Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease worldwide, and progressive NAFLD can develop into non-alcoholic steatohepatitis (NASH), liver cirrhosis, or hepatocellular carcinoma (HCC). NAFLD is a kind of metabolic disordered disease, which is commonly associated with lipid metabolism, insulin resistance, oxidative stress, inflammation, and fibrogenesis, as well as autophagy. Growing studies have shown Notch signaling pathway plays a pivotal role in the regulation of NAFLD progression. Here, we review the profile of the Notch signaling pathway, new evidence of Notch signaling involvement in NAFLD, and describe the potential of Notch as a biomarker and therapeutic target for NAFLD treatment.
Introduction
Non-alcoholic fatty liver disease (NAFLD), affecting over a quarter of the global population, has emerged as the highest prevalent type of chronic liver disease (Younossi et al., 2016). NAFLD encompasses a spectrum of progressive liver diseases including simple steatosis (SS), non-alcoholic fatty steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (D. Q. Huang et al., 2021; Powell et al., 2021). NAFLD is defined by the presence of steatosis in >5% of hepatocytes in histological analysis and exclusion of excessive alcohol consumption daily (≥30 g for men and ≥20 g for women) (EASL et al., 2016). Evidence suggests that NAFLD is related to liver manifestations of metabolic syndrome such as obesity, diabetes, insulin resistance (IR), and dyslipidemia (Younossi et al., 2018; Jarvis et al., 2020).
The individual clinical outcomes of patients with NAFLD are highly variable. For the majority of patients with simple steatosis, their liver disease is in non- or slow-progression. A prospective cohort study reported in a three-year period, over 20% of patients with simple steatosis developed into NASH (Wong et al., 2010), a more severe stage in which fatty liver is accompanied by necroinflammatory changes like hepatocyte ballooning and lobular inflammation (Vernon et al., 2011). In the final stages, collagen deposition and subsequent vascular remodeling result in fibrosis and cirrhosis (EASL et al., 2016). Thus far, there is no accurate non-invasive diagnostic biomarker and effective treatment toward NAFLD (Francque and Vonghia, 2019; Younossi, 2019), and current therapy is mainly focused on lifestyle changes (EASL et al., 2016; Chalasani et al., 2018).
Studies have shown that NAFLD is mainly characterized by hepatocyte inflammation and steatosis in the early stage and fibrosis and/or cirrhosis in the late stage (Wang et al., 2020). However, the pathogenesis of NAFLD has not been fully understood. In 1998, scientists first proposed the “two-hit” hypothesis to explain that steatosis (the first “hit”) and other factors associated with free radicals (the second ‘‘hit”) are necessary for NASH progression (Day and James, 1998). In recent years, based on animal models and descriptive clinical trials, the “multiple hits” hypothesis is widely accepted (Tilg and Moschen, 2010; Buzzetti et al., 2016; Tilg et al., 2021). The primary hit is the infiltration and pro-inflammatory state of macrophages in the visceral adipose tissue, resulting in IR. Meanwhile, the abnormal lipolysis increases the delivery of fatty acids to the liver, and along with steatosis, aggravates the lipid metabolic burden. The imbalance results in the formation of lipotoxic lipids that generate a series of multiple hits, including oxidative and/or endoplasmic reticulum (ER) stress, inflammasome activation, and apoptotic damage, followed by inflammation, tissue regeneration, and fibrogenesis (Tilg and Moschen, 2010; Bessone et al., 2019; Sanyal, 2019). Besides, mitochondrial dysfunctions, lifestyle, and epigenetic and genetic factors also jointly affect the occurrence and progression of the NAFLD (Loomba et al., 2021) (Figure 1).
FIGURE 1
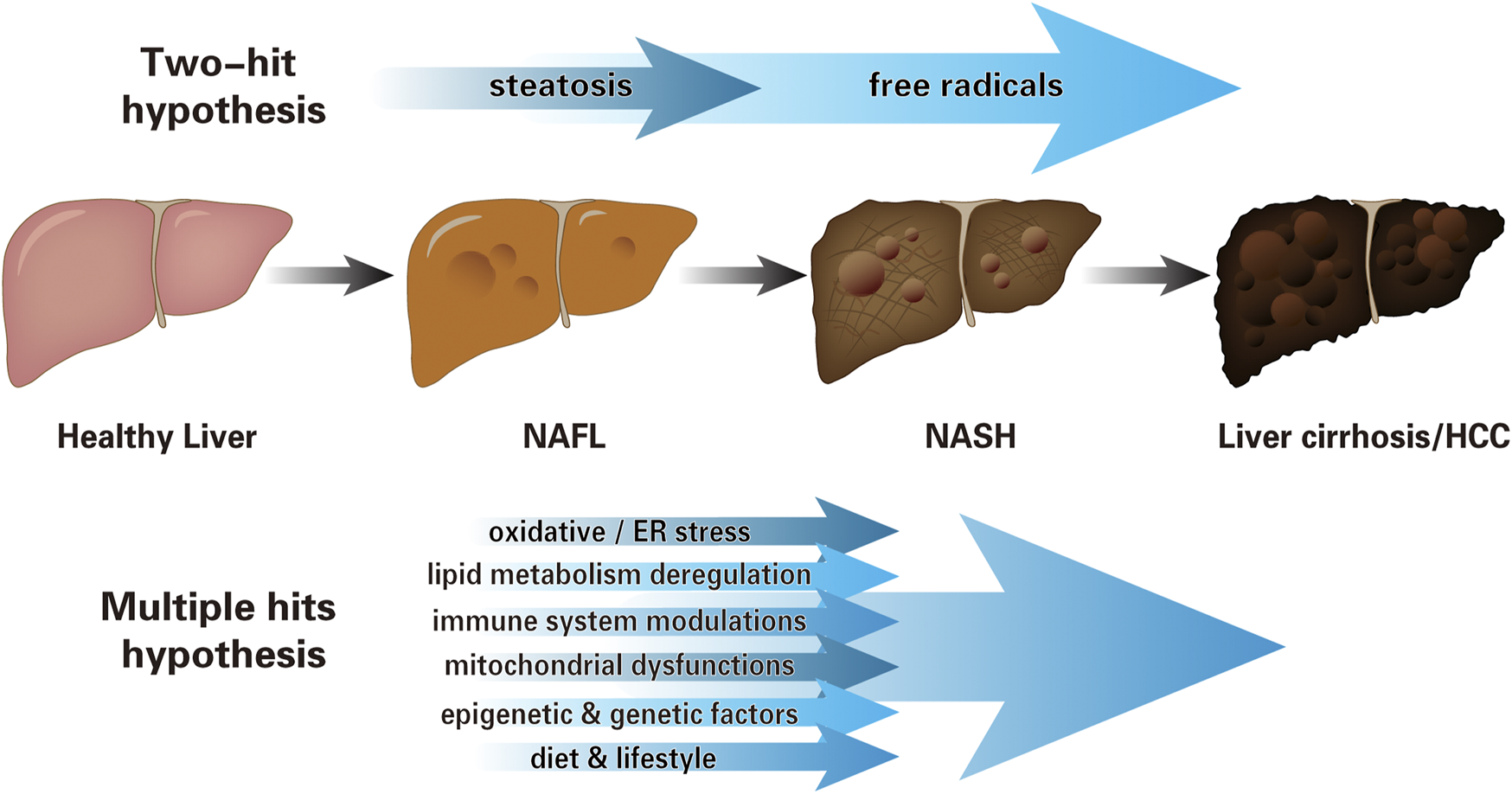
The pathogenesis of NAFLD. Schematic representation illustrating cognition toward the progression of NAFLD: from “two-hit” hypothesis to “multiple hits” hypothesis. The early “two-hit” hypothesis, in which the first “hit” is steatosis, leads to the second “hit”: oxidative stress, endotoxin, etc. The “multiple hits” hypothesis considers several parallel hits jointly affect the NAFLD pathogenesis, which includes, but is not limited to oxidative and/or ER stress, lipid metabolism deregulation, immune system modulations, mitochondrial dysfunctions, lifestyle, and epigenetic and genetic factors (NAFL, non-alcoholic fatty liver, i.e., simple steatosis without hepatocellular injury; NASH, non-alcoholic steatohepatitis; HCC, hepatocellular carcinoma; ER, endoplasmic reticulum).
Notch signaling pathway plays a crucial role in cell differentiation (Amsen et al., 2009; Amsen et al., 2015), proliferation (Bartolome et al., 2019), and apoptosis (Guruharsha et al., 2012). Recently, it has also been demonstrated that Notch is involved in liver development, homeostasis, and metabolism (Bi and Kuang, 2015; Geisler and Strazzabosco, 2015; Adams and Jafar-Nejad, 2019). However, the association of the Notch signaling with NAFLD has rarely been reported. Here we review the recent advances in Notch signaling in liver pathophysiology and analyze the Notch signaling pathway as a potential target to prevent and treat NAFLD.
Overview of Notch Signaling Pathway
Notch signaling is a juxtracrine signal transduction mechanism that enables cell-cell communication directly (Artavanis-Tsakonas et al., 1999). In mammals, four receptors (Notch1-4) and five ligands [Jagged (JAG) 1-2, Delta-like ligand (DLL) 1, 3, and 4] have been identified in canonical Notch signaling (D'Souza et al., 2010). In the liver of adults, four Notch receptors are expressed, while only two Notch ligands (JAG1 and DLL4) are expressed (Y. Chen et al., 2012). The ligand-receptor interaction is the initiation of Notch signaling pathway, making various cellular regulations more precise and orderly (Bray, 2016).
