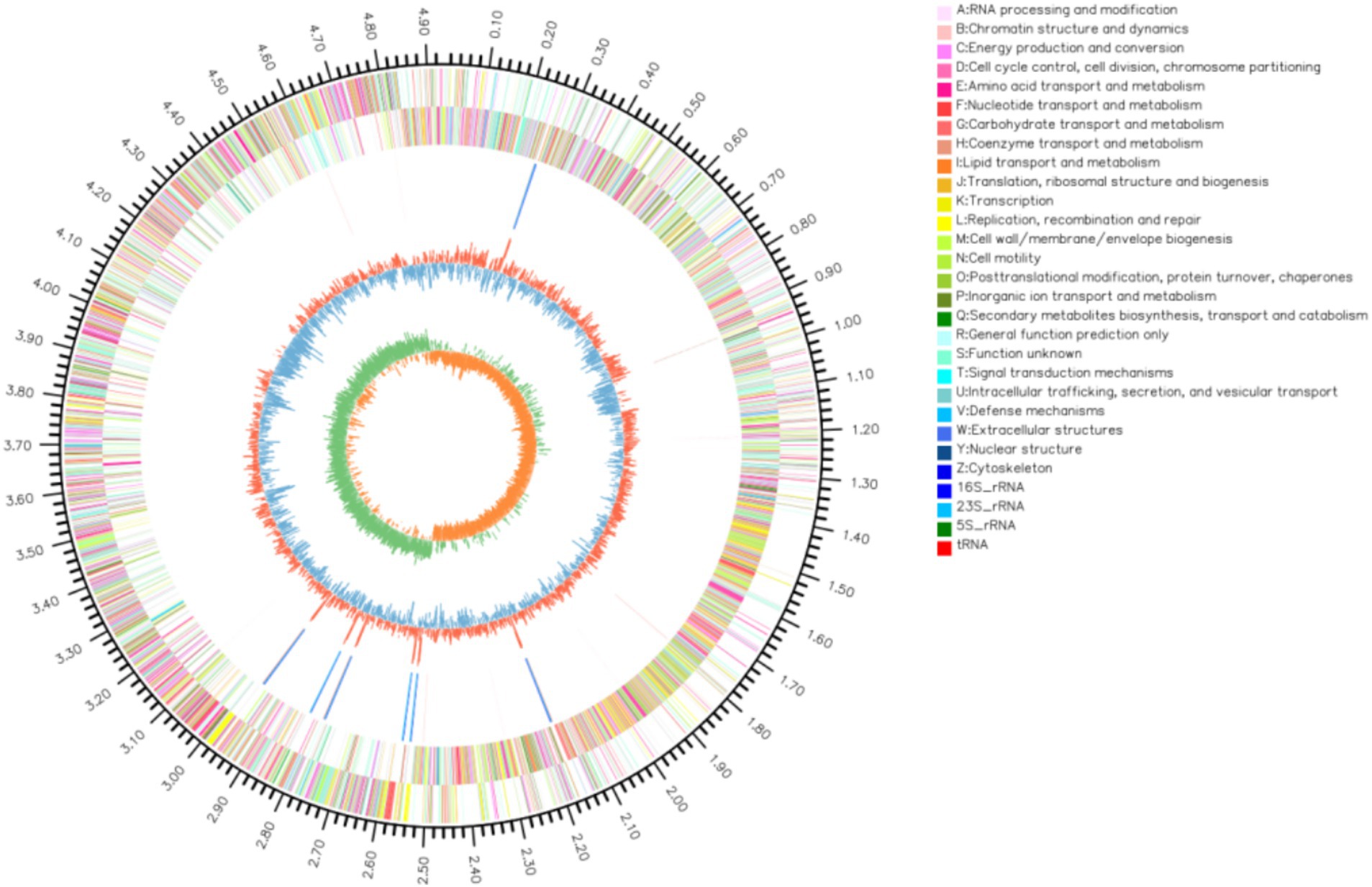
Figure 3. Genome map of B. velezensis LMY3-5. The circle diagram from outside to inside is: (1) the identification of genome size; (2) CDS on the positive chain; (3) CDS on the negative chain; (4) rRNA and tRNA; (5) GC content (6) GC-Skew.
3.4 Functional annotation of the LMY3-5 genome
We performed functional annotation of the B. velezensis LMY3-5 genome using six gene annotation databases. We found that 3,909 genes were annotated in the NR database, accounting for 99.87% of all genes. In addition, we found that 3,563 (99.87%), 3,412 (87.17%), 3,109 (79.43%), 1801 (46.01%), and 2,847 (72.73%) genes were annotated in the Swiss-Prot, Pfam, COG, GO, and KEGG databases, respectively (Figure 4A).
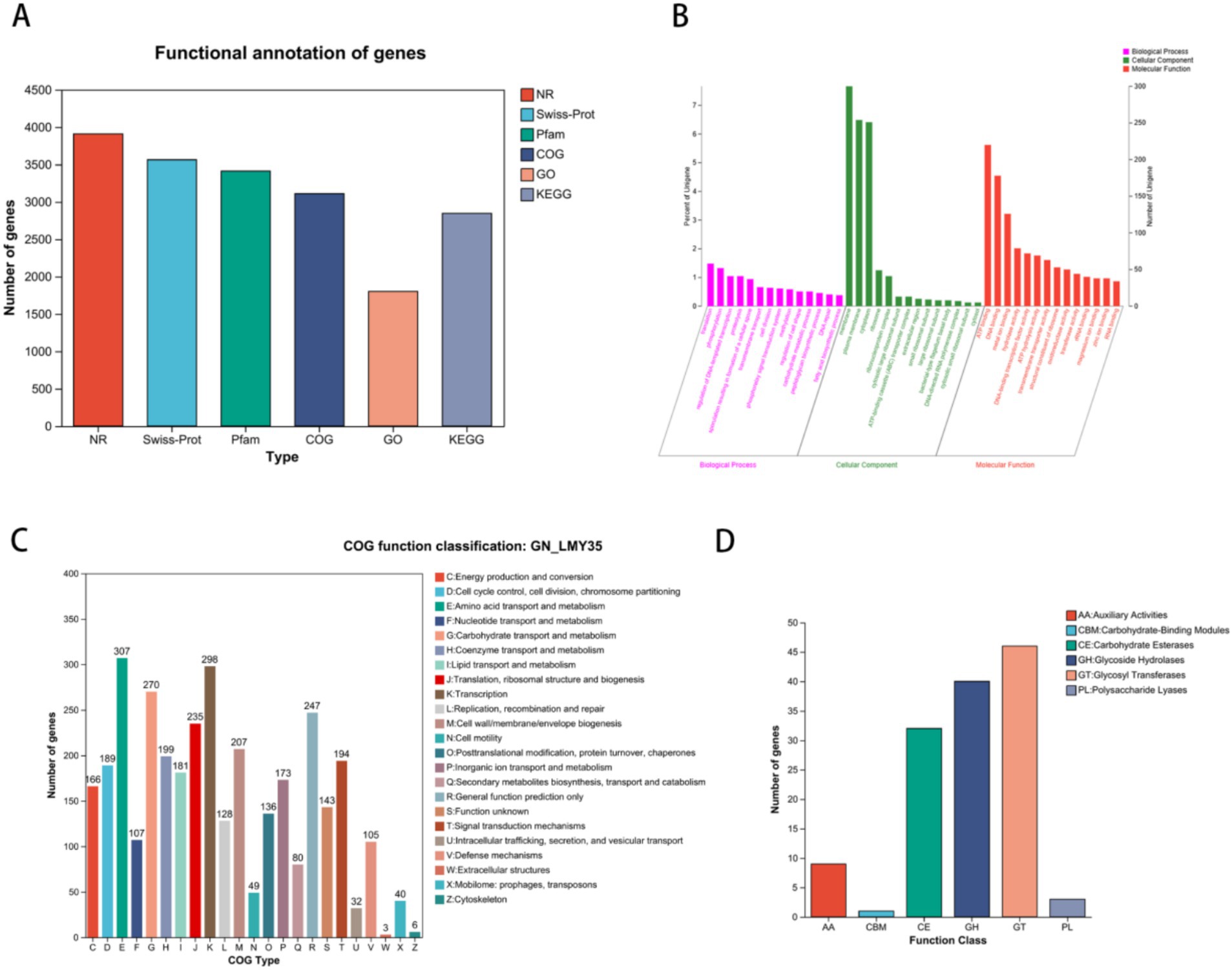
Figure 4. Functional analysis of gene and protein sequence annotations from B. velezensis LMY3-5. (A) Annotation summary. (B) B. velezensis LMY3-5 genome GO annotation functional classification. (C) B. velezensis LMY3-5 genome (COG) analysis. (D) Comparison of carbohydrate active enzyme.
