 Mingzhu Zheng1,2
Mingzhu Zheng1,2 Can Zhong
Can Zhong Gen Pan
Gen Pan Shuihan Zhang
Shuihan Zhang Jian Jin
Jian Jin- 1Institute of Chinese Medicine Resources, Hunan Academy of Chinese Medicine, Changsha, China
- 2Hunan Academy of Chinese Medicine, Hunan University of Chinese Medicine, Changsha, China
- 3Management Committee, Hunan Xiangxi High-Tech Industrial Development Zone, Jishou, China
Introduction: Poria cocos (P. cocos) is a medicinal fungus renowned for its bioactive triterpenoids, particularly pachymic acid. However, the relationship between its specialized microbiota and the accumulation of this key metabolite remains poorly understood.
Methods: This study systematically compared the microbial communities and pachymic acid distribution patterns between P. cocos and different soil microenvironments using integrated 16S rRNA/ITS1 sequencing and HPLC-QTOF-MS/MS analysis.
Results: The results revealed significantly lower microbial diversity in P. cocos compared to the surrounding soil, with a dominance of Proteobacteria and Ascomycota, along with specific enrichments of Burkholderia-Caballeronia-Paraburkholderia and Scytalidium. Pachymic acid was found to accumulate predominantly within the sclerotia, with trace amounts detectable in adjacent soils. Significant positive correlations were identified between pachymic acid and these enriched microbial taxa.
Discussion: These findings indicate that P. cocos forms a specialized microenvironment characterized by selective microbial enrichment associated with pachymic acid accumulation, offering valuable insights for optimizing cultivation strategies to improve its medicinal quality.
1 Introduction
Poria cocos (P. cocos), a medicinal and edible macrofungus with over 2000 years of documented therapeutic use, is renowned for its diuretic, sedative, and immunomodulatory properties (Zhang et al., 2019; He et al., 2025; Guo et al., 2025). Pachymic acid is one of its principal bioactive components and has attracted considerable attention due to its remarkable antitumor (Yang et al., 2022; Jiang and Fan, 2020), anti-inflammatory, and immunoregulatory effects (Li et al., 2015). Recent advances in omics technologies have enabled preliminary elucidation of the biosynthetic pathways of triterpenoids in P. cocos through genomic and metabolomic studies (Liu et al., 2024; Noushahi et al., 2024; Liu et al., 2023). Nevertheless, the microbial regulatory mechanisms involved in sclerotium formation, particularly the relationship between pachymic acid biosynthesis and microbial community interactions, remain poorly understood.
In studies of biological-environmental interactions, both plants and macrofungi exhibit complex interactions with microbial communities. Research on plants has demonstrated that terpenoid compounds can resist pest infestation and enhance plant resistance (Pétriacq et al., 2017a; Huang et al., 2019; Zhong et al., 2022), while also functioning as signaling molecules to regulate rhizosphere microbial communities (Sugiyama et al., 2016; Bouwmeester et al., 2025). Distinct from plant root systems, most fungi establish unique microecosystems through their mycelial networks, facilitating material exchange and signal transmission with the surrounding substrate (Xu et al., 2023). In edible mushroom cultivation systems, Pleurotus ostreatus cultivation relies on Actinobacteria and Firmicutes to degrade lignocellulose, providing carbon sources for mycelial growth (Suarez et al., 2020). Stropharia rugosoannulata can enrich beneficial fungi such as Mortierella and Cladosporium, which not only suppress pathogens but also enhance soil nutrient levels through secondary metabolite production (Tang et al., 2022). Additionally, studies on Morchella have revealed a close correlation between the enrichment of carbon-fixing Chloroflexi and yield improvement, potentially through providing carbon skeleton precursors for host secondary metabolism (Tan et al., 2021). Regarding medicinal fungi, the synthesis of triterpenoids in Ganoderma lucidum shows significant correlation with rhizosphere microorganisms (Ren et al., 2020a; Chang et al., 2018). These findings provide important references for understanding the microbial regulation mechanisms in medicinal fungi.
Unlike typical mushrooms or polypores, the medicinal part of P. cocos is not the fruiting body but rather the sclerotium formed by tightly aggregated mycelia. This unique structure likely relies on distinct microbial interaction patterns. While numerous studies have investigated microorganism-secondary metabolite relationships in medicinal plants such as Astragalus (Lin et al., 2022; Zhiyong et al., 2024; Li et al., 2021), Salvia (Zhang et al., 2023) and Ginkgo (Jia et al., 2025), Astragalus, similar research on medicinal fungi - particularly regarding P. cocos - remains completely unexplored.
By integrating high-throughput sequencing with high-performance liquid chromatography-quadrupole/time-of-flight mass spectrometry (HPLC-QTOF-MS/MS), this study systematically characterizes the structural features of the P. cocos sclerotium microecosystem and its correlation with pachymic acid synthesis. The findings provide novel theoretical insights into the correlation mechanisms between medicinal fungi and associated microbiota, while offering scientific support for ecological cultivation techniques aimed at enhancing P. cocos quality through microecological regulation.
2 Materials and methods
2.1 Cultivation of Poria cocos
The present study was conducted at the Chinese Medicine Resources Experimental Base of the Hunan Academy of Chinese Medicine, Changsha, Hunan Province, China (N 28°12′09.10″, E 112°45′03.05″). P. cocos spawn was provided by the Jingzhou P. cocos Association of Hunan, China, and cultivation was carried out using pine wood segments (60 cm in length) as the substrate. Four experimental groups were established: spawn & woods, woods-only, spawn-only, and bare soil as the control, with six replicates per group. For cultivation, trenches (50 cm deep × 90 cm wide) were dug, and wood segments were horizontally placed at 2-meter intervals. In the spawn + woods group, the spawn bags were cross-cut with three incisions each vertically and horizontally before being placed between two tightly adjacent pine wood segments and secured with polyethylene film. The woods-only and spawn-only groups contained only wood segments or spawn bags, respectively, while the bare soil group remained untreated. All trenches were backfilled with topsoil.
2.2 Harvesting of samples
After 9 months of cultivation, sampling was conducted by carefully excavating the topsoil to collect mature P. cocos specimens. For the P. cocos samples, the sclerotia were carefully unearthed, rinsed with distilled water to remove surface soil, and then wiped with 75% ethanol. The outer peel was aseptically excised using a sterilized scalpel as the representative sample. Soil samples were collected from each treatment group at a distance of 10 cm from either the P. cocos, spawn bags, or wood segments, as illustrated in Figure 1. Following collection, all soil and fungal samples were systematically divided into two aliquots: the first was immediately flash-frozen in liquid nitrogen and stored at −80°C for subsequent microbial community analysis through ITS and 16S rRNA gene sequencing, while the second aliquot was preserved for precise quantification of pachymic acid content.
