 Chanjuan Huangfu
Chanjuan Huangfu Xiujun Ma2†
Xiujun Ma2† Kuo Han
Kuo Han Ti Liu
Ti Liu- 1School of Public Health and Health Management, Shandong First Medical University, Jinan, China
- 2Shandong Centre for Disease Control and Prevention, Jinan, China
- 3Weifang Centre for Disease Control and Prevention, Weifang, China
Introduction: The recent identification of Brucella abortus in human clinical samples from Shandong, China, highlights an ongoing zoonotic threat.
Methods: We characterized 12 B. abortus strains isolated from human patients since 2021 using a combination of conventional biotyping, Multiple Locus Variable-number Tandem Repeat Analysis (MLVA), and core-genome SNP (cgSNP) analysis.
Results: Epidemiological data indicated that infections primarily occurred in middle-aged men with occupational livestock exposure. Molecular typing revealed biovar 3 as the predominant type (91.7%), dominated by MLVA-8 genotype 36 and its corresponding MLVA-11 genotype 72 (66.7%). MLVA-16 distinguished 12 unique genotypes. The phylogeny based on cgSNP classified the strains into clades C1 (11 bv. 3 strains) and C2 (one bv. 1 strain). Within clade C1, nine strains in subclade C1-III exhibited ≤119 SNP differences, eight of which formed a local clonal transmission chain (≤52 SNPs) and shared MLVA-11 genotype 72. Subclade C1-I contained two strains with novel genotypes resulting from variations at the Bruce18 and Bruce43 loci. The sole C2 strain differed by only 3 SNPs from the A19 vaccine strain, suggesting a potential vaccine-related origin. Genetic links were also identified with strains from other Chinese provinces, among them Heilongjiang and Inner Mongolia, as well as from several countries, including Mongolia and Russia.
Discussion: These findings revealed a complex epidemiological pattern in Shandong, primarily characterized by local transmission chains with occasional external introductions, provided a scientific basis for targeted brucellosis control strategies.
1 Introduction
Brucellosis, caused by the genus Brucella, is a globally significant bacterial zoonosis and is listed as a priority for control by the Food and Agriculture Organization (FAO) and the World Health Organization (WHO) (Cutler et al., 2005). In China, human brucellosis remains a major public health problem, causing substantial economic losses and threatening both physical and mental health. The prevention and control of brucellosis is a national strategic priority. The Ministry of Agriculture and Rural Affairs included it in its plan for infectious diseases requiring control and eradication in 2012. Furthermore, the Chinese government launched a five-year control action plan in 2022, underscoring the urgency of its prevention and control efforts. Shandong Province, as a major livestock production region in China, serves as a crucial frontline defence in the national control of brucellosis. The province has reported a persistently high number of human cases in recent years, with an annual average exceeding 2,700 (Yu et al., 2023). Between 2021 and 2024, cumulative reported cases reached 13,096, with the annual incidence rate consistently maintaining a high level between 3.13 per 100,000 and 3.32 per 100,000. This presents a severe and persistent challenge to local public health authorities.
Brucella spp. are pathogenic bacteria with cross-host adaptability, capable of spreading among natural hosts through direct or indirect contact and infecting other susceptible animals (Pappas et al., 2006; De Massis et al., 2019). Among the 12 known Brucella species, the B.melitensis, B.abortus, and B.suis pose the greatest threat to human health, but their pathogenicity varies. B.melitensis and B.suis strains are the most virulent, while B.abortus has relatively milder pathogenicity (Alton and Jones, 1967). Cattle brucellosis is primarily caused by B. abortus, leading to abortion, infertility, and reduced milk production in cattle, and causing significant economic losses to farmers by reducing livestock productivity and reproductive capacity (Tian et al., 2024). In humans, infection manifests as irregular fever, arthritis, and other clinical symptoms (Dean et al., 2012). Through biotyping schemes, B. abortus has been classified into seven distinct biovars. Based on multi-locus sequence typing (MLST-21) research, B. abortus can be divided into three major evolutionary clades globally. The early-branching clade A/B strains are entirely of African origin, while the widely distributed clade C is further divided into two major clades, C1 and C2, which represent the main drivers of recent global spread of B. abortus (Whatmore et al., 2016). Among these, C1 is highly associated with bv. 3 and is primarily distributed across the Eurasian continent; C2 is dominated by bv. 1 and exhibits global widespread distribution. This MLST-based clade framework has become a crucial foundation for studying the population genetics and geographical distribution of this bacterial species.
Although the macro-epidemiological profile of human brucellosis in Shandong Province has been outlined, the molecular characteristics of the causative Brucella pathogens remain largely unexplored (Yu et al., 2023). Specifically, in contrast to the relatively well-studied B. melitensis, data on the population structure, genetic diversity, and transmission dynamics of B. abortus isolates from human clinical cases in this region are strikingly scarce (Li et al., 2024). This lack of high-resolution molecular data impedes a thorough understanding of the origins of local outbreaks and hinders the development of targeted control strategies.
Effective control of brucellosis relies on precise monitoring and high-resolution methods to identify sources of infection and transmission pathways. Although MLST provides an important foundation for population studies, its limited resolution still limits its application in molecular epidemiology. MLVA provides higher resolution and standardized typing schemes by detecting copy number variations in tandem repeat loci, making it suitable for tracking regional transmission chains. However, its resolution remains limited in precisely tracing the origin and transmission of outbreak strains. Advances in molecular technology have driven the development of typing methods toward genome-based technologies. In recent years, molecular typing methods based on WGS technology (such as cgSNP and wgSNP) have become the mainstream direction due to their ability to provide the highest resolution of genotypic information and their applicability to detailed genetic evolution and transmission studies (Holzer et al., 2021; Edao et al., 2023).
Therefore, this study investigated the molecular epidemiology of B. abortus from human cases in Shandong Province between 2021 and 2024. We used MLVA-16 and cgSNP analysis to investigate the genetic characteristics, population structure, and geographical distribution of clinical isolates. This work provides the first comprehensive molecular dataset for B. abortus in the region, offering scientific evidence to improve surveillance and enable targeted interventions for brucellosis control in Shandong and throughout China.
2 Materials and methods
2.1 Strain sources and patient inclusion criteria
All Brucella isolates in this study were obtained from the human brucellosis surveillance system in Shandong Province, China, between 2021 and 2024. Strain inclusion was based on the following criteria: isolates were derived from patients meeting the confirmed case definition of brucellosis according to the Chinese Health Industry Standard (WS 269–2019), which requires a clear epidemiological history, typical clinical manifestations, a positive Rose Bengal plate test (RBT), and a standard tube agglutination test (SAT) titer of ≥1:100++ (China’s National Health Commission, 2019).
Blood samples from eligible patients were cultured using biphasic blood culture bottles. Blood culture bottles that signaled positive but from which no Brucella colonies could be isolated on Columbia blood agar plates, or that were identified as non-Brucella species, were excluded from the study. Samples that yielded no visible bacterial growth on the solid agar phase were excluded. Culture bottles with visible growth were transported under standardized protocols by local CDC Centres to the Shandong Provincial CDC for further identification and bacterial isolation. All isolates were confirmed as B. abortus by the provincial reference laboratory through biochemical profiling, PCR assays, and molecular typing. Following this protocol, a total of 12 clinical B. abortus isolates were successfully collected and included in this study.
2.2 Brucella isolation and DNA extraction
After enrichment of 5–10 mL blood samples in blood culture bottles, a 100 μL aliquot of the enriched culture was inoculated onto Columbia blood agar plates and incubated at 37 °C under 5% CO₂ for 48–72 h. Typical Brucella colonies, characterized by being moist, round, slightly raised, smooth, and opalescent, were observed on Columbia blood agar plates (Supplementary Figure S1). Brucella colonies were subcultured to obtain pure isolates. A single colony was suspended in 200 μL of sterile physiological saline and inactivated at 80 °C for 90 min. The genomic DNA, extracted with the TIANamp Bacterial DNA Kit (TianGen, Beijing, China), was used for subsequent molecular identification and whole-genome sequencing.