The core signaling pathway most commonly used to describe Notch-dependent processes is named the canonical Notch signaling pathway (Andersson et al., 2011; Guruharsha et al., 2012). The ligand presented by the Notch signal sending cell binds to the receptor on the signal-receiving cell. The endocytosis of the ligand leads to a conformational change of the Notch receptor, exposing the cleavage site of the ADAM10. Subsequent cleavage of the γ-secretase complex releases the Notch intracellular domain (NICD) (Kopan and Ilagan, 2009). NICD then migrates to the nucleus, binds to the transcription factor RBP-Jκ (also called CSL) (Kovall and Blacklow, 2010), and recruits the co-activator Mastermind-like (MAML) to initiate downstream gene transcription, including the hairy enhancer of split (HES) and HES-related (HEY) family genes (Nam et al., 2006; Wilson and Kovall, 2006; Bolos et al., 2007; Guruharsha et al., 2012) (Figure 2).
FIGURE 2
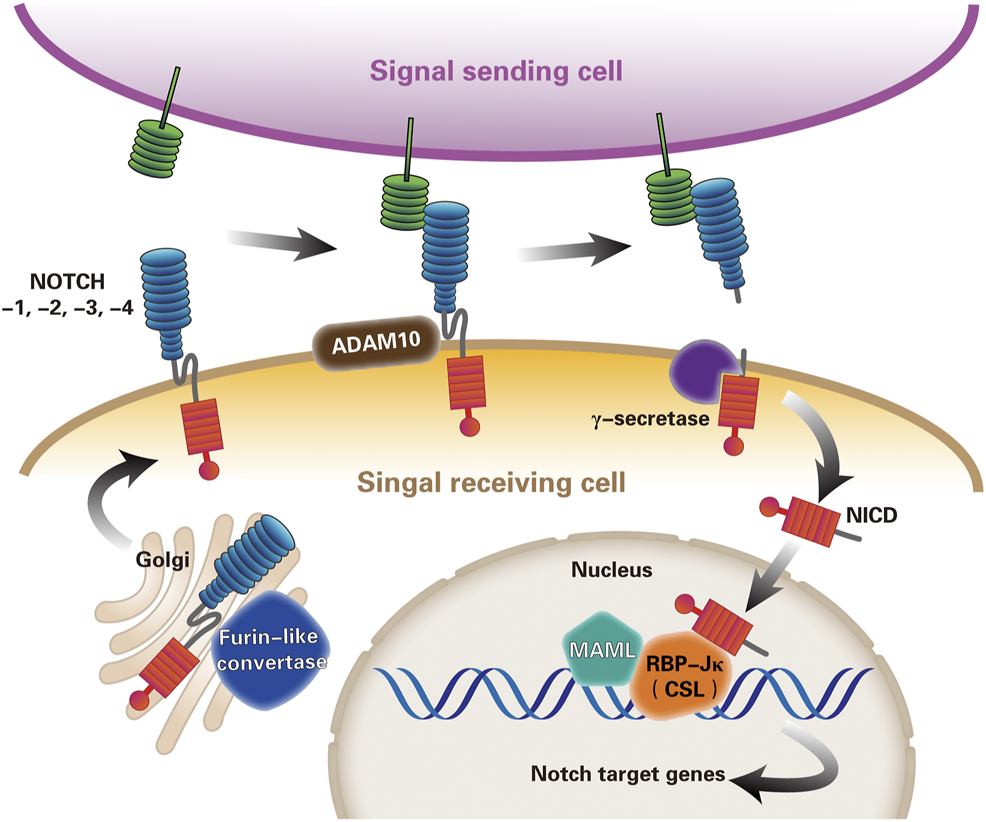
The canonical Notch signaling pathway. Notch signaling pathway is currently thought to be activated by three steps of proteolysis. First, the mammalian Notch receptors are cleaved by a furin-like convertase in the Golgi compartment. After digestion, the extracellular subunits and transmembrane subunits formed by Ca2+ dependent non-covalent bonding to form heterodimers, and exocytosed to the cell membrane become mature Notch receptors. Second, Notch ligand-receptor binding enables proteolytic cleavage of the Notch extracellular domain by ADAM10 metalloprotease, and Notch receptor releases extracellular subunits. Third, γ-secretase complex cleaves the remnant receptor to allow the release and nuclear translocation of the NICD, where NICD forms a trimeric complex with transcription factor RBP-Jκ(or CSL) and the co-activator MAML, imitating the expression of Notch target genes transcription. (ADAM, a disintegrin and metalloprotease10; RBP-Jκ, recombination signal binding protein immunoglobulin kappa J; NICD, Notch intracellular domain; CSL, CBF1–suppressor of hairless–LAG1; MAML, mastermind-like).
Different from other classical signal transduction processes, the canonical Notch signaling pathway is characterized by the lack of cascade amplification in the transduction process, and only NICD is generated after a Notch receptor is consumed. Therefore, its signal intensity is crucial for generating the corresponding cellular response, and any deviation in the expression level of any molecular component in the Notch signaling pathway may have a vital impact (Andersson et al., 2011). For example, Alagille syndrome (AGS) is caused by mutations in the gene for the Notch ligand JAG1 and NOTCH2 receptor (McDaniell et al., 2006). Currently, various Notch signaling pathway modulation approaches have been explored, including inhibition of the ligand-receptor interaction and interference with the proteolytic process of the receptor (Groth and Fortini, 2012; Shao et al., 2012; Andersson and Lendahl, 2014). Several Notch inhibitors are demonstrated effects in the NAFLD (Table 1).
TABLE 1
| Inhibitor | Target | Function | Object | References |
|---|---|---|---|---|
| Peroxiredoxin 6 (PRDX6) | Notch1 | Improve lipid accumulation through induction of mitophagy | Mice | Lee et al. (2019) |
| Notch1 decoy | Notch1 | Decrease hepatic glucose production | Mice | Funahashi et al. (2008) Pajvani et al. (2013) |
| N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) | Notch1 | Alleviate lipid accumulation and hepatocyte injury | Mice | Zhang et al. (2021) |
| Delta-like1 homolog (DLK1) | Notch1 | Reduce hepatic steatosis and improve glucose and insulin tolerance | Mice | Lee et al. (2016) |
| Triptolide (TP) | Notch1 | Initiate oxidative stress in hepatocyte | Mice | Shen et al. (2019) |
| Hepatocyte Toll-like receptor 4 (TLR4) | Jag1/JAG1 | Reduce NASH related liver fibrosis | Mice/Human | Yu et al. (2021) |
| Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) | NICD | Ameliorate hepatic lipogenesis dyslipidemia and insulin resistance | Mice | Chartoumpekis et al. (2018) |
| γ-secretase inhibitor (GSI) | γ-secretase | Improve glucose metabolism and ameliorate liver fibrosis | Mice | Richter et al. (2020) |
| Liver-specific Rbp-jκ knockout (L-RBP-Jκ) | RBP-Jκ | Protect from obesity-induced insulin resistance | Mice | Pajvani et al. (2011) |
| Silybin (SIL) | NOTCH1 | Hepatoprotective and antitumorigenic effect in HCC cells | Human | Zhang et al. (2013) |
| Delta-tocotrienol (δ-T) | NOTCH1 | Reduce biochemical markers of hepatocellular injury and steatosis | Human | Pervez et al. (2020) |
Major Notch signaling inhibitors in NAFLD
Notch in Lipid Metabolism
As the central hub of lipid homeostasis, the liver is responsible for coordinating the whole process of lipid circulation, including the synthesis, export, redistribution, and utilization of free fatty acids (Nguyen et al., 2008). The main pathways that constitute hepatic lipid homeostasis, including uptake of circulating lipids, de novo lipogenesis (DNL), fatty acid oxidation (FAO), and export as very-low-density lipoprotein (VLDL) particles (Gluchowski et al., 2017; Ipsen et al., 2018). Hallmarked by hepatic steatosis, NAFLD is connected with lipid metabolism. When lipid acquisition exceeds lipid disposal in the liver, that is, the uptake of fatty acids and DNL covering oxidation and output of fatty acids, hepatic steatosis occurred (Ipsen et al., 2018). Feng et al. (2017) proposed that no significant differences between free fatty acids (FFAs) in lean or obese patients with NAFLD were observed, and the value of serum FFAs in early diagnosis of NAFLD.
The studies suggested that nutrition-induced activation of mammalian target of rapamycin (mTOR) may cause an increase in liver lipid content, which also increases the activity of basal serine/threonine kinases, leading to a self-perpetuating lipogenic cycle (Lamming and Sabatini, 2013; Caron et al., 2015; Han and Wang, 2018). Pajvani et al. (Pajvani et al., 2013) demonstrated that inhibition of Notch signaling prevented hepatic steatosis by blocking mTOR complex 1 (mTORC1) activity, which could be reversed by rapamycin treatment. They also showed that Notch signaling augmented mTORC1 function and SREBP1c-mediated lipogenesis and that inhibition of hepatic Notch signaling protects from the fatty liver by reducing DNL.
Although the specific pathogenesis of lipid metabolism disorder in NAFLD patients is still not completely clear, studies have shown it may be associated with Notch pathway regulation (Li et al., 2019). Ding et al. (2020) investigated the dynamic role of Notch gene expression in the development of NAFLD in vitro and in vivo. They used palmitic acid (PA) and methionine-choline-deficient (MCD) models to assess notch signaling genes expression changes at different time points. Based on the characteristics of Notch mRNA expression levels, they evaluated that expression of Notch3 mRNA has been dynamically changed significantly in the development of hepatic steatosis during NAFLD (Ding et al., 2020). Furthermore, Auguet et al. (2020) explored the association between the Notch transcriptional repressor and hepatic expression of lipid metabolism-related genes in a cohort of women with NAFLD. They found a negative relationship between hepatic HEY2 expression and low-density lipoprotein (LDL) cholesterol (Auguet et al., 2020).
