 Simon T. L. Amoikon1,2
Simon T. L. Amoikon1,2 Kanny Diallo1*
Kanny Diallo1* Jeremie K. Tuo1,3
Jeremie K. Tuo1,3 Naima Nasir4,5Vitalis F. Feteh4,5Grace Mzumara4,5Adeniyi Aderoba4,5Rebecca Jacques5Hansini Mandal5
Naima Nasir4,5Vitalis F. Feteh4,5Grace Mzumara4,5Adeniyi Aderoba4,5Rebecca Jacques5Hansini Mandal5 Keith A. Jolley4
Keith A. Jolley4 James E. Bray4Odile B. Harrison4,5
James E. Bray4Odile B. Harrison4,5 Martin C. J. Maiden4
Martin C. J. Maiden4- 1Centre Suisse de Recherches Scientifiques en Côte d’Ivoire (CSRS), Abidjan, Côte d’Ivoire
- 2Institut Pierre Richet (IPR), Institut national de santé publique (INSP), Bouaké, Côte d’Ivoire
- 3Institut National Polytechnique Felix Houphouët-Boigny, Yamoussoukro, Côte d’Ivoire
- 4Department of Biology, University of Oxford, Oxford, United Kingdom
- 5Nuffield Department of Population Health, University of Oxford, Oxford, United Kingdom
Context: Diagnosing meningitis remains challenging with etiological agents frequently unidentified. Using both in silico and in vitro approaches, this study evaluated published and novel genetic targets for the detection of common bacterial species known to cause meningitis: Neisseria meningitidis, Streptococcus agalactiae, Streptococcus pneumoniae, and Haemophilus influenzae.
Methods: A total of 29 genetic targets were investigated for the detection of N. meningitidis, S. agalactiae, S. pneumoniae, and H. influenzae, using the Gene Presence tool and whole genome sequence data (WGS) found in the genomics platform, PubMLST. These targets were further tested in silico by screening WGS using the PCR tool hosted on PubMLST allowing the sensitivity, specificity, Negative Predicted Values (NPV) and Positive Predictive Values (PPV) to be determined. Ten targets were then further evaluated in vitro by real-time PCR against a panel of 44 bacterial isolates representative of the genera evaluated.
Results: The best performing in silico genetic determinants targeted: N. meningitidis, sodC (NEIS1339) (sensitivity 99.7%, specificity, 99.4%, PPV, 99.6% and NPV, 99.6%); S. pneumoniae, SP2020 (99.5%, 99.9%, 99.9%, and 81.5%) and H. influenzae, dmsA (HAEM1183) (98%, 100%, 99.6%, and 77.4%). All three of these targets also had the best in vitro sensitivity (100%), specificity [91.7% sodC (NEIS1339), 100% SP2020 and 97.3% dmsA (HAEM1183), PPV (72.7% sodC (NEIS1339), 100% SP2020 and 87.5% dmsA (HAEM1183)] and NPV (100% for all targets). The gene sip (SAG0032) encoding the surface immunogenic protein (sip) exhibited the best sensitivity (99.6%) and NPV (96.9%) for S. agalactiae compared to 99.3% and 94.8% for cfb (SAG2043), respectively in silico. However, in vitro, cfb showed the best sensitivity (100% vs. 85.7%) and NPV (100% vs. 97.4%) when compared to sip.
Conclusion: SodC, cfb, SP2020, and dmsA have the potential to enhance the accuracy of molecular diagnostics for the four most common bacterial species causing meningitis. Moreover, a combined in silico and in vitro approach that leverages WGS deposited in databases such as PubMLST, offers an efficient and cost-effective means for the preliminary evaluation of diagnostic targets.
1 Introduction
Bacterial meningitis and related invasive infections, including pneumonia, bacteraemia and septicaemia, are devastating diseases that represent a major public health concern worldwide (Rodgers et al., 2020), the main bacterial aetiological agents being: Neisseria meningitidis (the meningococcus); Streptococcus pneumoniae (the pneumococcus); Haemophilus influenzae; and Streptococcus agalactiae (group B streptococcus, GBS). Despite the availability of vaccines against many variants of these pathogens (Tsang, 2021), with the exception of S. agalactiae for which intrapartum antibiotic prophylaxis is used (Hughes et al., 2017), meningitis continues to affect populations globally, particularly in sub-Saharan Africa.
In sub-Saharan Africa, meningitis is endemic, with seasonal epidemics occurring unpredictably every 5–12 years (Hughes et al., 2017). A recent example being the meningitis outbreak among miners in the northeastern cities of the Democratic Republic of Congo, declared on 8 September 2021 (WHO Africa, 2022). These epidemics are associated with a high case fatality rate of 50% and many cases remain undiagnosed, increasing the delay in appropriate public health interventions. Control of meningitis requires improved access to and efficiency of diagnostic methods; indeed, diagnosis is one of the five pillars of the World Health Organisation (WHO) roadmap to defeating meningitis by 2030 (World Health Organization [WHO], 2021).
Efficient surveillance, outbreak investigation and clinical management of meningitis depends on laboratory confirmation of the causative pathogen from sterile sites, such as cerebrospinal fluid (CSF) and blood. The “gold standard” methods for confirmation of meningitis remain: (i) culture and (ii) polymerase chain reaction (PCR). Culture allows an isolate to be retained for further use; however, this can take at least 24 h and is often more challenging in sub-Saharan Africa due to long transportation time and/or previous antimicrobial treatment (Diallo et al., 2021). PCR is a rapid molecular diagnostic method that enables identification within a few hours. While PCR is sensitive, specific and does not depend on the presence of viable bacteria, it requires expensive equipment, reagents and expertise (Diallo et al., 2021; Feagins et al., 2020; Griffiths et al., 2018). The success of PCR depends on the presence of genomic regions specific to each pathogen. The genetic diversity of meningitis-associated pathogens (Spratt and Maiden, 1999) and their genomic variability indicates there is an on-going need to monitor the effectiveness of existing molecular diagnostic tests targeting pathogens associated with meningitis. It is also important to continue searching for novel genetic targets that are more sensitive and specific than those currently used, while also considering their genetic variability.
A narrative review identified 25 genetic targets used in the detection of H. influenzae, N. meningitidis, S. pneumoniae, and S. agalactiae (Diallo et al., 2021). Testing these targets in vitro is costly and time-consuming. An alternative is to identify suitable targets in silico through bioinformatic analyses using large genome datasets and then confirming their appropriateness in vitro using a reduced panel of bacterial isolates (van Weezep et al., 2019).
The development of high-throughput whole genome sequencing has led to the creation of genome databases such as PubMLST, which contain bacterial population sequence data and provenance metadata for over 100 species and genera (Jolley et al., 2018). This platform receives thousands of yearly submissions including new sequences, allele profiles and isolate records (Jolley et al., 2018). N. meningitidis, S. pneumoniae, H. influenzae, and S. agalactiae WGS deposited in PubMLST include data from healthy carriers, invasive disease cases and other clinical sources.
This study aimed to evaluate in silico published genetic targets used in PCR assays for the detection of N. meningitidis, S. pneumoniae, H. influenzae, and S. agalactiae and identify optimal genetic targets. These were then tested in vitro using a panel of bacteria strains representative of the genera present.
2 Materials and methods
2.1 In silico analyses
2.1.1 Whole genome sequence data (WGS)
In silico analyses were performed on WGS belonging to 70,697 isolate records stored in PubMLST1 (Jolley et al., 2018): 1964 H. influenzae (Hi); 146 WGS from other Haemophilus species (non-Hi); 8,793 S. agalactiae (GBS); 1,181 WGS from other streptococci including 463 S. pneumoniae (non-GBS); 14,401 N. meningitidis (Nm); 10,186 from other Neisseria species (non-Nm); 33,267 of S. pneumoniae (Sp); and 761 WGS from other streptococci including 44 S. agalactiae (non-Sp). The average genome lengths were: H. influenzae, 1.8 Mb, S. agalactiae, 2.2 Mb; N. meningitidis, 2.1 Mb; and S. pneumoniae, 2.1 Mb, with contig lengths averaging 400 bp.
A library of type strain genomes (n = 18,500) was annotated in the PubMLST Ribosomal MLST database2 (Jolley et al., 2012) to provide a comprehensive reference for species identification and facilitate accurate genome comparisons. This extensive library enables standardized comparisons between query genomes and a well-defined set of reference genomes, minimizing ambiguity during species identification. It complements species-specific databases by allowing efficient identification of unknown or mixed-species samples. To facilitate efficient genome comparison, the FastANI program (Jain et al., 2018) was employed to scan query genomes against this library. Initially, the MASH algorithm (Ondov et al., 2016) was utilized to identify the 10 nearest type strains. Subsequently, FastANI was applied to this subset to calculate the Average Nucleotide Identity (ANI) values. The type strain genome exhibiting the highest identity percentage was reported. While it is possible that the query genome may contain two or more species, only the top match was documented.
The in silico PCR, Gene Presence, and Field Breakdown plugins of the BIGSdb software (Jolley and Maiden, 2010) were used to analyze WGS data. In silico PCR was performed with a stringent criterion of no-mismatch for all sets of primers (primer sequences are listed in Supplementary Table 1) and the results were used to calculate the specificity, sensitivity, positive predictive value (PPV) and negative predictive value (NPV) for each assay. The Gene Presence tool, using default settings, was used to detect whole genome sequence data lacking any of the genes examined (genes analyzed are listed in Supplementary Table 1). This was undertaken in each pathogen-specific database using annotated full coding sequences of genes of interest as defined in PubMLST. A low detection rate despite high gene presence was taken as evidence of sequence divergence at primer binding sites. Only targets with consistent detection were retained. The Field Breakdown tool was used to assess results in association with available metadata with a focus on clinical sources, i.e., bacteremia, meningitis, other invasive diseases, carriage or not specified. We defined the best target as one that exhibited the highest sensitivity and specificity, detected in the majority of WGS. All genes defined in the PubMLST database are assigned a unique locus name, starting with “HAEM” for Haemophilus, “NEIS” for Neisseria, “SPNE” for S. pneumoniae, and “SAG” for S. agalactiae, followed by an arbitrary number. Additionally, each locus may also be associated with a common gene name. For example, NEIS1339 corresponds to the sodC gene.
In PubMLST there are multiple fields that can provide information on the bacterial capsule type: the isolates fields including for Nm, “serogroup” and “capsule group,” for GBS, “capsular serotype,” for Hi “serotype” and for Sp “submitted serotype” are filled by the submitter based on confirmatory tests done in their lab, serological or PCR tests. When genomes are available, the fields “genogroup” for Nm or “genotype,” “capsular genotype” for GBS, “genotype” for Hi and “serotype” for Sp indicate the capsule type based on the analysis of the cps genes involved in capsule synthesis, identified through the submitted whole genome sequences. The analysis of serotype/genotype done in the study were based on the genomic typing fields for all four pathogens. Isolates were categorized by PubMLST as non-typeable (NT) when they had non-functional or absent capsule genes (for example, Haemophilus influenzae non-typeable strains where the cps genes are absent). In the occasion were the genes were truncated at the end of a contig, the isolates were classified as undetermined, indicating that serotypes’ assignment could not be done.
2.1.2 Identification of improved targets for H. influenzae detection
The Genome Comparator tool (Jolley and Maiden, 2010) in the PubMLST database3 was used to identify improved genetic targets for the detection of H. influenzae that achieved a sensitivity greater than 96.3% and a specificity greater than 94.7% compared with published genetic targets (Diallo et al., 2021). Genome Comparator analysis was conducted using the 1,898 loci defined in the database with a set core presence threshold of 97%, meaning that only genes present in at least 97% of H. influenzae WGS were considered for further analyses. Pairwise allelic differences between isolates were calculated using default settings: a minimum sequence identity of 70%, a minimum alignment coverage of 50%, and a BLASTN word size of 20. Nucleotide sequences of identified loci were compared to sequences deposited in GenBank using the BLAST program (Altschul et al., 1990) to confirm species specificity. In parallel, a BLAST search of targets identified from the Genome Comparator analysis, was undertaken in a collection of non influenzae Haemophilus genomes (non-Hi), using WGS stored in the Ribosomal Multilocus Sequence Typing (rMLST: see text footnote 2) database (Jolley et al., 2012) to confirm the absence of those targets in non-Hi species. A gene was considered absent if the length of the aligned sequence was less than half of the total length of the sequence of that gene. Primers and probes were designed for each selected target using Primer 3 (Untergasser et al., 2012) with default settings and tested in silico before applying in vitro.