2.3 Identification and typing of strains
Phenotypic characterization and biotyping were performed using standard Brucella identification methods according to the Chinese Health Industry Standard (China’s National Health Commission, 2019). The CO₂ requirement was assessed by comparing bacterial growth on Columbia blood agar after 48 h of incubation at 37 °C under both aerobic and 5% CO₂ conditions. Slide agglutination tests were carried out using monospecific antisera against A, M, and R antigens to evaluate antigenic reactivity. Dye inhibition susceptibility was determined using discs impregnated with thionin (20 μg/mL) and basic fuchsin (20 μg/mL); inhibition zones were recorded following incubation at 37 °C under 5% CO₂ for 48 h.
Molecular identification of all 12 isolates was performed on the extracted DNA using the BCSP31-PCR assay for genus confirmation (Fekete et al., 1990), and AMOS-PCR for species and biovar discrimination (Bricker and Halling, 1994). The primer sequences, detailed PCR reaction mixtures, and thermal cycling conditions used in this study are provided in Supplementary Table S1. The amplified products were analyzed by 1.5% agarose gel electrophoresis and imaging (Supplementary Figure S2).
2.4 Whole genome sequencing
Following molecular identification, the genomic DNA of 12 isolates was subjected to paired-end sequencing (PE150) on the Illumina NovaSeq X Plus platform. The raw data output for each sample exceeded 1 Gb, with an average sequencing depth of 100×. Raw reads were first evaluated comprehensively using FastQC v0.11.9 for quality assessment. Subsequently, Fastp v0.21.0 was employed for strict quality control and filtering to generate high-quality clean data along with relevant quality metrics. De novo genome assembly was performed using SPAdes v3.15.5 based on the cleaned reads. The assembly quality was evaluated using key assembly metrics, including N50, N90, GC content, and number of scaffolds. The genome assembly metrics for all 12 isolates are summarized in Supplementary Table S2.
2.5 Core genome SNP genotyping
A core-genome phylogenetic analysis was conducted using the Snippy v4.6.0. Following alignment of all isolate sequences to the B. abortus 2308 reference genome (GenBank accession numbers NC_007618 and NC_007624), a high-quality core-genome alignment was generated.
2.6 Phylogenetic and diversity analysis
Molecular characterization was performed based on whole-genome sequencing data. The assembled genomes of the 12 isolates were imported into CLC Genomics Workbench 23.0.3 (QIAGEN, Germany) for MLVA-16 analysis. Using standard Brucella MLVA primer sequences as probes, target loci were identified and extracted from the whole genomes. The copy numbers of tandem repeats at the 16 VNTR loci were precisely determined by comparing the amplified fragment lengths with the known sizes of repeat units. The resulting MLVA-16 profiles were analyzed using BioNumerics 8.0 (Applied Maths, Belgium). An Unweighted Pair Group Method with Arithmetic Mean (UPGMA) dendrogram was constructed by clustering the study isolates with 112 B. abortus strains originating from China. In addition, a Minimum Spanning Tree (MST) was generated incorporating the study isolates and 286 B. abortus strains isolated globally since 2015.
For higher-resolution evolutionary analysis, a cgSNP phylogenetic tree was constructed. The 12 Shandong isolates were analyzed together with 390 globally representative B. abortus reference genomes obtained from the NCBI database. The core-genome alignment and SNP extraction were performed using the workflow described in Section 2.5, yielding a core genome SNP alignment file. A maximum-likelihood (ML) phylogenetic tree was constructed from this alignment using MEGA X (Mega Limited, New Zealand), with nodal support assessed by 1,000 bootstrap replicates. To enhance the clarity and phylogenetic resolution of the tree, strains within the same clade that exhibited identical core-genome sequences were filtered out. This process resulted in a refined, high-quality dataset comprising the 12 local Shandong isolates and 141 globally representative strains, which was used to infer the final SNP-based phylogeny.
2.7 Geographic information mapping
The geographic distribution of the studied strains was visualized using ArcGIS 10.8 (ESRI, USA). The map was projected in the WGS 1984 geographic coordinate system. Geographic coordinates (longitude and latitude) for each isolate, representing the residential locations of the patients they originated from, were obtained from epidemiological records. The resulting point layer map clearly displays the locations of the 12 isolates, with points symbolized according to key attributes such as biovar, MLVA genotype, and year of isolation.
3 Results
3.1 Epidemiological and clinical characteristics of patients infected with Brucella abortus
Epidemiological characteristics were analyzed for 12 laboratory-confirmed human cases of B. abortus infection identified in Shandong Province between 2021 and 2024 (Table 1). Most patients were male (9/12, 75.0%) with a median age of 48 years (range: 25–58); the majority (10/12, 83.3%) were between 40 and 59 years old. The annual number of cases increased over time, with one case each in 2021 and 2022, and five cases in both 2023 and 2024. Geographically, cases were distributed across seven cities (Figure 1), with the highest number reported in Weifang (4/12, 33.3%), followed by Dezhou and Dongying (2 cases each, 16.7%), and single cases in Linyi, Liaocheng, Rizhao, and Yantai. Occupational exposure was the primary transmission route (11/12, 91.7%), predominantly through cattle or sheep rearing (9 cases). Only one case was attributed to foodborne transmission. The most characteristic clinical symptoms were arthralgia (91.7%), fever (75.0%), and hyperhidrosis (66.7%), consistent with the classic presentation of brucellosis.
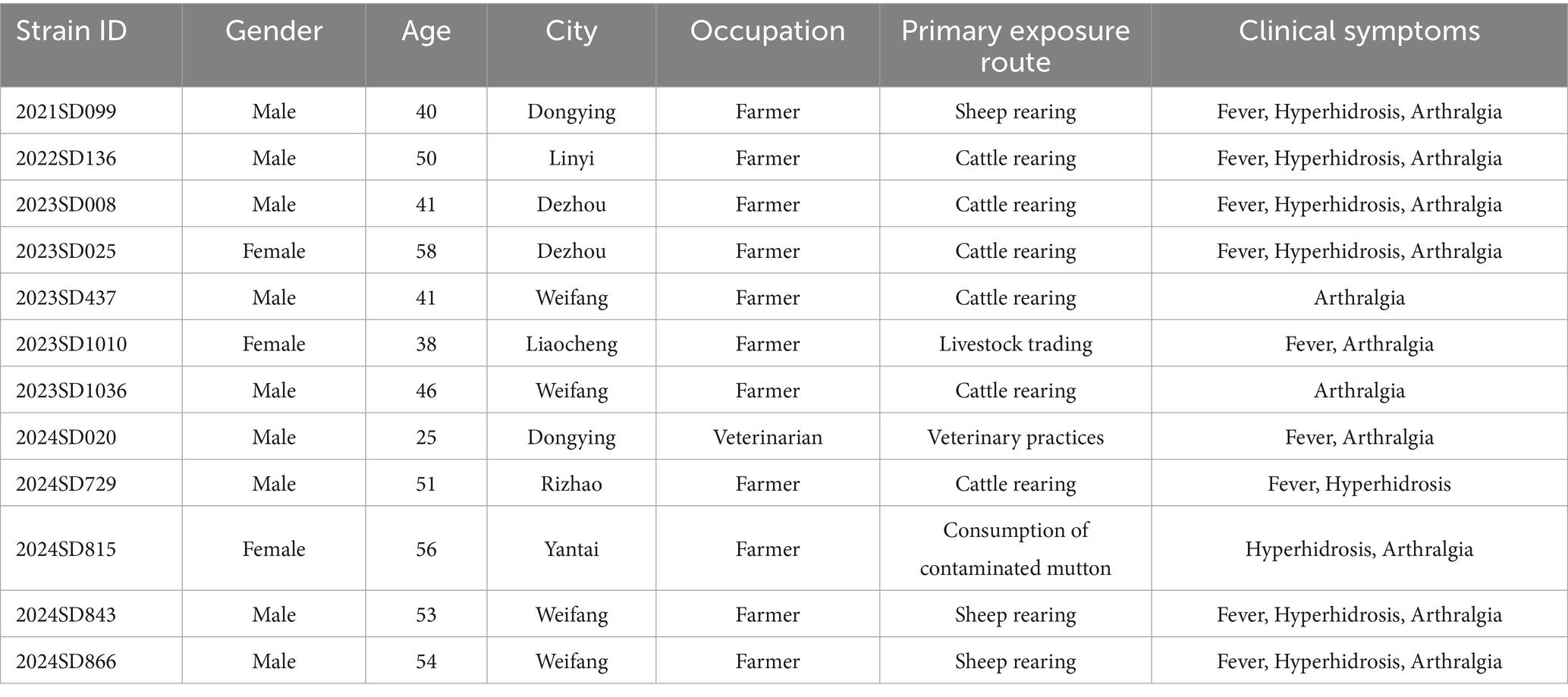
Table 1. Epidemiological and clinical characteristics of patients infected with B. abortus in Shandong Province, China.