Notch in Insulin Resistance
It is generally recognized that IR is pivotal in the pathogenesis and progression of NAFLD (Lomonaco et al., 2012). IR is essentially a decrease in the sensitivity of whole-body, liver, and adipose tissue to insulin, which is involved in the development of hepatic steatosis (E. Bugianesi, 2010). In NAFLD patients, increases in circulating glucose and insulin associated with IR promote hepatic DNL (Smith et al., 2020). Specifically, when IR occurs, it causes an impaired ability of insulin to inhibit adipose tissue lipolysis, resulting in increased delivery of FFAs to the liver (Bugianesi et al., 2005). Meanwhile, large lipid deposition promotes IR, which leads to fasting hyperglycemia and compensatory hyperinsulinemia, further contributing to the pathophysiology of NAFLD via exacerbating DNL (Donnelly et al., 2005).
The abnormal activation of Notch signaling pathway and IR are closely linked. It is recognized factor forkhead box protein O1 (FOXO1) has a beneficial effect on insulin-mediated glucose homeostasis (Matsumoto et al., 2007; O-Sullivan et al., 2015). Notch signal mainly affects hepatic glucose via the synergistic effect of NICD and FoxO1 transcription. Glucose-6-phosphatase catalytic subunit (G6PC) and phosphoenolpyruvate carboxykinase (PCK1) are both rate-limiting enzymes of hepatic glycogenolysis and gluconeogenesis, which would be correlated with Notch activation (Valenti et al., 2013; Dongiovanni et al., 2016). Pajvani et al. (2011) reported that combined haploinsufficiency of FoxO1 and Notch1 notably improves insulin sensitivity in diet-induced IR. Hepatic overexpression of Notch1 regulates hepatic gluconeogenesis by inducing G6PC in a FoxO1-dependent mode, in turn, aggravates insulin resistance (Pajvani et al., 2011; Bernsmeier et al., 2016). Additionally, the reduction of metabolic activity in brown adipose tissue (BAT) has been found connected with IR in human (Stanford et al., 2013; Mottillo et al., 2016). Bi et al. (2014) revealed mice in which Notch1 or Rbp-jκ selectively deleted in adipocytes show upregulated expression of BAT-specific genes and improvement in glucose tolerance and insulin sensitivity.
Based on the close relation between IR and Notch, several possible pharmacological targets of NAFLD are identified. Blocking the abnormal expression of Notch at the gene level can inhibit the accumulation of liver gluconeogenesis and triglycerides (TGs), thereby reducing the risk of NAFLD. The cleavage of NICD by γ-secretase inhibitor (GSI) exhibited an improvement of glucose homeostasis and insulin sensitivity in diet-induced obese (DIO) mice (Pajvani et al., 2011). Lee et al. (2016) demonstrated that Delta-like 1 homolog (DLK1), an inhibitory regulator of Notch signaling, would reduce hepatic steatosis and hyperglycemia via exogenous administration. Chartoumpekis et al. (2018) showed nuclear factor (erythroid-derived 2)-like 2 (Nrf2) could profoundly ameliorate hepatic lipogenesis and IR by repressing NICD. Besides, researchers also found plant extracts (such as curcumin) have been shown to suppress NOTCH1, which could ameliorate fatty liver and enhance insulin sensitivity in the high-fat diet (HFD) model (Zhao et al., 2017; Saadati et al., 2019; El et al., 2021).
Notch in Oxidative Stress
Oxidative stress (OS) is a concept used to describe an imbalance between pro-oxidants and antioxidants, leading to cellular damage and tissue injury (Sies, 2015). The chronic high-calorie diet causes lipid accumulation in hepatocytes and excessive generation of reactive oxygen species (ROS) (Sahini and Borlak, 2014). Meanwhile, affected by lipotoxicity from high levels of lipid metabolites, OS inhibits insulin sensitivity and facilitates DNL (Gehrke and Schattenberg, 2020).
In the pathophysiological process of NAFLD, OS is considered a pivotal mediator of the inflammatory response (Koek et al., 2011). Notch signaling has been reported to be associated with steatosis and OS. It has been proposed that ROS like H2O2 regulates the expression of Notch (Marinho et al., 2014). Notch1 regulates the expression of lipid oxidation genes and exhibited an obvious lipid accumulation reduction in Notch1 deficient antisense transgenic (NAS) mice (Song et al., 2016). Similarly, Notch1 inhibitor reduces ethanol-induced OS and lipid accumulation in HepG2 cells (Wang et al., 2014).
Among the multiple mechanisms that accelerate the progression of NAFLD to NASH, mitochondrial dysfunction is the prime one (Caldwell et al., 1999). Mitochondrial abnormalities disrupt the balance between pro-oxidants and antioxidants, leading to an increase of FFAs (Begriche et al., 2013). Peroxiredoxin 6 (PRDX6) is a mitochondrial antioxidant enzyme and is highly expressed in the liver (Fisher, 2011; Arriga et al., 2019). Lee et al. (2019) demonstrated that PRDX6 induces effects of maintaining mitochondrial integrity and inhibits OS-induced Notch signaling, thereby reducing ROS production and lipid accumulation. They pointed out that PRDX6 mitophagy-mediated mechanisms offer endogenous protection against NAFLD (Lee et al., 2019).
Moreover, triptolide (TP) is the main ingredient of the medicinal herb Tripterygium wilfordii Hook f (TWHF) (Ziaei and Halaby, 2016). TP caused hepatotoxicity through initiating OS. Shen et al. (2019) investigated TP inhibited the protein expression of Notch1 and NICD, and the activation of Notch signaling has the potential to protect against TP-induced live injury. Interestingly, Huang et al. (2021) demonstrated that dose-related TP as an allosteric AMPK agonist alleviates NAFLD. Combined, the regulation of Notch signaling pathway may better enable TP to play a protective role in NAFLD.
Notch in Inflammation and Fibrogenesis
Liver fibrosis is a decisive factor of liver disease progression, particularly as it is associated with adverse prognosis and mortality in patients with NASH (Vilar-Gomez et al., 2018; Powell et al., 2021). Even in the early stage of fibrosis, it is shown a series of adverse liver-related events are gradually increasing (Angulo et al., 2015; Dulai et al., 2017; Hagstrom et al., 2017). In advanced NASH, hepatocytes are partially replaced by fibrotic scar tissue, the severe pathological change makes it difficult to treat NASH by correcting the underlying metabolic abnormality. Therefore, anti-fibrosis has become the focus of NASH therapy.
Notch activity is almost absent in healthy adult hepatocytes, mildly elevated in simple steatosis, and significantly increased in NASH (Valenti et al., 2013; Zhu et al., 2018). In various mouse models of fibrosis, over 80% of collagenous myofibroblasts are caused by hepatic stellate cell (HSC) (Mederacke et al., 2013). Notch-activated hepatocytes facilitate liver profibrogenic in NASH by both osteopontin (Opn) secretion mediated HSC activation in vitro and in vivo (Zhu et al., 2018), leading to a continuous extracellular matrix (ECM) accumulation and liver parenchyma gradually replaced by fibrous tissue (Mederacke et al., 2013). Conversely, in Notch loss-of-function mouse models, hepatocyte-specific liver inflammation and fibrosis are reduced, suggesting maladaptive hepatocytic Notch response to NASH-associated liver fibrosis (Zhu et al., 2018).
Sawitza et al. (2009) explored Jag1 as one of the cell surface ligands in Notch signaling activates HSC to stimulate α-SMA and collagen production. Yu et al. (2021) proved increasing Jag1 is responsible for fibrosis-inducing Notch reactivation. Also, other hepatic non-parenchymal cells could activate the Notch pathway to promote NASH latently through various mechanisms. Duan et al. (2018) investigated Notch activation in liver sinusoids endothelial cell (LSEC), which leads to HSC activation and the subsequent hepatic fibrosis, by downregulating eNOS-sGC signaling. Besides, researchers found that inhibitors inactivate M1 polarization of macrophage by regulating Notch signaling could reduce the secretion of inflammatory cytokine and fibrogenesis in CCl4-induced liver injury mice (Bansal et al., 2015; Xu et al., 2015; Sheng et al., 2020). Additionally, γ-secretase inhibitor (Chen et al., 2012) and Notch3 siRNA (Y. X. Chen et al., 2012) suppressed the myofibroblastic gene expression of rat HSC line by blocking Notch signaling. Therefore, selective interruption of these Notch-related targets may provide more anti-fibrosis strategies for NAFLD (Romeo, 2019).
Notch in Autophagy
Autophagy is a process in which cells degrade and metabolize their own damaged organelles or protein aggregation (Wang et al., 2019), which plays a vital role in regulating multiple liver functions and maintaining hepatic homeostasis (Ueno and Komatsu, 2017). Accumulating evidence suggests autophagy regulates liver-mediated systemic glucose and lipid metabolism (Singh et al., 2009; Galluzzi et al., 2014). Meanwhile, the liver is surrounded by exogenous substances from the portal vein circulation, including potential inflammatory mediators, in which autophagy has major cell-protective and anti-inflammatory effects (Deretic et al., 2013; Deretic and Levine, 2018; Hazari et al., 2020). All of the above suggests autophagy is associated with the occurrence and development of various liver diseases such as NAFLD.
The lipid droplets (LDs) are specialized cytosolic organelles in which some organs including the liver store neutral lipids (such as TGs) to protect from lipotoxicity (Gross and Silver, 2014). The progression of LDs degradation is regarded as a specific form of autophagy, also known as lipophagy (Garcia et al., 2018). Recent studies have revealed that disturbances in lipophagy have been linked to hepatic lipid accumulation, the process of lipophagy could be regarded as a new way of controlling NAFLD development (Grefhorst et al., 2021).
