 Christian Mammen1
Christian Mammen1 Julia Gottstein1Pablo Cea2
Julia Gottstein1Pablo Cea2 Kira Tantsur1
Kira Tantsur1 Jens Reiners3Michele Bonus2Holger Gohlke2,4
Jens Reiners3Michele Bonus2Holger Gohlke2,4 Sander H. J. Smits1,3*
Sander H. J. Smits1,3*- 1Institute of Biochemistry, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 2Institute for Pharmaceutical and Medicinal Chemistry, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany
- 3Center for Structural Studies, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 4Institute of Bio- and Geosciences (IBG-4: Bioinformatics), Forschungszentrum Jülich, Jülich, Germany
Peptidoglycan serves as the first permeability barrier of Gram-positive bacteria. Intermediates of the peptidoglycan synthesis cycle are typical targets of antimicrobial compounds, including the peptide antibiotics nisin and bacitracin. In human pathogenic bacteria, gene clusters have been identified that are upregulated to confer resistance against these compounds. One such cluster found in Streptococcus agalactiae encodes a Bacitracin efflux (BceAB)-type ATP binding cassette transporter, SaNsrFP, and an associated two-component system, SaNsrRK. SaNsrFP has been shown to confer resistance against multiple antimicrobial peptides in vivo, with highest activity against bacitracin. Like other BceAB-type ABC-transporters, SaNsrFP features a large extracellular domain (ECD) that determines the substrate spectrum. Here, we report the purification and in vitro characterization of the SaNsrFP complex. Measuring the ATPase activity in the presence and absence of bacitracin showed that the binding of bacitracin allosterically modulates ATPase activity. By expressing and purifying only the soluble ECD of SaNsrP, we could show through two in vitro binding assays that this segment alone is responsible for bacitracin binding and then explored the putative binding mechanism using molecular docking. Additionally, we assessed the structural conservation of the ECD across 24 BceAB-type ABC-transporters with the AlphaFold database. Enabling us to create a first classification within this superfamily based on the structural fold of the ECD.
1 Introduction
Adaptation is one of the key factors that enable bacteria to conquer and thrive in a rapidly changing environment. As the cell wall is the first point of contact, its maintenance, modification, and protection are crucial to shield bacteria from harmful environmental factors, such as ions, pH, or toxic compounds. Peptidoglycan constitutes the first permeability barrier in Gram-positive bacteria, and its synthesis relies on lipid II. Therefore, the lipid II cycle is a vital process and target for many antimicrobial compounds. Inhibiting this cycle leads to cells incapable of extending the peptidoglycan layer, inhibits cell division, and subsequently leads to cell death. Thus, intermediates of the lipid II cycle are well-studied targets for antibiotics such as bacitracin, nisin, or vancomycin (Muller et al., 2017). To survive antimicrobial exposure, the bacterial cell needs to accurately assess the severity of the situation to react with a minimized metabolic cost. Here, the presence of antimicrobials is sensed by two-component systems (TCS), which induce the expression of resistance proteins resulting in different adaptive responses, for example, modulation of the membrane composition (Fritz et al., 2015, Tollerson and Ibba, 2020).
Moreover, mainly in soil and human pathogenic Gram-positive bacteria, gene clusters were identified encoding for a resistance system containing membrane-embedded proteins, such as the Bacitracin efflux (BceAB)-type ATP-binding cassette (ABC)-transporter family (Orlando, 2024; Dintner et al., 2011).
Recently, the cryo-electron microscopy structure of BceAB from B. subtilis was solved (George et al., 2022; George and Orlando, 2023) and revealed that this transporter consists of two nucleotide-binding domains, which hydrolyze ATP to generate energy, and a single transmembrane domain, consisting of 10 transmembrane helices (TMH). TMH 1 to 4 and TMH 7 to 10 form bundles that are related by two-fold pseudosymmetry, representing an FtsX-domain fold similar to type VII ABC transporters involved in mechanotransmission (Thomas et al., 2020). The overall arrangement of the TMHs of BceB is asymmetrical due to the closer position of TMH 5 and 6 to TMH 7 to 10 than to the other TMH bundle (George et al., 2022). TMH 7 and 8 form longer and extended stalk helices with a 200–250 amino acid large extracellular domain (ECD) in between, which is the hallmark of BceAB-type transporters (Figure 1) (Khosa et al., 2013). Since BceAB confers resistance against bacitracin, it is proposed that it detects the complex of bacitracin with undecaprenylpyrosphate (UPP) (Kobras et al., 2020). In the structure of BceAB, between TMH 5, 6 and TMH 7, 9, a hydrophobic lipid-binding pocket with a suggested bound UPP derivate 4-amino-4-deoxy-L-arabinopyranosyl undecaprenyl phosphate (AUP) was identified, which is situated directly underneath the ECD (George et al., 2022). It is proposed that native UPP and other UPP-lipid derivates might bind to this binding site. Mutation studies replacing the ECD of VraG in S. aureus with its counterpart from VraE, responsible for bacitracin resistance, led to enhanced bacitracin resistance in VraG and increased sensitivity to colistin (Falord et al., 2012; Hiron et al., 2011; Cho et al., 2022). This suggests that the ECD is involved in substrate sensing, particularly, that it binds bacitracin when bound to UPP (Kobras et al., 2020).
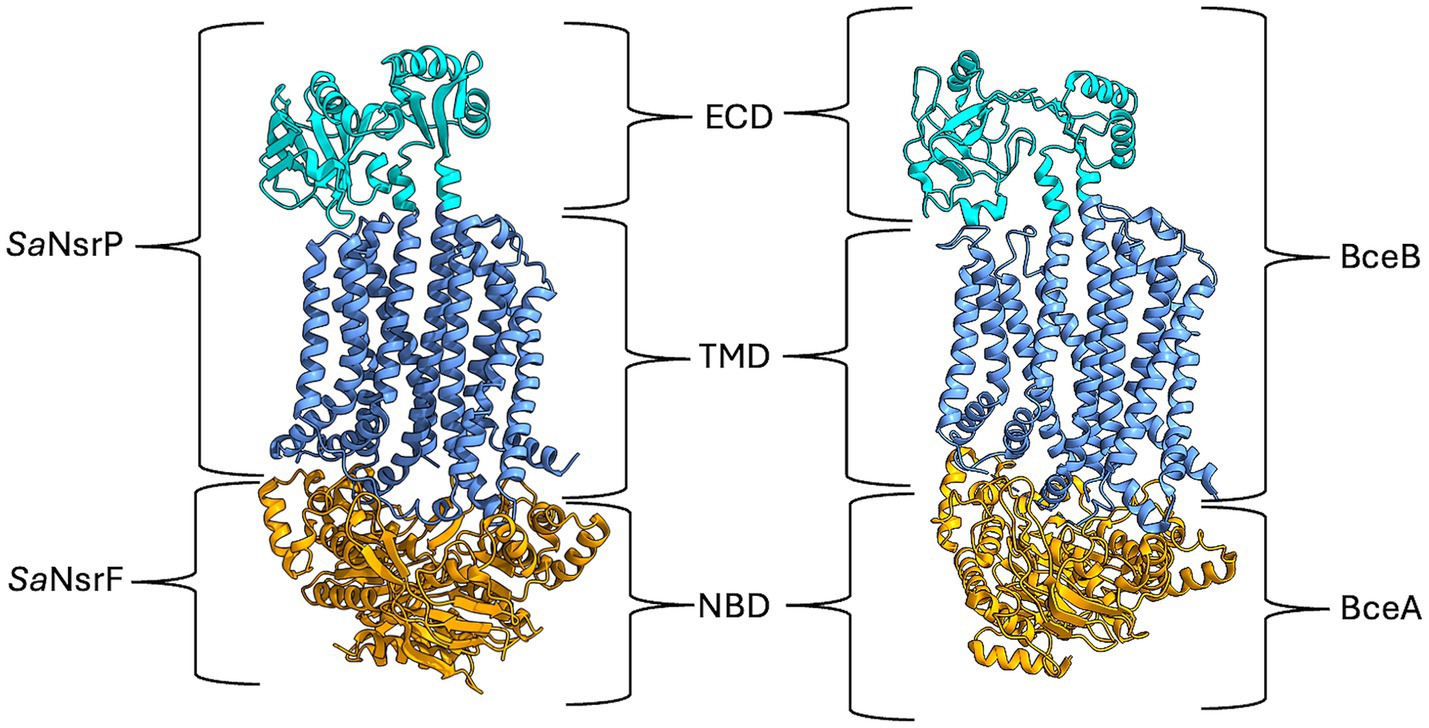
Figure 1. AlphaFold model of SaNsrFP and cryo-EM structure of BceAB from B. subtilis [7TCG, (George et al., 2022)]. The ECD of SaNsrP and BceB is highlighted in cyan, while the TMD is depicted in cornflower blue. Lastly, the dimeric NBD SaNsrF and BceA are marked in orange. Image was created with ChimeraX version 1.8.
Within most bce operons, a co-evolved BceRS-type TCS is present consisting of a response regulator (BceR) and a transmembrane histidine kinase (HK, BceS) sensor. In contrast to other HK proteins, BceS has a notoriously short extracellular domain of roughly 25 amino acids, which is likely too small to allow an effective binding to the target lantibiotic (Rietkötter et al., 2008; Mascher et al., 2003; Mascher, 2006; Clemens et al., 2017; Khosa et al., 2013). Thus, the currently proposed signal transduction pathway is that BceAB senses the presence of an antimicrobial peptide and activates HK through contacts between the transmembrane regions (Gebhard, 2012; Revilla-Guarinos et al., 2014). This process was described for the detoxification system GraRS-VraFG in S. aureus (Cho et al., 2021) and various TCS-ABC transporters in B. subtilis (BceRS-AB, YxdJK-LM, and YvcPQ-RS) (Dintner et al., 2011; Fritz et al., 2015). Furthermore, it has been shown that the antimicrobial activity of the transporter depends on ATP hydrolysis through the ATPase BceA (Rietkötter et al., 2008) as well as the complex formation with BceRS (Dintner et al., 2014). Major efforts have been made to unravel the mechanism of BceAB-type transporters. Proposals ranged from AMP removal from the membrane (Gebhard and Mascher, 2011) over functioning as an exporter (Reiners et al., 2017) to flipping the UPP (Kingston et al., 2014). A more recent study postulated a target-AMP-dissociative, ATP hydrolysis-driven mechanism for BceAB-type transporters, in which the target-AMP complex is recognized and UPP is physically released from the grip of bacitracin (Kobras et al., 2020).