The GO database annotation results for protein-coding genes in the LMY3-5 genome are shown in Figure 4B. In the Biological Process category, the largest number of genes was annotated in phosphorylation (58) and transmembrane transport (52). In the Cellular Function category, the largest number of genes were annotated for ATP binding (220) and DNA binding (178). COG annotation results showed that 3,109 genes were annotated (Figure 4C), accounting for 79.43% of all genes, including genes involved in amino acid transport and metabolism (307); transcription (298); carbohydrate transport and metabolism (270); general function prediction only (247); translation, ribosomal structure, and biogenesis (235); and cell wall/membrane/envelope biogenesis (207). In addition, genes related to secondary metabolite biosynthesis, transport, and catabolism (80) and defense mechanisms (105) were also observed, indicating that LMY3-5 has the ability to biosynthesize secondary metabolites and resist the influence of external unfavorable factors, providing guaranteed biological control. Concomitantly, we identified 143 genes with unknown functions that may be unique to LMY3-5; however, the characteristics and functions of these remain undetermined.
Analysis of carbohydrate-active enzymes (CAZymes) showed that B. velezensis LMY3-5 encodes 131 CAZyme gene families (Figure 4D), which are divided into six protein categories: glycoside hydrolase, glycosyltransferase, polysaccharide lyase, carbohydrate esterase, carbohydrate-binding module, and auxiliary active family (Figure 5). Among them, the glycoside hydrolase category was found to contain 40 genes related to glycosidic bond hydrolysis, including chitinase and β-1,3-glucanase, which are used for the hydrolysis of carbohydrates and their derivatives, accounting for approximately 30.5% of CAZymes.
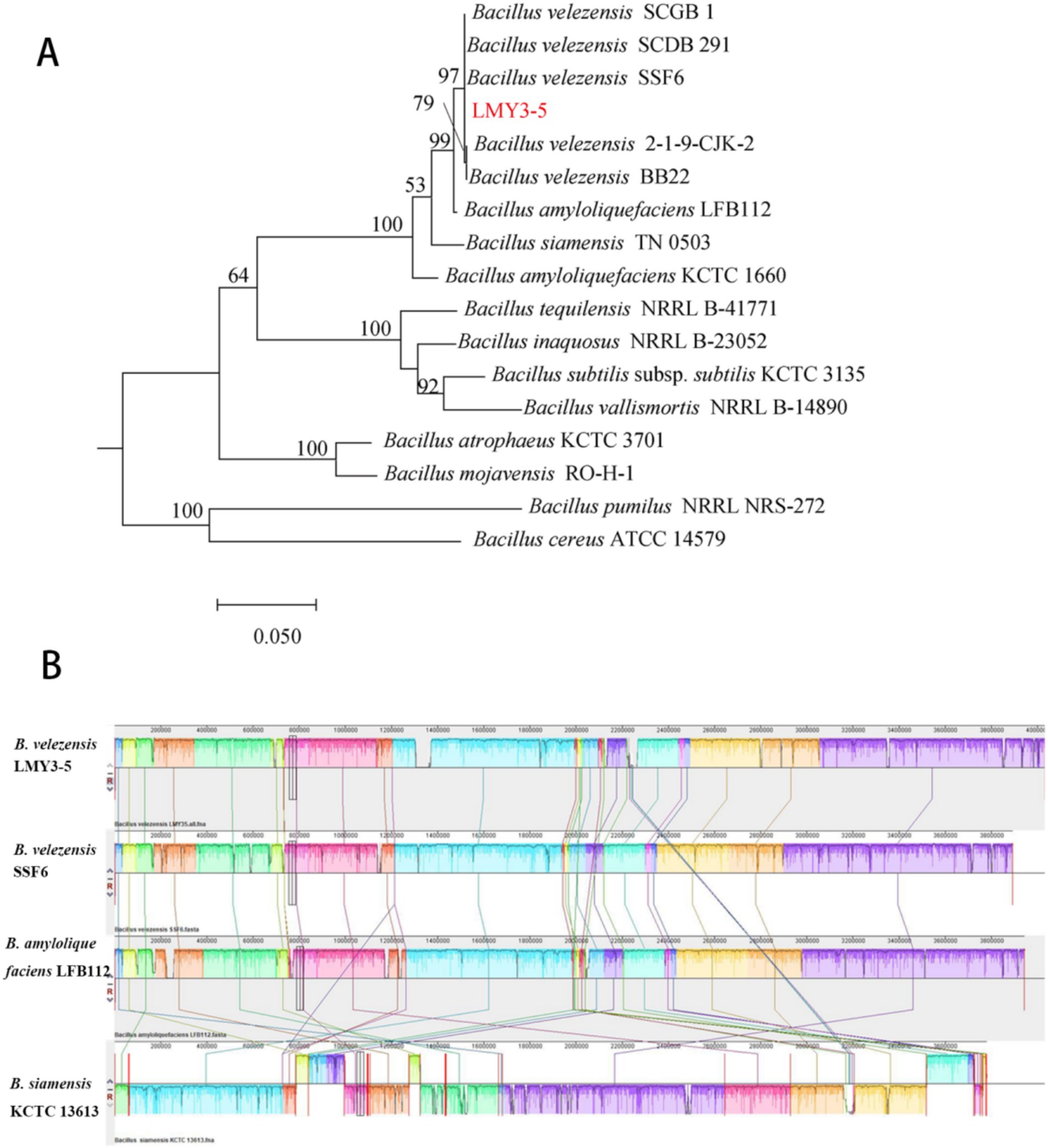
Figure 5. Species identification analysis of strain LMY3-5. (A) Phylogenetic tree constructed by ML clustering method, based on gyrA. (B) Comparison of H2 genome sequences against other four Bacillus genome sequences. Collinearity analysis of the LMY3-5 and SSF6, LFB112, and KCTC13613 genomes. LMY3-5 genome as the reference genome. Pairwise alignments of genomes were generated using Mauve. Boxes with the same colors indicate syntenic regions. Boxes below the horizontal strain line indicate inverted regions. Rearrangements are shown with colored lines. The scale is in nucleotides.
3.5 Comparative genomics analysis of B. velezensis LMY3-5
Phylogenetic analysis confirmed B. velezensis LMY3-5’s closest kinship to B. velezensis SSF6 (ANI/DDH = 100%) and SCDB291 (99.32%/93.4%) (Figure 5A) and Table 1), with gyrA-based phylogeny and ANI/dDDH thresholds (95%/70%) solidifying its taxonomic assignment. Collinearity analysis (Mauve; Figure 5B) revealed >95% genome synteny with SSF6, while exposing three functional hotspots underpinning superior biocontrol: (1) A 118-kb NRPS-PKS hybrid cluster (1.82–1.94 Mb; 78% paenibactin similarity) adjacent to hyperexpressed surfactin operons (+32% vs. SSF6); (2) GH18 chitinase expansion (18 genes vs. SCDB291’s 13) in collinearity break zones, correlating with enhanced cell wall degradation; (3) A 28-kb defense island harboring β-glucanase gluB and ABC transporters.