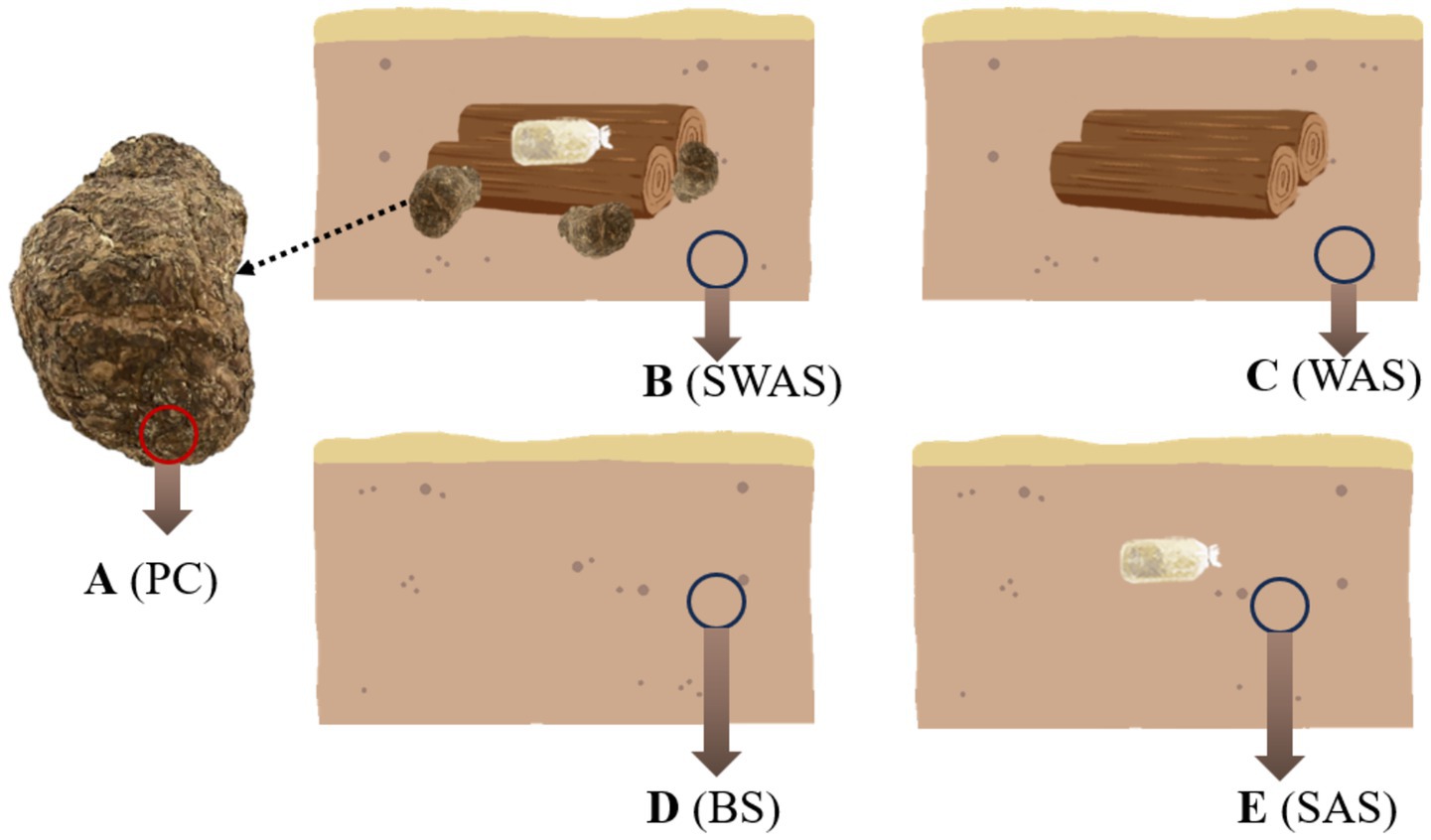
Figure 1. Schematic diagram of sampling locations in Poria cocos and the surrounding soil. (A) (PC), Poria cocos; (B) (SWAS), Spawn’ and Wood Adiacent Soil; (C) (WAS), Wood Adiacent Soil; (D) (BS), Bare Soil; (E) (SAS), Spawn Adiacent Soil.
2.3 DNA extraction, PCR amplification and sequencing
According to the manufacturer’s instructions, DNA extraction was performed using a DNA extraction kit. For bacterial 16S rRNA amplification, the primers 341F (CCTAYGGGRBGCASCAG) and 806R (GGACTACNNGGGTATCTAAT) (Ferreira et al., 2024) were used. For fungal ITS1 region amplification, the primers ITS1F (CTTGGTCATTTAGAGGAAGTAA) and ITS2R (GCTGCGTTCTTCATCGATGC) (Bukavina et al., 2022) were employed. After purification, quantification, and normalization of the PCR products, 16S rRNA/ITS amplicon sequencing was performed on an Illumina NovaSeq 6,000 platform by Wuhan Frasergen Bioinformatics Co., Ltd. (Wuhan, China). The raw image data files were then converted into raw sequencing reads through base calling. The results were saved in FASTQ format, which includes sequence data and corresponding quality scores. All sequences were deposited at the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under project accession number PRJNA1310015.
2.4 Bioinformatics analysis
The QIIME 2 software platform was used to process the data obtained from Illumina HiSeq amplicon sequencing. Briefly, the reads were processed and applied to DADA2 pipeline for the assignment of amplicon sequence variants (ASVs). Before application to DADA2, the fungal sequences were trimmed by the plugin of ITSxpress. The DADA2 workflow includes filtering, dereplication, chimera identification, and merging of the paired-end sequences. Taxonomic assignment of the ASVs was performed using the SILVA 128 database for bacteria and the UNITE database for fungi. The RDP Classifier algorithm was employed for sequence alignment analysis. To assess sequencing depth sufficiency and sampling completeness, Good’s coverage estimator was computed. Alpha diversity metrics (including observed ASVs, Shannon index, and Faith’s phylogenetic diversity) and beta diversity analyses (using UniFrac distances) were subsequently calculated to evaluate within-sample and between-sample microbial diversity patterns, respectively. Beta diversity was visualized by principal coordinate analysis (PCoA). In order to further explore the differences in microbial community structure among different groups, partial least squares discrimination analysis (PLS-DA) was applied.