Recently, a high level of resistance against bacitracin was observed for the BceAB transporter from S. agalactiae COH1, SaNsrFP, which was previously found to also mediate resistance against nisin, an antimicrobial peptide belonging to the lantibiotic family (Gottstein et al., 2022; Reiners et al., 2020) even in the absence of the TCS. SaNsrFP consists of a homodimeric nucleotide-binding domain, NsrF, that can hydrolyze ATP and the monomeric transmembrane protein NsrP. It was shown that the observed resistances mediated by SaNsrFP are ATP hydrolysis-dependent by using an ATP hydrolysis-deficient mutant of SaNsrFP, termed SaNsrFH202AP (Furtmann et al., 2020; Gottstein et al., 2022).
To analyze the interplay between the ECD and its substrate bacitracin, we investigated the influence of substrate binding on the in vitro ATPase activity of SaNsrFP. We further set out to measure the binding affinity of bacitracin and Zn-bacitracin towards the purified ECD and identified a putative binding mode for bacitracin through molecular docking experiments. To translate our findings to other BceAB-type ABC-transporters, we searched for and identified new members of this family using structural homology and could classify them by the distinct architecture of the ECD into five distinct groups.
2 Materials and methods
2.1 Heterologous expression and purification of SaNsrFP in E. coli (DE3) C41 dd
SaNsrFP (Uniprot ID: X5KGL2 + Q8DZX0) was cloned from pIL-sv-NsrFP into pET16b for heterologous expression of SaNsrFP with an N-terminal 10x-His-tag at SaNsrF in E. coli (DE3) C41 dd cells (Kanonenberg et al., 2019). Expression was performed at 18 °C overnight in LB. Resuspended cells were lysed at 1.5 kbar with a cell disruptor (Microfluidics) and membranes were collected after two centrifugation steps at 20,000 × g and 150,000 × g, respectively. Membranes were homogenized with resuspension buffer and stored at −80 °C. For purification, membranes were solubilized with 1% (w/V) LMNG and subjected to IMAC followed by SEC. The detailed procedures are described in the Supplementary material.
2.2 ATPase activity measurements of SaNsrFP in LMNG micelles
Purified SaNsrFP was diluted in purification buffer (100 mM NaCl, 50 mM HEPES pH 8) supplemented with 0.005% (w/V) LMNG to 0.1 mg/mL. For the ATPase kinetic measurements, ATP stock concentrations ranging from 0.5–50 mM were prepared. For each sample, 21 μL SaNsrFP (2.1 μg per sample) was supplemented with 3 μL 100 mM MgCL2 (final concentration 10 mM) or bidest H2O (background measurement) and subsequently, the reaction was started with 6 μL of the respective ATP stock. Reaction mixes were incubated at 30 °C for 30 min. A 96-well plate featuring a phosphate standard ranging from 0 to 12.5 nmol phosphate was prepared with 175 μL 40 mM H2SO4. To stop the reaction, 25 μL of the reaction mix was added to the H2SO4 solution. Next, the solution was stained with staining solution (15.7% H2SO4, 0.1% Malachite green, 1.5% Ammonium molybdate, 0.17% Tween 20) for 8 min at room temperature. Afterwards, the absorbance of each well was measured at 595 nm and the resulting values were analyzed with GraphPad Prism version 10.3.1 for MacOS, GraphPad Software, San Diego, California USA, www.graphpad.com and the Michaelis Menten fit. For the ATPase kinetics containing 0.1 mM bacitracin, SaNsrFP was spiked with either 10 mM MgCl2 and 0.1 mM bacitracin or bidest H2O and 0.1 mM bacitracin. All positive and background measurements were done in at least duplicates.
2.3 Heterologous expression and purification of SaNsrP-ECD in E. coli (DE3) BL21
SaNsrP-ECD (aminoacids 311 to 512) was cloned from pIl-sv-NsrFP vector into pET28b with an N-terminal 6xHis tag. SaNsrP-ECD was expressed in E. coli (DE3) BL21 in LB supplemented with 30 μg/mL kanamycin overnight at 18 °CAfter overnight expression, cells were harvested at 5,000 × g for 15 min at 4 °C and resuspended in resuspension buffer (50 mM Tris pH 8.0, 50 mM NaCl, 10% Glycerol) supplemented with 1,000 U of DNAse, RNAse and one protease inhibitor tablet (Roche). After cell disruption at 1.5 kbar, cell debris and membranes were removed by ultracentrifugation at 150,000 × g for 1 h at 4 °C. The supernatant was purified by IMAC, followed by thrombin cleavage, reverse IMAC and SEC. Detailed procedures are stated in the Supplementary material.
2.4 Multi-angle light scattering (MALS)
Purified ECD was concentrated to 2 mg/mL using centrifugal filters with a 3-kDa cut-off (Amicon Ultra-0.5 MERCK/ Millipore) and the samples were centrifuged at 100,000 × g at 4 °C for 30 min. For the measurement with Zn-bacitracin, the protein sample was preincubated with 1 mM Zn-bacitracin. Superdex 75 Increase 10/300 GL column (GE Healthcare) was pre-equilibrated overnight at 0.1 mL/min flow rate with buffer (20 mM Tris pH 8.0, 500 mM NaCl). For each analysis, 200 μL of a protein sample at 2.0 mg/mL concentration was loaded onto the column at 0.6 mL/min flow rate using a 1,260 binary pump (Agilent Technologies). The scattered light was measured with a miniDAWN TREOS II light scatterer, (Wyatt Technologies), and the refractive index was measured with an Optilab T-rEX refractometer (Wyatt Technologies). Data analysis was performed with ASTRA 7.3.2.21 (Wyatt Technologies) (Slotboom et al., 2008).
2.5 Tyrosine fluorescence quenching
The measurements were conducted using a Fluorolog Jobin Yvon FL-3-11. 1 mL of NsrP-ECD solution in SEC buffer (25 mM Tris pH 8, 500 mM NaCl) with concentrations of NsrP-ECD in the range from 1.3 to 1.7 nM was prepared in a Hellma Macro-cuvette 100-QS. 10–50 μL of 20 mM bacitracin stock solution in H2O was added to the cuvette. The cuvette was placed into the fluorolog sample holder and the reaction mixture was incubated for 2 min while stirring before measuring the fluorescence intensity. The measurements were conducted for bacitracin concentrations in the range from 0 to 2.8 mM. For measurements with Zn2+, 100 mM of ZnCl2 was added to the 20 mM bacitracin stock solution.
In this experiment, the fluorescence of tyrosine was monitored because SaNsrP ECD lacks tryptophan residues. 260 nm and 304 nm were used as excitation and emission wavelengths, respectively. The resulting fluorescence signal peak maximum was normalized and showed a decrease upon bacitracin binding. This difference was plotted as 1-F(%) against the bacitracin/Zn-bacitracin concentration. Finally, the KD was determined with GraphPad Prism version 9.5.1 for MacOS, GraphPad Software, San Diego, California USA. Measurements were performed in triplicates.
2.6 Small angle X-ray scattering of SaNsrP ECD and SaNsrFP
We collected the data of SaNsrP ECD and SaNsrFP on the beamline BM29 at the ESRF Grenoble (Tully et al., 2023). The BM29 beamline was equipped with a PILATUS 3 × 2 M detector (Dectris) at a fixed distance of 2.813 m. The measurements were performed with a SaNsrP ECD (without His-tag) concentration of 1.65 mg/mL and SaNsrFP concentration of 0.5 mg/mL, respectively, at 10 °C with the corresponding buffers as background. We collected 10 frames with an exposer time of 1 second per frame and scaled the data to absolute intensity against water. We checked each frame for radiation damage using CorMap/χ2 test, implemented in PRIMUS (Konarev et al., 2003).
All used programs for data processing were part of the ATSAS Software package (Version 3.1.3) (Manalastas-Cantos et al., 2021). Primary data reduction was performed with the program PRIMUS (Konarev et al., 2003). With the Guinier approximation (Guinier, 1939), we determine the forward scattering I(0) and the radius of gyration (Rg). The program GNOM (Svergun, 1992) was used to estimate the maximum particle dimension (Dmax) with the pair-distribution function p(r). We calculated an ab initio model of SaNsrP ECD with GASBOR (Svergun et al., 2001) and superimposed it with an AlphaFold3 (Abramson et al., 2024) model using SUPCOMB (Kozin and Svergun, 2001). All results are displayed in Supplementary Table 1.
2.7 Computational predictions of the bacitracin binding mode
A full model of the SaNsrFP protein was generated with ColabFold (Mirdita et al., 2022) using 12 recycling cycles and creating four independent models. A sequence search for MSA construction was performed against Mgnify, UniRef, and PDB70 databases. The final model was chosen based on the resulting pLDDT score (Supplementary Figure 5). The extracellular domain comprising the residues between the amino acids N307 and L517 was taken for further experiments. The final model has been uploaded to https://www.modelarchive.org/doi/10.5452/ma-m0kcc.
Extensive conformational sampling of bacitracin was carried out using the MacroModel tool in Schrödinger (Watts et al., 2014) based on the protocol used in previous macrocycle sampling benchmarks (Alogheli et al., 2017). The initial structure of bacitracin was taken from the PDB entry 4K7T. Protonation states for side chains were assigned using Propka (Rostkowski et al., 2011). 1,000,000 steps of the Monte Carlo Multiple Minimum (MCMM) search were performed incorporating distance restraints between the peptide and the bound zinc ion. Extended sampling was allowed by incorporating the sampling of torsional angles of amides, esters, C-N and N-N single bonds, as well as C=N and N=N double bonds. A wide-opening ring criterion was used (0–100 Å) avoiding atoms adjacent to stereocenters. 50,000 steps of truncated Newton’s conjugate gradient (TNCG) method were performed for energy minimization. Redundancy within the resulting conformers was removed using an RMSD criterion of 0.5 Å for heavy atoms. This resulted in 67,949 unique conformations. The zinc ion was removed before moving on to docking experiments to prevent steric clashes with the receptor.
A putative binding region within the extracellular loop domain was identified using Schrodinger’s SiteMap (Halgren, 2009). A 46 Å Grid was manually placed within the site. Rigid docking was carried out using Glide in standard precision (Repasky et al., 2007) mode, including a reward for intramolecular hydrogen bonds. The resulting poses were filtered according to their docking energy score. Poses with energies below −6.0 kcal mol−1 were kept for further analyses. The final poses were clustered with cpptraj (Roe and Cheatham, 2013) using the DBScan algorithm with an RMSD-based cut-off of 2 Å for heavy atoms and three minimum points. Statistical assessments were performed using SciPy and NumPy in Python 3.10.