2.2 In vitro analyses
2.2.1 Bacterial strains and growth conditions
Reference strains were obtained from the National Collection of Type Cultures (NCTC). These were H. influenzae NCTC8143, Haemophilus aegyptius NCTC8502, Haemophilus haemolyticus NCTC10659, S. pneumoniae NCTC7465, S. agalactiae NCTC8181, Streptococcus mitis NCTC12261 and Neisseria lactamica NCTC10617 (Supplementary Table 2). H. influenzae NCTC8143, H. aegyptius NCTC8502 and H. haemolyticus NCTC10659 were cultured on Chocolate agar plate with sheep blood with the remaining species cultured on blood agar with sheep blood. Plates were incubated at 37 °C in 5% CO2 for 24 h.
2.2.2 DNA samples
A total of 44 DNA samples were used for real-time PCR assays (Supplementary Table 2). Seven DNA samples were extracted from the reference strains using the Wizard® Genomic DNA Purification Kit, following manufacturer’s instructions (Promega, United States). Twenty-eight additional DNA samples extracted from pure cultures of H. influenzae, S. pneumoniae, S. agalactiae, N. meningitidis, N. gonorrhoeae, N. lactamica, and H. haemolyticus, were kindly donated by Dr. Mignon du Plessis from the National Institute for Communicable Diseases of South Africa for this study. Among these 28 DNA extracts, four were from control strains and the others from specimens isolated from blood cultures, CSF, pleural fluid and patient tissue (Supplementary Table 2). In addition, nine DNA samples extracted from N. lactamica, Neisseria sp., N. meningitidis, and Moraxella catarrhalis isolates from a collection at Centre Suisse de Recherches Scientifiques in Côte d’Ivoire (CSRS) were used. These have been cultured from oropharyngeal swabs and saliva samples collected from healthy carriers as part of a carriage study conducted in a cohort of school children in Côte d’Ivoire (Missa et al., 2024) and their identity confirmed by WGS (Data not shown).
2.2.3 Real-time PCR
Real-time PCR amplifications were carried out targeting the two genetic determinants most prevalent in WGS and exhibiting the highest sensitivities and specificities with at least 95% identified in in silico analyses (top two best target genes). In cases where the PCR did not work, a third gene, was tested (Table 1). For H. influenzae, two additional high-scoring genes were also tested. Assays were performed on a CFX96 Touch™ Real-Time PCR Detection system (Bio-Rad) using the TaqMan® Gene Expression Master Mix (Applied Biosystems). The reaction mixture consisted of 7.5 μL of 2× Master Mix, 0.5 μM of each primer (forward and reverse), 0.5 μM of probe (Table 1), template DNA (2 μL) and UltraPure DNase/RNase-Free Distilled Water for a final volume of 15 μL. Positive controls and no-template control were included in each experiment. The cycling parameters consisted of 2 min at 50 °C, 10 min at 95 °C, 45 cycles of 95 °C for 15 s and 60 °C for 1 min, and then a holding stage at 4 °C. Samples with Ct values below 35 were considered positive, those above 40 negative, and values between 35 and 40 were classified as equivocal, unless otherwise specified. Equivocal samples were diluted 1:10 to reduce potential inhibitors and retested (Pouladfar et al., 2022).
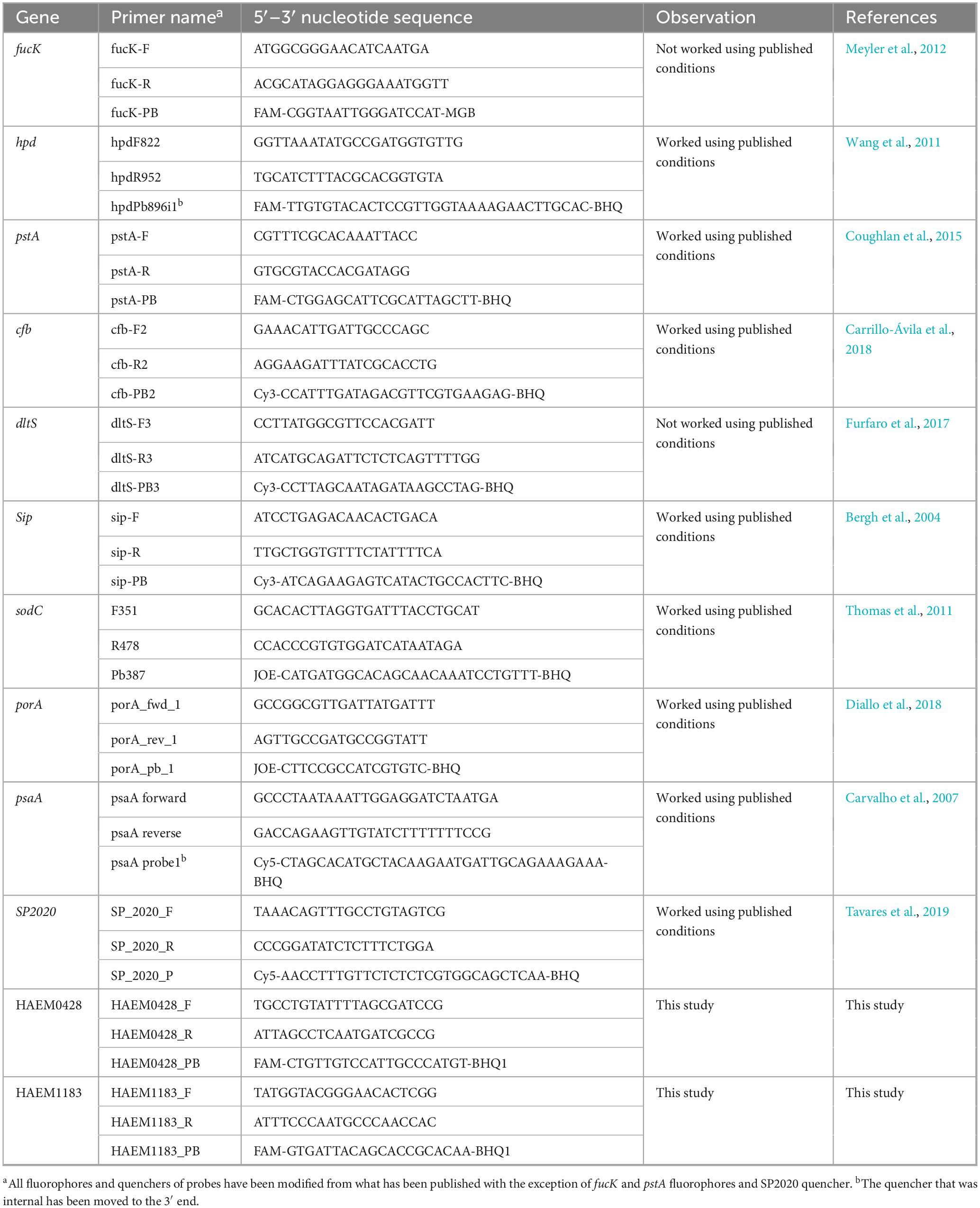
Table 1. Primer sequences used for real-time polymerase chain reaction (PCR).
2.3 Statistical analysis
The sensitivity and specificity of assays were determined using the following formula:
and
Positive predictive value (PPV) and negative predictive value (NPV) were determined according to the following formula:
and
3 Results
3.1 Geographic and temporal distribution of datasets
H. influenzae sequences dated from 1941 to 2020 and originated from Europe (917/1964, 46.7%), North America (805/1964, 41%), Oceania (115/1964, 5.9%), Africa (83/1964, 4.2%), Asia (32/1964, 1.6%), Unknown (10/1964, 0.5%), and South America (2/1964, 0.1%). S. agalactiae sequences dated from 1953 to 2018 and originated from North America (4,881/8793, 55.5%), Europe (1,652/8793, 18.8%), Africa (449/8793, 16.5%), Unknow (389/8793, 4.4%), Asia (239/8793, 2.7%), Oceania (174/8793, 2%), and South America (9/8793, 0.1%). N. meningitidis sequences dated from 1915 to 2021 and originated from Europe (9,872/14,401, 68.5%), North America (1,927/14,401, 13.4%), Africa (1,350/14,401, 9.4%), Asia (617/14,401, 4.3%), South America (244/14,401, 1.7%), Oceania (377/14,401, 2.6%) and Unknown (14/14,401, 0.1%). S. pneumoniae sequences dated from 1916 to 2018 and originated from Africa (10,754/33,267, 32.3%), Europe (8,274/33,267, 24.9%), Asia (7,621/33,267, 22.9%), North America (4,785/33,267, 14.4%), South America (1,390/33,267, 4.2%), Unknown (343/33,267, 1%) and Oceania (100/33,267, 0.3%). ANI analysis confirmed all isolates had values greater than 95% (Supplementary Table 5).
3.2 In silico analysis: gene presence, sensitivity and specificity of existing targets
A total of five genes were identified and tested for their presence in N. meningitidis WGS [ctrA (NEIS0055), sodC (NEIS1339), crgA (NEIS0362), nspA (NEIS0612), and porA (NEIS1364)]. Gene presence ranged from 98.8% (ctrA) to 100% (nspA), with primer/probe sensitivities ranging from 0.5% (crgA) to 99.7% (sodC) and specificities from 99.4% (sodC) to 100% (crgA). Overall, the best N. meningitidis candidate primer sequences targeted sodC with a sensitivity of 99.7%, specificity of 99.4%, PPV of 99.6% and NPV of 99.6% closely followed by porA (sensitivity: 99.1%, specificity: 99.9%, PPV: 99.8% and NPV: 98.8%) (Table 2).
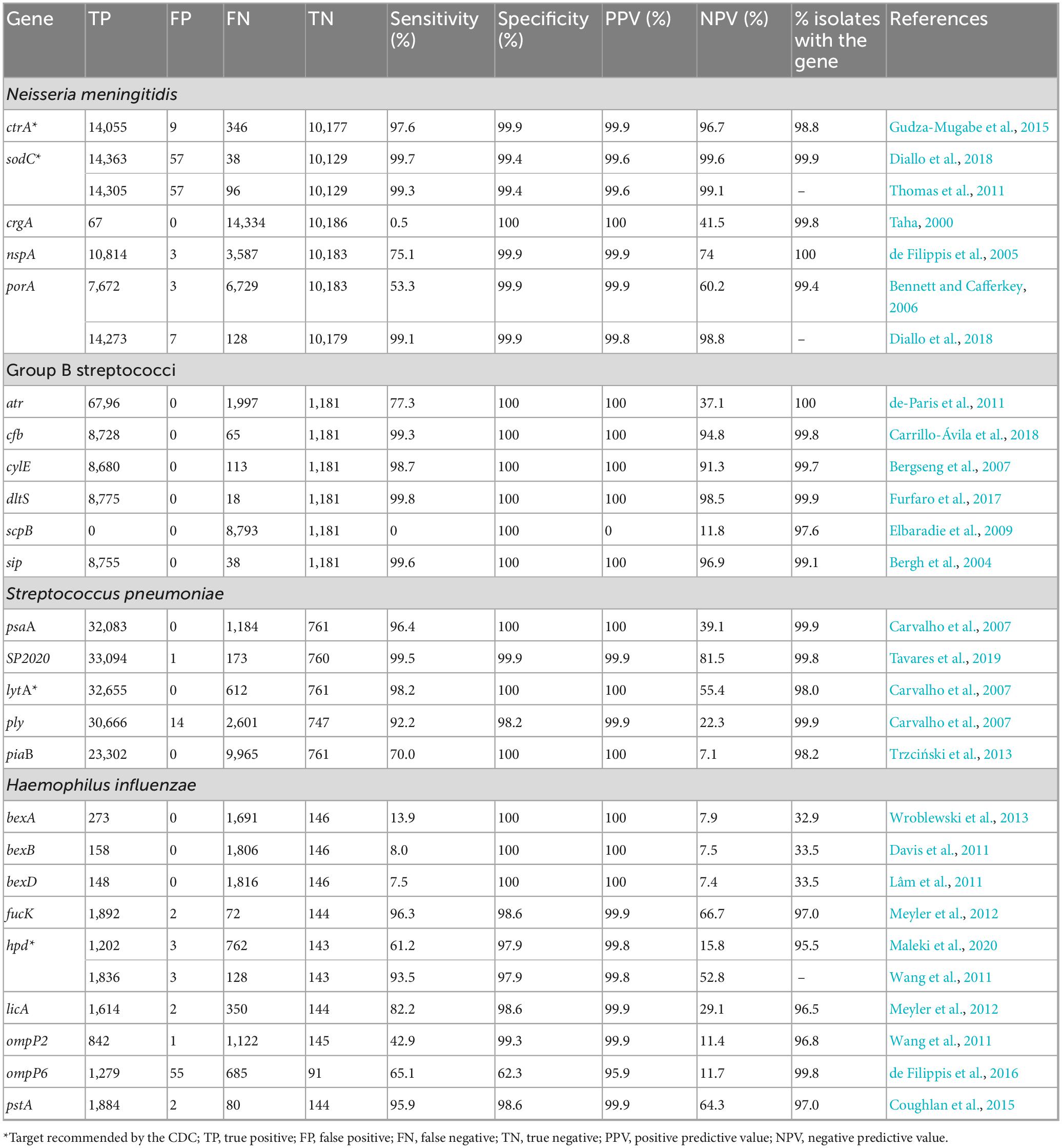
Table 2. In silico deduced specificity, sensitivity for polymerase chain reaction (PCR) primers and gene presence values for complete coding sequences.
