 Austin C. Surphlis1,2
Austin C. Surphlis1,2 An-Chi Cheng3Morgan C. Metrailer2,4Andrew P. Bluhm2,4Treenate Jiranantasak2,4
An-Chi Cheng3Morgan C. Metrailer2,4Andrew P. Bluhm2,4Treenate Jiranantasak2,4 Frank Tuozzo II2,4Enrique Doster5
Frank Tuozzo II2,4Enrique Doster5 Jason K. Blackburn2,4
Jason K. Blackburn2,4 Kwangcheol C. Jeong2,6
Kwangcheol C. Jeong2,6 Christina Boucher7Juan M. Campos-Krauer3,8
Christina Boucher7Juan M. Campos-Krauer3,8 Samantha M. Wisely2,8
Samantha M. Wisely2,8 Kuttichantran Subramaniam1,2*
Kuttichantran Subramaniam1,2*- 1Department of Infectious Diseases and Immunology, College of Veterinary Medicine, University of Florida, Gainesville, FL, United States
- 2Emerging Pathogens Institute, University of Florida, Gainesville, FL, United States
- 3Department of Large Animal Clinical Sciences, College of Veterinary Medicine, University of Florida, Gainesville, FL, United States
- 4Spatial Epidemiology and Ecology Research Laboratory (SEER Lab), Department of Geography, University of Florida, Gainesville, FL, United States
- 5VERO Program, Texas A&M University, Canyon, TX, United States
- 6Department of Animal Sciences, Institute of Food and Agricultural Sciences University of Florida, Gainesville, FL, United States
- 7Department of Computer and Information Science and Engineering, University of Florida, Gainesville, FL, United States
- 8Department of Wildlife Ecology and Conservation, Institute of Food and Agricultural Sciences University of Florida, Gainesville, FL, United States
Background: Antimicrobial resistance (AMR) is a critical public health issue; with many experts suggesting we are already in a post-antibiotic era. The widespread use of antibiotics in agriculture, human, and veterinary medicine influences the spread of antibiotic resistance genes (ARGs) among humans, animals, and the environment. In Florida, white-tailed deer (WTD; Odocoileus virginianus) farming plays a vital role in the economy and environment, but the use of antimicrobials in farmed WTD, along with their proximity to urban and agricultural areas, increases the pressure for AMR development. Understanding the resistance patterns in these deer populations is crucial for their health, as well as for wildlife and ecosystems. This research aimed to investigate the resistome of Florida-farmed WTD. Escherichia coli, a commonly used indicator bacterium, was chosen to study AMR due to its pathogenicity and ease of culture.
Methods: Samples from various tissues were collected during necropsy. Escherichia coli was isolated and cultured, and whole-genome sequencing was performed using a high-throughput NovaSeq platform. The AMR++ v 3.0 pipeline and ResistoXplorer tool were employed for data normalization and analysis. Antimicrobial susceptibility testing of the E. coli isolates was conducted using the Kirby-Bauer disk diffusion method on Mueller-Hinton agar, based on the guidelines and recommendations in the CLSI VET01S.
Results: A total of 362 unique ARGs were identified, conferring resistance to 12 antimicrobial classes via 19 mechanisms. The most abundant classes were ß-lactams, multi-drug resistance, and bacitracin. Antimicrobial susceptibility testing showed that 30% of E. coli isolates were resistant to at least one drug under aerobic conditions, while 68% were resistant under anaerobic conditions. Moreover, 15% of isolates displayed multi-drug resistance in both conditions. The study also compared genotypic and phenotypic AMR profiles using kappa, revealing good to very good agreement for several drugs.
Conclusion: This is the first study to characterize the resistome of farmed WTD in Florida, providing valuable data for better management of antimicrobial use in these populations.
1 Introduction
The resistome, first conceptualized by D'Costa et al. (2006) in response to the discovery of antimicrobial resistance (AMR) traits in soil bacteria, encompasses “the collection of all antibiotic resistance genes and their precursors in both pathogenic and non-pathogenic bacteria” (Wright, 2007). As neither antimicrobial resistance genes (ARGs) nor their bacterial hosts are confined by physical or operational boundaries, this broad definition underscores AMR’s significance as a global public health concern (Figure 1). Resistome characterization is critical to understanding ARG distribution, variation, and abundance across hosts and habitats. Early studies relied on culture-dependent techniques (Benveniste and Davies, 1973), but next-generation sequencing (NGS) and whole-genome sequencing (WGS) have since revolutionized resistome analysis (Ellington et al., 2017; Witney et al., 2016; Crofts et al., 2017). While metagenomics offers a powerful analytical tool, challenges such as sensitivity limitations, sequencing depth, and host DNA contamination persist (Abayasekara et al., 2017; Zhou et al., 2015). In contrast, WGS-based approaches for AMR analysis also present several limitations, including the need for pure cultures, an incomplete representation of the resistome, difficulties in detecting mobile genetic elements (e.g., plasmids and transposons) due to short-read sequencing, gaps in predicting phenotypic resistance, and the labor- and time-intensive nature of the process (Sadovska et al., 2024; Zwe et al., 2020; Weinmaier et al., 2023; Tagami et al., 2024; McDermott et al., 2016; Fauzia et al., 2023).
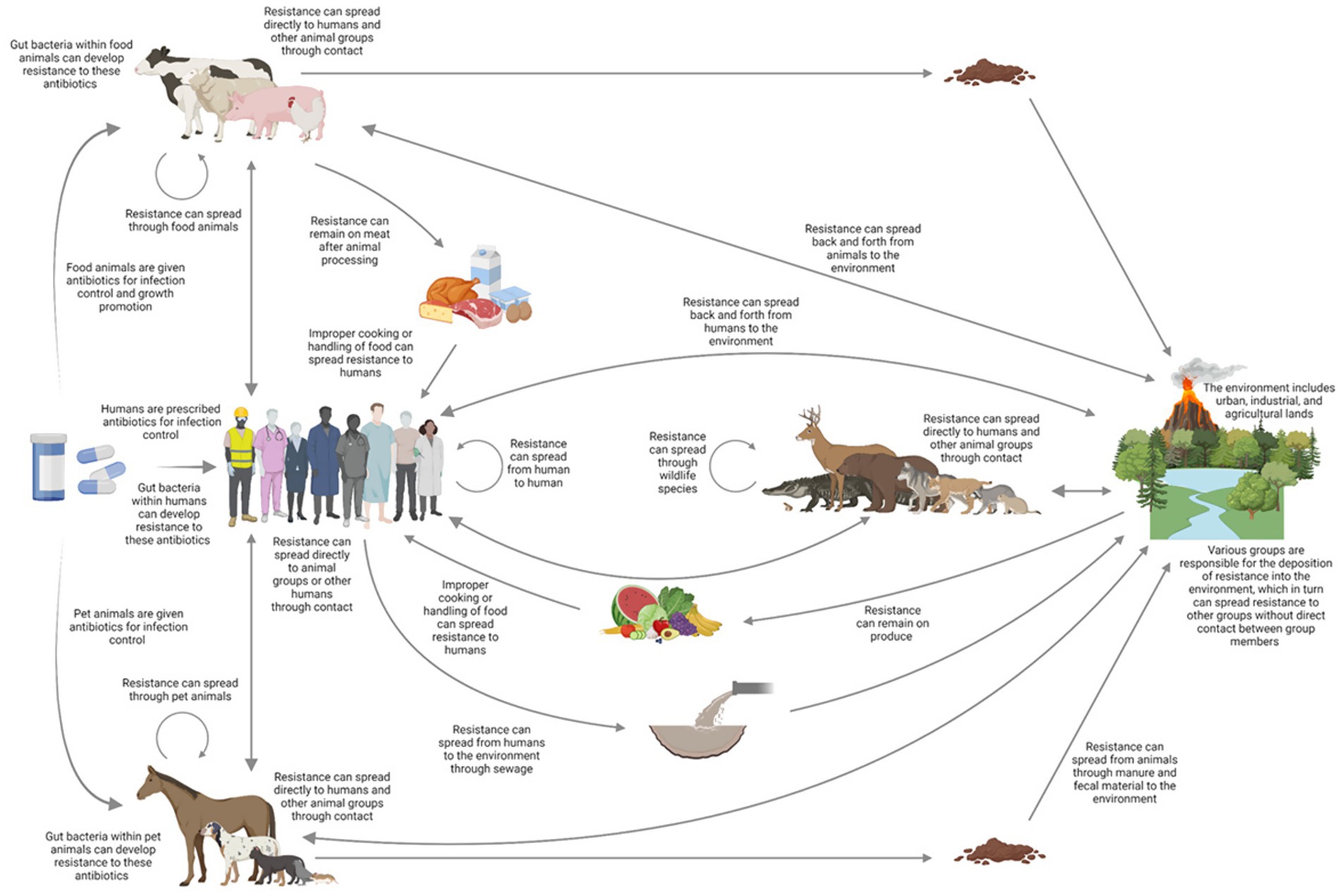
Figure 1. Routes of AMR may be transmitted through various direct and indirect routes to humans, livestock, companion animals, the environment, and wildlife shown by arrows. The overall effectiveness of these channels will vary significantly depending on the type of microbe and resistance mechanism, as well as the environment and location. Adapted from a diagram of the intricate process presented by Woolhouse and Ward (2013).
The prevalence of AMR bacteria is increasingly common in companion and food-producing animals, which is likely linked to the selective pressure from prolonged antimicrobial use in livestock production, veterinary care, and direct human contact (Agersø and Aarestrup, 2013; Durso and Cook, 2014). Peridomestic wildlife comprises species that have adapted to human-modified environments and persist in close proximity to humans, including those deliberately introduced for consumption (Haider et al., 2020). These animals often serve as reservoirs of antimicrobial-resistant microorganisms, although resistance prevalence varies across taxa and geographic regions (Arnold et al., 2016). This variation is influenced by several factors, including environmental contamination primarily from anthropogenic sources, which drives acquired resistance in wildlife (Vittecoq et al., 2016). Although clinically used antimicrobials are not typically found in wild environments, AMR can still occur. Resistance factors have been identified in remote and historically isolated settings, including ancient permafrost layers dating back 30,000 years (D'Costa et al., 2011), secluded cave ecosystems (Bhullar et al., 2012), and even frozen human remains from the Copper Age (Lugli et al., 2017). Nevertheless, numerous environmental exposure pathways—such as manure, wastewater, and pollution from areas with intense anthropogenic activity—can contribute to the selection and dissemination of resistance (Martinez, 2009).
For example, cervids often forage on croplands fertilized with compost, a known hotspot for ARGs, AMR bacteria, and antimicrobials (Rogers et al., 2018; Lima et al., 2020). Additionally, freshwater sources, critical to wildlife hydration, are similarly vulnerable to antimicrobial contamination from sewage and agricultural runoff (Zhu et al., 2017; Zhou et al., 2018; Cacace et al., 2019; Nnadozie and Odume, 2019). Evidence also supports the significant role migratory birds play in the spread of AMR (Pinto et al., 2010; Hasan et al., 2014). Furthermore, the host diet can influence gut microbiota dynamics, affecting the prevalence of AMR microorganisms among commensal bacteria (Williams et al., 2011). A predominantly herbivorous diet may account for the generally low levels of AMR observed in many wildlife species that graze on vegetation, in contrast to the higher levels seen in species with omnivorous or carnivorous diets (Vittecoq et al., 2016).
White-tailed deer (WTD; Odocoileus virginianus), widely distributed across the Americas, inhabit diverse environments, from natural ecosystems to urban areas (Ballash et al., 2022). As a keystone species, WTD influence food web dynamics, notably as prey for many species including the endangered Florida panther (Boughton et al., 2020). Cervid hunting and farming in the U. S. contribute significantly to both the economy and ecosystem management (Anderson et al., 2017; Brooks et al., 2015; Lantz, 1908; Hewitt, 2015; Conover, 2011). Deer hunting generates over $13 billion annually, supporting 209,000 jobs in the southeastern U. S. alone (National Deer Association, 2022). Ecologically, hunting helps regulate deer populations, mitigating overbrowsing that disrupts understory vegetation (Marquis and Brenneman, 1981; Warren, 1991). WTD, the most commonly farmed cervid, also provide environmental benefits by thriving on brushlands unsuitable for cattle or horses (Lantz, 1908).
Cervid farming, which is currently expanding in Florida, is situated in a state where AMR bacteria are prevalent in hospitals, livestock, companion animals, and wildlife (Sy-Trias, 2023; Gutierrez et al., 2020; Giguère et al., 2010; White and Forrester, 1979; Greig et al., 2007). However, antimicrobial use in farmed WTD, coupled with proximity to urban and agricultural areas, may drive AMR development. While the implications for Florida’s cervid farming remain unclear, AMR-associated morbidity and mortality could pose risks for WTD farmers. Assessing resistance patterns is crucial for managing cervid health and preserving ecosystem integrity.
However, the AMR in Florida farmed WTD is unknown. To date, AMR research in cervids has primarily focused on antimicrobial susceptibility of indicator bacteria such as Escherichia coli and Enterococcus spp., as well as pathogens like Campylobacter spp. and Salmonella spp. (Dias et al., 2015; Plaza-Rodríguez et al., 2021; Turchi et al., 2019). High-throughput metagenomic sequencing enables comprehensive ARG profiling, yet studies on ARGs in cervids remain scarce (Rogers et al., 2018). Hence, this study aims to characterize the genotypic and phenotypic profiles of ARGs in E. coli isolates from these cervid populations using high-throughput sequencing, ARG annotation through publicly available databases, and antimicrobial susceptibility testing.
E. coli is widely recognized as a valuable indicator of AMR in wildlife and environmental monitoring (Anjum et al., 2021). Studies have demonstrated a significant correlation between the presence of AMR in E. coli and the detection of resistant pathogenic strains within the same sample (Nyirabahizi et al., 2020). Due to its role as an indicator organism, strain-specific pathogenicity, and ease of culture (Anjum et al., 2021), E. coli was selected in this study for AMR characterization from farmed WTD in Florida. In addition, to address the limitations of metagenomic approaches—namely sensitivity constraints, limited sequencing depth, and host DNA contamination—this study employs WGS of E. coli.
