 Bo Wang1,2,3Yang Liu4Xinhua Wu5
Bo Wang1,2,3Yang Liu4Xinhua Wu5 Yunfei Liu5Ziying Li5Jian Wang1,3Yingli Lian1,3Jiayi Tang1,3Biao Yun1,3
Yunfei Liu5Ziying Li5Jian Wang1,3Yingli Lian1,3Jiayi Tang1,3Biao Yun1,3 Xiangli Tian4*
Xiangli Tian4*- 1Animal Husbandry and Fisheries Research Center of Guangdong Haid Group Co., Ltd., Guangzhou, China
- 2State Key Laboratory of Tropical Oceanography, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 3Key Laboratory of Microecological Resources and Utilization in Breeding Industry, Ministry of Agriculture and Rural Affairs, Guangdong Haid Group Co., Ltd., Guangzhou, China
- 4The Key Laboratory of Mariculture, Ocean University of China, Ministry of Education, Qingdao, China
- 5Bohai Seafoods Co., Ltd., Binzhou, China
An increasing number of studies have evaluated the effects of host, dietary, and environmental factors on the gut microbial community of Penaeus vannamei. However, the characteristics of the gut microbial community of this species in hypersaline aquaculture environments have not yet been clarified. Our findings demonstrate that salinity has a strong impact on the gut bacterial community of shrimp. The alpha diversity of the gut bacterial community of shrimp decreased with salinity. Significant differences in the composition and abundance of the core gut bacterial taxa were observed among ponds with varying salinity, and only 13 shared core operational taxonomic units (OTUs) were identified; the abundance of potential opportunistic pathogens decreased significantly in hypersaline environments. Salinity is identified as a critically important environmental factor affecting the structure of the gut bacterial community of shrimp in hypersaline environments. The structure of the gut bacterial community of shrimp was distinct at salinities of 31–39 and 47–55, and the predicted functions differed at salinities of 31–47 and 55 based on 16S rRNA gene prediction using PICRUSt2 and principal coordinate analysis. Network analysis showed that higher salinity was associated with less connectivity and cooperation among species. Neutral Community Model analysis and the normalized stochasticity ratio revealed that stochastic processes were dominant at lower salinity; however, deterministic processes became more important as salinity increased. In addition, the community-level habitat niche breadths of the gut bacterial community decreased with salinity, which further confirmed this trend. These findings provide new insights into the characteristics of the gut bacterial community of shrimp in hypersaline environments and would contribute to the improvement of farming health management of shrimp in hypersaline ponds aquaculture practices.
1 Introduction
Gut microbiota are critical for maintaining host health by regulating growth performance, nutrient absorption, metabolic processes, immune responses, and the maintenance of homeostasis (Hooper et al., 2002; Hooper and Macpherson, 2010; Boulangé et al., 2016; Cornejo-Granados et al., 2017; Cani et al., 2019). Studies of the microbial characteristics of the host gut and the factors affecting their characteristics are thus critically important. Previous studies have revealed a large number of diseases associated with dysbiosis of the host intestinal microbiota (Dai et al., 2020; de Souza Valente et al., 2020; Huang et al., 2020; Zhang et al., 2021). Many studies have reported that environmental factors, such as the salinity, ammonia nitrogen, temperature, and pH of culture water, have significant effects on the intestinal microbiota of aquatic animals (Sullam et al., 2012; Huang et al., 2018; Hou et al., 2020; Wang et al., 2021). In recent years, the differential effects of seawater and freshwater on the intestinal microbiota of aquatic animals have been investigated; however, the effects of high salinity on the intestinal microbiota of aquatic animals have not been widely examined.
The Pacific white shrimp (Penaeus vannamei), a native species inhabiting the eastern Pacific coast from Mexico to Peru (FAO, 2018), can grow and survive at a wide salinity range from 0.5 to 60. This species is cultivated throughout the world for its rapid growth rate, tender flesh, and high nutritional value (Jin et al., 2018). China is currently the world’s largest white shrimp producer. The Pacific white shrimp seawater cultivation area was 1.63 × 105 ha in 2022, and 1.34 million tons of Pacific white shrimp were produced in this same year (China Fishery Statistical Yearbook, 2023). P. vannamei can adapt to a wide range of environmental salinities; thus, clarifying the responses of P. vannamei to various salinities is critically important for ensuring the efficacy of aquaculture management. Understanding the effects of salinity on the gut microbial community might shed light on the mechanisms underlying differences in the growth performance of P. vannamei at different salinities. Most previous studies have focused on characterizing differences in the gut bacterial community of shrimp cultured in freshwater and seawater (Roy et al., 2010; Hou et al., 2020); however, few studies have determined the characteristics of the gut bacterial community of shrimp cultured in hypersaline environments (salinity more than 35).
In northern China, especially in Liaoning, Tianjin, Shandong, and Hebei Provinces, which comprise the coasts along the Yellow and Bohai Seas, there is a vast area of primary salt evaporation ponds for salterns, which cover an area greater than 1.33 × 105 ha (unpublished data), and shrimp aquaculture in this area has greatly increased the economic benefits of salterns. In these types of ponds, the farming area is large (these are commonly referred to as “Dawangzi” in Chinese, which means large ponds in salt pan), and the salinity is high; the stocking density and survival rate of shrimp are low in these areas, but the flavor of shrimp is improved. Few studies have examined the ecological characteristics of shrimp and environments in these aquaculture ecosystems. There is thus a need for more studies of the gut microbiota of P. vannamei cultured in hypersaline environments. Our study was conducted in Binzhou, Shandong Province, which has a large number of primary salt evaporation ponds that cover an area of more than 5.33 × 104 ha, and the salinity ranges from 31 to 65. The average production of shrimp per ha in such ponds is lower than that in conventional culture ponds (Supplementary Table S1); there is thus much room for improvement in the natural resource utilization and economic benefits of salterns.
Here, we investigated the primary salt evaporation ponds for shrimp culture in Bohai Seafoods Co., Ltd., (Binzhou, China) to characterize the responses of the gut bacterial community of P. vannamei to various hypersaline environments. The findings from the present study would provide new insights into the characteristics of the gut bacterial community of shrimp in hypersaline environments and contribute to the improvement of farming health management in hypersaline ponds.
2 Materials and methods
2.1 Sample collection and physicochemical analysis
A total of 120 shrimp were collected from four culture ponds [six sampling positions per pond, five shrimp per position were collected from Bohai Seafoods Co., Ltd., (Binzhou, China)] (38.03°–38.06°N, 117.56°–118.01°E), in September 2020, and the 5 shrimp per replicate pooled into a single sample. The shrimp in the four ponds came from the same batch of postlarvae and their rearing days (~100 days) were consistent. Four shrimp culture ponds were randomly sampled, A (106.7 ha in area), B (112.7 ha in area), C (133 ha in area), and D (167 ha in area); the depth of the water in these ponds ranged between 0.5 and 3 m, and the salinity was 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74, respectively. Intestinal samples were aseptically dissected from each shrimp using sterile surgical instruments and immediately transferred to individual 2-mL sterile centrifuge tubes containing. All samples were flash-frozen in liquid nitrogen and stored at −80 °C until genomic DNA extraction. The average body weight of shrimp ranged from 14 to 15 g. For each pond, six water samples (1.0 L water of each sample) were collected at depths of 0.5 m below the surface using sterile bottles, and samples were immediately placed on ice before being filtered through 0.45 μm glass fiber filter membranes using peristaltic pumps; the filtered water was immediately stored at 4 °C and then transported to the laboratory for water quality analysis. Temperature, pH, DO, and salinity were measured on-site using a YSI handheld multi-parameter instrument (Model YSI ProPlus, YSI Incorporated, United States). Total nitrogen (TN), total phosphorus (TP), ammonia nitrogen (NH4+-N), nitrate nitrogen (NO3−-N), nitrite nitrogen (NO2−-N), and sulfide and active phosphorus (PO43−-P) were measured using an automatic discrete analyzer (Model CleverChem 380, DeChem-Tech, Germany). The complete water quality analysis results for all four ponds are provided in the Supplementary Table S2.
2.2 DNA extraction, amplification, and sequencing
Genomic DNA from shrimp guts was extracted using a PowerSoil DNA Isolation Kit (Mobio, Carlsbad, CA, United States) according to the manufacturer’s protocol. The V4 region of the 16S rRNA gene from bacteria and archaea was amplified using the primers 515FmodF (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806RmodR (5′-GGACTACNVGGGTWTCTAAT-3′). PCR reactions were performed in 20 μL mixtures containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu polymerase, and 10 ng of purified DNA as a template. The thermal cycling parameters were as follows: 95 °C for 3 min; 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s; and a final extension at 72 °C for 10 min. The quality of the PCR products was detected using 1.5% agarose gel electrophoresis. Subsequently, the purified PCR products were sequenced on an Illumina MiSeq platform by Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). Raw 16S rRNA sequence data were submitted to the NCBI Sequence Read Archive (SRA) database under accession number PRJNA833737.
2.3 Sequence data processing
Raw sequencing data generated from the Illumina MiSeq platform were merged with FLASH (version 1.2.11) (Mago and Salzberg, 2011). For quality control, merged sequences were processed using the Quantitative Insights into Microbial Ecology software (QIIME version 1.9.1) (Caporaso et al., 2010). All chimeric and normal-quality reads were removed using the UCHIME algorithm (Edgar et al., 2011), and the qualified sequences were clustered into operational taxonomic units (OTUs) using a 97% similarity threshold via UPARSE (version 7.0.1090) (Edgar, 2013). The most abundant sequence of each OTU was used as the representative sequence; taxonomic annotations of the sequences were obtained using the RDP Classifier algorithm (Wang et al., 2007) and the SILVA database 138, and a close relative could be identified for each OTU. Rarefaction curves based on the observed features and Shannon indexes were drawn to determine the sequencing depth. QIIME software (version 1.9.1) was used to analyze alpha diversity, including community richness indexes (ACE and Chao1), community diversity indexes (Shannon and Simpson), and Good’s coverage index. The Shannon and Simpson indexes are commonly used to quantify diversity, and the Chao1 and ACE indexes are used to quantify richness. Principal coordinate analysis (PCoA) and PERMANOVA analysis were conducted to evaluate differences in bacterial community structure based on Bray-Curtis distance metrics in R software, and QIIME was used to construct phylogenetic trees based on Bray-Curtis distances to investigate the relationships among samples. Linear discriminant analysis (LDA) effect size (LEfSe) was used to determine statistically differential taxa (biomarkers) (Segata et al., 2011). To investigate the relationships between microbial community structure and environmental factors, redundancy analysis (RDA) was performed using CANOCO 5.0 software. Analysis of variance (ANOVA) followed by stepwise ordination was used to determine the significance of the overall model and perform RDA; the significance of each of the environmental variables was then determined based on the p-values. The concept of the core microbiome considers persistent and highly abundant microbes in a microbial community, which is a stable community (Shade and Handelsman, 2012). The core gut microbiome of shrimp comprised the microbes that were present at all regional sites and in ≥80% of all samples with average relative abundances ≥0.1% (Wu et al., 2019).
2.4 Co-occurrence pattern analysis
Co-occurrence network analysis was performed on the 100 most abundant bacterial genera. The CoNet plug-in of Cytoscape 3.9.0 was used to construct the network (Faust et al., 2012). The network was constructed using four different algorithms: Pearson correlation, Spearman correlation, Kullback–Leibler distance, and Bray-Curtis distance. The use of multiple algorithms effectively reduces the likelihood of constructing erroneous network relationships, which yields more realistic and accurate network relationships. The final network was obtained through several steps such as Permutations, Bootstrapping, and Restoring using Brown’s p-value merging method and Benjamini-Hochberg’s multiple test correction method. The topological properties of the network were analyzed by Network Analyzer in Cytoscape 3.9.0.