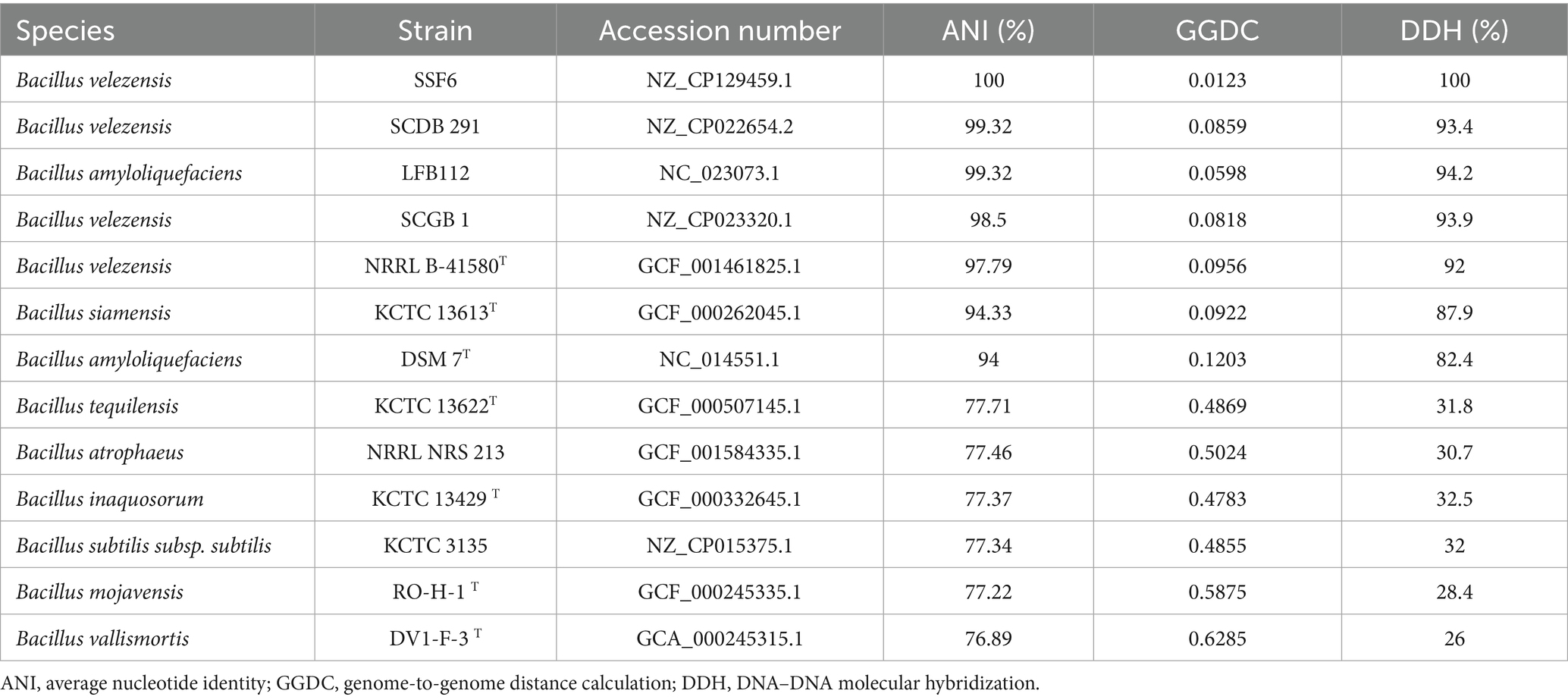
Table 1. ANI and GGDC values between selected Bacillus strains and strain LMY3-5.
3.6 Analysis of CAZymes and secondary metabolite gene clusters of LMY3-5
Using the antiSMASH 4.0 database, we identified 13 secondary metabolite biosynthetic gene clusters (BGCs) in the genome of B. velezensis LMY3-5 (Table 2). Among them, three encoded non-ribosomal peptide synthetases (NRPSs) (Clusters 1, 7, and 11); three encoded trans-acyltransferase polyketide synthases (trans-AT PKSs) (Clusters 5, 6, and 10); two encoded terpene synthases (TPSs) (Clusters 4 and 8); one encoded a polyketide synthase (PKS) (Cluster 3), one encoded a ladderane (Cluster 2); one encoded a lanthipeptide class II (Cluster 12); one encoded a T3 polyketide synthase (T3PKS) (Cluster 9); and one encoded an unknown synthase (Cluster 13). These secondary metabolites included lipopeptides such as surfactin and fengycin and siderophores such as bacillibactin. Macrolactin H, plantazolicin, butirosin, bacillaene, difficidin, and bacilysin are all polyketides. Comparative analysis of key metabolites relative to reference strain FZB42 is visualized in Figure 6. These compounds exert antifungal activity by inhibiting fungal and bacterial pathogens, nutrient uptake, induced systemic response (ISR), and plant colonization (Table 2) (Ongena et al., 2007; Stein, 2005). These BGCs identified in the genome of LMY3-5 are potentially required for its application in the biological control of plant diseases. These findings indicated that B. velezensis LMY3-5 contains a variety of biocontrol-related gene clusters and metabolites, exhibiting strong biocontrol potential.
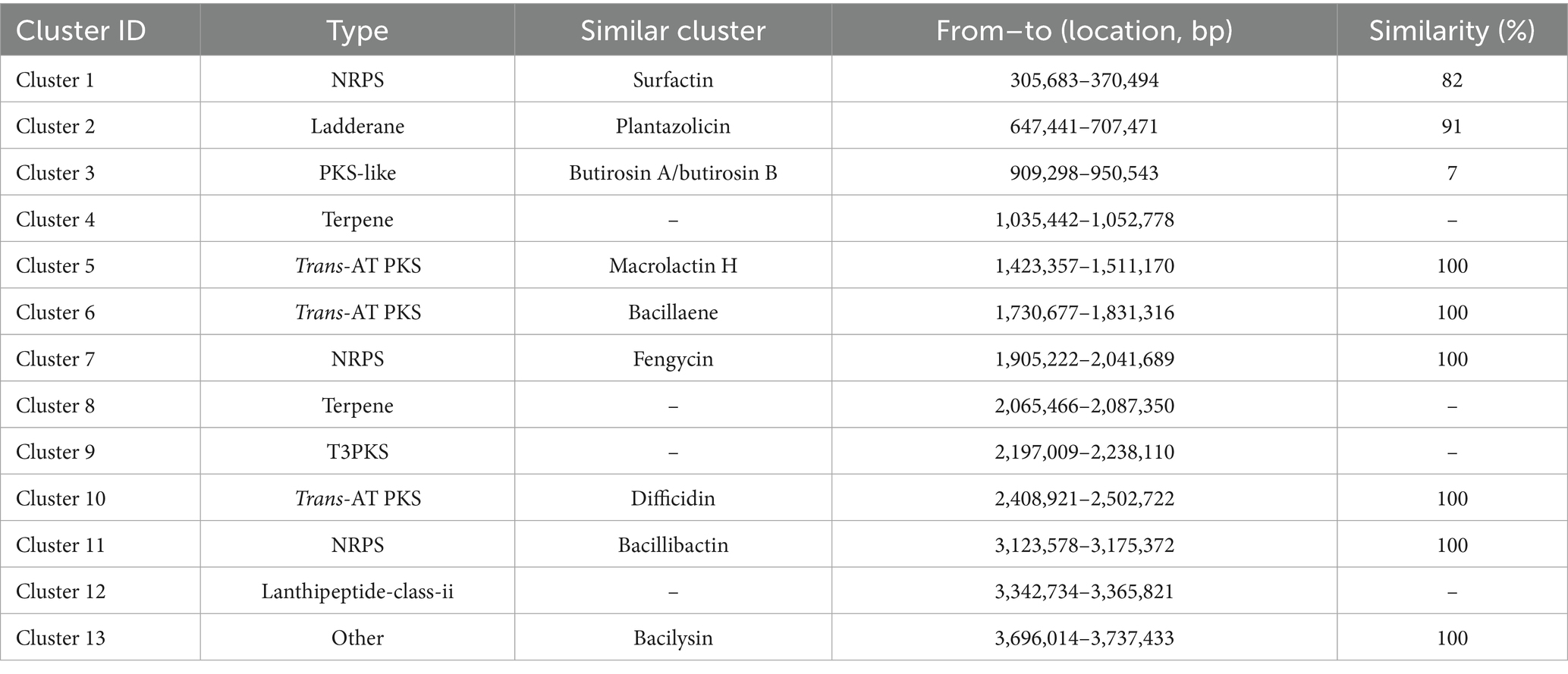
Table 2. Secondary metabolite biosynthetic gene clusters in LMY3-5, identified using antiSMASH 4.0.

Figure 6. Secondary metabolites gene clusters in B. velezensis LMY3-5, identified by antiSMASH 6.0.
3.7 Analysis of DEGs during confrontation culture of LMY3-5
We screened for DEGs between the treatment (T) and control (CK) groups. The results are shown in Figure 7A. We observed that compared to the CK group, 2,870 DEGs were identified in the T group: 31 upregulated and 113 downregulated genes. Cluster heat map analysis showed significant differences in gene expression profiles between the T and CK groups (Figure 7B).
