 Alua Gusmaulemova
Alua Gusmaulemova Botakoz Kurentay
Botakoz Kurentay Gulmira Kulmambetova
Gulmira Kulmambetova- Department of Genomics, National Center for Biotechnology, Astana, Kazakhstan
Fusobacterium nucleatum and Helicobacter pylori are two microbial species increasingly recognized for their roles in gastrointestinal (GI) carcinogenesis, particularly in colorectal cancer (CRC) and gastric cancer (GC), respectively. While H. pylori has been long classified as a Group 1 carcinogen due to its well-characterized pathogenic mechanisms, F. nucleatum has more recently emerged as a key microbial contributor to CRC, with growing evidence linking it to tumor progression, immune evasion, and poor clinical outcomes. Despite occupying anatomically distinct niches within the GI tract, both bacteria converge on similar oncogenic pathways, including the activation of NF-κB signaling, β-catenin pathway dysregulation, and epithelial barrier disruption. In parallel, dietary factors – particularly the consumption of red and processed meats – contribute additional oncogenic pressure via carcinogenic compounds such as heme iron, N-nitroso compounds, and polycyclic aromatic hydrocarbons. These dietary components not only damage host tissue but may also potentiate bacterial virulence and promote microbial persistence. This review provides a comparative analysis of the oncogenic strategies employed by F. nucleatum and H. pylori, with an emphasis on their interactions with diet-derived carcinogens and implications for therapeutic interventions targeting the microbiota–diet–host axis in GI cancers.
1 Introduction
Gastrointestinal (GI) cancers, notably colorectal cancer (CRC) and gastric cancer (GC), remain among the most prevalent and lethal malignancies globally, accounting for over 1.5 million deaths annually (Singh, 2024). While genetic susceptibility and environmental exposures such as smoking and obesity are established contributors to GI tumorigenesis, increasing attention has turned to the gut microbiome as a dynamic and potentially modifiable factor in cancer development (Shen et al., 2025).
Among the vast array of microbial species inhabiting the GI tract, Fusobacterium nucleatum and Helicobacter pylori have emerged as key pathogens with oncogenic potential. H. pylori has been extensively studied and is classified by the International Agency for Research on Cancer (IARC) as a Group 1 carcinogen, responsible for the majority of non-cardia gastric cancer cases worldwide (Moss, 2017; Wroblewski et al., 2010). In contrast, F. nucleatum, an oral anaerobe frequently detected in colorectal tumors, has gained attention only in recent years due to its association with advanced disease stages, immune evasion, and poor response to therapy (Galasso et al., 2025).
Despite residing in different anatomical compartments – H. pylori in the acidic environment of the stomach and F. nucleatum in the anaerobic colon – both bacteria employ strikingly similar pathogenic strategies. These include the induction of chronic inflammation, disruption of epithelial junctions, activation of proliferative signaling pathways, and modulation of host immune responses (Ito et al., 2020; Ou et al., 2022). At the molecular level, both species manipulate key signaling hubs such as NF-κB and β-catenin, which serve as convergence points for microbial virulence and host cell transformation (Moss, 2017; Neagu et al., 2025).
Beyond microbial mechanisms, dietary exposures play a pivotal role in shaping both cancer risk and microbial dynamics. High intake of red and processed meats, for example, is consistently associated with elevated CRC and GC risk in epidemiological studies (Di et al., 2023). This association is partially attributed to the formation of procarcinogenic compounds such as polycyclic aromatic hydrocarbons (PAHs), heterocyclic amines (HCAs), heme iron, and N-nitroso compounds (NOCs) during meat processing and digestion. Emerging evidence suggests that these dietary carcinogens can act synergistically with microbial factors–either by enhancing virulence gene expression, promoting reactive oxygen species (ROS) production, or creating a tissue microenvironment conducive to bacterial persistence (Malesza et al., 2022; Momal et al., 2025).
In preparing this review, a targeted literature search was conducted to explore the oncogenic mechanisms of Fusobacterium nucleatum and Helicobacter pylori in gastrointestinal carcinogenesis. Relevant peer-reviewed publications were identified using databases such as PubMed, Scopus, and Google Scholar, covering the period from 2000 to 2025. The search focused on studies containing key terms including Fusobacterium nucleatum, Helicobacter pylori, colorectal cancer, gastric cancer, CagA, FadA, VacA, NF-κB, immune evasion, red meat, processed meat, dietary carcinogens, reactive oxygen species, gut microbiota, and tumor microenvironment. Articles were included if they were written in English and provided molecular, clinical, or epidemiological insights into inflammation-driven tumorigenesis, immune modulation, or diet-microbiota interactions. Studies unrelated to gastrointestinal cancers, outdated reviews lacking mechanistic relevance, or non-English publications were excluded. A total of 61 high-quality studies were selected based on relevance, scientific rigor, and thematic alignment with the aims of this review.
In this review, we systematically compare the oncogenic mechanisms of F. nucleatum and H. pylori, with a focus on their interactions with diet-derived carcinogens. We examine how chronic inflammation, immune modulation, and barrier dysfunction collectively drive carcinogenesis, and how microbial–dietary synergy exacerbates these processes. By delineating both converging and diverging pathways of microbial carcinogenesis, we aim to identify potential targets for microbiota-based prevention and treatment strategies in GI cancers.
2 Oncogenic mechanisms of F. nucleatum in colorectal cancer
The role of Fusobacterium nucleatum in colorectal cancer (CRC) has become increasingly evident over the past decade. Initially identified as an oral commensal, F. nucleatum is now recognized as a recurrent component of the CRC microbiota, frequently enriched in tumor tissues compared to adjacent normal mucosa. Its presence correlates with advanced tumor stage, lymph node metastasis, microsatellite instability, and poor patient outcomes (Dadgar-Zankbar et al., 2024; Kulmambetova et al., 2023; McIlvanna et al., 2021). Mechanistically, F. nucleatum promotes tumorigenesis via three interconnected pathways: chronic inflammation, dysregulation of epithelial signaling, and suppression of antitumor immune responses. These effects are largely mediated by specific virulence factors, including lipopolysaccharide (LPS), FadA adhesin, and Fap2 protein (Rubinstein et al., 2013).
2.1 Chronic inflammation and TLR4/NF-kB signaling
A central mechanism by which F. nucleatum contributes to colorectal tumor development is through the induction of chronic inflammation. Its LPS component engages Toll-like receptor 4 (TLR4) on colonic epithelial cells and innate immune cells, triggering a MyD88-dependent signaling cascade that culminates in the activation of the NF-κB transcription factor (Yang et al., 2017). Once activated, NF-κB translocates to the nucleus and drives the expression of pro-inflammatory cytokines such as IL-6, IL-1β, TNF-α, and IL-17. These cytokines collectively foster a microenvironment conducive to tumor initiation and progression by promoting epithelial proliferation, angiogenesis, and immune cell recruitment (Wei et al., 2023).
This inflammatory signaling is further amplified by dietary factors. Heme iron, abundant in red meat, promotes the formation of reactive oxygen species (ROS), which not only cause direct DNA damage in colonocytes but also serve as secondary activators of NF-κB. The resulting oxidative stress reinforces the inflammatory loop initiated by F. nucleatum, establishing a chronic, tumor-promoting environment (Kang et al., 2019; Lee et al., 2023; Malesza et al., 2022; Figure 1).