Because autophagy can remove damaged organelles, autophagy may alleviate hepatocellular injury during NASH. The protective effects of carbamazepine-induced autophagy could reduce steatosis and improve IR in the NAFLD model (Lin et al., 2013). Indeed, modulating autophagy may prevent the progression of NAFLD. Zhang et al. (2021) investigated that Notch1 is an activated intensity of autophagy in FFA-treated HepG2 cells, and decreased Notch1 levels may alleviate hepatocyte damage by enhancing autophagy, which could be reversed by autophagy inhibitor chloroquine. Niture et al. (2018) demonstrated that inhibition of Notch reduced the expression of autophagy biomarker and serotonin-mediated liver cell steatosis. These findings provide helpful clues for the strategy of Notch signaling pathway to regulate autophagy and thereby remit the progression of NAFLD.
Notch in NAFLD-Related HCC
HCC is the fourth leading cause of cancer-related deaths worldwide and occurs in patients with various chronic liver diseases (Bray et al., 2018; Llovet et al., 2021). Although hepatitis B virus (HBV) infection has been the prominent risk factor of HCC, NAFLD has become the most rapidly growing driver of HCC in many countries (Younossi et al., 2019; Hester et al., 2020). The incidence in patients with NAFLD-related HCC increases with the histological stage, which is highest in patients with NAFLD-cirrhosis (Ioannou, 2021).
Thus far, the exact pathogenesis underlying NAFLD-induced HCC is only incompletely understood but mainly focuses on the effects of DNA damage response, inflammation, autophagy, and intestinal microbiota (Anstee et al., 2019; Behary et al., 2021). In addition, the chronic activation of metabolic pathways seems to play a critical role (Baffy et al., 2012). These pathways may provoke infinite hepatocyte proliferation and genomic instability, and on the other hand, provide a microenvironment conducive to malignant transformation and tumor growth.
Recent studies suggest that Notch signaling pathway is frequently associated with tumorigenesis (Nowell and Radtke, 2017). Selective blocking of Notch1 inhibits cancer cell growth and deregulates angiogenesis (Wu et al., 2010). By performing RNA sequencing of hepatocyte populations HFD-fed reporter mice, Zhu et al. (2021) illustrated that Notch-active hepatocytes showed transcriptional enrichment of ECM-related genes, which may represent a mechanism that persists in the tumorigenic process. Furthermore, they found HFD-diet mice with Notch-active mutation spontaneously formed fully developed liver tumors (Zhu et al., 2021). Therefore, it can be inferred that the continuous activation of Notch signaling pathway promotes the occurrence of NAFLD-related HCC.
Conclusion and Perspective
NAFLD is a manifestation of metabolic syndrome in the liver. With the changes in lifestyle and dietary habits, the incidence of NAFLD is rising rapidly. The previous studies have revealed the significance of the Notch signaling pathway in metabolism. The abnormal expression of Notch may lead to several metabolic disorders, thus inducing NAFLD. Although the relation between NAFLD and Notch signaling has been observed both in vitro and in vivo, most of the research findings are based on phenotypic studies and the underlying mechanisms and potential associations between different Notch molecules, and require further in-depth research.
The development of liver-specific Notch inhibitors is pivotal for the treatment of NAFLD-related hepatic lipid accumulation, IR, OS, fibrogenesis, and autophagy progression (Figure 3). But until now, most intervention studies are conducted in animal models (especially mice), the potential role of Notch regulators in human NAFLD needs to be explored extensively. Recently, the rising field of “hepatokines” biology would help reveal the complex molecular regulation in NAFLD (Watt et al., 2019). If so, it would promote the development of more non-invasive diagnostic tests to improve early diagnosis rates.
FIGURE 3
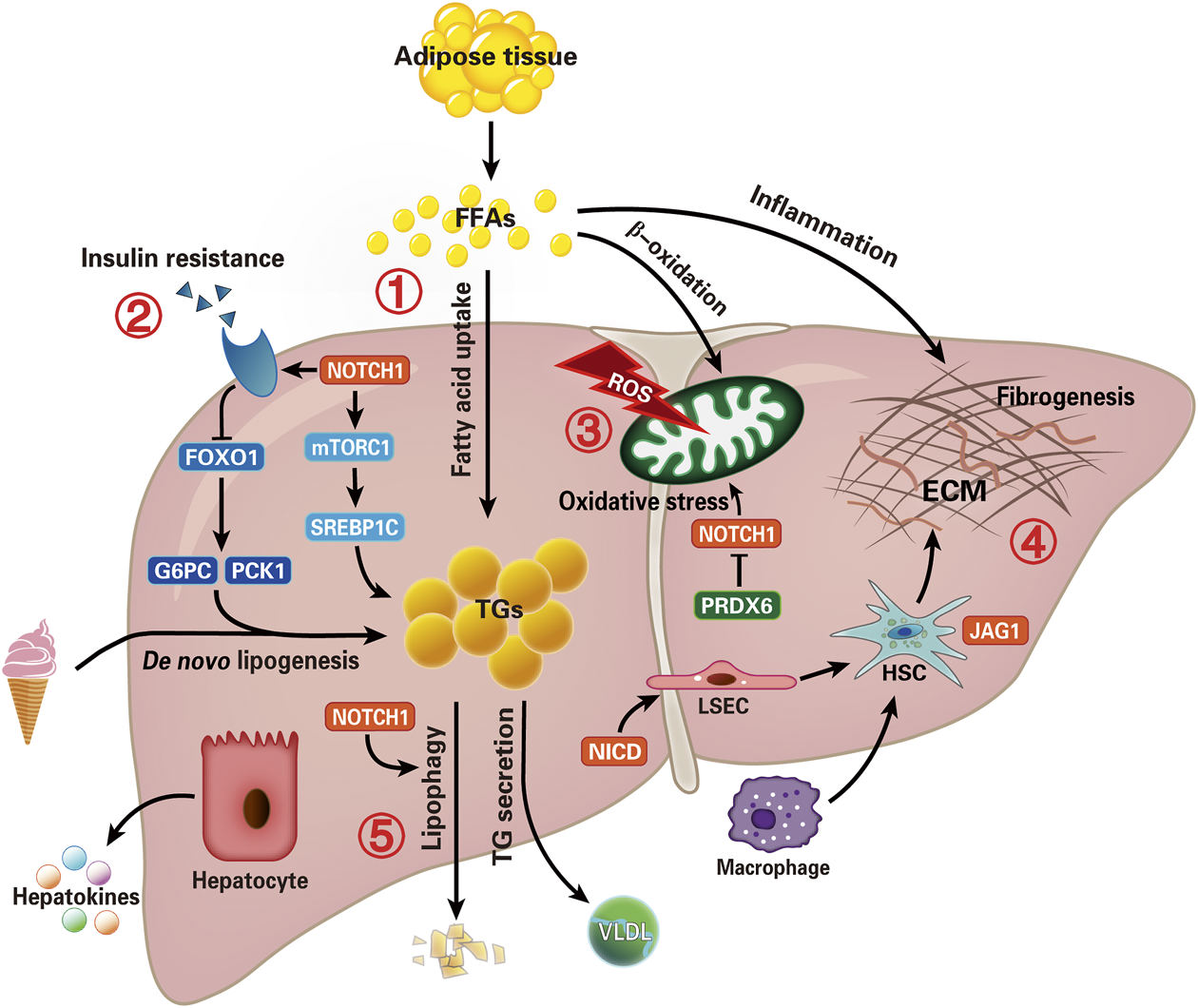
Overview of potential therapeutic targets of Notch signal pathway in NAFLD. Summarize the recent critical advances evolving Notch signaling in the NAFLD. Intrahepatic lipid levels depend on the balance between lipid acquisition and disposal. Therefore, lipid accumulation is the result of uptake of fatty acids and de novo lipogenesis exceeding export as VLDL and oxidation of fatty acid, which is also a main pathophysiological change of NAFLD. The five potential pathways to intervene in NAFLD are focused on (1) hepatic lipid accumulation, (2) insulin resistance, (3) oxidative stress, (4) inflammation and fibrogenesis, and (5) autophagy (lipophagy) progression. Although several aspects of these functions remain to be fully understood, these findings offer an intriguing rationale for investigating Notch-based therapies in patients with NAFLD. Furthermore, the “hepatokines” secreted by hepatocytes may help reveal the complex molecular regulation in NAFLD. (NICD, Notch intracellular domain; FFAs, free fatty acids; mTORC1, mammalian target of rapamycin complex one; FOXO1, factor forkhead box protein O1; G6PC, glucose-6-phosphatase catalytic subunit; PCK1, phosphoenolpyruvate carboxy kinase; TGs, triglycerides; VLDL, very-low-density lipoprotein; ROS, reactive oxygen species; PRDX6, peroxiredoxin 6; ECM, extracellular matrix; HSC, hepatic stellate cell; LSEC, liver sinusoids endothelial cell).
There is no specific and effective pharmacotherapy toward NAFLD, however, some drugs have shown therapeutic potential by regulating a Notch signal pathway. Vitamin E (α-tocopherol) is a dietary antioxidant recommended as a treatment for NASH (Yakaryilmaz et al., 2007; Chalasani et al., 2018). Recent clinical research supports vitamin E use brought obvious histological benefits and improved prognosis in patients with NASH (Sato et al., 2015; Brunt et al., 2019; Vilar-Gomez et al., 2020). δ-tocotrienol, an isomer of vitamin E, has been explored to inhibit tumor invasion and metastasis via downregulating the NOTCH1 signaling pathway (Rajasinghe et al., 2018). Notably, Pervez et al. (2020) launched a randomized, double-blind, placebo-controlled trial of 71 patients with NAFLD. Compared with placebo, δ-tocotrienol significantly reduced biochemical markers of hepatocellular injury and steatosis in patients (Pervez et al., 2020). Silybin (SIL), a hepatoprotective drug, could be an inhibitor targeting the NICD, RBP-Jκ, and Hes1 proteins in HCC cells and exert antitumorigenic effects (Zhang et al., 2013).