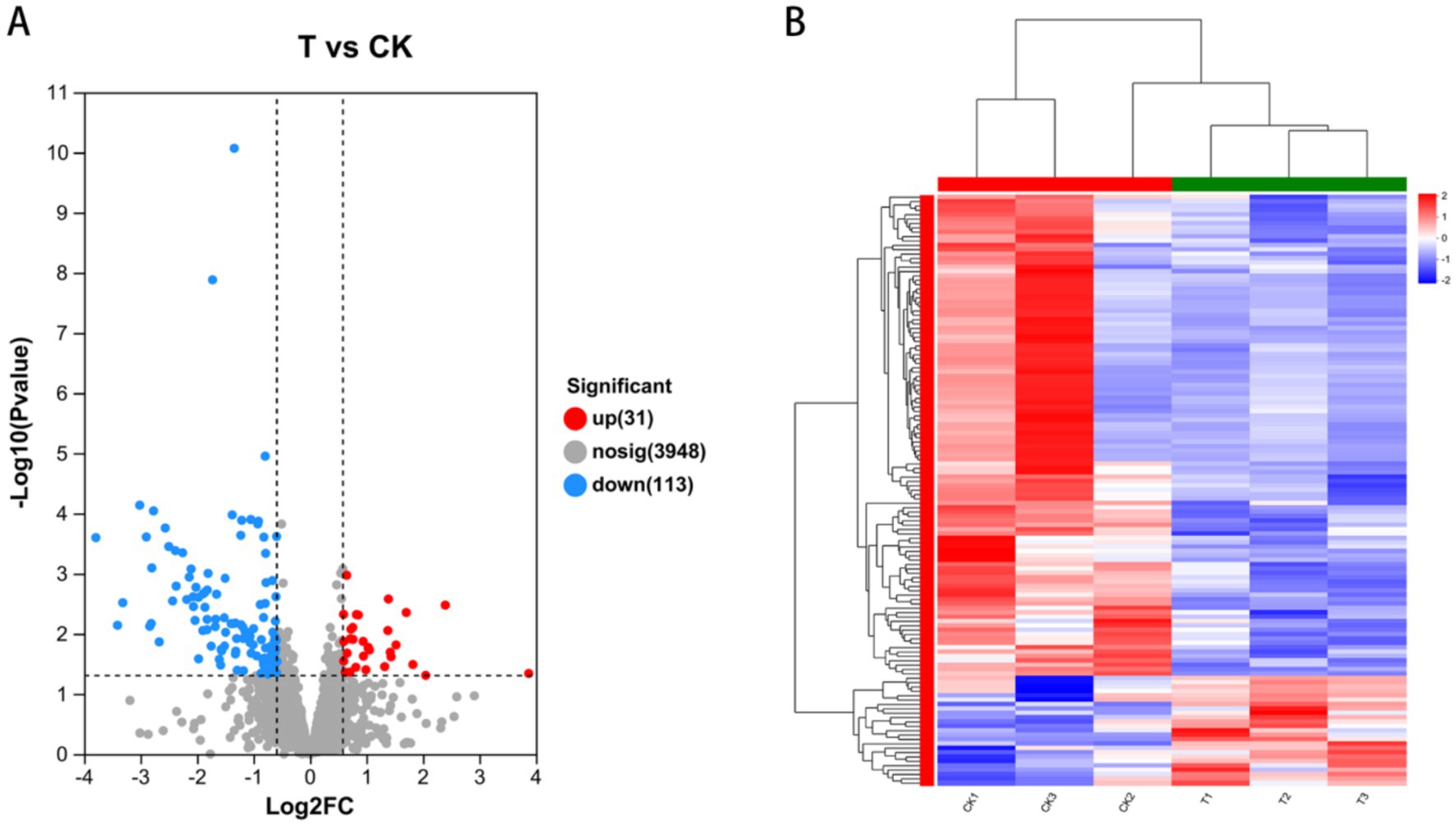
Figure 7. Phytopathogenic genes that were differentially expressed after co-cultured with LMY3-5 strain. (A) Volcano plot of differentially expressed genes between T vs. CK. (B) Heat map of differential expressed genes between T vs. CK.
3.8 Functional enrichment analysis of DEGs related to the antifungal process of LMY3-5
We performed GO function enrichment analysis on DEGs (first 20 entries). We found that in molecular functions, DEGs were mainly enriched in the protein-N (PI)-phosphohistidine-sugar phosphotransferase and oxidoreductase activities, acting as peroxide acceptors (Figure 8). In biological processes such as peroxidase, oxidoreductase, and antioxidant activities, DEGs were mainly enriched in capsule organization and polysaccharide biosynthetic processes. However, the number of DEGs enriched in cellular components was small.
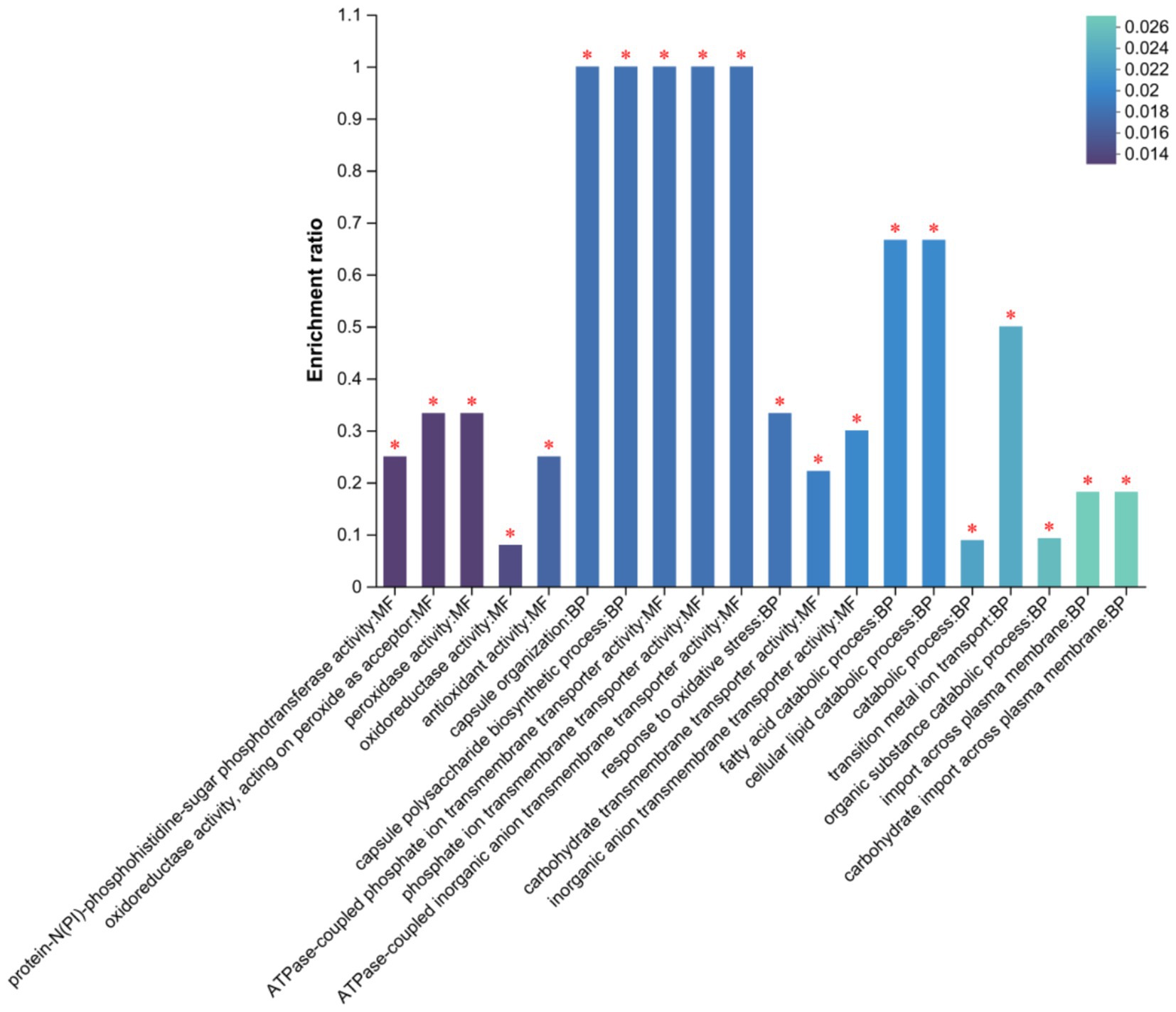
Figure 8. GO Secondary classification statistics plot of differentially expressed genes. FDR < 0.05 marked as *.
According to the KEGG enrichment results, 80 DEGs were enriched in 57 metabolic pathways. We mapped the first 20 KEGG pathways with the smallest default Q values, showing the enrichment of KEGG pathways in three dimensions (Figure 9). These DEGs were significantly concentrated in four related metabolic pathways: the phosphotransferase system (PTS), starch and sucrose metabolism, biosynthesis of various other secondary metabolites, and benzoate degradation. Among them, scrA (gene3748), celB (gene3792), celA (gene3793), and lacE (gene1198) were downregulated in the two metabolic pathways related to the PTS system and starch and sucrose metabolism. The expression of fadN (gene3226) and fadB (gene2792) in the benzoate degradation pathway was also downregulated. Likewise, the expression of iucD (gene0985) was downregulated in the biosynthesis of various other secondary metabolites pathway (Figure 9).