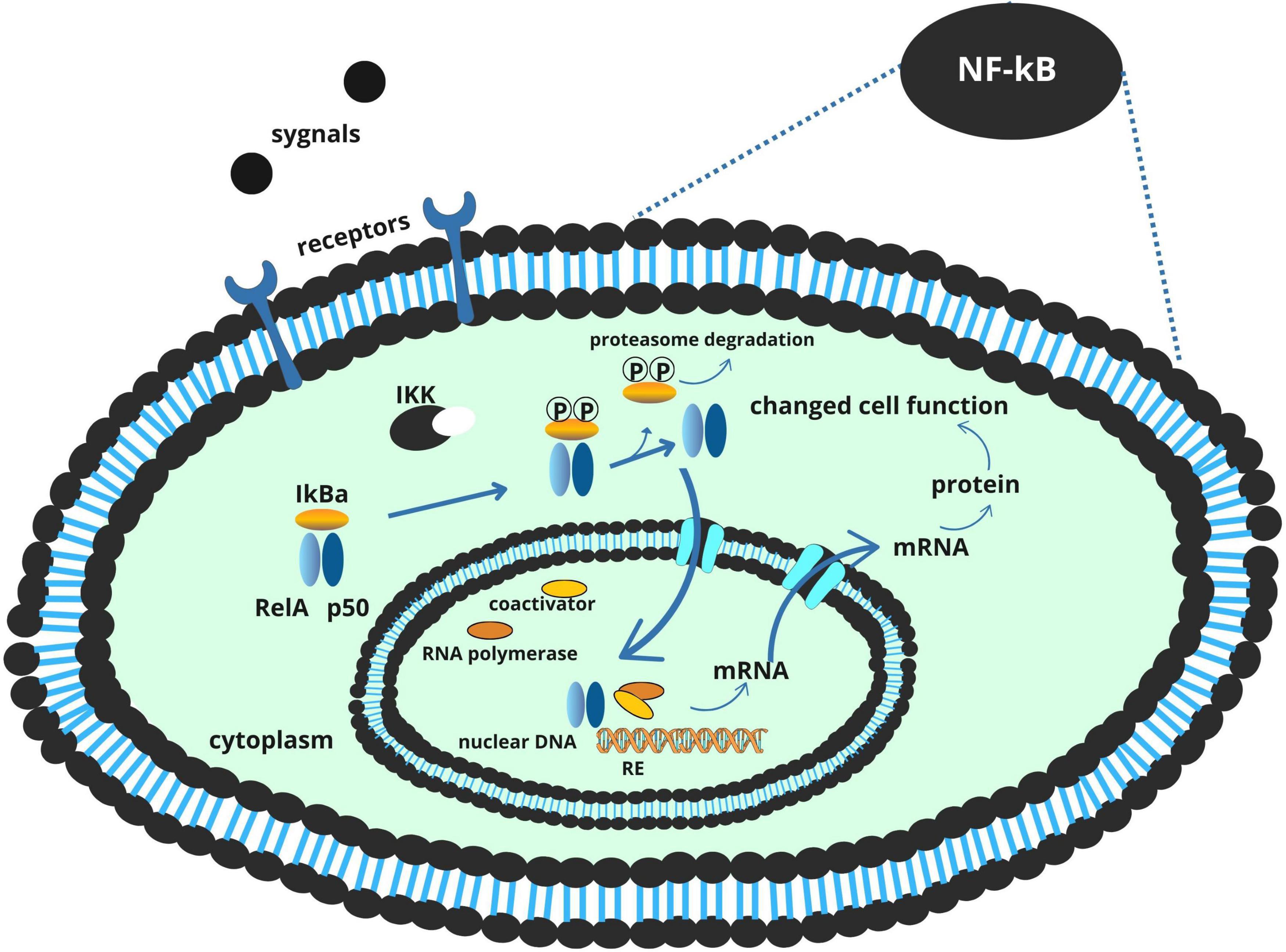
Figure 1. NF-κB signaling pathway and its role in inflammation and tumorigenesis.
2.2 Proliferation and β-catenin pathway disruption via FadA
In addition to promoting inflammation, F. nucleatum directly perturbs epithelial cell signaling through the action of FadA, an adhesin expressed on its surface. FadA binds to E-cadherin on the surface of colonic epithelial cells, leading to the disassembly of the E-cadherin–β-catenin complex (Figure 2). This interaction results in the nuclear translocation of β-catenin, a transcriptional co-activator implicated in Wnt signaling. Within the nucleus, β-catenin induces the expression of genes associated with cell proliferation and survival, including c-Myc and cyclin D1. This aberrant activation of proliferative pathways facilitates uncontrolled cell division and contributes to adenoma-to-carcinoma progression (Fardini et al., 2011; Rubinstein et al., 2013).
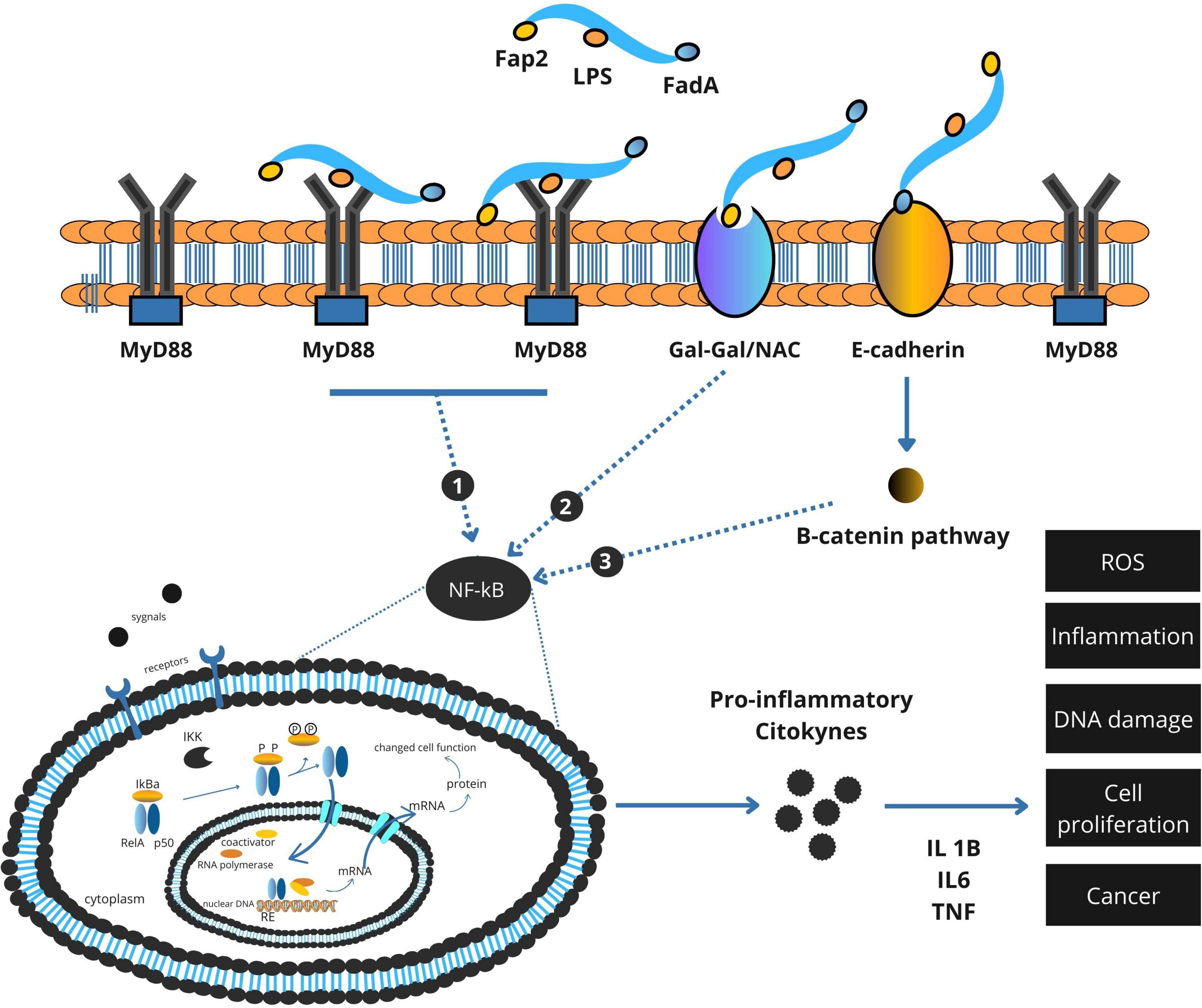
Figure 2. Integrated model of Fusobacterium nucleatum virulence factor–mediated activation of NF-κB and β-catenin signaling in colorectal cancer.
Importantly, the epithelial-disruptive effects of FadA can be potentiated by dietary carcinogens. N-nitroso compounds (NOCs), formed during the digestion of processed meats or endogenously in the colon, have been shown to cause DNA alkylation and mutation of tumor suppressor genes. In combination with FadA-induced β-catenin signaling, these mutagenic insults may accelerate cellular transformation (Galasso et al., 2025).
2.3 Immune evasion via Fap2-TIGIT interaction
Effective antitumor immunity relies on the activity of cytotoxic lymphocytes, including natural killer (NK) cells and CD8+ T cells. F. nucleatum circumvents immune surveillance by expressing Fap2, a surface protein that binds to the T cell immunoreceptor with Ig and ITIM domains (TIGIT), an inhibitory receptor on NK and T cells. This interaction mimics immune checkpoint signaling, suppressing cytotoxic activity and allowing tumor cells to evade immune-mediated destruction (Coppenhagen-Glazer et al., 2015; Gur et al., 2015).
Furthermore, dietary patterns that favor F. nucleatum persistence may exacerbate this immune evasion. High consumption of processed meats is associated with a depletion of beneficial gut commensals such as Bifidobacterium, and a corresponding enrichment of pro-inflammatory or opportunistic microbes, creating a microbiota environment that supports F. nucleatum colonization and immune resistance (Wang Y. et al., 2023).
2.4 Integrated model of F. nucleatum –mediated carcinogenesis
Taken together, the oncogenic potential of F. nucleatum in CRC is mediated by a coordinated network of molecular interactions. Through LPS-TLR4 signaling, it establishes a chronic inflammatory state. Via FadA, it disrupts epithelial signaling and promotes hyperproliferation. And through Fap2, it silences antitumor immune responses (Coppenhagen-Glazer et al., 2015; Rubinstein et al., 2013). These effects are further modulated by host dietary exposures, particularly red and processed meat intake, which not only provide pro-carcinogenic compounds but may also enhance bacterial virulence and survival (Ungvari et al., 2025).