2 Materials and methods
2.1 Sample collection and initial processing
A total of 60 tissue samples—including kidney, lung, liver, and heart—were collected during necropsies of 51 individual WTD. Of these, 33 samples were obtained from 30 clinically ill deer, which were either euthanized (2/30) or found dead (28/30). The 2 animals were euthanized by the farm owner due to bacterial pneumonia. When clinical signs such as lameness or upper respiratory illness are observed, owners may choose to initiate treatment, typically with antibiotics or supportive care, or proceed with euthanasia. Because deer, as prey animals, often mask signs of illness until advanced stages, treatment outcomes are frequently limited. Some owners attempt treatment until the animal is no longer viable, whereas others elect euthanasia earlier to reduce suffering. Consequently, there is no standardized threshold for this decision, and practices vary on a case-by-case basis. The remaining 27 samples were collected from deer that were found dead without prior clinical signs and necropsied within 24 h of death. These samples were gathered from 16 counties in Florida, U. S., between September 16, 2020, and November 14, 2022. The organs were grossly examined, and tissue was collected and placed in 5 mL sterile Eppendorf snap cap tubes (Thermo Fisher Scientific) in a cooler with ice packs while in the field and subsequently transferred to a 4 °C refrigerator. The WTD tissue samples were submitted to the Clinical Microbiology Laboratory at the University of Florida (UF) College of Veterinary Medicine (CVM) for aerobic or anaerobic bacterial culture, depending on the sample type and diagnostic suspicion. CNA agar (Columbia Nalidixic Acid) is a selective medium designed for the isolation of Gram-positive bacteria. Since E. coli is a Gram-negative organism and the primary target of this study, no growth was expected or observed on CNA agar. The choice between aerobic and anaerobic culture conditions was guided by both sample type and the clinical or diagnostic context. For example, lung tissues were generally cultured under aerobic conditions due to their natural exposure to oxygen, whereas pus or lesion specimens were more often cultured anaerobically, reflecting their origin in enclosed or oxygen-limited environments. In addition, the suspected characteristics of the pathogen influenced this decision. Importantly, as a facultative anaerobe, E. coli was successfully isolated under both aerobic and anaerobic conditions. This work was approved by the UF Institutional Animal Care and Use Committee (IACUC Protocol Numbers 201609390 and 201909390).
2.2 Bacterial isolation and identification
Tissue samples were cultured specimens were cultured in their native form, directly incubated on differential media, including MacConkey agar (MAC), Columbia Nalidixic Acid agar (CNA), chocolate agar (CHOC), and general blood agar plates for bacterial isolation. No growth was observed on CNA agar. MALDI-TOF (MALDI Biotyper sirius one System from Bruker) was used for the identification of bacteria from the MAC, CHOC, and blood agar plates. Single colonies were then picked from E. coli positive plates and placed into CryoSavers tubes consisting of Brucella broth and 10% glycerol (Hardy Diagnostics) utilizing disposable inoculation wands (Thermo Fisher Scientific). The CryoSavers tubes were then stored in a −80 °C freezer until they were reinoculated into brain heart infusion (BHI) (DB Difco 237500) broth at 37 °C with shaking overnight at 220 revolutions per minute (RPM) for WGS and antimicrobial susceptibility testing (AST).
2.3 DNA extraction and whole genome sequencing
One milliliter of the overnight culture was pelleted at 10,000× relative centrifugal force (RCF) for one minute. Then, the genomic DNA of the E. coli was extracted using the Wizard Genomic DNA Purification Kit (Promega #A1120), according to the manufacturer’s protocol. The sequencing libraries were prepared using the Illumina ILMN DNA LP Kit (#20060059) and IDT for Illumina DNA/RNA UD Indexes Set A (#20027213). The DNA libraries were then sequenced at the UF Interdisciplinary Center for Biotechnology Research (ICBR) NextGen DNA Sequencing core (RRID: SCR_019152) using an Illumina NovaSeq sequencer with 2 × 150 cycle S4 kit (Bruinsma et al., 2018). The E. coli isolates were categorized into distinct phylogroups using the EZclermont web-based tool, allowing for a clear illustration of the collection’s diversity (Waters et al., 2020).
2.4 Data management and resistance gene analyses
To characterize the resistome of the E. coli isolates, the AMR++ Pipeline (version 3.0) (Bonin et al., 2023) was utilized. The AMR++ Pipeline trims the raw paired-end reads (FASTQ) for quality control and then aligns them to approximately 9,000 hand-curated ARGs from the MEGARes database 3.0 (https://www.meglab.org) using Burrows-Wheeler-Aligner (BWA) to produce Sequence Alignment/Map (SAM) format (Lakin et al., 2017). The Single Nucleotide Polymorphism (SNP) confirmation and “deduped” functions of the pipeline were implemented to confirm SNPs and deduplicate counts. Sequences from public databases that have been published and represent distinct accession numbers were referred to as individual ARGs. The main function of the structural genes in the MEGARes database is regulatory activity (e.g., efflux system), even though they are typically encoded on the chromosome and do not dictate phenotypic resistance. Three levels of hierarchical classification were established by aggregating ARGs: class (e.g., phenicols), mechanism (e.g., Phenicol resistance MFS efflux pumps), and group (e.g., floR) (Lakin et al., 2017). In addition to maintaining reasonable biological categories throughout the database, the group annotation provides insights into the primary gene classification while preserving nucleotide arrangements. Only ARGs that were more than 80 percent covered by sample reads and for which a single nucleotide polymorphism (SNP) did not confer resistance were taken into consideration for further examination.
ResistoXplorer (https://www.resistoxplorer.no) was implemented for data normalization, analysis, and visualization (Dhariwal et al., 2021). The low-count and variance filters of the web-based tool were used to filter poor quality or non-informative ARGs to improve downstream comparative analyses. The low count filters were adjusted so that the software only included features present in at least one count and were prevalent in at least 10% of samples. The low variance filter was modified so that zero percentage variance was removed, and the calculation was based on the standard deviation. The Cumulative Sum Scaling (CSS) (Paulson et al., 2013) function was applied to remove any possible sampling or sequencing biases and to normalize the ARG counts before converting for relative abundance and core resistome analyses. CSS is similar to total sum scaling (TSS), which calculates the ratio of the read count for each ARG compared to the total read count per sample. However, to reduce the impact of highly abundant variable genes, the denominator is calculated by summing the total read counts, starting with low-abundance genes and continuing up to a predefined threshold. In a comparative analysis of nine normalization techniques for count data, CSS emerged as one of the most effective methods for handling large metagenomic datasets (Pereira et al., 2018). The relative abundance and core resistome matrices for different levels (e.g., class and mechanism) were then produced after CSS counts were aggregated for varying resistance levels (e.g., class and mechanism). These matrices were used to generate the plots in RStudio (R version 4.0.3), which illustrate the class and mechanism level relative abundance as well as the core resistome for all E. coli isolates.
2.5 Statistical analysis: genotype
Alpha diversity of the resistome was assessed using the Shannon diversity index, which accounts for both richness and evenness but does not capture compositional differences and is not normally distributed. Shannon indices were calculated for each sample across resistance profiles classified at the class, mechanism, group, and gene levels. To evaluate the influence of metadata variables—including phylogroup, isolate origin, and farm—on resistome diversity, analysis of variance (ANOVA) was applied, acknowledging its assumptions of normality and homoscedasticity. This approach facilitated the identification of potential associations between environmental or biological factors and the complexity of antimicrobial resistance patterns.
2.6 Antimicrobial susceptibility testing
Antimicrobial susceptibility testing of the E. coli isolates was conducted using the Kirby-Bauer disk diffusion method on Mueller-Hinton agar, based on the guidelines and recommendations in the CLSI VET01S (CLSI, 2024). Thirteen antimicrobials were selected for testing based on their frequent use in Florida deer farms and their relevance in both human and veterinary medicine. These include: ampicillin (AMP, 10 μg) (Becton Dickinson and Company, Sparks, MD, United States), penicillin (PEN, 10 μg) (Becton Dickinson and Company, Sparks, MD, United States), ceftiofur (CEFT, 30 μg) (Becton Dickinson and Company, Sparks, MD, United States), tetracycline (TET, 30 μg) (Becton Dickinson and Company, Sparks, MD, United States), oxytetracycline (OXY, 30 μg) (Becton Dickinson and Company, Sparks, MD, United States), gentamicin (GENT, 10 μg) (Becton Dickinson and Company, Sparks, MD, United States), neomycin (NEOM, 30 μg) (Becton Dickinson and Company, Sparks, MD, United States), NUFLOR® (florfenicol, FLU, 300 mg ml−1) (Merck & Co., Inc., Rahway, NJ, United States), Resflor Gold® (florfenicol + flunixin meglumine, FF, 300/16.5 mg ml−1) (Merck & Co., Inc., Rahway, NJ, United States), trimethoprim-sulfamethoxazole (SXT, 1.25/23.7 μg) (Becton Dickinson and Company, Sparks, MD, United States), enrofloxacin (ENRO, 5 μg) (Becton Dickinson and Company, Sparks, MD, United States), Draxxin® (tulathromycin, TUL, 100 mg ml−1) (Zoetis Inc. Kalamazoo, MI, United States), and ZACTRAN® (gamithromycin, GAM, 150 mg ml−1) (Boehringer Ingelheim Animal Health United States Inc., Duluth, GA, United States). Fifteen microliters of the commercial antimicrobials—NUFLOR®, Resflor Gold®, Draxxin®, ZACTRAN®—were applied to blank disks and left overnight covered in petri dishes while in a biosafety cabinet. The final antimicrobial amounts per disk were 4.5 μg, 4.5/0.425 μg, 1.5 μg, and 2.25 μg, respectively. Susceptibility results were interpreted using zone of inhibition breakpoints established by the CLSI for AMP, CEFT, TET, OXY, GENT, and SXT, and by the NCCLS for NEOM. Multi-drug resistance was defined as resistance to at least three antimicrobial agents as described by Begum et al. (2018).
The isolates stored in CryoSavers tubes, consisting of Brucella broth and 10% glycerol (Hardy Diagnostics), at −80 °C were reinoculated into brain heart infusion (BHI) (BD Difco 237500; Becton, Dickinson and Company) broth at 37 °C with shaking overnight at 220 revolutions per minute (RPM). The culture was adjusted to an optical density (OD) of 0.1 (approximately 0.5 McFarland standards) in BHI. Sterile swabs were dipped into the adjusted bacterial culture and gently rolled on the inside wall of the tube to squeeze out any excess culture. The entire surface of the Mueller-Hinton agar plate was swabbed in three directions, with the plate being rotated 60° after each pass to ensure even distribution and create a uniform bacterial lawn. The plates were then allowed to dry for no more than 15 min. A Sensi-Disc Dispenser (BD 260660; Becton, Dickinson and Company) was used to place 8 disks, while the remaining 5 were applied manually using sterile forceps. A minimum distance of 24 mm between the centers of adjacent disks and 15 mm from the edge of the plate was maintained. Each disk was gently pressed to ensure firm contact with the agar surface. The plates were inverted and incubated at 37 °C for 16–18 h under aerobic or anaerobic conditions. Following incubation, the diameter of the clear zone around each antimicrobial disk was measured in millimeters. To account for potential irregularities in the shape of the zone of inhibition (ZOI), measurements were taken at three different points along the edge and then averaged. The CLSI and NCCLS breakpoints were applied to classify each isolate as resistant, intermediate, or susceptible.
2.7 Statistical analysis: phenotype
A generalized linear model (GLM) with a Poisson distribution and log link function was used to assess the effects of phylogroup, isolate, and farm on phenotypic resistance counts. The GLM was constructed using phenotypic resistance counts as the dependent variable and the tested factors—phylogroup, isolate, and farm—as independent variables. This approach allowed for the assessment of potential associations between these factors and the observed resistance phenotypes while accounting for variability within the data. Statistical significance was determined by calculating p-values for each factor, with a threshold of 0.05 indicating significance.
2.8 Correlation among antimicrobial resistant genotypes and phenotypes
The resistant genotype of the E. coli isolates was identified through whole genome sequencing, and the use of the AMR++ pipeline to align the sequencing reads to a curated ARG database. A comprehensive workflow for genotype and phenotype determination of the E. coli isolates is outlined in the above sections. Cohen’s kappa statistics were used to assess the agreement between genotype and phenotype. The presence of the cmy, ctx, or tem gene groups was classified as a resistant genotype for AMP and CEFT, while the tetA, tetB, or tetD gene groups were considered a resistant genotype for TET and OXY. For GENT and NEOM, a resistant genotype was determined by the existence of the aac3 gene group, and for FLU, the floR or cmlA, gene groups. A resistant genotype to ENRO was linked to the qnrS gene group. Due to the complex interaction of SXT, which inhibits two pathways in folate synthesis, the simultaneous presence of the sulII, sulIII, and dfrA gene groups was considered a resistant genotype (Projan, 2002). The ZOI breakpoints for Enterobacterales, as outlined by CLSI and NCCLS, were used to classify phenotypic resistance. For statistical purposes, isolates in the intermediate category were treated as susceptible (Schwan et al., 2021). Kappa coefficient (κ) values were interpreted as follows: <0.2 = poor, 0.21–0.4 = fair, 0.41–0.6 = moderate, 0.61–0.8 = good, and 0.81–1.0 = very good agreement (Azen and Walker, 2021). A two-sided z-test with a p-value < 0.05 was considered statistically significant.
3 Results
3.1 Animal information and phylogroup
The 60 E. coli strains were collected from tissue samples obtained during the necropsy of 51 individual WTD. The bacteria were initially isolated, and identified using MALDI-TOF, followed by DNA extraction and whole genome sequencing. Following sequencing, the E. coli isolates were typed by phylogroup to assess their diversity, employing methods described by Metrailer et al. (2024). The isolates were grouped into seven distinct phylotypes: A, B1, B2, C, D, E, and F (Table 1). Of the 60 isolates, 75% (45/60) grouped into phylogroup B1, while 11.67% (7/60), 0.05% (3/60), 0.03% (2/60), 0.01% (1/60), 0.01% (1/60), 0.01% (1/60), grouped into phylogroups A, D, E, B2, C, and F, respectively.