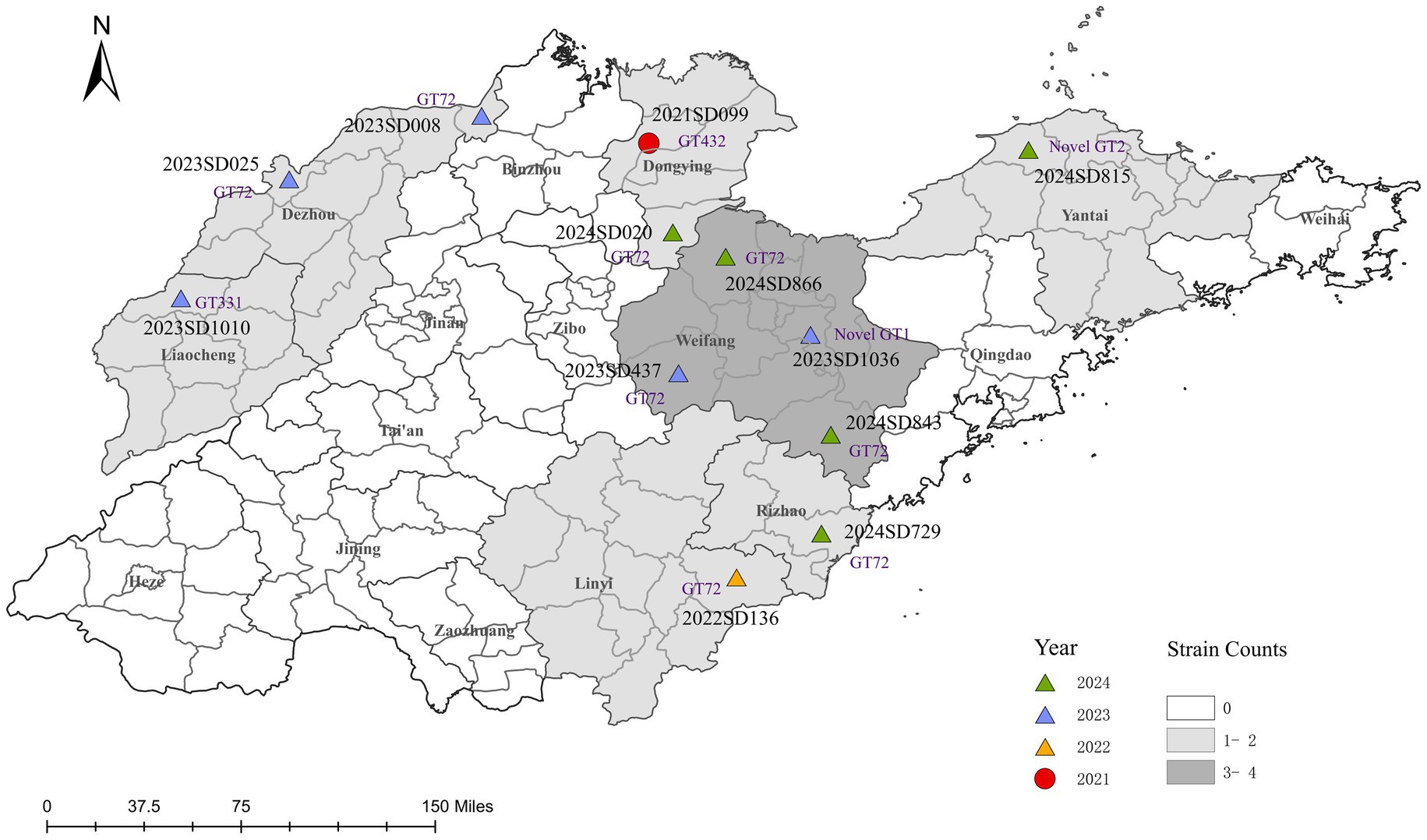
Figure 1. Spatiotemporal distribution of clinical B. abortus isolates from human cases in Shandong Province, China, 2021–2024. The map shows the geographical distribution of 12 B. abortus clinical isolates. Points are colored according to the year of isolation and shaped according to the biovar. Each point is labeled with the corresponding strain ID and its MLVA-11 genotype (novel genotypes are denoted as Novel GT1 and Novel GT2). The background colour of prefecture-level administrative regions is graded according to the total number of isolates collected. The base map was created using ArcGIS 10.8 software.
3.2 Strain identification and biovar distribution
Through traditional phenotypic identification combined with BCSP31-PCR and AMOS-PCR molecular identification, it was confirmed that all 12 isolates analyzed in this study were B. abortus. (Supplementary Figure S2). Biotyping revealed a clear temporal and geographical pattern (Figure 1). Only one isolate, recovered from Dongying in 2021, was identified as B. abortus bv. 1. The remaining eleven isolates (91.7%), collected from the other six cities between 2022 and 2024, were all identified as bv. 3, confirming it as the predominant biovar. To date, no B. abortus strains have been reported from the other nine prefecture-level cities in Shandong Province.
3.3 Results of MLVA-16 genotyping
MLVA genotyping analysis was performed on 12 strains of B. abortus in Shandong. The results showed that among the MLVA-8 genotypes, 11 strains (91.7%) were known genotypes, with genotype 36 (4-5-3-12-2-2-3-1) being the predominant genotype, accounting for 8 strains (66.7%). Other genotypes included 112 (n = 1), 116 (n = 1), and 235 (n = 1). One strain (2024SD729, Yantai) was identified as a novel genotype due to variations in the number of repeats at the Bruce43 locus. In MLVA-11 typing, 10 strains (83.3%) were known genotypes, with genotype 72 (corresponding to genotype 36 in MLVA-8) also accounting for 66.7% (8/12). Other genotypes included genotype 331 (n = 1) and genotype 432 (n = 1). Two strains (2023SD1036, Weifang; 2024SD815, Yantai) were identified as novel strains due to variations in the number of repeats at the Bruce18 locus. MLVA-16 further improved resolution, with 12 strains identified as 12 unique genotypes, demonstrating high genetic diversity among the strains.