A total of six genes were identified and tested for their presence in S. agalactiae WGS [atr, cfb (SAG2043), cylE (SAG0669), dltS (SAG1791), scpB (SAG1236) and sip (SAG0032)]. Gene presence ranged from 97.6% (scpB) to 100% (atr); primer sensitivities ranged from 0% (scpB) to 99.8% (dltS); specificities and PPV were 100% for all targets tested except scpB which had PPV of 0%. NPV ranged from 11.8% (scpB) to 98.5% (dltS). The best S. agalactiae primer sequences targeted dltS (with a sensitivity of 99.8%, a specificity of 100%, PPV of 100% and NPV of 98.5%) followed by sip (with a sensitivity of 99.6%, specificity of 100%, PPV of 100% and NPV of 96.9%) and cfb (sensitivity of 99.3%, specificity of 100%; PPV of 100% and NPV of 94.8%) (Table 2).
A total of five genes were identified and tested for their presence in S. pneumoniae WGS [psaA (SPNE00983), SP2020, lytA, ply (SPNE01149) and piaB]. Gene presence ranged from 98.0% (lytA) to 99.9% (psaA and ply), with primer sensitivities ranging from 70.0% (piaB) to 99.5% (SP2020) and specificities from 98.2% (ply) to 100% (psaA, lytA and piaB). PPVs were 99.9% (ply and SP2020) and 100% (psaA, lytA, and piaB) with NPVs ranging from 7.1% (piaB) to 81.5% (SP2020). In these analyses, the best candidate primers targeted SP2020 (with a sensitivity of 99.5%, specificity of 99.9%, PPV of 99.9% and NPV of 81.5%) followed by lytA (with a sensitivity of 98.2%, specificity of 100%, PPV of 100% and NPV of 55.4%) and psaA (with a sensitivity of 96.4%, specificity of 100%, PPV of 100% and NPV of 39.1%) (Table 2).
A total of nine genes were identified and tested for their presence in H. influenzae WGS [bexA (HAEM1156), bexB (HAEM1155), bexD (HAEM1153), fucK, hpd (HAEM0810), licA (HAEM1656), ompP2 (HAEM0191), ompP6 (HAEM0484) and pstA (HAEM1519)]. Gene presence ranged from 32.9% (bexA) to 99.8% (ompP6), with primer sensitivities from 7.5% (bexD) to 96.3% (fucK) and specificities from 62.3% (ompP6) to 100% (bexA, bexB, bexD). PPVs ranged from 95.9% (ompP6) to 100% (bexA, bexB, bexD) and NPVs from 7.4% (bexD) to 66.7% (fucK). Overall, the best candidate genetic target for molecular detection of H. influenzae was fucK (with a sensitivity of 96.3%, specificity of 98.6%, PPV of 99.9% and NPV of 66.7%), followed by pstA (with a sensitivity of 95.9%, specificity of 98.6%, PPV of 99.9% and NPV of 64.3%) and hpd (with a sensitivity of 93.5%, specificity of 97.9%, PPV of 99.8% and NPV of 52.8%) (Table 2).
3.3 Undetected targets following in silico PCR analysis
The gene sodC did not detect 38/14401 (0.3%) of the N. meningitidis tested. These genomes were either genogroup B, C, W and non-groupable (NG) isolates (7/38, 18.4% each), or genogroup Y (6/38, 15.8%), capsule null (cnl) (1/38, 2.6%) or with an undetermined genogroup (3/38, 7.9%) (Figure 1a). The porA gene did not detect 128/14401 (0.9%) strains. These were genogroup B isolates (49/128, 38.3%), genogroup C (47/128, 36.7%) or genogroup W (18/128, 14.1%) (Figure 1b).
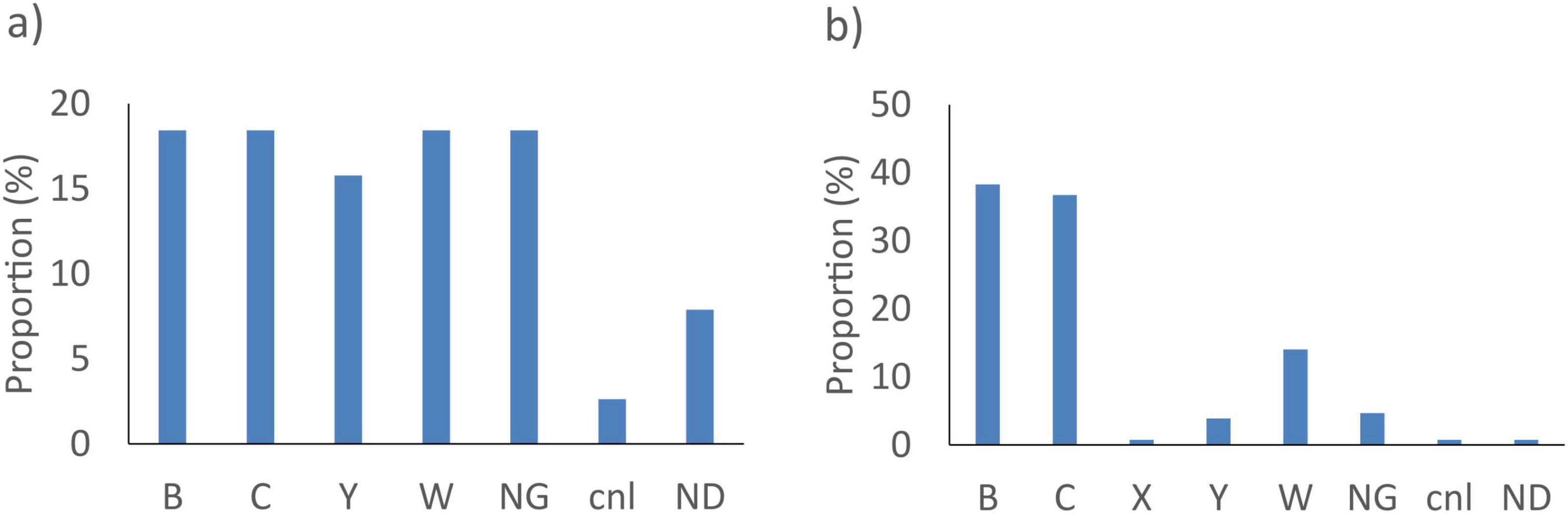
Figure 1. Proportion of genogroup of N. meningitis not detected by the best targets: (a) sodC and (b) porA in silico. NG, non-groupable; cnl, capsule null locus; ND, not determined.
The dltS gene did not detect 18/8793 (0.2%) S. agalactiae genomes. These were isolates with an undetermined serotype (8/18, 44.4%) (Figure 2b). The sip gene did not detect 38/8793 (0.4%) of the S. agalactiae tested. They were predominantly from serotype III isolates (14/38, 36.8%) (Figure 2a). As for cfb, it did not detect 65/8793 (0.7%) S. agalactiae. These isolates had undetermined serotypes (33/65, 50.8%) (Figure 2c).
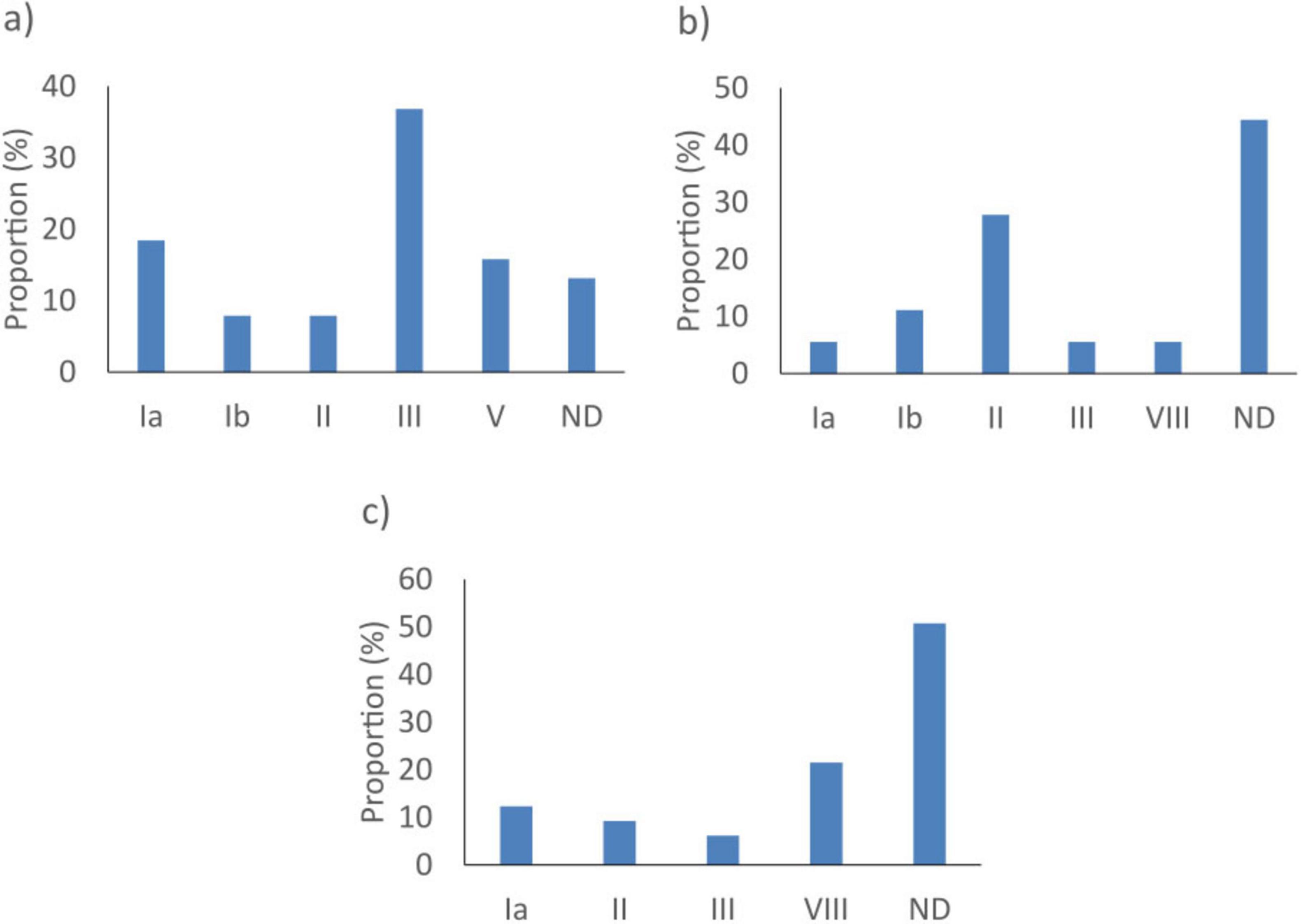
Figure 2. Proportion of serotypes of Streptococcus agalactiae not detected by the best targets: (a) sip, (b) dltS, and (c) cfb in silico. ND, not determined.