2.5 Prediction of gut bacterial community functions
The OTU table was normalized by dividing their abundances by their known or predicted 16S rRNA gene copy number abundances prior to making final metagenomic predictions. Bacterial community functions were predicted from 16S rRNA sequencing data using PICRUSt2, and the predicted functions were annotated at levels 1, 2, and 3 using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Langille et al., 2013). A principal coordinate analysis (PCoA) was performed to analyze the similarity in the content of level 3 predicted functions for the gut bacterial community based on the Bray-Curtis distance; PERMANOVA was used to evaluate the significance of differences between groups. One-way ANOVA was performed to evaluate the significance of differences in level 2 and level 3 predicted functions.
2.6 Bacterial community assembly analysis
A Neutral Community Model (NCM) was used to determine the potential contribution of stochastic processes to gut bacterial community assembly by predicting the relationship between the occurrence frequency of OTUs and their relative abundance (Sloan et al., 2010). The R2 value represents the goodness of fit of the model, which ranges between 0 to 1. The higher R2 indicates that the community assembly is closer to stochastic process. NCM was performed in R version 4.2.2 using the “Hmisc,” “minpack.lm,” and “stats4” packages. We also calculated the normalized stochasticity ratio (NST) to quantify the relative importance of stochastic and deterministic processes in community assembly using 50% as the threshold for inferring whether assembly was more deterministic (<50%) or stochastic (>50%) (Ning et al., 2019). This analysis was performed in R version 4.2.2 using the “NST” package. Habitat niche breadth was calculated using Levin’s niche breadth index, which was determined as follows:
where Bj represents the habitat niche breadth of OTUj in a metacommunity, N represents the total number of communities of each metacommunity, and Pij represents the proportion of OTU j in community I (Pandit et al., 2009; Wu et al., 2017). A high B-value indicates that the OTU is widespread and uniform at a variety of sites, which indicates a wide habitat niche breadth. We calculated the average B values of all taxa in a single community (Bcom) as an indicator of habitat niche breadth at the community level. The analysis was performed in R version 4.2.2 using the “niche.width” function and “spaa” package (Zhang, 2013; Zhang M. et al., 2016).
2.7 Statistical analysis
Results were expressed as mean ± standard error of the mean. One-way ANOVA followed by Duncan’s multiple-range test was used to compare the significance of differences in alpha diversity indexes among gut samples using SPSS22.0. A value of p < 0.05 was considered statistically significant.
3 Results
3.1 16S rRNA gene sequencing and analysis of bacterial diversity in shrimp gut
A total of 1,232,430 high-quality sequences with an average of 61,296 sequences (35,873 to 148,098) were obtained (Supplementary Table S3); after subsampling 35,873 minimum sequences per sample, 717,460 sequences were retained. Based on analysis of the rarefaction curve and Shannon-Wiener curve, the sequencing depth was sufficient for sampling all bacterial diversity (Supplementary Figure S1). Sequences with 97% similarity were clustered into a class, and a total of 23,167 OTUs were identified. The numbers of OTUs detected in each sample ranged from 350 to 1,476 (Supplementary Table S3).
Bacterial community complexity was estimated and compared by calculating the diversity index and richness index for all samples. The diversity (Shannon and Simpson) of the shrimp gut bacterial community at salinities A (31 ± 0.85) and B (39 ± 1.23) was significantly higher than that at salinities C (47 ± 0.62) and D (55 ± 1.74) (p < 0.05) (Table 1). The richness (Chao1 and ACE) of the gut bacterial community of shrimp at salinities A and B was also significantly higher than that at salinities C and D (p < 0.05). Although the differences were not always statistically significant, the richness and diversity of gut bacterial community of shrimp were highest at salinity A, followed by salinity B, C, and D. The Good’s coverage index for each sample was greater than 99.30% (99.308 to 99.679%), suggesting that the sequencing depth was sufficient for microbial community analysis. A PCoA, based on the Bray-Curtis distance algorithm at the OTU level, was performed to characterize the beta diversity of the gut bacterial community of shrimp. Significant separation was observed between the structure of the gut bacterial community at salinities A and B and that at salinities C and D along the PC1 axis, (p < 0.05) (Figure 1). The PC1 and PC2 axes explained 40.65% of the variation between samples (Figure 1a). To investigate whether there are significant differences in the bacterial community between samples in specific evolutionary lineages, a hierarchical cluster tree based on the Bray-Curtis distance algorithm at the OTU level was constructed (Figure 1b). The gut bacterial community samples at salinities A and B were clustered on the same branch, suggesting that the gut bacterial samples at salinities A and B were closely related; the gut bacterial community samples at salinities C and D were clustered on the same branch with the exception of the DSc_6, suggesting that the gut bacteria samples from salinities C and D were closely related.

Table 1. Richness and diversity indexes for gut bacterial community in shrimp at different salinity.
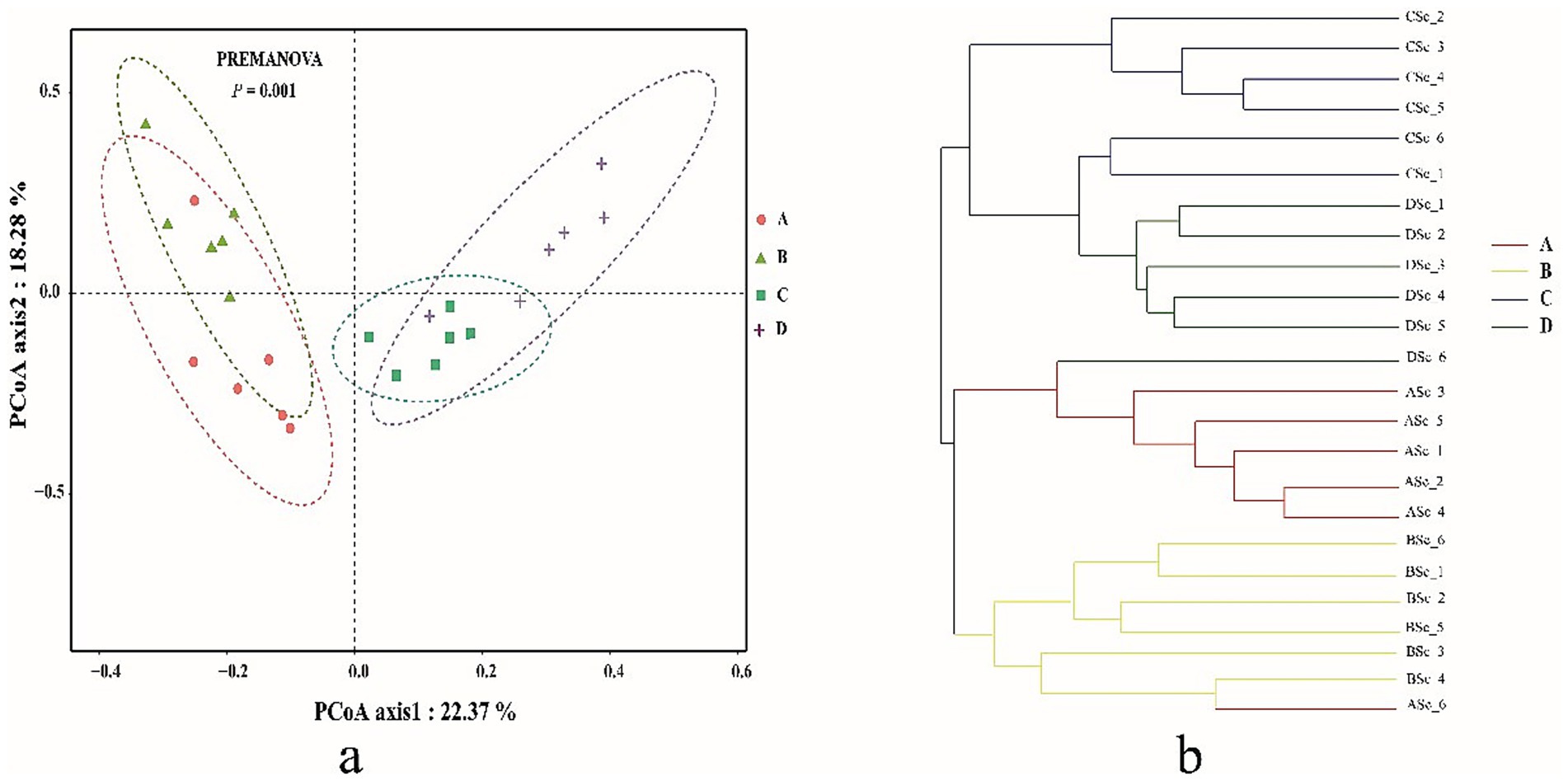
Figure 1. The principal co-ordinates analysis (PCoA) (a) and the hierarchical clustering tree (b) of gut bacterial community in shrimp. A, B, C, D mean salinity 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74.
3.2 Taxonomic composition of the gut bacterial community and biomarker analysis
To compare the gut bacterial community composition of shrimp at the four salinities, the bacterial profiles at the phylum, family, and genus levels were evaluated. Cyanobacteria, Proteobacteria, Planctomycetes, Firmicutes, and Actinobacteria were the five predominant phyla (Figure 2a). The relative abundance of Cyanobacteria, Firmicutes, Actinobacteria, and Bacteroidetes increased with salinity (Figure 2a). The relative abundance of Proteobacteria, norank_d_Bacteria, and Chloroflexi first increased and then decreased with salinity (Figure 2a). The relative abundance of Planctomycetes and Verrucomicrobia decreased with salinity (Figure 2a). Pirellulaceae, Cyanobiaceae, Phormidiaceae, and Mycoplasmataceae were the most abundant families (Figure 2b). The relative abundance of Cyanobiaceae, Mycoplasmataceae, Rhodobacteraceae, Microbacteriaceae, and Burkholderiaceae increased with salinity, and they were significantly enriched in the gut of shrimp at salinities C and D (p < 0.05) (Figure 2b). The relative abundance of some potential opportunistic pathogens such as Vibrio, Pseudomonas, Candidatus_Bacilloplasma, and Photobacterium was significantly higher at salinities A and B than at salinities C and D (Figure 2c). Synechococcus_CC9902, norank_f__Mycoplasmataceae, and Arthrospira_PCC-7345 were most abundant at salinities C and D (Figure 2c).