The precise drug delivery without toxicity brings a wide application prospect for the treatment of NAFLD. A nanoparticle-mediated delivery system to target GSI in the liver (GSI NPs) has been developed (Richter et al., 2020), which avoids goblet cell metaplasia caused by intestinal Notch inhibition (van Es et al., 2005). Based on similar studies above would advance clinical therapy research, thereby optimizing therapies for various NAFLD subtypes to increase the cure rate while complications can be decreased. In a word, findings on Notch signaling pathway research could bring NAFLD patients a hopeful future with ever more promising targets for prevention and treatment.
Statements
Author contributions
HX designed the outline of the review and drafted the manuscript. LW contributed his scientific advice and revision of the manuscript. All authors read and approved the submitted version.
Funding
This study was supported by the MOST 2016YFA0102100 (The National Key Research and Development Program of China: Stem Cell and Translational Research), and NSFC 81670863, 81422009, 81401940, 81770560, 81800533.
Acknowledgments
We thank Dr. Hua Han and Dr. Kefeng Dou from the Fourth Military Medical University, for their constructive scientific advice.
Conflict of interest
The authors declare that the research was conducted in the absence of any potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
- AGS
Alagille syndrome
- DIO
diet-induced obese
- DLK1
Delta-like 1 homolog
- DNL
de novo lipogenesis
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- FAO
fatty acid oxidation
- FFAs
free fatty acids
- FOXO1
factor forkhead box protein O1
- G6PC
glucose-6-phosphatase catalytic subunit
- GSI
γ-secretase inhibitor
- HCC
hepatocellular carcinoma
- HFD
high-fat diet
- HSC
hepatic stellate cell
- IR
insulin resistance
insulin resistance
- IR
insulin resistance
insulin resistance
- LDs
lipid droplets
- LSEC
liver sinusoids endothelial cell
- MCD
methionine-choline-deficient
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- NAFL
non-alcoholic fatty liver
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic fatty steatohepatitis
- NAS
Notch1 deficient antisense transgenic
- NICD
Notch intracellular domain
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- Opn
osteopontin
- OS
oxidative stress
- PA
palmitic acid
- PCK1
phosphoenolpyruvate carboxy kinase
- PRDX6
Peroxiredoxin 6
- ROS
reactive oxygen species
- SIL
Silybin; δ-T(Delta-tocotrienol)
- SS
simple steatosis
- TGs
triglycerides
- TP
triptolide
- TWHF
tripterygium wilfordii Hook f
- VLDL
very-low-density lipoprotein
References
1
Adams J. M. Jafar-Nejad H. (2019). The Roles of Notch Signaling in Liver Development and Disease. Biomolecules (Basel, Switzerland)9 (10), 608. 10.3390/biom9100608
2
Amsen D. Antov A. Flavell R. A. (2009). The Different Faces of Notch in T-Helper-Cell Differentiation. Nat. Rev. Immunol.9 (2), 116–124. 10.1038/nri2488
3
Amsen D. Helbig C. Backer R. A. (2015). Notch in T Cell Differentiation: All Things Considered. Trends Immunol.36 (12), 802–814. 10.1016/j.it.2015.10.007
4
Andersson E. R. Lendahl U. (2014). Therapeutic Modulation of Notch Signalling — Are We There yet?Nat. Rev. Drug Discov.13 (5), 357–378. 10.1038/nrd4252
5
Andersson E. R. Sandberg R. Lendahl U. (2011). Notch Signaling: Simplicity in Design, Versatility in Function. Development138 (17), 3593–3612. 10.1242/dev.063610
6
Angulo P. Kleiner D. E. Dam-Larsen S. Adams L. A. Bjornsson E. S. Charatcharoenwitthaya P. et al (2015). Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-Term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology149 (2), 389–397. 10.1053/j.gastro.2015.04.043
7
Anstee Q. M. Reeves H. L. Kotsiliti E. Govaere O. Heikenwalder M. (2019). From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol.16 (7), 411–428. 10.1038/s41575-019-0145-7
8
Arriga R. Pacifici F. Capuani B. Coppola A. Orlandi A. Scioli M. G. et al (2019). Peroxiredoxin 6 Is a Key Antioxidant Enzyme in Modulating the Link between Glycemic and Lipogenic Metabolism. Oxid Med. Cel Longev2019, 9685607. 10.1155/2019/9685607
9
Artavanis-Tsakonas S. Rand M. D. Lake R. J. (1999). Notch Signaling: Cell Fate Control and Signal Integration in Development. Science284 (5415), 770–776. 10.1126/science.284.5415.770
10
Auguet T. Bertran L. Binetti J. Aguilar C. Martínez S. Guiu Jurado E. et al (2020). Hepatocyte Notch Signaling Deregulation Related to Lipid Metabolism in Women with Obesity and Nonalcoholic Fatty Liver. Obesity28 (8), 1487–1493. 10.1002/oby.22873
11
Baffy G. Brunt E. M. Caldwell S. H. (2012). Hepatocellular Carcinoma in Non-alcoholic Fatty Liver Disease: an Emerging Menace. J. Hepatol.56 (6), 1384–1391. 10.1016/j.jhep.2011.10.027
12
Bansal R. van Baarlen J. Storm G. Prakash J. (2015). The Interplay of the Notch Signaling in Hepatic Stellate Cells and Macrophages Determines the Fate of Liver Fibrogenesis. Sci. Rep.5, 18272. 10.1038/srep18272
13
Bartolome A. Zhu C. Sussel L. Pajvani U. B. (2019). Notch Signaling Dynamically Regulates Adult Beta Cell Proliferation and Maturity. J. Clin. Invest.129 (1), 268–280. 10.1172/JCI98098
14
Begriche K. Massart J. Robin M. A. Bonnet F. Fromenty B. (2013). Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology58 (4), 1497–1507. 10.1002/hep.26226
15
Behary J. Amorim N. Jiang X. T. Raposo A. Gong L. McGovern E. et al (2021). Gut Microbiota Impact on the Peripheral Immune Response in Non-alcoholic Fatty Liver Disease Related Hepatocellular Carcinoma. Nat. Commun.12 (1), 187. 10.1038/s41467-020-20422-7
16
Bernsmeier C. Dill M. T. Provenzano A. Makowska Z. Krol I. Muscogiuri G. et al (2016). Hepatic Notch1 Deletion Predisposes to Diabetes and Steatosis via Glucose-6-Phosphatase and Perilipin-5 Upregulation. Lab. Invest.96 (9), 972–980. 10.1038/labinvest.2016.75
17
Bessone F. Razori M. V. Roma M. G. (2019). Molecular Pathways of Nonalcoholic Fatty Liver Disease Development and Progression. Cell Mol. Life Sci.76 (1), 99–128. 10.1007/s00018-018-2947-0
18
Bi P. Kuang S. (2015). Notch Signaling as a Novel Regulator of Metabolism. Trends Endocrinology Metabolism26 (5), 248–255. 10.1016/j.tem.2015.02.006
19
Bi P. Shan T. Liu W. Yue F. Yang X. Liang X. et al (2014). Inhibition of Notch Signaling Promotes browning of white Adipose Tissue and Ameliorates Obesity. Nat. Med.20 (8), 911–918. 10.1038/nm.3615
20
Bolos V. Grego-Bessa J. de la Pompa J. L. (2007). Notch Signaling in Development and Cancer.
21
Bray F. Ferlay J. Soerjomataram I. Siegel R. L. Torre L. A. Jemal A. (2018). Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin.68 (6), 394–424. 10.3322/caac.21492
22
Bray S. J. (2016). Notch Signalling in Context. Nat. Rev. Mol. Cel Biol.17 (11), 722–735. 10.1038/nrm.2016.94
23
Brunt E. M. Kleiner D. E. Wilson L. A. Sanyal A. J. Neuschwander-Tetri B. A. (2019). Improvements in Histologic Features and Diagnosis Associated with Improvement in Fibrosis in Nonalcoholic Steatohepatitis: Results from the Nonalcoholic Steatohepatitis Clinical Research Network Treatment Trials. Hepatology70 (2), 522–531. 10.1002/hep.30418
24
Bugianesi E. Gastaldelli A. Vanni E. Gambino R. Cassader M. Baldi S. et al (2005). Insulin Resistance in Non-diabetic Patients with Non-alcoholic Fatty Liver Disease: Sites and Mechanisms. Diabetologia48 (4), 634–642. 10.1007/s00125-005-1682-x
25
Bugianesi E. (2010). Insulin Resistance in Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des.16 (17).