2.5 Extraction and HPLC-QTOF-MS/MS analysis of pachymic acid
The collected samples were placed in a constant-temperature drying oven and continuously dried at 55°C until reaching constant weight, then ground into uniformly-sized powder. Precisely 1.00 ± 0.01 g of the powdered sample was weighed and mixed with 5 mL of methanol for ultrasound-assisted extraction. The extraction process was conducted in an ultrasonic bath with 220 W power and 50 kHz frequency for 15 min. After allowing the extract to stand for 20 min, the supernatant was filtered through a 0.22 μm organic phase microporous membrane. Chromatographic analysis was performed using HPLC-QTOF-MS/MS. Acetonitrile and pure water were used as mobile phases A and B, respectively. The injection volume was 10 μL with a solvent flow rate of 1 mL/min, using an InterSustain C18 column (5 μm, 4.6 × 250 mm, GL Sciences Inc., Tokyo, Japan) maintained at 20°C. The solvent gradient program was set as follows:0–10 min: 51% A; 10–15 min: 51–86% A; 15–25 min: 86–100% A; 25–35 min: 100% A; 35–40 min: 100–51% A; 40–50 min: 51% A. Mass spectrometry conditions employed negative ion mode with a scan range of m/z 100–1,500.
2.6 Statistical analysis
The quantitative determination of pachymic acid was done in six repetitions and expressed as mean ± standard deviation. Differences among groups were analyzed statistically using one-way analysis of variance. Statistical analyses of different samples were performed using the Origin statistical software. Pearson’s correlation coefficient was utilized to analyze the correlation. p < 0.05 was considered to indicate a statistically significant difference.
3 Results
3.1 Microbial community composition
This study conducted a systematic taxonomic analysis of microbial communities across five sample groups. A total of 39 bacterial phyla and 820 genera were identified, along with 16 fungal phyla and 587 genera. Significant differences were observed in the relative abundance of various taxonomic units among sample groups, with their phylogenetic relationships illustrated in Figure 2a (bacterial genus level) and Figure 2b (fungal genus level).

Figure 2. Phylogenetic trees of fungal (a) and bacterial (b) genera at the genus level. Venn diagrams showing shared and unique fungal (c) and bacterial (d) ASVs among samples. A, Poria cocos; B, spawn and wood adjacent soil; C, wood adjacent soil; D, bare soil; E, spawn adjacent soil.
High-throughput sequencing yielded 17,578 high-quality ASVs. Among these, bacterial ASVs accounted for 13,175 (75.0%), while fungal ASVs comprised 4,403 (25.0%). Microbial community analysis revealed that the core bacterial and fungal ASVs shared across all samples numbered only 75 and 64, respectively (Figures 2c,d), indicating significant compositional differences among microenvironments.
Microbial composition analysis across sample groups revealed that P. cocos (A) contained 925 bacterial ASVs and 244 fungal ASVs, while the spawn & wood adjacent soil (B) harbored 2,966 bacterial ASVs and 1,501 fungal ASVs. The wood-adjacent soil (C) showed 3,148 bacterial ASVs and 1,726 fungal ASVs, with bare soil (D) containing 3,248 bacterial ASVs and 1,605 fungal ASVs. The spawn adjacent soil (E) demonstrated the highest microbial diversity with 3,286 bacterial ASVs and 1,690 fungal ASVs (Figures 2c,d). These results demonstrate that P. cocos harbors a distinct microbial community composition compared to surrounding soil environments, with significantly lower microbial diversity.
3.2 Beta diversity analysis of microbial community
Multivariate statistical analysis revealed significant differences in microbial communities among sample groups. Principal coordinate analysis (PCoA) based on Bray-Curtis distance showed that the first two axes (PCoA1 and PCoA2) of bacterial communities cumulatively explained 31.79% of the total variance (PCoA1: 20.66%; PCoA2: 8.61%), while those of fungal communities explained 29.65% (PCoA1: 21.36%; PCoA2: 8.29%). The analysis demonstrated that P. cocos samples (A) were separated from soil sample groups (B-E) in the coordinate space, while different soil groups showed partial overlap but still exhibited clustering tendencies (Figures 3a,b). To validate this pattern, partial least squares-discriminant analysis (PLS-DA) was performed to examine inter-group differences among soil samples. The results showed significant separation of all groups under the supervised model (Figures 3c,d). The consistency between these two analytical approaches indicates that P. cocos possesses a distinct microbial community structure, while differences among soil groups exist but with relatively blurred boundaries.

Figure 3. Multivariate analysis of microbial beta diversity. (a) Principal coordinates analysis (PCoA) of bacterial communities; (b) PCoA of fungal communities; (c) partial least squares-discriminant analysis (PLS-DA) of bacterial communities; (d) PLS-DA of fungal communities. A, Poria cocos; B, spawn and wood adjacent soil; C, wood adjacent soil; D, bare soil; E, spawn adjacent soil.
3.3 Alpha diversity analysis of microbial community
The alpha diversity analysis reflects microbial community richness and diversity within samples. Shannon, Simpson, and Chao1 indices collectively revealed that all soil groups (B-E) exhibited substantially higher microbial richness and diversity compared to P. cocos samples (A) (p < 0.001), while no significant differences were observed among the various soil groups themselves. The high Goods coverage values (>99.9%) across all samples confirmed adequate sequencing depth, ensuring comprehensive detection of predominant microbial taxa and reliable representation of both bacterial and fungal community compositions. Notably, the boxplot visualization (Figure 4) clearly illustrated these patterns, with P. cocos (A) consistently showing the lowest diversity values for both bacteria (Figure 4a) and fungi (Figure 4b), while the four soil microenvironment groups (B-E) formed a cluster of higher diversity values without statistically significant inter-group variations. These results further corroborate the distinct nature of the P. cocos microbiome relative to its surrounding soil environments.

Figure 4. Alpha diversity of microbial communities. (a) bacteria; (b) fungi. A, Poria cocos; B, spawn and wood adjacent soil; C, wood adjacent soil; D, bare soil; E, spawn adjacent soil. Statistical significance was determined by Student’s t-test. *p<0.05; **p<0.01; ***p<0.001.
3.4 Bacterial community composition analysis
The bacterial community analysis revealed that Proteobacteria, Actinobacteriota, and Acidobacteriota were the dominant phyla (relative abundance >5% in all groups) (Figure 5a; Supplementary Table S1). Notably, Proteobacteria was the most abundant phylum across all samples, with its relative abundance in P. cocos (A) being more than double that in soil groups (approximately 75% vs. 30–35%). The P. cocos group exhibited distinct bacterial community characteristics, showing significantly higher relative abundances of Proteobacteria and Firmicutes, but lower abundances of Actinobacteriota, Bacteriodota, Chloroflexi, and Gemmatimonadota compared to soil groups. Interestingly, soil samples containing pine wood segments (B and C) demonstrated higher relative abundances of Proteobacteria, Actinobacteriota, and Gemmatimonadota, but lower abundances of Acidobacteriota and Chloroflexi compared to wood-free soil samples (D and E).

