 Stanislav I. Svetlov1,2*
Stanislav I. Svetlov1,2* Victor Prima1*
Victor Prima1* Olena Glushakova1 Artem Svetlov1 Daniel R. Kirk3 Hector Gutierrez3 Victor L. Serebruany4 Kenneth C. Curley5
Olena Glushakova1 Artem Svetlov1 Daniel R. Kirk3 Hector Gutierrez3 Victor L. Serebruany4 Kenneth C. Curley5 Kevin K. W. Wang6
Kevin K. W. Wang6 Ronald L. Hayes1
Ronald L. Hayes1
- 1 Banyan Laboratories, Inc, Alachua, FL, USA
- 2 Department of Medicine, University of Florida, Gainesville, FL, USA
- 3 Department of Mechanical and Aerospace Engineering, Florida Institute of Technology, Melbourne, FL, USA
- 4 Heart Drug Research LLC, Towson, MD, USA
- 5 United States Army Medical Research and Materiel Command, Fort Detrick, MD, USA
- 6 Department of Psychiatry, University of Florida, Gainesville, FL, USA
A number of experimental models of blast brain injury have been implemented in rodents and larger animals. However, the variety of blast sources and the complexity of blast wave biophysics have made data on injury mechanisms and biomarkers difficult to analyze and compare. Recently, we showed the importance of rat position toward blast generated by an external shock tube. In this study, we further characterized blast producing moderate traumatic brain injury and defined “composite” blast and primary blast exposure set-ups. Schlieren optics visualized interaction between the head and a shock wave generated by external shock tube, revealing strong head acceleration upon positioning the rat on-axis with the shock tube (composite blast), but negligible skull movement upon peak overpressure exposure off-axis (primary blast). Brain injury signatures of a primary blast hitting the frontal head were assessed and compared to damage produced by composite blast. Low to negligible levels of neurodegeneration were found following primary blast compared to composite blast by silver staining. However, persistent gliosis in hippocampus and accumulation of GFAP/CNPase in circulation was detected after both primary and composite blast. Also, markers of vascular/endothelial inflammation integrin alpha/beta, soluble intercellular adhesion molecule-1, and L-selectin along with neurotrophic factor nerve growth factor-beta were increased in serum within 6 h post-blasts and persisted for 7 days thereafter. In contrast, systemic IL-1, IL-10, fractalkine, neuroendocrine peptide Orexin A, and VEGF receptor Neuropilin-2 (NRP-2) were raised predominantly after primary blast exposure. In conclusion, biomarkers of major pathological pathways were elevated at all blast set-ups. The most significant and persistent changes in neuro-glial markers were found after composite blast, while primary blast instigated prominent systemic cytokine/chemokine, Orexin A, and Neuropilin-2 release, particularly when primary blast impacted rats with unprotected body.
Introduction
The nature of twenty-first century warfare has led to a significant increase in human exposure to blast overpressure (OP) impulses, which result in a complex of neuro-somatic disorders, including traumatic brain injury (TBI). Blast-related casualties outnumber conventional injuries during the last several years in Iraq and Afghanistan, while blast itself is being termed “the fourth weapon of mass destruction” (Born, 2005). Moreover, for every blast-related fatality, many more soldiers suffer multiple, low level non-lethal blast exposures. This often leads to mild traumatic brain injury (mTBI), which is rarely recognized in a timely manner and has become a signature injury of the Iraq and Afghanistan conflicts (Warden, 2006; Jones et al., 2007; Terrio et al., 2009). Symptoms of mild or moderate blast brain injury often do not manifest themselves until sometime after the injury has occurred (Cernak et al., 1999, 2011; Yilmaz and Pekdemir, 2007; Cernak and Noble-Haeusslein, 2010) and go undiagnosed and untreated because emergency medical attention is directed toward more visible injuries, such as penetrating flesh wounds (Belanger et al., 2005; Nelson et al., 2006; Wolf et al., 2009). However, even mild and moderate brain injuries can produce significant deficits and, particularly when repeated, can lead to sustained neuro-somatic damage and neurodegeneration (Cernak and Noble-Haeusslein, 2010). Thus, identifying pathogenic mechanisms and biochemical markers of blast brain injury in relevant experimental models is vital to the development of diagnostics for mTBI through severe TBI.
However, because of the design inconsistency of blast/shock generators used in the different studies, incomplete understanding of blast wave biophysics associated with real explosives vs. those produced by air or gas-driven shock tubes, and the details of wave interaction with model animals, disparities between laboratory models and data on brain injury mechanisms and putative biomarkers have been difficult to analyze and compare (Jaffin et al., 1987; Elsayed, 1997; Guy et al., 1998b; Chavko et al., 2009; Gyorgy et al., 2011, see Bass et al., 2012 for review). Moreover, pathogenic pathways and molecular signatures of neural responses and injurious effects of blast exposures remain elusive. Recently, we developed and employed a model of “composite” blast exposure with controlled parameters of blast wave impact and brain injury in rats (Svetlov et al., 2010). Our studies demonstrated the importance of positional orientation of the head and whole body of rats toward a blast wave generated from an external shock tube (Svetlov et al., 2011). Data from several laboratories including our studies (Svetlov et al., 2010, 2011) suggest that the mechanisms underlying blast-induced injuries, particularly mild/moderate, appear to be distinct from those imposed by mechanical impact or acceleration, and may involve the prominent systemic response (please see Cernak, 2010 for review).
The main objective of this study was to compare the effects of moderate peak overpressure exposure (primary blast) with brain injury produced by a severe/moderate blast accompanied by strong head acceleration (composite blast). The high speed imaging using Schlieren optic demonstrated blast wave interaction with the animal’s head/body and revealed a negligible degree of acceleration at a position “off-axis” with the shock tube (primary blast wave exposure) compared to the “on-axis” experimental setup, which was accompanied by strong head/cervical acceleration generated by peak OP + venting gas (composite blast, or primary blast wave plus gas jetting phenomena). The specific dynamics of systemic, vascular inflammatory, and neuro-glial injury signatures, including neuron-specific enolase (NSE)/ubiquitin C-terminal hydrolase L1 (UCH-L1), GFAP, and CNPase biomarkers in serum, were established and characterized. For major pathway signatures and biomarkers, the detected levels raised at all the set-ups studied. However, the most significant and persistent changes in neuro-glial markers were found after composite blast, while primary blast instigated prominent systemic/vascular reactions, particularly when the whole animal body was subjected to blast wave.
Materials and Methods
Hardware Design and Setup
The compressed air-driven shock tube capable of generating a wide range of controlled blast waves was described in details previously (Svetlov et al., 2010). The tube consists of two sections: high pressure (driver) and low-pressure (driven) separated by a diaphragm. Peak overpressure, composition, and duration of the generated high pressure shockwaves are determined by the shock tube configuration including thickness, type of diaphragm material, driver/driven ratio, and the initial driver pressure at the moment of diaphragm rupture. In the presented series of experiments to explore the effects of different components of the blast/shock waves on the targeted animal brain we employed different spatial set-ups as described below. The blast pressure data was acquired using PCB piezoelectric blast pressure transducers and LabView 8.2 software. A National Instruments 1.25 M samples/s data acquisition card was used to acquire data from multiple channels. The rat head images during the blast event were captured at 40,000 frames/s using a high speed video camera (Phantom V310, Vision Research, Wayne, NJ, USA) and mirror-based Schlieren optics.