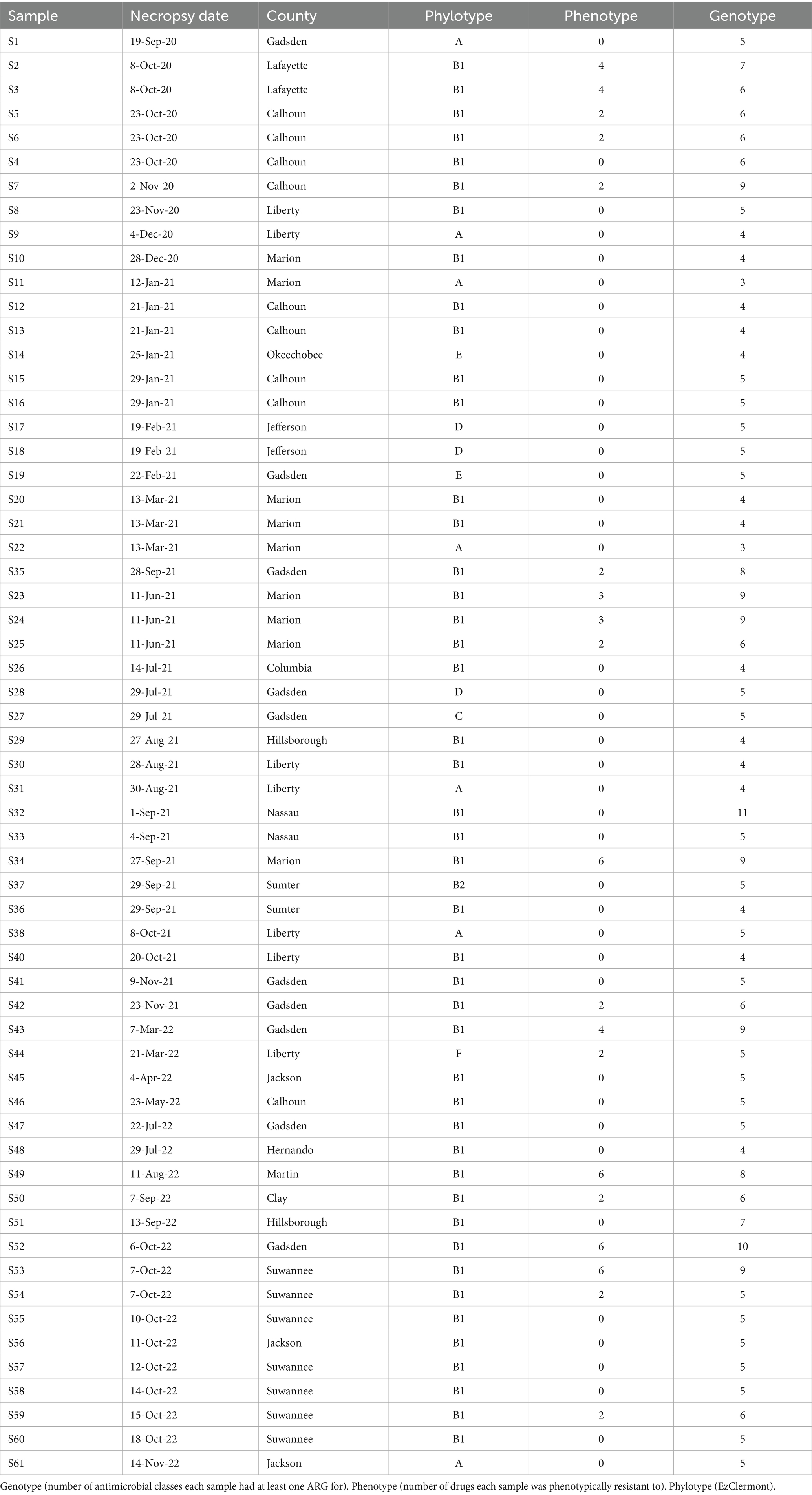
Table 1. Typing information for animals and E. coli isolates collected from farmed WTD in Florida between September 2020 and November 2022 (n = 60).
3.2 Core resistome and abundance profiles
The core resistome refers to the set of ARGs, gene groups, mechanisms, or classes that are consistently detected in a large proportion of a population, exceeding a defined abundance threshold. In E. coli isolates from farmed WTD in Florida, the core resistome comprises ARGs associated with five major antimicrobial classes: β-lactams, multidrug resistance (MDR), bacitracin, macrolide-lincosamide-streptogramin (MLS), and cationic antimicrobial peptides (CAP). These ARGs are both abundant and broadly distributed across the sampled population (Figure 2). The heat map of the core resistome illustrates five classes to be prevalent in at least 10% of the samples. For example, the prevalence of the bacitracin class having a relative abundance (or detection threshold) of 0.001%, is 90%. As the detection threshold becomes more stringent, with a relative abundance of at least 0.100%, the prevalence drops to approximately 0.20 (only 20% of samples will contain bacitracin resistance at a relative abundance of 0.100%).
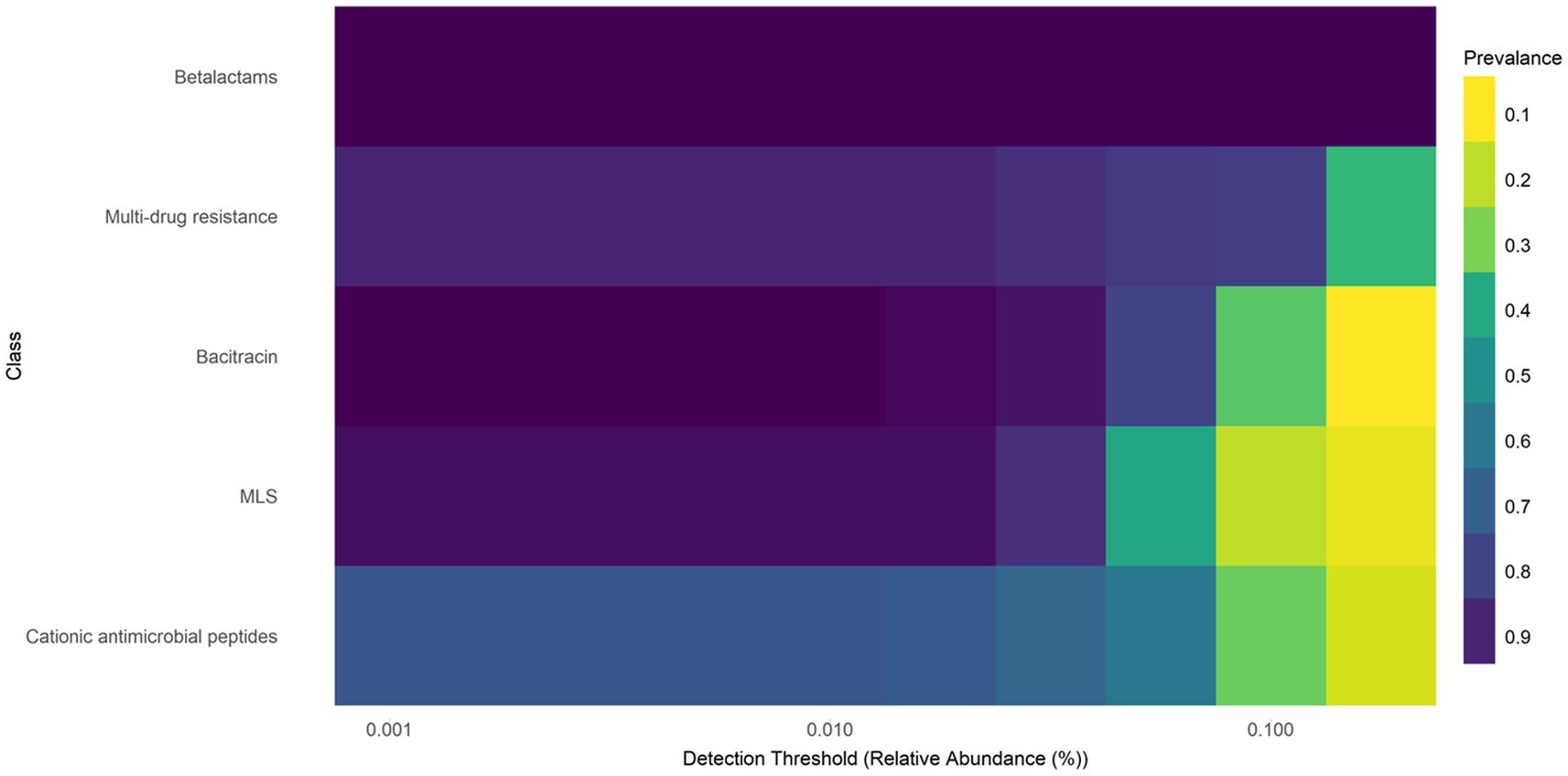
Figure 2. Heatmap of core resistome analysis of E. coli isolates from white-tailed deer revealed five classes to be prevalent in at least 10% of the samples. Macrolide, lacosamide, Streptogramin (MLS).
Among the 60 E. coli samples analyzed, 362 unique ARGs were identified as resistant to 12 antimicrobial classes. Overall, the ARGs identified confer resistance to the ß-lactam (49.06%), MDR (14.82%), CAP (8.1%), bacitracin (8.1%), MLS (6.02%), aminoglycoside (4.55%), tetracycline (3.76%), phenicol (3.17%), sulfonamide (2.12%), trimethoprim (0.2%), fluoroquinolone (0.06%), and fosfomycin (0.03%) antimicrobial class (Figure 3A). ß-lactam and MLS resistance were abundant in every sample tested, and MDR was abundant in 90% (54/60) of the samples tested (Figure 3B).
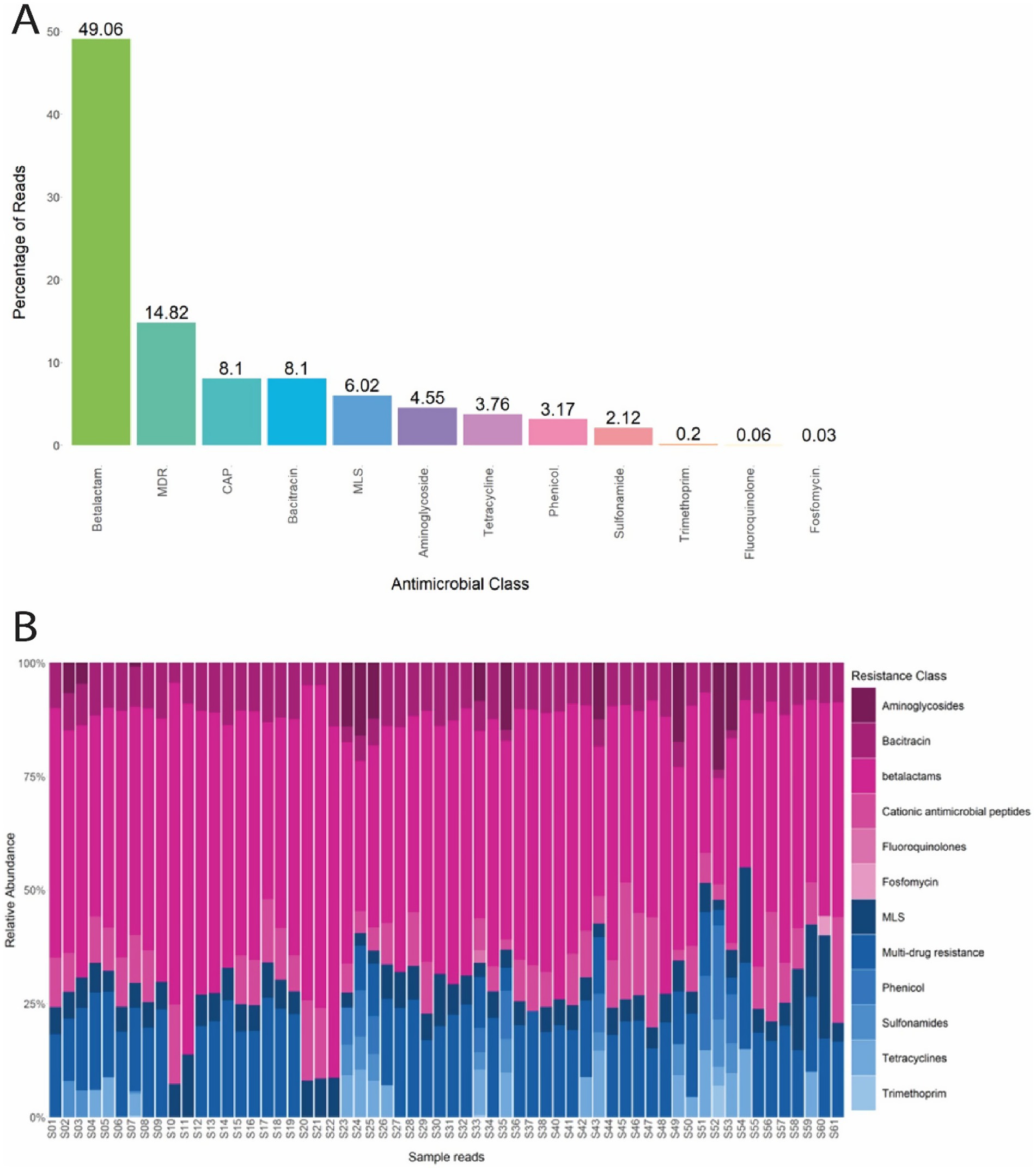
Figure 3. (A) Percentage of reads from E. coli isolates that confer resistance to each antimicrobial class. (B) Normalized relative abundance (percentage) of ARGs in E. coli isolates grouped by resistance class for all 60 samples. Multi drug resistance (MDR); Cationic antimicrobial peptides (CAP); Macrolide, lacosamide, Streptogramin (MLS).
The 12 antimicrobial classes confer resistance by 19 mechanisms, including Penicillin binding protein (28.1%), Multi-drug RND efflux regulator (14.82%), Undecaprenyl pyrophosphate phosphatase (8.1%), Lipid A modification (8.1%), Class C β-lactamases (7.49%), Mutant porin proteins (7.1%), Class A β-lactamases (6.38%), Macrolide phosphotransferases (5.51%), Aminoglycoside O-phosphotransferases (3.82%), Tetracycline resistance MFS efflux pumps (3.76%), Phenicol resistance MFS efflux pumps (3.17%), and Sulfonamide- resistant dihydropteroate synthase (2.12%). The remaining seven mechanisms had an overall abundance of less than 1 % and are summarized in Figure 4A. Penicillin binding protein was abundant in every sample tested and mechanisms associated with MLS resistance and MDR were observed in 98.3% (59/60) and 90% (54/60) of samples, respectively (Figure 4B).