Figure 5. Bacteria abundance in different samples: (a) at phylum level, top 30 phylum shown, rest as others; (c) at genus level, top 30 genus shown, rest as others; Fungi abundance in different samples: (b) at phylum level; (d) at genus level, top 30 genus shown, rest as others. A, Poria cocos; B, spawn & wood adjacent soil; C, wood adjacent soil; D, bare soil; E, spawn adjacent soil.
At the genus level (Figure 5b; Supplementary Table S2), P. cocos (A) displayed a unique profile dominated by Burkholderia-Caballeronia-Paraburkholderia (50.02%), Acidisoma (6.93%), and Mycobacterium (5.10%). In contrast, the four soil groups shared more similar community structures with greater overall richness. For instance, Group B was characterized by distinct dominant genera including Sphingomonas (5.82%), RB41, Gemmatimonas (3.59%), and several uncultured taxa from Gemmatimonadaceae, Xanthobacteraceae, and Vicinamibacteraceae families. The higher relative abundance of Sphingomonas and Gemmatimonas in Group B compared to other soil groups suggests potential microenvironment-specific bacterial adaptations.
3.5 Fungal community composition analysis
The fungal community characteristics (Figure 5c) mirrored those of the bacterial communities: P. cocos samples (A) exhibited distinct characteristics at the phylum level, with Ascomycota dominating (98.29%) while Basidiomycota and Mortierellomycota were present in minimal abundance (<1%). This contrasted sharply with soil groups (B-E), where Ascomycota accounted for 58–60% of the community (Supplementary Table S3). Beyond the dominant phyla, soil samples harbored various low-abundance phyla including Mortierellomycota, Chytridiomycota, and Fungi_phy_Incertae_sedis. Notably, bare soil (D) and spawn-only adjacent soil (E) showed significantly higher relative abundance of Basidiomycota (19–20%), potentially reflecting substrate-specific influences.
At the genus level (Figure 5d; Supplementary Table S4), Group A was characterized by dominant genera Scytalidium (58.37%), uncultured Ascomycota (15.11%), and Talaromyces (8.86%). In contrast, soil groups demonstrated clear substrate-dependent community patterns: Group B featured Polyschema as its characteristic genus, Group C was dominated by Peziza, Group D showed enrichment of Humicola, while Group E was represented by Corynascella. These differences further substantiate the significant impact of growth substrates on fungal community structure.
3.6 Pachymic acid distribution and chemical profiling
After 9 months of cultivation, quantitative analysis of pachymic acid content across all samples (Figure 6) revealed distinct accumulation patterns. P. cocos samples (Group A) exhibited substantial pachymic acid accumulation, while only trace amounts were detected in the spawn & wood adjacent soil (B). No pachymic acid was detected in other soil groups (C-E). These findings demonstrate that pachymic acid is a specific metabolite of P. cocos, primarily synthesized through its endogenous metabolic activity rather than by environmental microorganisms. The trace detection in Group B likely originates from either active secretion or passive diffusion from P. cocos.
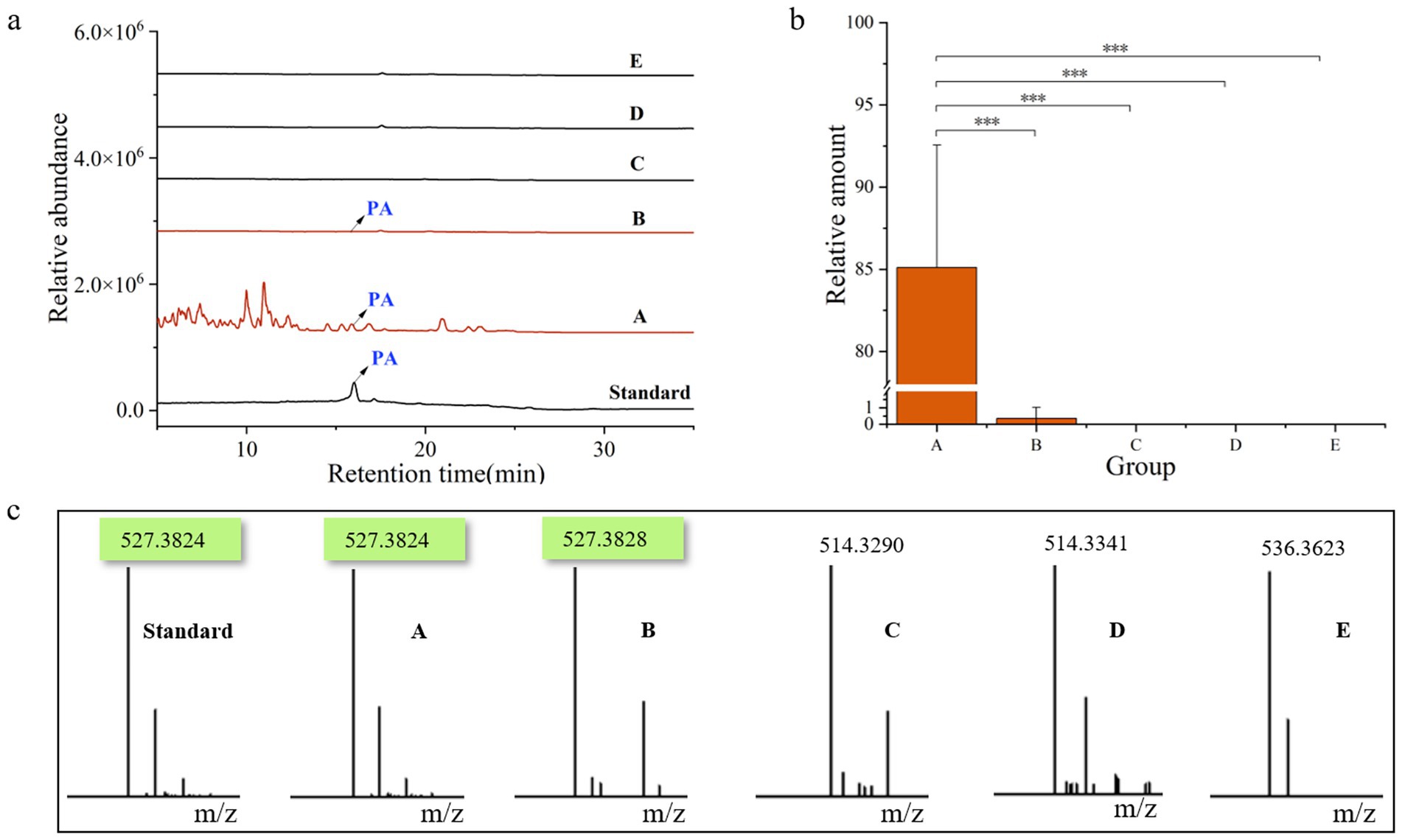
Figure 6. Chemical profiling of pachymic acid in different sample groups. (a) Total ion chromatograms of the six samples. (b) Relative amount of pachymic acid in each group of samples. (c) Corresponding mass spectrometry profiles of the six samples. Standard, pachymic acid; A, Poria cocos; B, spawn & wood adjacent soil; C, wood adjacent soil; D, bare soil; E, spawn adjacent soil. *p<0.05; **p<0.01; ***p<0.001.