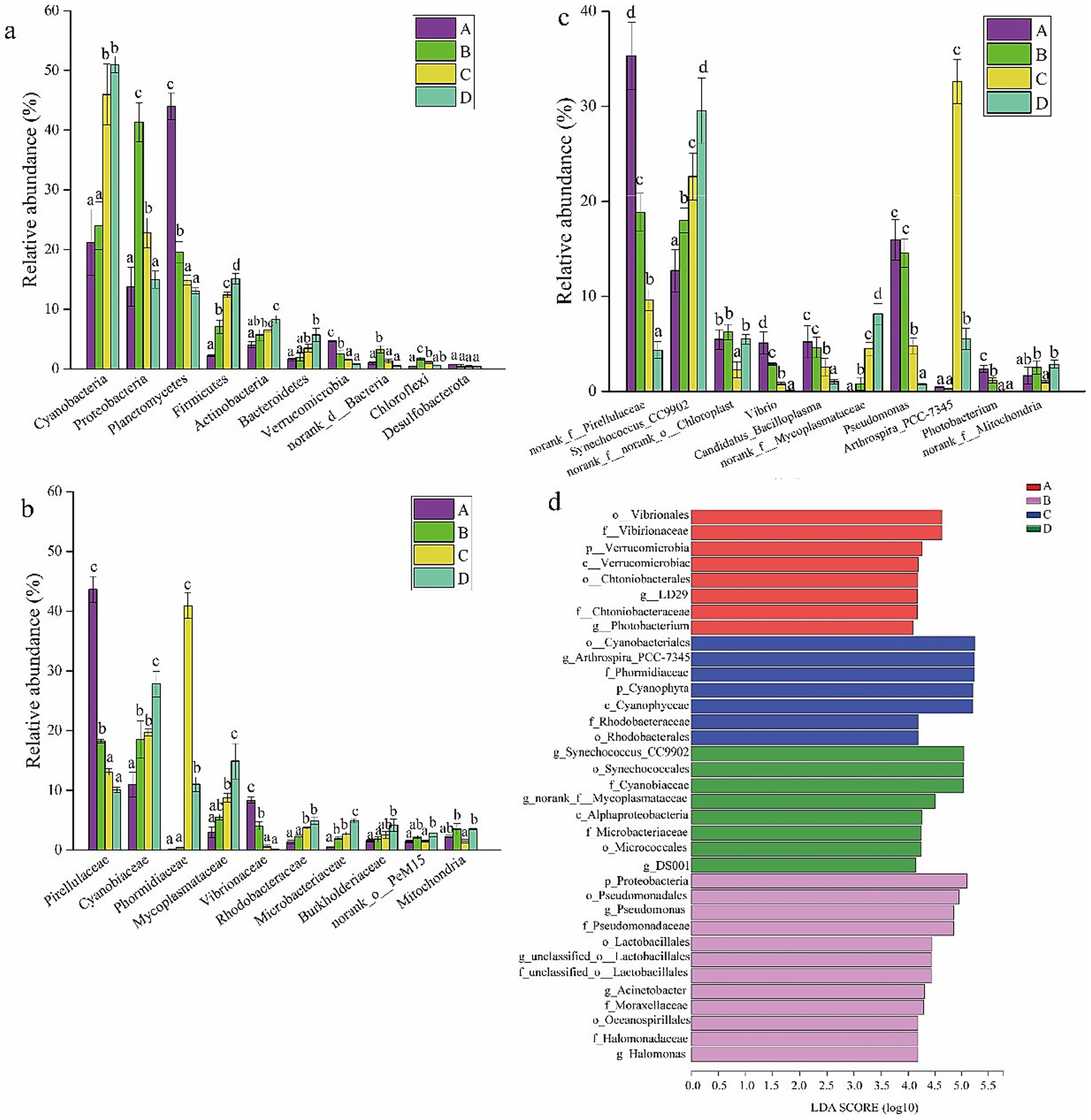
Figure 2. Microbial composition of gut bacterial taxa at phylum level (a), at family level (b), at genus level (c). Data in the same column sharing the different superscript letter are significantly different as determined by one-way ANOVA test (p < 0.05). The linear discriminant analysis (LDA) effect size (LEFSe) identified the differentially abundant taxa in four ponds (d). Only taxa meeting an LDA significant threshold of > 4.0 are shown. A, B, C, D mean salinity 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74.
To identify the classified bacterial taxa showing significant differences in abundance among gut samples at the four salinities, a biomarker analysis using the linear discriminant analysis (LDA) effect size (LEFSE) method was performed. Statistically significant differences were observed for 35 bacterial taxa among the four salinities at an LDA threshold of 4.0 (Figure 2d). Eight taxa were highly abundant in the shrimp gut samples at salinity A, including Vibrionales (order), Vibrionaceae (family), Verrucomicrobia (phylum), Verrucomicrobiae (class), Chthoniobacterales (order), Sphingobium yanoikuyae (genus), Chthoniobacteraceae (family), and Photobacterium (genus). Twelve taxa were highly abundant in the shrimp gut samples at salinity B, including Proteobacteria (phylum), Pseudomonadales (order), Pseudomonas (genus), Pseudomonadaceae (family), Lactobacillales (order), unclassified_o__Lactobacillales (genus), unclassified_o__Lactobacillales (family), Acinetobacter (genus), Moraxellaceae (family), Oceanospirillales (order), Halomonadaceae (family), and Halomonas (genus). Seven taxa were highly abundant in shrimp gut samples at salinity C, including Cyanobacteriales (order), Arthrospira_PCC-7345 (genus), Phormidiaceae (family), Cyanophyta (phylum), Cyanophyceae (class), Rhodobacteraceae (family), and Rhodobacterales (order). Eight taxa were highly abundant in shrimp gut samples at salinity D, including Synechococcus_CC9902 (genus), Synechococcales (order), Cyanobiaceae (family), norank_f__Mycoplasmataceae (genus), Alphaproteobacteria (class), Microbacteriaceae (family), Micrococcales (order), and DS001 (genus). Some characters at higher taxonomic levels are unlikely to be effective biomarkers because they include various genera and species. Therefore, characters at the genus level can potentially be more effective biomarkers. Photobacterium (genus) and Sphingobium yanoikuyae (genus) can serve as biomarkers at salinity A (31 ± 0.85); Pseudomonas (genus), unclassified_o__Lactobacillales (genus), Acinetobacter (genus), and Halomonas (genus) can serve as biomarkers at salinity B; Arthrospira_PCC-7345 (genus) can serve as a biomarker at salinity C; and Synechococcus_CC9902 (genus), norank_f__Mycoplasmataceae (genus), and DS001 (genus) can serve as biomarkers at salinity D.
3.3 Core bacterial taxa and environmental drivers of gut bacterial community
The core taxa of the gut bacterial community of shrimp at the four salinities were identified based on the frequency and abundance of OTUs. Approximately 1.35, 1.72, 1.45, and 1.97% of OTUs comprised core OTUs at salinities A, B, C, and D, which accounted for 79.68, 78.99, 83.59, and 78.70% of all obtained gut bacterial sequences, respectively (Figure 3; Supplementary Table S5). The core gut bacterial OTUs of shrimp at salinity A were in the phyla Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, and Bdellovibrionota. The core gut bacterial OTUs of shrimp at salinity B were in the phyla Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Chloroflexi, and norank_d_Bacteria. The core gut bacterial OTUs of shrimp at salinity C were in the phyla Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Bacteroidetes, Desulfobacteria, and Chloroflexi. The core gut core bacterial OTUs of shrimp at salinity D were in the phyla Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Bacteroidetes, and Dependentiae. There were 13 shared core taxa among the core bacterial community of shrimp at salinities A, B, C, and D (Supplementary Figure S2). The 13 shared core OTUs were affiliated with Rhodococcus, Pseudomonas, Mycobacterium, uncultured Planctomycetaceae bacterium, uncultured Actinomycetales bacterium, Delftia tsuruhatensis, Planctomycetaceae bacterium D2, Blastopirellula, Synechococcus_CC9902, uncultured Pirellulaceae bacterium, Ralstonia, Sphingobium yanoikuyae, and Microbacteriaceae bacterium CL-Dokdo102 (Supplementary Table S4). In addition, Vibrio OTU1121 (4.98%), Photobacterium OTU1254 (2.34%), Candidatus Bacilloplasma OTU18492 (0.75%), Vibrio OTU267 (0.58%), Mycobacterium OTU16545 (0.4%), Ralstonia OTU1370 (2.69%), and Pseudomonas OTU47 (0.23%) were identified as core OTUs at salinity A; Pseudomonas OTU18484 (16.94%), Candidatus Bacilloplasma OTU75 (4.43%), Photobacterium (1.11%), Candidatus Bacilloplasma OTU18492 (0.74%), Vibrio OTU1121 (0.65%), Pseudomonas OTU24521 (0.27%), Vibrio OTU4771 (0.58%), Ralstonia OTU1370 (1.08), and Pseudomonas OTU18020 (0.23%) were identified as core OTUs at salinity B. Candidatus Bacilloplasma OTU360 (1.80%), Candidatus Bacilloplasma OTU75 (1.20%), Pseudomonas OTU18484 (0.74%), Candidatus Bacilloplasma OTU18492 (0.46%) Ralstonia OTU1370 (1.34), and Vibrio OTU1121 (0.44%) were identified as core OTUs at salinity C. Pseudomonas OTU18484 (0.49%), Ralstonia OTU1370 (2.86%), and Aeromonas OTU3599 (0.23%) were identified as core OTUs at salinity D (Supplementary Table S5), which indicated that the core gut bacterial taxa of shrimp comprised more opportunistic pathogens under lower salinity culture conditions.

Figure 3. Composition and abundance of the core OTUs in gut bacterial community of shrimp. (A) Thirty-three OTUs were identified as the core OTUs in salinity A, accounting for 79.68% of all gut bacterial sequences obtained, which were affiliated to Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, and Bdellovibrionota. (B) Fifty-one OTUs were identified as the core OTUs in salinity B, accounting for 78.99% of all intestine bacterial sequences obtained, which were affiliated to Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Chloroflexi, and norank_d_Bacteria. (C) Forty-three OTUs were identified as the core OTUs in salinity C, accounting for 83.59% of all intestine bacterial sequences obtained, which were affiliated to Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Bacteroidetes, Desulfobacteria, and Chloroflexi. (D) Fifty OTUs were identified as the core OTUs in salinity D, accounting for 78.70% of all intestine bacterial sequences obtained, which were affiliated to Planctomycetes, Cyanobacteria, Proteobacteria, Verrucomicrobia, Actinobacteria, Firmicutes, Bacteroidetes, and Dependentiae.
An RDA diagram was constructed (Figure 4a), and axis 1 and axis 2 explained 43.67 and 21.91% of the variation in the data, respectively. After removing the redundant variables, 11 environmental characteristics were selected for RDA. Salinity (R2 = 0.6448, p = 0.002), T (temperature) (R2 = 0.3937, p = 0.012), pH (R2 = 0.33324, p = 0.029), NO2 −-N (R2 = 0.4636, p = 0.041), and NO3−-N (R2 = 0.245, p = 0.051) were significantly correlated with the gut bacterial community structure of shrimp. The results indicated that salinity was the most important environmental factor affecting the structure of the gut bacterial community. A heat map revealed correlations between the 20 most abundant genera and environmental factors (Figure 4b). NO3− - N was significantly positively correlated with Ralstonia, norank_f_Mycoplasmataceae, and DS001 (p < 0.05) and significantly negatively correlated with norank_f_Pirellulaceae and Sphingobium yanoikuyae (p < 0.05). Salinity was significantly positively correlated with Synechococcus_CC9902, norank_f_Mycoplasmataceae, DS001, Arthrospira_PCC-7345, unclassified_f_Rhodobacteraceae, and Blastopirellulaceae (p < 0.05) and significantly negatively correlated with norank_f_Pirellulaceae, Sphingobium yanoikuyae, Vibrio, Photobacterium, Candidatus_Bacilloplasma, and Pseudomonas (p < 0.05). DO was significantly positively correlated with Sphingobium yanoikuyae (p < 0.05) and significantly negatively correlated with norank_f_Mycoplasmataceae (p < 0.05). TN was significantly positively correlated with norank_f_Mitochondria and unclassified_o_Latobacillales (p > 0.05). NO2− - N was significantly negatively correlated with Photobacterium (p < 0.05). PO43− - P was significantly negatively correlated with Staphylococcus (p < 0.05). pH was only significantly positively correlated with Sphingobium yanoikuyae (p < 0.05) and significantly negatively correlated with DS001, Arthrospira_PCC-7345, unclassified_f_Rhodobacteraceae, and Blastopirellula (p < 0.05). TP was significantly positively correlated with unclassified_k_norank_d_Bacteria and Pseudomonas (p < 0.05) and significantly negatively correlated with Synechococcus_CC9902, unclassified_f_Rhodobacteraceae, and Blastopirellula (p < 0.05). T (temperature) was significantly positively correlated with unclassified_k_norank_d_Bacteria and Pseudomonas (p < 0.05) and significantly negatively correlated with Synechococcus_CC9902, norank_f_Mycoplasmataceae, DS001, Arthrospira_PCC-7345, and Blastopirellula (p < 0.05).