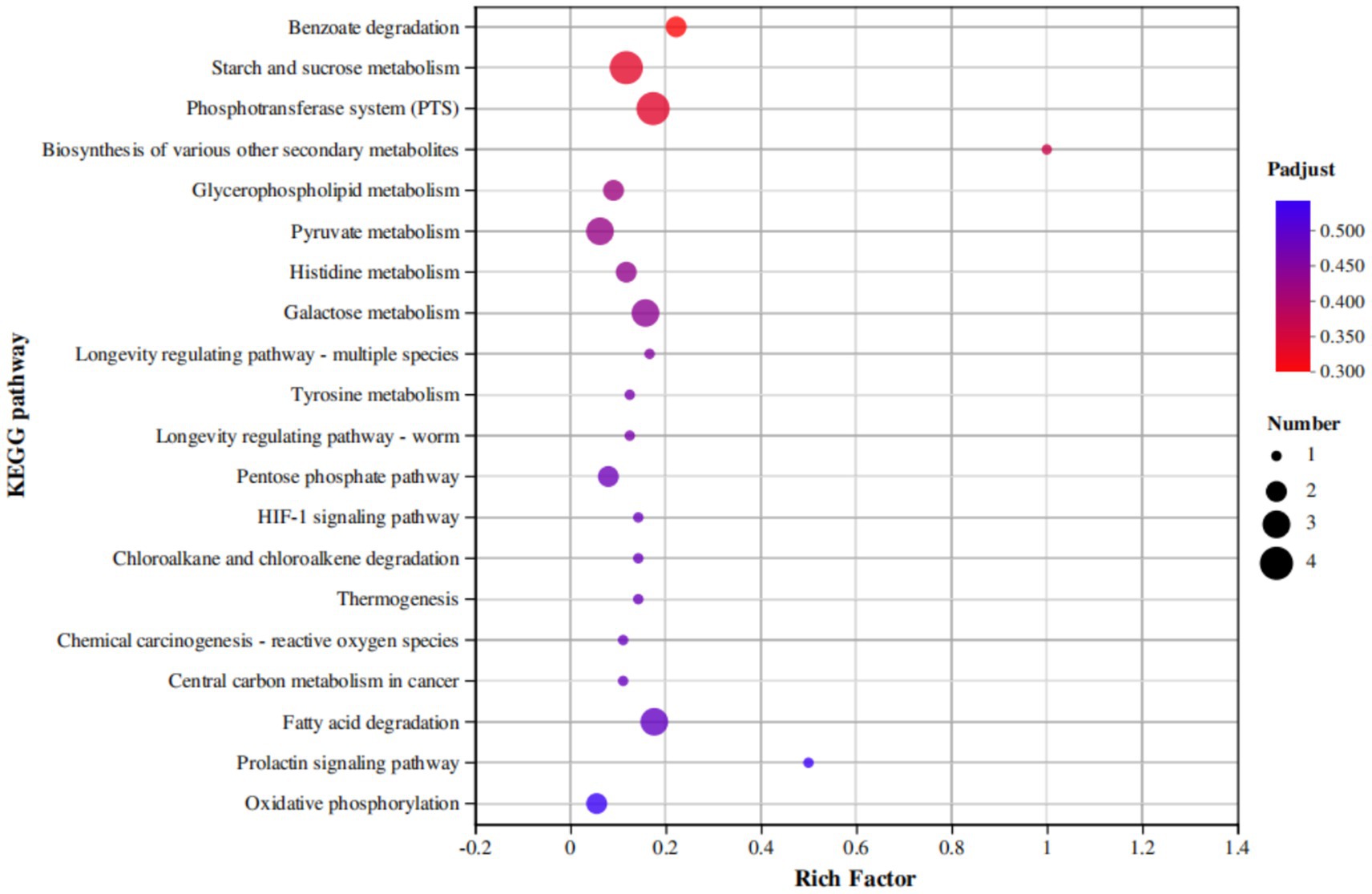
Figure 9. KEGG secondary classification statistics plot of differentially expressed genes.
3.9 Real-time PCR (qRT-PCR) validation
We selected eight DEGs that were mainly involved in microbial metabolism and non-ribosomal peptide synthesis. We found that the expression trends of the eight DEGs were consistent. In general, the reverse transcription quantitative PCR results confirmed that the expression pattern of DEGs was the same as that obtained in the transcriptome sequencing analysis (Figure 10).
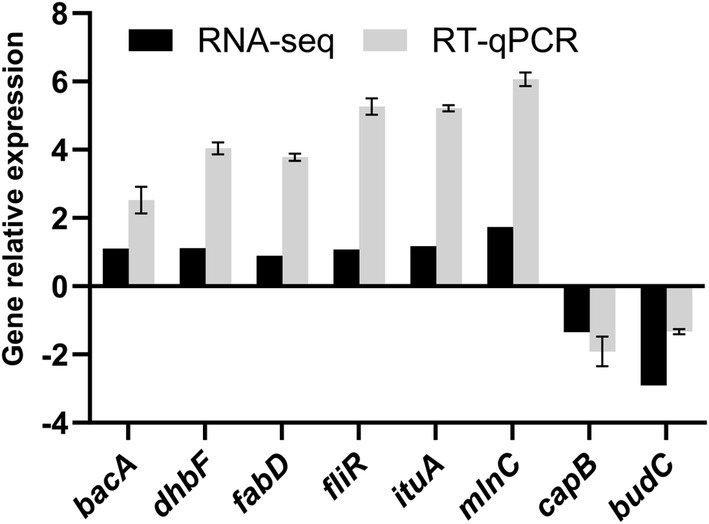
Figure 10. RT-qPCR validation of differentially expressed genes (DEGs). Relative expression levels of target genes (bacA, dhbF, fabD, fliR, ituA, mlnC, capB, budC) are shown. Error bars represent ± standard deviation (SD) of three biological replicates.
4 Discussion
4.1 Analysis of the biocontrol function of B. velezensis LMY3-5
Compared with chemical preventive and control methods, biological control and prevention have attracted increasing attention because they offer green environmental protection and high safety to humans. Pantoea (Nunes et al., 2010; Usall et al., 2001), Pseudomonas (Lam et al., 1987), Bacillus amyloliquefaciens (Chen et al., 2016), Trichoderma harzianum (Zhang H. F. et al., 2022; Zhang Y. H. et al., 2022), Fusicolla violacea (Li et al., 2021) and B. velezensis (Ren et al., 2025) can inhibit the growth of plant pathogens.
Some of these bacteria have been successfully used for the biological control of soybean, grape, and other plant diseases. Compared with other beneficial antagonistic strains, Bacillus has better biocontrol application advantages. Coincidently, Bacillus is also the dominant flora in many plants. Notably, some Bacillus strains play a significant role in the prevention and control of plant pathogenic fungi or bacteria, such as Fusarium graminearum (Chan et al., 2003; Christopher et al., 2013), Sclerotinia sclerotiorum (Hou et al., 2006; Sun et al., 2017), Xanthomonas oryzae (Lin et al., 2002), Bacillus vallismortis (Xu et al., 2023), Bacillus amyloliquefaciens (Aleinikova et al., 2023) and Bacillus velezensis (Ren et al., 2025). In this study, LMY3-5 significantly inhibited the growth of B. dothidea and nd caused morphological alterations (e.g., mycelial distortion and fracture), thereby reducing the incidence of kiwifruit soft rot. These results suggest its potential as a biocontrol agent against this disease.
4.2 Genome mining analysis
Whole-genome sequencing can help determine the biocontrol potential of bacterial strains in agriculture (Grady et al., 2019., Pan et al., 2023). Extensive studies have demonstrated that whole-genome sequencing, proteomics, transcriptomics, metabolomics, and multi-omics integrated analysis have been increasingly applied in the field of biocontrol agents (Ding et al., 2021; Zhang H. F. et al., 2022; Zhang Y. H. et al., 2022). Exploring and predicting the types, structures, and biosynthetic pathways of microbial metabolites at the molecular level has become a mainstream approach for mining functional genes related to antimicrobial activity, discovering bioactive compounds, and investigating antimicrobial mechanisms. For instance, Li L. et al. (2023) and Li W. et al. (2023) utilized whole-genome and transcriptome analyses to predict 146 genes associated with carbohydrate-active enzymes (e.g., chitinases, proteases) and 35 putative gene clusters encoding antifungal metabolites in Bacillus velezensis Htq6, while also elucidating the stress response mechanisms of Botrytis cinerea to this biocontrol strain.