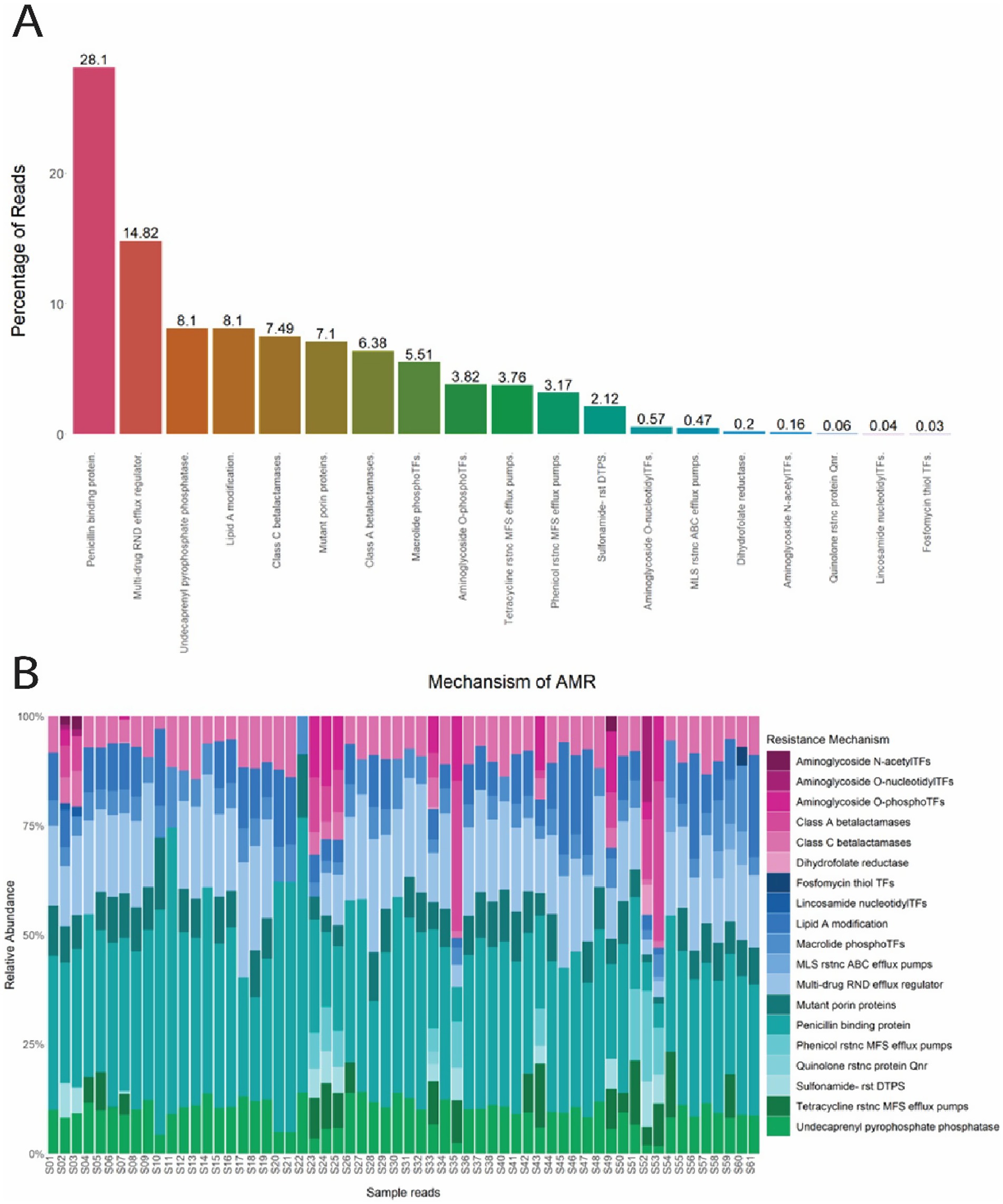
Figure 4. (A) Percentage of reads from E. coli isolates for each mechanism of antimicrobial resistance. (B) Normalized relative abundance (percentage) of ARGs in E. coli isolates grouped by resistance mechanism for all 60 samples. N-acetylTFs (N-acetyltransferases); O-nucleotidylTFs (O-nucleotidyltransferases); O-phosphoTFs (O-Phosphotransferases); Transferases (TFs); rstnc (Resistance); ABC (ATP- binding cassette); RND (Resistance-Nodulation-Division); MFS (major facilitator superfamily); rst (resistant); DTPS (dihydropteroate synthase).
In addition to the class and mechanism abundance profiles, various level 1 high-risk AMR genes were discovered in relatively high abundances in the current study (Figure 5). A total of five ARG groups that are considered “present hazards” (Zhang et al., 2021) including aac3, aph6, floR, mphA, and mphB were found in the E. coli isolated from the WTD samples. ARG groups: aac3, aph6, floR, mphA also contain genes in multiple categories (e.g., Level 3, Level 4, and unassigned). Among the rank 1 ARGs, mphB was identified in over 98% (59/60) of the samples tested. Notably, samples S11 and S30 exhibited the highest relative abundances, with mphB representing 13.8% (551/527233) and 11.5% (229/52733) of the total reads, respectively. Additionally, floR, which is known to confer resistance to chloramphenicol and florfenicol (White et al., 2000), showed a relative abundance of 16.3% (4,122/32,389), the highest percentage among all samples and ARG groups.
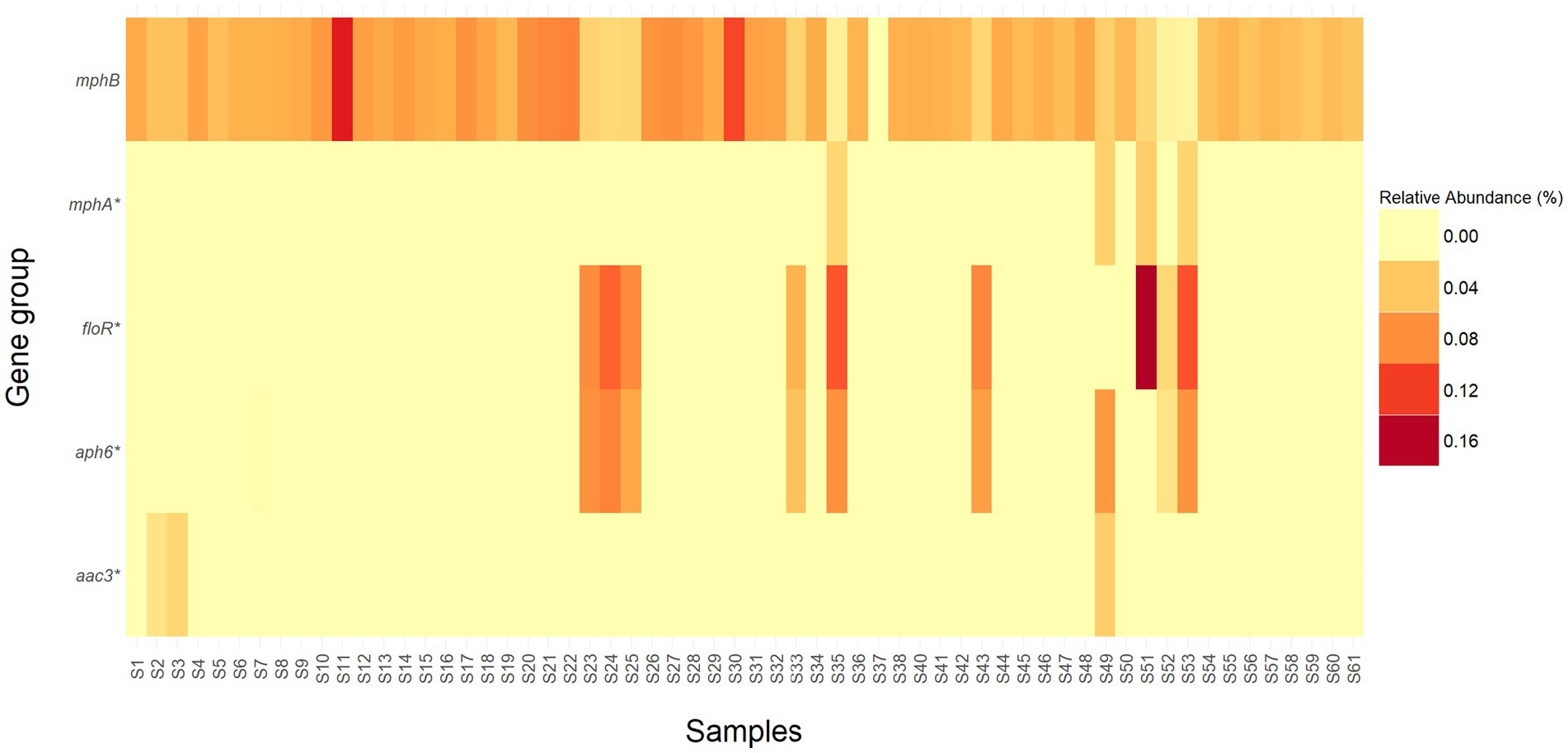
Figure 5. Heatmap representing the relative abundance (percentage) of high-risk ARG groups (Rank 1) detected in each sample. ARG groups mphA, floR, aph6, and aac3 contain genes that are classified in different risk levels and are denoted by an *.
Alpha diversity analyses were conducted using analysis of variance (ANOVA) with the Shannon diversity index to assess whether any metadata factors influenced resistance profile levels. Prior to conducting the ANOVA, assumptions of normality and homogeneity of variance were verified. No statistically significant differences were observed between the levels of resistance profiles (class, mechanism, group, and gene) across the experimental factors (region, farm, and phylogroup) (p-value > 0.05).
3.3 Antimicrobial susceptibility testing
The Kirby-Bauer disk diffusion method was conducted on 60 E. coli isolates from farmed WTD in Florida. The ZOI for each antimicrobial agent varied between aerobic and anaerobic conditions. Under aerobic conditions, the following resistance rates were observed: 14.75% of the isolates were resistant to AMP (Ampicillin); 8.20% to CEFT (Ceftiofur); 6.56% to ENRO (Enrofloxacin); 14.75% to FLU (Florfenicol); 4.92% to GENT (Gentamicin); 1.64% to NEO (Neomycin); 29.51% to OXY (Oxytetracycline); 26.23% to TET (Tetracycline); and 1.64% to SXT (Trimethoprim–sulfamethoxazole). Additionally, intermediate resistance was detected in 1.64, 4.92, 3.28, and 3.28% of isolates for CEFT, ENRO, FLU, and TET, respectively (Figure 6A). Under anaerobic conditions, the following resistance rates were observed: 11.48% of the isolates were resistant to AMP (Ampicillin); 8.20% to CEFT (Ceftiofur); 6.56% to ENRO (Enrofloxacin); 11.48% to FLU (Florfenicol); 32.79% to GENT (Gentamicin); 39.34% to NEO (Neomycin); 29.51% to OXY (Oxytetracycline); 8.20% to TET (Tetracycline); and 3.28% to SXT (Trimethoprim–sulfamethoxazole). Additionally, intermediate resistance was detected in 4.92, 1.64, 4.92, 60.66, 57.38, 1.64, 1.64, 21.31, and 6.56% of isolates for AMP, CEFT, ENRO, GENT, NEO, OXY, SXT, TET, and FLU, respectively (Figure 6B). The CLSI veterinary guidelines do not specify ZOI breakpoints for Florfenicol + Flunixin meglumine (FF), Gamithromycin (GAM), Penicillin (PEN), and Tulathromycin (TUL) in the context of E. coli. Consequently, it is not possible to categorize these antimicrobials as resistant, intermediate, or susceptible. To aid in interpretation, Figure 7 presents a boxplot depicting the mean ZOI values for all tested drugs under both aerobic and anaerobic conditions. Based on clinical breakpoints for the order Enterobacterales, 30% (18/60) of the E. coli isolates displayed AMR to no less than one drug tested under aerobic conditions, while 68% (41/60) showed resistance under anaerobic conditions. In contrast, nine E. coli isolates (15%) exhibited an MDR phenotype under both aerobic and anaerobic conditions (Figures 8A,B).
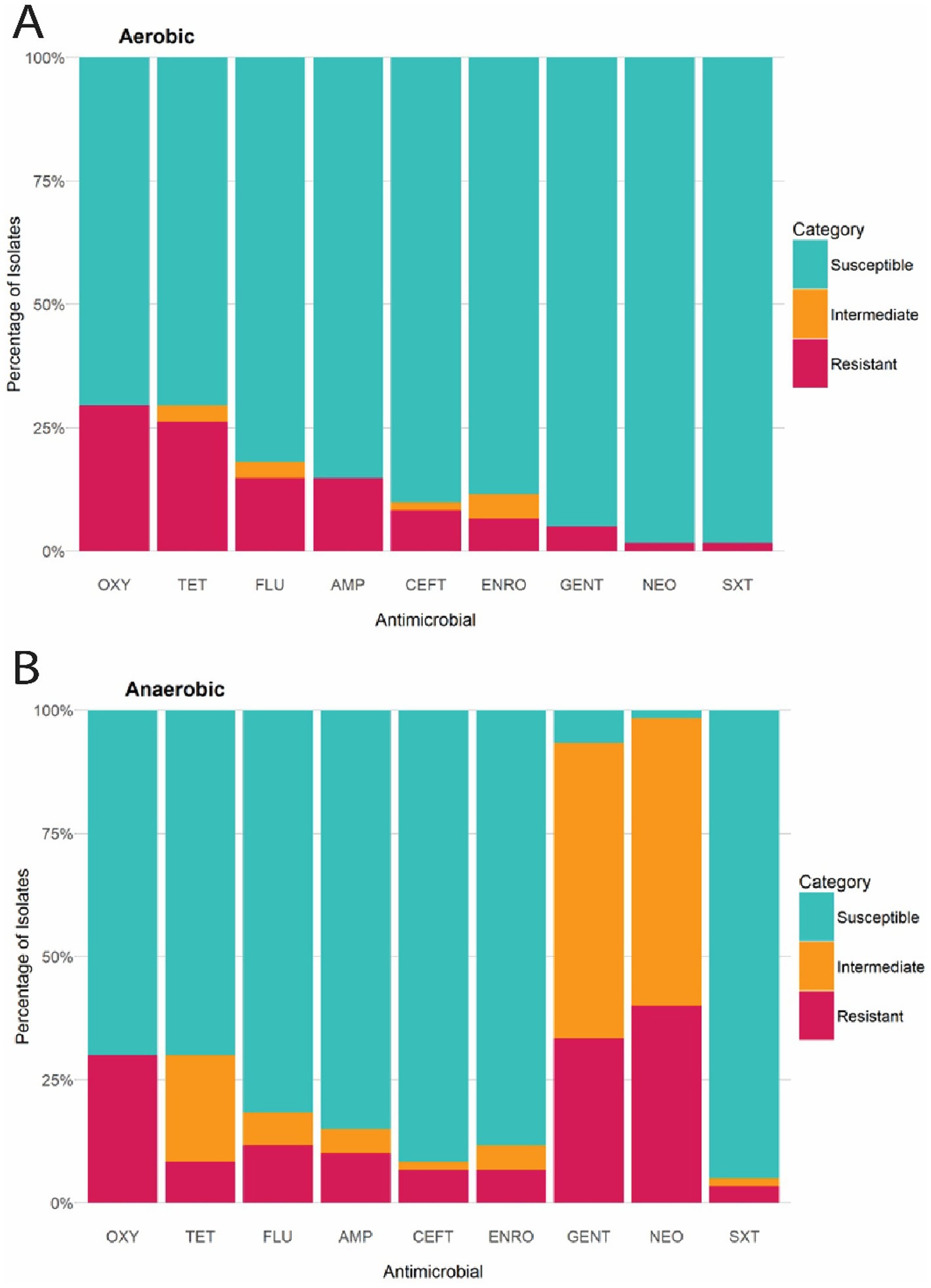
Figure 6. (A) Bar chart illustrating the percentage of E. coli isolates from WTD classified as Susceptible, Intermediate, or Resistant to each antimicrobial compound, according to CLSI breakpoints under aerobic conditions. (B) Bar chart illustrating percentage of E. coli isolates from WTD classified as Susceptible, Intermediate, or Resistant to each antimicrobial compound, according to CLSI breakpoints under aerobic conditions. AMP (Ampicillin); CEFT (Ceftiofur); FLU (Florfenicol); NEO (Neomycin); GENT (Gentamicin); ENRO (Enrofloxacin); TET (Tetracycline); OXY (Oxytetracycline); SXT (Trimethoprim–sulfamethoxazole).

Figure 7. Boxplots and individual points representing the mean zone of inhibition (in millimeters) for E. coli isolates from WTD, grouped by antimicrobial agent under aerobic and anaerobic conditions. AMP (Ampicillin); CEFT (Ceftiofur); FLU (Florfenicol); NEO (Neomycin); GENT (Gentamicin); ENRO (Enrofloxacin); TET (Tetracycline); OXY (Oxytetracycline); SXT (Trimethoprim–sulfamethoxazole); FF (Florfenicol + Flunixin meglumine); GAM (Gamithromycin); PEN (Penicillin); TUL (Tulathromycin). “an” indicates anaerobic condition.