UPGMA clustering analysis was performed on 112 representative strains of B. abortus from other regions of China downloaded from MLVAbank (Figure 2; Supplementary Table S3). One strain (2023SD437, Weifang) shared the same MLVA-16 genotype with three domestic strains (two from Hebei). One strain (2021SD099, Dongying) shared the same MLVA-16 genotype with one A19 strain isolated domestically. The remaining 10 Shandong strains had independent MLVA-16 genotypes and did not share any with other domestic strains. In the domestic database, genotype 36 was the predominant type, with genotypes 112, 116, and 235 also present, among which 235 was the least common.

Figure 2. UPGMA cluster analysis of B. abortus isolates from China based on MLVA-16 genotyping. The dendrogram was constructed using UPGMA, analyzing the genetic relationships among 124 strains. This set included 12 isolates from this study and 112 reference strains from various regions across China. The tree illustrates the distribution of the Shandong isolates within the national genetic context.
The MLVA-16 data from this study were analyzed using MST with global strains from the MLVAbank database since 2015 (Figure 3; Supplementary Table S3). Ten strains from Shandong (MLVA-8: 8 strains of genotype 36, 1 strain of genotype 116, and 1 strain of a novel genotype) were distributed in the clade C1 and clustered with strains from Kazakhstan and other countries. The remaining 2 strains (MLVA-8: 1 strain of genotype 112 and 1 strain of genotype 235) were distributed in the clade C2, with the genotype 112 strain clustering most closely with strains from Qinghai, China, and the genotype 235 strain exhibiting the closest genetic affinity to strains from South Africa.
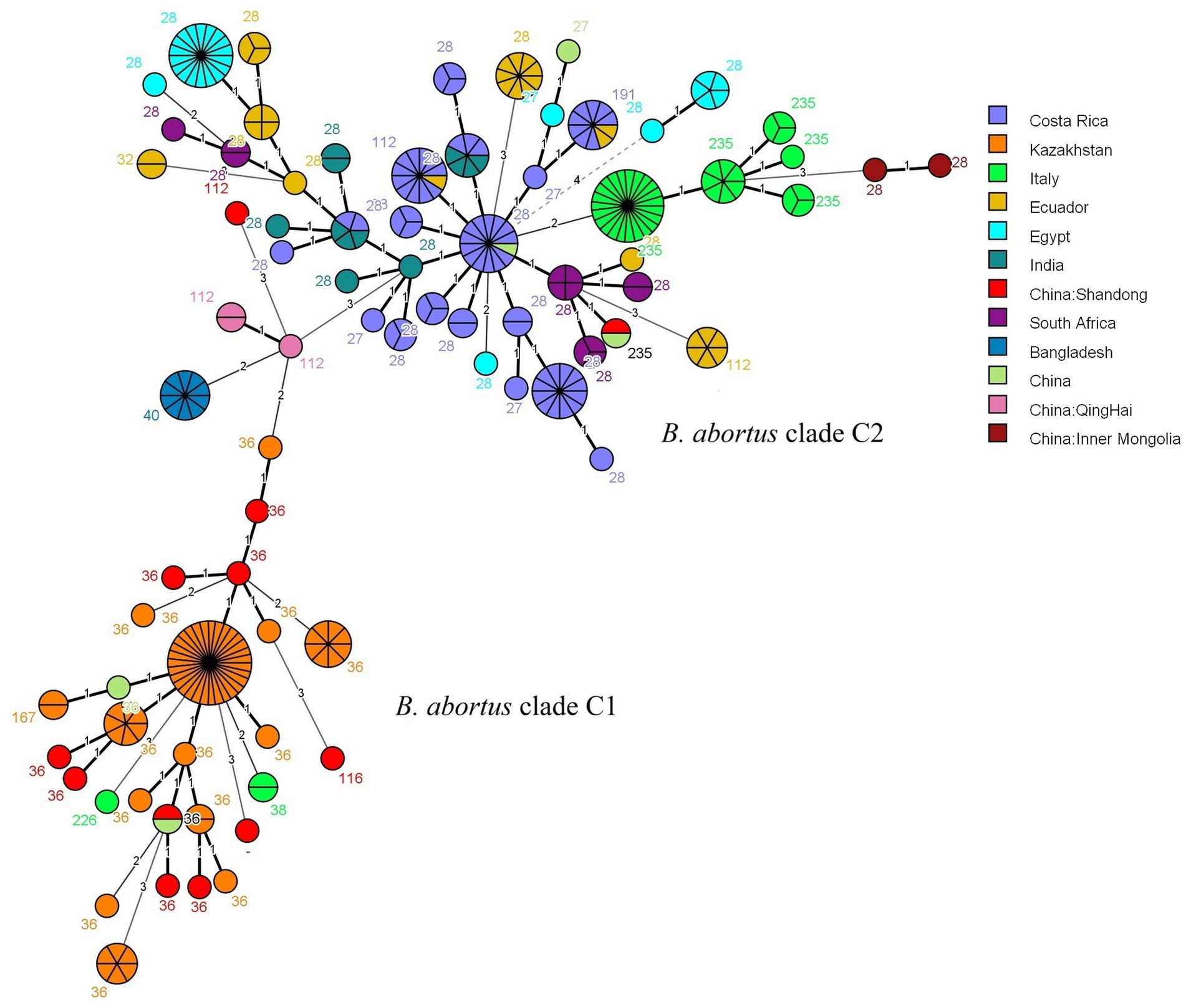
Figure 3. MST of B. abortus isolates from worldwide based on MLVA-16 genotyping. The analysis included 286 strains: 12 isolates from this study (highlighted in red) and 274 global reference strains collected since 2015. The reference strains are coloured by their country of origin. Based on the MLST typing information available in the MLVAbank database for the reference strains, the two main clusters are labeled as the B. abortus clade C1 and B. abortus clade C2 evolutionary clades to indicate the phylogenetic position of the Shandong isolates. Each node represents a unique MLVA-16 genotype, with its size proportional to the number of isolates. The number adjacent to each node corresponds to its MLVA-8 genotype code.
3.4 cgSNP analysis results
The MST analysis based on MLVA-16 initially divided the strains into two main clusters. According to the MLST typing information of reference strains in the MLVAbank database, these two clusters were correspondingly labeled as the C1 and C2 evolutionary clades of B. abortus; the subsequent cgSNP phylogenetic analysis adhered to this clade framework and performed higher-resolution analysis within it. To validate and further resolve this population structure, cgSNP analysis was performed based on WGS data from the 12 Shandong isolates and global B. abortus reference strains from NCBI, and a phylogenetic tree was constructed (Figure 4). The results showed that all strains were clearly divided into two major clades, C1 and C2. Clade C1 included 11 Shandong isolates (all bv. 3), which were further divided into three subclades: C1-I, C1-II, and C1-III. Subclade C1-I contained two Shandong strains (2023SD1036, Weifang; 2024SD815, Yantai), which differed by 186 SNPs. These two strains were classified as novel genotypes by MLVA-11 typing and clustered with a Tibetan yak-derived strain isolated in Tibet, China, in 2016 (SNP differences: 323 and 345, respectively). Their closest foreign relative was a cattle-derived strain from Greece (SNP differences: 310 and 332, respectively), forming a cluster with Italian and Russian strains. Subclade C1-III included 75% of Shandong strains (9/12), with 1 strain from 2022, 4 strains from 2023, and 4 strains from 2024. The maximum pairwise SNP distance among these nine strains was less than 119, which, in the context of the global phylogeny, indicates a close genetic relationship. Among them, the Liaocheng strain 2023SD1010 was genetically closest to the Russian human-derived strain from 2012, with only 87 SNP differences. The remaining 8 strains were genetically closest to human- or cattle-derived strains from Heilongjiang, Ningxia, Hebei, Inner Mongolia, and Gansu provinces in China, with SNP differences within 66 SNPs, and also showed close phylogenetic relationships with cattle-derived strains from Mongolia. Notably, the Linyi strain (2022SD136) and Rizhao strain (2024SD729) differed by only 3 SNPs, suggesting they may represent the same clone or share a common infection source. Subclade C1-II was not detected among the Shandong isolates in this study.