SP2020 did not detect 173/33267 (0.5%) of the S. pneumoniae tested. These were predominantly serotypes 6A (57/173, 32.9%), 19F (40/173, 23.1%) and 31 (25/173, 14.5%) (Figure 3a). The lytA gene did not detect 612/33267 (1.8%) of the samples tested. These were serotypes 14 (112/173, 18.3%), 23F (77/173, 12.6%) and non-typeable (52/173, 8.5%) and undetermined serotype (58/173, 9.5%) isolates (Figure 3b). The psaA gene did not detect 1184/33267 (3.6%) bacterial genomes. These were predominantly from undetermined serotypes (252/1184, 21.3%), serotypes 22F (239/1184, 20.2%) and 1 (150/1184, 12.7%) (Figure 3c).
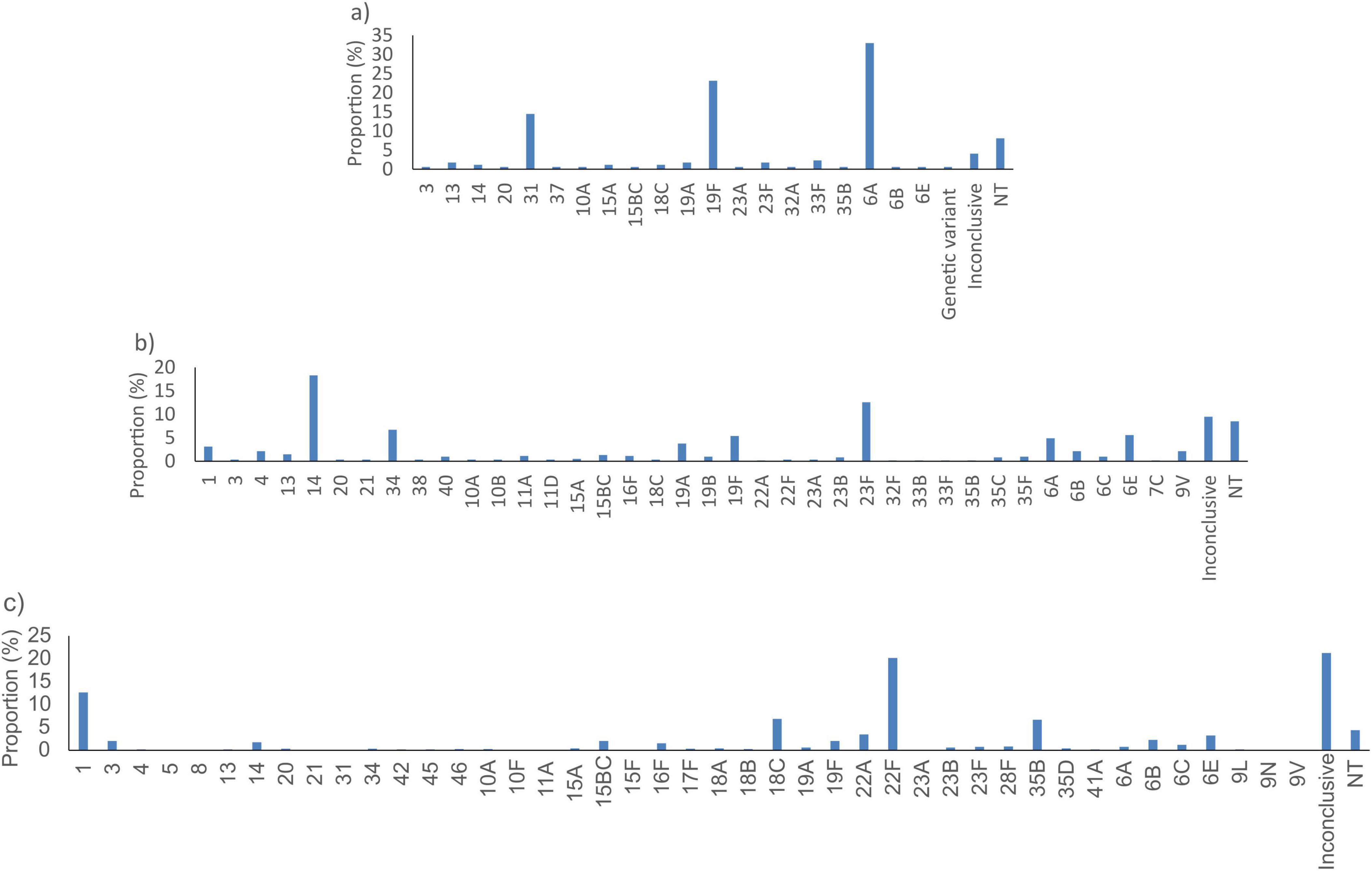
Figure 3. Proportion of serotypes of S. pneumoniae not detected by the best targets: (a) SP2020, (b) lytA, and (c) psaA in silico. NT, non-typeable.
The fucK gene did not detect 72/1964 (3.7%) H. influenzae. These genomes were predominantly from isolates with serotype e (36/72, 50%) and Non-typeable serotype (NT) (35/72, 48.6%) (Figure 4a). The pstA gene did not detect 80/1964 (4.1%) bacterial genomes. These were mainly from isolates with Non-typeable serotype (NT) (75/80, 93.8%) (Figure 4c). The hpd gene did not detect 128/1964 (6.5%) bacterial genomes. These genomes were predominantly from isolates with Non-typeable serotype (NT) (111/128, 86.7%) (Figure 4b).
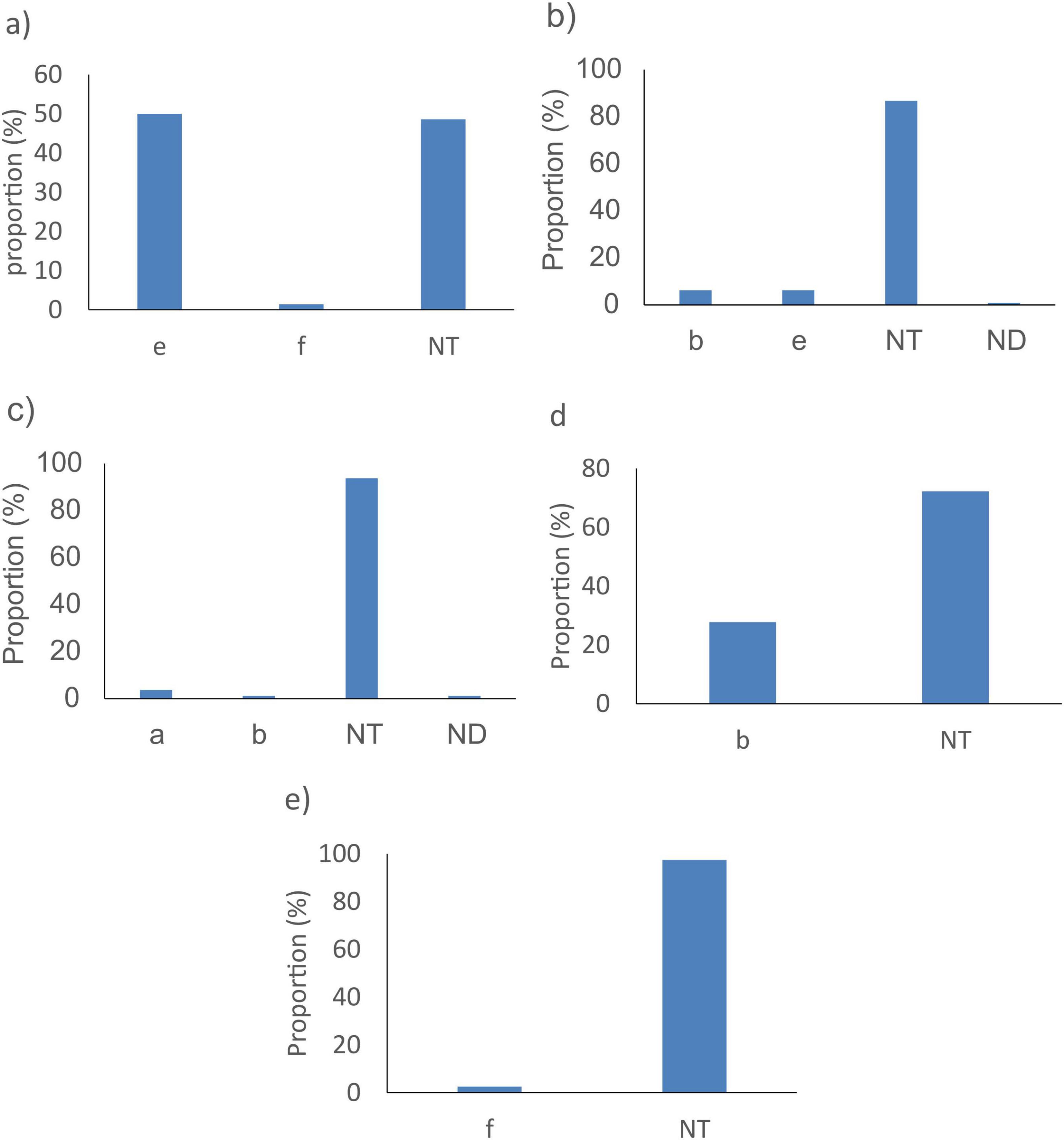
Figure 4. Proportion of serotypes of H. influenzae not detected by the best targets: (a) fucK, (b) hpd, (c) pstA, (d) HAEM0428, and (e) HAEM1183 in silico. NT, non-typeable, ND, not determined.
3.4 Novel targets for detection of H. influenzae
Given the sub-optimal in silico performance observed for all published H. influenzae targets, additional analyses were performed to identify better targets. Comparative genome analyses identified 327 loci that were present in 97% of the H. influenzae WGS investigated of which four (HAEM0428, HAEM1179, HAEM1181, and HAEM1183) were absent or had significantly lower presence in a dataset of 152 other Haemophilus species (Supplementary Table 3). HAEM0428 (ICMT gene) encodes protein-S-isoprenylcysteine methyltransferase, HAEM1179 (dmsD) encodes the Tat proofreading chaperone, HAEM1181 (dmsC) encodes Anaerobic dimethyl sulfoxide reductase chain C and HAEM1183 (dmsA) encodes an anaerobic dimethyl sulfoxide reductase chain A. Of these four genes, HAEM0428 and HAEM1183 showed better or identical sensitivity and specificities as fucK. Indeed, compared to fucK, HAEM0428 showed similar sensitivity (96.3% vs. 96.3%) but lower specificity (95.9% vs. 98.6%). In contrast, HAEM1183 showed a better sensitivity (98.0%) and specificity (100%) than fucK and HAEM0428 (Table 3). In silico PCR analyses revealed that HAEM0428 did not detect 72/1964 (3.7%) H. influenzae. These isolates were either with Non-typeable serotype (NT) (52/72, 72.2%) or serotype b isolates (20/72, 27.8%) (Figure 4d). HAEM1183 did not detect 39/1964 (2%) H. influenzae. These sequences were in isolates from serotype NT (38/39, 97.4%) (Figure 4e).