Parameters for the non-canonical residues of bacitracin were derived for the AMBER ff14SB force field using a fragmentation approach. Custom parameters were developed for D-ornithine (ORN), an isopeptide-linked lysine (LYX), and the N-terminal thiazoline-isoleucine moiety in its neutral (ICN) and protonated (ICP) states. For each residue, partial atomic charges were obtained via a multi-conformer Restrained Electrostatic Potential (RESP) (Wang et al., 2000) fitting protocol using capped model compounds. Low-energy conformers were generated with GOAT (De Souza, 2025) (ORCA 6.1), optimized in Gaussian 16 at the HF/6-31G(d) level of theory, and validated as true minima via frequency calculations. After excluding structures with intramolecular hydrogen bonds, Merz-Singh-Kollman electrostatic potentials were calculated [HF/6-31G(d)] and fitted using the respgen and resp programs in AmberTools22.
The specific models and constraints were tailored to each residue. For LYX, an N-acetyl-Nε-acetyl-l-lysine-N′-methylamide model was used to represent the isopeptide environment, with charges for five conformers fitted under the constraint that both capping groups and the total residue charge were zero. The fitting for ORN (N-acetyl-D-ornithine-N′-methylamide) used four conformers with a total charge constraint of +1. The ICN (charge 0) and ICP (charge +1) residues each used a single low-energy conformer for fitting. While most bonded parameters were assigned by analogy from ff14SB and GAFF2, a custom frcmod file was created to define specific parameters for the novel thiazoline ring structure. These terms were derived using the parmcal utility, transferred from the GAFF2 force field, or estimated based on chemical similarity. The final bacitracin molecule was assembled in tLEaP, where the isopeptide bond between the LYX6 side chain and the Asn12 C-terminus was manually defined.
All atom molecular dynamics simulations were performed using the Amber22 suite of molecular simulation software (Case et al., 2005). Topology files were generated with tleap (Case et al., 2023), using ff14SB as protein forcefield. The N and C terminus of the ECD were capped to reproduce the local environment of a non-truncated protein. The complexes were solvated in a TIP3P truncated octahedron box extending 12 Å from the protein. Cl− counter ions were added to maintain electroneutrality of the simulation box. Three rounds of minimization were performed, first placing heavy positional restraints (5 kcal mol−1 Å−2) on all solute heavy atoms, then it was reduced to 0.1 kcal mol−1 Å−2, and then with no restraints. Each minimization round consisted in 500 steps of steepest descend, followed by 2000 steps of conjugate gradient. The systems were heated to 100 K in a timeframe of 25 ps in NVT conditions, using the Langevin thermostat with a collision coefficient of 1 ps−1. The systems were pressurized under NPT conditions using the Berendsen barostat with isotropic position and brought to a final temperature of 300 K over a time of 250 ps. All simulations were performed with a timestep of 2 fs, constraining bonds with hydrogen atoms using SHAKE (Kräutler et al., 2001) applying the GPU-accelerated implementation of the Particle Mesh Ewald Method (Salomon-Ferrer et al., 2013). Production runs of 0.5 μs were performed for the best scoring pose of each of the five best docking solution clusters. Post processing of trajectories was done using cpptraj. The RMSD of the macrocyclic region and the N-terminal region were calculated by fitting only the ECD with the rms command. We calculated the RMSD only for the center of mass of the atoms that make up the macrocycle backbone, and the backbone and thiazoline atoms that make up the linear N-terminus of bacitracin. Ligand occupation density was calculated using the volmap tool in VMD (Humphrey et al., 1996), calculating the average across all replicas. The macrocyclic region and N-terminal regions were defined using only their backbone heavy atoms. Representative poses of the C0 and C1 trajectories, respectively, were obtained by performing a hierarchical agglomerative clustering with an epsilon value of 2.5 Å in cpptraj.
2.8 Searching the AlphaFold protein structure database
The AlphaFold database (Jumper et al., 2021; Varadi et al., 2022; Varadi et al., 2024) was screened for BceAB-type ABC transporters. Parameters looked for were the characteristic domain architecture found for BceAB transporters of 10 transmembrane helices and a large extracellular domain (150–250 amino acids) located in between transmembrane helices 7 and 8 (Collins et al., 2010). Furthermore, the nucleotide-binding domain BceA should be located next to the gene encoding the transmembrane protein. BceB permease models were found under different names such as BceB, Bacitracin-export permease, FtsX-like permease, and FtsX domain-containing protein.
2.9 Structural alignments
Permeases of the different BceAB transporters from the AlphaFold database were cropped so that only the SABRE and Porter domain of the ECD with the first helical turn of the stalk helices remained. The structural alignment was performed by using the cealign tool of Pymol Version 2.5.4. The sequence alignments were performed by clustalO EMBL server (Madeira et al., 2024) and the sequence identity matrix was calculated with BioEdit V7.7.
3 Results
3.1 In vitro characterization of SaNsrFP
To characterize the transporter in vitro, we expressed and purified SaNsrFP from E. coli. After membrane isolation, membranes were solubilized with LMNG and subjected to an IMAC and SEC. Analyzing the elution fraction by SDS-PAGE analysis revealed pure bands above 55 kDa for SaNsrP and 25 kDa for SaNsrF (Supplemenatry Figure 1a). The results from SAXS (Figures 2a–d) show that SaNsrFP is folded and contains no aggregates, as shown by the kratky plot (Figure 2d). The molecular weight calculated from the SAXS scattering is 148.30 kDa and suggests a fully assembled SaNsrFP (135 kDa + detergent micelle) protein sample in a stoichiometry of 2:1 (Supplementary Table 1). ATPase assays revealed that SaNsrFP displays a typical ATPase kinetic with a Bmax of 113 ± 28 nmol/min/mg, a KD of 5.2 ± 2.0 mM, and a Hill coefficient of 1.05 ± 0.08 (Figure 2e). Next, we set out to test whether bacitracin is capable of stimulating ATPase activity of SaNsrFP. The addition of 100 μM bacitracin resulted in a Bmax of 127 ± 8.2 nmol/min/mg, a KD of 1.7 ± 0.32 mM, and a Hill coefficient of 1.25 ± 0.14 (Figure 2e). Highlighting, that the addition of bacitracin results in positive cooperative binding of ATP in the NBD, while it decreases the KD by 3.1-fold. To confirm that the activity observed directly correlates to SaNsrFP, we expressed, purified, and tested the ATPase-deficient mutant SaNsrFH202AP (Furtmann et al., 2020), which showed only marginal ATPase activity (Figure 2e).

Figure 2. Characterization of SaNsrFP. (a) Scattering data of SaNsrFP. Experimental data are shown in black dots, with grey error bars. The GNOM fit is shown as red line and below is the residual plot of the data. (b) The Guinier plot of SaNsrFP. (c) The P(R) function of SaNsrFP indicates an elongated particle. (d) Dimensionless Kratky plots of SaNsrFP. (e) Shown is the ATPase kinetic recorded for SaNsrFP (WT) (dark blue), the ATP-deficient mutant SaNsrFH202AP (red), and the WT with 100 μM bacitracin (light blue). All measurements were done at least in duplicates and background measurements without added magnesium were subtracted from all absorbance values. A final MgCl2 concentration of 10 μM and 2.1 μg SaNsrFP was mainted in all samples. Error bars denote standard deviation. Graphs were fitted using GraphPad Prism version 10.3.1 for MacOS, GraphPad Software, San Diego, California USA, www.graphpad.com.
3.2 In vitro characterization of the ECD of SaNsrP
To verify that the SaNsrP-ECD is capable of binding bacitracin, we cloned and subsequently expressed the SaNsrP-ECD without stalk helices (amino acids 307–517) in E. coli. Subsequently, purified SaNsrP-ECD subjected to size exclusion chromatography (SEC) revealed a single homogenous peak (Supplementary Figure 1b). The purification was monitored via SDS-PAGE analysis (Supplementary Figure 1b).
SaNsrFP ECD showed a band lower than 25 kDa, which is in line with the theoretical mass of 23.8 kDa (Supplementary Figure 1b). Thus, we can show the stable and successful expression and purification of the ECD of a BceAB-type ABC transporter. To corroborate the molecular weight and oligomeric state of the protein sample, SAXS was performed, confirming that the ECD of SaNsrP is a monomer in solution (Figures 3a–f; Supplementary Table 1 gives an overview of the results obtained by the SAXS measurement).