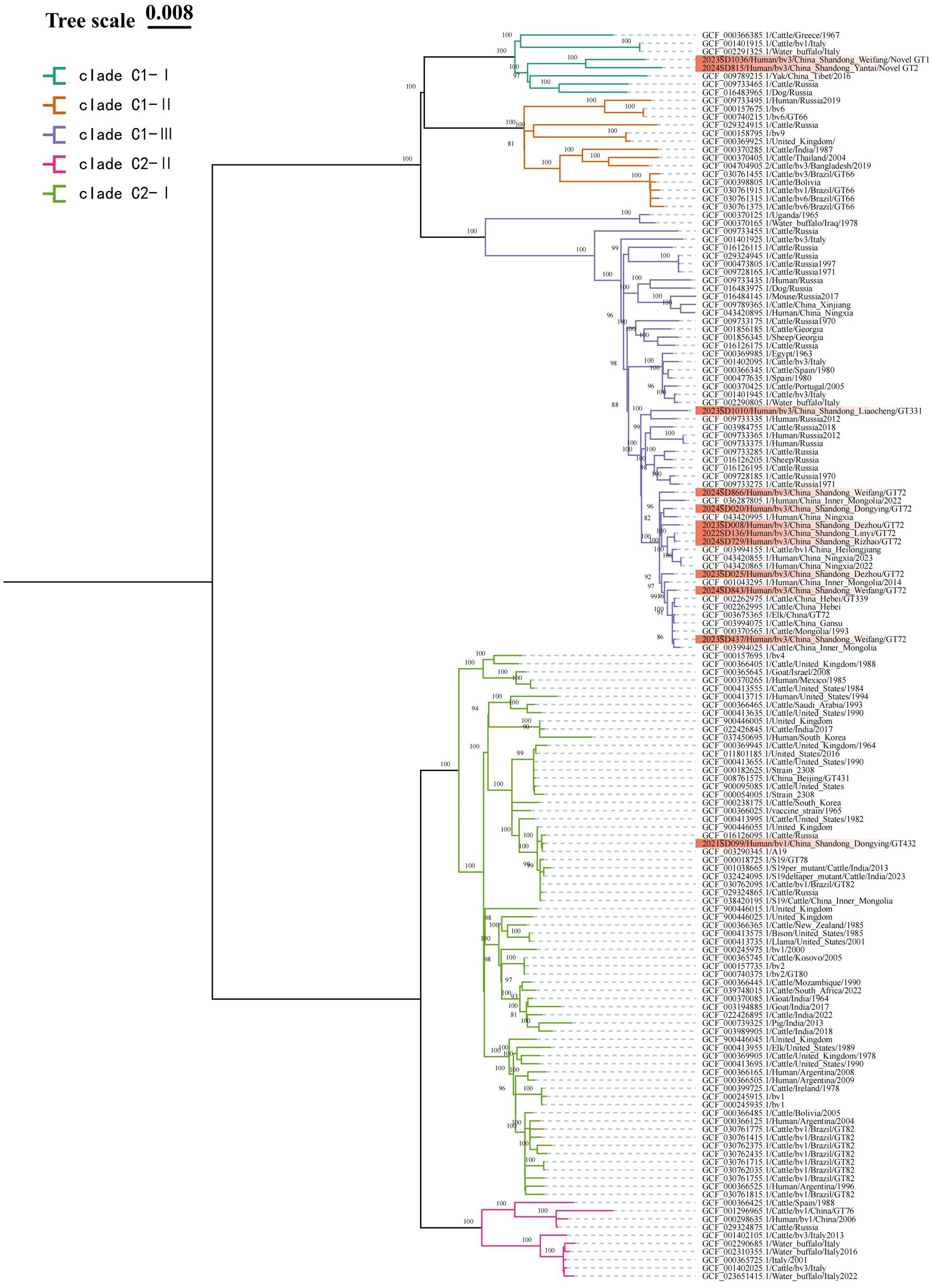
Figure 4. ML phylogenetic tree of B. abortus strains based on cgSNP. To enhance phylogenetic resolution, strains within the same clade possessing identical core-genome sequences are filtered out. The final dataset used for phylogenetic reconstruction consist of 12 local Shandong isolates (highlighted in red) and 141 globally representative strains. Nodal support is assessed with 1,000 bootstrap replicates.
Clade C2 contained only one Shandong isolate (2021SD099, bv. 1, Dongying), which further branches into two subclades: C2-I, and C2-II. This Shandong strain resides within C2-I, differing from the A19 vaccine strain by only 3 SNPs, indicating extremely close genetic proximity. Its SNP differences with other vaccine strains (such as S19 and 2308) are also within 30 sites. In the phylogenetic tree, strains clustered around this isolate were predominantly bv. 1 strains. Subclade C2-II was not detected among the Shandong isolates in this study.
4 Discussion
This study provided the first systematic analysis of the epidemiological patterns of B. abortus derived from human cases in Shandong Province. By integrating epidemiological and molecular genotyping data, we revealed that its transmission dynamics are characterized by a core of local transmission chains, along with extensive genetic linkages to strains from other Chinese provinces and internationally. These findings provide key molecular epidemiological evidence for enhanced disease surveillance and precise source tracing in the region. In 2021, B. abortus was first isolated from human cases in Shandong, and a total of 12 isolates have been obtained to date. This number is significantly lower than that of B. melitensis isolates, consistent with previous studies indicating that B. abortus accounts for a very low proportion of human brucellosis outbreaks, and that B. melitensis remains the dominant species in human cases in Asian countries such as Iran (Vergnaud et al., 2018; Khan et al., 2021; Dadar et al., 2023). This phenomenon may be attributed to the relatively lower potential virulence of B. abortus compared to B. melitensis (Kneipp et al., 2019).
All 12 patients with B. abortus infection in this study had clear epidemiological exposure histories, indicating a primarily local transmission pattern. Patients were predominantly male (75.0%) and aged 40–59 years (83.3%), consistent with the demographics of livestock workers (Liang et al., 2020). In terms of exposure routes, except for one foodborne case, all other cases were related to occupational contact with livestock, including nine cases of family farming, one veterinarian, and one livestock trader. Our findings indicated multiple pathways of transmission in Shandong Province: family farming serves as the main transmission route, infection in a veterinarian reflects occupational risk, livestock trading may expand the spread, and the foodborne case indicates a potential threat in the local food chain. These findings are consistent with the known transmission routes of brucellosis through direct contact or consumption of contaminated animal products (Gwida et al., 2010; Pappas, 2010). The main clinical symptoms were arthralgia (91.7%), fever (75.0%), and hyperhidrosis (66.7%), matching the typical manifestations of brucellosis. Spatiotemporal analysis showed a recent clustering trend, with 83.3% (10/12) of isolates obtained between 2023 and 2024, and 33.3% (4/12) of cases concentrated in Weifang. This clustering suggested an active local transmission chain in Weifang and surrounding areas. The developed livestock industry and high-density cattle and sheep farming in this region are likely contributing factors. High-density farming increases contact among animals, raising the likelihood of Brucella spread within herds, and thereby increasing the risk of human exposure to the source of infection (Yu et al., 2023).