Animal Exposure to a Controlled Blast Wave
All experimental procedures in rats, including post-blast euthanasia, tissue, and blood collection were performed under guidelines and upon approval by the IACUC of the University of Florida and the ACURO office of the Department of Defense. Modeling of the primary blast and the “composite” overpressure load was achieved by variable positioning of the target vs. blast generator. All rats were anesthetized with isoflurane inhalations described previously in detail. After reaching a deep plane of anesthesia, they were placed into a holder exposing either only their head (body-armored setup) or whole body at the distance 5 cm below the exit nozzle of the shock tube. Rats were positioned either directly on the shock tube axis or at the 45° angle to it to expose them correspondingly to the “composite” blast including the compressed air jet or only to the primary blast wave (Figure 1D). Animals were then subjected to a single blast with a mean peak overpressure at the target of 230–380 kPa (Figures 1A,B). The exact static and dynamic overpressure values depending on the angle and distance of rat head from the nozzle of shock tube were established during the prior calibration tests (Figure 1C). The control group of animals underwent the same treatment (anesthesia, handling, recovery) except they were not exposed to blast.
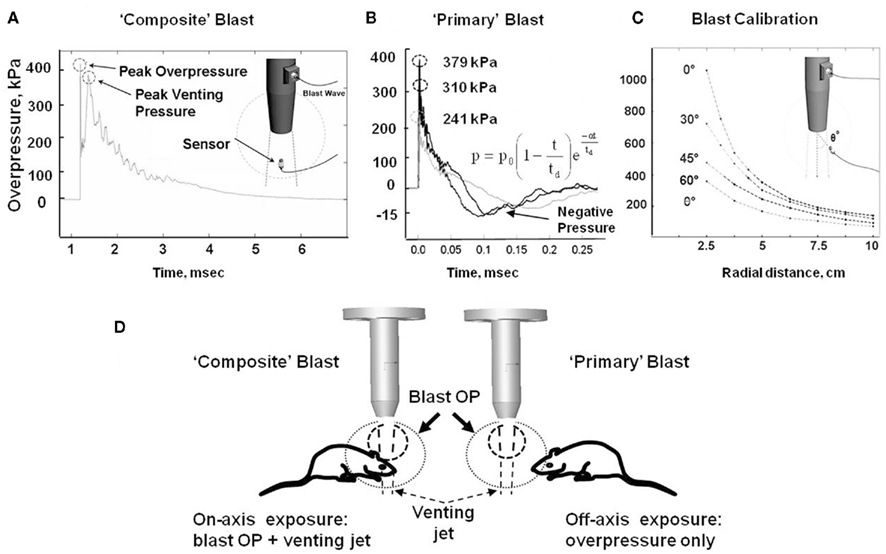
Figure 1. Schematic presentation of blast exposure modeling in rats. (A) Overpressure recording on shock tube axis at 5 cm from the nozzle. (B) Overpressure recording with external “pencil” PCB at 5 cm and 45° from shock tube nozzle at three different diaphragm configurations. (C) Calibration of pressure on rat head depending on the angle and distance from the nozzle of shock tube. (D) Different shock tube set-ups to model “primary” and “composite” blast. Inset formula in (B) an empirical expression for the pressure decay with time at a fixed distance is characterized by a decay parameter α Kinney (1985).
Blood and Tissues Collection
At the required time points following blast exposure, animals were euthanized, blood was withdrawn directly from the heart under isoflurane anesthesia and brain tissue samples were collected, snap-frozen in liquid nitrogen and stored at −70°C until further analysis. Immunohistochemical analysis. At 1 and 7 days after TBI (primary, head-only blast) animals were euthanized with lethal dose of pentobarbital, transcardially perfused with 4% paraformaldehyde and whole brains were removed, processed, and embedded in paraffin. Immunohistochemistry analysis was performed on paraffin-embedded 6 μm brain sections. Slides were de-paraffinized, incubated for 10 min at 95°C in Trilogy solution (Cell Marque, Rocklin, CA, USA) for antigen retrieval, blocked for endogenous peroxides, and incubated with primary antibodies for GFAP (Cell Signaling Technology, Danvers, MA, USA) or CNPase (Abcam, Cambridge, MA, USA) overnight at 4°C followed by treatments with secondary antibodies. The staining was visualized with 3,3′-diaminobenzidine (DAB; Dako, Carpinteria, CA, USA) for brown color development. Sections were counterstained with Hematoxylin (Dako). Negative controls were performed by treatment with species-matched secondary antibodies only (not shown). The slides were scanned and examined using Aperio ScanScope GL system with either 5× or 20× objective and ScanScope software.
Silver Staining Assessment of Neurodegeneration in Rat Brain
Neuroinjury and neurodegeneration was examined in the perfused and fixed brains using silver staining histochemical procedures according to Neuroscience Associates (Knoxville, TN, USA) utilizing the de Olmos Amino Cupric Silver Stain as previously described in detail (Svetlov et al., 2010). In addition, silver staining Kit from FD NeuroTechnologies (Ellicott City, MD, USA) was used where indicated. Rats were subjected to (i) “composite” head-directed severe blast exposure (358 kPa/10 ms total) on-axis (body protected); (ii) primary blast-off-axis exposure to peak overpressure only (233 kPa/113 μs total); and (iii) controlled cortical impact (CCI) of 2.0 mm depth performed as described previously (Liu et al., 2010).
Western Blot Analysis of Brain Tissues
For Western blot analyses tissue samples were prepared, separated by SDS-polyacrylamide gel electrophoresis and electro-blotted onto polyvinylidene difluoride membranes as described previously in detail (Svetlov et al., 2010). After overnight incubation with primary antibodies for CNPase or Neuropilin-2 (Cell Signaling Technology, Danvers, MA, USA) proteins were incubated with conjugated secondary antibodies and detected by either colorimetric or chemiluminescent (ECL) detection system. Actin was used as a loading control and bands of interest were normalized for actin expression. Semi-quantitative assessment of protein levels by western blot densitometry was conducted using NIH ImageJ image processing program.
Protein ELISA assays. Commercially available Sandwich ELISA (SW ELISA) kits for GFAP (BioVendor, Candler, NC, USA), NSE (Life Sci. Advanced Tech., St. Petersburg, FL, USA), β-NGF; Abnova, Walnut, CA, USA), Orexin A (Uscn Life Sci., Wuhan, P. R. China), L-selectin (CUSABIO Biotech, Wuhan, P. R. China) and soluble intercellular adhesion molecule-1 (sICAM-1; CUSABIO Biotech) were used according to the manufacturer’s instructions. UCH-L1 in CSF and plasma was quantitatively detected using proprietary SW ELISA (Banyan Biomarkers, Inc.) and recombinant UCH-L1 as standard.
Antibody Array Assays
Custom Biotin Label-based (L-series) Rat Antibody array (RayBiotech, Norcross, GA, USA) was used to assess relative levels of Interleukin-1, Interleukin-10, Neuropilin-2, Fractalkine, and Integrin α/β in rat serum following blast exposure.
Statistics
Statistical analyses were performed using GraphPad Prism 5 software. Values are means ± SEM. Data were evaluated by two-tailed unpaired t-test with or without Welch corrections where indicated.