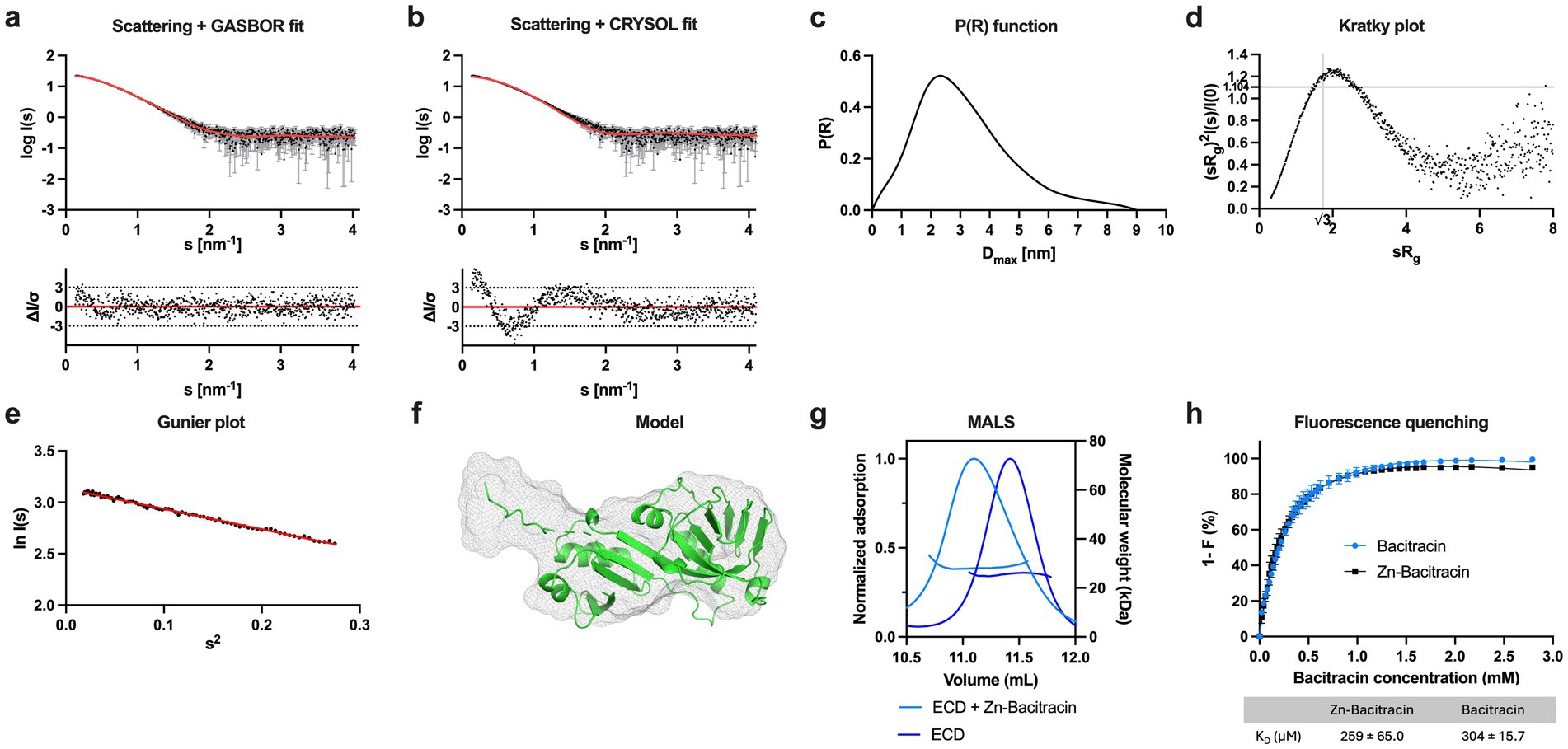
Figure 3. Characterization of the ECD of SaNsrP. (a) SAXS scattering data for monomeric SaNsrP-ECD without His-tag. Experimental data are shown in black dots, with grey error bars. The GASBOR ab initio fit is show as a red line and below is the residual plot of the data. (b) Scattering data of SaNsrP-ECD. Experimental data are shown in black dots, with grey error bars. The CRYSOL comparison fit from the AlphaFold3 model is shown as red line and below is the residual plot of the data. (c) The P(R) function of SaNsrP-ECD indicates a globular particle with an extension. (d) Dimensionless Kratky plots of SaNsrP-ECD. (e) The Guinier plot of SaNsrP-ECD. (f) AlphaFold3 model of SaNsrP-ECD superimposed with the GASBOR ab-initio model. The AlphaFold3 model is shown in green cartoon representation and the GASBOR ab-initio model is shown in grey mesh representation. (g) MALS measurement of SaNsrP-ECD (blue) and SaNsrP-ECD with the addition of Zn-Bacitracin (light blue). The normalized UV absorption at 280 nm and molecular mass were plotted against the elution volume. (h) Relative quenching of tyrosine fluorescence intensity of SaNsrP-ECD with bacitracin (blue) and Zn-bacitracin (black). The measurements were conducted in the range of 304 nm and 0–2.79 mM bacitracin was added subsequently to the protein. Dissociation constants were calculated and graphs were fitted using GraphPad Prism version 9.5.1 for MacOS, GraphPad Software, San Diego, California USA, www.graphpad.com.
To confirm the interaction between bacitracin and the ECD of S. agalactiae, we used two independent methods: (1) Multi-Angle Light Scattering (MALS) and (2) intrinsic tyrosine fluorescence of the SaNsrP-ECD (containing 9 tyrosines), which is altered upon bacitracin binding. The peak of the SaNsrP-ECD without Zn-bacitracin (Figure 3g, blue) showed a molecular weight of 25.8 kDa in the MALS measurement, whereas the peak of SaNsrP-ECD with Zn-bacitracin (1.5 kDa) (Figure 3g, light blue) shifted and contains particles of 28 kDa. Thus, the difference and the peak shift indicates binding of bacitracin (Figure 3g). Monitoring the tyrosine fluorescence of SaNsrP-ECD upon the addition of bacitracin or Zn-bacitracin reveals a decrease in fluorescence with increasing concentrations (Figure 3h). For Zn-bacitracin, a KD of 259 ± 65.0 μM was obtained and for bacitracin, a KD of 304 ± 15.7 μM was obtained. This is significantly higher than the observed interaction of bacitracin with the full-length BceAB transporter from B. subtilis where bacitracin is bound with a KD of 60 nM (Dintner et al., 2014). This difference might be due to the in vitro approach of our setup, where the transmembrane domain of SaNsrP embedded in a lipid environment is lacking.
3.3 Molecular modeling prediction of bacitracin binding to SaNsrP-ECD
In order to propose a bacitracin binding mode for SaNsrP, we first used SiteMap to identify putative binding sites within it. This revealed two sites: one located within the transmembrane region near the extracellular side, and a second one placed in the central cleft of the ECD, between its two constituent subdomains, the SABRE and Porter subdomains (Supplementary Figure 2). Since the fluorescence quenching experiments showed that the ECD can bind on its own to bacitracin, all the key interactions between bacitracin and the protein must occur through the second site. Therefore, we performed molecular docking centered at the ECD cleft.
Given the large size and many rotatable bonds of bacitracin, conventional flexible docking is unviable. Thus, we first generated over 60,000 unique ligand conformations with MacroModel and then docked them into the SaNsrP-ECD using rigid docking in Glide. The resulting poses were filtered according to their docking score (< −6 kcal mol−1) and clustered according to their RMSD. Only the five most populated clusters were considered for further analysis (Supplementary Figure 3).
In two of these clusters, the macrocyclic ring of bacitracin is placed within the central cleft of the ECD, while the thiazoline-containing N-terminus is placed at the edge, pointing towards the transmembrane domain (Supplementary Figure 6a, C0 and C3). In the three remaining clusters, the placement of bacitracin is reversed, with the N-terminus placed at the center of the ECD and the ring pointing toward the membrane surface (Supplementary Figure 6a, C1, C2 and C4). In all five clusters, bacitracin is in an extended conformation, with the N- and C-terminus spread apart from each other. These conformational states differ from the one in which bacitracin binds its lipidic target, where both ends come close together to surround the pyrophosphate moiety in the target, leading to a compact globular conformation (Supplementary Figure 4).
The most populated cluster (cluster C0), where bacitracin binds with its macrocyclic C-terminus placed at the central cleft of the protein, neither yields the best median binding energy nor the best scoring single pose. The best median binding energy is found in cluster C4 (−8.05 kcal·mol−1) and the single best scoring pose is in cluster C1 (−10.8 kcal·mol−1). In contrast to C0, C1 and C4 contain poses where the N-terminus is at the central cleft of the protein and the macrocycle is situated at the edge, suggesting that such poses are energetically more favorable. To further corroborate whether there are systematic binding energy differences between the two possible macrocycle orientations, we divided all the poses of the top five clusters into the two groups (Supplementary Figure 6b). The resulting distributions of binding energies differ significantly (p = 1.02·10−23, non-parametric paired Mann–Whitney U test), with poses where the ring is at the edge and the free tail is at the center being more favorable.
To further assess the robustness of the predicted poses, we performed molecular dynamics simulations taking as a starting point the best pose of each cluster. To evaluate the degree of structural variance throughout the simulations, we calculated the RMSD of the center of mass of the macrocyclic ring and the RMSD of the backbone atoms of the tail. The results show some conformational variability. Nonetheless, in poses starting with the macrocycle at the center (C0 and C3, Supplementary Figure 7), the macrocycle remained within the starting site in most of the replicas (9 out of 10 and 8 out of 10, respectively), while the N-terminus fluctuates widely (Supplementary Figure 8). The opposite trend was observed for poses with the N-terminus at the cleft, (C1, C2 and C4), albeit with an even larger fluctuation margin. Among the assessed poses, the least structural variance for the poses with the macrocycle at the center was observed starting from the binding pose C0, and for the poses with N-terminus at the center, it was observed for C1.
To analyze where on the protein surface each segment of bacitracin spent most of the simulation time, we performed a density analysis over both simulations sets. This is in essence a 3D histogram that shows the most occupied regions during an aggregated simulation time of 5 μs. For the simulations starting from C0, the macrocycle spends most of the time at the central cleft, close to where it was initially placed by molecular docking (yellow blob, Figure 4a), whereas the tail occupies two regions preferentially, one tucked under the ring near the SABRE subdomain, and the other one extended over the Porter subdomain (orange blob, Figure 4a). Similarly, the simulation of C1 shows the N-terminus occupying preferentially a wide region within the central cleft, slightly closer to the Porter subdomain (yellow blob, Figure 4b). However, the macrocyclic ring shows a less defined preferential region, with sparse densities observed above and below the position of the N-terminus. Finally, we derived a representative pose for the conformational ensembles sampled during the MD simulations by clustering according to the RMSD of the bacitracin backbone atoms; the resulting poses for the simulations started from C0 and C1 are shown in Figures 4c,d, respectively. Compared to the starting poses, bacitracin adopts a more compact conformation after MD simulation. For C0, the macrocyclic ring sits in a dominantly hydrophobic region of the SABRE domain, while the N-terminus points downwards, where the membrane would be located. On the other hand, C1 shows the N-terminus placed in a region close to the Porter subdomain rich in polar amino acids, while the ring remains detached from the protein, exposed to the solvent.