A comprehensive summary of these oncogenic mechanisms is provided in Figure 3, which integrates F. nucleatum’s roles in inflammation, epithelial disruption, immune evasion, and cell death within the tumor microenvironment.
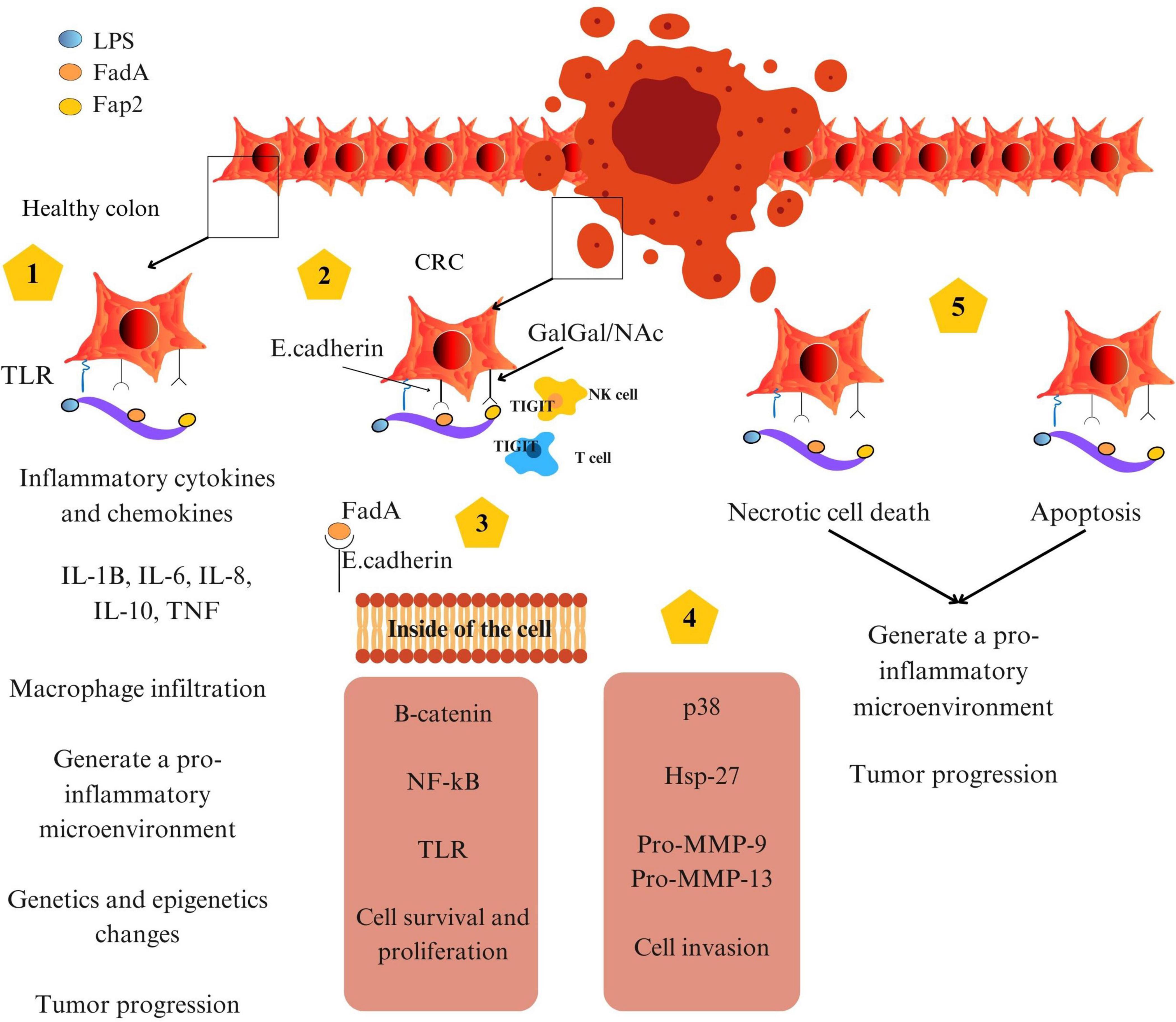
Figure 3. Schematic representation of CagA-induced signaling pathway in gastric epithelial cells.
3 Oncogenic mechanisms of Helicobacter pylori in gastric cancer
Helicobacter pylori is a Gram-negative, spiral-shaped bacterium that colonizes the gastric mucosa of nearly half the global population (Wroblewski et al., 2010). Its persistent infection is the primary etiological factor in non-cardia gastric cancer and has earned its classification as a Group 1 carcinogen by the IARC (Ono et al., 2025). The oncogenic capacity of H. pylori arises from its unique ability to colonize the harsh gastric environment, disrupt epithelial integrity, hijack proliferative signaling pathways, and evade immune surveillance. These effects are mediated by a suite of well-characterized virulence factors, including urease, cytotoxin-associated gene A (CagA), and vacuolating cytotoxin A (VacA) (Alipour, 2021; Cooke et al., 2013).
3.1 Colonization, mucosal damage and role of urease
The survival of H. pylori in the highly acidic environment of the stomach depends on its production of urease, an enzyme that hydrolyzes urea into ammonia and carbon dioxide. This local neutralization of gastric acid creates a hospitable niche within the mucus layer of the gastric epithelium, facilitating bacterial colonization. However, the process of colonization itself contributes to mucosal injury. Ammonia, a byproduct of urease activity, is cytotoxic at high concentrations and disrupts tight junction integrity, enhancing paracellular permeability. This damage promotes further bacterial adherence and translocation of virulence factors into epithelial cells (Ansari and Yamaoka, 2017).
Notably, high dietary salt intake exacerbates this mucosal vulnerability. Hyperosmolar conditions induced by excess salt have been shown to upregulate the expression of H. pylori virulence genes, including cagA and vacA, while simultaneously weakening mucosal defenses. Salt-induced epithelial damage thus acts as both an enabler and amplifier of bacterial pathogenesis, particularly in the context of diets rich in processed meats (Ansari and Yamaoka, 2017; Jones et al., 2010; Kim et al., 2019).
3.2 CagA-dependent pathogenesis: manipulation of proliferative pathways
The CagA oncoprotein is the most extensively studied virulence factor of H. pylori (Kim et al., 2015). Delivered directly into gastric epithelial cells via a type IV secretion system (T4SS), CagA exerts multifaceted effects on host cell behavior depending on its phosphorylation status (Wang H. et al., 2023).
Once translocated, CagA is phosphorylated by host Src and Abl tyrosine kinases. The phosphorylated form interacts with SHP-2, a protein tyrosine phosphatase that activates the Ras–ERK signaling cascade (Figure 4). This results in increased cell proliferation, morphological transformation, and disruption of cellular polarity (Alipour, 2021; Tsutsumi et al., 2003).