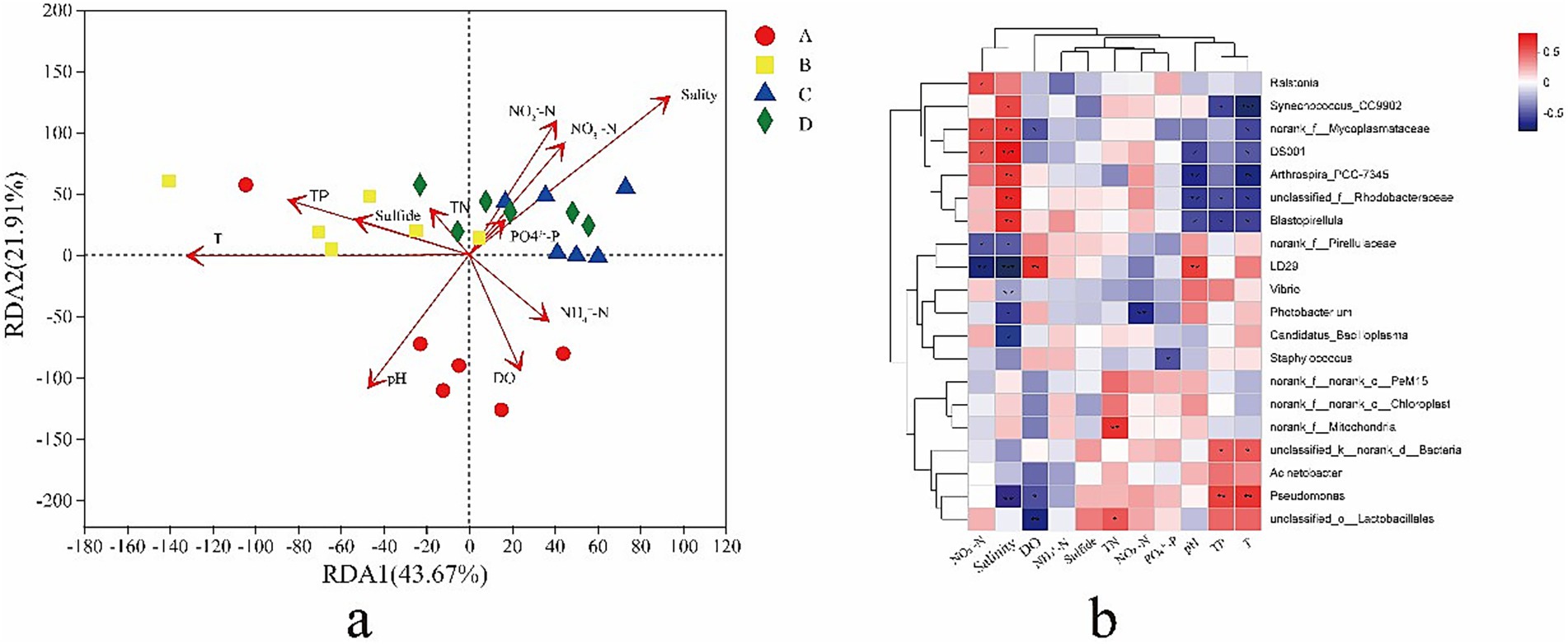
Figure 4. Redundancy analysis (RDA) based on the phylum data and environmental factors (a). Arrows indicate the direction and magnitude of environmental factors related to microbial communities. The length of an arrow-line indicates the strength of relationship between microbial community and environmental variable. Dots of the same color represent the microbial communities in one group. A heat map showing the correlations between the top 20 bacterial genus in relative abundance and environmental factors (b). A, B, C, D mean salinity 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74.
3.4 Functional differences in the gut bacterial community of shrimp
PICRUSt2 analysis was performed to predict the functions of the gut bacterial community of shrimp. Differences in the relative abundance of level 2 and level 3 functional pathways of the gut bacterial community based on the KEGG database are shown in Figure 5. As shown in Figure 5a, a total of 15 level 2 functional pathways were compared, and significant differences (p < 0.05) were observed in 14 functional pathways of the gut bacterial community of shrimp. The relative abundance of six functional pathways (Cellular community—prokaryotes, Amino acid metabolism, Metabolism of other amino acids, Xenobiotics biodegradation and metabolism, Signal transduction, and Membrane transport) was significantly higher at salinity D than at the other salinities (p < 0.05). No significant differences were observed among the three groups (p > 0.05). The relative abundance of seven functional pathways (Energy metabolism, Metabolism of cofactors and vitamins, Biosynthesis of other secondary metabolites, Lipid metabolism, Global and overview maps, Folding sorting and degradation, and Translation), was significantly lower at salinity D than at the other three salinities (p < 0.05). No significant differences were observed among the three other salinity groups (p > 0.05). A total of 18 level 3 functional pathways were compared, and significant differences (p < 0.05) were observed in 16 functional pathways of the gut bacterial community of shrimp (Figure 5b). The relative abundance of six functional pathways (Two-component system, ABC transporters, Microbial metabolism in diverse environments, Quorum sensing, Butanoate metabolism, and Valine leucine and isoleucine degradation) was significantly higher at salinity D than at the other three salinities (p < 0.05), and no significant differences were observed among the three salinity groups (p > 0.05). The abundance of seven functional pathways (Ribosome, Aminoacyl-tRNA biosynthesis, Purine metabolism, Metabolic pathways, Biosynthesis of secondary metabolites, Biosynthesis of amino acids, and Amino sugar and nucleotide sugar metabolism) was significantly lower at salinity D than at the other three salinities (p < 0.05); no significant differences were observed among the three groups (p > 0.05). The above results showed that the functions of the gut bacterial community significantly differed when the salinity was 55 ± 1.74. A PCoA analysis was conducted to analyze the functional content similarity of all samples (i.e., data explained 91.39% of the variation) (Figure 6). Significant separation was observed among samples at different salinities (p < 0.05). Salinity D samples were most clearly separated from the samples at the other three salinities. The results suggested that salinity is an important environmental factor affecting the functions of the gut bacterial community of shrimp.
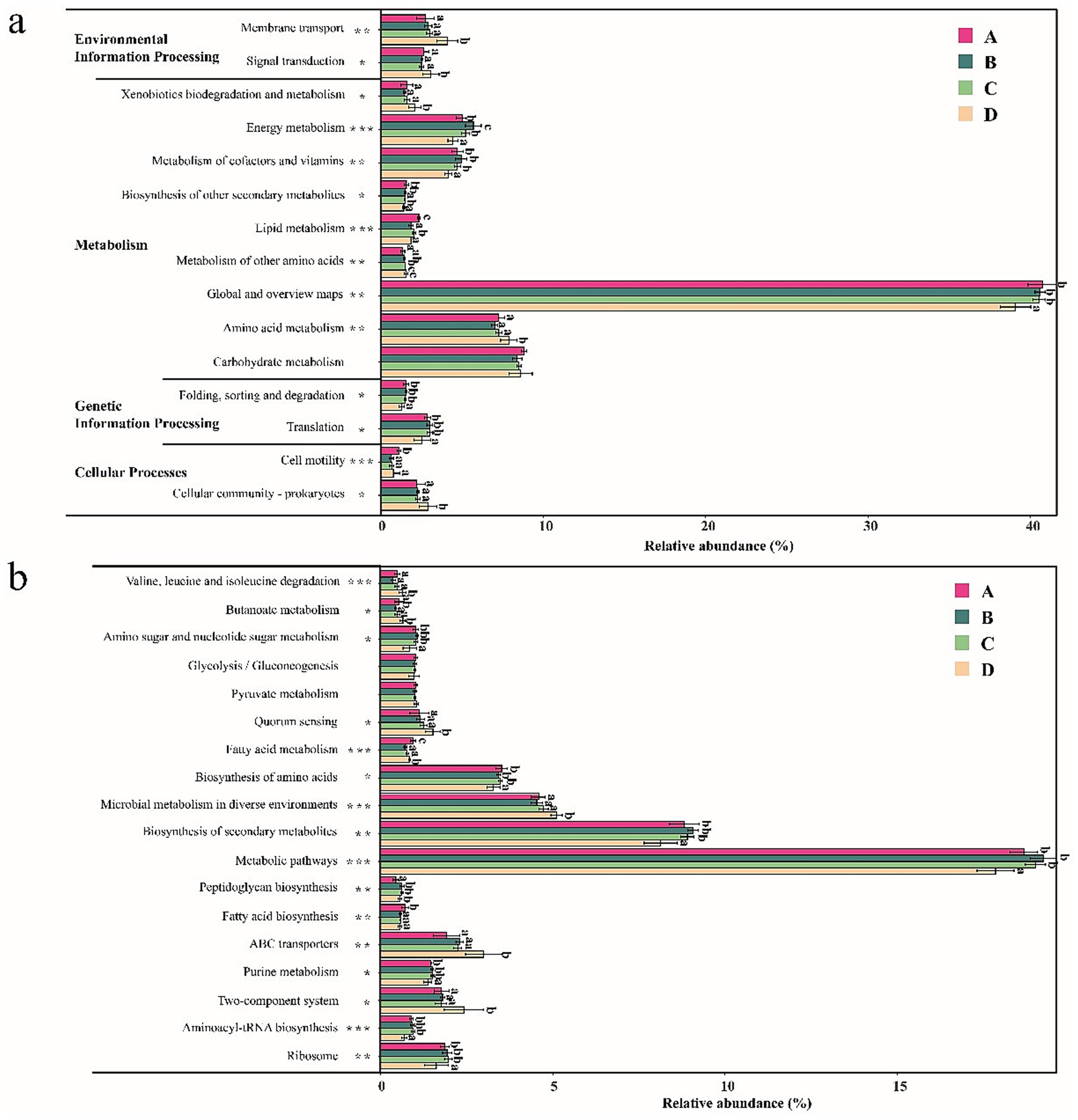
Figure 5. The difference in the abundance of KEGG functional pathways of gut bacterial community at level-2 (a) and level-3 (b). A, B, C, D mean salinity 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74. Different superscript letters indicate significant differences between the same column (p < 0.05).
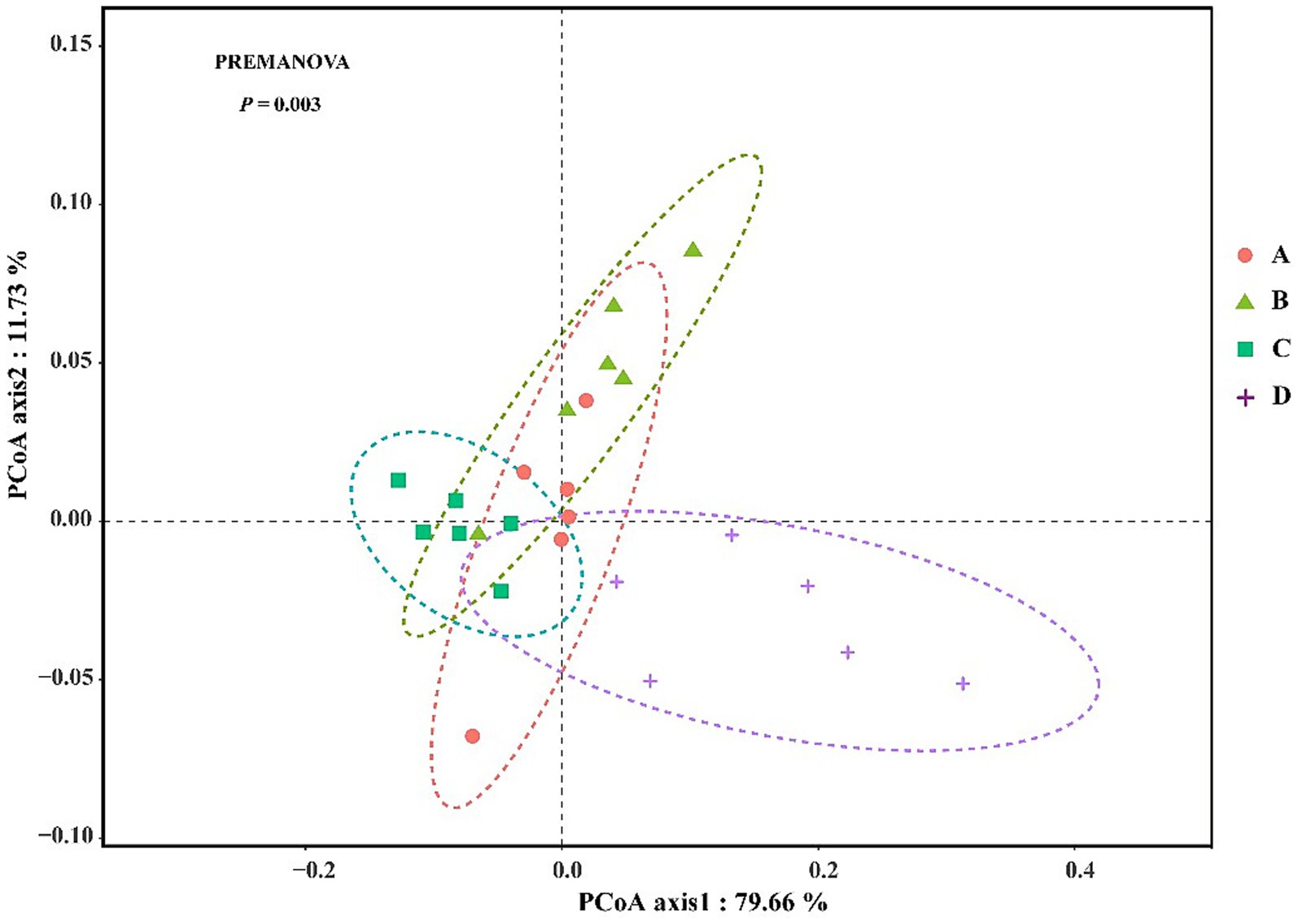
Figure 6. Principal coordinated analysis (PCoA) of the gut bacterial community function content similarity based on metagenomic functional predictions at level-3.