26
Buzzetti E. Pinzani M. Tsochatzis E. A. (2016). The Multiple-Hit Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD). Metabolism65 (8), 1038–1048. 10.1016/j.metabol.2015.12.012
27
Caldwell S. H. Swerdlow R. H. Khan E. M. Iezzoni J. C. Hespenheide E. E. Parks J. K. et al (1999). Mitochondrial Abnormalities in Non-alcoholic Steatohepatitis. J. Hepatol.31 (3), 430–434. 10.1016/s0168-8278(99)80033-6
28
Caron A. Richard D. Laplante M. (2015). The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr.35, 321–348. 10.1146/annurev-nutr-071714-034355
29
Chalasani N. Younossi Z. Lavine J. E. Charlton M. Cusi K. Rinella M. et al (2018). The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology67 (1), 328–357. 10.1002/hep.29367
30
Chartoumpekis D. V. Yagishita Y. Fazzari M. Palliyaguru D. L. Rao U. N. M. Zaravinos A. et al (2018). Nrf2 Prevents Notch-Induced Insulin Resistance and Tumorigenesis in Mice. JCI Insight3 (5), 97735. 10.1172/jci.insight.97735
31
Chen Y. X. Weng Z. H. Zhang S. L. (2012). Notch3 Regulates the Activation of Hepatic Stellate Cells. World J. Gastroenterol.18 (12), 1397–1403. 10.3748/wjg.v18.i12.1397
32
Chen Y. Zheng S. Qi D. Zheng S. Guo J. Zhang S. et al (2012). Inhibition of Notch Signaling by a Gamma-Secretase Inhibitor Attenuates Hepatic Fibrosis in Rats. PLoS One7 (10), e46512. 10.1371/journal.pone.0046512
33
D'Souza B. Meloty-Kapella L. Weinmaster G. (2010). Canonical and Non-canonical Notch Ligands. Curr. Top. Dev. Biol.92, 73–129. 10.1016/S0070-2153(10)92003-6
34
Day C. P. James O. F. W. (19981943). Steatohepatitis: A Tale of Two “Hits”. Gastroenterol. (N.Y, N.Y.114 (4), 842–845. 10.1016/S0016-5085(98)70599-2
35
Deretic V. Levine B. (2018). Autophagy Balances Inflammation in Innate Immunity. Autophagy14 (2), 243–251. 10.1080/15548627.2017.1402992
36
Deretic V. Saitoh T. Akira S. (2013). Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol.13 (10), 722–737. 10.1038/nri3532
37
Ding W. Wu W. Chen Y. Chen H. Fan J. Qiao L. (2020). Expression of Notch Family Is Altered in Non-alcoholic Fatty Liver Disease. Mol. Med. Rep.22 (3), 1702–1708. 10.3892/mmr.2020.11249
38
Dongiovanni P. Rametta R. Meroni M. Valenti L. (2016). The Role of Insulin Resistance in Nonalcoholic Steatohepatitis and Liver Disease Development-Aa Potential Therapeutic Target?Expert Rev. Gastroenterol. Hepatol.10 (2), 229–242. 10.1586/17474124.2016.1110018
39
Donnelly K. L. Smith C. I. Schwarzenberg S. J. Jessurun J. Boldt M. D. Parks E. J. (2005). Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Invest.115 (5), 1343–1351. 10.1172/JCI23621
40
Duan J. L. Ruan B. Yan X. C. Liang L. Song P. Yang Z. Y. et al (2018). Endothelial Notch Activation Reshapes the Angiocrine of Sinusoidal Endothelia to Aggravate Liver Fibrosis and blunt Regeneration in Mice. Hepatology68 (2), 677–690. 10.1002/hep.29834
41
Dulai P. S. Singh S. Patel J. Soni M. Prokop L. J. Younossi Z. et al (2017). Increased Risk of Mortality by Fibrosis Stage in Nonalcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Hepatology65 (5), 1557–1565. 10.1002/hep.29085
42
Easl E. A. F. T. Easd E. A. F. T. Easo E. A. F. T. (2016). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-alcoholic Fatty Liver Disease. J. Hepatol.64 (6), 1388–1402. 10.1016/j.jhep.2015.11.004
43
El S. G. El-Shafey E. S. Elsherbiny E. S. (2021). Ziziphus Spina-Christi (L.) Fortified with Camellia Sinensis Mediates Apoptosis, Notch-1 Signaling, and Mitigates Obesity-Induced Non-alcoholic Fatty Liver. J. Food Biochem., e13849. 10.1111/jfbc.13849
44
Feng R. Luo C. Li C. Du S. Okekunle A. P. Li Y. et al (2017). Free Fatty Acids Profile Among Lean, Overweight and Obese Non-alcoholic Fatty Liver Disease Patients: a Case - Control Study. Lipids Health Dis.16 (1), 165. 10.1186/s12944-017-0551-1
45
Fisher A. B. (2011). Peroxiredoxin 6: a Bifunctional Enzyme with Glutathione Peroxidase and Phospholipase A(2) Activities. Antioxid. Redox Signal.15 (3), 831–844. 10.1089/ars.2010.3412
46
Francque S. Vonghia L. (2019). Pharmacological Treatment for Non-alcoholic Fatty Liver Disease. Adv. Ther.36 (5), 1052–1074. 10.1007/s12325-019-00898-6
47
Funahashi Y. Hernandez S. L. Das I. Ahn A. Huang J. Vorontchikhina M. et al (2008). A Notch1 Ectodomain Construct Inhibits Endothelial Notch Signaling, Tumor Growth, and Angiogenesis. Cancer Res.68 (12), 4727–4735. 10.1158/0008-5472.CAN-07-6499
48
Galluzzi L. Pietrocola F. Levine B. Kroemer G. (2014). Metabolic Control of Autophagy. Cell159 (6), 1263–1276. 10.1016/j.cell.2014.11.006
49
Garcia E. J. Vevea J. D. Pon L. A. (2018). Lipid Droplet Autophagy during Energy Mobilization, Lipid Homeostasis and Protein Quality Control. Front. Biosci. (Landmark Ed.23, 1552–1563. 10.2741/4660
50
Gehrke N. Schattenberg J. M. (2020). Metabolic Inflammation-A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease?Gastroenterology158 (7), 1929–1947. 10.1053/j.gastro.2020.02.020
51
Geisler F. Strazzabosco M. (2015). Emerging Roles of Notch Signaling in Liver Disease. Hepatology61 (1), 382–392. 10.1002/hep.27268
52
Gluchowski N. L. Becuwe M. Walther T. C. Farese R. V. (2017). Lipid Droplets and Liver Disease: from Basic Biology to Clinical Implications. Nat. Rev. Gastroenterol. Hepatol.14 (6), 343–355. 10.1038/nrgastro.2017.32
53
Grefhorst A. van de Peppel I. P. Larsen L. E. Jonker J. W. Holleboom A. G. (2021). The Role of Lipophagy in the Development and Treatment of Non-alcoholic Fatty Liver Disease. Front. Endocrinol.11. 10.3389/fendo.2020.601627
54
Gross D. A. Silver D. L. (2014). Cytosolic Lipid Droplets: from Mechanisms of Fat Storage to Disease. Crit. Rev. Biochem. Mol. Biol.49 (4), 304–326. 10.3109/10409238.2014.931337
55
Groth C. Fortini M. E. (2012). Therapeutic Approaches to Modulating Notch Signaling: Current Challenges and Future Prospects. Semin. Cel Dev Biol23 (4), 465–472. 10.1016/j.semcdb.2012.01.016
56
Guruharsha K. G. Kankel M. W. Artavanis-Tsakonas S. (2012). The Notch Signalling System: Recent Insights into the Complexity of a Conserved Pathway. Nat. Rev. Genet.13 (9), 654–666. 10.1038/nrg3272
57
Hagstrom H. Nasr P. Ekstedt M. Hammar U. Stal P. Hultcrantz R. et al (2017). Fibrosis Stage but Not NASH Predicts Mortality and Time to Development of Severe Liver Disease in Biopsy-Proven NAFLD. J. Hepatol.67 (6), 1265–1273. 10.1016/j.jhep.2017.07.027
58
Han J. Wang Y. (2018). mTORC1 Signaling in Hepatic Lipid Metabolism. Protein Cell9 (2), 145–151. 10.1007/s13238-017-0409-3
59
Hazari Y. Bravo-San P. J. Hetz C. Galluzzi L. Kroemer G. (2020). Autophagy in Hepatic Adaptation to Stress. J. Hepatol.72 (1), 183–196. 10.1016/j.jhep.2019.08.026
60
Hester D. Golabi P. Paik J. Younossi I. Mishra A. Younossi Z. M. (2020). Among Medicare Patients with Hepatocellular Carcinoma, Non-alcoholic Fatty Liver Disease Is the Most Common Etiology and Cause of Mortality. J. Clin. Gastroenterol.54 (5), 459–467. 10.1097/MCG.0000000000001172
61
Huang D. Q. El-Serag H. B. Loomba R. (2021). Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol.18 (4), 223–238. 10.1038/s41575-020-00381-6
62
Huang R. Guo F. Li Y. Liang Y. Li G. Fu P. et al (2021). Activation of AMPK by Triptolide Alleviates Nonalcoholic Fatty Liver Disease by Improving Hepatic Lipid Metabolism, Inflammation and Fibrosis. Phytomedicine92, 153739. 10.1016/j.phymed.2021.153739