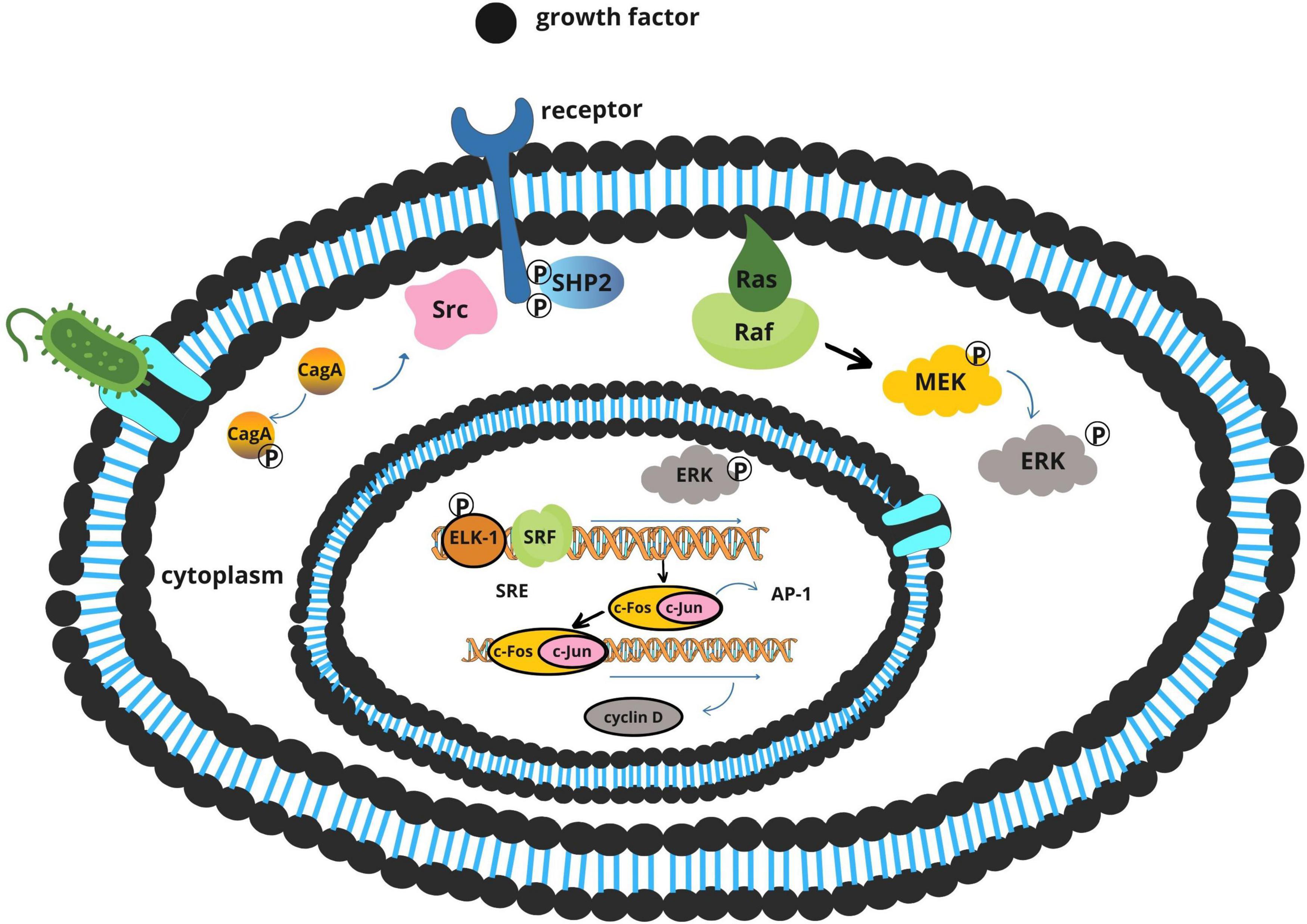
Figure 4. Proposed mechanisms of Fusobacterium nucleatum in colorectal carcinogenesis.
Simultaneously, the non-phosphorylated form of CagA perturbs adherens junctions by binding to E-cadherin, destabilizing the β-catenin complex (Figure 5). Released β-catenin translocates into the nucleus, where it activates Wnt target genes such as cyclin D1 and c-Myc, promoting uncontrolled cellular proliferation (Jones et al., 2010; Wang H. et al., 2023).
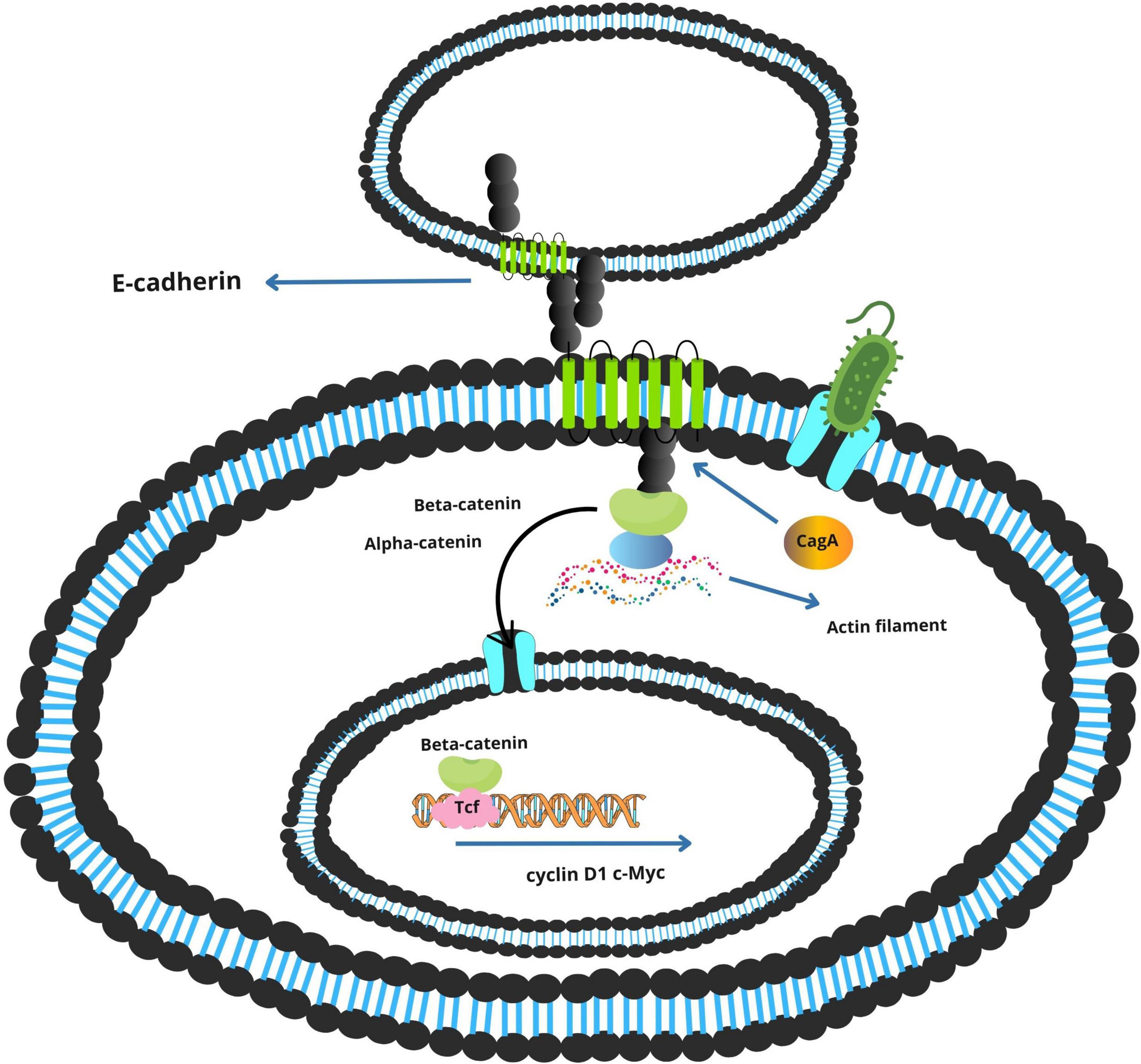
Figure 5. Activation of the E-cadherin/β-catenin signaling pathway by Helicobacter pylori CagA.
The tumorigenic potential of CagA is further enhanced by dietary carcinogens. Polycyclic aromatic hydrocarbons (PAHs), commonly formed during high-temperature cooking of meats, can independently activate the ERK pathway and synergize with CagA-mediated signaling, intensifying proliferative and anti-apoptotic responses (Balendra et al., 2023; Jones et al., 2010; Twarda-Clapa et al., 2022).
3.3 VacA-mediated immune modulation and persistence
VacA, a pore-forming exotoxin secreted by H. pylori, contributes to both immune evasion and tissue damage. Once internalized, VacA forms anion-selective channels in host membranes, leading to cellular vacuolization and mitochondrial dysfunction (Jones et al., 2010).
In immune cells, VacA inhibits T-cell activation by interfering with the calcineurin–NFAT signaling axis, reducing IL-2 production and impairing cytotoxic responses. It also induces apoptosis in T lymphocytes and other immune cell subsets, contributing to long-term immune suppression and bacterial persistence (Boncristiano et al., 2003; Gebert et al., 2003).
Interestingly, the genotoxic effects of dietary nitrosamines–common in processed meats–appear to parallel and enhance VacA activity. These compounds selectively damage normal gastric epithelial cells while allowing pre-malignant cells to evade apoptosis, fostering clonal expansion in a dysregulated environment (Szabò et al., 1999; Zhao et al., 2017).
3.4 Integrated model of H. pylori–induced gastric carcinogenesis
The pathogenesis of H. pylori-associated gastric cancer involves a stepwise progression from chronic gastritis to atrophic gastritis, intestinal metaplasia, dysplasia, and ultimately adenocarcinoma. This cascade is driven by a persistent cycle of mucosal damage, immune modulation, and aberrant proliferation (Cooke et al., 2013; Moss, 2017).
Urease enables colonization, CagA activates proliferative and survival pathways, and VacA disrupts immune surveillance and epithelial integrity. When these bacterial effects are compounded by dietary factors – particularly salt, PAHs, and nitrosamines – the risk of malignant transformation increases substantially (Diggs et al., 2011; Zhu et al., 2013).
Figure 5 presents a schematic overview of these mechanisms, highlighting the interplay between H. pylori and virulence factors in the gastric tumor microenvironment.
4 Comparative analysis: converging and diverging oncogenic strategies
Despite inhabiting distinct regions of the gastrointestinal (GI) tract and employing different mechanisms of host interaction, Fusobacterium nucleatum and Helicobacter pylori exhibit remarkable convergence in their capacity to drive tumorigenesis. Both bacteria exploit the host’s immune and signaling pathways to promote chronic inflammation, disrupt epithelial homeostasis, and enhance cellular proliferation. Understanding these converging and diverging mechanisms is critical for identifying shared therapeutic targets and pathogen-specific intervention strategies.