3.7 Correlation analysis between Pachymic acid content and microbial communities
To investigate the potential associations between pachymic acid accumulation and microbial communities, we performed correlation analysis between pachymic acid content and the top 10 most abundant bacterial and fungal genera across samples (Figure 7). The analysis revealed significant positive correlations between pachymic acid content and Burkholderia-Caballeronia-Paraburkholderia (p < 0.001) among bacterial communities, while showing significant negative correlations with Gemmatimonadaceae, RB41, Vicinamibacteraceae, Vicinamibacterales, Ellin6067 and MND1 (p < 0.05). Notably, these negatively correlated bacterial genera exhibited strong positive inter-genus relationships. For fungal communities, pachymic acid content demonstrated significant positive correlations with both Scytalidium and Talaromyces (p < 0.01), which themselves showed an exceptionally strong positive correlation (p < 0.001). These results suggest that pachymic acid accumulation is closely associated with selective enrichment of specific microbial taxa in the sclerotium microenvironment, suggesting a potential association between host metabolite production and microbial community composition.

Figure 7. Correlations between pachymic acid content and top 10 microbial genera: (a) bacterial, (b) fungal. Red indicates positive correlations while blue shows negative correlations, with color intensity representing correlation strength. Circle size corresponds to statistical significance: *p < 0.05; **p < 0.01; ***p < 0.001.
4 Discussion
This study reveals the unique microbial composition of P. cocos and its interaction mechanisms with the environment. Alpha and beta diversity analyses demonstrated significantly lower microbial diversity (Shannon, Simpson and Chao1 index, p < 0.001) in P. cocos samples (Group A) compared to each soil group. PCoA analysis (31.79% variance explained for bacteria, 29.65% for fungi) showed distinct clustering of P. cocos communities, a pattern strongly correlated with the specific distribution of pachymic acid. This suggests that host secondary metabolites may play a crucial role in microbial community selection. Similar mechanisms have been demonstrated in various plant-microbe interaction systems. For instance, Arabidopsis thaliana shapes specific rhizosphere microbiomes through secreted triterpenoids (Huang et al., 2019; Pétriacq et al., 2017b), while tomatoes recruit actinobacteria to suppress pathogens by secreting riboflavin and 3-hydroxyflavones (Yang et al., 2023).
Analysis of microbial community composition revealed that the dominant bacterial phyla across all samples included Proteobacteria, Actinobacteria, and Acidobacteria, consistent with their widespread distribution in soil ecosystems (Maestre et al., 2015). Notably, P. cocos exhibited significant enrichment of Proteobacteria and Firmicutes, while Actinobacteriota, Bacteroidota, and Chloroflexi were more abundant in soil groups. This differential distribution pattern implies host-mediated active selection, potentially through suppression of actinobacterial resource competition or antibiotic effects to optimize its microecological environment (Janssen, 2006; Yun et al., 1997). At the genus level, Burkholderia-Caballeronia-Paraburkholderia showed prominent enrichment and significant correlation with pachymic acid content. These lignin-degrading microbes can produce secondary metabolic precursors such as ferulic acid, vanillic acid, and malic acid, which provide carbon sources for P. cocos growth (Smith et al., 2007; Akita et al., 2016; Morya et al., 2021). Paraburkholderia, a monophyletic clade derived from Burkholderia, has been shown to promote plant growth and enhance disease resistance (Cao et al., 2025; Mitter et al., 2017; Ledger et al., 2016). Similar to its role in inducing the accumulation of flavonoids and hydroxycinnamic acid in broccoli (Jeon et al., 2021), the strain may further affect the synthesis of medicinal active ingredients by regulating the secondary metabolism of P. cocos. Additionally, acid-tolerant Acidisoma may thrive in P. cocos ‘s acidic microenvironment rich in triterpenoids (Yu et al., 2023). The selective enrichment of functional microbes like nitrogen-fixing Sphingomonas collectively constitutes the unique microbiome signature of P. cocos (Li et al., 2023; Li et al., 2025; Kämpfer et al., 2025). Particularly noteworthy is a recent discovery that Streptomyces in citrus rhizospheres harbors abundant terpenoid synthesis gene clusters, potentially promoting host monoterpene accumulation by gene encoding providing 1-deoxy-D-xylulose-5-phosphate synthase pathway intermediates (Su et al., 2023), offering new theoretical insights into the interaction between pachymic acid synthesis and microbial communities.
Wood-containing groups (B and C) showed increased Proteobacteria, Actinobacteriota, and Gemmatimonadota abundance with concurrent reduction of Acidobacteriota and Chloroflexi, potentially reflecting carbon partitioning from lignocellulose degradation (Mendes et al., 2021; Gavande et al., 2021; Koubová et al., 2023). The phosphorus-solubilizing Gemmatimonas (Kang et al., 2015; Zhu et al., 2024) may gain a competitive advantage in the high-carbon, low-phosphorus wood substrate environment, consistent with observations in Ganoderma cultivation systems (Ren et al., 2020b). However, these correlations require validation through metabolomic approaches to elucidate direct carbon utilization mechanisms.
The fungal communities were dominated by Ascomycota across all samples, with significantly higher relative abundance in P. cocos compared to soil groups. In contrast, Basidiomycota, Mortierellomycota, and Chytridiomycota were more prevalent in soils. This distinct distribution pattern may stem from antibiotic secretion by P. cocos suppressing competitors, analogous to the antifungal strategies employed by Tricholoma matsutake (Oh et al., 2016; Liu et al., 2021). The dominant fungal genera in P. cocos, Scytalidium and Talaromyces, showed significant correlations with pachymic acid content. Scytalidium is a thermophilic fungal genus. Co-culture of Scytalidium parasiticum and Ganoderma boninense induces the production of antimicrobial metabolites (e.g., alkaloids and flavonoids) active against Ganoderma (Ahmad et al., 2020), which may help suppress Ganoderma-induced basal stem rot (Goh et al., 2016). Talaromyces has been reported to possess abundant secondary metabolite gene clusters including terpene synthases (Zhai et al., 2016; Weber et al., 2015), while some Ascomycetes, like Bulgaria inquinans, can synthesize triterpenoid acids, such as betulinic and ursolic acids. While some Ascomycetes (e.g., Bulgaria inquinans) have been shown to produce triterpenoid acids (Zhang et al., 2005). This suggests potential direct involvement of P. cocos-associated fungi in triterpenoid acid biosynthesis. Soil groups exhibited substrate-dependent variations at the genus level, with distinct dominant taxa (Polyschema in B, Peziza in C, Humicola in D, and Corynascella in E), reflecting the differential impacts of fungal inoculation, spawn bags and wood segments.