Results
Rat Models of Blast Exposure Using External Shock Tube: Primary Blast Load vs. “Composite” Blast Exposure
Our shock tube was designed and built to model a freely expanding blast wave as generated by a typical explosion. Both static and dynamic (total) pressures were measured as functions of angle and radial distance from shock tube exit using piezoelectric blast pressure transducers positioned at the target (Figure 1C). The pressure transducers registered three distinct events: (i) peak OP, (ii) gas venting jet-on-axis only, and (iii) negative pressure phase-off-axis only (Figures 1A,B). The exhaust of venting gas apparently distorted propagation of the blast wave and no negative phase was registered when dynamic pressure was measured on-axis of shock tube (Figure 1A), while a distinct and substantial negative phase (15–20 kPa) was detected off-axis (Figure 1B). Peak OP, positive phase duration, and impulse appear to be the key parameters that correlate to injury and likelihood of fatality in animals and humans, for various orientations of the specimen relative to the blast wave. A schematic of a shock tube nozzle and the alternative rat locations relative to the shock tube axis, blast OP wave, and gas venting cone is shown in Figure 1D. Shock tubes produce a “venting gas jet” immediately after the blast wave forms, substantially contaminating the blast wave in the direction of the shock tube axis (Figure 1D). In a composite blast setup, venting gas jet lasts the longest (up to ∼3–5 ms), albeit lower in magnitude than peak overpressure, represents the bulk of blast impulse, and possibly produces the most devastating impact. Schlieren optics (Figure 2A) demonstrated a strong downward head acceleration following the passage of peak overpressure which lasts 50–100 μs. However, cranial deformation was more severe during the gas venting phase, lasting up to 5 ms. This effect was eliminated by placing rats off-axis from the venting jet in a way that the main effect acting on the specimen is the peak overpressure event. The high speed recording coupled with Schlieren optical system visualized interaction of the blast wave with the animal head/body and revealed a negligible degree of acceleration at rat positioning “off-axis” toward shock tube (primary blast; Figure 2B). The pressure on the surface of rats was calibrated depending on the distance and angle from the nozzle of shock tube (Figure 1C).

Figure 2. Visualization of blast wave interaction with head on-axis (“composite” blast) and off-axis (primary blast) using Schlieren optics. High speed recording with Schlieren optics: (A) “composite blast”; (B) “primary blast.” Black arrows indicate formation, traveling, and interaction of blast wave with rat head (accomplished within ∼0.1 ms). White arrows show gas venting jet hitting rat head after blast wave passed through (persists for milliseconds). The solid contour line in (A) outlines the shape of animal head at time point 0; the dotted line-current shape. Please see Section “Materials and Methods” for details.
Neural Injury and Gliosis in Rat Brain after Different Blast Exposures Assessed by Silver Staining and Immunohistochemistry
As can be seen in Figure 3, composite blast (on-axis) produces silver accumulation at the seventh day post-blast (Figures 3A,D), particularly in the hippocampus (indicated by arrows). CCI also results in positive staining in ipsilateral cortex and hippocampus (Figures 3C,F). In contrast, there was a rare occurrence of silver accumulation observed in the cortex or hippocampus after exposure to primary blast (Figures 3B,E; indicated by arrowheads).
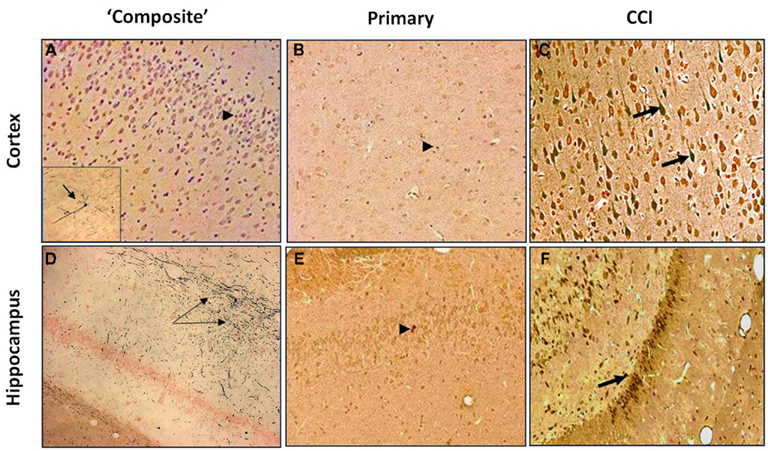
Figure 3. Silver Staining of coronal brain sections following primary or “composite” blast exposure. Corresponding tissue staining 7 days after “composite blast,” primary blast, and CCI is shown in (A–C) for cortex, and in (D–F) for hippocampus. Arrowheads indicate occasional silver accumulation in the cells of non-neuronal origin. Arrows indicate diffuse silver accumulation in neurons. Figure 3A inset: a very rare accumulation of silver in a cortical neuron. Please see Section “Materials and Methods” for details.
Time-dependent expression of GFAP and CNPase characteristic for astrocytes and oligodendrocytes, respectively was studied by IHC after moderate composite on-axis blast (358 kPa/∼10 ms) with strong head acceleration or moderate primary off-axis blast (234 kPa/113.8 μs positive phase) with minor head acceleration (Figure 4). These data suggest that both primary and “composite” blasts strongly induce astrogliosis (GFAP, Figure 4: upper panel) and oligodendrocytosis (CNPase, Figure 4: lower panel) in rat hippocampus evident as early as 1 day and lasting up to 7 days post-blast.
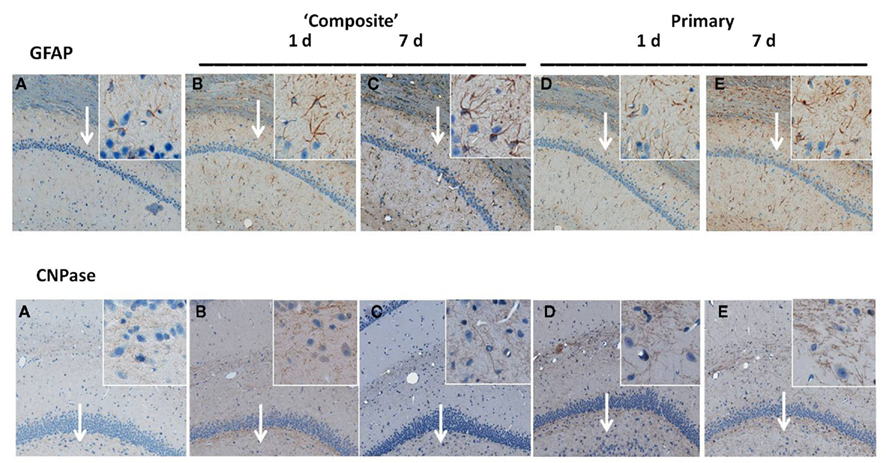
Figure 4. Immunohistochemical analysis of astrocyte and oligodendrocyte markers in hippocampus after blast. Time-dependent GFAP and CNPase expression was studied by IHC on paraffin-embedded 6 μm brain sections after blast exposure at different set-ups. (A) Naive; (B) “composite” blast, 1 day; (C) “composite,” 7 days; (D) “primary,” 1 day; and (E) “primary,” 7 days. Magnifications 5× and 20× (insets) are shown. Arrows indicate inset locations for CA1 region (GFAP) and DG region (CNPase) in hippocampus. Please see Section “Materials and Methods” for details.