4.1 Converging mechanisms: common pathways to oncogenesis
Both F. nucleatum and H. pylori co-opt host signaling pathways that are central to inflammation and tumor promotion (Table 1). Specifically, they converge on two key molecular axes: the NF-κB pathway and the Wnt/β-catenin signaling cascade (Galasso et al., 2025; Ito et al., 2020).
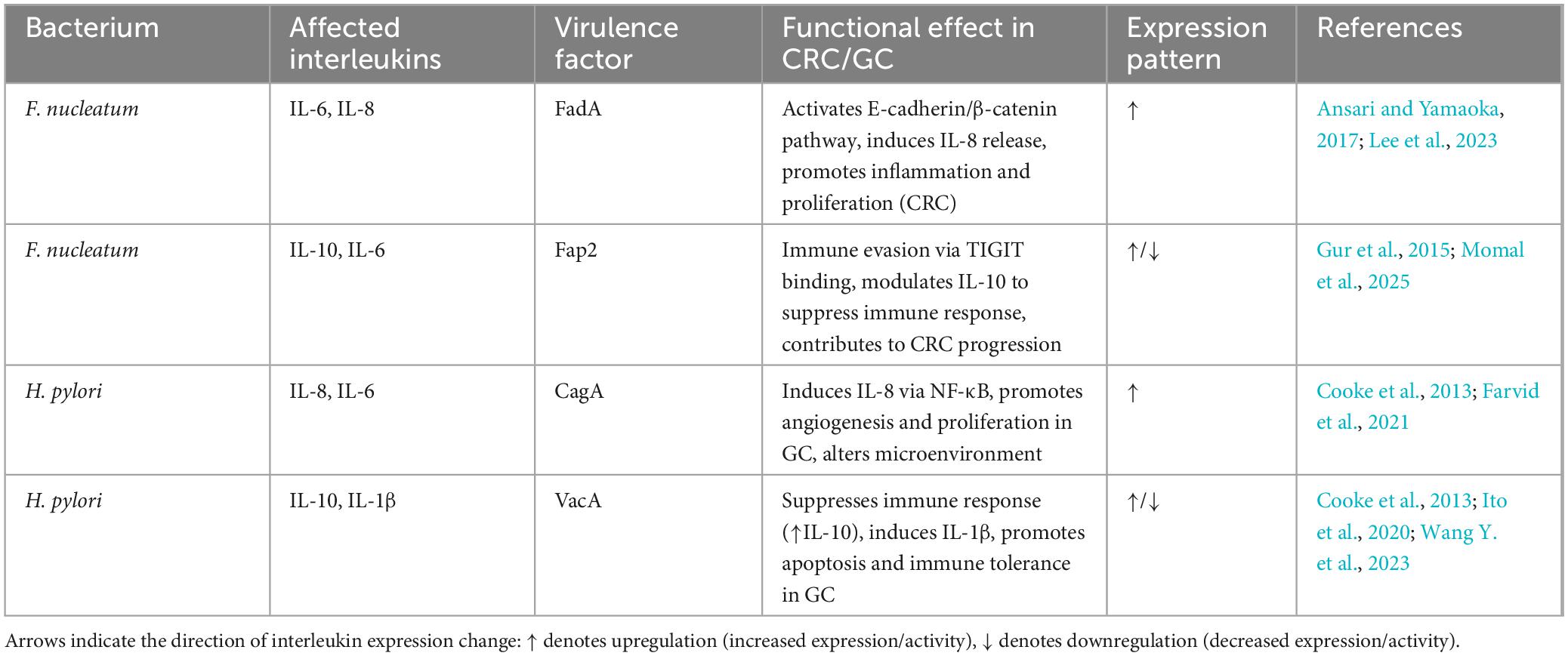
Table 1. Summary of bacterial virulence factors and interleukin interactions in CRC and GC.
In F. nucleatum, activation of NF-κB is initiated by lipopolysaccharide (LPS) recognition via TLR4 on epithelial and immune cells, resulting in the transcription of pro-inflammatory cytokines (e.g., IL-6, TNF-α) (Kang et al., 2019). H. pylori, although lacking classical LPS-TLR4 stimulation, activates NF-κB through intracellular pattern recognition receptors such as NOD1, which senses bacterial peptidoglycan following CagA-mediated cell manipulation. In both cases, chronic NF-κB signaling creates a pro-tumorigenic inflammatory microenvironment characterized by immune cell infiltration, angiogenesis, and oxidative stress (Wang H. et al., 2023).
Both pathogens disrupt E-cadherin-mediated cell-cell adhesion to activate β-catenin-dependent transcription. F. nucleatum achieves this through its FadA adhesin binding to E-cadherin, while H. pylori employs the non-phosphorylated form of CagA to interfere with the same complex. The downstream consequences are similar: nuclear accumulation of β-catenin, upregulation of c-Myc and cyclin D1, and enhanced epithelial proliferation (Fardini et al., 2011; Tsutsumi et al., 2003).
A critical point of convergence is the interaction with dietary carcinogens. Both bacteria operate within microenvironments shaped by red and processed meat consumption. Dietary heme iron, PAHs, and nitrosamines potentiate ROS production, which in turn amplifies NF-κB and Wnt pathway signaling. This synergy escalates DNA damage, promotes epithelial turnover, and facilitates clonal selection of transformed cells (Ferro et al., 2020; Lee et al., 2023; Oostindjer et al., 2014).
These overlapping mechanisms suggest the existence of shared “oncogenic chokepoints” in the GI mucosa – molecular nodes that are repeatedly exploited by different pathogens and dietary carcinogens to drive malignancy.
4.2 Diverging strategies: niche adaptation and toxin delivery
While F. nucleatum and H. pylori share several functional outcomes, their strategies of achieving these effects are fundamentally distinct, reflecting their adaptation to specific anatomical and microbial contexts.
Helicobacter pylori uses a type IV secretion system (T4SS) to inject the CagA protein directly into gastric epithelial cells. This allows for precise manipulation of host signaling in a contact-dependent and cell-specific manner (Tsutsumi et al., 2003). In contrast, F. nucleatum exerts its effects extracellularly, relying on surface adhesins like FadA and Fap2 to bind host receptors and modulate signaling cascades indirectly (Dadgar-Zankbar et al., 2024). This difference in delivery likely contributes to the more chronic and opportunistic nature of F. nucleatum-mediated pathogenesis.
Fusobacterium nucleatum subverts antitumor immunity primarily through immune checkpoint mimicry. Its Fap2 protein binds TIGIT, an inhibitory receptor on NK and T cells, suppressing cytotoxic activity and enabling immune evasion (Gur et al., 2015). H. pylori, on the other hand, employs the VacA toxin to directly impair T-cell activation by targeting intracellular calcium signaling and inducing apoptosis (Boncristiano et al., 2003). These divergent strategies result in distinct patterns of immune tolerance and inflammation within their respective tissue microenvironments.
5 Synergistic role of dietary and environmental factors
Diet plays a critical role in shaping the risk landscape for gastrointestinal (GI) cancers, both by directly influencing host physiology and by modulating the composition and behavior of the gut microbiota (Bouvard et al., 2015). Among dietary exposures, the consumption of red and processed meats is consistently linked to increased incidence of colorectal cancer (CRC) and, to a lesser extent, gastric cancer (GC). These associations are supported by robust epidemiological data and underpinned by well-characterized molecular mechanisms involving diet-derived carcinogens, host inflammation, and microbial synergy (Alisson-Silva et al., 2016; Benarba, 2018; Cross et al., 2007).
5.1 Epidemiological evidence linking meat consumption to GI cancers
5.1.1 Data on colorectal cancer
Numerous large-scale epidemiological studies and meta-analyses consistently associate the consumption of red and processed meats with an increased risk of CRC (Larsson and Wolk, 2006). Evidence from major cohort studies, like the Million Women Study involving over 540,000 participants, first highlighted this significant association (Papier et al., 2025). These findings have been powerfully reinforced by comprehensive meta-analyses (Ungvari et al., 2025). For instance, a review of 148 prospective studies confirmed elevated risks for colorectal (RR = 1.17), colon (RR = 1.21), and rectal cancers (Farvid et al., 2021), a conclusion mirrored in another analysis of nearly 4 million people that underscored the consistency of this link across global populations (Di et al., 2023; Kulmambetova et al., 2024; Ono et al., 2025).
Furthermore, the risk appears to be dose-dependent, with research establishing that consuming as little as 100 g/day of red meat or 50 g/day of processed meat leads to a measurable increase in CRC risk (Chan et al., 2011). Collectively, these extensive data provide the scientific basis for the IARC’s classification of processed meat as a definite (Group 1) and red meat as a probable (Group 2A) carcinogen (Moss, 2017).