63
Ioannou G. N. (2021). Epidemiology and Risk-Stratification of NAFLD-Associated HCC. J. Hepatol.8278. 02007. 10.1016/j.jhep.2021.08.012
64
Ipsen D. H. Lykkesfeldt J. Tveden-Nyborg P. (2018). Molecular Mechanisms of Hepatic Lipid Accumulation in Non-alcoholic Fatty Liver Disease. Cell Mol. Life Sci.75 (18), 3313–3327. 10.1007/s00018-018-2860-6
65
Jarvis H. Craig D. Barker R. Spiers G. Stow D. Anstee Q. M. et al (2020). Metabolic Risk Factors and Incident Advanced Liver Disease in Non-alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis of Population-Based Observational Studies. PLOS Med.17 (4), e1003100. 10.1371/journal.pmed.1003100
66
Koek G. H. Liedorp P. R. Bast A. (2011). The Role of Oxidative Stress in Non-alcoholic Steatohepatitis. Clin. Chim. Acta412 (15-16), 1297–1305. 10.1016/j.cca.2011.04.013
67
Kopan R. Ilagan M. X. (2009). The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell137 (2), 216–233. 10.1016/j.cell.2009.03.045
68
Kovall R. A. Blacklow S. C. (2010). Mechanistic Insights into Notch Receptor Signaling from Structural and Biochemical Studies. Curr. Top. Dev. Biol.92, 31–71. 10.1016/S0070-2153(10)92002-4
69
Lamming D. W. Sabatini D. M. (2013). A Central Role for mTOR in Lipid Homeostasis. Cel Metab.18 (4), 465–469. 10.1016/j.cmet.2013.08.002
70
Lee D. H. Jung Y. Y. Park M. H. Jo M. R. Han S. B. Yoon D. Y. et al (2019). Peroxiredoxin 6 Confers Protection against Nonalcoholic Fatty Liver Disease through Maintaining Mitochondrial Function. Antioxid. Redox Signaling31 (5), 387–402. 10.1089/ars.2018.7544
71
Lee Y. H. Yun M. R. Kim H. M. Jeon B. H. Park B. C. Lee B. W. et al (2016). Exogenous Administration of DLK1 Ameliorates Hepatic Steatosis and Regulates Gluconeogenesis via Activation of AMPK. Int. J. Obes. (Lond)40 (2), 356–365. 10.1038/ijo.2015.173
72
Li T. Yan H. Geng Y. Shi H. Li H. Wang S. et al (2019). Target Genes Associated with Lipid and Glucose Metabolism in Non-alcoholic Fatty Liver Disease. Lipids Health Dis.18 (1), 211. 10.1186/s12944-019-1154-9
73
Lin C. W. Zhang H. Li M. Xiong X. Chen X. Chen X. et al (2013). Pharmacological Promotion of Autophagy Alleviates Steatosis and Injury in Alcoholic and Non-alcoholic Fatty Liver Conditions in Mice. J. Hepatol.58 (5), 993–999. 10.1016/j.jhep.2013.01.011
74
Llovet J. M. Kelley R. K. Villanueva A. Singal A. G. Pikarsky E. Roayaie S. et al (2021). Hepatocellular Carcinoma. Nat. Rev. Dis. Primers7 (1), 6. 10.1038/s41572-020-00240-3
75
Lomonaco R. Ortiz-Lopez C. Orsak B. Webb A. Hardies J. Darland C. et al (2012). Effect of Adipose Tissue Insulin Resistance on Metabolic Parameters and Liver Histology in Obese Patients with Nonalcoholic Fatty Liver Disease. Hepatology55 (5), 1389–1397. 10.1002/hep.25539
76
Loomba R. Friedman S. L. Shulman G. I. (2021). Mechanisms and Disease Consequences of Nonalcoholic Fatty Liver Disease. Cell184 (10), 2537–2564. 10.1016/j.cell.2021.04.015
77
Marinho H. S. Real C. Cyrne L. Soares H. Antunes F. (2014). Hydrogen Peroxide Sensing, Signaling and Regulation of Transcription Factors. Redox Biol.2, 535–562. 10.1016/j.redox.2014.02.006
78
Matsumoto M. Pocai A. Rossetti L. Depinho R. A. Accili D. (2007). Impaired Regulation of Hepatic Glucose Production in Mice Lacking the Forkhead Transcription Factor Foxo1 in Liver. Cell Metab6 (3), 208–216. 10.1016/j.cmet.2007.08.006
79
McDaniell R. Warthen D. M. Sanchez-Lara P. A. Pai A. Krantz I. D. Piccoli D. A. et al (2006). NOTCH2 Mutations Cause Alagille Syndrome, a Heterogeneous Disorder of the Notch Signaling Pathway. Am. J. Hum. Genet.79 (1), 169–173. 10.1086/505332
80
Mederacke I. Hsu C. C. Troeger J. S. Huebener P. Mu X. Dapito D. H. et al (2013). Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of its Aetiology. Nat. Commun.4, 2823. 10.1038/ncomms3823
81
Mottillo E. P. Desjardins E. M. Crane J. D. Smith B. K. Green A. E. Ducommun S. et al (2016). Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cel Metab.24 (1), 118–129. 10.1016/j.cmet.2016.06.006
82
Nam Y. Sliz P. Song L. Aster J. C. Blacklow S. C. (2006). Structural Basis for Cooperativity in Recruitment of MAML Coactivators to Notch Transcription Complexes. Cell124 (5), 973–983. 10.1016/j.cell.2005.12.037
83
Nguyen P. Leray V. Diez M. Serisier S. Bloc H. J. L. Siliart B. et al (2008). Liver Lipid Metabolism. J. Anim. Physiol. Anim. Nutr.92 (3), 272–283. 10.1111/j.1439-0396.2007.00752.x
84
Niture S. Gyamfi M. A. Kedir H. Arthur E. Ressom H. Deep G. et al (2018). Serotonin Induced Hepatic Steatosis Is Associated with Modulation of Autophagy and Notch Signaling Pathway. Cell Commun. Signaling16 (1), 6. 10.1186/s12964-018-0282-6
85
Nowell C. S. Radtke F. (2017). Notch as a Tumour Suppressor. Nat. Rev. Cancer17 (3), 145–159. 10.1038/nrc.2016.145
86
O-Sullivan I. Zhang W. Wasserman D. H. Liew C. W. Liu J. Paik J. et al (2015). FoxO1 Integrates Direct and Indirect Effects of Insulin on Hepatic Glucose Production and Glucose Utilization. Nat. Commun.6, 7079. 10.1038/ncomms8079
87
Pajvani U. B. Qiang L. Kangsamaksin T. Kitajewski J. Ginsberg H. N. Accili D. (2013). Inhibition of Notch Uncouples Akt Activation from Hepatic Lipid Accumulation by Decreasing mTorc1 Stability. Nat. Med.19 (8), 1054–1060. 10.1038/nm.3259
88
Pajvani U. B. Shawber C. J. Samuel V. T. Birkenfeld A. L. Shulman G. I. Kitajewski J. et al (2011). Inhibition of Notch Signaling Ameliorates Insulin Resistance in a FoxO1-dependent Manner. Nat. Med.17 (8), 961–967. 10.1038/nm.2378
89
Pervez M. A. Khan D. A. Slehria A. U. R. Ijaz A. (2020). Delta-tocotrienol Supplementation Improves Biochemical Markers of Hepatocellular Injury and Steatosis in Patients with Nonalcoholic Fatty Liver Disease: A Randomized, Placebo-Controlled Trial. Complement. Therapies Med.52, 102494. 10.1016/j.ctim.2020.102494
90
Powell E. E. Wong V. W. Rinella M. (2021). Non-alcoholic Fatty Liver Disease. The Lancet. 10.1016/S0140-6736(20)32511-3
91
Rajasinghe L. D. Pindiprolu R. H. Gupta S. V. (2018). Delta-tocotrienol Inhibits Non-small-cell Lung Cancer Cell Invasion via the Inhibition of NF-kappaB, uPA Activator, and MMP-9. Onco Targets Ther.11, 4301–4314. 10.2147/OTT.S160163
92
Richter L. R. Wan Q. Wen D. Zhang Y. Yu J. Kang J. K. et al (2020). Targeted Delivery of Notch Inhibitor Attenuates Obesity-Induced Glucose Intolerance and Liver Fibrosis. ACS Nano14 (6), 6878–6886. 10.1021/acsnano.0c01007
93
Romeo S. (2019). Notch and Nonalcoholic Fatty Liver and Fibrosis. N. Engl. J. Med.380 (7), 681–683. 10.1056/NEJMcibr1815636
94
Saadati S. Hatami B. Yari Z. Shahrbaf M. A. Eghtesad S. Mansour A. et al (2019). The Effects of Curcumin Supplementation on Liver Enzymes, Lipid Profile, Glucose Homeostasis, and Hepatic Steatosis and Fibrosis in Patients with Non-alcoholic Fatty Liver Disease. Eur. J. Clin. Nutr.73 (3), 441–449. 10.1038/s41430-018-0382-9
95
Sahini N. Borlak J. (2014). Recent Insights into the Molecular Pathophysiology of Lipid Droplet Formation in Hepatocytes. Prog. Lipid Res.54, 86–112. 10.1016/j.plipres.2014.02.002
96
Sanyal A. J. (2019). Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol.16, 377–386.