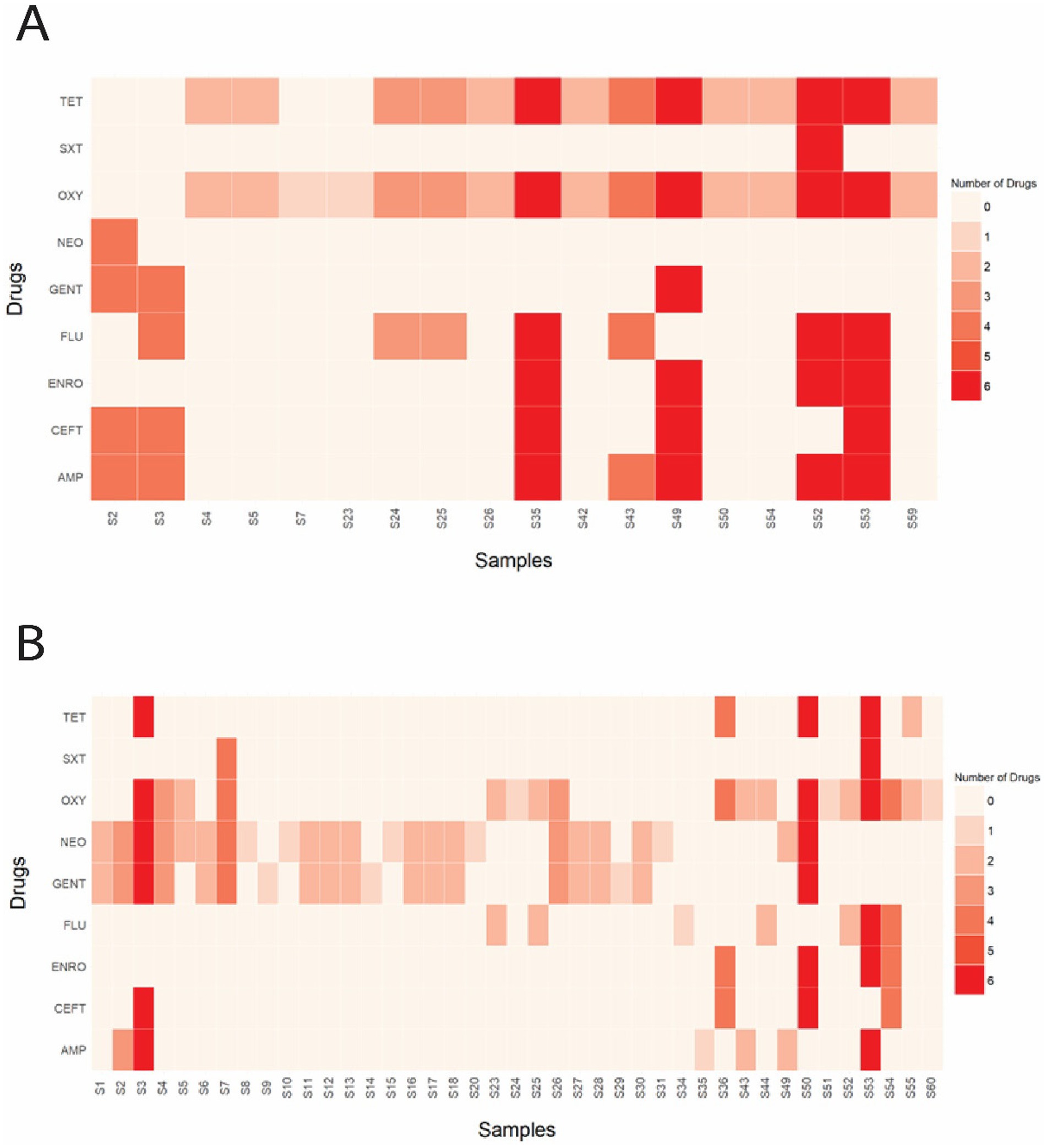
Figure 8. (A) Heatmap illustrating the total number and specific drug(s) to which each sample displayed phenotypic resistance under aerobic conditions. (B) Heatmap illustrating the total number and specific drug(s) to which each sample displayed phenotypic resistance under anaerobic conditions. AMP (Ampicillin); CEFT (Ceftiofur); FLU (Florfenicol); NEO (Neomycin); GENT (Gentamicin); ENRO (Enrofloxacin); TET (Tetracycline); OXY (Oxytetracycline); SXT (Trimethoprim–sulfamethoxazole).
Notably, all isolates exhibiting phenotypic resistance to at least one tested antimicrobial belonged to the B1 phylogroup. However, none of the tested factors—phylogroup, isolate, or farm—had a significant effect on phenotypic resistance counts (p-value > 0.05).
3.4 Comparison between antimicrobial susceptibility genotypes and phenotypes
To assess the potential of WGS in predicting AMR profiles, we compared genotypic data (gene presence) with phenotypic resistance behaviors. Understanding the concordance between these two factors is essential for evaluating the accuracy of genetic predictions in reflecting observed resistance patterns. For instance, a strong agreement (Kappa > 0.80) was observed between genotype-based resistance (presence of tetA, tetB, or tetD genes) and phenotypic resistance (determined by a ZOI diameter ≤10 mm) for both tetracyclines tested (tetracycline and oxytetracycline). This finding is particularly important, as E. coli exhibiting high concordance between genotype and phenotype could be utilized in AMR surveillance programs, where gene presence is strongly correlated with the resistant phenotype. However, the comparison between genotypic and phenotypic AMR profiles can be complex due to the varying resistance mechanisms across different antimicrobial agents and bacterial species. While the genotype represents the genetic “potential” for resistance, it does not always correspond fully to the phenotype expressed by the bacterium. The consonance between genotypic and phenotypic drug resistance was good for AMP (kappa = 0.762) and very good for GENT (kappa = 1), TET (kappa = 0.875), OXY (kappa = 0.96), and SXT (kappa = 1), as demonstrated in Table 2. In contrast, CEFT (kappa = 0), ENRO (kappa = −0.0274) displayed poor agreement, while FLU (kappa = 0.36) demonstrated fair agreement and NEO (kappa = 0.487) showed moderate agreement (Table 2).
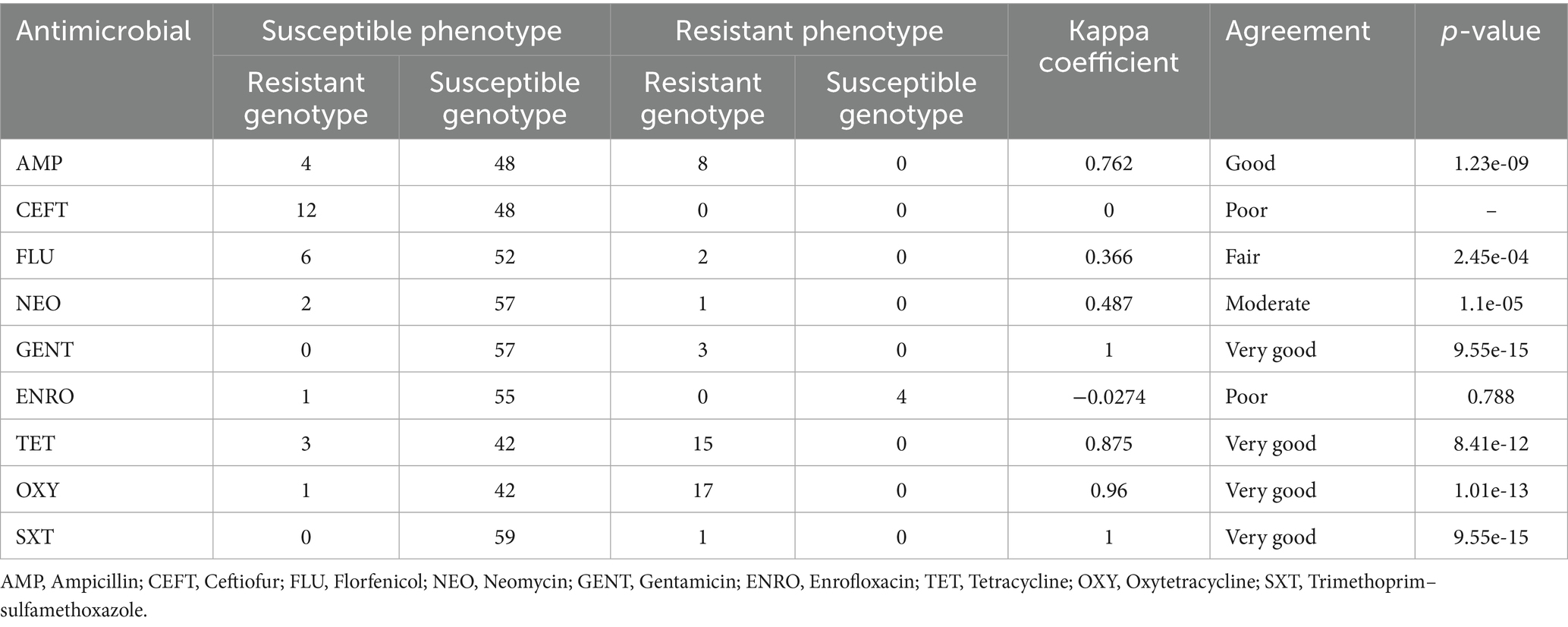
Table 2. Contingency table comparing genotypic resistance to phenotype resistance.
4 Discussion
4.1 Resistome profiling and phylogroup distribution in Escherichia coli from Florida farmed WTD
In this study, whole genome sequencing of E. coli genomic DNA from various tissue samples of WTD was employed to quantify ARGs and report the relative abundance of the various genetic determinants attributed to AMR. A total of 362 distinct ARGs were identified, conferring resistance to 12 antimicrobial classes through 19 different mechanisms. These results exceed those of similar studies using different matrices, such as roe deer feces (41 ARGs from 7 antimicrobial classes) (Dias et al., 2022), soils treated with bovid and swine manure (77 ARGs from 8 antimicrobial classes) (Chen et al., 2019), and porcine excrement (146 ARGs from 9 antimicrobial classes) (Zhao R. et al., 2018; Zhao Y. et al., 2018).
In our analysis of E. coli isolated from WTD tissue samples, we identified a range of ARGs that align with documented antimicrobial resistance trends across wildlife and agricultural species. Two classes of the core resistome, MLS and MDR, are especially concerning as these classes are less specific and are responsible for conferring resistance to numerous antimicrobials or classes. Overall, ARGs associated with β-lactam resistance were highly abundant, along with those conferring MDR and resistance to individual classes like bacitracin, cationic antimicrobial peptides (CAP), macrolide-lincosamide-streptogramin (MLS), aminoglycoside, and tetracycline. These antimicrobial classes mirror those commonly used in global livestock production systems (He et al., 2020) as well as ice core samples without anthropogenic influence (Paun et al., 2021). Their widespread presence has also been documented in wildlife, particularly near anthropogenic sources such as manure and biosolid application sites. For example, tetracycline resistance genes (tetQ) were frequently detected in wild deer in such environments, highlighting the impact of human activity on wildlife resistomes (Rogers et al., 2018). Comparable ARG profiles have been observed in farmed sika deer in China, where tetracycline resistance genes were similarly predominant (Huang et al., 2016), and in wildlife gut microbiota across species in Poland, where tetQ was the most prevalent ARG (Skarżyńska et al., 2020).
Our findings also corroborate above mentioned patterns, with mphB—an ARG conferring resistance to MLS—identified in over 98% (59/60) of the samples. The highest relative abundance of mphB was observed in samples S11 and S30, where it represented 13.8% (551/527,233) and 11.5% (229/52,733) of total reads, respectively. Additionally, floR, associated with chloramphenicol and florfenicol resistance (White et al., 2000), exhibited the highest relative abundance among all samples and ARG groups, reaching 16.3% (4,122/32,389). This prevalence echoes findings in cattle farming where, β-lactam, MLS, aminoglycoside, and tetracycline ARGs to be dominant (Muurinen et al., 2017; Qian et al., 2018; Zhao R. et al., 2018; Zhao Y. et al., 2018; Chen et al., 2019; Wu et al., 2020). Recent studies have also reported a rising prevalence of β-lactam resistance, including cephalosporin- and carbapenem-resistant genotypes, highlighting the widespread dissemination of these ARGs in companion animals, dairy cattle, and wastewater (Daniels et al., 2018; Mollenkopf et al., 2018; Mathys et al., 2019). This trend is also consistent with a linear increase in β-lactam resistance, including cephalosporins, observed in E. coli from WTD in Ohio, United States (Ballash et al., 2021).
Beyond our specific findings, we identified ARGs such as sulII (sulfonamide resistance), qnrS (fluoroquinolone resistance), blaTEM (class A β-lactamase), and various aph genes (aminoglycoside O-phosphotransferases), which are recognized as environmental resistance markers (Berendonk et al., 2015). These findings reveal a complex ARG profile in farmed WTD from Florida, influenced by both environmental factors and human activities related to antimicrobial resistance. This study is the first to characterize the resistome of farmed WTD in Florida, identifying five high-risk antimicrobial resistance genes (ARGs) present at relatively high abundances. These ARGs are considered ‘present hazards’ due to their association with ESKAPE pathogens—Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species—which pose a global threat to human health and their potential for horizontal gene transfer (HGT) (Zhang et al., 2021). These insights provide valuable guidance for managers, farm owners, and veterinarians, supporting informed decisions on medication use in Florida deer farms.
The distribution of E. coli phylogroups observed in this study aligns with prior research (Touchon et al., 2020; Munkhdelger et al., 2017). The most prevalent phylogroups were B1, A, and D, while C, E, and F were the least common. Phylogroups A and B1, commonly linked to commensal strains, are generally less virulent but exhibit notable AMR in specific contexts, particularly in agricultural environments. These phylogroups often harbor ARGs for tetracyclines, sulfonamides, and aminoglycosides (Monroy-Pérez et al., 2020; Pais et al., 2022). Although phylogroup A showed an overall lack of resistance, ARGs for tetracyclines, sulfonamides, and aminoglycosides were detected in phylogroup B1. In our study, phylogroup B1 is predominant and demonstrates high resistance to tetracyclines, β-lactams, and sulfonamides and MDR, which is lower than in clinical isolates (Pais et al., 2022). While the prevalence of MDR and ESBL genes were lower than in clinical isolates, their presence still poses a significant risk of zoonotic transmission (Pais et al., 2022).
Interestingly, although phylogroup D was the third most prevalent group in this study, it accounted for only 0.05% (3/60) of total isolates—a markedly lower proportion than the 26 and 28.4% reported in previous studies in humans, domestic and wild animals and the environment (Touchon et al., 2020; Munkhdelger et al., 2017). Phylogroup D is generally considered more virulent than phylogroups A and B1, typically associated with extraintestinal pathogenic E. coli (ExPEC) in humans rather than with commensal strains (Clermont et al., 2000; Da Silva and Mendonça, 2012; Picard et al., 1999).