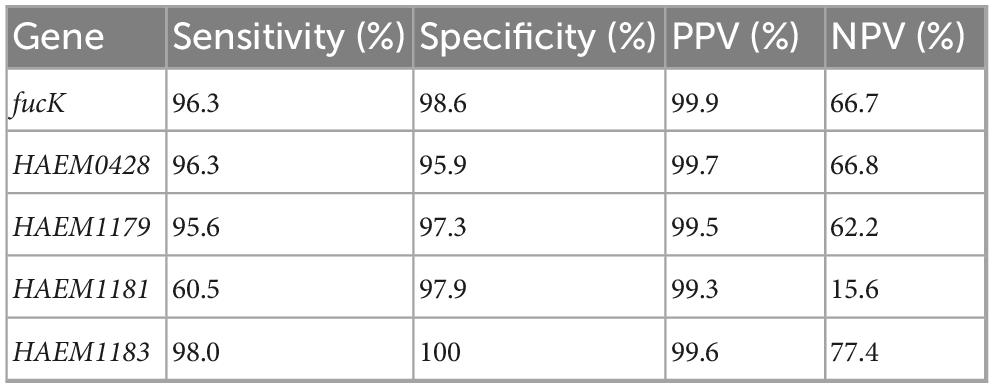
Table 3. Specificity and sensitivity of new assays and fucK for detection of H. influenzae obtained in silico.
3.5 Efficiency of the in silico assays by reported isolation source clinical sources
According to the available provenance and phenotype information, 12,241/14,401 (85%) N. meningitidis WGS originated from invasive meningococcal disease (IMD), 291/14,401 (2%) were from asymptomatic carriage and 1,869/14,401 (13%) had no information on their isolation source (Figure 5A). The best target genes showed high sensitivity for WGS associated with IMD (92.3%–99.9%) and, specifically, from meningitis cases (94.7%–100%) (Table 4). Indeed, sodC detected 12,207/12,241 (99.7%) IMD N. meningitidis WGS with 540/542 (99.6%) associated with meningitis only. The porA gene detected 12,135/12,241 (99.1%) IMD WGS with 539/542 (99.4%) from meningitis cases only.
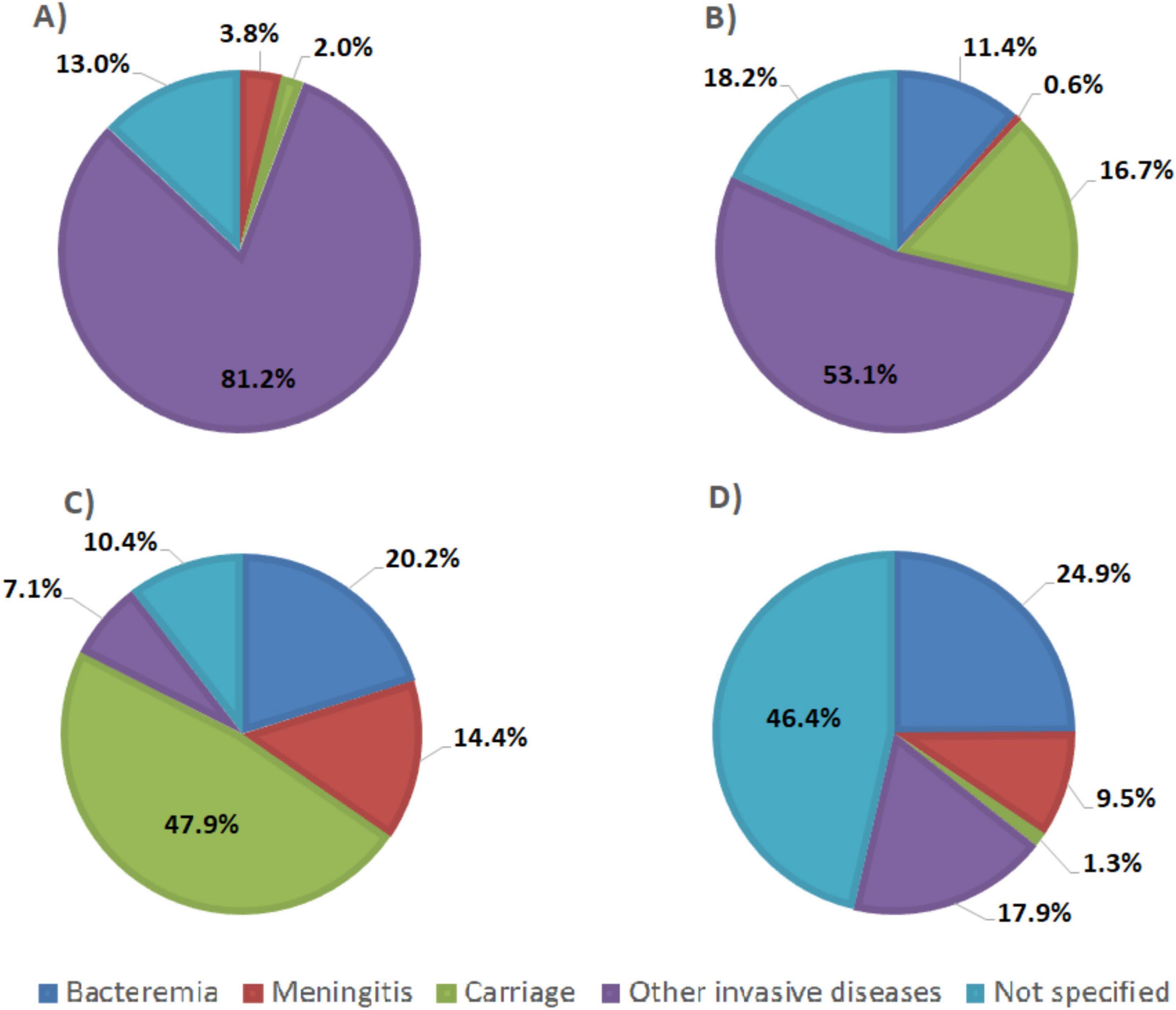
Figure 5. Pie chart representing clinical sources of genomic sequences of (A) N. meningitidis, (B) Streptococcus agalactiae, (C) S. pneumoniae, and (D) H. influenzae from PubMLST database.
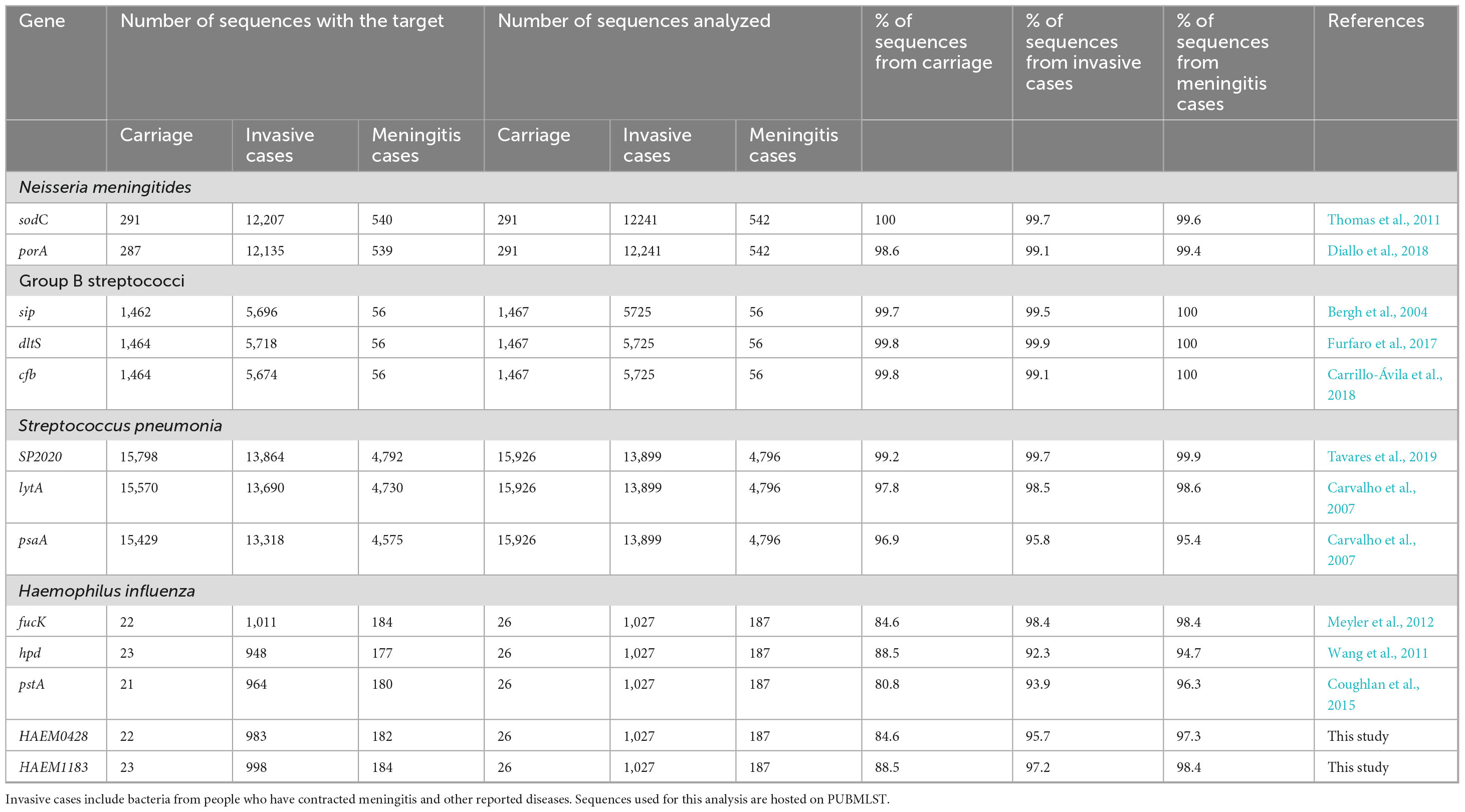
Table 4. Proportion of positives reported as coming from carriage and invasive bacteria including meningitis cases detected by the best targets in silico.
A total of 5,725/8,793 (65.1%) S. agalactiae WGS were from invasive disease (Figure 5B), 1,467/8,793 (16.7%) were from carriage and 1,601/8,793 (18.2%) were from unspecified sources (Figure 5B). The genetic target, dltS, detected 5,718/5,725 (99.9%) of S. agalactiae WGS associated with invasive disease and 56/56 (100%) from meningitis cases. The gene sip detected 5,696/5,725 (99.5%) WGS associated with invasive disease and 56/56 (100%) from meningitis cases. The cfb gene detected 5,674/5,725 (99.1%) invasive disease WGS and 56/56 (100%) from meningitis cases (Table 4).
A total of 13,899/33,267 (41.8%) of S. pneumoniae genomes were associated with invasive disease, 15,926/33,267 (47.9%) from carriage and 3,442/33,267 (10.3%) from unspecified sources (Figure 5C). SP2020 detected 13,864/13,899 (99.7%) of S. pneumoniae isolated from invasive cases and 4,792/4,796 (99.9%) from meningitis cases while lytA detected 13,690/13,899 (98.5%) of sequences coming from invasive cases and 4,730/4,796 (98.6%) from meningitis cases. The psaA gene detected 13,318/13,899 (95.8%) of sequences coming from invasive cases and 4,575/4,796 (95.4%) from meningitis cases (Table 4).
H. influenzae genome sequence database included 1,027/1,964 (52.3%) sequences came from invasive diseases, 28/1,964 (1.3%) from carriage and 911/1,964 (46.4%) from unspecified sources (Figure 5D). The fucK gene detected 1,011/1,027 (98.4%) of H. influenzae from invasive cases and 184/187 (98.4%) from meningitis cases whereas HAEM1183 detected 998/1,027 (97.2%) of sequences from invasive cases and 184/187 (98.4%) from meningitis cases, and HAEM 0428 detected 983/1,027 (95.7%) of sequences from invasive cases and 182/187 (97.3%) from meningitis cases. The pstA gene detected 964/1,027 (93.9%) of sequences from invasive cases and 180/187 (96.3%) from meningitis cases. The hpd gene detected 948/1,027 (92.3%) of sequences from invasive cases and 177/187 (94.7%) from meningitis cases (Table 4).
Sensitivity of targets in sequences from carriage isolates was 291/291 (100%) for sodC and 287/291 (98.6%) for porA in N. meningitidis; 1,462/1,467 (99.7%) for sip, 1,464/1,467 (99.8%) for dltS and 1,464/1,467 (99.8%) for cfb in S. agalactiae;15,798/15,926 (99.2%) for SP2020, 15,570/15,926 (97.8%) for lytA and 15,429/15,926 (96.9%) for psaA in S. pneumoniae; and 22/26 (84.6%) for fucK, 23/26 (88.5%) for hpd, 21/26 (80.8%) for pstA, 22/26 (84.6%) for HAEM0428 and 23/26 (88.5%) for HAEM 1183 in H. influenzae (Table 4).