The unique spatial distribution of pachymic acid - abundant in P. cocos, only a small amount in the surrounding soil, but it is not detected in the control soil at all. This strongly supports active host selection of specific microbiota, consistent with known roles of terpenoids in microbial community regulation (Pichersky and Raguso, 2018). While this study has revealed significant associations between the microbial community and metabolic products in P. cocos, the underlying functional mechanisms require further validation. Future investigations should employ strain isolation and multi-omics approaches to systematically elucidate the P. cocos-microbiota-triterpenoid interaction network. These studies will provide more robust theoretical foundations for microbiome-mediated cultivation optimization of medicinal fungi.
5 Conclusion
This study revealed that P. cocos establishes a unique microecosystem by enriching specific microbial taxa, particularly Proteobacteria and Ascomycota, with the Burkholderia-Caballeronia-Paraburkholderia and Scytalidium showing significant correlations with the specific accumulation of pachymic acid. In contrast, surrounding soil groups exhibited higher microbial diversity with distinct compositional differences observed among various treatment groups. The findings suggest that the secretion of triterpenoid compounds in P. cocos coincides with the presence of specific microbial taxa, while pine wood substrates further optimize this metabolic microenvironment by promoting lignin-degrading microbial communities. These results advance our understanding of fungus-microbe interactions in medicinal species and provide insights for cultivation optimization.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
MZ: Writing – original draft, Visualization. CZ: Methodology, Writing – original draft, Conceptualization. GP: Software, Writing – original draft, Data curation. JX: Writing – original draft, Formal analysis, Validation. SZ: Methodology, Conceptualization, Writing – original draft. JJ: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by The National Natural Science Foundation of China (Nos. 82474039 and 82404805); Hunan Natural Science Foundation (Nos. 2024JJ8161 and 2023JJ50074); 2024 Innovative Project of Postgraduates in Hunan University of Chinese Medicine (No. 2024CX138); 2023 National Traditional Chinese Medicine Specialty Technology Inheritance Talent Training Project (No. T20234832005); the First-Class Discipline of Pharmaceutical Science of Hunan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1674216/full#supplementary-material
References
Ahmad, R., Lim, C. K., Marzuki, N. F., Goh, Y.-K., Azizan, K. A., Goh, Y. K., et al. (2020). Metabolic profile of Scytalidium parasiticum-Ganoderma boninense co-cultures revealed the alkaloids, flavonoids and fatty acids that contribute to anti-Ganoderma activity. Molecules 25:965. doi: 10.3390/molecules25245965
Akita, H., Kimura, Z.-i., Mohd Yusoff, M. Z., Nakashima, N., and Hoshino, T. (2016). Isolation and characterization of Burkholderia sp. strain CCA53 exhibiting ligninolytic potential. Springerplus 5:596. doi: 10.1186/s40064-016-2237-y
Bouwmeester, H., Dong, L., Wippel, K., Hofland, T., and Smilde, A. (2025). The chemical interaction between plants and the rhizosphere microbiome. Trends Plant Sci. 2025:6. doi: 10.1016/j.tplants.2025.06.001
Bukavina, L., Prunty, M., Calaway, A. C., Isali, I., Retuerto, M., Ghannoum, M., et al. (2022). Characterization of fungal mycobiome in bladder cancer. J. Clin. Oncol. 40:542. doi: 10.1200/JCO.2022.40.6_suppl.542
Cao, X., Yuan, Q., Hu, C., Zhang, H., Sun, X., Yan, B., et al. (2025). Wild wisdom meets cultivation: comparative rhizomicrobiome analysis unveils the key role of Paraburkholderia in growth promotion and disease suppression in Coptis chinensis. Microbiome 13:150. doi: 10.1186/s40168-025-02136-4
Chang, T.-S., Ko, H.-H., Wang, T.-Y., Lee, C.-H., and Wu, J.-Y. (2018). Biotransformation of ganoderic acid a to 3-O-acetyl ganoderic acid a by soil-isolated Streptomyces sp. Fermentation 4:101. doi: 10.3390/fermentation4040101
Ferreira, I. M. M., De Inácio Abreu, M. J. I. J., Rodrigues, A. N. N., De Souza, G. A. P., Marcon, H. J., Viquez-Umana, F. L., et al. (2024). PSXII-18 nitrogen balance and ruminal bacterial diversity indexes of Nellore cattle fed with amylolytic and fibrolytic enzymes in feedlot. J. Anim. Sci. 102, 782–783. doi: 10.1093/jas/skae234.881 J Journal of Animal Science
Gavande, P. V., Basak, A., Sen, S., Lepcha, K., Murmu, N., Rai, V., et al. (2021). Functional characterization of thermotolerant microbial consortium for lignocellulolytic enzymes with central role of Firmicutes in rice straw depolymerization. Sci. Rep. 11:3032. doi: 10.1038/s41598-021-82163-x
Goh, Y. K., Marzuki, N. F., Goh, T. K., Tan, S. Y., Goh, Y. K., Goh, K. J. J. B. S., et al. (2016). Mycoparasitic Scytalidium parasiticum as a potential biocontrol agent against Ganoderma boninense basal stem rot in oil palm. Biocontrol Sci. Tech. 26, 1352–1365. doi: 10.1080/09583157.2016.1202192
Guo, Z.-y., Wu, X., Zhang, S.-j., Yang, J.-h., Miao, H., and Zhao, Y.-y. (2025). Poria cocos: traditional uses, triterpenoid components and their renoprotective pharmacology. Acta Pharmacol. Sin. 46, 836–851. doi: 10.1038/s41401-024-01404-7
He, X., Tao, Y., Song, C., He, J., Gong, D., Yu, W., et al. (2025). Novel parkin agonists from Poria cocos against dyslipidemia. Fitoterapia 182:106469. doi: 10.1016/j.fitote.2025.106469
Huang, A. C., Jiang, T., Liu, Y. X., Bai, Y. C., Reed, J., Qu, B., et al. (2019). A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364:389. doi: 10.1126/science.aau6389