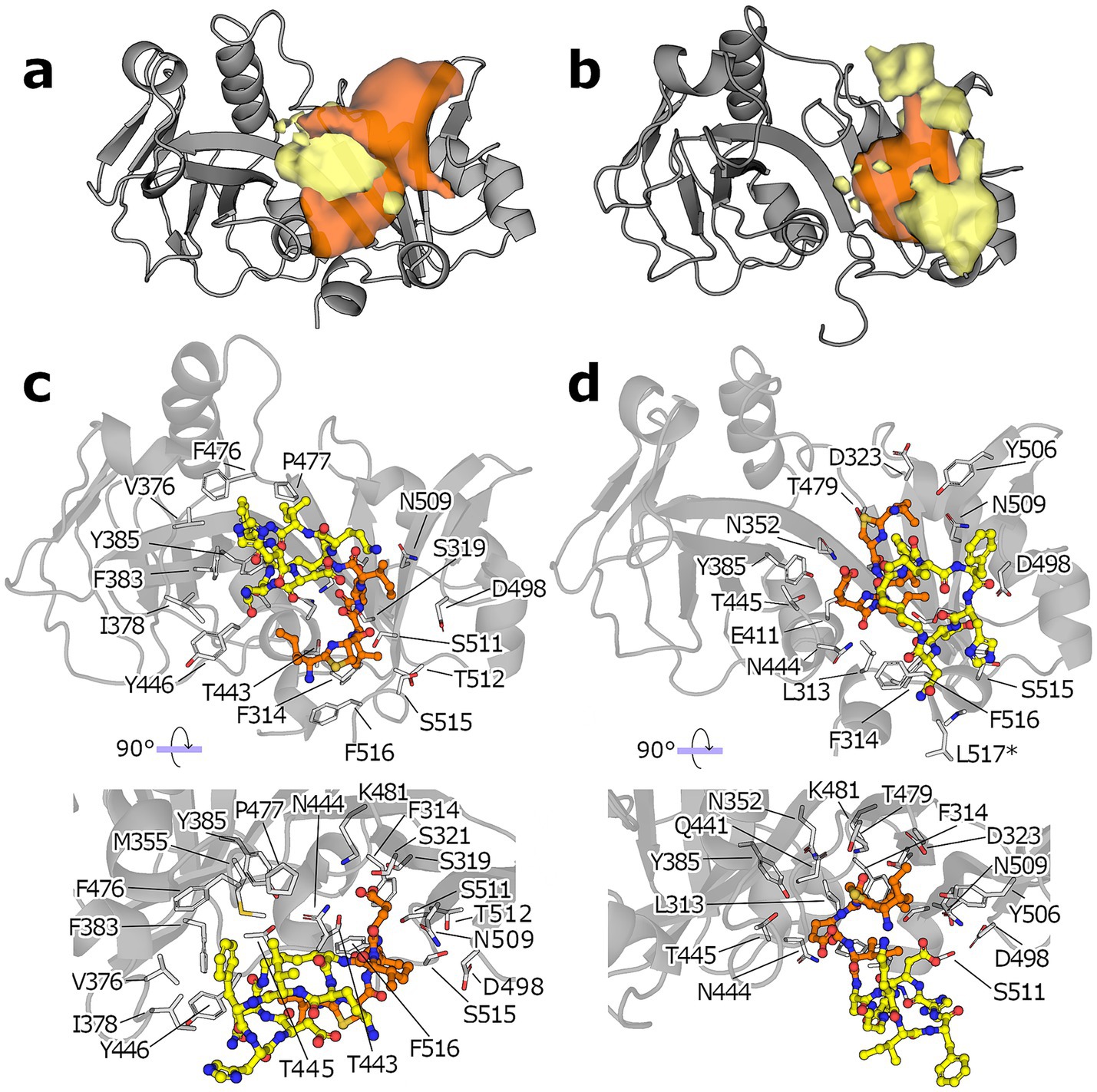
Figure 4. MD simulations of the ECD: bacitracin complex. (a) Occupancy of the macrocyclic region (yellow blob) and the N-terminus (orange blob) of bacitracin throughout the MD simulations starting from the pose C0. (b) Same as in (a) but for simulations starting from pose C1 (protein shown a gray cartoon). All isosurfaces are shown at the same sigma level (0.07). (c) Binding mode of the most populated cluster derived from the MD ensemble started from C0. (d) The protein is shown as gray cartoon, the amino acid side chains closer than 4 Å to any bacitracin atom are shown as white sticks and numbered according to the full length NsrP protein, the macrocyclic region of bacitracin is shown as yellow.
3.4 Structural identification of other BceAB-type transporters
We then investigated whether our results could be extended to other members of the BceAB transporter family. BceAB-type transporters are mainly found in Firmicutes bacteria (Dintner et al., 2011; Kobras et al., 2020). Since sequence conservation is low (Figure 5a), standard BLAST searches based on the SaNsrFP sequence did not yield any hits. Hence, we used the AlphaFold Protein Structure Database (Jumper et al., 2021; Varadi et al., 2022) to search for other BceAB-type transporters applying structural alignments. We identified several BceAB-type ABC transporters in the genomes of different organisms including opportunistic pathogens as well as medically relevant human pathogens and ESKAPE organisms listed by the WHO (Tacconelli et al., 2018), such as S. pneumonia, MRSA, and E. faecium (Supplementary Table 2). A comparison of the predicted structures showed that the transmembrane domain of all BceB is structurally conserved. In contrast, many different structures are found for the ECDs (Supplementary Figure 9).
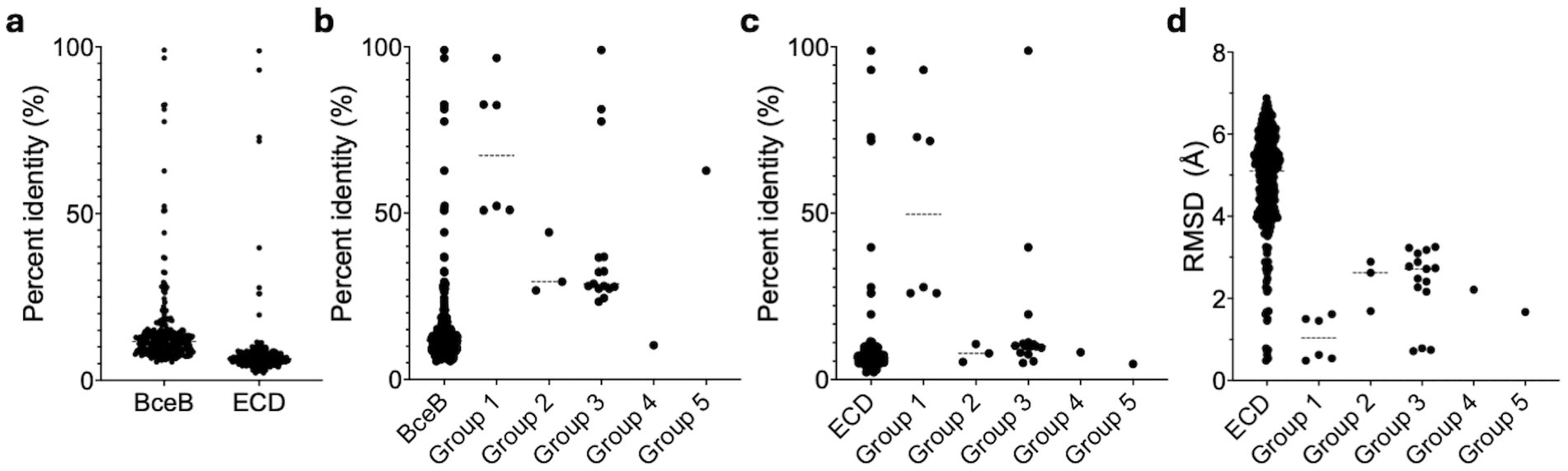
Figure 5. Statistical evaluation of sequence and structure conservation among BceBs and ECDs. (a) Represented is the sequence identity of the mutual alignment of all 24 BceBs/ECDs. The group-specific alignments for the whole BceB and the ECD are highlighted in (b,c) respectively. (d) The mutual RMSD values obtained with the cealign tool of Pymol are displayed for all ECDs and the respective groups. Dashed lines represent averages. Graphs were fitted using GraphPad Prism version 10.3.1 for MacOS, GraphPad Software, San Diego, California USA, www.graphpad.com. n = 276 (all BceBs/ECDs), 6 (Group 1), 3 (Group 2), 15 (Group 3) and 1 (Group 4 and 5).
3.5 Structural alignments of ECDs reveal five distinct groups
The sequence identity observed for the ECD is lower than that of the whole BceB (Figure 5a). Therefore, we constructed structure-based alignments of only the ECD by removing the TMDs of the different BceAB-type transporters found in the AlphaFold database, leaving only the SABRE and Porter subdomains of the ECD with the first helical turn of the stalk helices. ECDs with approximately 180–230 amino acids were used for structural alignments. We determined RMSD values of the ECDs over 110–200 C α atoms with the cealign tool of PyMol.
From all 24 ECDs, we were able to classify 17 ECDs into five groups (Supplementary Figures 9, 10). Based on this grouping, the sequence identity on the whole BceB level increases around 3.5-fold from below 20% to around 70% for group 1 and 5, while groups 2 and 3 show an 2-fold increase to a sequence identity below 40% (Figure 5b). For group 4 a sequence identity of below 20% is maintained. In contrast, the sequence identity of the ECD level is not affected and stays below 20% for all groups except group 1, which shows elevated sequence identity values around 50% (Figure 5c; Supplementary Figure 10). This however, does not correlate to the structural conservation, as the average RMSD within each group is 2-fold lower than the average of all RMSDs from all ECD to ECD comparisons. A average RMSD < 2.5 Å is maintained in all groups. This also correlates visually to the grouping of the AlphaFold models depicted in Figure 6. Group 1 is formed by four, group 2 by three, group 3 by six and group 4 and 5 by two ECDs each.

Figure 6. Superimposition of all ECDs. (a) Shown is the structural alignment of ECDs from BceB (S. pneumoniae, Group 1, red), BceB (S. suis, Group 2, green), BceB (B. subtilis, Group 3, blue), YvcS (B. cereus, Group 4, violet), YxdM (B. subtilis, Group 5, gold). Additional structural alignments are depicted for Group 1 (b), Group 2 (c), Group 3 (d), Group 4 (e) and Group 5 (f). Image was created using ChimeraX Version 1.8.
Overall, conserved secondary structures are observed among all ECDs. In all ECDs, two β-strands connect the SABRE and Porter domains. The Porter subdomain is formed by two conserved β-sheets and two α-helices, while the SABRE subdomain contains three β-strands, a β-β-loop, and one conserved α-helix in front of one of the two domain-connecting β-strands (Figure 7, left). This core structure can then be elongated by a short stretch of α-helices and β-strand segments, which seem to be specific insertions for each group. These group-specific secondary structure elements can be classified into one of three categories.