3.6 In vitro analyses: performance of real-time PCR assays
The genes sodC and porA were tested for their ability to detect N. meningitidis. The two genes showed a sensitivity of 100%, a specificity of 91.7%, a PPV of 72.7% and a NPV of 100% (Table 5 and Supplementary Table 4).
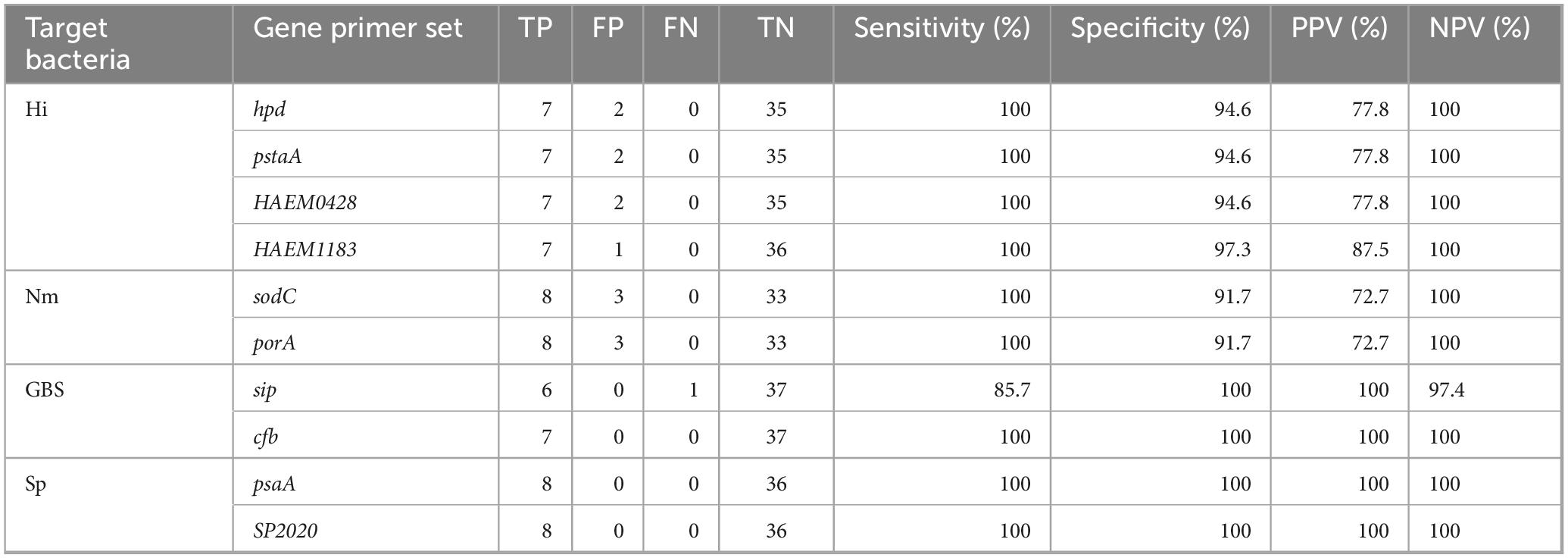
Table 5. In vitro results for real-time polymerase chain reaction (PCR) assays.
The gene dltS, one of the top S. agalactiae in silico targets did not perform well in our study using published conditions (Furfaro et al., 2017). This target was therefore not considered further. cfb and sip had a specificity of 100% and a PPV of 100% each. In addition, cfb showed a sensitivity and a NPV of 100% while sip showed a sensitivity of 85.7% and a NPV of 97.4% (Table 5 and Supplementary Table 4).
The lytA gene target was not tested in vitro, due to the presence of lytA homologues in pneumococcal prophages (Carvalho et al., 2007). Therefore, psaA and SP2020 were tested for their ability to detect S. pneumoniae. These genes had a sensitivity and specificity of 100%. Also, the PPV and NPV of the real-time PCR tests were 100% for psaA and SP2020 (Table 5 and Supplementary Table 4).
The gene, fucK, one of the top in silico H. influenzae genetic determinants did not work using the reaction conditions described in the original paper (Meyler et al., 2012). This target was therefore not considered further. The remaining targets, hpd, pstA, HAEM0428 and HAEM1183, showed a sensitivity of 100% and a NPV of 100% each. The specificity of HAEM1183 was 97.3%, hpd, pstA and HAEM0428 were identical (94.6%). PPV was 87.5% for HAEM1183, 77.8% for hpd, pstA and HAEM0428 (Table 5 and Supplementary Table 4).
4 Discussion
Bacterial meningitis remains a major public health threat, particularly in sub-Saharan Africa due to unpredictable epidemics and the urgent need to improve diagnostic methods for the rapid and accurate detection of the causative pathogens. This study sought to address these challenges using in silico approaches with PubMLST, a large nucleotide sequence database, providing a preliminary assessment of the specificity and sensitivity of diagnostic targets to guide in vitro validation tests. This approach enables a preliminary assessment of the specificity and sensitivity of diagnostic targets before extensive laboratory testing, significantly reducing the time, effort and costs associated with assay development (Santa Lucia et al., 2020; van Weezep et al., 2019). Promising assays identified in silico can then be validated by various laboratories, including those led by citizen scientists, using their available local strains. This collective effort increases the variety of isolates tested and reduces issues related to sample shipments. Additionally, using PubMLST to both select targets and evaluate their performance for H. influenzae may introduce a risk of overfitting, since the same dataset informs both steps. PubMLST is the largest and most diverse publicly available database for H. influenza genomes. However, it remains important to validate promising targets using independent genomic datasets or clinical isolates to ensure broader applicability and robustness.
Based on our findings, we recommend using sodC for N. meningitidis, cfb for S. agalactiae, SP2020 for S. pneumoniae, and dmsA for H. influenzae due to their high sensitivity, specificity, and consistent prevalence in WGS data. The gene sodC, also recommended by WHO/CDC, is highly specific and sensitive for detecting meningococci (Thomas et al., 2011). This gene, which encodes Cu-Zn superoxide dismutase, is ubiquitous in N. meningitidis and less likely to undergo antigenic variation due to selective pressure (Thomas et al., 2011). This study also revealed the equivalent efficacy of porA (an outer membrane porin). However, rectal and pharyngeal N. gonorrhoeae isolates from Australian and Swedish patients have been found to harbor an N. meningitidis porA sequence, presumably acquired through horizontal genetic exchange and recombination (Golparian et al., 2012; Whiley et al., 2011). Thus, caution is advised when using porA as a target, although detecting N. gonorrhoeae in invasive meningococcal cases would be unlikely.
The gene cfb, which encodes the extracellular pore-forming toxin (CAMP factor), has been demonstrated as an effective target for GBS detection (Carrillo-Ávila et al., 2018). It is noteworthy that other rtPCR tests such as (i) the Becton Dickinson MAX GBS assay; (ii) the ARIES GBS assay from Luminex Corporation; and (iii) the Xpert GBS LB assay produced by Cepheid Inc. also prioritize cfb as the primary target gene (Diallo et al., 2021). However, there is no WHO/CDC recommendation for this gene as GBS is not routinely tested in surveillance.
Despite lytA (the major autolysin of pneumococcus) being widely recommended by WHO/CDC and routinely used in surveillance of S. pneumoniae (Satzke et al., 2013), concerns arise due to its homologs in closely related Streptococcus species (Tavares et al., 2019), potentially increasing the false positivity rate (Ganaie et al., 2021; Llull et al., 2006; Simões et al., 2016). Furthermore, the work of Martín-Galiano and García (2021) revealed that pneumococcal prophages harbor lytA-like genes homologous to S. pneumoniae lytA, and that there were recombination events between the pneumococcal and phage lytA homologs, further questioning its reliability. In contrast, SP2020 (a putative transcriptional regulator gene of the GntR family) has been shown to be a better target than lytA for S. pneumoniae diagnosis, consistent with the findings of Tavares et al. (2019). In this study, lytA was included in the in silico analysis to enable direct comparison with SP2020 due to its common use. Given its limitations, lytA was not evaluated further in vitro. The results support the use of SP2020 as a preferred diagnostic target.
The gene fucK (gene encoding fuculokinase) demonstrated the best overall performance in silico for the identification of H. influenzae. However, this target gene performed poorly in comparison to the ones used for the three other pathogens, suggesting a need for improvement of H. influenzae diagnostic determinants. Furthermore, more than 1% of non-Hi sequences had the fucK gene and this gene did not detect all H. influenzae, as reported by de Gier et al. (2015). We were unable to amplify fucK in vitro using the published conditions (Meyler et al., 2012). Therefore, although in silico analyses suggest that it is a promising target, we cannot confidently assert that it is the best target without experimental validation. It would thus be beneficial to optimize the existing primers or design new ones. In contrast, dmsA performed better than hpd, the gene recommended by WHO/CDC (Wang et al., 2011), suggesting its efficacy for H. influenzae identification. The dmsA gene is required for fitness of H. influenzae (Dhouib et al., 2021) and appears to be the most efficient test for the identification of H. influenzae in our study. Nasreen et al. (2024) showed that the DmsABC complex protects H. influenzae against oxidative stress, particularly from host-derived hypochlorite. Expression of dmsA increases under such stress, and its deletion impairs bacterial survival and intracellular persistence. These findings suggest that dmsA contributes to both stress adaptation and host interaction.
The PubMLST database includes isolates from various sources, some known to be from asymptomatic carriage and others from unspecified sources. Our assays are able to detect isolates from these different sources. Additional analysis was performed to evaluate the efficacy of the targets for bacteria isolated from cases of invasive disease and/or meningitis. All four targets exhibited high sensitivity for the target bacteria from invasive disease (97.2%–99.7%) and for bacteria from meningitis specimens (98.4%–100%). Further evaluation with more specimens from invasive diseases from diverse geographical regions, which was beyond the scope of the present work, would be useful to confirm the results presented here. Although some bacterial variants are currently more prevalent than others, the inclusion of non-invasive isolates in the different test panels remains important. Indeed, these strains have no intrinsic factors that prevent them from causing disease, and also need to be monitored because they can acquire virulent genes especially as they are exposed to new vaccine pressures such as for N. meningitidis and potentially for S. agalactiae. This has been shown with the non-virulent N. meningitidis carriage strain that acquired both a serogroup C capsule and the filamentous bacteriophage MDAΦ, which has been shown to enhance colonization of nasopharyngeal epithelial cells, increasing virulence, and leading to epidemics first reported in 2013 in the Tambuwal area of Nigeria, with the strain spreading to different regions of Niger (Brynildsrud et al., 2018). Similarly, in S. pneumoniae, non-encapsulated strains (NESp) typically cause non-invasive pneumococcal diseases. However, NESp strains have recently been identified as causative agents of invasive disease. Bradshaw et al. (2020) demonstrated that NESp are highly transformable, capable of acquiring large DNA segments that increase their persistence and virulence during invasive disease. Group B streptococci (GBS), commensals of the vagina and gastrointestinal tract, can become invasive, particularly in newborns, through GBS adaptation to environmental changes under the control of the CovRS two-component regulatory system (Patras et al., 2013). Genomic mutations, including those affecting capsule synthesis regulator (CovR), also appear to influence the transition of GBS from a commensal state to a pathogen and its ability to persist in mothers before and after delivery (Shabayek and Spellerberg, 2018). The assays failed to detect the sequences of the target genes analyzed in isolates belonging to certain serotypes or capsules in silico due to complete or partial deletion of these genes (Khatami et al., 2018; Whyte et al., 2020) in some strains and due to the stringent conditions applied (no mismatches allowed in the primers). This issue needs to be monitored in real life, as some missed genotypes, such as serogroups B and C in N. meningitidis, serotypes Ia, Ib, III, and V in S. agalactiae, serotypes 4, 14, 7F, 9V, and 18C in S. pneumoniae, and non-typeable H. influenzae (NTHi), can cause disease (Lambertsen et al., 2010; Levy et al., 2010; Moreno et al., 2020; Resman et al., 2011; Sleeman et al., 2006; Tazi et al., 2011).