Serum Levels of Biomarkers of Neuro-Gial Injury Following Blast Exposure
To asses if markers of neuronal injury are released into circulation, we assayed serum levels of NSE and UCH-L1 after different blast exposures (Figure 5). As shown in Figure 5A, remarkable accumulation of NSE in serum occurred within 6 h following exposure to either “composite” or primary blast, and persisted up to 7 days post-blast. Average serum UCH-L1 level was also elevated during 1–7 days after “primary” blast (Figure 5B), though its difference from controls was statistically significant only at 1 day post-blast.
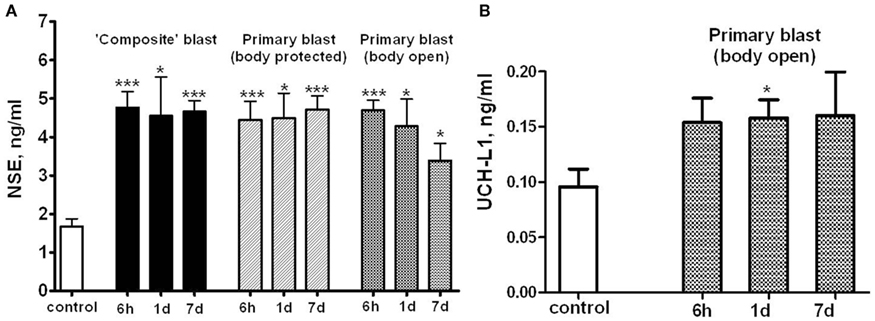
Figure 5. Blast-induced accumulation of NSE and UCHL-1 in rat serum. Blood was collected from overpressure-exposed rats at different shock tube set-ups and assayed by NSE (A) and UCHL-1 (B) SW ELISA Kits. Unpaired t-test was used to analyze statistical significance of values. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 vs. naïve. Please see Section “Materials and Methods” for details.
Glia cell-specific up-regulation of GFAP and CNPase in brain after either “composite” or primary blast was accompanied by a significant serum accumulation of GFAP and CNPase biomarkers measured by SW ELISA for GFAP (Figure 6A) and semi-quantitative western blot densitometry for serum CNPase (Figure 6B). These biomarkers persisted in blood up to 7 days post-blast at both blast set-ups employed.
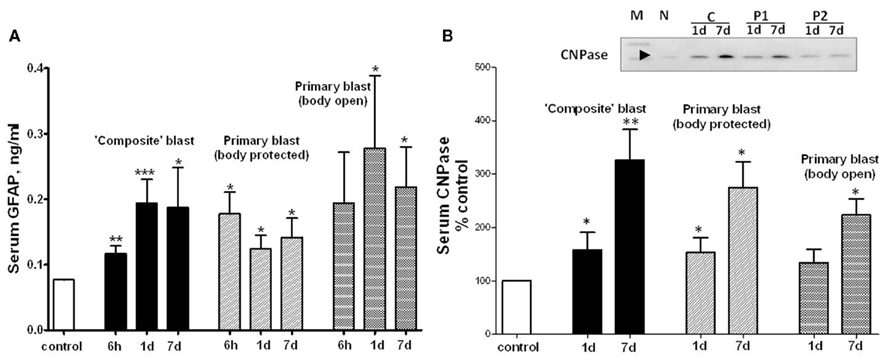
Figure 6. GFAP and CNPase levels in blood after different blast exposures. Blood was collected after OP exposure at different shock tube set-ups. (A) Serum GFAP detection by SW ELISA; (B) semi-quantitative serum CNPase detection by western blot densitometry. Inset: representative western blot. (N, naïve; C, “composite”; P1, primary/head; P2, primary/body). t-Test with Welch correction was done. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 vs. naïve. Please see Section “Materials and Methods” for details.
Systemic, Vascular Inflammatory, Neuroendocrine and Growth Factor Responses Following Different Blast Exposures
Based on our previous global and targeted proteomic data, the following molecular components and injury biomarkers were assessed in rat serum. Systemic/vascular responses: interleukin-1 and interleukin-10 (IL-1, IL-10), adipo-chemokine Fractalkine/CX3CL1, Integrin α/β, a complement receptor composed of CD11c/CD18, sICAM-1, and L-, E-selectin.
Cytokine/Chemokine Levels after Blast Exposures
We hypothesized that systemic responses and neuroinflammmation together with impaired vascular reaction in the brain, result in enhancement of endothelial permeability/leakage, infiltration of macrophages from circulation and activation of brain-resident microglia cells. As can be seen in Figures 7A,B, both pro-inflammatory (IL-1) and counteracting anti-inflammatory molecules (IL-10) accumulate in circulation at 24 h after open body exposure to frontal (off-axis) blast. These results are in agreement with data obtained using non-blast TBI models (Dietrich et al., 2004; Maegele et al., 2007). Moreover, CX3CL1 chemokine Fractalkine was also significantly elevated after primary blast further suggesting a systemic component in response to blast (Figure 7C) consistent with reports on the level of this chemokine in patients with TBI and in mouse model of closed head injury (Rancan et al., 2004; Ralay Ranaivo et al., 2011). While immune cell-derived IL-1, IL-10, and fractalkine were significantly increased predominantly after primary blast exposure, integrin alpha/beta levels were elevated at all set-ups indicating that blast is triggering microcirculatory disorders whether it produces head hyperacceleration or not.
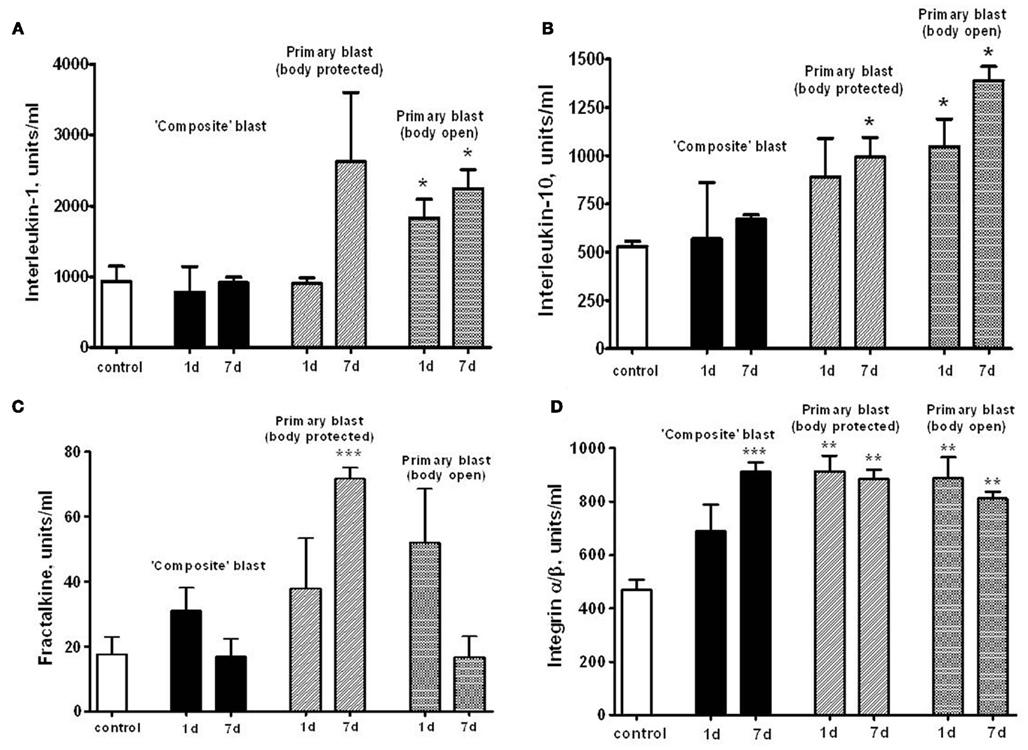
Figure 7. Serum levels of interleukins, fractalkine, and integrin α/β after blast. Concentrations of interleukin-1 (A), interleukin-10 (B), fractalkine (C), and integrin α/β (D) were assessed in rat serum by RayBiotech antibody arrays. Please see Section “Materials and Methods” for details. Blood was collected from OP-exposed rats at different shock tube set-ups. Unpaired t-test was used to analyze statistical significance of values. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005.
