 Veronica Ghiglieri
Veronica Ghiglieri Valeria Calabrese
Valeria Calabrese Paolo Calabresi
Paolo Calabresi- 1Dipartimento di Filosofia, Scienze Sociali, Umane e della Formazione, Università degli Studi di Perugia, Perugia, Italy
- 2Laboratorio di Neurofisiologia, Fondazione Santa Lucia, IRCCS, Rome, Italy
- 3Clinica Neurologica, Dipartimento di Medicina, Università degli Studi di Perugia, Ospedale Santa Maria della Misericordia di Perugia, Perugia, Italy
Over the last two decades, many experimental and clinical studies have provided solid evidence that alpha-synuclein (α-syn), a small, natively unfolded protein, is closely related to Parkinson’s disease (PD) pathology. To provide an overview on the different roles of this protein, here we propose a synopsis of seminal and recent studies that explored the many aspects of α-syn. Ranging from the physiological functions to its neurodegenerative potential, the relationship with the possible pathogenesis of PD will be discussed. Close attention will be paid on early cellular and molecular alterations associated with the presence of α-syn aggregates.
The Many Roles of Alpha-Synuclein (α-syn)
Alpha-synuclein is a 140 aminoacid protein, encoded by the SNCA gene on human chromosome 4. This protein is mainly expressed in presynaptic sites at several neurotransmitter systems in the central nervous system (CNS) (1). Despite its ubiquitous distribution through many areas involved in complex behaviors, α-syn pathology does not impact on all brain sites of expression, but rather shows a prevalent effect in selective vulnerable sites (1, 2). Moreover, α-syn is highly present in red blood cells (3) and in other extra CNS tissues (4, 5), indicating a wide range of actions of this protein throughout the body.
Although α-syn is gaining increasing consideration as a critical factor in Parkinson’s disease (PD) pathophysiology and 20 years of research have been spent in the attempt to unravel the physiological roles of this protein, its mechanisms of action are still unclear and so are the complex dynamics that characterize its flexibility to adapt and the tendency to become toxic.
α-syn exists in a dynamic balance between monomeric and oligomeric states, which are not easily prone to form fibrils in physiological conditions. Interestingly, its structure predicts the multifunctional properties that have been attributed to this protein (6). As a result, this structural flexibility allows α-syn to adopt a wide range of conformations depending on the environment and binding partners (7, 8). In fact, α-syn can either relate to intracellular and membrane proteins with its enzymatic activity or interact with lipid surfaces and organize membrane activities through steric mechanisms.
Given its prevalent localization at presynaptic sites, the first function described for α-syn was its chaperone function and in particular its ability in controlling exocytosis through management of synaptic vesicle pool and trafficking. Accordingly, mutations of the SNCA gene coding for α-syn leads to functional alterations of SNAP REceptor (SNARE) proteins, a family of receptors that binds the soluble N-ethylmaleimide sensitive fusion attachment proteins (SNAP) receptor (SNARE) proteins and regulates their assembly (9). Another presynaptic target for α-syn is the DA active trasporter (DAT) (10, 11).
Upon interaction with lipidic surfaces α-syn binding causes the formation of an amphipathic alpha-helix that in physiologic conditions does not cross the bilayer. Under specific stimulations, oligomers of α-syn may form membrane pores that may dissipate the transmembrane potential, dysregulating ion gradients (12, 13).
Several strategies exist to ensure the prevention of α-syn oligomerization (14–17), including complex hydrophobic interactions between C- and N-tails of the protein (16, 18, 19). Interestingly, α-syn possesses a polar C terminal tail able to interact with the hydrophobic region of a separate denatured protein, sharing structural and functional homology with other molecular chaperones. Thus, the extreme flexibility of this protein also relies on the ability of α-syn to auto assemble and act as an intramolecular chaperone (20). In agreement, α-syn truncated at the C-terminus lacks this auto-chaperone property (21) and aggregates at an increased rate compared with the full-length counterpart (14, 15, 21, 22). Despite its crucial contribution to ensure a good orchestration of processes at the active zones, α-syn translocates late to the terminals during development (23) and its absence seems to be not detrimental for synaptogenesis, indicating that its function is rather essential for stressful and sustained activity over time during the long life of a neuron (24, 25). All these characteristics strongly argue for a critical role in neurotransmitter release and synaptic plasticity. A feature that makes multifunctional α-syn as much enigmatic as difficult to counteract in pathological settings is that, like many other disease-associated misfolding proteins, its absence is less detrimental than its accumulation (26, 27). In physiological conditions, during proteins translation, polypeptides fold under control of chaperones. Errors in assembly are frequent and become more common with aging but they are usually limited by several quality control mechanisms that target denatured and misfolded proteins to degradation (28–30). Given the complex management of this protein expression and the high versatility of its functions, failures in these homeostatic steps do not simply bring to an abnormal gain of function but rather to a potent trigger for a series of neurodegenerative cascades in the intracellular environment. The possibility that residual physiological functions and compensative mechanisms are in act during degeneration, complicates therapeutic approaches and adds unpredictability to possible manipulation of α-syn functions.