Strain identification revealed that 91.7% (11/12) of the isolates were B. abortus bv. 3, consistent with the global predominance of this biovar (Ma et al., 2020). To unravel the genetic characteristics, we employed MLVA, a key technique in Brucella molecular epidemiology that effectively identifies subtle genetic variations among strains and is suitable for analyzing transmission chains at regional and even global levels (Wareth et al., 2020; Holzer et al., 2022). In recent years, the in silico MLVA typing based on whole-genome sequences has also been widely adopted. MLVA analysis revealed the genetic characteristics of the Shandong strains at multiple loci (Dadar and Alamian, 2025). On one hand, a widely disseminated dominant clone was identified, with genotype 36 (MLVA-8) and its corresponding genotype 72 (MLVA-11) being predominant (8/12, 66.7%). This genotype has been persistently prevalent in China since the 1980s and has also been reported in Eurasian countries such as Kazakhstan and Portugal, suggesting close genetic links between the Shandong epidemic strains and Eurasian strains (Ferreira et al., 2012; Jiang et al., 2013; Shevtsov et al., 2015). On the other hand, the population exhibited high diversity. Higher-resolution MLVA-16 distinguished the 12 strains into 12 unique genotypes. Furthermore, novel genotypes were detected arising from variations in the number of repeats at the Bruce43 and Bruce18 loci. These sites were previously considered highly stable in Chinese B. melitensis; their mutation suggests ongoing region-specific microevolution within the local endemic population (Kattar et al., 2008; Jiang et al., 2011; Mirkalantari et al., 2021).
Clustering analyses based on domestic and international strains provided multidimensional insights into the origins and genetic background of B. abortus in Shandong Province (Sayour et al., 2020). UPGMA clustering using a domestic strain database showed that Shandong strains possessed both unique and shared characteristics with strains from other regions. Most isolates (10/12, 83.3%) exhibited distinct MLVA-16 genotypes, indicating a significant local genetic background. However, a few strains shared genotypes with isolates from other provinces; for example, the Weifang strain 2023SD437 shared an identical genotype with two strains from Hebei, suggesting inter-regional transmission. The Dongying strain 2021SD099 matched a domestic A19 vaccine strain, providing a direction for further traceback investigation. MST analysis based on global MLVA data further clarified the population structure of Shandong strains on a broader evolutionary scale. All strains clustered within clade C, which could be divided into two subclades, C1 and C2. Subclade C1 was the dominant group, comprising ten bv. 3 strains. These strains formed a tight cluster with isolates from Kazakhstan and other countries, indicating close genetic relationships. This genetic connection was likely driven by the combined effects of geographical proximity and historical ruminant trade along routes akin to the grassland silk road, factors jointly known to facilitate the regional spread of Brucella (Liu et al., 2020; Shevtsov et al., 2023). In contrast, subclade C2 contained only two Shandong strains but demonstrated a complex genetic background, with members originating from various regions in China, such as Inner Mongolia and Qinghai, as well as other countries including South Africa and Italy. This pattern suggests a history of transregional or even international transmission for this subclade. The presence of the rare bv. 1 strain in Shandong is of particular importance for surveillance and early warning.
Phylogenetic analysis based on cgSNP provided higher-resolution validation and refinement of the genetic structure revealed by MST. Both methods classified the Shandong strains into two primary lineages, clade C1 and clade C2, with no detection of clade A or B. This population structure is consistent with findings from studies in Kazakhstan and other Asian countries. However, minor discrepancies in the classification of individual strains were observed between the two methods (MST: C1 = 10, C2 = 2; cgSNP: C1 = 11, C2 = 1), underscoring the superior resolution of cgSNP and highlighting the necessity and complementarity of high-resolution methods in molecular epidemiological investigations. cgSNP analysis confirmed clade C1 as the dominant lineage. The nine B. abortus bv. 3 strains within this clade exhibited a maximum pairwise SNP distance of less than 119, indicating their close genetic relatedness within the context of the global phylogeny and suggesting descent from a common ancestor. Notably, eight strains from Liaocheng and other areas showed ≤52 SNP differences and shared the MLVA-11 genotype 72, strongly supporting their belonging to an active clonal transmission chain (Jackson et al., 2016). This clone showed close phylogenetic relationships with strains from Heilongjiang and Inner Mongolia in China, as well as from Mongolia and Russia, implying a possible common regional origin. This pattern of regional transmission is consistent with findings from Sichuan Province, where Brucella strains frequently share genetic links with those from neighboring provinces and countries (Li et al., 2025). The sole B. abortus bv. 1 strain was unambiguously assigned to clade C2 by cgSNP analysis. This strain differed from the standard A19 vaccine strain by only 3 SNPs. When combined with its MLVA-16 profile and the patient’s occupation as a livestock farmer, these findings suggested that the strain is probably related to the A19 vaccine, likely resulting from exposure during vaccination procedures. This finding underscored that although the A19 vaccine has been widely and safely used in China since the 1940s, strict adherence to biosafety protocols by personnel is essential to prevent accidental infection (Wang et al., 2020; Zhou et al., 2022).
Our results clearly demonstrated the complementary value of MLVA and cgSNP typing techniques. MLVA is effective for the initial identification of dominant genotypes and potential transmission clusters on a regional scale (Zhao et al., 2021), such as the successful identification of the genotype 36 clonal group widely distributed across the province. In contrast, cgSNP provides higher resolution for analyzing finer genetic variation, offering superior capability in confirming genetic relatedness within clones, enabling precise traceback, and correcting biases inherent in conventional typing methods (Holzer et al., 2021; Kydyshov et al., 2022; Zhu et al., 2024). For instance, although the Linyi strain 2023SD136 and the Rizhao strain 2024SD729 had different MLVA-16 genotypes, they showed only 3 SNP differences, suggesting they likely originated from the same transmission event and providing conclusive evidence of inter-city spread within the province. Furthermore, one strain initially classified into C2 by MST analysis was reassigned to clade C1 based on cgSNP, more accurately reflecting its true genetic ancestry. The two novel MLVA-11 genotypes were independently distributed within subclade C1-I in the cgSNP analysis, confirming them as genuine genetic variants potentially representing either new introductions or local evolutionary events. Therefore, combining the broad screening capacity of MLVA with the high-resolution traceback capability of cgSNP provided a more comprehensive molecular epidemiological basis for elucidating pathogen transmission dynamics (Li et al., 2024).
5 Conclusion
This study presented the first comprehensive molecular epidemiological investigation of B. abortus in Shandong Province, China, by integrating high-resolution MLVA-16 and cgSNP analysis. Our results confirmed the dominant prevalence of B. abortus bv. 3 and revealed the sporadic occurrence of a bv. 1 strain genetically related to the A19 vaccine strain, indicating the coexistence of natural transmission and potential vaccine-related events. cgSNP analysis provided high-resolution genetic insights, revealing closely related clusters among the predominant bv. 3 strains. This finding indicated the presence of localized transmission chains within the broader context of sporadic distribution, which is crucial for understanding the micro-evolution and spread of B. abortus in endemic regions.
The successful application of the combined MLVA-cgSNP approach offered a powerful strategy for bacterial genotyping and traceback investigations. From a public health perspective, the genetic linkages identified between Shandong strains and those from other provinces and countries underscore the transboundary nature of brucellosis transmission. This highlighted the necessity for enhanced regional collaboration and coordinated cross-jurisdictional control measures. Furthermore, the detection of a vaccine-like strain emphasized the importance of continuous monitoring and prudent vaccine management in the field.
In summary, this study filled a critical knowledge gap regarding the molecular epidemiology of B. abortus in Shandong Province. The findings provided a scientific foundation for evidence-based prevention and control strategies, contributing to the optimization of brucellosis surveillance networks and the development of more effective public health interventions in China, ultimately aiming to reduce the disease burden in both animal and human populations.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1310245.
Ethics statement
This study was approved by the Ethics Committee of Shandong Provincial Center for Disease Control and Prevention (Approval no. SDJK(K)2025-065-01).