Serum Accumulation of SICAM-1 and L-Selectin Connecting Vascular Inflammatory and Tissue Damage
Soluble intercellular adhesion molecule, E-selectin and L-selectin are adhesion molecules which reflect the activation of the vascular component of inflammation and interaction of circulatory cells with the endothelial component of blood–brain-barrier (BBB; Nottet, 1999; Whalen et al., 1999, 2000). sICAM levels in serum raised nearly fourfold within 6 h post-blast, followed by a decline to lower, but still significantly higher than control levels, values at day 7 after exposure to both composite and primary blast (Figure 8A). In contrast, serum L-selectin content increased remarkably at 1 and 7 days, but not 6 h following blast (Figure 8B). Thus, the prominent activation of the L-selectin component of blast responses occurs when peak overpressure interacts with the frontal part of the head without significant acceleration, reflecting a somewhat delayed involvement of leukocytes compared with earlier (6 h) vascular endothelial activation indicated by sICAM-1 and, to some extent, serum integrin alpha/beta increases.
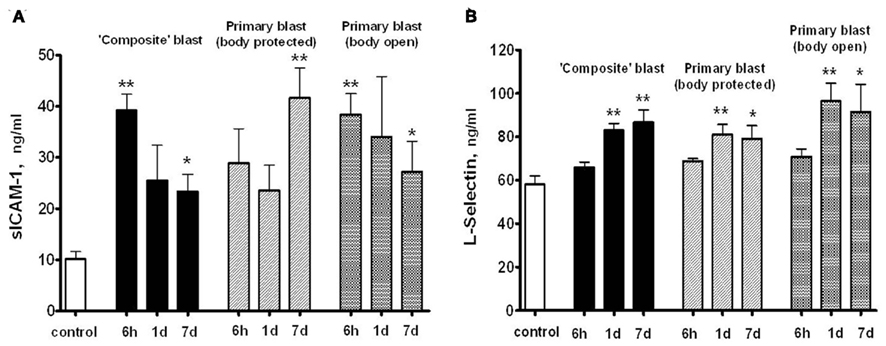
Figure 8. Soluble intercellular adhesion molecule -1 (A) and L-selectin (B) concentrations in rat serum following blast exposure. Blood was collected after blast at different shock tube set-ups and assayed by SW ELISA. Unpaired t-test was done to analyze statistical significance of values. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 vs. naïve. Please see Section “Materials and Methods” for details.
Neuroendocrine, Neurotrophic, and Growth Factor Responses after Blast Exposure
Orexin A is a neuropeptide secreted by the hypothalamus, which promotes food intake, wakefulness, and metabolic activity/energy consumption. As seen in Figure 9, a nearly threefold increase in serum Orexin A occurs 1 and 7 days after primary blast with open body, but not after composite blast, at least within the 7 day interval.
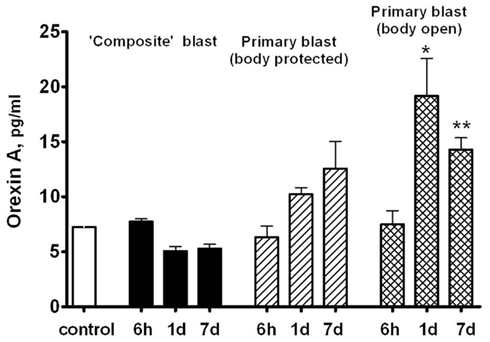
Figure 9. Orexin A content in rat serum after blast. Blood was collected from overpressure-exposed rats at different shock tube set-ups and assayed by SW ELISA. Unpaired t-test was done to analyze statistical significance of values. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 vs. naïve. Please see Section “Materials and Methods” for details.
Using a targeted approach, we identified additional components of neurotrophic response to blast exposure – nerve growth factor beta (NGF-beta) and Neuropilin-2 (NRP-2). NGF-beta has been suggested to play a neurotrophic role in several neurodegenerative diseases (Li et al., 2007; Syed et al., 2007; Calissano et al., 2010). Neuropilin-2 is a receptor for VEGF and semaphorins, a large family of secreted and transmembrane signaling proteins that regulate axonal guidance in the developing CNS (Cloutier et al., 2002; Bannerman et al., 2008; Roffers-Agarwal and Gammill, 2009). Serum levels of NGF-beta were assessed using SW ELISA and NRP-2 by antibody array (Ray Biotech) and semi-quantitative western blot after blast exposure at different set-ups (Figure 10). Generally, exposures to composite and primary blast resulted in a significant increase of NGF-beta in serum 1 and 7 day after challenge, however the magnitude of increase was much higher after primary blast hit open body compared to composite blast exposure (Figure 10A). Likewise, high levels of NRP-2 were found in circulation at 1 and 7 day in rats exposed to primary blast with unprotected body, as compared to composite blast together with NRP-2 up-regulation in hippocampus at all blast set-ups. These data suggest that predominantly primary blast activates neuroregeneration and that NRP-2 may be involved in this process. In addition, these results indicate that NGF-beta and NRP-2 may have neuroprotective functions and be involved in adaptive responses/neurorepair after blast-induced TBI.
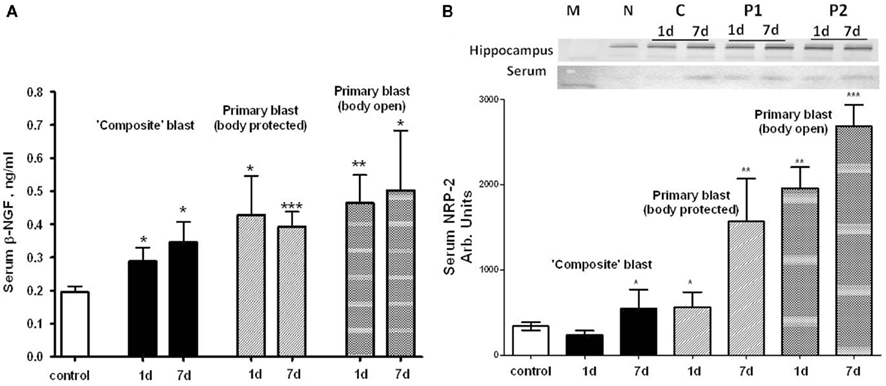
Figure 10. Blast-induced accumulation of β-NGF and Neuropilin-2 in rat serum. (A) Serum β-NGF after different types of blast (SW ELISA); (B) serum Neuropilin-2 detection by antibody arrays. Inset: representative western blots for hippocampus and serum. (N, naïve; C, “composite”; P1, primary/head; P2, primary/body). t-Test with Welch correction was done. Data shown are mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 vs. naïve. Please see Section “Materials and Methods” for details.