5.1.2 Data on gastric cancer
Although epidemiological evidence is more robust for colorectal cancer, multiple studies also implicate red and processed meat consumption as risk factors for gastric cancer (GC). A meta-analysis by Zhu et al. (2013) reported that high intake of red or processed meat was associated with a 45% increased risk of gastric cancer. Dietary analyses from a broader cohort corroborate these findings, indicating that processed meat intake is positively associated with non-cardia GC, though associations with red meat alone may be weaker or inconsistent (Ferro et al., 2020).
In aggregate, Kim et al. (2019) conducted a meta-analysis including 5 cohorts and 19 case–control studies (9,726 cases), revealing that subjects with high red meat intake had a 41% higher risk of gastric cancer, while high processed meat intake conferred a 57% increased risk. Notably, dose–response analysis showed a 26% increased GC risk per 100 g/day increment in red meat consumption (Kim et al., 2010, 2019).
These associations appear particularly strong in case–control studies, whereas many cohort investigations yield null or weaker findings, especially for gastric cardia and non-cardia subtypes, highlighting heterogeneity across study designs and geographic regions.
5.2 Molecular mechanisms of dietary carcinogens
Mechanistically, the carcinogenic potential of red and processed meats is driven by several bioactive compounds formed during cooking and processing. High-temperature cooking methods generate heterocyclic amines (HCAs) and polycyclic aromatic hydrocarbons (PAHs) – both of which are metabolically activated into DNA-damaging agents. HCAs such as MeIQx and PhIP are hydroxylated by cytochrome P450 enzymes into reactive intermediates that form DNA adducts, particularly in colon epithelial cells, leading to mutagenesis (Cascella et al., 2018; Twarda-Clapa et al., 2022). PAHs like benzo[a]pyrene follow similar metabolic routes, producing genotoxic metabolites that interact with DNA and upregulate carcinogenic signaling pathways such as NF-κB and aryl hydrocarbon receptor signaling (Diggs et al., 2011; Huderson et al., 2013).
Heme iron, abundant in red meat, further exacerbates this risk. Its redox activity promotes the formation of reactive oxygen species (ROS), which trigger lipid peroxidation and produce mutagenic aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), both implicated in colonocyte damage and tumor initiation (Szabò et al., 1999). In addition, heme iron facilitates the endogenous formation of N-nitroso compounds (NOCs), which alkylate DNA bases and have been shown to induce KRAS mutations, a hallmark of CRC (Cascella et al., 2018; Malesza et al., 2022). Nitrite and nitrate preservatives in processed meats also contribute to NOC formation, particularly under acidic gastric conditions, enhancing genotoxicity (Crowe et al., 2019).
A more recently discussed factor is N-glycolylneuraminic acid (Neu5Gc), a non-human sialic acid prevalent in red meat. Humans can incorporate Neu5Gc into their cell membranes, where it is recognized as foreign by circulating anti-Neu5Gc antibodies. This immune recognition may trigger chronic inflammation (xenosialitis), a proposed mechanism linking Neu5Gc accumulation to tumor progression in the colon (Cascella et al., 2018). Notably, poultry and fish-lacking Neu5Gc do not show the same carcinogenic profile, further strengthening this dietary specificity (Alisson-Silva et al., 2016).
5.3 Mechanistic synergy: How dietary factors potentiate bacterial virulence?
One of the most compelling emerging concepts in GI oncology is the functional synergy between dietary carcinogens and microbial pathogens. Both F. nucleatum and H. pylori rely on host inflammation, oxidative stress, and disrupted epithelial signaling to promote carcinogenesis conditions that are exacerbated by diet (Galasso et al., 2025; Ito et al., 2020).
Red meat–derived heme iron enhances ROS production in the gut, reinforcing NF-κB activation initiated by bacterial LPS (in F. nucleatum) or NOD1 signaling (in H. pylori). The resulting oxidative stress contributes to DNA damage and creates a pro-inflammatory niche favorable to bacterial persistence (Macho-González et al., 2020).
N-nitroso compounds and PAHs potentiate Wnt/β-catenin signaling, mirroring the effects of FadA (from F. nucleatum) and CagA (from H. pylori) on epithelial cells. This convergence amplifies the expression of oncogenes and anti-apoptotic factors, accelerating the progression from dysplasia to carcinoma (Huderson et al., 2013; Lee et al., 2023).
Processed meats alter the gut microbiome by depleting protective species such as Bifidobacterium and expanding pro-inflammatory taxa. These changes may promote colonization or overgrowth of F. nucleatum, which thrives in low-diversity, inflammation-prone environments (Lee et al., 2023; Ma et al., 2017).
5.4 The impact of other lifestyle factors: obesity, smoking, and alcohol
In addition to bacterial virulence and dietary carcinogens, obesity, smoking, and alcohol consumption are significant risk factors for gastrointestinal cancers. A Mendelian randomization study demonstrated a causal association between increased waist-to-hip ratio and colorectal cancer (OR ≈ 1.38), as well as a suggestive link to smoking (Li et al., 2024). A meta-analysis found that alcohol consumption elevated gastric cancer risk by approximately 39 % (OR = 1.39) (Ma et al., 2017). Moreover, obesity in combination with smoking or heavy alcohol use further increases gastric cancer susceptibility by up to 19 % (Lim et al., 2022). At a population level, smoking accounts for 43 % of GI cancer mortality, alcohol for 21 %, and elevated BMI for approximately 20 % (Danpanichkul et al., 2025). These host and lifestyle factors likely interact with microbial and dietary mechanisms to exacerbate oncogenesis and should be considered in multifactorial prevention models.
6 Discussion
This review has examined the distinct yet converging roles of Fusobacterium nucleatum and Helicobacter pylori in gastrointestinal (GI) carcinogenesis, highlighting their shared molecular targets, niche-specific adaptations, and synergistic interactions with dietary carcinogens. A central theme emerging from this comparative analysis is the functional redundancy by which unrelated bacterial species exploit common vulnerabilities in the host to promote malignant transformation.
Despite significant differences in ecological localization and virulence delivery, both pathogens converge on two key signaling hubs: NF-κB and β-catenin/Wnt. These pathways not only mediate inflammatory and proliferative responses but also serve as integration points for dietary carcinogens such as heme iron, polycyclic aromatic hydrocarbons (PAHs), and N-nitroso compounds (NOCs) (Kang et al., 2019; Rubinstein et al., 2013). The resulting molecular crosstalk creates a self-reinforcing loop of microbial virulence, host tissue damage, and environmental exposure – an axis that significantly accelerates tumor initiation and progression.
Importantly, the synergy between bacterial factors and diet is not merely additive but mechanistically intertwined. For example, FadA- or CagA-mediated disruption of epithelial integrity increases cellular vulnerability to ROS and DNA alkylation induced by red meat–derived compounds. Conversely, dietary elements can enhance microbial virulence: salt exposure upregulates H. pylori’s expression of CagA and VacA, while iron-rich diets promote F. nucleatum persistence through microbiota remodeling and oxidative stress. These findings support a “triad model” of carcinogenesis, wherein pathogen, host, and diet form an interactive network rather than operating as independent risk factors (Alisson-Silva et al., 2016; Balendra et al., 2023; Cooke et al., 2013; Dadgar-Zankbar et al., 2024).
From a clinical perspective, these insights present both challenges and opportunities. The overlapping pathways exploited by F. nucleatum and H. pylori underscore the potential of shared therapeutic targets, such as inhibitors of NF-κB or modulators of β-catenin activity. Such interventions could, in theory, be effective across multiple cancer types and microbial contexts. Second, the divergence in delivery systems and immune evasion tactics suggests that pathogen-specific strategies – e.g., T4SS blockers for H. pylori, TIGIT inhibitors for F. nucleatum – may enhance therapeutic precision and reduce off-target effects (Gur et al., 2015; Larsson and Wolk, 2006). Third, the strong interaction between dietary carcinogens and microbial virulence highlights the importance of integrated prevention models. Modifying meat intake, especially processed and high-heat-cooked products, may not only reduce direct carcinogen exposure but also attenuate microbial-driven pathogenesis (Crowe et al., 2019; Lee et al., 2023). Finally, the role of the microbiome as a dynamic and modifiable risk factor suggests avenues for prebiotic, probiotic, or vaccine-based interventions. For example, reducing colonization by virulent F. nucleatum strains or altering microbial community structures that support H. pylori persistence could represent adjunctive strategies in high-risk populations.
Nevertheless, several key research gaps remain. Most studies evaluating microbial–diet interactions are either cross-sectional or based on animal models, limiting causal inference. Future work should prioritize longitudinal cohort studies incorporating microbiome sequencing, dietary profiling, and host genomics; functional analyses of virulence gene expression in response to dietary components; and evaluation of interventional strategies that simultaneously target microbial and dietary drivers of carcinogenesis.