Our in silico analysis was conducted using WGS databases, which only include culturable bacteria. All available genome sequences are derived from cultured bacteria, which can limit the diversity captured, especially for strains that are difficult to grow. This approach may introduce bias as the need to culture pathogens for whole genome sequencing (WGS) limits the representativeness of the data. Primer design depends on the available sequence data, which is currently mostly from cultured bacteria. As a result, the diversity of uncultivable strains may be overlooked and PCR may lack the sensitivity to detect them. One solution to improve representativeness would be to use culture-independent sequencing methods, such as metagenomics, which can explore a wider bacterial diversity without relying on specific primers. These allow detection of both culturable and unculturable strains and may reveal additional genetic targets for more sensitive molecular diagnostics. Furthermore, according to the WHO report, laboratory data from weeks 1 to 30 of 2024 (January 1 to July 28) indicated that 4,926 cerebrospinal fluid (CSF) samples tested by PCR out of 7,468 suspected cases were negative, despite strong clinical suspicions of meningitis (World Health Organization [WHO], 2024). These results suggest either the presence of other pathogens that current tests do not detect, or a lack of sensitivity in the current diagnostic methods. In this context, our study is particularly relevant. By improving diagnostic tools, we aim to enhance the detection capacity of the four most virulent meningitis pathogens. However, this approach must be expanded to identify other genes for diagnosing additional pathogens responsible for meningitis, such as viruses or other infectious agents known to be difficult to detect with current methods. WHO data highlights the critical need to develop more sensitive diagnostic tests adapted to the contexts of low- and middle-income countries (LMICs). Given the healthcare challenges posed by the burden of infectious diseases in these regions, implementing tools suited to local conditions is essential.
In conclusion, the genes sodC, cfb, SP2020, and dmsA have allowed for the in silico identification of N. meningitidis, S. agalactiae, S. pneumoniae, and H. influenzae, respectively, from various clinical sources, including invasive cases, and specifically in cases reported clinically as meningitis, and have shown promising results in vitro despite the limited number of samples tested. The diagnostic measures should nevertheless be interpreted with caution given the absence of confidence intervals and formal statistical testing. These genes thus have the potential to significantly enhance the precision of molecular diagnostics for meningitis. However, laboratory confirmation with a larger number of samples, including patient samples such as CSF or blood, remain necessary. Additionally, the performance of these targets in cases of co-infection or samples with low pathogen loads was not assessed in this study due to limited data. Future work should evaluate diagnostic accuracy under these conditions to ensure reliability in diverse clinical scenarios. The in silico approach, utilizing extensive WGS databases such as PubMLST, combined with the in vitro approach, enables efficient and cost-effective preliminary evaluation of diagnostic targets. This can be particularly beneficial in situations characterized by variability in etiological agents and potential changes in their relative prevalence due to collective immunity induced by vaccines, especially in resource-constrained environments.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
SA: Conceptualization, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. KD: Conceptualization, Investigation, Methodology, Project Administration, Resources, Supervision, Validation, Writing – review & editing. JT: Formal analysis, Investigation, Writing – original draft. NN: Formal analysis, Investigation, Writing – original draft. VF: Formal analysis, Investigation, Writing – original draft. GM: Formal analysis, Investigation, Writing – original draft. AA: Formal analysis, Investigation, Writing – original draft. RJ: Formal analysis, Investigation, Writing – original draft. HM: Formal analysis, Investigation, Writing – original draft. KJ: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Validation, Writing – review & editing. JB: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Validation, Writing – review & editing. OH: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Validation, Writing – review & editing. MM: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Department of Health and Social Care using UK Aid funding as part of the UK Vaccine Network and is managed by NIHR. This work made use of the PubMLST database and analysis tools developed by Keith Jolley. James Bray and Keith Jolley are funded by a Wellcome Trust Biomedical Resources Grant (218205/Z/19/Z): “PubMLST: Disseminating and exploiting bacterial diversity data for public health benefit.” Diallo Kanny was supported by a Crick African Network Fellowship and the DELTAS Africa Initiative (Afrique One-ASPIRE/DEL-15-008).
Acknowledgments
We thank Mignon du Plessis, National Institute for Communicable Diseases of South Africa, for the donation of DNA samples. We also thank the Research Support Unit (UAR) of Centre Suisse de Recherches Scientifiques in Cote d’Ivoire, for its contribution to improving this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed in this publication are those of the author(s) and not necessarily those of the Department of Health and Social Care.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1655490/full#supplementary-material
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bennett, D. E., and Cafferkey, M. T. (2006). Consecutive use of two multiplex PCR-based assays for simultaneous identification and determination of capsular status of nine common Neisseria meningitidis serogroups associated with invasive disease. J. Clin. Microbiol. 44, 1127–1131. doi: 10.1128/JCM.44.3.1127-1131.2006
Bergh, K., Stoelhaug, A., Loeseth, K., and Bevanger, L. (2004). Detection of group B streptococci (GBS) in vaginal swabs using real-time PCR with TaqMan probe hybridization. Indian J. Med. Res. 119(Suppl.), 221–223.
Bergseng, H., Bevanger, L., Rygg, M., and Bergh, K. (2007). Real-time PCR targeting the sip gene for detection of group B Streptococcus colonization in pregnant women at delivery. J. Med. Microbiol. 56, 223–228. doi: 10.1099/jmm.0.46731-0
Bradshaw, J. L., Rafiqullah, I. M., Robinson, D. A., and McDaniel, L. S. (2020). Transformation of non-encapsulated Streptococcus pneumoniae during systemic infection. Sci. Rep. 10:18932. doi: 10.1038/s41598-020-75988-5
Brynildsrud, O. B., Eldholm, V., Bohlin, J., Uadiale, K., Obaro, S., and Caugant, D. A. (2018). Acquisition of virulence genes by a carrier strain gave rise to the ongoing epidemics of meningococcal disease in West Africa. Proc. Natl. Acad. Sci. U. S. A. 115:5510. doi: 10.1073/pnas.1802298115
Carrillo-Ávila, J. A., Gutiérrez-Fernández, J., González-Espín, A. I., García-Triviño, E., and Giménez-Lirola, L. G. (2018). Comparison of qPCR and culture methods for group B Streptococcus colonization detection in pregnant women: Evaluation of a new qPCR assay. BMC Infect. Dis. 18:305. doi: 10.1186/s12879-018-3208-4
Carvalho, M., da, G. S., Tondella, M. L., McCaustland, K., Weidlich, L., McGee, L., et al. (2007). Evaluation and improvement of real-time PCR assays targeting lytA, ply, and psaA genes for detection of pneumococcal DNA. J. Clin. Microbiol. 45, 2460–2466. doi: 10.1128/JCM.02498-06
Coughlan, H., Reddington, K., Tuite, N., Boo, T. W., Cormican, M., Barrett, L., et al. (2015). Comparative genome analysis identifies novel nucleic acid diagnostic targets for use in the specific detection of Haemophilus influenzae. Diagn Microbiol. Infect. Dis. 83, 112–116. doi: 10.1016/j.diagmicrobio.2015.06.013
Davis, G. S., Sandstedt, S. A., Patel, M., Marrs, C. F., and Gilsdorf, J. R. (2011). Use of bexB To Detect the Capsule Locus in Haemophilus influenzae. J. Clin. Microbiol. 49, 2594–2601. doi: 10.1128/JCM.02509-10
de Filippis, I., de Andrade, C. F., Caldeira, N., de Azevedo, A. C., and de Almeida, A. E. (2016). Comparison of PCR-based methods for the simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in clinical samples. Braz. J. Infect. Dis. 20, 335–341. doi: 10.1016/j.bjid.2016.04.005
de Filippis, I., do Nascimento, C. R. S., Clementino, M. B. M., Sereno, A. B., Rebelo, C., Souza, N. N. F., et al. (2005). Rapid detection of Neisseria meningitidis in cerebrospinal fluid by one-step polymerase chain reaction of the nspA gene. Diagn. Microbiol. Infect. Dis. 51, 85–90. doi: 10.1016/j.diagmicrobio.2004.10.004
de Gier, C., Kirkham, L.-A. S., and Nørskov-Lauritsen, N. (2015). Complete deletion of the fucose operon in Haemophilus influenzae is associated with a cluster in multilocus sequence analysis-based phylogenetic group II Related to Haemophilus haemolyticus: Implications for Identification and Typing. J. Clin. Microbiol. 53, 3773–3778. doi: 10.1128/JCM.01969-15
de-Paris, F., Pinheiro Machado, A. B. M., Gheno, T. C., Ascoli, B. M., de Oliveira, K. R. P., and Barth, A. L. (2011). Group B Streptococcus detection: Comparison of PCR assay and culture as a screening method for pregnant women. Braz. J. Infect. Dis. 15, 323–327. doi: 10.1016/S1413-8670(11)70199-4
Dhouib, R., Nasreen, M., Othman, D. S. M. P., Ellis, D., Lee, S., Essilfie, A.-T., et al. (2021). The DmsABC sulfoxide reductase supports virulence in Non-typeable Haemophilus influenzae. Front. Microbiol. 12:686833. doi: 10.3389/fmicb.2021.686833
Diallo, K., Coulibaly, M. D., Rebbetts, L. S., Harrison, O. B., Lucidarme, J., Gamougam, K., et al. (2018). Development of a PCR algorithm to detect and characterize Neisseria meningitidis carriage isolates in the African meningitis belt. PLoS One 13:e0206453. doi: 10.1371/journal.pone.0206453
Diallo, K., Feteh, V. F., Ibe, L., Antonio, M., Caugant, D. A., Plessis, M., et al. (2021). Molecular diagnostic assays for the detection of common bacterial meningitis pathogens: A narrative review. eBioMedicine 65:103274. doi: 10.1016/j.ebiom.2021.103274
Elbaradie, S. M. Y., Mahmoud, M., and Farid, M. (2009). Maternal and neonatal screening for Group B streptococci by SCP B gene based PCR: A preliminary study. Indian J. Med. Microbiol. 27, 17–21. doi: 10.1016/S0255-0857(21)01746-1
Feagins, A. R., Ronveaux, O., Taha, M.-K., Caugant, D. A., Smith, V., Fernandez, K., et al. (2020). Next generation rapid diagnostic tests for meningitis diagnosis. J. Infect. 81, 712–718. doi: 10.1016/j.jinf.2020.08.049
Furfaro, L. L., Chang, B. J., and Payne, M. S. (2017). A novel one-step real-time multiplex PCR assay to detect Streptococcus agalactiae presence and serotypes Ia, Ib, and III. Diagn. Microbiol. Infect. Dis. 89, 7–12. doi: 10.1016/j.diagmicrobio.2017.06.003
Ganaie, F., Branche, A. R., Peasley, M., Rosch, J. W., and Nahm, M. H. (2021). Effect of oral streptococci expressing Pneumococcus-like cross-reactive capsule types on world health organization recommended pneumococcal carriage detection procedure. Clin Infect Dis. 75, 647–656. doi: 10.1093/cid/ciab1003