Discussion
Over the last several decades, a number of experimental animal models to study blast wave effects have been implemented, including rodents and larger animals, such as sheep (Savic et al., 1991; Stuhmiller et al., 1996). Shock tubes have been used as the fundamental research tool for the last several decades (Jaffin et al., 1987; Elsayed, 1997; Guy et al., 1998a,b). There is still concern whether a blast waves generated by shock tubes using compressed gas accurately reflect real explosive blast. In our study, dynamic pressure measured by a PCB “pencil” sensor indicated that shock tubes produce a “venting gas jet” immediately after blast wave formation (see the shoulder at Figure 1A), substantially contaminating the blast wave in the direction of shock tube axis (Figure 1A). In addition, the exhaust “venting gas” apparently masked the negative phase of the shock wave, which was present when the dynamic pressure was recorded at an angle to the shock tube nozzle (Figure 1B). Schlieren optics techniques clearly defined the areas of pressure, either peak OP or venting gas jet (Figure 2).
This pattern is characteristic of “external” shock tube models where the target/animal is placed outside rather than within the tube. Placing animals within the tube also can produce confounding effects when the animal is very large relative to the tube diameter or when the animal is suspended and or shielded inappropriately. The shape of the blast wave and the development of constructive or destructive secondary waves as the primary wave exits the tube can be affected by the size and shape of the exit as well. This can be visualized with Schlieren optics. By placing rats off-axis from the shock tube nozzle, we eliminated the venting gas in a way that the main effect acting on the rat is the peak overpressure event and negative phase of the blast wave. Thus, we examined the pathological impact of two different types of blast with precisely controlled magnitude, duration, and impulse at the surface of the rat, different orientations of the head to the blast wave, and open or armored body: (i) primary blast/peak overpressure only with rats located off-axis with the shock tube and (ii) composite blast with rats located on-axis, accompanied by linear and, to a lesser extent, rotational head hyperacceleration (Figure 2). It should be noted that any blast produced in the laboratory models only a particular component of a complex blast that might be experienced on the battlefield. The detonation of real explosives in the field does not produce the “venting gas,” but can result in significant bulk flow of air and debris. This makes the separation of the effects of primary and particularly tertiary blast (the target being displaced by the blast) difficult to separate in most existing testing regimes. Although the blast generated in our on-axis model is a single blast event, the type of blast load observed resembles the complex effect produced by multiple blasts, such as in a confined space where the blast waves reverberate and overlap, hence the effect of displaced air mass flow on the resultant wave structure and magnitude can be important.
There was a substantial difference in the effects of composite vs. primary blast on neurodegenerative processes in the cortex and, particularly, hippocampus at 7 day post-blast (Figure 3). Silver accumulation in the cortex after composite blast was modest, with a very rare finding of “classical type” neurodegeneration (Figure 3A, inset). On the other hand, the hippocampus significantly accumulated silver in fiber-like structures after composite blast (Figure 3D), while very occasional silver staining was observed in both cortex and hippocampus after primary blast (Figures 3B,E). As expected and in accordance with data reported previously, CCI evoked a distinct cellular neurodegeneration in both cortical and hippocampal tissue (Figures 3C,F). The most common types of closed head impact TBI are diffuse axonal injury, contusion, and subdural hemorrhage as an overall result of rotational acceleration (Vander Vorst et al., 2007). Diffuse axonal injuries are very common following closed head injuries. They result when shearing, stretching, and/or angular forces pull on axons and small vessels. Impaired axonal transport leads to focal axonal swelling and, after several hours, may result in axonal disconnection (Hurley et al., 2004). The typical locations are the corticomedullary (gray matter-white matter) junction, internal capsule, deep gray matter, upper brainstem, and corpus callosum. Multifocal axonal degeneration, as evidenced by amino cupric silver staining is characteristic also for shock wave insult as was shown in a study with head-only exposed rats inside a shock tube (Garman et al., 2011). Our recent (Svetlov et al., 2010) and present studies clearly demonstrate the presence of neural degeneration in deeper structures of the brain, specifically hippocampus after composite blast producing linear and rotational head acceleration, which is lacking or negligible following primary blast.
Exposure to a single moderate blast, both composite and primary, led to prominent gliosis in the hippocampus, evidenced by expression of GFAP and CNPase (Figure 4). Markers of activated astrocytes GFAP and oligodendrocytes CNPase were strongly up-regulated in CA1 and DG regions of hippocampus, respectively, at 1 day and sustained up to 7 days post-blast. These findings are in strict accordance with many previous reports, including from our group, supporting the notion that gliosis represents a common and rapid response to brain insult regardless of the nature-mechanical or blast-induced exposure (Urrea et al., 2007; Svetlov et al., 2010; Kwon et al., 2011).
NSE was significantly elevated in serum within 6 h after both composite and primary blast (Figure 5A), and the increased levels generally persisted up to 7 days, although was not statistically significant upon open body primary blast exposure. In these experiments, we used NSE SW ELISA Kit from Life Sciences Advanced Technologies designed to detect specifically rat NSE. However, several reports indicate that NSE may not be highly specific for the CNS and is present in platelets and red blood cells (see Svetlov et al., 2009 for review). In previous studies, we reported a slight UCH-L1 increase after “composite” blast, followed by a rapid decline (Svetlov et al., 2010). The UCH-L1 SW ELISA used in early experiments had low specificity and sensitivity for rat samples, thus many serum substances interfered and masked the UCH-L1 content. In this study, an improved version of the UCH-L1 assay was employed, still not particularly specific for rats (data not shown). Increases in serum UCH-L1 were statistically significant only at day 1 after a single primary blast exposure (n = 4), although an elevation trend could be detected (Figure 5B). In contrast, a rat-specific GFAP SW ELISA has been generated and employed in these studies. Serum GFAP increase was prominent within 6 h after composite and primary blast with body protected (Figure 6A), and elevated levels persisted up to 7 days post-blast, consistent with up-regulation in hippocampus. The CNPase content assessed by semi-quantitative western blot was raised at day 1 after blast exposure (except primary blast with open body) and further substantially increased at 7 day post-blast (Figure 6B). It remains to be examined whether CNPase up-regulation reflects a long-term disorder of myelination following blast exposure and whether CNPase can be a biomarker of chronic injury.
We postulated that impaired vascular reactions, systemic responses, and neuroinflammmation, result in enhancement of endothelial permeability/leakage, recruitment of immune/inflammatory cells from circulation, and activation of brain-resident glial cells. This paradigm is in line with the previous hypothesis set forth by Cernak (2010) and is further supported by present data.
As can be seen in Figures 7A,B, both pro-inflammatory (IL-1) and counteracting anti-inflammatory molecules (IL-10) accumulate in circulation at 1 and 7 days post-blast, predominantly after primary blast exposure with open body (Figures 7A,B). These results are in agreement with data obtained using non-blast TBI models (Dietrich et al., 2004; Maegele et al., 2007). Moreover, CX3CL1 chemokine Fractalkine was also significantly elevated after primary blast (mostly with protected body), further suggesting a systemic component in response to blast (Figure 7C), consistent with reports on the level of this chemokine in patients with TBI and in mouse model of closed head injury (Rancan et al., 2004; Ralay Ranaivo et al., 2011). Most intriguing is that serum IL-1, IL-10, and Fractalkine did not rise significantly after composite blast at 1 and 7 days post-blast. In contrast, integrin alpha/beta, a complement receptor composed of CD11c/CD18, was increased substantially at all set-ups (Figure 7D), further supporting the important roles for a microcirculatory component of neuroinflammation in brain injury shown in rat models of fluid percussion injury (Utagawa et al., 2008).