Author contributions
CH: Conceptualization, Validation, Writing – original draft. XM: Conceptualization, Methodology, Writing – original draft. JF: Formal analysis, Software, Writing – original draft. KH: Investigation, Validation, Writing – original draft. TL: Data curation, Visualization, Writing – original draft. ZK: Funding acquisition, Project administration, Writing – review & editing. YL: Funding acquisition, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Shandong Provincial Medical and Health Science and Technology Project (grant number 202412061299) and the Shandong Traditional Chinese Medicine Science and Technology Project (grant number Q-2022091).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1695815/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Typical colony morphology of Brucella abortus on Columbia blood agar after 48-72 hours of incubation.
SUPPLEMENTARY FIGURE S2 | Gel electrophoresis results of BCSP31-PCR and AMOS-PCR for the identification and biotyping of Brucella isolates.
SUPPLEMENTARY TABLE S1 | Oligonucleotide primers, PCR reaction mixtures, and thermal cycling conditions used in this study. This table is provided as a separate Excel file (Supplementary Table S1.xlsx) containing the following three sheets: Sheet 1 (Primers): Oligonucleotide primers used in this study. Sheet 2 (Reaction_Mixture): Detailed PCR reaction components and their final concentrations. Sheet 3 (Cycling_Conditions): Thermal cycling parameters for both PCR assays.
SUPPLEMENTARY TABLE S2 | Quality metrics and assembly statistics for the whole-genome sequencing data. The detailed content of Table S2 is provided as a separate Excel file (Supplementary Table S2.xlsx).
SUPPLEMENTARY TABLE S3 | Complete MLVA-16 profiles for the strains analyzed in this study. Sheet 1 (UPGMA): Contains the Shandong isolates and Chinese reference strains used for UPGMA analysis (related to Figure 2 in the main text). Sheet 2 (MST): Contains the Shandong isolates and global reference strains used for MST analysis (related to Figure 3 in the main text). The detailed content is provided as a separate Excel file (Supplementary Table S3.xlsx).
SUPPLEMENTARY TABLE S4 | Molecular and genomic characteristics of Brucella abortus strains from human cases in Shandong Province, China (2021-2024). The detailed content of Table S4 is provided as a separate Excel file (Supplementary Table S4.xlsx). The table includes strain identifiers, genomic accession numbers, geographical origins, collection years, biovar classification, and complete MLVA genotyping profiles.
References
Alton, G. G., and Jones, L. M. (1967). Laboratory techniques in brucellosis. Monogr. Ser. World Health Organ. 55, 1–92.
Bricker, B. J., and Halling, S. M. (1994). Differentiation of Brucella abortus bv. 1, 2, and 4, Brucella melitensis, Brucella ovis, and Brucella suis bv. 1 by PCR. J. Clin. Microbiol. 32, 2660–2666. doi: 10.1128/jcm.32.11.2660-2666.1994
China’s National Health Commission (2019). Diagnosis for brucellosis: WS 269–2019. Beijing: China Standards Press.
Cutler, S. J., Whatmore, A. M., and Commander, N. J. (2005). Brucellosis--new aspects of an old disease. J. Appl. Microbiol. 98, 1270–1281. doi: 10.1111/j.1365-2672.2005.02622.x
Dadar, M., and Alamian, S. (2025). In silico MLVA analysis of Brucella melitensis from human and livestock in Iran. Curr. Microbiol. 82:74. doi: 10.1007/s00284-024-03940-1
Dadar, M., Brangsch, H., Alamian, S., Neubauer, H., and Wareth, G. (2023). Whole-genome sequencing for genetic diversity analysis of Iranian Brucella spp. isolated from humans and livestock. One Health 16:100483. doi: 10.1016/j.onehlt.2023.100483
De Massis, F., Zilli, K., Di Donato, G., Nuvoloni, R., Pelini, S., Sacchini, L., et al. (2019). Distribution of Brucella field strains isolated from livestock, wildlife populations, and humans in Italy from 2007 to 2015. PLoS One 14:e0213689. doi: 10.1371/journal.pone.0213689
Dean, A. S., Crump, L., Greter, H., Hattendorf, J., Schelling, E., and Zinsstag, J. (2012). Clinical manifestations of human brucellosis: a systematic review and meta-analysis. PLoS Negl. Trop. Dis. 6:e1929. doi: 10.1371/journal.pntd.0001929
Edao, B. M., Ameni, G., Berg, S., Tekle, M., Whatmore, A. M., Wood, J. L. N., et al. (2023). Whole genome sequencing of Ethiopian Brucella abortus isolates expands the known diversity of an early branching sub-Saharan African lineage. Front. Microbiol. 14:1128966. doi: 10.3389/fmicb.2023.1128966
Fekete, A., Bantle, J. A., Halling, S. M., and Sanborn, M. R. (1990). Preliminary development of a diagnostic test for Brucella using polymerase chain reaction. J. Appl. Bacteriol. 69, 216–227. doi: 10.1111/j.1365-2672.1990.tb01512.x
Ferreira, A. C., Chambel, L., Tenreiro, T., Cardoso, R., Flor, L., Dias, I. T., et al. (2012). MLVA16 typing of Portuguese human and animal Brucella melitensis and Brucella abortus isolates. PLoS One 7:e42514. doi: 10.1371/journal.pone.0042514
Gwida, M., Al Dahouk, S., Melzer, F., Rösler, U., Neubauer, H., and Tomaso, H. (2010). Brucellosis - regionally emerging zoonotic disease? Croat. Med. J. 51, 289–295. doi: 10.3325/cmj.2010.51.289
Holzer, K., El-Diasty, M., Wareth, G., Abdel-Hamid, N. H., Hamdy, M. E. R., Moustafa, S. A., et al. (2021). Tracking the distribution of Brucella abortus in Egypt based on Core genome SNP analysis and in silico MLVA-16. Microorganisms 9:1942. doi: 10.3390/microorganisms9091942
Holzer, K., Wareth, G., El-Diasty, M., Abdel-Hamid, N. H., Hamdy, M. E. R., Moustafa, S. A., et al. (2022). Tracking the distribution, genetic diversity and lineage of Brucella melitensis recovered from humans and animals in Egypt based on core-genome SNP analysis and in silico MLVA-16. Transbound. Emerg. Dis. 69, 3952–3963. doi: 10.1111/tbed.14768
Jackson, B. R., Tarr, C., Strain, E., Jackson, K. A., Conrad, A., Carleton, H., et al. (2016). Implementation of Nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin. Infect. Dis. 63, 380–386. doi: 10.1093/cid/ciw242
Jiang, H., Fan, M., Chen, J., Mi, J., Yu, R., Zhao, H., et al. (2011). MLVA genotyping of Chinese human Brucella melitensis biovar 1, 2 and 3 isolates. BMC Microbiol. 11:256. doi: 10.1186/1471-2180-11-256
Jiang, H., Wang, H., Xu, L., Hu, G., Ma, J., Xiao, P., et al. (2013). MLVA genotyping of Brucella melitensis and Brucella abortus isolates from different animal species and humans and identification of Brucella suis vaccine strain S2 from cattle in China. PLoS One 8:e76332. doi: 10.1371/journal.pone.0076332
Kattar, M. M., Jaafar, R. F., Araj, G. F., Le Flèche, P., Matar, G. M., Abi Rached, R., et al. (2008). Evaluation of a multilocus variable-number tandem-repeat analysis scheme for typing human Brucella isolates in a region of brucellosis endemicity. J. Clin. Microbiol. 46, 3935–3940. doi: 10.1128/jcm.00464-08