Moreover, the broader context of microbial consortia and community-level interactions – beyond F. nucleatum and H. pylori – remains underexplored. GI cancers likely result from the cumulative effects of microbial networks, not single pathogens, and future models should account for these complex ecological dynamics.
7 Conclusion
Comparative insights into the oncogenic mechanisms of Fusobacterium nucleatum and Helicobacter pylori reveal a shared ability to integrate microbial virulence with dietary and host-derived factors, amplifying gastrointestinal cancer risk. Recognition of these parallels underscores the importance of microbiome-targeted diagnostics and therapeutics. By leveraging existing strategies developed for H. pylori, novel interventions against F. nucleatum-associated colorectal cancer may be developed, offering promising avenues for integrated cancer prevention approaches.
Author contributions
AG: Resources, Writing – review & editing, Writing – original draft, Visualization, Conceptualization. BK: Writing – review & editing, Resources, Formal analysis, Conceptualization. DB: Writing – review & editing, Resources, Formal analysis, Conceptualization. GK: Writing – review & editing, Supervision, Data curation, Conceptualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the grant number AP23488977, titled “Proteotranscriptomic Analysis of Colorectal Cancer Factors,” from the Ministry of Science and Higher Education of the Republic of Kazakhstan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alipour, M. (2021). Molecular mechanism of Helicobacter pylori-induced gastric cancer. J. Gastrointest. Cancer 52, 23–30. doi: 10.1007/s12029-020-00518-5
Alisson-Silva, F., Kawanishi, K., and Varki, A. (2016). Human risk of diseases associated with red meat intake: Analysis of current theories and proposed role for metabolic incorporation of a non-human sialic acid. Mol. Aspects Med. 51, 16–30. doi: 10.1016/j.mam.2016.07.002
Ansari, S., and Yamaoka, Y. (2017). Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 22:10.1111/hel.12386. doi: 10.1111/hel.12386
Balendra, V., Amoroso, C., Galassi, B., Esposto, J., Bareggi, C., Luu, J., et al. (2023). High-salt diet exacerbates H. pylori infection and increases gastric cancer risks. J. Pers. Med. 13:1325. doi: 10.3390/jpm13091325
Benarba, B. (2018). Red and processed meat and risk of colorectal cancer: An update. Excli J. 17, 792–797. doi: 10.17179/excli2018-1554
Boncristiano, M., Paccani, S., Barone, S., Ulivieri, C., Patrussi, L., Ilver, D., et al. (2003). The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198, 1887–1897. doi: 10.1084/jem.20030621
Bouvard, V., Loomis, D., Guyton, K., Grosse, Y., Ghissassi, F., Benbrahim-Tallaa, L., et al. (2015). Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 16, 1599–1600. doi: 10.1016/S1470-2045(15)00444-1
Cascella, M., Bimonte, S., Barbieri, A., Del Vecchio, V., Caliendo, D., Schiavone, V., et al. (2018). Dissecting the mechanisms and molecules underlying the potential carcinogenicity of red and processed meat in colorectal cancer (CRC): An overview on the current state of knowledge. Infect. Agent Cancer 13:3. doi: 10.1186/s13027-018-0174-9
Chan, D., Lau, R., Aune, D., Vieira, R., Greenwood, D., Kampman, E., et al. (2011). Red and processed meat and colorectal cancer incidence: Meta-analysis of prospective studies. PLoS One 6:e20456. doi: 10.1371/journal.pone.0020456
Cooke, C., Torres, J., and Solnick, J. (2013). Biomarkers of Helicobacter pylori-associated gastric cancer. Gut Microbes 4, 532–540. doi: 10.4161/gmic.25720
Coppenhagen-Glazer, S., Sol, A., Abed, J., Naor, R., Zhang, X., Han, Y., et al. (2015). Fap2 of Fusobacterium nucleatum is a galactose-inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect. Immun. 83, 1104–1113. doi: 10.1128/IAI.02838-14
Cross, A., Leitzmann, M., Gail, M., Hollenbeck, A., Schatzkin, A., and Sinha, R. A. (2007). prospective study of red and processed meat intake in relation to cancer risk. PLoS Med. 4:e325. doi: 10.1371/journal.pmed.0040325
Crowe, W., Elliott, C., and Green, B. D. A. (2019). Review of the in vivo evidence investigating the role of nitrite exposure from processed meat consumption in the development of colorectal cancer. Nutrients 11:2673. doi: 10.3390/nu11112673
Dadgar-Zankbar, L., Elahi, Z., Shariati, A., Khaledi, A., Razavi, S., and Khoshbayan, A. (2024). Exploring the role of Fusobacterium nucleatum in colorectal cancer: Implications for tumor proliferation and chemoresistance. Cell Commun. Signal. 22:547. doi: 10.1186/s12964-024-01909-y
Danpanichkul, P., Suparan, K., Pang, Y., Auttapracha, T., Tham, E., Vuthithammee, C., et al. (2025). Mortality of gastrointestinal cancers attributable to smoking, alcohol, and metabolic risk factors, and its association with socioeconomic development status 2000-2021. Am. J. Med. 138, 800–808.e2. doi: 10.1016/j.amjmed.2024.12.019
Di, Y., Ding, L., Gao, L., and Huang, H. (2023). Association of meat consumption with the risk of gastrointestinal cancers: A systematic review and meta-analysis. BMC Cancer 23:782. doi: 10.1186/s12885-023-11218-1
Diggs, D., Huderson, A., Harris, K., Myers, J., Banks, L., Rekhadevi, P., et al. (2011). Polycyclic aromatic hydrocarbons and digestive tract cancers: A perspective. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 29, 324–357. doi: 10.1080/10590501.2011.629974
Fardini, Y., Wang, X., Témoin, S., Nithianantham, S., Lee, D., Shoham, M., et al. (2011). Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol. Microbiol. 82, 1468–1480. doi: 10.1111/j.1365-2958.2011.07905.x
Farvid, M., Sidahmed, E., Spence, N., Mante Angua, K., Rosner, B., and Barnett, J. (2021). Consumption of red meat and processed meat and cancer incidence: A systematic review and meta-analysis of prospective studies. Eur. J. Epidemiol. 36, 937–951. doi: 10.1007/s10654-021-00741-9
Ferro, A., Rosato, V., Rota, M., Costa, A., Morais, S., Pelucchi, C., et al. (2020). Meat intake and risk of gastric cancer in the Stomach cancer Pooling (StoP) project. Int. J. Cancer 147, 45–55. doi: 10.1002/ijc.32707
Galasso, L., Termite, F., Mignini, I., Esposto, G., Borriello, R., Vitale, F., et al. (2025). Unraveling the role of Fusobacterium nucleatum in Colorectal cancer: Molecular mechanisms and pathogenic insights. Cancers 17:368. doi: 10.3390/cancers17030368
Gebert, B., Fischer, W., Weiss, E., Hoffmann, R., and Haas, R. (2003). Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301, 1099–1102. doi: 10.1126/science.1086871
Gur, C., Ibrahim, Y., Isaacson, B., Yamin, R., Abed, J., Gamliel, M., et al. (2015). Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355. doi: 10.1016/j.immuni.2015.01.010
Huderson, A., Myers, J., Niaz, M., Washington, M., and Ramesh, A. (2013). Chemoprevention of benzo(a)pyrene-induced colon polyps in ApcMin mice by resveratrol. J. Nutr. Biochem. 24, 713–724. doi: 10.1016/j.jnutbio.2012.04.005
Ito, N., Tsujimoto, H., Ueno, H., Xie, Q., and Shinomiya, N. (2020). Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer. J. Clin. Med. 9:3699. doi: 10.3390/jcm9113699
Jones, K., Whitmire, J., and Merrell, D. S. (2010). A tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front. Microbiol. 1:115. doi: 10.3389/fmicb.2010.00115
Kang, W., Jia, Z., Tang, D., Zhang, Z., Gao, H., He, K., et al. (2019). Fusobacterium nucleatum facilitates apoptosis, ROS generation, and inflammatory cytokine production by activating AKT/MAPK and NF- κ B signaling pathways in human gingival fibroblasts. Oxid. Med. Cell. Longev. 2019:1681972. doi: 10.1155/2019/1681972
Kim, H., Lim, S., Lee, J., Park, S., Shin, A., Choi, B., et al. (2010). Fresh and pickled vegetable consumption and gastric cancer in Japanese and Korean populations: A meta-analysis of observational studies. Cancer Sci. 101, 508–516. doi: 10.1111/j.1349-7006.2009.01374.x