3.5 Interspecific interactions of the gut microbiota in shrimp
Analysis of the structure of co-occurrence networks can provide insights into interactions among microbes (Barberán et al., 2012). To evaluate the effect of salinity on interspecific interactions of the gut bacterial community, the interspecies interaction network of the 100 most abundant genera was established. The network plots showed that the bacterial networks in the shrimp gut at salinities A and B were more complex and had higher interconnectedness than those at salinities C and D, as indicated by the greater number of connections and larger network size (Figure 7). This pattern was further confirmed by the topological properties, as the number of edges, the average number of neighbors, network density, and network diameter were higher in the shrimp gut at salinities A and B than at salinities C and D (Table 2). The percentage of positive associations decreased with salinity (Table 2).
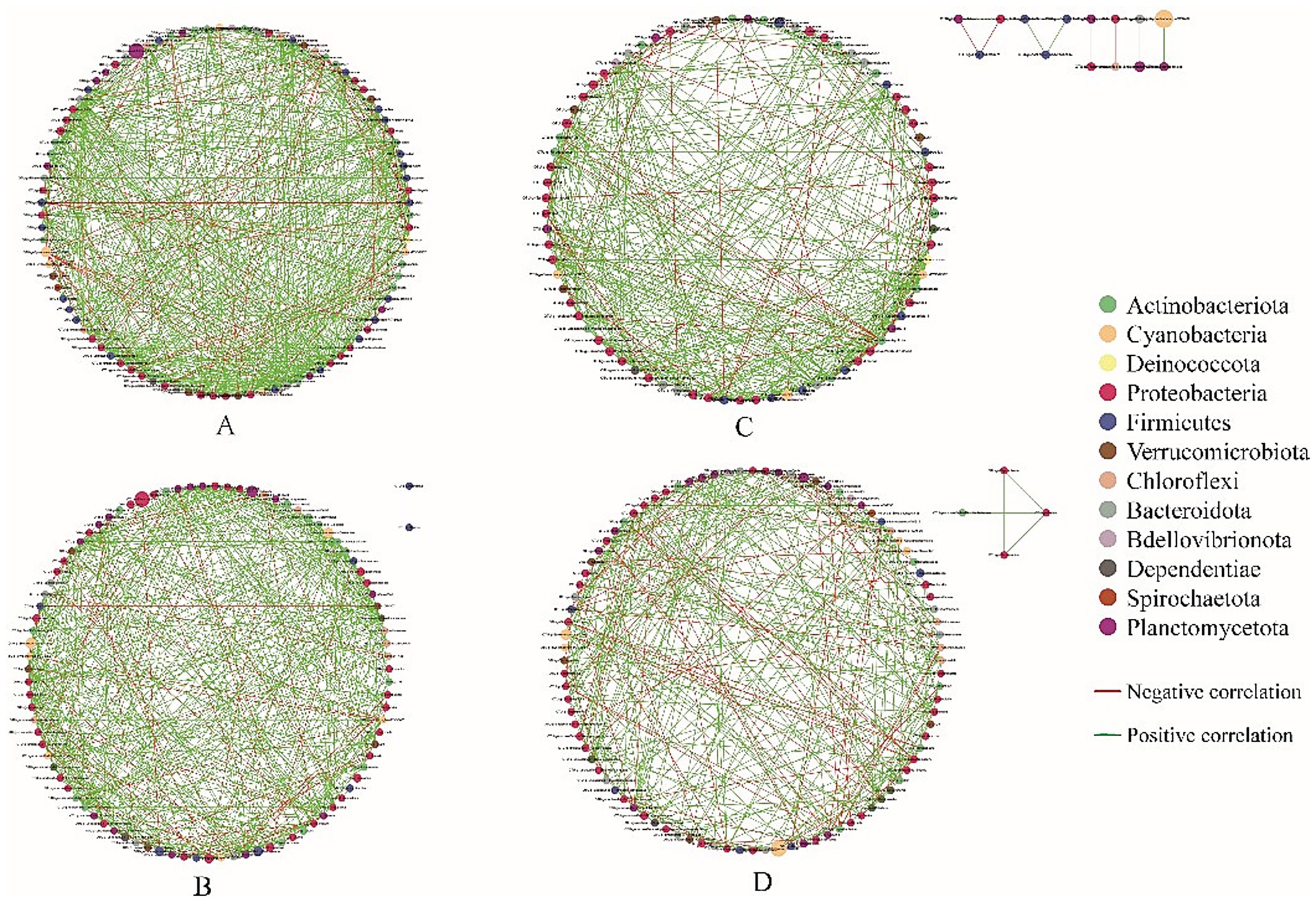
Figure 7. Co-occurrence networks analysis of the gut microbial community under different salinity levels. Each node represents one genus. Node colors indicate genus affiliated to different major phylum. The green edge indicates positive relationship between two individual nodes, whereas the red edge indicates negative relationship. The (A–D) represent the salinities 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74, respectively.
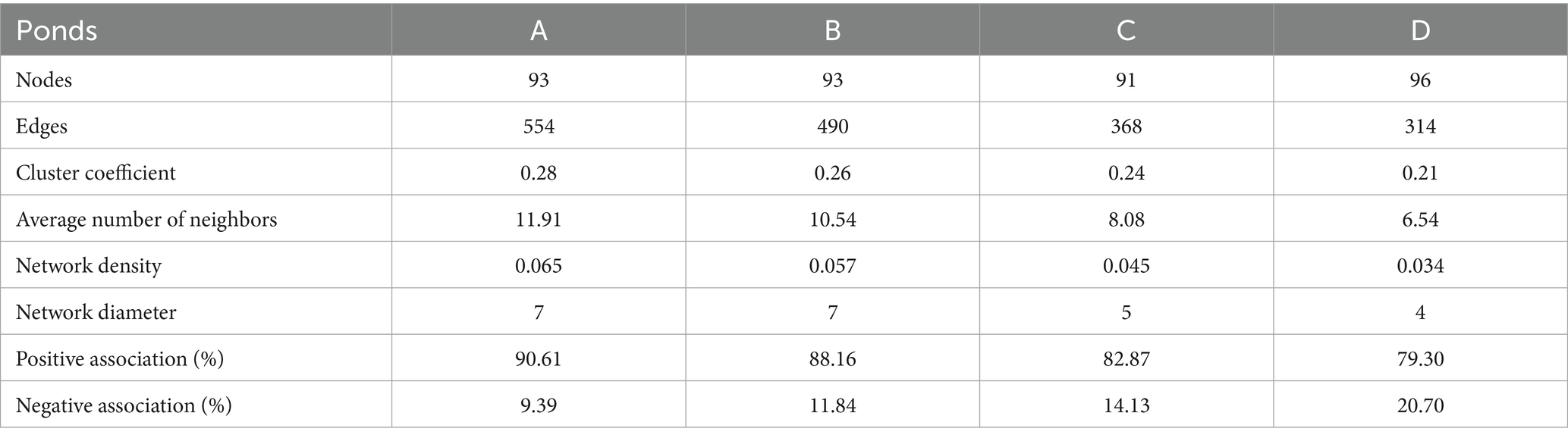
Table 2. Topological parameters of co-occurrence network for gut microbial community at different salinity levels.
3.6 Assembly of the gut microbial community in shrimp
The NCM quantified the relationship between the occurrence frequency of OTUs and their relative abundance (Figure 8a). The relative contribution of stochastic processes gradually decreased with salinity and explained 51.8, 43.6, 37.7, and 29.5% of the community variance at salinities A, B, C, and D, respectively. The NST was also used to quantify the relative contribution of stochastic and deterministic processes in gut bacterial community assembly (Figure 8b). The NST average value was above the 50% boundary point for the gut bacterial community at salinities A, B, and C, which was 85.18, 76.29, and 68.49%, respectively. This result indicated that the gut bacterial community at salinities A, B, and C was predominately regulated by stochastic processes. However, the NST average value of the gut bacterial community at salinity D was 46.26%, suggesting that deterministic processes had a marginally stronger effect than stochastic processes on the gut bacterial community at salinity D. These observations suggested that the strength of the effect of deterministic processes on the gut bacterial community of shrimp increased with salinity whereas that of stochastic processes decreased with salinity. In addition, community-level habitat niche breadths (Bcom) were also calculated to explore the relative importance of deterministic and stochastic processes in gut bacterial community assembly. The gut bacterial community had significantly wider niche breadths at salinity A than at other salinities (p < 0.05) (Figure 8c). The community-level habitat niche breadths (Bcom) decreased with salinity, which suggested that gut bacterial community assembly was more strongly affected by deterministic processes at higher salinity levels.

Figure 8. Relative contribution ratios of deterministic and stochastic processes on microbial community assembly in gut of shrimp. A, B, C, D mean salinity 31 ± 0.85, 39 ± 1.23, 47 ± 0.62, and 55 ± 1.74. The letters (a-c) indicate significant differences between the data groups.
4 Discussion
The gut microbiota of shrimp plays a key role in maintaining host health and promoting disease resistance, and an increasing number of studies have examined the factors affecting the structure and function of the gut microbiota (Rooks and Garrett, 2016). The gut microbiota of shrimp is influenced not only by biological factors (Dai et al., 2017; Zheng et al., 2020; Zhou et al., 2020; Zhang et al., 2021) but also by abiotic factors such as salinity, temperature, and pH (Schmidt et al., 2015; Zhang M. et al., 2016; Hou et al., 2017; Huang et al., 2018). Therefore, understanding the composition and functions of the gut bacterial community in aquaculture systems and their relationships with environmental factors is critically important for improving the management of shrimp culture. The extensive farming of shrimp in the primary salt evaporation ponds of the salterns in the coastal areas of the Yellow and Bohai Seas is critically important for increasing the natural resource utilization and economic benefits of these regions. However, few studies have reported the characteristics of the gut bacterial community of shrimp cultured in these hypersaline environments.