L-selectin and ICAM-1 are adhesion molecules which characterize the activation of a vascular component of inflammation and interaction of circulatory cells with the endothelial component of the (BBB; Nottet, 1999; Whalen et al., 1999, 2000). As can be seen in Figure 8, prominent activation of the L-selectin after blast occurs when peak overpressure interacts with the frontal part of head without significant acceleration, reflecting a somewhat delayed involvement of leukocytes compared with earlier (6 h) vascular endothelial activation reflected by sICAM-1 and, to some extent, serum integrin alpha/beta increases. Thus, the sustained activation of vascular components of blast responses occurs when peak overpressure interacts with the frontal part of the head without significant acceleration: “flowing blast inside the brain” (blast off-axis open body).
Orexin A, a neuroendocrine component of rat response to blast exposure, exhibited the most prominent pattern of difference between composite and primary blast (Figure 9). Serum Orexin A levels raised gradually within 1–7 days after primary blast and were significantly elevated in rats subjected to blast with open body. Although at present the precise mechanisms are not clear, this suggests that several systemic factors affected by primary blast wave in the whole body other than brain structures directly or indirectly stimulate hypothalamic release of Orexin A as well as interleukins/chemokines in circulation. We speculate that the presence of a distinct negative phase in primary blast wave is capable of producing cavitation-induced secondary microblasts. This could partially explain the different pattern in systemic/vascular responses to primary vs. composite blast exposure which lacks the negative phase. Further in-depth studies are needed to explore this hypothesis and elucidate potential roles for blast cavitation in damage, particularly at the interface of gas, liquid, and tissue.
Beta-NGF has been suggested to play a neurotrophic role in several neurodegenerative diseases (Li et al., 2007; Syed et al., 2007; Calissano et al., 2010). Our data indicate that NGF may also have neuroprotective functions and be involved in adaptive responses/neurorepair after blast-induced TBI. Exposure of whole body to primary overpressure blast instigated a rapid and sustained accumulation of beta-NGF in serum. Neuropilin-2 is the receptor for VEGF and semaphorins, a large family of secreted and transmembrane signaling proteins that regulate axonal guidance in the developing CNS (Cloutier et al., 2002; Bannerman et al., 2008; Roffers-Agarwal and Gammill, 2009). Our present data (Figure 10B) suggest that predominantly primary blast activates neuroregeneration and that NRP-2 may be involved in this process.
In conclusion, the specific dynamics of systemic, vascular inflammatory, neuroendocrine, growth factor, and neuro-glial biomarkers in serum were established and characterized. For major pathway signatures and biomarkers, the detected levels raised at all the set-ups studied. However, the most significant and persistent changes in neuro-glial injury markers were found after composite blast, while primary blast instigated the most prominent systemic/vascular, neuroendocrine, and growth factor responses, particularly when the rat was subjected to frontal, head-directed, open body exposure. We suggest that the mechanisms underlying primary blast brain injuries, particularly mild and moderate, are different from blast accompanied by head acceleration and may be triggered by systemic, cerebrovascular, and neuro-glia responses as overlapping events.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Mr. Danny Johnson for his excellent technical assistance. This work was supported by grants W81XWH-8-1-0376 and W81XWH-07-01-0701 from Department of Defense. The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The views and opinions expressed in this manuscript are those of the author(s) and do not reflect official policy or position of the Department of Defense or the U.S. Government.
References
Bannerman, P., Ara, J., Hahn, A., Hong, L., McCauley, E., Friesen, K., and Pleasure, D. (2008). Peripheral nerve regeneration is delayed in neuropilin 2-deficient mice. J. Neurosci. Res. 86, 3163–3169.
Bass, C. R., Panzer, M. B., Rafaels, K. A., Wood, G., Shridharani, J., and Capehart, B. (2012). Brain injuries from blast. Ann. Biomed. Eng. 40, 185–202.
Belanger, H. G., Scott, S. G., Scholten, J., Curtiss, G., and Vanderploeg, R. D. (2005). Utility of mechanism-of-injury-based assessment and treatment: Blast Injury Program case illustration. J. Rehabil. Res. Dev. 42, 403–412.
Born, C. T. (2005). Blast trauma: the fourth weapon of mass destruction. Scand. J. Surg. 94, 279–285.
Calissano, P., Matrone, C., and Amadoro, G. (2010). Nerve growth factor as a paradigm of neurotrophins related to Alzheimer’s disease. Dev. Neurobiol. 70, 372–383.
Cernak, I. (2010). The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front. Neurol. 1:151. doi:10.3389/fneur.2010.00151
Cernak, I., Merkle, A. C., Koliatsos, V. E., Bilik, J. M., Luong, Q. T., Mahota, T. M., Xu, L., Slack, N., Windle, D., and Ahmed, F. A. (2011). The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 41, 538–551.
Cernak, I., and Noble-Haeusslein, L. J. (2010). Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J. Cereb. Blood Flow Metab. 30, 255–266.
Cernak, I., Savic, J., Ignjatovic, D., and Jevtic, M. (1999). Blast injury from explosive munitions. J. Trauma. 47, 96–103; discussion 103–104.
Chavko, M., Adeeb, S., Ahlers, S. T., and McCarron, R. M. (2009). Attenuation of pulmonary inflammation after exposure to blast overpressure by N-acetylcysteine amide. Shock 32, 325–331.
Cloutier, J. F., Giger, R. J., Koentges, G., Dulac, C., Kolodkin, A. L., and Ginty, D. D. (2002). Neuropilin-2 mediates axonal fasciculation, zonal segregation, but not axonal convergence, of primary accessory olfactory neurons. Neuron 33, 877–892.
Dietrich, W. D., Chatzipanteli, K., Vitarbo, E., Wada, K., and Kinoshita, K. (2004). The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir. Suppl. 89, 69–74.
Garman, R. H., Jenkins, L. W., Switzer, R. C., Bauman, R. A., Tong, L. C., Swauger, P. W., Parks, S. A., Ritzel, D. V., Dixon, C. E., Clark, R. S. B., Bayir, H., Kagan, V., Jackson, E. K., and Kochanek, P. M. (2011). Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J. Neurotrauma 28, 947–959.
Guy, R. J., Glover, M. A., and Cripps, N. P. (1998a). The pathophysiology of primary blast injury and its implications for treatment. Part I: the thorax. J. R. Nav. Med. Serv. 84, 79–86.
Guy, R. J., Kirkman, E., Watkins, P. E., and Cooper, G. J. (1998b). Physiologic responses to primary blast. J. Trauma. 45, 983–987.
Gyorgy, A., Ling, G., Wingo, D., Walker, J., Tong, L., Parks, S., Januszkiewicz, A., Baumann, R., and Agoston, D. V. (2011). Time-dependent changes in serum biomarker levels after blast traumatic brain injury. J. Neurotrauma 28, 1121–1126.
Hurley, R. A., McGowan, J. C., Arfanakis, K., and Taber, K. H. (2004). Traumatic axonal injury: novel insights into evolution and identification. J. Neuropsychiatry Clin. Neurosci. 16, 1–7.
Jaffin, J. H., McKinney, L., Kinney, R. C., Cunningham, J. A., Moritz, D. M., Kraimer, J. M., Graeber, G. M., Moe, J. B., Salander, J. M., and Harmon, J. W. (1987). A laboratory model for studying blast overpressure injury. J. Trauma. 27, 349–356.