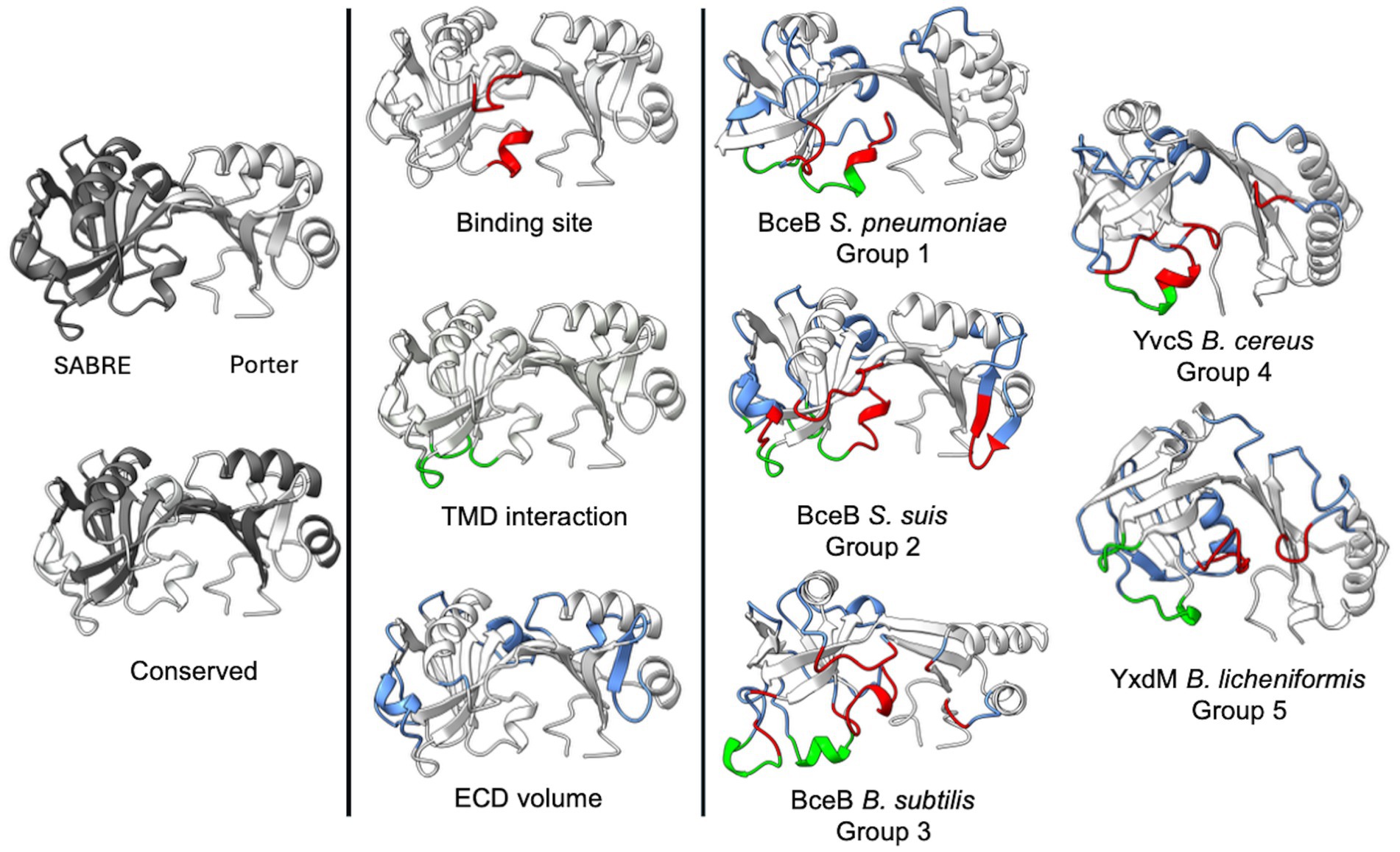
Figure 7. Structural classification of BceAB-type permeases. Depiction of the AlphaFold models of SaNsrP and one representative for each of the five groups. The models show the conserved secondary structure elements (grey/white), as well as elements of category one (red), two (green), and three (blue). Image was created using ChimeraX Version 1.8.
The first category (Figure 7, red) includes α-helices and unstructured loops, which are directly involved in forming the binding interface of the ECD located below the domain-connecting β-strands, between the SABRE and Porter subdomain, where the ligand binding site is predicted to be. Typically, these elements originate from the conserved three β-strands in the SABRE subdomain and reach into the binding interface.
The second category (Figure 7, green) contains α-helices and unstructured loops originating below the conserved β-strands of the SABRE subdomain. These structures are most likely directly involved in interactions with TMH 5–6, helix 9–10, and the binding interface of the ECD and might play a role in signal transduction of a binding event. Thus, the size and position might be directly related to TMH 5–6 and helix 9–10 architecture.
α-helices and β-strands of the third category (Figure 7, blue) expand the overall shape of the ECD but do not influence the binding site and do not contribute to a possible interaction with the TMD.
As the size and position of these elements are group-specific, they might explain how substrates are recognized and how substrate binding can differently affect the full-size transporter.
This highlights that the structure of the ECD might provide a scaffold that restricts the volume of the binding interface of the ECD and, therefore, the bulkiness of substrates recognized, while the specific sequence is responsible for substrate interactions and the overall charge of the binding interface.
4 Discussion
In recent work, L. lactis NZ9000 cells expressing SaNsrFP showed the highest resistance against bacitracin while conferring lower resistance against cationic antimicrobial peptides such as nisin in comparison to the sensitive empty vector control strain (Gottstein et al., 2022). It was concluded in the same study that there is a first-line resistance mechanism against bacitracin by directly binding it or shielding the target UPP. This gives the cell time to react and to rebuild the peptidoglycan layer which becomes impermeable for several structurally diverse cationic antimicrobial peptides. This appears to be a second line of defense against antimicrobial peptides and is coupled to the binding of bacitracin to SaNsrFP and subsequent ATP hydrolysis since the ATP-deficient variant does not show this phenotype.
To gain further insight into how SaNsrFP operates, we expressed and purified the BceAB-type ABC-transporter in LMNG micelles and investigated its ATPase activity. The results show that the WT transporter is active and has a maximal ATPase activity of 135 ± 32.7 nmol/min/mg, while the mutant SaNsrFH202AP is showing only marginal activity. This is in line with the in vivo data for SaNsrFP, where the ATPase-deficient mutant lacks resistance (Reiners et al., 2017), as well as the in vitro characterization of SaNsrF (Furtmann et al., 2020). Next, we probed the influence of bacitracin binding on the ATPase activity. Here, our results show that bacitracin stimulates SaNsrFP ATPase by inducing cooperativity for ATP in the NDB and decreasing the KD by 2.8-fold, while the Bmax remains the same. This stimulation is in line with the work of Diagne et al. (2022), which shows ATPase stimulation of BceAB from S. pneumonia by bacitracin. In this study, the addition of bacitracin increased ATPase levels, but no comment was made regarding KD or possible cooperativity. In contrast, BceAB from B. subtilis (George et al., 2022) showed ATPase inhibition by bacitracin. This suggests that there does not seem to be a uniform response to bacitracin in terms of ATPase activity.
The study of Cho et al. (2021) showed that by swapping the ECD the substrate specificity is altered, which suggests that the ECD is involved in substrate recognition. To confirm this, we successfully expressed and purified the SaNsrP-ECD and demonstrated via SEC-MALS and via measuring intrinsic tyrosine fluorescence that it binds bacitracin. The observed KD shows minimal variation in the presence or absence of zinc ions, implying that the interaction is not influenced by bacitracin conformations.
Putative binding site prediction by SiteMap revealed two distinctive sites at each region of SaNsrP: One at the transmembrane region and another one at the ECD. The presence of two binding sites in NsrP is consistent with the mechanistic model proposed for the homologous protein BceAB (George et al., 2022), which postulates that this transporter detaches the lantibiotic from its lipidic target by first sequestering the complex through interactions with the transmembrane domain, followed by a direct interaction with the lantibiotic occurring through the ECD.
The docking results revealed two main possible binding orientations for bacitracin: Either the macrocyclic ring or the N-terminus of bacitracin is at the center of the ECD (between the Porter and SABRE domains) of which the latter orientation has significantly better scores. Performing MD simulations of the complexes showed some structural variation for bacitracin. Poses in which the macrocycle was at the central cleft showed a more firmly bound macrocyclic region and a highly variable N-terminus region and vice versa. Overall, non-tight binding of bacitracin to the ECD with high conformational fluctuations is not surprising given the binding affinity observed for the complex in the micromolar range. That we observe two putative binding modes for the ligand could be either related to the fact that SaNsrP is not a selective protein and confers resistance against structurally different peptides such as nisin and bacitracin, indicating that it can bind different peptidic moieties. Alternatively, it is also possible that the ECD binds to bacitracin in different ways through the catalytic cycle required to detach it and release it from LIPID II. It is also worth noting, that the significant difference between the KD values observed during our characterization of the ECD compared to the previously reported values for the whole complex (Dintner et al., 2014) suggests that there may be an allosteric contribution of the transmembrane regions to the conformational landscape of the ECD, indicating that the conformation of the ECD may significantly change during the entire catalytic cycle. Nonetheless, the results provide a forward step into constructing an atomistic model of the mechanism of action of SaNsrFP.
Bacitracin resistance has been attributed to several other BceAB-type transporters in B. subtilis, L. monocytogenes, S. mutans, S. aureus (Gebhard, 2012), and S. pneumoniae (Diagne et al., 2022). We further showed that BceAB transporters can be found in opportunistic pathogenic and clinically pathogenic bacterial strains when using structural searches rather than sequence alignments. The presence of these transporters might be the cause of resistance against bacitracin and or other antimicrobial peptides.
Also, we could classify ECDs from BceB models from different bacteria into five groups, identifying conserved secondary structures in all (a set of two β-strands over a set of three β-strands in the SABRE domain, two conserved β-strands and α-helices in the porter domain, as well as two domain connecting β-strands) and individual secondary structure elements unique for each group. Comparing the latter ones between all ECDs enabled us to classify which secondary structures are responsible for generating the binding interface, forming interactions with the TMD, or modulating the ECD shape.
As resistance data for all categorized BceABs is limited (8 out of 17 are partially characterized), a direct correlation between the grouping and mediated resistance is not possible. However, the available data shows that overall BceABs respond to LL-37 (8 of 17 positive) and bacitracin (7 of 17 positive, VraFG negative) (Table 1).