Microbial diversity, especially gut microbial diversity, is a key characteristic affecting host health (Ren et al., 2019). In this study, alpha diversity (Shannon index, Simpson index, Chao1 index, and ACE index) decreased with salinity. This result is consistent with the findings of previous studies (Cornejo-Granados et al., 2018; Hou et al., 2020). The results of PCoA and a hierarchical clustering tree showed that the threshold effects of salinity on bacterial community structure and phylogenetic relationships were observed when the salinity exceeded 39; these findings indicate that the salinity is a critical factor influencing the bacterial community structure. Previous studies have shown that Proteobacteria, Firmicutes, Bacteroides, and Actinobacteria are the predominant phyla in the gut bacterial community of P. vannamei in hyposaline environments (Duan et al., 2018; Li et al., 2018; Gao et al., 2019). Proteobacteria, as a highly adaptable phylum, plays a key role in nutrient acquisition and osmotic regulation. For instance, certain members of Gammaproteobacteria can synthesize compatible solutes (e.g., betaine and ectoine) to help the host maintain cellular osmotic balance under high salinity stress (Imhoff et al., 2021). Bacteroidetes contribute to nutrient acquisition via polysaccharide degradation enzymes. For instance, Bacteroides thetaiotaomicron is considered as the best degrader of polysaccharides (Li et al., 2021). Firmicutes, particularly lactic acid bacteria (LAB) belonging to this phylum, contribute significantly to intestinal health. LAB can produce lactic acid and bacteriocins, which inhibit the colonization of pathogenic bacteria (e.g., Vibrio spp.) by lowering the intestinal pH and competing for ecological niches (Yang et al., 2008). Actinobacteria, as a phylum with diverse metabolic capabilities, contribute to both nutrient metabolism and immune enhancement. Many Actinobacteria species can synthesize antibiotics and enzymes (e.g., proteases and lipases) that inhibit pathogenic microorganisms and aid in the digestion of proteins and lipids, which are essential for shrimp growth under stressful hypersaline conditions. For instance, Streptomyces are soil dwelling bacteria and characterized by their remarkable capacity for sporulation and biosynthesis of diverse secondary metabolites, particularly antibiotics (Jones, 2023). Salinity plays a dominant role in shaping the intestinal microbiota of shrimp which change the osmotic pressure and ion concentrations in the gut, determining the survival of microorganisms and enhancing digestion and immunity (Gou et al., 2018; Chen et al., 2022). In our study, the predominant phyla in the gut bacterial community of P. vannamei under hypersaline environments were Cyanobacteria, Proteobacteria, Planctomycetes, Firmicutes, and Actinobacteria. This suggests that salinity plays an important role in the gut bacterial composition of P. vannamei. The relative abundances of some opportunistic pathogens were significantly lower at salinities of 47 (C) and 55 (D), such as Vibrio, Pseudomonas, Candidatus_Bacilloplasma, and Photobacterium, than at 31 (A) and 39 (B). The excessive growth of opportunistic pathogens in the gut of aquatic animals can contribute to disease (Pérez et al., 2010; Xing et al., 2013; Huang et al., 2020). For example, the overgrowth of Vibrio, Pseudomonas, Candidatus_Bacilloplasma, and Photobacterium can have pathogenic effects on aquatic animals (Soto-Rodriguez et al., 2015; Liu et al., 2016; Hou et al., 2018a; Xin et al., 2018). Bacterial diseases, mainly caused by the Vibrio genus, are considered as the most serious threat to shrimp aquaculture. For instance, the common pathogenic Vibrio species: Vibrio alginolyticus, Vibrio harveyi, Vibrio parahaemolyticus, usually induced the pale hepatopancreas, empty gut, and sloughing of the epithelial cells of the hepatopancreas tubule and massive hemolytic infiltration (Baker-Austin et al., 2018; Lee et al., 2015; Zhang et al., 2023). Pseudomonas aeruginosa threatens whiteleg shrimp by causing black gill disease and shell ulcers via toxin production (LasB, pyocyanin), suppressing immunity (Ramalingam and Ramarani, 2007). Photobacterium may cause symptoms such as pale hepatopancreas and whitish body coloration in shrimp, ultimately leading to mortality (Liu et al., 2016). Study has revealed significant intestinal proliferation of Candidatus Bacilloplasma in P. vannamei affected by White Feces Syndrome (WFS), suggesting its potential role as a key etiological agent in WFS pathogenesis (Hou et al., 2018b). This means that the hypersaline environment could reduce the mortality rate of shrimp caused by opportunistic bacterial pathogens to some degree.
The core microbiota and their compositions are the characteristic microbiome of a microbial community and play a key role in bacterial community functions (Shade and Handelsman, 2012). Core microbiota in shrimp intestines may aid nutrient digestion by secreting enzymes to break down complex carbohydrates and proteins (Tao et al., 2020), enhance immunity via competing with pathogens and stimulating antimicrobial peptide production and maintain gut homeostasis by regulating pH and redox balance, supporting host health and environmental adaptation (Macke et al., 2017). Identification of the core microbiota associated with a host is essential for clarifying the key microbial metabolic functions of the host (Mueller and Sachs, 2015). In general, polysaccharide digestion, essential amino acid biosynthesis, SCFA production and lipid metabolism are all essential for hosts (Song et al., 2021). Some studys foud that microorganisms such as some species of Vibrio and Flavobacterium are often present in the early larval stage of shrimp. For example, studies by Holt et al. (2020) have shown that in the gastrointestinal tract of giant tiger prawns and Pacific white shrimp, γ-Proteobacteria, including Vibrio, are dominant in the early stages. As the shrimp grow into the juvenile stage, the proportion of some beneficial bacteria like Lactobacillus may gradually increase, which can help with the digestion and immune function of the shrimp (Xia et al., 2020). During the adult stage, the core microbiome may be more complex, with the co-existence of a variety of bacteria involved in nutrient metabolism, such as certain members of the Bacteroidetes and Actinobacteria (Li et al., 2021, Jones, 2023). Studies have shown that variation in core gut microbes largely depends on the host environment (Mortzfeld et al., 2016). In our study, the composition of the core gut microbes of shrimp cultured at different salinities was compared. The core microbes significantly differed at the four salinity levels, and only 13 core microorganisms were shared among groups at various salinities. Part of the core gut microbes of the host can be identified early in life because of their significant contribution to basic gut microbial functions (Shade and Handelsman, 2012). The other part may be acquired through the deterministic process of colonization of the gut with specific microbes from the environment, which perform specific functions in the host (Weigel, 2020). The above results indicated that salinity strongly affected the composition of the core microorganisms in the gut of shrimp. In general, the core gut microbes are considered beneficial to host health (Walter and Ley, 2011). However, the results of this study showed that the core gut microbes of shrimp cultured at salinities of 31 and 39 3 contained more opportunistic pathogens than those at salinities of 47 and 55.
Salinity and temperature are key environmental factors that shape gut microbiota composition and function through direct and host-mediated mechanisms (Staley et al., 2015; Sunagawa et al., 2015; Li et al., 2018). Salinity influences gut microbiota by altering osmotic pressure in the intestinal lumen. High salinity selects for osmotolerant taxa (e.g., Vibrio, Pseudorhodobacter) while suppressing osmosensitive species like Cetobacterium in Gymnocypris przewalskii (Wang et al., 2022). This shift reduces diversity and disrupts metabolic pathways, such as osmoregulation via compatible solute accumulation. Temperature directly impacts microbial enzyme activity and membrane fluidity. In ectotherms like European sea bass (Dicentrarchus labrax), seasonal temperature drops correlate with increased pathogenic Vibrio abundance and altered skin/intestinal microbiota structure (Mokrani et al., 2019). Temperature also modulates host metabolism, affecting nutrient availability and indirectly driving shifts in fermentation pathways (e.g., short-chain fatty acid production). Extreme temperatures induce heat-shock proteins and immune activation, compromising gut barrier integrity and promoting dysbiosis. These factors often act synergistically. For instance, in aquaculture, combined salinity and temperature fluctuations exacerbate microbial imbalance, increasing disease susceptibility. Collectively, salinity and temperature serve as ecological filters, reshaping gut microbiota to influence host health and adaptive fitness. Some researchers indicate that salinity was more important environmental factor affecting the global microbial community distribution patterns than temperature (Lozupone and Knight, 2007; Auguet et al., 2010; Wang et al., 2011). Similarly, salinity was more important than temperature on the gut bacterial community of shrimp in our study. There was a negative correlation between salinity and the abundance of some opportunistic pathogens, such as Vibrio, Photobacterium, Candidatus_Bacilloplasma, and Pseudomonas (Figure 4b), which was consistent with the results of a previous study (Bauer et al., 2021). In addition to salinity and temperature, pH, NO2− - N, and NO3− - N concentrations were significantly correlated with gut bacterial community structure, and similar observations were made in other studies (Hou et al., 2017; Huang et al., 2018). However, the gut bacterial community of shrimp is affected not only by the surrounding environment but also by the hosts (Pérez et al., 2010; Sullam et al., 2012). Additional in-depth studies are needed to clarify the interactions between environmental pressures and host selective mechanisms. Salinity and temperature have a significant effect on microbial community functions (Székely et al., 2013; Sunagawa et al., 2015). Some previous studies have shown that temperature plays a greater role than salinity in affecting the functional composition of microorganisms in the marine environment (Sunagawa et al., 2015). In our study, salinity was a key factor affecting the functions of the gut bacterial community in shrimp. The threshold effects of salinity on the functions of the gut bacterial community of shrimp were observed at the salinity of 55 had a major effect on the functions of the gut bacterial community of shrimp. Similar results were obtained by PCoA. This finding indicated that salinity affected the functions of the bacterial community of shrimp in hypersaline environments. The gut microbiota comprises a variety of species that interact with each other and form a complex ecological network characterized by different types of interactions, such as cooperative, competitive, and predatory interactions (Deng et al., 2012). Co-occurrence networks can provide valuable insights into biological interactions within the microbial community (Williams et al., 2014). Generally, complex networks with high connectivity are more robust against external disturbances and more active than simple networks (Lu et al., 2013; Santolini and Barabási, 2018). In this study, the gut bacterial networks of shrimp were more complex and better connected in lower salinity environments than those in higher salinity environments, suggesting that the gut microbial ecosystem is more stable and active in hyposaline environments than in hypersaline environments. In addition, the percentage of positive associations decreased with salinity, which was consistent with the results of a previous study (Hou et al., 2020), suggesting that there was a higher degree of cooperative activities in the shrimp gut at lower salinity (Montoya et al., 2006).
In general, the microbial community assembly is driven by deterministic and stochastic process (Hanson et al., 2012). Deterministic processes refer to ecological assembly mechanisms driven by environmental filtering (e.g., salinity, pH, nutrients) and biological interactions (e.g., competition, predation), leading to predictable community structures and stochastic processes involve random events (e.g., dispersal limitation, ecological drift) that contribute to unpredictable variations in microbial communities (Dini-Andreote et al., 2015). Deterministic processes regulate the fitness of microbial communities, thereby determining species composition and relative abundance, whereas stochastic processes induce unpredictable alterations in community structure. Ultimately, these two distinct processes collectively govern the functional roles of microbial communities in biogeochemical cycles (Tilman, 2004; Zhang W. et al., 2016). In the present study, salinity had an important effect on the assembly of the gut bacterial community of shrimp, mainly by affecting the balance between deterministic and stochastic processes. The R2 values of the NCMs decreased with salinity, suggesting that the relative contribution of stochastic processes in the gut bacterial community of shrimp decreased with salinity. The NST index also revealed that the relative contribution of deterministic processes compared with stochastic processes in the gut bacterial community of shrimp increased with salinity, likely because the greater environmental heterogeneity at high salinity exposes the gut bacterial community to a wider range of filters, which increases the strength of deterministic processes of environmental selection (Vellend et al., 2014). Species with a wider niche range are considered generalists that exhibit greater metabolic plasticity and are less affected by environmental factors due to their higher environmental tolerances (Pandit et al., 2009; Li et al., 2019). Therefore, we characterized the habitat niche breadths of the gut bacterial community of shrimp and found that niche breadths decreased with salinity, suggesting that the assembly of the gut bacterial community in shrimp was more strongly affected by deterministic processes as salinity increased. A niche refers to the functional role and habitat conditions that a microorganism occupies within an ecosystem, including its resource use and environmental tolerances. In the gut microbiota, niche differentiation (e.g., oxygen gradients, nutrient availability) shapes microbial diversity and stability. In this study, the gut bacterial community had significantly wider niche breadths at salinity A than at other higher salinities, which suggested salinity fluctuations may alter niche availability, favoring halotolerant taxa and restructuring bacterial community structure. The results further demonstrated that the gut bacterial community in shrimp was increasingly governed by deterministic processes with elevated salinity levels.