A notable finding in the present study was that only one isolate (S36) belonged to phylogroup B2, a strikingly low proportion compared to the 39 and 33.8% reported by Touchon et al. (2020) and Munkhdelger et al. (2017), respectively. This discrepancy is surprising given the dominance of phylogroup B2 in isolates from Asia (Zhao et al., 2015; Lee et al., 2016; Bashir et al., 2012), Europe (Ejrnæs et al., 2011; Dubois et al., 2010), Africa (Dadi et al., 2020), and North America (Paniagua-Contreras et al., 2017). In these studies, B2 isolates frequently harbored ESBL genes and MDR profiles, particularly against cephalosporins and fluoroquinolones (Monroy-Pérez et al., 2020; Hemati et al., 2024). In contrast, no resistance determinants were identified in the phylogroup B2 isolate from our study. These differences highlight the variability in phylogroup distribution across geographic regions and underscore the need for further investigation to understand the factors driving these patterns.
4.2 AMR profiles of Escherichia coli isolated from farmed WTD in Florida
The bacterial AMR profile of various vertebrate species and the environment is still primarily assessed through culture-dependent methods (Guitor et al., 2019). In this study, we assessed the prevalence of AMR by evaluating the susceptibility of E. coli isolated from farmed WTD in Florida to 14 commonly used farm-associated antimicrobials. Based on clinical breakpoints for the family Enterobacteriaceae, 30% (18/60) of the E. coli isolates displayed resistance under aerobic conditions, while 68% (41/60) showed resistance under anaerobic conditions. These values exceed the resistance rates reported in venison from Germany (9%) and red deer in Spain (7, 23%) (Mateus-Vargas et al., 2017; Alonso et al., 2016; Dias et al., 2022). In the U. S., 16.7% of E. coli isolates obtained from bison (Bison bison) carcasses exhibited resistance to at least one antimicrobial agent (Li et al., 2007).
In our study, E. coli isolates predominantly exhibited phenotypic resistance to tetracyclines under aerobic conditions. A high proportion of isolates also demonstrated phenotypic resistance to penicillin when interpreted using ZOI breakpoints reported in previous studies (Bughe et al., 2020). However, as the CLSI does not provide breakpoints for penicillin in E. coli, we excluded penicillin from formal phenotypic categorization. This observation is consistent with the findings of wild roe deer (Mayrhofer et al., 2006) and farmed red deer (Alonso et al., 2016), but contrasts with those of wild red deer (Dias et al., 2022) and wild WTD (Ballash et al., 2021), where resistance to ß-lactams and sulfonamides was most prevalent. The tetA and tetB gene groups, responsible for encoding an active efflux pump, were present in all isolates with phenotypic resistance to tetracyclines, indicating these gene groups play a key role in tetracycline resistance observed in our study. Under anaerobic conditions, aminoglycosides, specifically GENT and NEO, demonstrated the highest resistance rates, with 32.8 and 39.3%, respectively. This is expected, as aminoglycosides require oxygen to cross the cell membrane (Whelton, 1984), and the accumulation of reactive metabolic byproducts has been noted in cells treated with these antibiotics (Wong et al., 2022). Penicillin was included in the AST, despite the lack of ZOI breakpoints for the medication in the CLSI VET01S. It is important to note that if the ZOI breakpoints from previous studies were applied for resistant, intermediate, or susceptible categorization, 95% (57/60) of isolates would have been resistant to penicillin under aerobic conditions (Bughe et al., 2020).
In the present study, isolates exhibiting phenotypic resistance to three or more antimicrobials were considered MDR, following the criteria by Begum et al. (2018). Under aerobic conditions, nine E. coli isolates (15%) exhibited a MDR phenotype, a significantly higher proportion compared to farmed red deer (1/72, 1.3%) (Alonso et al., 2016), wild red deer (2/101, 1.9%) (Dias et al., 2022), and wild roe deer (1/76, 1.3%) (Mayrhofer et al., 2006). Similarly, under anaerobic conditions, nine E. coli isolates (15%) also exhibited a MDR phenotype. The high prevalence of multidrug-resistant (MDR) strains may be attributed to the indiscriminate use of antimicrobial agents (Van Den Bogaard and Stobberingh, 2000). Nevertheless, the MDR patterns identified in this study offer valuable insights for farm managers, owners, and veterinarians in selecting appropriate treatments for use in deer farms—for instance, by choosing antimicrobials that retain efficacy and avoiding those associated with high resistance rates.
Our findings underscore the advantages of utilizing both culture-dependent and culture-independent methods to investigate drug resistant bacteria, as they complement each other. For example, ß-lactam resistance-associated ARGs were the most abundant across all samples, yet only seven of the isolates showed phenotypic resistance to two antimicrobials commonly used on deer farms: CEFT and AMP. Moreover, the presence of gene groups: cmy, ctx, or tem could serve as a proxy for phenotypic resistance for AMP, which exhibited good agreement (kappa = 0.762), but not for CEFT (kappa = 0). However, the presence of ARGS in our analysis was in good agreement with an intermediate phenotype for CEFT. In addition, tetA, tetB, or tetD gene groups showed very good agreement with the phenotypic resistance in isolates from farmed WTD in Florida. These findings are encouraging, especially as tetracyclines are among the most widely used antimicrobial classes in veterinary (Daghrir and Drogui, 2013) and agricultural production (Chang et al., 2023). Despite the challenges in drawing direct comparisons, both approaches need globally standardized methodologies. Software like the AMR++ pipeline, coupled with the ResistoXplorer platform, performed exceptionally well in standardizing metagenomics-based results, despite the diversity of upstream approaches.
The results of this study showed that none of the evaluated factors—phylogroup, isolate, or farm—significantly affected phenotypic resistance counts (p > 0.05). Thus, no associations were detected between resistance phenotypes and either genetic background or environmental origin within the dataset. Nevertheless, the observation that phenotypic resistance occurred exclusively in phylogroup B1 suggests an intriguing trend that merits further investigation.
While our study provides valuable information on resistance profiles from Florida’s farmed WTD, it is important to consider the limitations of our study design. The overrepresentation of phylogroups A and B1 in the dataset may have introduced bias, potentially underestimating the role of other phylogroups, such as B2 and D, which are known to frequently harbor multidrug-resistant strains (Hemati et al., 2024; Tohmaz et al., 2022). Phylogroup B1, commonly associated with commensal strains, has also been linked to resistance determinants, especially in agricultural and environmental settings. The diverse resistance profiles observed within this phylogroup highlight its adaptability to selective pressures, such as the use of antibiotics in livestock (Hemati et al., 2024; Raimondi et al., 2019). The absence of significant associations might also be explained by a limited sample size or insufficient diversity in metadata factors. Future studies with a more balanced representation of phylogroups and broader environmental and clinical contexts may uncover nuanced relationships between genetic background and resistance patterns. This underscores the importance of expanding datasets to capture a more comprehensive view of how resistance determinants are distributed across E. coli populations.
This study has several limitations. First, the samples were obtained from necropsied farmed WTD, which may not fully represent the broader farmed or wild deer populations across Florida. The use of E. coli as an indicator organism, while informative, captures only the culturable fraction of the microbial community and may overlook additional ARGs present in unculturable taxa. Although whole-genome sequencing provided valuable genotypic insights, it has limited ability to resolve mobile genetic elements such as plasmids and transposons, which are central to AMR dissemination. Furthermore, discrepancies between genotypic predictions and phenotypic resistance highlight the need for functional validation beyond genomic data. Finally, the cross-sectional design and lack of detailed antimicrobial usage records restricted our ability to assess temporal dynamics or directly link farm management practices with resistance outcomes.
5 Conclusion and future directions
The detection of ARGs in E. coli isolated from farmed WTD in Florida underscores the presence of resistance to multiple antimicrobial classes. In total, 362 unique ARGs were identified, conferring resistance to 12 antimicrobial classes through 19 distinct mechanisms. Among the E. coli isolates, 30% (18/60) exhibited resistance to at least one antimicrobial agent under aerobic conditions, while 15% (9/60) demonstrated a MDR phenotype. Notably, five high-risk ARG groups—aac3, aph6, floR, mphA, and mphB—were found in high abundance, with mphB and floR particularly prevalent, comprising up to 13.8 and 16.3% of total ARG reads, respectively.
These findings highlight an urgent need for targeted management practices to limit disease transmission and mitigate the development and spread of antimicrobial resistance. Certain ARGs may serve as effective bioindicators for environmental health monitoring, resistance quantification, strategy evaluation, and predictive modeling of ARG distribution (Ishii, 2020). To address these risks, robust compartmentalization practices—including the implementation of barrier fencing—are critical for preventing direct and indirect transmission of ARGs between farmed WTD and surrounding free-ranging populations.
The high prevalence of resistance genes—particularly those associated with β-lactam antibiotics (49%) and MDR phenotypes (14.8%)—emphasizes the potential for horizontal gene transfer between wildlife and livestock. Routine surveillance of ARGs and pathogenic organisms in farmed WTD is essential to guide antimicrobial stewardship and anticipate emerging resistance trends. Monitoring key indicator genes such as sulI, sulII, aadA, bacA, oqxA, ermB, and mexE may provide early warnings of ARG proliferation (Zhao R. et al., 2018; Zhao Y. et al., 2018; Tarek and Garner, 2023).
Tracking the progression of resistance, particularly to drugs like ceftiofur and enrofloxacin—both associated with intermediate resistance phenotypes—can inform timely updates to treatment protocols. This study also confirmed strong genotype–phenotype agreement for several resistance genes, such as cmy, ctx, and tem for ampicillin; tetA, tetB, and tetD for tetracycline and oxytetracycline; aac3 for gentamicin and neomycin; and sulII, sulIII, and dfrA for sulfamethoxazole-trimethoprim. These genes should be prioritized in resistance monitoring due to their predictive power for phenotypic resistance.
Antimicrobial treatment in farmed WTD should be tailored to resistance profiles to ensure therapeutic efficacy and manage potential co-infections. Drugs with low genotype–phenotype agreement—such as ceftiofur, florfenicol, and enrofloxacin—may be more suitable for targeted use, whereas antimicrobials with high resistance rates and strong genotype–phenotype agreement (e.g., tetracycline, oxytetracycline, and ampicillin) should be avoided. Tetracycline and oxytetracycline exhibited the highest rates of resistance (26.2 and 29.5%, respectively), making them unsuitable for prophylactic or routine treatment on Florida’s WTD farms.
Although the absence of ZOI breakpoints for penicillin in the CLSI manual limited definitive conclusions, reference to prior literature suggests that approximately 95% of isolates would be classified as resistant—further supporting its exclusion from treatment protocols (Bughe et al., 2020). In summary, farmed WTD may serve as reservoirs for ARGs with the potential to impact both agricultural and ecological systems. By integrating ARG surveillance, antimicrobial susceptibility data, and tailored management strategies, stakeholders can more effectively control disease spread, limit resistance development, and safeguard both animal and environmental health.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/PRJNA1183872.
Ethics statement
The animal studies were approved by University of Florida Institutional Animal Care and Use Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
AS: Conceptualization, Data curation, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. A-CC: Investigation, Methodology, Resources, Writing – review & editing. MM: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. AB: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. TJ: Data curation, Investigation, Methodology, Writing – review & editing. FT: Data curation, Investigation, Methodology, Writing – review & editing. ED: Data curation, Investigation, Methodology, Writing – review & editing. JB: Project administration, Resources, Writing – review & editing. KJ: Investigation, Methodology, Project administration, Resources, Writing – review & editing. CB: Investigation, Methodology, Project administration, Resources, Writing – review & editing. JC-K: Investigation, Methodology, Project administration, Resources, Writing – review & editing. SW: Funding acquisition, Investigation, Methodology, Project administration, Writing – review & editing. KS: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the University of Florida, Cervidae Health Research Initiative, with funds provided by the State of Florida legislature.
Acknowledgments
We extend our gratitude to the Florida deer farms for providing specimens, all UF CHeRI necropsy technicians for conducting fieldwork, and the UF CVM Clinical Microbiology Diagnostic Laboratory for the bacterial pathogen identification.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abayasekara, L. M., Perera, J., Chandrasekharan, V., Gnanam, V. S., Udunuwara, N. A., Liyanage, D. S., et al. (2017). Detection of bacterial pathogens from clinical specimens using conventional microbial culture and 16S metagenomics: a comparative study. BMC Infect. Dis. 17, 1–11. doi: 10.1186/s12879-017-2362-9
Agersø, Y., and Aarestrup, F. M. (2013). Voluntary ban on cephalosporin use in Danish pig production has effectively reduced extended-spectrum cephalosporinase-producing Escherichia coli in slaughter pigs. J. Antimicrob. Chemother. 68, 569–572. doi: 10.1093/jac/dks427
Alonso, C. A., González-Barrio, D., Tenorio, C., Ruiz-Fons, F., and Torres, C. (2016). Antimicrobial resistance in faecal Escherichia coli isolates from farmed red deer and wild small mammals: detection of a multiresistant E. coli producing extended-spectrum beta-lactamase. Comp. Immunol. Microbiol. Infect. Dis. 45, 34–39. doi: 10.1016/j.cimid.2016.02.003
Anderson, D. P., Outlaw, J. L., Earle, M., and Richardson, J. W. (2017). Economic impact of US deer breeding and hunting operations : Texas A&M University Agricultural and Food Policy Center, 17–24.
Anjum, M. F., Schmitt, H., Börjesson, S., Berendonk, T. U., Donner, E., Stehling, E. G., et al. (2021). The potential of using E. coli as an indicator for the surveillance of antimicrobial resistance (AMR) in the environment. Curr. Opin. Microbiol. 64, 152–158. doi: 10.1016/j.mib.2021.09.011
Arnold, K. E., Williams, N. J., and Bennett, M. (2016). ‘Disperse abroad in the land’: the role of wildlife in the dissemination of antimicrobial resistance. Biol. Lett. 12:20160137. doi: 10.1098/rsbl.2016.0137
Azen, R., and Walker, C. M. (2021). Categorical data analysis for the behavioral and social sciences. New York, NY: Routledge.
Ballash, G. A., Dennis, P. M., Mollenkopf, D. F., Albers, A. L., Robison, T. L., Adams, R. J., et al. (2022). Colonization of white-tailed deer (Odocoileus virginianus) from urban and suburban environments with cephalosporinase- and carbapenemase-producing Enterobacterales. Appl. Environ. Microbiol. 88, e00465–e00422. doi: 10.1128/AEM.00465-22
Ballash, G. A., Munoz-Vargas, L., Albers, A., Dennis, P. M., LeJeune, J. T., Mollenkopf, D. F., et al. (2021). Temporal trends in antimicrobial resistance of fecal Escherichia coli from deer. EcoHealth 18, 288–296. doi: 10.1007/s10393-021-01559-3
Bashir, S., Haque, A., Sarwar, Y., Ali, A., and Anwar, M. I. (2012). Virulence profile of different phylogenetic groups of locally isolated community-acquired uropathogenic Escherichia coli from Faisalabad region of Pakistan. Ann. Clin. Microbiol. Antimicrob. 11, 1–6. doi: 10.1186/1476-0711-11-19
Begum, R., Sarker, M. S., Ngamsanga, P., Pulsrikarn, C., Pichpol, D., Meeyam, T., et al. (2018). Prevalence and antimicrobial resistance of Salmonella isolated from meat and eggs in Muang District in Chiang Mai Province, Thailand. In The 5th food safety and Zoonoses symposium for Asia Pacific, Chiang Mai, Thailand (6–7 July), p. 73–79.