Jones, E., Fear, N. T., and Wessely, S. (2007). Shell shock and mild traumatic brain injury: a historical review. Am. J. Psychiatry 164, 1641–1645.
Kwon, S. K., Kovesdi, E., Gyorgy, A. B., Wingo, D., Kamnaksh, A., Walker, J., Long, J. B., and Agoston, D. V. (2011). Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front. Neurol. 2:12. doi:10.3389/fneur.2011.00012
Li, Y., Zhang, S. F., Zou, S. E., Xia, X., and Bao, L. (2007). Accumulation of nerve growth factor and its receptors in the uterus and dorsal root ganglia in a mouse model of adenomyosis. Reprod. Biol. Endocrinol. 9, 30.
Liu, M. C., Akinyi, L., Scharf, D., Mo, J., Larner, S. F., Muller, U., Oli, M. W., Zheng, W., Kobeissy, F., Papa, L., Lu, X. C., Dave, J. R., Tortella, F. C., Hayes, R. L., and Wang, K. K. (2010). Ubiquitin C-terminal hydrolase-L1 as a biomarker for ischemic and traumatic brain injury in rats. Eur. J. Neurosci. 31, 722–732.
Maegele, M., Sauerland, S., Bouillon, B., Schafer, U., Trubel, H., Riess, P., and Neugebauer, E. A. (2007). Differential immunoresponses following experimental traumatic brain injury, bone fracture and “two-hit”-combined neurotrauma. Inflamm. Res. 56, 318–323.
Nelson, T. J., Wall, D. B., Stedje-Larsen, E. T., Clark, R. T., Chambers, L. W., and Bohman, H. R. (2006). Predictors of mortality in close proximity blast injuries during Operation Iraqi Freedom. J. Am. Coll. Surg. 202, 418–422.
Nottet, H. S. (1999). Interactions between macrophages and brain microvascular endothelial cells: role in pathogenesis of HIV-1 infection and blood – brain barrier function. J. Neurovirol. 5, 659–669.
Ralay Ranaivo, H., Zunich, S., Choi, N., Hodge, J., and Wainwright, M. (2011). Mild stretch-induced injury increases susceptibility to interleukin-1beta-induced release of matrix metalloproteinase-9 from astrocytes. J Neurotrauma. 28, 1757–1766.
Rancan, M., Bye, N., Otto, V. I., Trentz, O., Kossmann, T., Frentzel, S., and Morganti-Kossmann, M. C. (2004). The chemokine fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J. Cereb. Blood Flow Metab. 24, 1110–1118.
Roffers-Agarwal, J., and Gammill, L. S. (2009). Neuropilin receptors guide distinct phases of sensory and motor neuronal segmentation. Development 136, 1879–1888.
Savic, J., Tatic, V., Ignjatovic, D., Mrda, V., Erdeljan, D., Cernak, I., Vujnov, S., Simovic, M., Andelic, G., and Duknic, M. (1991). Pathophysiologic reactions in sheep to blast waves from detonation of aerosol explosives. Vojnosanit. Pregl. 48, 499–506.
Stuhmiller, J. H., Ho, K. H., Vander Vorst, M. J., Dodd, K. T., Fitzpatrick, T., and Mayorga, M. (1996). A model of blast overpressure injury to the lung. J. Biomech. 29, 227–234.
Svetlov, S. I., Prima, V., Kirk, D. R., Gutierrez, H., Curley, K. C., Hayes, R. L., and Wang, K. K. W. (2011). “Neuro-glial and systemic mechanisms of pathological responses to primary blast overpressure (OP) compared to “composite” blast accompanied by head acceleration in rats,” in Proceeding of NATO conference ’A Survey of Blast Injury across the Full Landscape of Military Science http://www.rto.nato.int/
Svetlov, S. I., Larner, S. F., Kirk, D. R., Atkinson, J., Hayes, R. L., and Wang, K. K. (2009). Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J. Neurotrauma 26, 913–921.
Svetlov, S. I., Prima, V., Kirk, D. R., Gutierrez, H., Curley, K. C., Hayes, R. L., and Wang, K. K. (2010). Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J. Trauma. 69, 795–804.
Syed, Z., Dudbridge, F., and Kent, L. (2007). An investigation of the neurotrophic factor genes GDNF, NGF, and NT3 in susceptibility to ADHD. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 375–378.
Terrio, H., Brenner, L. A., Ivins, B. J., Cho, J. M., Helmick, K., Schwab, K., Scally, K., Bretthauer, R., and Warden, D. (2009). Traumatic brain injury screening: preliminary findings in a US Army Brigade Combat Team. J. Head Trauma Rehabil. 24, 14–23.
Urrea, C., Castellanos, D. A., Sagen, J., Tsoulfas, P., Bramlett, H. M., and Dietrich, W. D. (2007). Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restor. Neurol. Neurosci. 25, 65–76.
Utagawa, A., Bramlett, H. M., Daniels, L., Lotocki, G., Dekaban, G. A., Weaver, L. C., and Dietrich, W. D. (2008). Transient blockage of the CD11d/CD18 integrin reduces contusion volume and macrophage infiltration after traumatic brain injury in rats. Brain Res. 1207, 155–163.
Vander Vorst, M., Ono, K., Chan, P., and Stuhmiller, J. (2007). Correlates to traumatic brain injury in nonhuman primates. J. Trauma 62, 199–206.
Warden, D. (2006). Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil. 21, 398–402.
Whalen, M. J., Carlos, T. M., Dixon, C. E., Robichaud, P., Clark, R. S., Marion, D. W., and Kochanek, P. M. (2000). Reduced brain edema after traumatic brain injury in mice deficient in P-selectin and intercellular adhesion molecule-1. J. Leukoc. Biol. 67, 160–168.
Whalen, M. J., Carlos, T. M., Kochanek, P. M., Clark, R. S., Heineman, S., Schiding, J. K., Franicola, D., Memarzadeh, F., Lo, W., Marion, D. W., and Dekosky, S. T. (1999). Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. J. Neurotrauma 16, 583–594.
Wolf, S. J., Bebarta, V. S., Bonnett, C. J., Pons, P. T., and Cantrill, S. V. (2009). Blast injuries. Lancet 374, 405–415.
Keywords: blast, brain injury, biomarkers, rat models, neuro-glia damage, systemic responses
Citation: Svetlov SI, Prima V, Glushakova O, Svetlov A, Kirk DR, Gutierrez H, Serebruany VL, Curley KC, Wang KKW and Hayes RL (2012) Neuro-glial and systemic mechanisms of pathological responses in rat models of primary blast overpressure compared to “composite” blast. Front. Neur. 3:15. doi: 10.3389/fneur.2012.00015
Received: 02 December 2011;
Accepted: 24 January 2012;
Published online: 09 February 2012.
Edited by:
Ibolja Cernak, Johns Hopkins University, USACopyright: © 2012 Svetlov, Prima, Glushakova, Svetlov, Kirk, Gutierrez, Serebruany, Curley, Wang and Hayes. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Stanislav I. Svetlov and Victor Prima, Banyan Laboratories, Inc., 12085 Research Drive, Alachua, FL 32615, USA. e-mail:c3N2ZXRsb3ZAYmFueWFuYmlvLmNvbQ==;dnByaW1hQGJhbnlhbmJpby5jb20=