This study highlights gut microbiota modulation as a viable strategy to improve shrimp health in hypersaline environment, with specific relevance to large ponds in salt pan. Future research should focus on: (1) developing locally-adapted probiotics from indigenous halotolerant strains, (2) deciphering host-microbiota metabolic crosstalk via multi-omics under hypersalinity, and (3) creating salinity-responsive management protocols. Practical implementation requires farmer-friendly delivery systems coupled with ecological monitoring. Such microbiome interventions may offer sustainable, chemical-free solutions for the shrimp farming in large ponds of salt pan.
5 Conclusion
Changes in the characteristics of the gut microbiota in P. vannamei with salinity were investigated in the primary salt evaporation ponds for the salterns in northern China. The results demonstrated that salinity significantly affected the gut bacterial community of shrimp. Alpha diversity indexes of the gut bacterial community in shrimp decreased with salinity. Potential opportunistic bacterial pathogens decreased significantly in hypersaline environments. Furthermore, the gut bacterial community structure differed significantly between salinities 31–39 and 47–55, the predicted functions were distinct at salinities of 31–47 and 55. Stochastic and deterministic processes jointly contributed to the assembly of the gut bacterial community of shrimp; however, the relative importance of stochastic processes decreased with salinity. Our findings provide new insights into the characteristics of the gut bacterial community of shrimp in hypersaline environments and contribute to the improvement of farming health management in hypersaline ponds.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The animal study was approved by All animal experiments were conducted in accordance with the guidelines and approval of the respective Animal Research and Ethics Committees of Ocean University of China (Permit Number: 20141201. http://www.gov.cn/gongbao/content/2011/content_1860757.htm). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
BW: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. YaL: Formal analysis, Investigation, Methodology, Writing – review & editing. XW: Investigation, Resources, Writing – review & editing. YuL: Investigation, Resources, Writing – review & editing. ZL: Investigation, Writing – review & editing. JW: Supervision, Writing – review & editing. YiL: Supervision, Writing – review & editing. JT: Supervision, Writing – review & editing. BY: Funding acquisition, Resources, Writing – review & editing. XT: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the National Key Research and Development Program of China (Grants 2020YFD0900200, 2023YFD2401705, and 2023YFD2402000). This work was also financially and personally supported by Animal Husbandry and Fisheries Research Center of Guangdong Haid Group Co., Ltd., Guangzhou, China, the Key Laboratory of Mariculture. State Key Laboratory of Tropical Oceanography, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China. Ocean University of China, Ministry of Education, Qingdao. China, Bohai Seafoods Co., Ltd., Binzhou, China and Key Laboratory of microecological resources and utilization in breeding industry, Ministry of Agriculture and Rural Affairs, Guangdong Haid Group Co., Ltd., Guangzhou, China.
Conflict of interest
BW, JW, YiL, JT, and BY were employed by Guangdong Haid Group Co., Ltd. XW, YuL, and ZL were employed by Bohai Seafoods Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1665547/full#supplementary-material
References
Auguet, J. C., Barberán, A., and Casamayor, E. O. (2010). Global ecological patterns in uncultured Archaea. ISME J. 4, 182–190. doi: 10.1038/ismej.2009.109
Baker-Austin, C., Oliver, J. D., Alam, M., Ali, A., Waldor, M. K., Qadri, F., et al. (2018). Vibrio spp. infections. Nat. Rev. Dis. Prim. 4, 1–19. doi: 10.1038/s41572-018-0005-8
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Bauer, J., Teitge, F., Neffe, L., Adamek, M., Jung, A., Peppler, C., et al. (2021). Impact of a reduced water salinity on the composition of Vibrio spp. in recirculating aquaculture systems for Pacific white shrimp (Litopenaeus vannamei) and its possible risks for shrimp health and food safety. J. Fish Dis. 44, 89–105. doi: 10.1111/jfd.13270
Boulangé, C. L., Neves, A. L., Chilloux, J., Nicholson, J. K., and Dumas, M. (2016). Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 8, 1–12. doi: 10.1186/s13073-016-0303-2
Cani, P. D., Van Hul, M., Lefort, C., Depommier, C., Rastelli, M., and Everard, A. (2019). Microbial regulation of organismal energy homeostasis. Nat. Metab. 1, 34–46. doi: 10.1038/s42255-018-0017-4
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, J., Wang, H. M., Yuan, H., Hu, N. J., Zou, F. Q., Li, C. Y., et al. (2022). Effects of dietary Clostridium autoethanogenum protein on the growth, disease resistance, intestinal digestion, immunity and microbiota structure of Litopenaeus vannamei reared at different water salinities. Front. Microbiol. 13, 1–18. doi: 10.3389/fimmu.2022.1034994
China Fishery Statistical Yearbook (2023). Fisheries Agency of China, agriculture ministry. Beijing: China Agriculture Press.
Cornejo-Granados, F., Gallardo-Becerra, L., Leonardo-Reza, M., Ochoa-Romo, J. P., and Ochoa-Leyva, A. (2018). A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 6:e5382. doi: 10.7717/peerj.5382
Cornejo-Granados, F., Lopez-Zavala, A. A., Gallardo-Becerra, L., Mendoza-Vargas, A., Sánchez, F., Vichido, R., et al. (2017). Microbiome of Pacific whiteleg shrimp reveals differential bacterial community composition between wild, aquacultured and AHPND/EMS outbreak conditions. Sci. Rep. 7, 1–15. doi: 10.1038/s41598-017-11805-8
Dai, W., Sheng, Z., Chen, J., and Xiong, J. (2020). Shrimp disease progression increases the gut bacterial network complexity and abundances of keystone taxa. Aquaculture 517:734802. doi: 10.1016/j.aquaculture.2019.734802
Dai, W., Yu, W., Zhang, J., Zhu, J., Tao, Z., and Xiong, J. (2017). The gut eukaryotic microbiota influences the growth performance among cohabitating shrimp. Appl. Microbiol. Biotechnol. 101, 6447–6457. doi: 10.1007/s00253-017-8388-0
de Souza Valente, C., Rodiles, A., Freire Marques, M. R., and Merrifield, D. L. (2020). White spot syndrome virus (WSSV) disturbs the intestinal microbiota of shrimp (Penaeus vannamei) reared in biofloc and clear seawater. Appl. Microbiol. Biotechnol. 104, 8007–8023. doi: 10.1007/s00253-020-10816-4
Deng, Y., Jiang, Y., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13, 1–20. doi: 10.1186/1471-2105-13-113
Dini-Andreote, F., Stegen, J. C., Elsas, J. D., and Salles, J. F. (2015). Disentangling mechanisms thatmediate the balance between stochastic and deterministic processes inmicrobial succession. Proc. Natl. Acad. Sci. 112:E1326–E 1332. doi: 10.1073/pnas.1414261112
Duan, Y., Liu, Q., Wang, Y., Zhang, J., and Xiong, D. (2018). Impairment of the intestine barrier function in Litopenaeus vannamei exposed to ammonia and nitrite stress. Fish Shellfish Immunol. 78, 279–288. doi: 10.1016/j.fsi.2018.04.050
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
FAO. (2018). The state of world fisheries and aquaculture 2018-meeting the sustainable development goals. FAO Rome, Italy.
Faust, K., Sathirapongsasuti, J. F., Izard, J., Segata, N., Gevers, D., Raes, J., et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8:e1002606. doi: 10.1371/journal.pcbi.1002606
Gao, S., Pan, L., Huang, F., Song, M., Tian, C., and Zhang, M. (2019). Metagenomic insights into the structure and function of intestinal microbiota of the farmed Pacific white shrimp (Litopenaeus vannamei). Aquaculture 499, 109–118. doi: 10.1016/j.aquaculture.2018.09.026
Gou, N. N., Wang, K. F., and Yang, X. C. (2018). Review of salinity levels and dietary protein on nutrition and digestion of Litopenaeus vannamei. Acta Agric. Boreali-Occident. Sin. 27, 306–315. doi: 10.7606/j.issn.1004-1389.2018.03.002
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Holt, C. C., Bass, D., Stentiford, G. D., and van der Giezen, M. (2020). Understanding the role of the shrimp gut microbiome in health and disease. J. Invertebr. Pathol. 186:107387. doi: 10.1016/j.jip.2020.107387
Hooper, L. V., and Macpherson, A. J. (2010). Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 10, 159–169. doi: 10.1038/nri2710
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Hou, D., Huang, Z., Zeng, S., Liu, J., Wei, D., Deng, X., et al. (2017). Environmental factors shape water microbial community structure and function in shrimp cultural enclosure ecosystems. Front. Microbiol. 8:2359. doi: 10.3389/fmicb.2017.02359
Hou, D., Huang, Z., Zeng, S., Liu, J., Wei, D., Deng, X., et al. (2018b). Intestinal bacterial signatures of white feces syndrome in shrimp. Appl. Microbiol. Biotechnol. 102, 3701–3709. doi: 10.1007/s00253-018-8855-2
Hou, D., Huang, Z., Zeng, S., Liu, J., Weng, S., and He, J. (2018a). Comparative analysis of the bacterial community compositions of the shrimp intestine, surrounding water and sediment. J. Appl. Microbiol. 125, 792–799. doi: 10.1111/jam.13919
Hou, D., Zhou, R., Zeng, S., Wei, D., Deng, X., Xing, C., et al. (2020). Intestine bacterial community composition of shrimp varies under low-and high-salinity culture conditions. Front. Microbiol. 11:2765. doi: 10.3389/fmicb.2020.576575
Huang, F., Pan, L., Song, M., Tian, C., and Gao, S. (2018). Microbiota assemblages of water, sediment, and intestine and their associations with environmental factors and shrimp physiological health. Appl. Microbiol. Biotechnol. 102, 8585–8598. doi: 10.1007/s00253-018-9229-5
Huang, Z., Zeng, S., Xiong, J., Hou, D., Zhou, R., Xing, C., et al. (2020). Microecological Koch’s postulates reveal that intestinal microbiota dysbiosis contributes to shrimp white feces syndrome. Microbiome 8, 1–13. doi: 10.1186/s40168-020-00802-3
Imhoff, J. F., Rahn, T., Künzel, S., Keller, A., and Neulinger, S. C. (2021). Osmotic adaptation and compatible solute biosynthesis of phototrophic Bacteria as revealed from genome analyses. Microorganisms 9:46. doi: 10.3390/microorganisms9010046
Jin, M., Xiong, J., Zhou, Q., Yuan, Y., Wang, X., and Sun, P. (2018). Dietary yeast hydrolysate and brewer's yeast supplementation could enhance growth performance, innate immunity capacity and ammonia nitrogen stress resistance ability of Pacific white shrimp (Litopenaeus vannamei). Fish Shellfish Immunol. 82, 121–129. doi: 10.1016/j.fsi.2018.08.020
Jones, G. H. (2023). Streptomyces rnases – function and impact on antibiotic synthesis. Front. Microbiol. 14:1096228. doi: 10.3389/fmicb.2023.1096228
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lee, C.-T., Chen, I. T., Yang, Y.-T., Ko, T.-P., Huang, Y.-T., Huang, J.-Y., et al. (2015). The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid that expresses a deadly toxin. Proc. Natl. Acad. Sci. USA 112, 10798–10803. doi: 10.1073/pnas.1503129112
Li, P., Liu, J., Jiang, C., Wu, M., Liu, M., and Li, Z. (2019). Distinct successions of common and rare bacteria in soil under humic acid amendment–a microcosm study. Front. Microbiol. 10:2271. doi: 10.3389/fmicb.2019.02271