Khan, A. U., Melzer, F., Sayour, A. E., Shell, W. S., Linde, J., Abdel-Glil, M., et al. (2021). Whole-genome sequencing for tracing the genetic diversity of Brucella abortus and Brucella melitensis isolated from livestock in Egypt. Pathogens 10:759. doi: 10.3390/pathogens10060759
Kneipp, C., Malik, R., Mor, S. M., and Wiethoelter, A. K. (2019). Commentary: retrospective and prospective perspectives on zoonotic brucellosis. Front. Microbiol. 10:1859. doi: 10.3389/fmicb.2019.01859
Kydyshov, K., Usenbaev, N., Berdiev, S., Dzhaparova, A., Abidova, A., Kebekbaeva, N., et al. (2022). First record of the human infection of Brucella melitensis in Kyrgyzstan: evidence from whole-genome sequencing-based analysis. Infect. Dis. Poverty 11:120. doi: 10.1186/s40249-022-01044-1
Li, Y., Yu, Y. F., Zhao, J., Ding, S. J., Zhang, G. Y., Yu, X. L., et al. (2024). Molecular epidemiological study of a human brucellosis outbreak - Weihai City, Shandong Province, China, 2022. China CDC Wkly. 6, 230–234. doi: 10.46234/ccdcw2024.046
Li, W., Zeng, L., Yuan, R., Qi, T., Liao, H., Cao, Y., et al. (2025). Genetic diversity atlas of Brucella melitensis strains from Sichuan Province, China. BMC Microbiol. 25:21. doi: 10.1186/s12866-024-03739-x
Liang, P. F., Zhao, Y., Zhao, J. H., Pan, D. F., and Guo, Z. Q. (2020). Human distribution and spatial-temporal clustering analysis of human brucellosis in China from 2012 to 2016. Infect. Dis. Poverty 9:142. doi: 10.1186/s40249-020-00754-8
Liu, Z. G., Wang, M., Ta, N., Fang, M. G., Mi, J. C., Yu, R. P., et al. (2020). Seroprevalence of human brucellosis and molecular characteristics of Brucella strains in Inner Mongolia autonomous region of China, from 2012 to 2016. Emerg. Microbes Infect. 9, 263–274. doi: 10.1080/22221751.2020.1720528
Ma, S. Y., Liu, Z. G., Zhu, X., Zhao, Z. Z., Guo, Z. W., Wang, M., et al. (2020). Molecular epidemiology of Brucella abortus strains from cattle in Inner Mongolia, China. Prev. Vet. Med. 183:105080. doi: 10.1016/j.prevetmed.2020.105080
Mirkalantari, S., Masjedian, F., and Fateme, A. (2021). Determination of investigation of the link between human and animal Brucella isolates in Iran using multiple-locus variable number tandem repeat method comprising 16 loci (MLVA-16). Braz. J. Infect. Dis. 25:101043. doi: 10.1016/j.bjid.2020.11.008
Pappas, G. (2010). The changing Brucella ecology: novel reservoirs, new threats. Int. J. Antimicrob. Agents 36, S8–S11. doi: 10.1016/j.ijantimicag.2010.06.013
Pappas, G., Papadimitriou, P., Akritidis, N., Christou, L., and Tsianos, E. V. (2006). The new global map of human brucellosis. Lancet Infect. Dis. 6, 91–99. doi: 10.1016/s1473-3099(06)70382-6
Sayour, A. E., Elbauomy, E., Abdel-Hamid, N. H., Mahrous, A., Carychao, D., Cooley, M. B., et al. (2020). MLVA fingerprinting of Brucella melitensis circulating among livestock and cases of sporadic human illness in Egypt. Transbound. Emerg. Dis. 67, 2435–2445. doi: 10.1111/tbed.13581
Shevtsov, A., Cloeckaert, A., Berdimuratova, K., Shevtsova, E., Shustov, A. V., Amirgazin, A., et al. (2023). Brucella abortus in Kazakhstan, population structure and comparison with worldwide genetic diversity. Front. Microbiol. 14:1106994. doi: 10.3389/fmicb.2023.1106994
Shevtsov, A., Ramanculov, E., Shevtsova, E., Kairzhanova, A., Tarlykov, P., Filipenko, M., et al. (2015). Genetic diversity of Brucella abortus and Brucella melitensis in Kazakhstan using MLVA-16. Infect. Genet. Evol. 34, 173–180. doi: 10.1016/j.meegid.2015.07.008
Tian, Z., Wan, L., Pei, J., Li, T., Wang, X., Yuan, P., et al. (2024). Brucellosis seroprevalence in cattle in China during 2014-2024: a systematic review and meta-analysis. Emerg. Microbes Infect. 13:2417859. doi: 10.1080/22221751.2024.2417859
Vergnaud, G., Hauck, Y., Christiany, D., Daoud, B., Pourcel, C., Jacques, I., et al. (2018). Genotypic expansion within the population structure of classical Brucella species revealed by MLVA16 typing of 1404 Brucella isolates from different animal and geographic origins, 1974-2006. Front. Microbiol. 9:1545. doi: 10.3389/fmicb.2018.01545
Wang, S., Wang, W., Sun, K., Bateer, H., and Zhao, X. (2020). Comparative genomic analysis between newly sequenced Brucella abortus vaccine strain A19 and another Brucella abortus vaccine S19. Genomics 112, 1444–1453. doi: 10.1016/j.ygeno.2019.08.015
Wareth, G., El-Diasty, M., Melzer, F., Schmoock, G., Moustafa, S. A., El-Beskawy, M., et al. (2020). MLVA-16 genotyping of Brucella abortus and Brucella melitensis isolates from different animal species in Egypt: geographical relatedness and the Mediterranean lineage. Pathogens 9:498. doi: 10.3390/pathogens9060498
Whatmore, A. M., Koylass, M. S., Muchowski, J., Edwards-Smallbone, J., Gopaul, K. K., and Perrett, L. L. (2016). Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: phylogeography and relationship to biovars. Front. Microbiol. 7:2049. doi: 10.3389/fmicb.2016.02049
Yu, X., Fang, M., Li, Y., Yu, J., Cheng, L., Ding, S., et al. (2023). Epidemiological characteristics and spatio-temporal analysis of brucellosis in Shandong province, 2015-2021. BMC Infect. Dis. 23:669. doi: 10.1186/s12879-023-08503-6
Zhao, Z. J., Li, J. Q., Ma, L., Xue, H. M., Yang, X. X., Zhao, Y. B., et al. (2021). Molecular characteristics of Brucella melitensis isolates from humans in Qinghai Province, China. Infect. Dis. Poverty 10:42. doi: 10.1186/s40249-021-00829-0
Zhou, C., Huang, W., Xiang, X., Qiu, J., Xiao, D., Yao, N., et al. (2022). Outbreak of occupational Brucella infection caused by live attenuated Brucella vaccine in a biological products company in Chongqing, China, 2020. Emerg. Microbes Infect. 11, 2544–2552. doi: 10.1080/22221751.2022.2130099
Zhu, L., Zhang, C., Liang, C., Peng, L., Yan, H., Liang, X., et al. (2024). Molecular epidemiological characteristics of osteoarthritis-associated Brucella melitensis in China: evidence from whole-genome sequencing-based analysis. Ann. Clin. Microbiol. Antimicrob. 23:18. doi: 10.1186/s12941-024-00671-w
Keywords: Brucella abortus, molecular epidemiology, genotyping, MLVA, cgSNP
Citation: Huangfu C, Ma X, Fan J, Han K, Liu T, Kou Z and Li Y (2025) Molecular epidemiology of Brucella abortus in Shandong, China: high-resolution insights from combined MLVA-16 and core genome SNP analysis. Front. Microbiol. 16:1695815. doi: 10.3389/fmicb.2025.1695815
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Maryam Dadar, Razi Vaccine and Serum Research Institute, IranGilles Vergnaud, Université Paris-Saclay, France
Copyright © 2025 Huangfu, Ma, Fan, Han, Liu, Kou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zengqiang Kou, amFjay1jb3VAMTYzLmNvbQ==; Yan Li, bGl5YW4yMzEzOTM1QDE2My5jb20=
†These authors have contributed equally to this work