In this study, the genome of the LMY3-5 strain was sequenced, greatly enriching the genomic database of biocontrol Bacillus strains. The ANI value and cluster analysis of the genomes of B. velezensis SSF6, SCDB 291, and B. amyloliquefaciens LFB112 showed that LMY3-5 belonged to B. velezensis and had the closest relationship with strain SSF6. The genome of Bacillus contains genes encoding secondary metabolites and proteins that participate in various metabolic pathways. By performing comparative genomic data analysis using the NJ13 strain, eight different gene clusters related to secondary metabolite synthesis were identified in strain LMY3-5. Non-ribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs), and hybrid enzymes synthesize cyclic lipopeptides such as surfactin, iturin, and fengycin, as well as polyketides and other antimicrobial metabolites. These compounds exert antifungal effects by inducing hyphal structural deformities, disrupting cell wall integrity and membrane permeability, and triggering oxidative stress responses, ultimately leading to pathogen death and disease suppression (Chen et al., 2009; Gong et al., 2015; Luo et al., 2015; Malik et al., 2024). Known surfactins exhibit antifungal and antifungal properties. In particular, surfactins can be embedded in bacterial cell membranes and dissolve phospholipid bilayers, resulting in the production of membrane pores and ion channels (Hamley, 2015; Stubbendieck and Straight, 2016). Surfactin exhibits significant antifungal activity against fungi. Its mechanism of action primarily involves the induction of intracellular ROS accumulation in fungal cells, leading to oxidative stress. Additionally, it reduces mitochondrial membrane potential, activates caspase-like proteases, and triggers chromatin condensation, indicating that fungal growth inhibition occurs via an apoptosis-like cell death pathways (Zhao et al., 2017; Cawoy et al., 2014; Chen et al., 2023). Notably, in one gene cluster, no homologous proteins with high similarity were identified, and it showed only 7% similarity with butirosin A or butirosin B synthesized in the saccharide pathway in B. velezensis FZB42. No homologous proteins were identified in the database for the other four gene clusters, indicating that LMY3-5 may produce new antifungal substances. In general, strain LMY3-5 has a rich antifungal substance synthesis gene cluster. Its antifungal activity against pathogenic fungi may be mainly derived from known substances with good antifungal activity; however, it produces other unknown metabolites that may also have antifungal activity.
4.3 Transcriptome analysis
Biocontrol strains regulate the growth of pathogens through the expression of antifungal-related genes and production of antifungal substances. Such as: Zhang H. F. et al. (2022) and Zhang Y. H. et al. (2022) obtained a high-quality genome sequence of B. velezensis GS-1 through multi-omics analysis and identified 13 gene clusters associated with secondary metabolite biosynthesis by integrating transcriptomic and metabolomic data. They predicted the potential production of antimicrobial lipopeptides, such as surfactin, iturin, and plantazolicin, and further confirmed the inhibitory effects of these lipopeptides against Magnaporthe oryzae along with their antagonistic mechanisms (Zhang H. F. et al., 2022; Zhang Y. H. et al., 2022). Su et al. (2021) analyzed the differentially expressed genes of Sporisorium scitamineum at different infection time points using transcriptomics and found that the number of differentially expressed genes peaked at 8 h post-infection. Fatty acid metabolism and peroxidases were identified as key metabolic pathways during the infection process of S. scitamineum (Su et al., 2021). Wang X. J. et al. (2021) performed whole-genome sequencing of Phomopsis phragmitis, the causative agent of kiwifruit soft rot, using the PacBio SMRT platform. They predicted 1,451 proteins associated with pathogen-host interactions and 558 virulence factors (Wang X. J. et al., 2021). To date, transcriptome sequencing has enabled the acquisition of extensive structural and functional information on genes. Although diverse secondary metabolites with antifungal activity have been discovered in various plant endophytic bacteria, and antimicrobial genes have been predicted, the regulatory mechanisms underlying their biosynthesis and their modes of antifungal action remain largely unknown. Further in-depth research is still required to elucidate their antimicrobial mechanisms.
In this study, we performed transcriptome sequencing after culturing B. velezensis LMY3-5 with B. dothidea for 72 h. Using the transcriptome data, we identified a total of 332 DEGs in the antagonistic LMY3-5 strain. The DEGs were mainly involved in benzoate degradation, starch and sucrose metabolism, PTS pathway, and biosynthesis of various other secondary metabolite pathways. The starch and sucrose metabolism pathways are mainly involved in the utilization of basic carbon and nitrogen sources in organisms. The biosynthesis of various antibiotics mainly involves the synthesis of related antifungal substances (such as lysozyme, dehydratase, and alanine-anti-capsular ligase). Thus, it can be deduced that this strain produces antifungal substances by initiating the metabolic synthesis pathways of related antifungal substances.
5 Conclusion
The biocontrol strain B. velezensis LMY3-5 demonstrates significant antagonistic activity against B. dothidea, the causal agent of kiwifruit soft rot, achieving 70.44% suppression efficacy in pathogen challenge assays. Genomic analysis revealed a 4.03 Mb circular chromosome harboring eight biosynthetic gene clusters (BGCs), including those encoding surfactin, fengycin, bacillaene, and bacillibactin, which collectively contribute to its antifungal potential. Transcriptomic profiling during coculture with B. dothidea identified 114 differentially expressed genes (DEGs), with upregulation of fengycin synthesis, siderophore production, and lysozyme-related genes, directly implicating these pathways in antifungal activity. Conversely, downregulation of flagellar assembly and chemotaxis genes suggests a metabolic shift toward secondary metabolite production during antagonism. Functional validation confirmed that lipopeptides (e.g., fengycin) and siderophores play pivotal roles in inhibiting fungal growth, likely through membrane disruption and iron competition. These findings provide molecular-level insights into LMY3-5’s biocontrol mechanisms and highlight its potential as a sustainable alternative to chemical fungicides for managing kiwifruit soft rot. Further field studies are warranted to translate these discoveries into practical agricultural applications.
Data availability statement
The data presented in the study are deposited in the NCBI GanBank repository, accession number CP196812 (https://www.ncbi.nlm.nih.gov/nuccore/CP196812.1).