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006
Jeon, J.-S., Carreno-Quintero, N., van Eekelen, H. D. L. M., De Vos, R. C. H., Raaijmakers, J. M., and Etalo, D. W. (2021). Impact of root-associated strains of three Paraburkholderia species on primary and secondary metabolism of Brassica oleracea. Sci. Rep. 11:2781. doi: 10.1038/s41598-021-82238-9
Jia, H.-M., Zhou, J., Zhao, W.-C., He, D.-M., and Yan, Z.-Y. (2025). β-Elemonic acid mediated enrichment of Paenibacillus to help Salvia miltiorrhiza Bunge alleviate drought stress. Microbiome 13:153. doi: 10.1186/s40168-025-02154-2
Jiang, Y., and Fan, L. (2020). Evaluation of anticancer activities of Poria cocos ethanol extract in breast cancer: in vivo and in vitro, identification and mechanism. J. Ethnopharmacol. 257:112851. doi: 10.1016/j.jep.2020.112851
Kämpfer, P., Lipski, A., Lawrence, K. S., Olive, W. R., Newman, M. M., McInroy, J. A., et al. (2025). Flavobacterium plantiphilum sp. nov., Flavobacterium rhizophilum sp. nov., Flavobacterium rhizosphaerae sp. nov., Chryseobacterium terrae sp. nov., and Sphingomonas plantiphila sp. nov. isolated from salty soil showing plant growth promoting potential. Syst. Appl. Microbiol. 48:126588. doi: 10.1016/j.syapm.2025.126588
Kang, S.-M., Ramalingam, R., Ko-Eun, L., Young-Hyun, Y., Jae-Hwan, K., Jin-Ho, K., et al. (2015). Mechanism of plant growth promotion elicited by Bacillus sp. LKE15 in oriental melon. Acta Agric. Scand. B Soil Plant Sci. 65, 637–647. doi: 10.1080/09064710.2015.1040830
Koubová, A., Lorenc, F., Horváthová, T., Chroňáková, A., and Šustr, V. (2023). Millipede gut-derived microbes as a potential source of cellulolytic enzymes. World J. Microbiol. Biotechnol. 39:169. doi: 10.1007/s11274-023-03620-5
Ledger, T., Rojas, S., Timmermann, T., Pinedo, I., Poupin, M. J., Garrido, T., et al. (2016). Volatile-mediated effects predominate in Paraburkholderia phytofirmans growth promotion and salt stress tolerance of Arabidopsis thaliana. Front. Microbiol. 7:1838. doi: 10.3389/fmicb.2016.01838
Li, C., Chen, X., Yang, J., Li, J., Wang, R., Xu, H., et al. (2025). Keystone root bacteria in Ambrosia artemisiifolia promote invasive growth by increasing the colonization rate of Funneliformis mosseae. Microbiol. Res. 293:128081. doi: 10.1016/j.micres.2025.128081
Li, Y., Liu, Y., Zhang, H., Yang, Y., Wei, G., and Li, Z. (2021). The composition of root-associated bacteria and fungi of Astragalus mongholicus and their relationship with the bioactive ingredients. Front. Microbiol. 12:642730. doi: 10.3389/fmicb.2021.642730
Li, J., Wu, H., Pu, Q., Zhang, C., Chen, Y., Lin, Z., et al. (2023). Complete genome of Sphingomonas paucimobilis ZJSH1, an endophytic bacterium from Dendrobium officinale with stress resistance and growth promotion potential. Arch. Microbiol. 205:132. doi: 10.1007/s00203-023-03459-2
Li, F. F., Yuan, Y., Liu, Y., Wu, Q. Q., Jiao, R., Yang, Z., et al. (2015). Pachymic acid protects H9c2 cardiomyocytes from lipopolysaccharide-induced inflammation and apoptosis by inhibiting the extracellular signal-regulated kinase 1/2 and p38 pathways. Mol. Med. Rep. 12, 2807–2813. doi: 10.3892/mmr.2015.3712
Lin, Y., Zhang, H., Li, P., Jin, J., and Li, Z. (2022). The bacterial consortia promote plant growth and secondary metabolite accumulation in Astragalus mongholicus under drought stress. BMC Plant Biol. 22:475. doi: 10.1186/s12870-022-03859-4
Liu, D., Perez-Moreno, J., Zhang, P., Wang, R., Chater, C. C. C., and Yu, F. (2021). Distinct compartmentalization of microbial community and potential metabolic function in the fruiting body of Tricholoma matsutake. J. Fungi 7:586. doi: 10.3390/jof7080586
Liu, X.-l., Xie, J., Xie, Z.-n., Zhong, C., Liu, H., Zhang, S.-h., et al. (2024). Identification of squalene epoxidase in triterpenes biosynthesis in Poria cocos by molecular docking and CRISPR-Cas9 gene editing. Microb. Cell Factories 23:34. doi: 10.1186/s12934-024-02306-3
Liu, X., Zhong, C., Xie, J., Liu, H., Xie, Z., Zhang, S., et al. (2023). Geographical region traceability of Poria cocos and correlation between environmental factors and biomarkers based on a metabolomic approach. Food Chem. 417:135817. doi: 10.1016/j.foodchem.2023.135817
Maestre, F. T., Delgado-Baquerizo, M., Jeffries, T. C., Eldridge, D. J., Ochoa, V., Gozalo, B., et al. (2015). Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. U. S. A. 112, 15684–15689. doi: 10.1073/pnas.1516684112
Mendes, I. V., Garcia, M. B., Bitencourt, A. C. A., Santana, R. H., Lins, P. C., Silveira, R., et al. (2021). Bacterial diversity dynamics in microbial consortia selected for lignin utilization. PLoS One 16:e0255083. doi: 10.1371/journal.pone.0255083
Mitter, B., Pfaffenbichler, N., Flavell, R., Compant, S., Antonielli, L., Petric, A., et al. (2017). A new approach to modify plant microbiomes and traits by introducing beneficial Bacteria at flowering into progeny seeds. Front. Microbiol. 8:11. doi: 10.3389/fmicb.2017.00011
Morya, R., Kumar, M., and Shekhar, T. I. (2021). Bioconversion of syringyl lignin into malic acid by Burkholderia sp. ISTR5. Bioresour. Technol. 330:124981. doi: 10.1016/j.biortech.2021.124981
Noushahi, H. A., Khan, A. H., Khan, H. A., Kiedrzyński, M., Akbar, A., Shahzad, R., et al. (2024). Optimizing liquid fermentation for Wolfiporia cocos: gene expression and biosynthesis of pachymic acid and mycelial biomass. Lett. Appl. Microbiol. 77:54. doi: 10.1093/lambio/ovae054
Oh, S. Y., Fong, J. J., Park, M. S., and Lim, Y. W. (2016). Distinctive feature of microbial communities and bacterial functional profiles in Tricholoma matsutake dominant soil. PLoS One 11:e0168573. doi: 10.1371/journal.pone.0168573