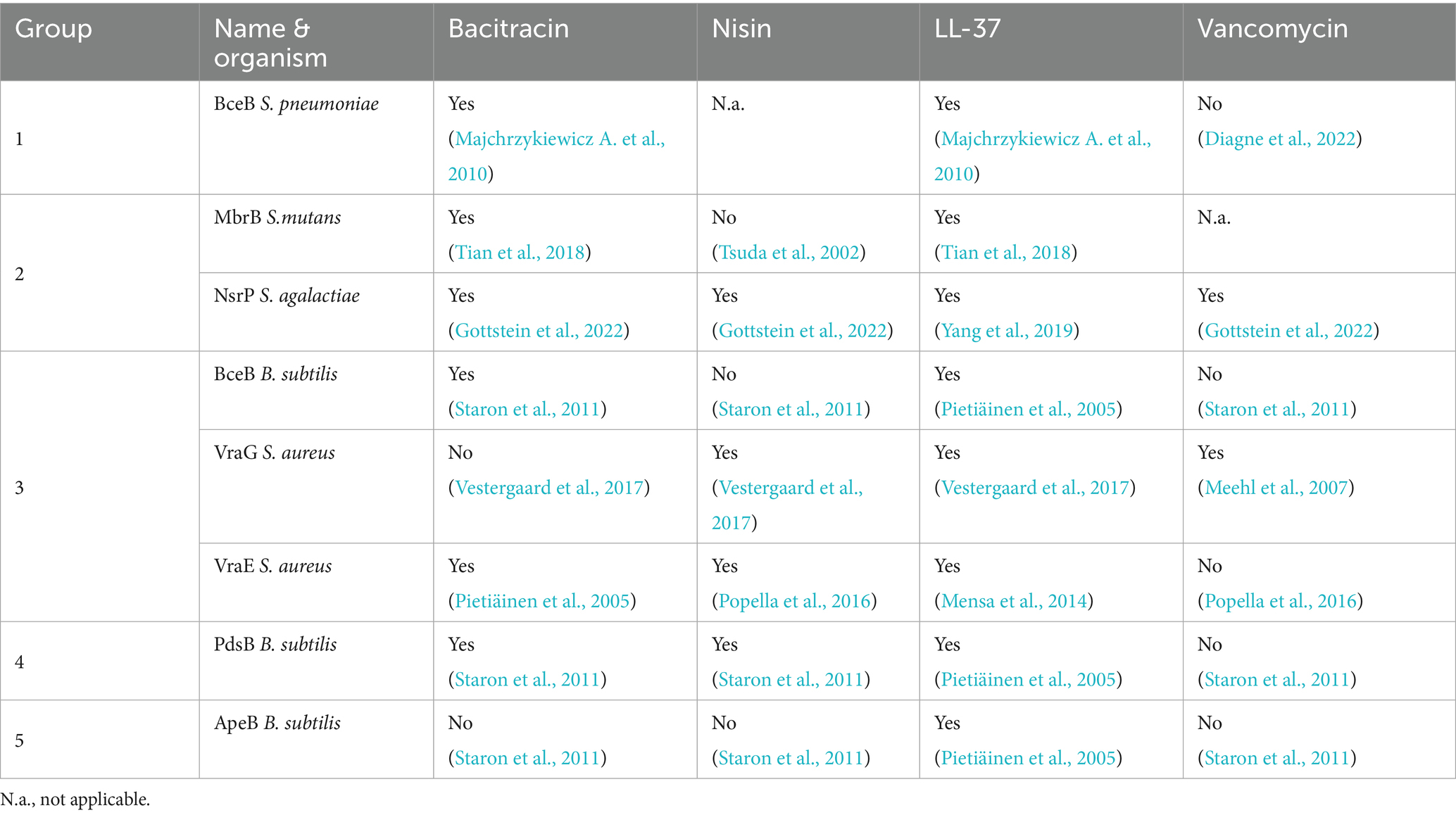
Table 1. Resistance profile of all categorized BceAB-type ABC-transporters.
In our grouping both VraFG and VraDE are placed in Group 3. This is interesting, as Cho et al. (2021) demonstrated that the different resistance profiles regarding bacitracin are inverted if the ECDs of both transporters are swapped. Together with the low sequence identity of around 10%, this suggests that the shape of the ECD might guide the width of the substrate spectrum by restricting the volume in the ECD, while the specific sequence is required for fine-tuning the substrate spectrum. To advance this classification based on structure and sequence identity, more functional resistance data, in vitro binding assays, and especially more substrate-bound structures of BceABs are needed.
Conclusively, BceAB-type transporters such as SaNsrFP are conserved in clinically relevant human pathogenic strains and nonpathogenic strains. Although not conserved at the sequence level, the topology of the protein and its cognate encoding operons are conserved. The results of this study are consistent with previous observations that SaNsrFP confers resistance against bacitracin, and we were able to show that the ECD is the binds bacitracin.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.modelarchive.org/doi/10.5452/ma-m0kcc, https://www.sasbdb.org/data/SASDWZ6/ and https://www.sasbdb.org/data/SASDW27/.
Author contributions
CM: Writing – review & editing, Formal analysis, Investigation, Validation, Writing – original draft, Data curation, Methodology. JG: Formal analysis, Methodology, Writing – review & editing, Investigation. PC: Software, Data curation, Investigation, Writing – review & editing, Methodology, Validation. KT: Investigation, Methodology, Writing – review & editing. JR: Validation, Methodology, Formal analysis, Writing – review & editing, Investigation, Conceptualization, Software. MB: Software, Data curation, Investigation, Writing – review & editing, Methodology. HG: Software, Resources, Project administration, Funding acquisition, Methodology, Investigation, Writing – review & editing, Validation, Supervision. SS: Formal analysis, Project administration, Validation, Supervision, Funding acquisition, Visualization, Conceptualization, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 270650915/GRK 2158 to SS and HG. The Center for Structural Studies is funded by the Deutsche Forschungsgemeinschaft (DFG Grant number 417919780 and INST 208/761-1 FUGG).
Acknowledgments
The authors thank Eymen Hachani for performing the MALS experiment. The authors thank the Institute of Biochemistry for fruitful discussions and are thankful to the CSS for performing SAXS measurements. We are grateful for computational support and infrastructure provided by the “Zentrum für Informations- und Medientechnologie” (ZIM) at the Heinrich-Heine University Düsseldorf. We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities and we would like to thank Anton Popov and Petra Pernot for assistance in using beamline BM29.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1662803/full#supplementary-material
References
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). Accurate structure prediction of biomolecular interactions with alpha fold 3. Nature 630, 493–500. doi: 10.1038/s41586-024-07487-w
Alogheli, H., Olanders, G., Schaal, W., Brandt, P., and Karlen, A. (2017). Docking of macrocycles: comparing rigid and flexible docking in glide. J. Chem. Inf. Model. 57, 190–202. doi: 10.1021/acs.jcim.6b00443
Case, D. A., Aktulga, H. M., Belfon, K., Cerutti, D. S., Cisneros, G. A., Cruzeiro, V. W. D., et al. (2023). AmberTools. J Chem Inf Model, 63, 6183–6191.doi: 10.1021/acs.jcim.3c01153
Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M.JR, et al. (2005). The Amber biomolecular simulation programs. J Comput Chem, 26, 1668–88. doi: 10.1002/jcc.20290
Cho, J., Costa, S. K., Wierzbicki, R. M., Rigby, W. F. C., and Cheung, A. L. (2021). The extracellular loop of the membrane permease VraG interacts with GraS to sense cationic antimicrobial peptides in Staphylococcus aureus. PLoS Pathog. 17:e1009338. doi: 10.1371/journal.ppat.1009338
Cho, J., Rigby, W. F. C., and Cheung, A. L. (2022). The thematic role of extracellular loop of VraG in activation of the membrane sensor GraS in a cystic fibrosis Mrsa strain differs in nuance from the ca-Mrsa strain Je2. PLoS One 17:e0270393. doi: 10.1371/journal.pone.0270393
Clemens, R., Zaschke-Kriesche, J., Khosa, S., and Smits, S. H. J. (2017). Insight into two Abc transporter families involved in lantibiotic resistance. Front. Mol. Biosci. 4:91. doi: 10.3389/fmolb.2017.00091
Collins, B., Curtis, N., Cotter, P. D., Hill, C., and Ross, R. P. (2010). The ABC transporter AnrAB contributes to the innate resistance of Listeria monocytogenes to nisin, bacitracin, and various beta-lactam antibiotics. Antimicrob Agents Chemother, 54, 4416–23. doi: 10.1128/AAC.00503-10
De Souza, B. (2025). GOAT: A Global Optimization Algorithm for Molecules and Atomic Clusters. Angew Chem Int Ed, 64:e202500393. doi: 10.1002/anie.202500393
Diagne, A. M., Pelletier, A., Durmort, C., Faure, A., Kanonenberg, K., Freton, C., et al. (2022). Identification of a two-component regulatory system involved in antimicrobial peptide resistance in Streptococcus pneumoniae. PLoS Pathog. 18:e1010458. doi: 10.1371/journal.ppat.1010458
Dintner, S., Heermann, R., Fang, C., Jung, K., and Gebhard, S. (2014). A sensory complex consisting of an Atp-binding cassette transporter and a two-component regulatory system controls bacitracin resistance in Bacillus subtilis. J. Biol. Chem. 289, 27899–27910. doi: 10.1074/jbc.M114.596221
Dintner, S., Staron, A., Berchtold, E., Petri, T., Mascher, T., and Gebhard, S. (2011). Coevolution of Abc transporters and two-component regulatory systems as resistance modules against antimicrobial peptides in Firmicutes Bacteria. J. Bacteriol. 193, 3851–3862. doi: 10.1128/JB.05175-11
Falord, M., Karimova, G., Hiron, A., and Msadek, T. (2012). Graxsr proteins interact with the Vrafg Abc transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 1047–1058. doi: 10.1128/AAC.05054-11
Fritz, G., Dintner, S., Treichel, N. S., Radeck, J., Gerland, U., Mascher, T., et al. (2015). A new way of sensing: need-based activation of antibiotic resistance by a flux-sensing mechanism. MBio 6:e00975. doi: 10.1128/mBio.00975-15
Furtmann, F., Porta, N., Hoang, D. T., Reiners, J., Schumacher, J., et al. (2020). Characterization of the nucleotide-binding domain NsrF from the Bceab-type Abc-transporter Nsrfp from the human pathogen Streptococcus agalactiae. Sci. Rep. 10:15208. doi: 10.1038/s41598-020-72237-7
Gebhard, S. (2012). Abc transporters of antimicrobial peptides in Firmicutes bacteria - phylogeny, function and regulation. Mol. Microbiol. 86, 1295–1317. doi: 10.1111/mmi.12078
Gebhard, S., and Mascher, T. (2011). Antimicrobial peptide sensing and detoxification modules: unravelling the regulatory circuitry of Staphylococcus aureus. Mol. Microbiol. 81, 581–587. doi: 10.1111/j.1365-2958.2011.07747.x
George, N. L., and Orlando, B. J. (2023). Architecture of a complete Bce-type antimicrobial peptide resistance module. Nat. Commun. 14:3896. doi: 10.1038/s41467-023-39678-w
George, N. L., Schilmiller, A. L., and Orlando, B. J. (2022). Conformational snapshots of the bacitracin sensing and resistance transporter Bceab. Proc. Natl. Acad. Sci. USA 119:e2123268119. doi: 10.1073/pnas.2123268119
Gottstein, J., Zaschke-Kriesche, J., Unsleber, S., Voitsekhovskaia, I., Kulik, A., Behrmann, L. V., et al. (2022). New insights into the resistance mechanism for the Bceab-type transporter SaNsrfp. Sci. Rep. 12:4232. doi: 10.1038/s41598-022-08095-2
Guinier, A. (1939). Small-angle X-ray diffraction: application to the study of ultramicroscopic phenomena. Ann. Phys. 11, 161–237.