Kim, S., Choi, D., and Chung, J. (2015). Antibiotic treatment for Helicobacter pylori: Is the end coming? World J. Gastrointest. Pharmacol. Ther. 6, 183–198. doi: 10.4292/wjgpt.v6.i4.183
Kim, S., Kim, K., Lee, S., Kwon, S., Lee, J., Keum, N., et al. (2019). Effect of red, processed, and white meat consumption on the risk of gastric cancer: An overall and dose?response meta-analysis. Nutrients 11:826. doi: 10.3390/nu11040826
Kulmambetova, G., Kurentay, B., Gusmaulemova, A., Utupov, T., Auganova, D., Tarlykov, P., et al. (2024). Association of Fusobacterium nucleatum infection with colorectal cancer in Kazakhstani patients. Front. Oncol. 14:1473575. doi: 10.3389/fonc.2024.1473575
Kulmambetova, G., Utupov, T., Kurentay, B., Auganova, D., Tarlykov, P., Shevtsov, A., et al. (2023). Draft genome sequence of a Fusobacterium nucleatum strain isolated from a patient in Kazakhstan with colorectal cancer. Microbiol. Resour. Announc. 12:e0036723. doi: 10.1128/MRA.00367-23
Larsson, S., and Wolk, A. (2006). Meat consumption and risk of colorectal cancer: A meta-analysis of prospective studies. Int. J. Cancer 119, 2657–2664. doi: 10.1002/ijc.22170
Lee, C., Lee, J., Eor, J., Kwak, M., Huh, C., and Kim, Y. (2023). Effect of consumption of animal products on the gut microbiome composition and gut health. Food Sci. Anim. Resour. 43, 723–750. doi: 10.5851/kosfa.2023.e44
Li, X., Chang, Z., Wang, J., Ding, K., Pan, S., Hu, H., et al. (2024). Unhealthy lifestyle factors and the risk of colorectal cancer: A Mendelian randomization study. Sci. Rep. 14:13825. doi: 10.1038/s41598-024-64813-y
Lim, J., Shin, C., Han, K., Lee, S., Jin, E., Choi, Y., et al. (2022). Association between the persistence of obesity and the risk of gastric cancer: A nationwide population-based study. Cancer Res. Treat. 54, 199–207. doi: 10.4143/crt.2021.130
Ma, K., Baloch, Z., He, T., and Xia, X. (2017). Alcohol consumption and gastric cancer risk: A meta-analysis. Med. Sci. Monit. 23, 238–246. doi: 10.12659/msm.899423
Macho-González, A., Garcimartín, A., López-Oliva, M., Bastida, S., Benedí, J., Ros, G., et al. (2020). Can meat and meat-products induce oxidative stress? Antioxidants 9:638. doi: 10.3390/antiox9070638
Malesza, I., Bartkowiak-Wieczorek, J., Winkler-Galicki, J., Nowicka, A., Dzięciołowska, D., Błaszczyk, M., et al. (2022). The dark side of iron: The relationship between iron, inflammation and gut microbiota in selected diseases associated with iron deficiency anaemia-a narrative review. Nutrients 14:3478. doi: 10.3390/nu14173478
McIlvanna, E., Linden, G., Craig, S., Lundy, F., and James, J. (2021). Fusobacterium nucleatum and oral cancer: A critical review. BMC Cancer 21:1212. doi: 10.1186/s12885-021-08903-4
Momal, U., Naeem, H., Aslam, F., Shahbaz, M., Imran, M., Hussain, M., et al. (2025). Recent perspectives on meat consumption and cancer proliferation. J. Food Processing Preserv. 2025, 1–27. doi: 10.1155/jfpp/6567543
Moss, S. (2017). The clinical evidence linking Helicobacter pylori to gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 3, 183–191. doi: 10.1016/j.jcmgh.2016.12.001
Neagu, A., Bostan, M., Ionescu, V., Gheorghe, G., Hotnog, C., Roman, V., et al. (2025). The impact of the microbiota on the immune response modulation in colorectal cancer. Biomolecules 15:1005. doi: 10.3390/biom15071005
Ono, A., Tanaka, S., Sawada, N., Goto, A., Tsugane, S., Muraki, I., et al. (2025). Helicobacter pylori eradication and gastric cancer prevention in a pooled analysis of large-scale cohort studies in Japan. Sci. Rep. 15:21307. doi: 10.1038/s41598-025-00713-z
Oostindjer, M., Alexander, J., Amdam, G., Andersen, G., Bryan, N., Chen, D., et al. (2014). The role of red and processed meat in colorectal cancer development: A perspective. Meat. Sci. 97, 583–596. doi: 10.1016/j.meatsci.2014.02.011
Ou, S., Wang, H., Tao, Y., Luo, K., Ye, J., Ran, S., et al. (2022). Fusobacterium nucleatum and colorectal cancer: From phenomenon to mechanism. Front. Cell. Infect. Microbiol. 12:1020583. doi: 10.3389/fcimb.2022.1020583
Papier, K., Bradbury, K., Balkwill, A., Barnes, I., Smith-Byrne, K., Gunter, M., et al. (2025). Diet-wide analyses for risk of colorectal cancer: Prospective study of 12,251 incident cases among 542,778 women in the UK. Nat. Commun. 16:375. doi: 10.1038/s41467-024-55219-5
Rubinstein, M., Wang, X., Liu, W., Hao, Y., Cai, G., and Han, Y. (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206. doi: 10.1016/j.chom.2013.07.012
Shen, Y., Fan, N., Ma, S., Cheng, X., Yang, X., and Wang, G. (2025). Gut microbiota dysbiosis: Pathogenesis, diseases, prevention, and therapy. MedComm 6:e70168. doi: 10.1002/mco2.70168
Singh, A. (2024). Global burden of five major types of gastrointestinal cancer. Prz. Gastroenterol. 19, 236–254. doi: 10.5114/pg.2024.141834
Szabò, I., Brutsche, S., Tombola, F., Moschioni, M., Satin, B., Telford, J., et al. (1999). Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18, 5517–5527. doi: 10.1093/emboj/18.20.5517
Tsutsumi, R., Higashi, H., Higuchi, M., Okada, M., and Hatakeyama, M. (2003). Attenuation of Helicobacter pylori CagA x SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J. Biol. Chem. 278, 3664–3670. doi: 10.1074/jbc.M208155200
Twarda-Clapa, A., Olczak, A., Białkowska, A., and Koziołkiewicz, M. (2022). Advanced glycation end-products (AGEs): Formation, chemistry, classification, receptors, and diseases related to AGEs. Cells 11:1312. doi: 10.3390/cells11081312
Ungvari, Z., Fekete, M., Varga, P., Lehoczki, A., Munkácsy, G., Fekete, J., et al. (2025). Association between red and processed meat consumption and colorectal cancer risk: A comprehensive meta-analysis of prospective studies. Geroscience 47, 5123–5140. doi: 10.1007/s11357-025-01646-1
Wang, H., Zhao, M., Shi, F., Zheng, S., Xiong, L., and Zheng, L. (2023). A review of signal pathway induced by virulent protein CagA of Helicobacter pylori. Front. Cell. Infect. Microbiol. 13:1062803. doi: 10.3389/fcimb.2023.1062803
Wang, Y., Uffelman, C., Bergia, R., Clark, C., Reed, J., Cross, T., et al. (2023). Meat consumption and gut microbiota: A scoping review of literature and systematic review of randomized controlled trials in adults. Adv Nutr. 14, 215–237. doi: 10.1016/j.advnut.2022.10.005
Wei, J., Zhang, Y., Li, H., Wang, F., and Yao, S. (2023). Toll-like receptor 4: A potential therapeutic target for multiple human diseases. Biomed. Pharmacother. 166:115338. doi: 10.1016/j.biopha.2023.115338
Wroblewski, L., Peek, R., and Wilson, K. (2010). Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 23, 713–739. doi: 10.1128/CMR.00011-10
Yang, Y., Weng, W., Peng, J., Hong, L., Yang, L., Toiyama, Y., et al. (2017). Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear Factor-κB, and up-regulating expression of MicroRNA-21. Gastroenterology 152, 851–866.e24. doi: 10.1053/j.gastro.2016.11.018
Zhao, Z., Yin, Z., and Zhao, Q. (2017). Red and processed meat consumption and gastric cancer risk: A systematic review and meta-analysis. Oncotarget 8, 30563–30575. doi: 10.18632/oncotarget.15699
Keywords: Fusobacterium nucleatum, Helicobacter pylori, colorectal cancer, gastric cancer, red meat, processed meat, virulence factors
Citation: Gusmaulemova A, Kurentay B, Bayanbek D and Kulmambetova G (2025) Comparative insights into Fusobacterium nucleatum and Helicobacter pylori in human cancers. Front. Microbiol. 16:1677795. doi: 10.3389/fmicb.2025.1677795
Received: 01 August 2025; Accepted: 30 September 2025;
Published: 21 October 2025.
Edited by:
Michal Letek, University of León, SpainReviewed by:
Munazzah Tasleem, University of Bisha, Saudi ArabiaMuhammad Nur Adam Hatta, University of Malaya, Malaysia
Copyright © 2025 Gusmaulemova, Kurentay, Bayanbek and Kulmambetova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gulmira Kulmambetova, a3VsbWFtYmV0b3ZhQGJpb2NlbnRlci5reg==