Pétriacq, P., Williams, A., Cotton, A., McFarlane, A. E., Rolfe, S. A., and Ton, J. (2017). Metabolite profiling of non-sterile rhizosphere soil. Plant J. 92, 147–162. doi: 10.1111/tpj.13639
Pichersky, E., and Raguso, R. A. (2018). Why do plants produce so many terpenoid compounds? New Phytol. 220, 692–702. doi: 10.1111/nph.14178
Ren, F., Zhang, Y., Yu, H., and Zhang, Y. A. (2020). Ganoderma lucidum cultivation affect microbial community structure of soil, wood segments and tree roots. Sci. Rep. 10:3435. doi: 10.1038/s41598-020-60362-2
Smith, D. J., Park, J., Tiedje, J. M., and Mohn, W. W. (2007). A large gene cluster in Burkholderia xenovorans encoding abietane diterpenoid catabolism. J. Bacteriol. 189, 6195–6204. doi: 10.1128/jb.00179-07
Su, J., Wang, Y., Bai, M., Peng, T., Li, H., Xu, H.-J., et al. (2023). Soil conditions and the plant microbiome boost the accumulation of monoterpenes in the fruit of Citrus reticulata ‘chachi’. Microbiome 11:61. doi: 10.1186/s40168-023-01504-2
Suarez, C., Ratering, S., Weigel, V., Sacharow, J., Bienhaus, J., Ebert, J., et al. (2020). Isolation of bacteria at different points of Pleurotus ostreatus cultivation and their influence in mycelial growth. Microbiol. Res. 234:126393. doi: 10.1016/j.micres.2019.126393
Sugiyama, A., Yamazaki, Y., Yamashita, K., Takahashi, S., Nakayama, T., and Yazaki, K. (2016). Developmental and nutritional regulation of isoflavone secretion from soybean roots. Biosci. Biotechnol. Biochem. 80, 89–94. doi: 10.1080/09168451.2015.1062714
Tan, H., Liu, T., Yu, Y., Tang, J., Jiang, L., Martin Francis, M., et al. (2021). Morel production related to soil microbial diversity and evenness. Microbiol. Spectr. 9, e00229–e00221. doi: 10.1128/Spectrum.00229-21
Tang, S., Fan, T., Jin, L., Lei, P., Shao, C., Wu, S., et al. (2022). Soil microbial diversity and functional capacity associated with the production of edible mushroom Stropharia rugosoannulata in croplands. PeerJ 10:e14130. doi: 10.7717/peerj.14130
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Xu, S., Li, F., Gao, J., Zhou, X., Li, M., Li, L., et al. (2023). Low GHG emissions and less nitrogen use in mushroom-based protein production from chitin-containing waste and cottonseed hull with two phase SSF. Ind. Crop. Prod. 201:116970. doi: 10.1016/j.indcrop.2023.116970
Yang, K., Fu, R., Feng, H., Jiang, G., Finkel, O., Sun, T., et al. (2023). RIN enhances plant disease resistance via root exudate-mediated assembly of disease-suppressive rhizosphere microbiota. Mol. Plant 16, 1379–1395. doi: 10.1016/j.molp.2023.08.004
Yang, T., Tian, S., Wang, Y., Ji, J., and Zhao, J. (2022). Antitumor activity of pachymic acid in cervical cancer through inducing endoplasmic reticulum stress, mitochondrial dysfunction, and activating the AMPK pathway. Environ. Toxicol. 37, 2121–2132. doi: 10.1002/tox.23555
Yu, F., Luo, W., Xie, W., Li, Y., Liu, Y., Ye, X., et al. (2023). The effects of long-term hexabromocyclododecanes contamination on microbial communities in the microcosms. Chemosphere 325:138412. doi: 10.1016/j.chemosphere.2023.138412
Yun, W., Hall, I. R., and Evans, L. A. (1997). Ectomycorrhizal fungi with edible fruiting bodies 1. Tricholoma matsutake and related fungi. Econ. Bot. 51, 311–327. doi: 10.1007/BF02862101
Zhai, M. M., Li, J., Jiang, C. X., Shi, Y. P., Di, D. L., Crews, P., et al. (2016). The bioactive secondary metabolites from Talaromyces species. Nat. Prod. Bioprospect. 6, 1–24. doi: 10.1007/s13659-015-0081-3
Zhang, P., Li, X., Li, N., Xu, J., Li, Z.-L., Wang, Y., et al. (2005). Antibacterial constituents from fruit bodies of ascomyce Bulgaria inquinans. Arch. Pharm. Res. 28, 889–891. doi: 10.1007/BF02973872
Zhang, S., Sun, C., Liu, X., and Liang, Y. (2023). Enriching the endophytic bacterial microbiota of Ginkgo roots. Front. Microbiol. 14:1163488. doi: 10.3389/fmicb.2023.1163488
Zhang, G., Wang, H., Xie, W., Wang, Q., Wang, X., Wang, C., et al. (2019). Comparison of triterpene compounds of four botanical parts from Poria cocos (Schw.) wolf using simultaneous qualitative and quantitative method and metabolomics approach. Food Res. Int. 121, 666–677. doi: 10.1016/j.foodres.2018.12.036
Zhiyong, S., Yaxuan, G., Yuanyuan, W., Xiang, Y., Xu, G., Zhenhong, L., et al. (2024). Nitrogen-fixing bacteria promote growth and bioactive components accumulation of Astragalus mongholicus by regulating plant metabolism and rhizosphere microbiota. BMC Microbiol. 24:261. doi: 10.1186/s12866-024-03409-y
Zhong, Y., Xun, W., Wang, X., Tian, S., Zhang, Y., Li, D., et al. (2022). Root-secreted bitter triterpene modulates the rhizosphere microbiota to improve plant fitness. Nat Plants 8, 887–896. doi: 10.1038/s41477-022-01201-2
Zhu, Y., Zhou, X., Li, J., Feng, J., Huang, Z., Chen, B., et al. (2024). Can functional micro-organisms associated with pumpkin sizes be sought out from the soil?—a comparison of soil microbial community structures in rhizospheres between giant- and small-sized pumpkin varieties. Plants 13:2258. doi: 10.3390/plants13162258
Keywords: Poria cocos , high-throughput sequencing, pachymic acid, microbiome, bacteria and fungi
Citation: Zheng M, Zhong C, Pan G, Xie J, Zhang S and Jin J (2025) Insights into microbiome-triterpenoid correlation in Poria cocos via comparative analysis of sclerotial and soil microenvironments. Front. Microbiol. 16:1674216. doi: 10.3389/fmicb.2025.1674216
Edited by:
Muhammad Usman, Lanzhou University, ChinaReviewed by:
Tianrui Liu, China Academy of Chinese Medical Sciences, ChinaMingbo Jing, Longdong University, China
Copyright © 2025 Zheng, Zhong, Pan, Xie, Zhang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Jin, amluamlhbjIwMTZAMTYzLmNvbQ==