Halgren, T. A. (2009). Identifying and characterizing binding sites and assessing Druggability. J. Chem. Inf. Model. 49, 377–389. doi: 10.1021/ci800324m
Hiron, A., Falord, M., Valle, J., Debarbouille, M., and Msadek, T. (2011). Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (Sa2417/Sa2418) and both the BraD/BraE and VraD/VraE Abc transporters. Mol. Microbiol. 81, 602–622. doi: 10.1111/j.1365-2958.2011.07735.x
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J Mol Graph, 14, 27–8. doi: 10.1016/0263-7855(96)00018-5
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kanonenberg, K., Royes, J., Kedrov, A., Poschmann, G., Angius, F., Solgadi, A., et al. (2019). Shaping the lipid composition of bacterial membranes for membrane protein production. Microb. Cell Factories 18:131. doi: 10.1186/s12934-019-1182-1
Khosa, S., Alkhatib, Z., and Smits, S. H. (2013). Nsr from Streptococcus agalactiae confers resistance against nisin and is encoded by a conserved nsr operon. Biol. Chem. 394, 1543–1549. doi: 10.1515/hsz-2013-0167
Kingston, A. W., Zhao, H., Cook, G. M., and Helmann, J. D. (2014). Accumulation of heptaprenyl diphosphate sensitizes Bacillus subtilis to bacitracin: implications for the mechanism of resistance mediated by the Bceab transporter. Mol. Microbiol. 93, 37–49. doi: 10.1111/mmi.12637
Kobras, C. M., Piepenbreier, H., Emenegger, J., Sim, A., Fritz, G., and Gebhard, S. (2020). Bceab-type antibiotic resistance transporters appear to act by target protection of Cell Wall synthesis. Antimicrob. Agents Chemother. 64:19. doi: 10.1128/AAC.02241-19
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003). Primus: a windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. doi: 10.1107/S0021889803012779
Kozin, M. B., and Svergun, D. I. (2001). Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41. doi: 10.1107/S0021889800014126
Kräutler, V., Van gunsteren, W. F., and Hünenberger, P. H. (2001). A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem., 22, 501–508. doi: 10.1002/1096-987X(20010415)22:5%3C501::AID-JCC1021%3E3.0.CO;2-V
Madeira, F., Madhusoodanan, N., Lee, J., Eusebi, A., Niewielska, A., Tivey, A. R. N., et al. (2024). The Embl-Ebi job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525. doi: 10.1093/nar/gkae241
Majchrzykiewicz, J. A., Kuipers, O. P., and Bijlsma, J. J. E. (2010). Generic and specific adaptive responses of Streptococcus pneumoniae to challenge with three distinct antimicrobial peptides, bacitracin, Ll-37, and nisin. Antimicrob. Agents Chemother. 54, 440–451. doi: 10.1128/AAC.00769-09
Manalastas-Cantos, K., Konarev, P. V., Hajizadeh, N. R., Kikhney, A. G., Petoukhov, M. V., Molodenskiy, D. S., et al. (2021). Atsas 3.0: expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 54, 343–355. doi: 10.1107/S1600576720013412
Mascher, T. (2006). Intramembrane-sensing histidine kinases: a new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol. Lett. 264, 133–144. doi: 10.1111/j.1574-6968.2006.00444.x
Mascher, T., Margulis, N. G., Wang, T., Ye, R. W., and Helmann, J. D. (2003). Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol. Microbiol. 50, 1591–1604. doi: 10.1046/j.1365-2958.2003.03786.x
Meehl, M., Herbert, S., Götz, F., and Cheung, A. (2007). Interaction of the Grars two-component system with the Vrafg Abc transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 51, 2679–2689. doi: 10.1128/AAC.00209-07
Mensa, B., Howell Gabriella, L., Scott, R., and Degrado William, F. (2014). Comparative mechanistic studies of Brilacidin, Daptomycin, and the antimicrobial peptide Ll16. Antimicrob. Agents Chemother. 58, 5136–5145. doi: 10.1128/AAC.02955-14
Mirdita, M., Schutze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., and Steinegger, M. (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. doi: 10.1038/s41592-022-01488-1
Muller, A., Klockner, A., and Schneider, T. (2017). Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep. 34, 909–932. doi: 10.1039/C7NP00012J
Orlando, B. J. (2024). Perception and protection: the role of Bce-modules in antimicrobial peptide resistance. Biochim. Biophys. Acta Biomembr. 1866:184309. doi: 10.1016/j.bbamem.2024.184309
Pietiäinen, M., Gardemeister, M., Mecklin, M., Leskelä, S., Sarvas, M., and Kontinen, V. P. (2005). Cationic antimicrobial peptides elicit a complex stress response in Bacillus subtilis that involves Ecf-type sigma factors and two-component signal transduction systems. Microbiology 151, 1577–1592. doi: 10.1099/mic.0.27761-0
Popella, P., Krauss, S., Ebner, P., Nega, M., Deibert, J., and Götz, F. (2016). VraH is the third component of the Staphylococcus aureus Vradeh system involved in Gallidermin and Daptomycin resistance and pathogenicity. Antimicrob. Agents Chemother. 60, 2391–2401. doi: 10.1128/AAC.02865-15
Reiners, J., Lagedroste, M., Ehlen, K., Leusch, S., Zaschke-Kriesche, J., and Smits, S. H. J. (2017). The N-terminal region of Nisin is important for the Bceab-type Abc transporter Nsrfp from Streptococcus agalactiae Coh1. Front. Microbiol. 8:1643. doi: 10.3389/fmicb.2017.01643
Reiners, J., Lagedroste, M., Gottstein, J., Adeniyi, E. T., Kalscheuer, R., Poschmann, G., et al. (2020). Insights in the antimicrobial potential of the natural Nisin variant Nisin H. Front. Microbiol. 11:573614. doi: 10.3389/fmicb.2020.573614
Repasky, M. P., Shelley, M., and Friesner, R. A. (2007). Flexible ligand docking with glide. Curr. Protoc. Bioinformatics 8:12. doi: 10.1002/0471250953.bi0812s18
Revilla-Guarinos, A., Gebhard, S., Mascher, T., and Zuniga, M. (2014). Defence against antimicrobial peptides: different strategies in Firmicutes. Environ. Microbiol. 16, 1225–1237. doi: 10.1111/1462-2920.12400
Rietkötter, E., Hoyer, D., and Mascher, T. (2008). Bacitracin sensing in Bacillus subtilis. Mol. Microbiol. 68, 768–785. doi: 10.1111/j.1365-2958.2008.06194.x
Roe, D. R., and Cheatham, T. E. (2013). Ptraj and cpptraj: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095. doi: 10.1021/ct400341p
Rostkowski, M., Olsson, M. H., Sondergaard, C. R., and Jensen, J. H. (2011). Graphical analysis of pH-dependent properties of proteins predicted using Propka. BMC Struct. Biol. 11:6. doi: 10.1186/1472-6807-11-6
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le grand, S., and Walker, R. C. (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput, 9, 3878–3888. doi: 10.1021/ct400314y
Slotboom, D. J., Duurkens, R. H., Olieman, K., and Erkens, G. B. (2008). Static light scattering to characterize membrane proteins in detergent solution. Methods 46, 73–82. doi: 10.1016/j.ymeth.2008.06.012
Staron, A., Finkeisen, D. E., and Mascher, T. (2011). Peptide antibiotic sensing and detoxification modules of Bacillus subtilis. Antimicrob. Agents Chemother. 55, 515–525. doi: 10.1128/AAC.00352-10
Svergun, D. I. (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503. doi: 10.1107/S0021889892001663
Svergun, D. I., Petoukhov, M. V., and Koch, M. H. (2001). Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953. doi: 10.1016/S0006-3495(01)76260-1
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: the who priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
Thomas, C., Aller, S. G., Beis, K., Carpenter, E. P., Chang, G., Chen, L., et al. (2020). Structural and functional diversity calls for a new classification of Abc transporters. FEBS Lett. 594, 3767–3775. doi: 10.1002/1873-3468.13935
Tian, X.-L., Salim, H., Dong, G., Parcells, M., and Li, Y.-H. (2018). The Bceabrs four-component system that is essential for cell envelope stress response is involved in sensing and response to host defence peptides and is required for the biofilm formation and fitness of Streptococcus mutans. J. Med. Microbiol. 67, 874–883. doi: 10.1099/jmm.0.000733
Tollerson, R., and Ibba, M. (2020). Translational regulation of environmental adaptation in bacteria. J. Biol. Chem. 295, 10434–10445. doi: 10.1074/jbc.REV120.012742
Tsuda, H., Yamashita, Y., Shibata, Y., Nakano, Y., and Koga, T. (2002). Genes involved in bacitracin resistance in Streptococcus mutans. Antimicrob. Agents Chemother. 46, 3756–3764. doi: 10.1128/AAC.46.12.3756-3764.2002
Tully, M. D., Kieffer, J., Brennich, M. E., Cohen Aberdam, R., Florial, J. B., Hutin, S., et al. (2023). Biosaxs at European Synchrotron Radiation Facility - extremely brilliant source: Bm29 with an upgraded source, detector, robot, sample environment, data collection and analysis software. J. Synchrotron Radiat. 30, 258–266. doi: 10.1107/S1600577522011286
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. doi: 10.1093/nar/gkab1061
Varadi, M., Bertoni, D., Magana, P., Paramval, U., Pidruchna, I., Radhakrishnan, M., et al. (2024). AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–d375. doi: 10.1093/nar/gkad1011
Vestergaard, M., Nøhr-Meldgaard, K., Bojer Martin, S., Krogsgård Nielsen, C., Meyer Rikke, L., et al. (2017). Inhibition of the Atp synthase eliminates the intrinsic resistance of Staphylococcus aureus towards polymyxins. MBio 8:e01114-17. doi: 10.1128/mbio.01114-17
Wang, J., Cieplak, P., and Kollman, P. A.. (2000). How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem, 21, 1049–1074. doi: 10.1002/1096-987X(200009)21:12%3C1049::AID-JCC3%3E3.0.CO;2-F
Watts, K. S., Dalal, P., Tebben, A. J., Cheney, D. L., and Shelley, J. C. (2014). Macrocycle conformational sampling with MacroModel. J. Chem. Inf. Model. 54, 2680–2696. doi: 10.1021/ci5001696
Keywords: BceAB, antimicrobial resistance, lantibiotic, ABC transporter, human pathogen
Citation: Mammen C, Gottstein J, Cea P, Tantsur K, Reiners J, Bonus M, Gohlke H and Smits SHJ (2025) The extracellular domain of SaNSrFP binds bacitracin and allows the identification of new members of the BceAB transporter family. Front. Microbiol. 16:1662803. doi: 10.3389/fmicb.2025.1662803
Edited by:
Manoj Kumawat, ICMR-National Institute for Research in Environmental Health, IndiaReviewed by:
Tahir Hussain, Iowa State University, United StatesJoão Paquete-Ferreira, New University of Lisbon, Portugal
Copyright © 2025 Mammen, Gottstein, Cea, Tantsur, Reiners, Bonus, Gohlke and Smits. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sander H. J. Smits, c2FuZGVyLnNtaXRzQGhodS5kZQ==