Author contributions
CR: Writing – original draft, Writing – review & editing. YL: Investigation, Validation, Writing – original draft. WS: Methodology, Writing – review & editing. ZH: Data curation, Writing – review & editing. DW: Methodology, Software, Writing – review & editing. WL: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the National Key Research and Development Program: 2021YFD1 100300, a post-subsidy fund.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Adnan, M., Morton, G., and Hadi, S. (2011). Analysis of rpoS and bolA gene expression under various stress-induced environments in planktonic and biofilm phase using 2 ΔΔCT method. Mol. Cell. Biochem. 357, 275–282. doi: 10.1007/s11010-011-0898-y
Aleinikova, N., Galkina, Y., Andreyev, V., Bolotianskaia, E., and Shaporenko, V. (2023). The prospects of using Bacillus amyloliquefaciens in the biological control of grape diseases. IOP Confer. Series Earth Environ. Sci. 1206:012025. doi: 10.1088/1755-1315/1206/1/012025
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., et al. (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Bakirci, G. T., Acay, D. B. Y., Bakirci, F., and Otles, S. (2014). Pesticide residues in fruits and vegetables from the Aegean region, Turkey. Food Chem. 160, 379–392. doi: 10.1016/j.foodchem.2014.02.051
Bolger, A. M., Marc, L., and Bjoern, U. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cawoy, H., Mariutto, M., Henry, G., Fisher, C., Vasilyeva, N., Thonart, P., et al. (2014). Plant defense stimulation by natural isolates of Bacillus depends on efficient surfactin production. Mol. Plant Microbe Interact. 27, 87–100. doi: 10.1094/MPMI-09-13-0262-R
Chan, Y. K., Mccormick, W. A., and Seifert, K. A. (2003). Characterization of an antifungal soil bacterium and its antagonistic activities against Fusarium species. Can. J. Microbiol. 49:253. doi: 10.1139/w03-033
Chen, X. H., Scholz, R., Borriss, M., Junge, H., Moegel, G., Kunz, S., et al. (2009). Difficidin and bacilysin produced by plant-associated Bacillus amyloliquefaciens are efficient in controlling fire blight disease. J. Biotechnol. 140, 38–44. doi: 10.1016/j.jbiotec.2008.10.015
Chen, L., Xu, X. X., Sun, Y. X., Xin, Q. H., Lv, Y. Y., Hu, Y. S., et al. (2023). Surfactin inhibits Fusarium graminearum by accumulating intracellular ROS and inducing apoptosis mechanisms. World J. Microbiol. Biotechnol. 39:340. doi: 10.1007/s11274-023-03790-2
Chen, X. Y., Zhang, Y. Y., Fu, X. C., Li, Y., and Wang, Q. (2016). Isolation and characterization of Bacillus amyloliquefaciens PG12 for the biological control of apple ring rot. Postharvest Biol. Technol. 115, 113–121. doi: 10.1016/j.postharvbio.2015.12.021
Christopher, A., Dunlap, M. J., Bowman, D. A., and Schisler, A. (2013). Genomic analysis and secondary metabolite production in Bacillus amyloliquefaciens AS 43.3: a biocontrol antagonist of Fusarium head blight. Biol. Control. 64, 166–175. doi: 10.1016/j.biocontrol.2012.11.002
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Ding, H. X., Mo, W. D., Yu, S., Cheng, H. H., Peng, L. J., and Liu, Z. Y. (2021). Whole genome sequence of Bacillus velezensis strain GUMT319: a potential biocontrol agent against tobacco black shank disease. Front. Microbiol. 12:658113. doi: 10.3389/fmicb.2021.658113
Fan, Y., Liu, K., Lu, R., Gao, J., Song, W., Zhu, H., et al. (2023). Cell-free supernatant of Bacillus subtilis reduces kiwifruit rot caused by Botryosphaeria dothidea through inducing oxidative stress in the pathogen. J. Fungi 9:127. doi: 10.3390/jof9010127
Gong, A. D., Li, H. P., Yuan, Q. S., Song, X. S., Yao, W., He, W. J., et al. (2015). Antagonistic mechanism of Iturin A and Plipastatin A from Bacillus amyloliquefaciens S76-3 from wheat spikes against Fusarium graminearum. PLoS One 10:e0116871. doi: 10.1371/journal.pone.0116871
Grady, E. N., Macdonald, J., Ho, M. T., Weselowski, B., Mcdowell, T., Solomon, O., et al. (2019). Characterization and complete genome analysis of the surfactin-producing, plant-protecting bacterium Bacillus velezensis 9D-6. BMC Microbiol. 19:5. doi: 10.1186/s12866-018-1380-8
Hamley, I. W. (2015). Lipopeptides: from self-assembly to bioactivity. Chem. Commun. 51, 8574–8583. doi: 10.1039/C5CC01535A
Hilario, L. A., and Artur, A. (2020). Botryosphaeriaceae species associated with blueberry stem blight and dieback in the Centre region of Portugal. Eur. J. Plant Pathol. 156, 31–44. doi: 10.1007/s10658-019-01860-6
Hou, X., Boyetchko, S. M., Brkic, M., Olson, D., Ross, A., and Hegedus, D. (2006). Characterization of the anti-fungal activity of a Bacillus spp. associated with sclerotia from Sclerotinia sclerotiorum. Appl. Microbiol. Biotechnol. 72, 644–653. doi: 10.1007/s00253-006-0315-8
Kanehisa, M. (2019). Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. doi: 10.1002/pro.3715
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., and Ishiguro-Watanabe, M. (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. doi: 10.1093/nar/gkac963
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim, G., Sung, J. J., Jae-Seoun, H., Jin, K. Y., and Young-seon, L. (2013). Optimal spray time, interval and number of preventive fungicides for the control of fruit rots of green and gold kiwifruit cultivars. Res. Plant Dis. 19, 1–6. doi: 10.5423/RPD.2013.19.1.001
Lam, B. S., Strobel, G. A., and Lam, H. S. T. (1987). Transposon mutagenesis and tagging of fluorescent pseudomonas: antimycotic production is necessary for control of Dutch Elm disease. Proc. Natl. Acad. Sci. U. S. A. 84, 6447–6451. doi: 10.1073/pnas.84.18.6447
Li, L., Chen, M. Y., Zhang, P., Han, F., Liu, X. L., and Zhong, C. H. (2016). Identification of the pathogen causing soft rot of kiwifruit. J. Plant Prot. 43, 527–528. doi: 10.13802/j.cnki.zwbhxb.2016.03.025
Li, W. Z., Long, Y. H., Mo, F. X., Shu, R., Yin, X. H., Wu, X. M., et al. (2021). Antifungal activity and biocontrol mechanism of Fusicolla violacea J-1 against soft rot in kiwifruit caused by Alternaria alternata. J. Fungi 7:937. doi: 10.3390/jof7110937
Li, W., Long, Y., Yin, X., Wang, W., Zhang, R., Mo, F., et al. (2023). Antifungal activity and mechanism of tetramycin against Alternaria alternata, the soft rot causing fungi in kiwifruit. Pestic. Biochem. Physiol. 192:105409. doi: 10.1016/j.pestbp.2023.105409
Li, L., Pan, H., Chen, M., Zhang, S., and Zhong, C. (2017). Isolation and identification of pathogenic fungi causing postharvest fruit rot of kiwifruit (Actinidia chinensis) in China. J. Phytopathol. 165, 782–790. doi: 10.1111/jph.12618
Li, L., Pan, H., Deng, L., Qian, G., Wang, Z., Li, W., et al. (2022). The antifungal activity and mechanism of silver nanoparticles against four pathogens causing kiwifruit post-harvest rot. Front. Microbiol. 13:8633. doi: 10.3389/fmicb.2022.988633
Li, L., Wang, R. J., Liang, X. X., Gai, Y. P., Jiao, C., and Wang, M. Q. (2023). Characterization of a Bacillus velezensis with antifungal activity and its inhibitory effect on gray mold germ. Agronomy 13:1553.
Lin, D., Qu, L. J., Gu, H., and Chen, Z. (2002). A 3.1-kb genomic fragment of Bacillus subtilis encodes the protein inhibiting growth of Xanthomonas oryzae pv. Oryzae. J. Appl. Microbiol. 91, 1044–1050. doi: 10.1046/j.1365-2672.2001.01475.x
Liu, F., Gao, R., Zhang, F., Ren, Y., Li, W., and He, B. (2023). Postharvest biocontrol of green mold (Penicillium digitatum) in citrus by Bacillus velezensis strain S161 and its mode of action. Biol. Control 187:105392. doi: 10.1016/j.biocontrol.2023.105392
Liu, J., Guo, X., Zhang, H., Cao, Y., and Sun, Q. (2021). First report of postharvest fruit rot disease of hardy kiwifruit caused by Diaporthe eres in China. Plant Dis. 105:3745. doi: 10.1094/PDIS-08-20-1705-PDN
Lu, Z., Chen, M., Long, X., Yang, H., and Zhu, D. (2023). Biological potential of Bacillus subtilis BS45 to inhibit the growth of Fusarium graminearum through oxidative damage and perturbing related protein synthesis. Front. Microbiol. 14:18. doi: 10.3389/fmicb.2023.1064838