97
Sato K. Gosho M. Yamamoto T. Kobayashi Y. Ishii N. Ohashi T. et al (2015). Vitamin E Has a Beneficial Effect on Nonalcoholic Fatty Liver Disease: a Meta-Analysis of Randomized Controlled Trials. Nutrition31 (7-8), 923–930. 10.1016/j.nut.2014.11.018
98
Sawitza I. Kordes C. Reister S. Haussinger D. (2009). The Niche of Stellate Cells within Rat Liver. Hepatology50 (5), 1617–1624. 10.1002/hep.23184
99
Shao H. Huang Q. Liu Z. J. (2012). Targeting Notch Signaling for Cancer Therapeutic Intervention. Adv. Pharmacol.65, 191–234. 10.1016/B978-0-12-397927-8.00007-5
100
Shen F. Xiong Z. Kong J. Wang L. Cheng Y. Jin J. et al (2019). Triptolide Impairs Thioredoxin System by Suppressing Notch1-Mediated PTEN/Akt/Txnip Signaling in Hepatocytes. Toxicol. Lett.300, 105–115. 10.1016/j.toxlet.2018.10.024
101
Sheng J. Zhang B. Chen Y. Yu F. (2020). Capsaicin Attenuates Liver Fibrosis by Targeting Notch Signaling to Inhibit TNF-Alpha Secretion from M1 Macrophages. Immunopharmacol Immunotoxicol42 (6), 556–563. 10.1080/08923973.2020.1811308
102
Sies H. (2015). Oxidative Stress: a Concept in Redox Biology and Medicine. Redox Biol.4, 180–183. 10.1016/j.redox.2015.01.002
103
Singh R. Kaushik S. Wang Y. Xiang Y. Novak I. Komatsu M. et al (2009). Autophagy Regulates Lipid Metabolism. Nature458 (7242), 1131–1135. 10.1038/nature07976
104
Smith G. I. Shankaran M. Yoshino M. Schweitzer G. G. Chondronikola M. Beals J. W. et al (2020). Insulin Resistance Drives Hepatic De Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Invest.130 (3), 1453–1460. 10.1172/JCI134165
105
Song N. J. Yun U. J. Yang S. Wu C. Seo C. R. Gwon A. R. et al (2016). Notch1 Deficiency Decreases Hepatic Lipid Accumulation by Induction of Fatty Acid Oxidation. Sci. Rep.6, 19377. 10.1038/srep19377
106
Stanford K. I. Middelbeek R. J. Townsend K. L. An D. Nygaard E. B. Hitchcox K. M. et al (2013). Brown Adipose Tissue Regulates Glucose Homeostasis and Insulin Sensitivity. J. Clin. Invest.123 (1), 215–223. 10.1172/JCI62308
107
Tilg H. Adolph T. E. Moschen A. R. (2021). Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited after a Decade. Hepatol. (Baltimore, Md73 (2), 833–842. 10.1002/hep.31518
108
Tilg H. Moschen A. R. (2010). Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatol. (Baltimore, Md52 (5), 1836–1846. 10.1002/hep.24001
109
Ueno T. Komatsu M. (2017). Autophagy in the Liver: Functions in Health and Disease. Nat. Rev. Gastroenterol. Hepatol.14 (3), 170–184. 10.1038/nrgastro.2016.185
110
Valenti L. Mendoza R. M. Rametta R. Maggioni M. Kitajewski C. Shawber C. J. et al (2013). Hepatic Notch Signaling Correlates with Insulin Resistance and Nonalcoholic Fatty Liver Disease. Diabetes62 (12), 4052–4062. 10.2337/db13-0769
111
van Es J. H. van Gijn M. E. Riccio O. van den Born M. Vooijs M. Begthel H. et al (2005). Notch/gamma-secretase Inhibition Turns Proliferative Cells in Intestinal Crypts and Adenomas into Goblet Cells. Nature435 (7044), 959–963. 10.1038/nature03659
112
Vernon G. Baranova A. Younossi Z. M. (2011). Systematic Review: the Epidemiology and Natural History of Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis in Adults. Aliment. Pharmacol. Ther.34 (3), 274–285. 10.1111/j.1365-2036.2011.04724.x
113
Vilar-Gomez E. Calzadilla-Bertot L. Wai-Sun W. V. Castellanos M. Aller-de L. F. R. Metwally M. et al (2018). Fibrosis Severity as a Determinant of Cause-specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology155 (2), 443–457. 10.1053/j.gastro.2018.04.034
114
Vilar-Gomez E. Vuppalanchi R. Gawrieh S. Ghabril M. Saxena R. Cummings O. W. et al (2020). Vitamin E Improves Transplant-free Survival and Hepatic Decompensation Among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology71 (2), 495–509. 10.1002/hep.30368
115
Wang C. Li X. Wang H. Xie Q. Xu Y. (2014). Notch1-nuclear Factor kappaB Involves in Oxidative Stress-Induced Alcoholic Steatohepatitis. Alcohol Alcohol49 (1), 10–16. 10.1093/alcalc/agt167
116
Wang H. Liu Y. Wang D. Xu Y. Dong R. Yang Y. et al (2019). The Upstream Pathway of mTOR-Mediated Autophagy in Liver Diseases. Cells8 (12), 1597. 10.3390/cells8121597
117
Wang Y. Wong G. L. He F. Sun J. Chan A. W. Yang J. et al (2020). Quantifying and Monitoring Fibrosis in Non-alcoholic Fatty Liver Disease Using Dual-Photon Microscopy. Gut69 (6), 1116–1126. 10.1136/gutjnl-2019-318841
118
Watt M. J. Miotto P. M. De Nardo W. Montgomery M. K. (2019). The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev.40 (5), 1367–1393. 10.1210/er.2019-00034
119
Wilson J. J. Kovall R. A. (2006). Crystal Structure of the CSL-Notch-Mastermind Ternary Complex Bound to DNA. Cell124 (5), 985–996. 10.1016/j.cell.2006.01.035
120
Wong V. W. Wong G. L. Choi P. C. Chan A. W. Li M. K. Chan H. et al (2010). Disease Progression of Non-alcoholic Fatty Liver Disease: a Prospective Study with Paired Liver Biopsies at 3years. Gut59 (7), 969–974. 10.1136/gut.2009.205088
121
Wu Y. Cain-Hom C. Choy L. Hagenbeek T. J. de Leon G. P. Chen Y. et al (2010). Therapeutic Antibody Targeting of Individual Notch Receptors. Nature464 (7291), 1052–1057. 10.1038/nature08878
122
Xu J. Chi F. Guo T. Punj V. Lee W. N. French S. W. et al (2015). NOTCH Reprograms Mitochondrial Metabolism for Proinflammatory Macrophage Activation. J. Clin. Invest.125 (4), 1579–1590. 10.1172/JCI76468
123
Yakaryilmaz F. Guliter S. Savas B. Erdem O. Ersoy R. Erden E. et al (2007). Effects of Vitamin E Treatment on Peroxisome Proliferator-Activated Receptor-Alpha Expression and Insulin Resistance in Patients with Non-alcoholic Steatohepatitis: Results of a Pilot Study. Intern. Med. J.37 (4), 229–235. 10.1111/j.1445-5994.2006.01295.x
124
Younossi Z. Anstee Q. M. Marietti M. Hardy T. Henry L. Eslam M. et al (2018). Global burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol.15 (1), 11–20. 10.1038/nrgastro.2017.109
125
Younossi Z. M. Koenig A. B. Abdelatif D. Fazel Y. Henry L. Wymer M. (2016). Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology64 (1), 73–84. 10.1002/hep.28431
126
Younossi Z. M. (2019). Non-alcoholic Fatty Liver Disease - A Global Public Health Perspective. J. Hepatol.70 (3), 531–544. 10.1016/j.jhep.2018.10.033
127
Younossi Z. Stepanova M. Ong J. P. Jacobson I. M. Bugianesi E. Duseja A. et al (2019). Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol.17 (4), 748–755. 10.1016/j.cgh.2018.05.057
128
Yu J. Zhu C. Wang X. Kim K. Bartolome A. Dongiovanni P. et al (2021). Hepatocyte TLR4 Triggers Inter-hepatocyte Jagged1/Notch Signaling to Determine NASH-Induced Fibrosis. Sci. Transl Med.13 (599), 1692. 10.1126/scitranslmed.abe1692
129
Zhang M. Wu P. Li M. Guo Y. Tian T. Liao X. et al (2021). Inhibition of Notch1 Signaling Reduces Hepatocyte Injury in Nonalcoholic Fatty Liver Disease via Autophagy. Biochem. Biophysical Res. Commun.547, 131–138. 10.1016/j.bbrc.2021.02.039
130
Zhang S. Yang Y. Liang Z. Duan W. Yang J. Yan J. et al (2013). Silybin-mediated Inhibition of Notch Signaling Exerts Antitumor Activity in Human Hepatocellular Carcinoma Cells. PLoS One8 (12), e83699. 10.1371/journal.pone.0083699
131
Zhao N. J. Liao M. J. Wu J. J. Chu K. X. (2017). Curcumin Suppresses Notch-1 Signaling: Improvements in Fatty Liver and Insulin Resistance in Rats. Mol. Med. Rep.17 (1), 819–826. 10.3892/mmr.2017.7980
132
Zhu C. Ho Y. Salomao M. A. Dapito D. H. Bartolome A. Schwabe R. F. et al (2021). Notch Activity Characterizes a Common Hepatocellular Carcinoma Subtype with Unique Molecular and Clinicopathologic Features. J. Hepatol.74 (3), 613–626. 10.1016/j.jhep.2020.09.032
133
Zhu C. Kim K. Wang X. Bartolome A. Salomao M. Dongiovanni P. et al (2018). Hepatocyte Notch Activation Induces Liver Fibrosis in Nonalcoholic Steatohepatitis. Sci. Transl Med.10 (468), 344. 10.1126/scitranslmed.aat0344
134
Ziaei S. Halaby R. (2016). Immunosuppressive, Anti-inflammatory and Anti-cancer Properties of Triptolide: A Mini Review. Avicenna J. Phytomed6 (2), 149–164.
Summary
Keywords
Notch signaling pathway, non-alcoholic fatty liver disease (NAFLD), steatohepatitis, lipid metabolism, insulin resistance (IR), fibrogenesis, autophagy
Citation
Xu H and Wang L (2021) The Role of Notch Signaling Pathway in Non-Alcoholic Fatty Liver Disease. Front. Mol. Biosci. 8:792667. doi: 10.3389/fmolb.2021.792667
Received
11 October 2021
Accepted
01 November 2021
Published
24 November 2021
Volume
8 - 2021
Edited by
Jinhang Gao, Sichuan University, China
Reviewed by
Huichang Bi, Sun Yat-sen University, China
Xiang Zhang, The Chinese University of Hong Kong, China
Lei Chen, Eastern Hepatobiliary Surgery Hospital, China
Updates
Copyright
© 2021 Xu and Wang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Wang, fierywang@163.com
This article was submitted to Molecular Diagnostics and Therapeutics, a section of the journal Frontiers in Molecular Biosciences
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.