Benveniste, R., and Davies, J. (1973). Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 70, 2276–2280. doi: 10.1073/pnas.70.8.2276
Berendonk, T. U., Manaia, C. M., Merlin, C., Fatta-Kassinos, D., Cytryn, E., Walsh, F., et al. (2015). Tackling antibiotic resistance: the environmental framework. Nat. Rev. Microbiol. 13, 310–317. doi: 10.1038/nrmicro3439
Bhullar, K., Waglechner, N., Pawlowski, A., Koteva, K., Banks, E. D., Johnston, M. D., et al. (2012). Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7:e34953. doi: 10.1371/journal.pone.0034953
Bonin, N., Doster, E., Worley, H., Pinnell, L. J., Bravo, J. E., Ferm, P., et al. (2023). MEGARes and AMR++, v3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res. 51, D744–D752. doi: 10.1093/nar/gkac1047
Boughton, R. K., Wight, B. R., Wisely, S., Hood, K., and Main, M. B. (2020). White-tailed deer of Florida: WEC133/UW121, rev. 04/2020. Edis 2020, 12.
Brooks, J. W., Wagner, D., Kime, L. F., Santini, E., Martin, K. F., Harper, J. K., et al. (2015). “White-tailed deer production” in PennState university agricultural alternatives. PennState College of Agricultural Sciences. EE0167 (State College, PA: The Pennsylvania State University).
Bruinsma, S., Burgess, J., Schlingman, D., Czyz, A., Morrell, N., Ballenger, C., et al. (2018). Bead-linked transposomes enable a normalization-free workflow for NGS library preparation. BMC Genomics 19, 1–16. doi: 10.1186/s12864-018-4603-4
Bughe, R. N., Oben, B. O., Nji, A. M., Chedjou, J. P., Mbange, A. E., Ali, I. M., et al. (2020). Occurrence and antibiotics susceptibility of Vibrio sp. from Penaeid shrimps from Kribi coastal water, Cameroon. Int. J. Adv. Res. Biol Sci 7, 96–111. doi: 10.22192/ijarbs.2020.07.07.012
Cacace, D., Fatta-Kassinos, D., Manaia, C. M., Cytryn, E., Kreuzinger, N., Rizzo, L., et al. (2019). Antibiotic resistance genes in treated wastewater and in the receiving water bodies: a pan-European survey of urban settings. Water Res., 162, 320–330. doi: 10.1016/j.watres.2019.06.039Chang D., Mao, Y., Qiu, W., Wu, Y., and Cai, B. 202 The source and distribution of tetracycline antibiotics in China: A review. Toxics, 11(3), 214. doi: 10.3390/toxics11030214
Chang, D., Mao, Y., Qiu, W., Wu, Y., and Cai, B. (2023). The source and distribution of tetracycline antibiotics in China: a review. Toxics 11:214. doi: 10.3390/toxics11030214
Chen, Z., Zhang, W., Yang, L., Stedtfeld, R. D., Peng, A., Gu, C., et al. (2019). Antibiotic resistance genes and bacterial communities in cornfield and pasture soils receiving swine and dairy manures. Environ. Pollut. 248, 947–957. doi: 10.1016/j.envpol.2019.02.093
Clermont, O., Bonacorsi, S., and Bingen, E. (2000). Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 66, 4555–4558. doi: 10.1128/AEM.66.10.4555-4558.2000
CLSI (2024). Performance standards for antimicrobial disk and dilution susceptibility tests for Bacteria isolated from animals. 7th Edn CLSI supplement VET015 (ISBN 978-1-68440-210-6 [Print]; ISBN 978-1-68440-211-3 [Electronic]): Clinical and Laboratory Standards Institute.
Conover, M. R. (2011). “Impacts of deer on society” in Biology and management of white-tailed deer. (Boca Raton, FL: CRC Press), 412–421.
Crofts, T. S., Gasparrini, A. J., and Dantas, G. (2017). Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 15, 422–434. doi: 10.1038/nrmicro.2017.28
Da Silva, G. J., and Mendonça, N. (2012). Association between antimicrobial resistance and virulence in Escherichia coli. Virulence 3, 18–28. doi: 10.4161/viru.3.1.18382
Dadi, B. R., Abebe, T., Zhang, L., Mihret, A., Abebe, W., and Amogne, W. (2020). Distribution of virulence genes and phylogenetics of uropathogenic Escherichia coli among urinary tract infection patients in Addis Ababa, Ethiopia. BMC Infect. Dis. 20, 1–12. doi: 10.1186/s12879-020-05309-5
Daghrir, R., and Drogui, P. (2013). Tetracycline antibiotics in the environment: a review. Environ. Chem. Lett. 11, 209–227. doi: 10.1007/s10311-013-0404-8
Daniels, J. B., Chen, L., Grooters, S. V., Mollenkopf, D. F., Mathys, D. A., Pancholi, P., et al. (2018). Enterobacter cloacae complex sequence type 171 isolates expressing KPC-4 carbapenemase recovered from canine patients in Ohio. Antimicrob. Agents Chemother. 62, 10–1128. doi: 10.1128/AAC.01028-18
D'Costa, V. M., King, C. E., Kalan, L., Morar, M., Sung, W. W. L., Schwarz, C., et al. (2011). Antibiotic resistance is ancient. Nature 477, 457–461. doi: 10.1038/nature10388
D'Costa, V. M., McGrann, K. M., Hughes, D. W., and Wright, G. D. (2006). Sampling the antibiotic resistome. Science 311, 374–377. doi: 10.1126/science.1120800
Dhariwal, A., Junges, R., Chen, T., and Petersen, F. C. (2021). ResistoXplorer: a web-based tool for visual, statistical and exploratory data analysis of resistome data. NAR Genom. Bioinformat. 3:lqab018. doi: 10.1093/nargab/lqab018
Dias, D., Fonseca, C., Caetano, T., and Mendo, S. (2022). Oh, deer! How worried should we be about the diversity and abundance of the faecal resistome of red deer? Sci. Total Environ. 825:153831. doi: 10.1016/j.scitotenv.2022.153831
Dias, D., Torres, R. T., Kronvall, G., Fonseca, C., Mendo, S., and Caetano, T. (2015). Assessment of antibiotic resistance of Escherichia coli isolates and screening of Salmonella spp. in wild ungulates from Portugal. Res. Microbiol. 166, 584–593. doi: 10.1016/j.resmic.2015.03.006
Dubois, D., Delmas, J., Cady, A., Robin, F., Sivignon, A., Oswald, E., et al. (2010). Cyclomodulins in urosepsis strains of Escherichia coli. J. Clin. Microbiol. 48, 2122–2129. doi: 10.1128/JCM.02365-09
Durso, L. M., and Cook, K. L. (2014). Impacts of antibiotic use in agriculture: what are the benefits and risks? Curr. Opin. Microbiol. 19, 37–44. doi: 10.1016/j.mib.2014.05.019
Ejrnæs, K., Stegger, M., Reisner, A., Ferry, S., Monsen, T., Holm, S. E., et al. (2011). Characteristics of Escherichia coli causing persistence or relapse of urinary tract infections: phylogenetic groups, virulence factors and biofilm formation. Virulence 2, 528–537. doi: 10.4161/viru.2.6.18189
Ellington, M. J., Ekelund, O., Aarestrup, F. M., Canton, R., Doumith, M., Giske, C., et al. (2017). The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST subcommittee. Clin. Microbiol. Infect. 23, 2–22. doi: 10.1016/j.cmi.2016.11.012
Fauzia, K. A., Alfaray, R. I., and Yamaoka, Y. (2023). Advantages of whole genome sequencing in mitigating the Helicobacter pylori antimicrobial resistance problem. Microorganisms 11:1239. doi: 10.3390/microorganisms11051239
Giguère, S., Lee, E., Williams, E., Cohen, N. D., Chaffin, M. K., Halbert, N., et al. (2010). Determination of the prevalence of antimicrobial resistance to macrolide antimicrobials or rifampin in Rhodococcus equi isolates and treatment outcome in foals infected with antimicrobial-resistant isolates of R. equi. J. Am. Vet. Med. Assoc. 237, 74–81. doi: 10.2460/javma.237.1.74
Greig, T. W., Bemiss, J. A., Lyon, B. R., Bossart, G. D., and Fair, P. A. (2007). Prevalence and diversity of antibiotic-resistant Escherichia coli in bottlenose dolphins (Tursiops truncatus) from the Indian River lagoon, Florida, and Charleston Harbor area, South Carolina. Aquat. Mamm. 33:185. doi: 10.1578/AM.33.2.2007.185
Guitor, A. K., Raphenya, A. R., Klunk, J., Kuch, M., Alcock, B., Surette, M. G., et al. (2019). Capturing the resistome: a targeted capture method to reveal antibiotic resistance determinants in metagenomes. Antimicrob. Agents Chemother. 64, 10–1128. doi: 10.1128/AAC.01073-19
Gutierrez, A., Jaysankar, D. E., and Schneider, K. R. (2020). Prevalence, concentration, and antimicrobial resistance profiles of Salmonella isolated from Florida poultry litter. J. Food Prot. 83, 2179–2186. doi: 10.4315/JFP-20-215
Haider, N., Rothman-Ostrow, P., Osman, A. Y., Arruda, L. B., Macfarlane-Berry, L., Elton, L., et al. (2020). COVID-19—zoonosis or emerging infectious disease? Front. Public Health 8:596944. doi: 10.3389/fpubh.2020.596944
Hasan, B., Melhus, Å., Sandegren, L., Alam, M., and Olsen, B. (2014). The gull (Chroicocephalus brunnicephalus) as an environmental bioindicator and reservoir for antibiotic resistance on the coastlines of the bay of Bengal. Microb. Drug Resist. 20, 466–471. doi: 10.1089/mdr.2013.0233
He, Y., Yuan, Q., Mathieu, J., Stadler, L., Senehi, N., Sun, R., et al. (2020). Antibiotic resistance genes from livestock waste: occurrence, dissemination, and treatment. NPJ Clean Water 3:4. doi: 10.1038/s41545-020-0051-0
Hemati, S., Halimi, S., Jabalameli, F., Emaneini, M., and Beigverdi, R. (2024). Phylogenetic group, antibiotic resistance, virulence gene, and genetic diversity of Escherichia coli causing bloodstream infections in Iran. Front. Microbiol. 15:1426510. doi: 10.3389/fmicb.2024.1426510
Hewitt, D. G. (2015). Hunters and the conservation and management of white-tailed deer (Odocoileus virginianus). Int. J. Environ. Stud. 72, 839–849. doi: 10.1080/00207233.2015.1073473
Huang, F. Y., An, X. L., Chen, Q. L., Ren, H. Y., and Su, J. Q. (2016). Distribution characteristics of antibiotic resistance genes in sika deer farm. Huan Jing Ke Xue =. Huanjing Kexue 37, 4402–4409. doi: 10.13227/j.hjkx.2016.11.043
Ishii, S. (2020). Quantification of antibiotic resistance genes for environmental monitoring: current methods and future directions. Curr. Opin. Environ. Sci. Health 16, 47–53. doi: 10.1016/j.coesh.2020.02.004
Lakin, S. M., Dean, C., Noyes, N. R., Dettenwanger, A., Ross, A. S., Doster, E., et al. (2017). MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 45, D574–D580. doi: 10.1093/nar/gkw1009
Lantz, D. E. (1908). Deer farming in the United States (No. 330). Washington, DC: U.S. Government Printing Office.
Lee, J. H., Subhadra, B., Son, Y. J., Kim, D. H., Park, H. S., Kim, J. M., et al. (2016). Phylogenetic group distributions, virulence factors and antimicrobial resistance properties of uropathogenic Escherichia coli strains isolated from patients with urinary tract infections in South Korea. Lett. Appl. Microbiol. 62, 84–90. doi: 10.1111/lam.12517
Li, Q., Sherwood, J. S., and Logue, C. M. (2007). Characterization of antimicrobial resistant Escherichia coli isolated from processed bison carcasses. J. Appl. Microbiol. 103, 2361–2369. doi: 10.1111/j.1365-2672.2007.03470.x
Lima, T., Domingues, S., and Da Silva, G. J. (2020). Manure as a potential hotspot for antibiotic resistance dissemination by horizontal gene transfer events. Vet. Sci. 7:110. doi: 10.3390/vetsci7030110
Lugli, G. A., Milani, C., Mancabelli, L., Turroni, F., Ferrario, C., Duranti, S., et al. (2017). Ancient bacteria of the Ötzi’s microbiome: a genomic tale from the copper age. Microbiome 5, 1–18. doi: 10.1186/s40168-017-0284-6
Marquis, D. A., and Brenneman, R. (1981). The impact of deer on forest vegetation in Pennsylvania. Gen. Tech. Rep. NE-65, vol. 7. Broomall, PA: US Department of Agriculture, Forest Service, northeastern Forest Experimental Station, 65.