Lu, Y., Ma, D., He, X., Wang, F., Wu, J., Liu, Y., et al. (2021). Bacillus subtilis KLBC BS6 induces resistance and defence-related response against Botrytis cinerea in blueberry fruit. Physiol. Mol. Plant Pathol. 114:1599. doi: 10.1016/j.pmpp.2020.101599
Malik, J., Moosa, A., Zulfiqar, F., Aslam, M. N., Albalawi, M. A., Almowallad, S., et al. (2024). Biocontrol potential of lipopeptides produced by the novel Bacillus altitudinis strain TM22A against postharvest Alternaria rot of tomato. LWT 191:115541. doi: 10.1016/j.lwt.2023.115541
Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Goeker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformat. 14:60. doi: 10.1186/1471-2105-14-60
Mo, F., Hu, X., Ding, Y., Li, R., Long, Y., Wu, X., et al. (2021). Naturally produced magnolol can significantly damage the plasma membrane of Rhizoctonia solani. Pestic. Biochem. Physiol. 178:104942. doi: 10.1016/j.pestbp.2021.104942
Mu, F., Chen, X., Fu, Z., Wang, X., Guo, J., Zhao, X., et al. (2023). Genome and transcriptome analysis to elucidate the biocontrol mechanism of Bacillus amyloliquefaciens XJ5 against Alternaria solani. Microorganisms 11:2055. doi: 10.3390/microorganisms11082055
Nunes, C., Usall, J., Teixidó, N., Fons, E., and Vias, I. (2010). Post-harvest biological control by Pantoea agglomerans (CPA-2) on Golden delicious apples. J. Appl. Microbiol. 92, 247–255. doi: 10.1046/j.1365-2672.2002.01524.x
Ongena, M., Jourdan, E., Adam, A., Paquot, M., Brans, A., Joris, B., et al. (2007). Surfactin and fengycin lipopeptides of Bacillus subtilis as elicitors of induced systemic resistance in plants. Environ Microbiolm 9,1084–90. doi: 10.1111/j.1462-2920.2006.01202.x
Pan, J. J., Meng, J., Zhang, D. M., Zeng, C. F., Wang, X. K., Wang, F., et al. (2023). Genome sequence resource of Botryosphaeria dothidea strain XNHG241, a causal agent of peach gummosis. Plant Dis. 107, 2205–2208. doi: 10.1094/PDIS-11-22-2539-A
Rabbee, M. F., Ali, M. S., Choi, J., Hwang, B. S., and Jeong, S. C. (2019). Bacillus velezensis: A valuable member of bioactive molecules within plant microbiomes. Molecules 24:1046. doi: 10.3390/molecules24061046
Ren, C. G., Liu, Y., Su, W. W., and Tian, B. (2025). Bacillus velezensis LMY3-5 for the biocontrol of soft rot in kiwifruit: antifungal action and underlying mechanisms. Front. Microbiol. 16:1562366. doi: 10.3389/fmicb.2025.1562366
Saxena, A. K., Kumar, M., Chakdar, H., Anuroopa, N., and Bagyaraj, D. J. (2020). Bacillus species in soil as a natural resource for plant health and nutrition. J. Appl. Microbiol. 128, 1583–1594. doi: 10.1111/jam.14506
Shin, Y. H., Ledesma, M., Whitman, S., Tyson, J., Zange, B., Kim, K. D., et al. (2021). Suppression of ripe rot on 'Zesy002' kiwifruit with commercial agrochemicals. Plant Pathol. J. 37:46. doi: 10.5423/PPJ.OA.03.2021.0046
Stein, T. (2005). Bacillus subtilis antibiotics: structures, syntheses and specific functions. Mol Microbiol 56, 845–57. doi: 10.1111/j.1365-2958.2005.04587.x
Stubbendieck, R. M., and Straight, P. D. (2016). Multifaceted interfaces of bacterial competition. J. Bacteriol. 198, 2145–2155. doi: 10.1128/JB.00275-16
Su, W. W., Wu, D., Han, Z. C., Li, L. L., Li, W. J., and Ren, C. G. (2021). Identification and analysis of pathogens causing fruit rot of kiwifruit in Guizhou Province. Journal of Henan agricultural sciences 50(3), 97–102.Effects on Brassica napus. Biol. Control 104, 35–43. doi: 10.15933/j.cnki.1004-3268.2021.03.013
Sun, G., Yao, T., Feng, C., Chen, L., Li, J., and Wang, L. (2017). Identification and biocontrol potential of antagonistic bacteria strains against Sclerotinia sclerotiorum and their growth-promoting. Biological Control. 104, 35–43. doi: 10.1016/j.biocontrol.2016.10.008
Tang, W., Ding, Z., Zhou, Z. Q., Wang, Y. Z., and Guo, L. Y. (2012). Phylogenetic and pathogenic analyses show that the causal agent of apple ring rot in China is Botryosphaeria dothidea. Plant Dis. 96, 486–496. doi: 10.1094/PDIS-08-11-0635
Usall, J., Teixidó, N., Viñas, I., and Smilanick, J. (2001). Biological control of Penicillium digitatum on citrus fruits with the antagonistic bacterium Pantoea agglomerans. Acta Hortic. 553, 377–382. doi: 10.17660/ActaHortic.2001.553.86
Wang, X. J., Dong, H. J., Lan, J. B., Liang, K., Liu, Y. Y., Lu, Q., et al. (2021). High-quality genome resource of the pathogen of Diaporthe (Phomopsis) phragmitis causing kiwifruit soft rot. Mol. Plant-Microbe Interact. 34, 218–221. doi: 10.1094/MPMI-08-20-0236-A
Wang, F., Xiao, J., Zhang, Y., Li, R., Liu, L., and Deng, J. (2021). Biocontrol ability and action mechanism of Bacillus halotolerans against Botrytis cinerea causing grey mould in postharvest strawberry fruit. Postharvest Biol. Technol. 174:1456. doi: 10.1016/j.postharvbio.2020.111456
Xu, L. S., Meng, Y. G., Liu, R. H., Xiao, Y., Wang, Y. Z., and Huang, L. L. (2023). Inhibitory effects of Bacillus vallismortis T27 against apple valsa canker caused by Valsa Mali. Pestic. Biochem. Physiol. 195:5564. doi: 10.1016/j.pestbp.2023.105564
Yanli, T., Yuqiang, Z., Ting, S., Lv, W., Jun, L., Xiaofeng, M., et al. (2018). Identification and characterization of Phomopsis amygdali and Botryosphaeria dothidea associated with peach shoot blight in Yangshan, China. Plant Dis. 102:2511. doi: 10.1094/PDIS-02-18-0243-RE
Yin, Y., Miao, J., Shao, W., Liu, X., Zhao, Y., and Ma, Z. (2023). Fungicide resistance: Progress in understanding mechanism, monitoring, and management. Phytopathology 113, 707–718. doi: 10.1094/PHYTO-10-22-0370-KD
Yoon, S.-H., Ha, S.-M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Zhang, H. F., Kong, N., Liu, B., Yang, Y. F., Li, C. H., Qi, J. Y., et al. (2022). Biocontrol potential of Trichoderma harzianum CGMCC20739 (Tha739) against postharvest bitter rot of apples. Microbiol. Res. 265:127182. doi: 10.1016/j.micres.2022.127182
Zhang, L. Z., Zhang, Q., Yang, P., Niu, Y., and Niu, W. (2019). First report of gummosis disease of sweet cherry caused by Botryosphaeria dothidea in China. Plant Dis. 12, 3283–3297. doi: 10.1094/PDIS-07-19-1418-PDN
Zhang, Y. H., Zhao, M. X., Chen, W., Yu, H. L., Jia, W. T., Pan, H. Y., et al. (2022). Multi-omics techniques for analysis antifungal mechanisms of lipopeptides produced by Bacillus velezensis GS-1 against Magnaporthe oryzae in vitro. Int. J. Mol. Sci. 23:3762. doi: 10.3390/ijms23073762
Zhao, H., Shao, D., Jiang, C., Shi, J., Li, Q., Huang, Q., et al. (2017). Biological activity of lipopeptides from Bacillus. Appl. Microbiol. Biotechnol. 101, 5951–5960. doi: 10.1007/s00253-017-8396-0
Zheng, L., Huang, S., Hsiang, T., Yu, G., Guo, D., Jiang, Z., et al. (2021). Biocontrol using Bacillus amyloliquefaciens PP19 against Litchi downy blight caused by Peronophythora litchii. Front. Microbiol. 11:3394. doi: 10.3389/fmicb.2020.619423
Zhi, Y., Wu, Q., and Xu, Y. (2017). Genome and transcriptome analysis of surfactin biosynthesis in Bacillus amyloliquefaciens MT45. Sci. Rep. 7:976. doi: 10.1038/srep40976
Zhou, D., Hu, F., Lin, J., Wang, W., and Li, S. (2019a). Genome and transcriptome analysis of Bacillus velezensis BS-37, an efficient surfactin producer from glycerol, in response to d−/l-leucine. Microbiology 8:794. doi: 10.1002/mbo3.794
Keywords: Bacillus velezensis, Botryosphaeria dothidea, kiwifruit soft rot, biocontrol, antimicrobial mechanisms
Citation: Ren C, Liu Y, Su W, Han Z, Wu D and Li W (2025) Omics insights into Bacillus velezensis LMY3-5 defense against Botryosphaeria dothidea in kiwifruit. Front. Microbiol. 16:1633015. doi: 10.3389/fmicb.2025.1633015
Edited by:
Mohammad Sayyar Khan, University of Agriculture, Peshawar, PakistanReviewed by:
Amjad Iqbal, Abdul Wali Khan University Mardan, PakistanSayed Usman Ali Shah, Guangxi Academy of Sciences, China
Ping Tang, Yunnan Agricultural University, China
Copyright © 2025 Ren, Liu, Su, Han, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijie Li, MTM3NjUwNzEzNThAMTYzLmNvbQ==