Golparian, D., Johansson, E., and Unemo, M. (2012). Clinical Neisseria gonorrhoeae isolate with a N. meningitidis porA gene and no prolyliminopeptidase activity, Sweden, 2011 – danger of false-negative genetic and culture diagnostic results. Eurosurveillance 17:20102. doi: 10.2807/ese.17.09.20102-en
Griffiths, M. J., McGill, F., and Solomon, T. (2018). Management of acute meningitis. Clin. Med. 18, 164–169. doi: 10.7861/clinmedicine.18-2-164
Gudza-Mugabe, M., Robertson, V., Mtapuri-Zinyowera, S., and d Mavenyengwa, R. (2015). Bacterial paediatric meningitis laboratory diagnosis. J. Cell Sci. Ther. 6:2. doi: 10.4172/2157-7013.1000204
Hughes, R., Brocklehurst, P., Steer, P., Heath, P., and Stenson, B. (2017). Prevention of early-onset neonatal group B streptococcal disease. BJOG 124, e280–e305. doi: 10.1111/1471-0528.14821
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015. doi: 10.1099/mic.0.055459-0
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Khatami, A., Randis, T. M., Chamby, A., Hooven, T. A., Gegick, M., Suzman, E., et al. (2018). Improving the sensitivity of real-time PCR detection of Group B Streptococcus using consensus sequence-derived oligonucleotides. Open Forum Infect. Dis. 5:ofy164. doi: 10.1093/ofid/ofy164
Lâm, T., Elias, J., Frosch, M., Vogel, U., and Claus, H. (2011). New diagnostic PCR for Haemophilus influenzae serotype e based on the cap locus of strain ATCC 8142. Int. J. Med. Microbiol. 301, 176–179. doi: 10.1016/j.ijmm.2010.07.004
Lambertsen, L., Ekelund, K., Skovsted, I. C., Liboriussen, A., and Slotved, H. (2010). Characterisation of invasive group B streptococci from adults in Denmark 1999 to 2004. Eur. J. Clin. Microbiol. Infect. Dis. 29, 1071–1077. doi: 10.1007/s10096-010-0941-z
Levy, C., Taha, M., Weil Olivier, C., Quinet, B., Lecuyer, A., Alonso, J. M., et al. (2010). Association of meningococcal phenotypes and genotypes with clinical characteristics and mortality of meningitis in children. Pediatr. Infect. Dis. J. 29:618. doi: 10.1097/INF.0b013e3181d3ce32
Llull, D., López, R., and García, E. (2006). Characteristic signatures of the lytA gene provide a basis for rapid and reliable diagnosis of Streptococcus pneumoniae infections. J. Clin. Microbiol. 44, 1250–1256. doi: 10.1128/JCM.44.4.1250-1256.2006
Maleki, A., Mansournia, F., Ghafourian, S., Taherikalani, M., Pakzad, I., Mohammadi, J., et al. (2020). Rapid and direct molecular detection of Streptococcus pneumoniae and Haemophilus influenzae isolated in oropharynx and nasal cavity of children. New Microbes New Infect. 33:100632. doi: 10.1016/j.nmni.2019.100632
Martín-Galiano, A. J., and García, E. (2021). Streptococcus pneumoniae: A plethora of temperate bacteriophages with a role in host genome rearrangement. Front. Cell. Infect. Microbiol. 11:775402. doi: 10.3389/fcimb.2021.775402
Meyler, K. L., Meehan, M., Bennett, D., Cunney, R., and Cafferkey, M. (2012). Development of a diagnostic real-time polymerase chain reaction assay for the detection of invasive Haemophilus influenzae in clinical samples. Diagn. Microbiol. Infect. Dis. 74, 356–362. doi: 10.1016/j.diagmicrobio.2012.08.018
Missa, K. F., Diallo, K., Bla, K. B., Tuo, K. J., Gboko, K. D. T., Tiémélé, L.-S., et al. (2024). Association of symptomatic upper respiratory tract infections with the alteration of the oropharyngeal microbiome in a cohort of school children in Côte d’Ivoire. Front. Microbiol. 15:1412923. doi: 10.3389/fmicb.2024.1412923
Moreno, J., Alarcon, Z., Parra, E., Duarte, C., Sanabria, O., Prada, D., et al. (2020). Molecular characterization of Neisseria meningitidis isolates recovered from patients with invasive meningococcal disease in Colombia from 2013 to 2016. PLoS One 15:e0234475. doi: 10.1371/journal.pone.0234475
Nasreen, M., Ellis, D., Hosmer, J., Essilfie, A. T., Fantino, E., Sly, P., et al. (2024). The DmsABC S-oxide reductase is an essential component of a novel, hypochlorite-inducible system of extracellular stress defense in Haemophilus influenzae. Front. Microbiol. 4:1359513. doi: 10.3389/fmicb.2024.1359513
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Patras, K. A., Wang, N., Fletcher, E. M., Cavaco, C. K., Jimenez, A., Garg, M., et al. (2013). Group B Streptococcus CovR regulation modulates host immune signaling pathways to promote vaginal colonization. Cell Microbiol. 15, 1154–1167. doi: 10.1111/cmi.12105
Pouladfar, G., Dashti, A. S., Kadivar, M. R., Jafari, M., Pourabbas, B., Jamalidoust, M., et al. (2022). Evaluation of multiplex real-time PCR and WHO criteria for diagnosing childhood bacterial meningitis in a tertiary referral hospital in Iran. Arch. Pediatr. Infect. Dis. 10:e101822. doi: 10.5812/pedinfect.101822
Resman, F., Ristovski, M., Ahl, J., Forsgren, A., Gilsdorf, J. R., Jasir, A., et al. (2011). Invasive disease caused by Haemophilus influenzae in Sweden 1997–2009; evidence of increasing incidence and clinical burden of non-type b strains. Clin. Microbiol. Infect. 17, 1638–1645. doi: 10.1111/j.1469-0691.2010.03417.x
Rodgers, E., Bentley, S. D., Borrow, R., Bratcher, H. B., Brisse, S., Brueggemann, A. B., et al. (2020). The global meningitis genome partnership. J. Infect. 81, 510–520. doi: 10.1016/j.jinf.2020.06.064
Santa Lucia, J. Jr., Sozhamannan, S., Gans, J. D., Koehler, J. W., Soong, R., Lin, N. J., et al. (2020). Appendix Q: Recommendations for developing molecular assays for microbial pathogen detection using modern In Silico approaches. J. Aoac Int. 103, 882–899. doi: 10.1093/jaoacint/qsaa045
Satzke, C., Turner, P., Virolainen-Julkunen, A., Adrian, P. V., Antonio, M., Hare, K. M., et al. (2013). Standard method for detecting upper respiratory carriage of Streptococcus pneumoniae: Updated recommendations from the World Health Organization Pneumococcal Carriage Working Group. Vaccine 32, 165–179. doi: 10.1016/j.vaccine.2013.08.062
Shabayek, S., and Spellerberg, B. (2018). Group B streptococcal colonization, molecular characteristics, and epidemiology. Front. Microbiol. 9:437. doi: 10.3389/fmicb.2018.00437
Simões, A. S., Tavares, D. A., Rolo, D., Ardanuy, C., Goossens, H., Henriques-Normark, B., et al. (2016). lytA-based identification methods can misidentify Streptococcus pneumoniae. Diagn. Microbiol. Infect. Dis. 85, 141–148. doi: 10.1016/j.diagmicrobio.2016.03.018
Sleeman, K. L., Griffiths, D., Shackley, F., Diggle, L., Gupta, S., Maiden, M. C., et al. (2006). Capsular serotype-specific attack rates and duration of carriage of Streptococcus pneumoniae in a population of children. J. Infect. Dis. 194, 682–688. doi: 10.1086/505710
Spratt, B. G., and Maiden, M. C. (1999). Bacterial population genetics, evolution and epidemiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 701–710. doi: 10.1098/rstb.1999.0423
Taha, M. K. (2000). Simultaneous approach for nonculture PCR-based identification and serogroup prediction of Neisseria meningitidis. J. Clin. Microbiol. 38, 855–857. doi: 10.1128/JCM.38.2.855-857.2000
Tavares, D. A., Handem, S., Carvalho, R. J., Paulo, A. C., de Lencastre, H., Hinds, J., et al. (2019). Identification of Streptococcus pneumoniae by a real-time PCR assay targeting SP2020. Sci. Rep. 9:3285. doi: 10.1038/s41598-019-39791-1
Tazi, A., Morand, P. C., Réglier-Poupet, H., Dmytruk, N., Billoët, A., Antona, D., et al. (2011). Invasive group B streptococcal infections in adults, France (2007–2010). Clin. Microbiol. Infect. 17, 1587–1589. doi: 10.1111/j.1469-0691.2011.03628.x
Thomas, J. D., Hatcher, C. P., Satterfield, D. A., Theodore, M. J., Bach, M. C., Linscott, K. B., et al. (2011). sodC-Based Real-Time PCR for Detection of Neisseria meningitidis. PLoS One 6:e19361. doi: 10.1371/journal.pone.0019361
Trzciński, K., Bogaert, D., Wyllie, A., Chu, M. L. J. N., Ende, A., van der Bruin, J. P., et al. (2013). Superiority of trans-oral over trans-nasal sampling in detecting Streptococcus pneumoniae Colonization in Adults. PLoS One 8:e60520. doi: 10.1371/journal.pone.0060520
Tsang, R. S. W. (2021). A narrative review of the molecular epidemiology and laboratory surveillance of vaccine preventable bacterial meningitis agents: Streptococcus pneumoniae, Neisseria meningitidis, Haemophilus influenzae and Streptococcus agalactiae. Microorganisms 9:449. doi: 10.3390/microorganisms9020449
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
van Weezep, E., Kooi, E. A., and van Rijn, P. A. (2019). PCR diagnostics: In silico validation by an automated tool using freely available software programs. J. Virol Methods 270:106. doi: 10.1016/j.jviromet.2019.05.002
Wang, X., Mair, R., Hatcher, C., Theodore, M. J., Edmond, K., Wu, H. M., et al. (2011). Detection of bacterial pathogens in Mongolia meningitis surveillance with a new real-time PCR assay to detect Haemophilus influenzae. Int. J. Med. Microbiol. 301, 303–309. doi: 10.1016/j.ijmm.2010.11.004
Whiley, D. M., Limnios, A., Moon, N. J., Gehrig, N., Goire, N., Hogan, T., et al. (2011). False-negative results using Neisseria gonorrhoeae porA pseudogene PCR - a clinical gonococcal isolate with an N. meningitidis porA sequence, Australia, March 2011. Eurosurveillance 16:19874. doi: 10.2807/ese.16.21.19874-ena
WHO Africa (2022). Province de la Tshopo, République démocratique du Congo : Epidémie de méningite - Rapport de situation N°055/2021 - Democratic Republic of the Congo | ReliefWeb [WWW Document]. Available online at: https://reliefweb.int/report/democratic-republic-congo/province-de-la-tshopo-r-publique-d-mocratique-du-congo-epid-mie-10 (accessed November 22, 2022).
Whyte, K. E., Hoang, L., Sekirov, I., Shuel, M. L., Hoang, W., and Tsang, R. S. (2020). Emergence of a clone of invasive fucK-negative serotype e Haemophilus influenzae in British Columbia. J. Assoc. Med. Microbiol. Infect. Dis. Can. 5, 29–34. doi: 10.3138/jammi.2019-0015
World Health Organization [WHO] (2021). Defeating Meningitis by 2030 [WWW Document]. Available online at: https://www.who.int/initiatives/defeating-meningitis-by-2030 (accessed November 22, 2022).
World Health Organization [WHO] (2024). Meningitis Weekly Bulletin 1 to 28 July 2024 [WWW Document]. Available online at: https://www.who.int/publications/m/item/meningitis-weekly-bulletin-1-to-28-july-2024 (accessed February 20, 2025).
Keywords: meningitis, molecular diagnostic, in silico analysis, sensitivity, specificity
Citation: Amoikon STL, Diallo K, Tuo JK, Nasir N, Feteh VF, Mzumara G, Aderoba A, Jacques R, Mandal H, Jolley KA, Bray JE, Harrison OB and Maiden MCJ (2025) In silico and in vitro analyses for the improved diagnosis of bacterial meningitis. Front. Microbiol. 16:1655490. doi: 10.3389/fmicb.2025.1655490
Received: 30 June 2025; Accepted: 08 September 2025;
Published: 26 September 2025.
Edited by:
Xiaoli Qin, Hunan Agricultural University, ChinaReviewed by:
Werner Solbach, University of Lübeck, GermanyZixu Wang, Bicycle Therapeutics, United Kingdom
Andrew, Peifer, Wadsworth Center, United States
Copyright © 2025 Amoikon, Diallo, Tuo, Nasir, Feteh, Mzumara, Aderoba, Jacques, Mandal, Jolley, Bray, Harrison and Maiden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kanny Diallo, a2FubnkuZGlhbGxvQGNzcnMuY2k=