Martinez, J. L. (2009). Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 157, 2893–2902. doi: 10.1016/j.envpol.2009.05.051
Mateus-Vargas, R. H., Atanassova, V., Reich, F., and Klein, G. (2017). Antimicrobial susceptibility and genetic characterization of Escherichia coli recovered from frozen game meat. Food Microbiol. 63, 164–169. doi: 10.1016/j.fm.2016.11.013
Mathys, D. A., Mollenkopf, D. F., Feicht, S. M., Adams, R. J., Albers, A. L., Stuever, D. M., et al. (2019). Carbapenemase-producing Enterobacteriaceae and Aeromonas spp. present in wastewater treatment plant effluent and nearby surface waters in the US. PLoS One 14:e0218650. doi: 10.1371/journal.pone.0218650
Mayrhofer, S., Paulsen, P., Smulders, F. J., and Hilbert, F. (2006). Antimicrobial resistance in commensal Escherichia coli isolated from muscle foods as related to the veterinary use of antimicrobial agents in food-producing animals in Austria. Microb. Drug Resist. 12, 278–283. doi: 10.1089/mdr.2006.12.278
McDermott, P. F., Tyson, G. H., Kabera, C., Chen, Y., Li, C., Folster, J. P., et al. (2016). Whole-genome sequencing for detecting antimicrobial resistance in nontyphoidal Salmonella. Antimicrob. Agents Chemother. 60, 5515–5520. doi: 10.1128/aac.01030-16
Metrailer, M. C., Cheng, A. C., Bluhm, A. P., Jiranantasak, T., Norris, M. H., Surphlis, A., et al. (2024). Phylogeographic patterns of Escherichia coli isolated from white-tailed deer, Odocoileus virginianus, during necropsy in a farmed deer surveillance program. bioRxiv. doi: 10.1101/2024.11.21.624738v1
Mollenkopf, D. F., Mathys, D. A., Feicht, S. M., Stull, J. W., Bowman, A. S., Daniels, J. B., et al. (2018). Maintenance of carbapenemase-producing Enterobacteriaceae in a farrow-to-finish swine production system. Foodborne Pathog. Dis. 15, 372–376. doi: 10.1089/fpd.2017.2355
Monroy-Pérez, E., Cerón, A. B., García Cortés, L. R., Alonso, N. N., Domínguez-Trejo, P., Hernández-Jaimes, T., et al. (2020). Virulence gene transcription, phylogroups, and antibiotic resistance of cervico-vaginal pathogenic E. coli in Mexico. PLoS One 15:e0234730. doi: 10.1371/journal.pone.0234730
Munkhdelger, Y., Gunregjav, N., Dorjpurev, A., Juniichiro, N., and Sarantuya, J. (2017). Detection of virulence genes, phylogenetic group and antibiotic resistance of uropathogenic Escherichia coli in Mongolia. J. Infect. Dev. Ctries. 11, 51–57. doi: 10.3855/jidc.7903
Muurinen, J., Stedtfeld, R., Karkman, A., Pärnänen, K., Tiedje, J., and Virta, M. (2017). Influence of manure application on the environmental resistome under Finnish agricultural practice with restricted antibiotic use. Environ. Sci. Technol. 51, 5989–5999. doi: 10.1021/acs.est.7b00551
National Deer Association. (2022). Economic, Social, and Conservation Benefits of Deer Hunting in the Southern United States. Available online at: https://deerassociation.com/wp-content/uploads/2022/09/SDP-Research-Report-2022-04-19.pdf (Accessed August 22, 2024).
Nnadozie, C. F., and Odume, O. N. (2019). Freshwater environments as reservoirs of antibiotic resistant bacteria and their role in the dissemination of antibiotic resistance genes. Environ. Pollut. 254:113067. doi: 10.1016/j.envpol.2019.113067
Nyirabahizi, E., Tyson, G. H., Dessai, U., Zhao, S., Kabera, C., Crarey, E., et al. (2020). Evaluation of Escherichia coli as an indicator for antimicrobial resistance in Salmonella recovered from the same food or animal ceca samples. Food Control 115:107280. doi: 10.1016/j.foodcont.2020.107280
Pais, S., Costa, M., Barata, A. R., Rodrigues, L., Afonso, I. M., and Almeida, G. (2022). Evaluation of antimicrobial resistance of different phylogroups of Escherichia coli isolates from feces of breeding and laying hens. Antibiotics 12:20. doi: 10.3390/antibiotics12010020
Paniagua-Contreras, G. L., Monroy-Pérez, E., Rodríguez-Moctezuma, J. R., Domínguez-Trejo, P., Vaca-Paniagua, F., and Vaca, S. (2017). Virulence factors, antibiotic resistance phenotypes and O-serogroups of Escherichia coli strains isolated from community-acquired urinary tract infection patients in Mexico. J. Microbiol. Immunol. Infect. 50, 478–485. doi: 10.1016/j.jmii.2015.08.005
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Paun, V. I., Lavin, P., Chifiriuc, M. C., and Purcarea, C. (2021). First report on antibiotic resistance and antimicrobial activity of bacterial isolates from 13,000-year old cave ice core. Sci. Rep. 11:514. doi: 10.1038/s41598-020-79754-5
Pereira, M. B., Wallroth, M., Jonsson, V., and Kristiansson, E. (2018). Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics 19, 274–217. doi: 10.1186/s12864-018-4637-6
Picard, B., Garcia, J. S., Gouriou, S., Duriez, P., Brahimi, N., Bingen, E., et al. (1999). The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 67, 546–553. doi: 10.1128/IAI.67.2.546-553.1999
Pinto, L., Radhouani, H., Coelho, C., Martins da Costa, P., Simões, R., Brandão, R. M., et al. (2010). Genetic detection of extended-spectrum β-lactamase-containing Escherichia coli isolates from birds of prey from Serra da Estrela natural reserve in Portugal. Appl. Environ. Microbiol. 76, 4118–4120. doi: 10.1128/AEM.02761-09
Plaza-Rodríguez, C., Alt, K., Grobbel, M., Hammerl, J. A., Irrgang, A., Szabo, I., et al. (2021). Wildlife as sentinels of antimicrobial resistance in Germany? Front. Vet. Sci. 7:627821. doi: 10.3389/fvets.2020.627821
Projan, S. J. (2002). New (and not so new) antibacterial targets–from where and when will the novel drugs come? Curr. Opin. Pharmacol. 2, 513–522. doi: 10.1016/s1471-4892(02)00197-2
Qian, X., Gu, J., Sun, W., Wang, X. J., Su, J. Q., and Stedfeld, R. (2018). Diversity, abundance, and persistence of antibiotic resistance genes in various types of animal manure following industrial composting. J. Hazard. Mater. 344, 716–722. doi: 10.1016/j.jhazmat.2017.11.020
Raimondi, S., Righini, L., Candeliere, F., Musmeci, E., Bonvicini, F., Gentilomi, G., et al. (2019). Antibiotic resistance, virulence factors, phenotyping, and genotyping of Escherichia coli isolated from the feces of healthy subjects. Microorganisms 7:251. doi: 10.3390/microorganisms7080251
Rogers, S. W., Shaffer, C. E., Langen, T. A., Jahne, M., and Welsh, R. (2018). Antibiotic-resistant genes and pathogens shed by wild deer correlate with land application of residuals. EcoHealth 15, 409–425. doi: 10.1007/s10393-018-1316-7
Sadovska, D., Nodieva, A., Pole, I., Vīksna, A., Ķimsis, J., Ozere, I., et al. (2024). Discordance between phenotypic and WGS-based drug susceptibility testing results for some anti-tuberculosis drugs: a snapshot study of paired Mycobacterium tuberculosis isolates with small genetic distance. Infection and Drug Resistance. 17, 3289–3307. doi: 10.2147/IDR.S468997
Schwan, C. L., Lomonaco, S., Bastos, L. M., Cook, P. W., Maher, J., Trinetta, V., et al. (2021). Genotypic and phenotypic characterization of antimicrobial resistance profiles in non-typhoidal Salmonella enterica strains isolated from Cambodian informal markets. Front. Microbiol. 12:711472. doi: 10.3389/fmicb.2021.711472
Skarżyńska, M., Leekitcharoenphon, P., Hendriksen, R. S., Aarestrup, F. M., and Wasyl, D. (2020). A metagenomic glimpse into the gut of wild and domestic animals: quantification of antimicrobial resistance and more. PLoS One 15:e0242987. doi: 10.1371/journal.pone.0242987
Sy-Trias, C. M. (2023). Laboratory practices and antimicrobial resistance in a Florida Hospital (Doctoral dissertation: Walden University.
Tagami, Y., Horita, N., Kaneko, M., Muraoka, S., Fukuda, N., Izawa, A., et al. (2024). Whole-genome sequencing predicting phenotypic antitubercular drug resistance: meta-analysis. J. Infect. Dis. 229, 1481–1492. doi: 10.1093/infdis/jiad480
Tarek, M. H., and Garner, E. (2023). A proposed framework for the identification of indicator genes for monitoring antibiotic resistance in wastewater: insights from metagenomic sequencing. Sci. Total Environ. 854:158698. doi: 10.1016/j.scitotenv.2022.158698
Tohmaz, M., Askari Badouei, M., Kalateh Rahmani, H., and Hashemi Tabar, G. (2022). Antimicrobial resistance, virulence-associated genes and phylogenetic background versus plasmid replicon types: the possible associations in avian pathogenic Escherichia coli (APEC). BMC Vet. Res. 18:421. doi: 10.1186/s12917-022-03496-x
Touchon, M., Perrin, A., De Sousa, J. A. M., Vangchhia, B., Burn, S., O’Brien, C. L., et al. (2020). Phylogenetic background and habitat drive the genetic diversification of Escherichia coli. PLoS Genet. 16:e1008866. doi: 10.1371/journal.pgen.1008866
Turchi, B., Dec, M., Bertelloni, F., Winiarczyk, S., Gnat, S., Bresciani, F., et al. (2019). Antibiotic susceptibility and virulence factors in Escherichia coli from sympatric wildlife of the Apuan Alps regional park (Tuscany, Italy). Microb. Drug Resist. 25, 772–780. doi: 10.1089/mdr.2018.0191
Van den Bogaard, A. E., and Stobberingh, E. E. (2000). Epidemiology of resistance to antibiotics: links between animals and humans. Int. J. Antimicrob. Agents 14, 327–335. doi: 10.1016/S0924-8579(00)00145-X
Vittecoq, M., Godreuil, S., Prugnolle, F., Durand, P., Brazier, L., Renaud, N., et al. (2016). Antimicrobial resistance in wildlife. J. Appl. Ecol. 53, 519–529. doi: 10.1111/1365-2664.12596
Warren, R. J. (1991). Ecological justification for controlling deer populations in eastern national parks. In Transactions of the North American Wildlife and Natural Resources Conference 56, 56–66.
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2020). Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2:e000143. doi: 10.1099/acmi.0.000143
Weinmaier, T., Conzemius, R., Bergman, Y., Lewis, S., Jacobs, E. B., Tamma, P. D., et al. (2023). Validation and application of long-read whole-genome sequencing for antimicrobial resistance gene detection and antimicrobial susceptibility testing. Antimicrob. Agents Chemother. 67:e0107222. doi: 10.1128/aac.01072-22
White, F. H., and Forrester, D. J. (1979). Antimicrobial resistant Salmonella spp. isolated from double-crested cormorants (Phalacrocorax auritus) and common loons (Gavia immer) in Florida. J. Wildl. Dis. 15, 235–237. doi: 10.7589/0090-3558-15.2.235
White, D. G., Hudson, C., Maurer, J. J., Ayers, S., Zhao, S., Lee, M. D., et al. (2000). Characterization of chloramphenicol and florfenicol resistance in Escherichia coli associated with bovine diarrhea. J. Clin. Microbiol. 38, 4593–4598. doi: 10.1128/JCM.38.12.4593-4598.2000
Williams, N. J., Sherlock, C., Jones, T. R., Clough, H. E., Telfer, S. E., Begon, M., et al. (2011). The prevalence of antimicrobial-resistant Escherichia coli in sympatric wild rodents varies by season and host. J. Appl. Microbiol. 110, 962–970. doi: 10.1111/j.1365-2672.2011.04952.x
Witney, A. A., Cosgrove, C. A., Arnold, A., Hinds, J., Stoker, N. G., and Butcher, P. D. (2016). Clinical use of whole genome sequencing for Mycobacterium tuberculosis. BMC Med. 14, 1–7. doi: 10.1186/s12916-016-0641-x
Wong, F., Stokes, J. M., Bening, S. C., Vidoudez, C., Trauger, S. A., and Collins, J. J. (2022). Reactive metabolic byproducts contribute to antibiotic lethality under anaerobic conditions. Mol. Cell 82, 3499–3512. doi: 10.1016/j.molcel.2022.07.009
Woolhouse, M. E., and Ward, M. J. (2013). Sources of antimicrobial resistance. Science 341, 1460–1461. doi: 10.1126/science.1243444
Wright, G. D. (2007). The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5, 175–186. doi: 10.1038/nrmicro1614
Wu, N., Xie, S., Zeng, M., Xu, X., Li, Y., Liu, X., et al. (2020). Impacts of pile temperature on antibiotic resistance, metal resistance, and microbial community during swine manure composting. Sci. Total Environ. 744:140920. doi: 10.1016/j.scitotenv.2020.140920
Zhang, A. N., Gaston, J. M., Dai, C. L., Zhao, S., Poyet, M., Groussin, M., et al. (2021). An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12:4765. doi: 10.1038/s41467-021-25096-3
Zhao, R., Feng, J., Yin, X., Liu, J., Fu, W., Berendonk, T. U., et al. (2018). Antibiotic resistome in landfill leachate from different cities of China deciphered by metagenomic analysis. Water Res. 134, 126–139. doi: 10.1016/j.watres.2018.01.063
Zhao, R., Shi, J., Shen, Y., Li, Y., Han, Q., Zhang, X., et al. (2015). Phylogenetic distribution of virulence genes among ESBL-producing uropathogenic Escherichia coli isolated from long-term hospitalized patients. J. Clin. Diagn. Res. 9, DC01–DC06. doi: 10.7860/JCDR/2015/13234.6157
Zhao, Y., Su, J. Q., An, X. L., Huang, F. Y., Rensing, C., Brandt, K. K., et al. (2018). Feed additives shift gut microbiota and enrich antibiotic resistance in swine gut. Sci. Total Environ. 621, 1224–1232. doi: 10.1016/j.scitotenv.2017.10.106
Zhou, Z. C., Feng, W. Q., Han, Y., Zheng, J., Chen, T., Wei, Y. Y., et al. (2018). Prevalence and transmission of antibiotic resistance and microbiota between humans and water environments. Environ. Int. 121, 1155–1161. doi: 10.1016/j.envint.2018.10.032
Zhou, J., He, Z., Yang, Y., Deng, Y., Tringe, S. G., and Alvarez-Cohen, L. (2015). High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. mBio 6:e02288-14. doi: 10.1128/mBio.02288-14
Zhu, Y. G., Zhao, Y. I., Li, B., Huang, C. L., Zhang, S. Y., Yu, S., et al. (2017). Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2, 1–7. doi: 10.1038/nmicrobiol.2017.57
Keywords: antimicrobial resistance, resistome, white-tailed deer, Escherichia coli, whole genome-sequencing, antimicrobial resistance genes, drug resistance
Citation: Surphlis AC, Cheng A-C, Metrailer MC, Bluhm AP, Jiranantasak T, Tuozzo F II, Doster E, Blackburn JK, Jeong KC, Boucher C, Campos-Krauer JM, Wisely SM and Subramaniam K (2025) Characterizing the resistome of Escherichia coli isolated from farmed white-tailed deer (Odocoileus virginianus) in Florida, United States. Front. Microbiol. 16:1681096. doi: 10.3389/fmicb.2025.1681096
Edited by:
Magdalena Popowska, University of Warsaw, PolandReviewed by:
Leonardo Gabriel Panunzi, CEA Saclay, FranceEmoke Pall, University of Agricultural Sciences and Veterinary Medicine of Cluj-Napoca, Romania
Copyright © 2025 Surphlis, Cheng, Metrailer, Bluhm, Jiranantasak, Tuozzo, Doster, Blackburn, Jeong, Boucher, Campos-Krauer, Wisely and Subramaniam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuttichantran Subramaniam, a3V0dGljaGFudHJhbkB1ZmwuZWR1