Li, E., Xu, C., Wang, X., Wang, S., Zhao, Q., Zhang, M., et al. (2018). Gut microbiota and its modulation for healthy farming of Pacific white shrimp Litopenaeus vannamei. Rev. Fish. Sci. Aquac. 26, 381–399. doi: 10.1080/23308249.2018.1440530
Li, S., Zhang, B. J., Hu, J. L., Zhong, Y. D., Sun, Y. G., and Nie, S. P. (2021). Utilization of four galactans by Bacteroides thetaiotaomicron A4 based on transcriptome. Food Front. 2, 218–231. doi: 10.1002/fft2.82
Liu, F., Liu, G. X., and Li, F. H. (2016). Characterization of two pathogenic Photobacterium strains isolated from Exopalaemon carinicauda causing mortality of shrimp. Aquaculture 464, 129–135. doi: 10.1016/j.aquaculture.2016.06.019
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 104, 11436–11440. doi: 10.1073/pnas.0611525104
Lu, L., Yin, S., Liu, X., Zhang, W., Gu, T., Shen, Q., et al. (2013). Fungal networks in yield-invigorating and-debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 65, 186–194. doi: 10.1016/j.soilbio.2013.05.025
Macke, E., Tasiemski, A., Massol, F., Callens, M., and Decaestecker, E. (2017). Life history and eco-evolutionary dynamics in light of the gut microbiota. Oikos 126, 508–531. doi: 10.1111/oik.03900
Mago, T., and Salzberg, S. L. (2011). Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mokrani, D., Cerezuela, R., Oumouna, M., Esteban, M. Á., and Cuesta, A. (2019). Bacteriological, metabolic and immunological evaluation of European sea bass reared in ponds with heated water under a natural vibriosis-like outbreak. Pak. Vet. J. 3, 329–334. doi: 10.29261/pakvetj/2019.082
Montoya, J. M., Pimm, S. L., and Solé, R. V. (2006). Ecological networks and their fragility. Nature 442, 259–264. doi: 10.1038/nature04927
Mortzfeld, B. M., Urbanski, S., Reitzel, A. M., Künzel, S., Technau, U., and Fraune, S. (2016). Response of bacterial colonization in Nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18, 1764–1781. doi: 10.1111/1462-2920.12926
Mueller, U. G., and Sachs, J. L. (2015). Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617. doi: 10.1016/j.tim.2015.07.009
Ning, D., Deng, Y., Tiedje, J. M., and Zhou, J. (2019). A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 116:201904623. doi: 10.1073/pnas.1904623116
Pandit, S. N., Kolasa, J., and Cottenie, K. (2009). Contrasts between habitat generalists and specialists: an empirical extension to the basic metacommunity framework. Ecology 90, 2253–2262. doi: 10.1890/08-0851.1
Pérez, T., Balcázar, J. L., Ruiz-Zarzuela, I., Halaihel, N., Vendrell, D., De Blas, I., et al. (2010). Host–microbiota interactions within the fish intestinal ecosystem. Mucosal Immunol. 3, 355–360. doi: 10.1038/mi.2010.12
Ramalingam, K., and Ramarani, S. (2007). Effect of Pseudomonas aeruginosa on the giant freshwater prawn, Macrobrachium rosenbergii: histopathological and electron microscopic study. J. Environ. Biol. 28, 627–635. doi: 10.2112/07-0836.1
Ren, Z., Li, A., Jiang, J., Zhou, L., Yu, Z., Lu, H., et al. (2019). Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 68, 1014–1023. doi: 10.1136/gutjnl-2017-315084
Rooks, M. G., and Garrett, W. S. (2016). Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352. doi: 10.1038/nri.2016.42
Roy, L. A., Davis, D. A., Saoud, I. P., Boyd, C. A., Pine, H. J., and Boyd, C. E. (2010). Shrimp culture in inland low salinity waters. Rev. Aquac. 2, 191–208. doi: 10.1111/j.1753-5131.2010.01036.x
Santolini, M., and Barabási, A. (2018). Predicting perturbation patterns from the topology of biological networks. Proc. Natl. Acad. Sci. USA 115, E6375–E6383. doi: 10.1073/pnas.1802930115
Schmidt, V. T., Smith, K. F., Melvin, D. W., and Amaral Zettler, L. A. (2015). Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol. Ecol. 24, 2537–2550. doi: 10.1111/mec.13177
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60–R18. doi: 10.1186/gb-2011-12-6-r60
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Sloan, W. T., Lunn, M., Woodcock, S., Head, I. M., Nee, S., and Curtis, T. P. (2010). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8, 732–740. doi: 10.1111/j.1462-2920.2009.02068.x
Song, Y., Shi, J., Xiong, Z., Shentu, X., and Yu, X. (2021). Three antimicrobials alter gut microbial communities and causing different mortality of brown planthopper, Nilaparvata lugens Stal. Pestic. Biochem. Physiol. 174:104806. doi: 10.1016/j.pestbp.2021.104806
Soto-Rodriguez, S. A., Gomez-Gil, B., Lozano-Olvera, R., Betancourt-Lozano, M., and Morales-Covarrubias, M. S. (2015). Field and experimental evidence of Vibrio parahaemolyticus as the causative agent of acute hepatopancreatic necrosis disease of cultured shrimp (Litopenaeus vannamei) in northwestern Mexico. Appl. Environ. Microbiol. 81, 1689–1699. doi: 10.1128/AEM.03610-14
Staley, C., Gould, T. J., Wang, P., Phillips, J., Cotner, J. B., and Sadowsky, M. J. (2015). Species sorting and seasonal dynamics primarily shape bacterial communities in the upper Mississippi River. Sci. Total Environ. 505, 435–445. doi: 10.1016/j.scitotenv.2014.10.012
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O'Connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359
Székely, A. J., Berga, M., and Langenheder, S. (2013). Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J. 7, 61–71. doi: 10.1038/ismej.2012.80
Tao, Y., Ersahin, M. E., Ghasimi, D. S. M., Ozgun, H., and Lier, J. B. V. (2020). Biogas productivity of anaerobic digestion process is governed by a core bacterial microbiota. Chem. Eng. J. 380:122425. doi: 10.1016/j.cej.2019.122425
Tilman, D. (2004). Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc. Natl. Acad. Sci. 101, 10854–10861. doi: 10.1073/pnas.0403458101
Vellend, M., Srivastava, D. S., Anderson, K. M., Brown, C. D., Jankowski, J. E., Kleynhans, E. J., et al. (2014). Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 123, 1420–1430. doi: 10.1111/oik.01493
Walter, J., and Ley, R. (2011). The human gut microbiome: ecology and recent evolutionary changes. Ann. Rev. Microbiol. 65, 411–429. doi: 10.1146/annurev-micro-090110-102830
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, L., Wei, C., Chang, Y., and Ding, J. (2021). Response of bacterial community in sea cucumber Apostichopus japonicus intestine, surrounding water and sediment subjected to high-temperature stress. Aquaculture 535:736353. doi: 10.1016/j.aquaculture.2021.736353
Wang, J., Yang, D., Zhang, Y., Shen, J., Van Der Gast, C., Hahn, M. W., et al. (2011). Do patterns of bacterial diversity along salinity gradients differ from those observed for macroorganisms? PLoS One 6:e27597. doi: 10.1371/journal.pone.0027597
Wang, F., Zhu, L., Wei, Y., Gao, P., Liu, Y., Zhou, K., et al. (2022). Intestinal ion regulation exhibits a daily rhythm in Gymnocypris przewalskii exposed to high saline and alkaline water. Sci. Rep. 12:807. doi: 10.1038/s41598-021-04472-5
Weigel, B. L. (2020). Sea cucumber intestinal regeneration reveals deterministic assembly of the gut microbiome. Appl. Environ. Microbiol. 86:e00489-20. doi: 10.1128/AEM.00489-20
Williams, R. J., Howe, A., and Hofmockel, K. S. (2014). Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front. Microbiol. 5:358. doi: 10.3389/fmicb.2014.00358
Wu, W., Lu, H. P., Sastri, A., Yeh, Y. C., Gong, G. C., Chou, W. C., et al. (2017). Contrasting the relative importance of species sorting and dispersal limitation in shaping marine bacterial versus protist communities. ISME J. 12, 485–494. doi: 10.1038/ismej.2017.183
Wu, L., Ning, D., Zhang, B., Li, Y., Zhang, P., Shan, X., et al. (2019). Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 4, 1183–1195. doi: 10.1038/s41564-019-0426-5
Xia, Y., Yi, H. X., Fan, R. B., Zhang, D., Liang, J. J., Cai, Y. Y., et al. (2020). Effects of dietary lactic acid bacteria on the meat quality of juvenile Litopenaeus vannamei. J. Fish. Sci. China 27, 74–82. doi: 10.3724/SP.J.1118.2020.19104
Xin, X., Kvitko, B., and He, S. Y. (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat. Rev. Microbiol. 16, 316–328. doi: 10.1038/nrmicro.2018.17
Xing, M., Hou, Z., Yuan, J., Liu, Y., Qu, Y., and Liu, B. (2013). Taxonomic and functional metagenomic profiling of gastrointestinal tract microbiome of the farmed adult turbot (Scophthalmus maximus). FEMS Microbiol. Ecol. 86, 432–443. doi: 10.1111/1574-6941.12174
Yang, H. L., Sun, Y. Z., Ye, J. D., Chang, J. B., and Chen, Z. Q. (2008). The antagonistic property of lactic acid bacteria derived from orange-spotted grouper (Epinephelus coioides) larvae. J. Shanghai Fish. Univ. 17, 344–349. doi: 10.3724/SP.J.1035.2008.00038
Zhang, J. L. (2013). Spaa: species association analysis. R package version 0.2.1. Available online at: https://cran.r-project.org/package=spaa
Zhang, W., Li, Y., Wang, C., Wang, P., Hou, J., Yu, Z., et al. (2016). Modeling the biodegradation of bacterial community assembly linked antibiotics in river sediment using a deterministic stochastic combined model. Environ. Sci. Technol. 50, 8788–8798. doi: 10.1021/acs.est.6b01573
Zhang, X., Sun, J., Han, Z., Chen, F., Lv, A., Hu, X., et al. (2021). Vibrio parahaemolyticus alters the community composition and function of intestinal microbiota in Pacific white shrimp, Penaeus vannamei. Aquaculture 544:737061. doi: 10.1016/j.aquaculture.2021.737061
Zhang, M., Sun, Y., Liu, Y., Qiao, F., Chen, L., Liu, W., et al. (2016). Response of gut microbiota to salinity change in two euryhaline aquatic animals with reverse salinity preference. Aquaculture 454, 72–80. doi: 10.1016/j.aquaculture.2015.12.014
Zhang, Q., Yu, Y., Luo, Z., and Li, F. (2023). Hepatopancreas color as a phenotype to indicate the infection process of Vibrio parahaemolyticus in Pacific white shrimp Litopenaeus vannamei. Aquaculture 572:739545. doi: 10.1016/j.aquaculture.2023.739545
Zheng, X., Duan, Y., Dong, H., and Zhang, J. (2020). The effect of Lactobacillus plantarum administration on the intestinal microbiota of whiteleg shrimp Penaeus vannamei. Aquaculture 526:735331. doi: 10.1016/j.aquaculture.2020.735331
Zhou, L., Li, H., Qin, J. G., Wang, X., Chen, L., Xu, C., et al. (2020). Dietary prebiotic inulin benefits on growth performance, antioxidant capacity, immune response and intestinal microbiota in Pacific white shrimp (Litopenaeus vannamei) at low salinity. Aquaculture 518:734847. doi: 10.1016/j.aquaculture.2019.734847
Keywords: co-occurrence network, community assembly, gut bacterial community, salinity, Penaeus vannamei
Citation: Wang B, Liu Y, Wu X, Liu Y, Li Z, Wang J, Lian Y, Tang J, Yun B and Tian X (2025) Characteristics of gut microbiota in Penaeus vannamei shifted with elevated salinity. Front. Microbiol. 16:1665547. doi: 10.3389/fmicb.2025.1665547
Edited by:
Marcia Saraiva, University of Porto, PortugalReviewed by:
Yafei Duan, South China Sea Fisheries Research Institute, ChinaChongqing Wen, Guangdong Ocean University, China
Ruth Escamilla Montes, Centro de Investigación Biológica del Noroeste (CIBNOR), Mexico
Copyright © 2025 Wang, Liu, Wu, Liu, Li, Wang, Lian, Tang, Yun and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangli Tian, eGlhbmdsaXRpYW5Ab3VjLmVkdS5jbg==