α-syn Oligomers and Fibrillary Aggregates: Pathological Implications
An increasing body of evidence from studies carried out in animal models and in patients support the hypothesis that the processes underlying α-syn proteostasis have central roles in the pathogenesis of PD. This concept dates back to 20 years ago when two discoveries provided support for a role of possible link between α-syn mutations and PD. The first report was the identification of a missense mutation of this gene (31) causing a form of early-onset familial PD by Polymeropoulos and his research team. In the same year, Spillantini’s group provided experimental evidence that α-syn is the primary structural component of Lewy bodies (LB), intracytoplasmatic inclusions of α-syn aggregates, which are considered the main pathological hallmark of PD (32). Shortly after, also sporadic idiopathic forms of PD were found associated with the presence of LB in the brain parenchyma (33).
In the last years, physiological and pathological functions of α-syn and other misfolding proteins have been investigated in relation with other known aspects of the disease, to explore possible causal relationships. For PD, many risk factors have been identified that include both environmental and genetic causes. Oxidative stress, mitochondrial dysfunctions, neuroinflammation, point mutations, multiplications, and specific polymorphisms are genetic determinants that may cooperate to create ideal conditions for developing PD.
Interestingly, these factors are also determinants that impact on the predisposition of α-syn to exert toxicity.
Despite the existence of redundant quality control systems to ensure a correct assembly of α-syn and the ability of other synucleins to inhibit and control oligomerization of α-syn, this protein may express its neurotoxic potential when soluble monomers initially form oligomers, then progressively combine to form small protofibrils and further aggregate in large, insoluble α-syn fibrils forming LB (34, 35). Although its natural propensity to balance between a soluble and membrane-bound state and its plasticity of conformation, acute triggers of accumulation and aggregation of α-syn can be manifold like overproduction of the protein, failure in the molecular system that cleave misfolded forms, exposure to pH changes, oxidative stress, and mitochondrial overwork.
More chance for aggregation is offered by a variety of post-translational covalent modifications (8) potentially promoting conformational changes that make α-syn more prone to aggregation. For example, tyrosine nitration (Tyr125) and truncation of α-syn at the C-terminus are frequently found in α-syn pathological aggregates and have been shown to promote fibrillation in vitro (36, 37).
Finally, a progressive, age-related decline of efficiency in the in proteolytic mechanisms might play a synergistic role in the accumulation of α-syn (38, 39). These observations are consistent with data showing increased levels of α-syn in nigral dopaminergic neurons during normal aging (40).
In the healthy brain, intracellular homeostasis of α-syn is ensured by the combined actions of the ubiquitin–proteasome (UP) system and the lysosomal autophagy system (LAS) with the latter more involved in the clearance of oligomeric assemblies (38). Any failure in these systems is a potential trigger to overproduction and accumulation of α-syn forms, although compensatory mechanisms and additional proteases can take control over the protein maturation (38, 41). An aspect that complicates the scenario is that accumulation of α-syn may itself inhibit these homeostatic systems (42, 43) and reduce chaperoning of misfolded forms, enrolling the whole compartment into a vicious cycle that rapidly and uncontrollably triggers multiple neurodegeneration pathways. Accordingly, several mutations associated with genetic forms of PD are associated with reduced LAS function.
Analysis of LB has been indicative of the post-translational modifications mostly associated with pathogenic forms of α-syn (44). Among them, phosphorylation is probably the most studied modification since Ser129 phosphorylated α-syn is thought to be the dominant form of α-syn in LB (45). In support of this prevalence, a recent proteomics study quantified cortical expression levels of various α-syn forms from PD cases and controls (46).
It remains unclear, however, whether phosphorylation of α-syn impacts the fibrillation process (47). The role of nitration and oxidation in favoring toxic species is more clearly demonstrated in decreasing the tendency of α-syn to form fibrils and stabilizing oligomers, leading to enhanced toxicity (48, 49). Nitration of α-syn at specific residues has been characterized in brains from patients with synucleinopathies (50). Oxidized α-syn may result by way of oxidized derivatives of DA leading to a decrease in fibril formation and a subsequent increase in protofibril accumulation (51).
Truncated α-syn species have been found in LB associated with an increased tendency to form fibrils in vitro and with increased toxicity in overexpressing laboratory animals (52, 53) even if evidence of correlation with human disease are scarce (46).
The pathological relevance of α-syn species is extensively debated (44) and stabilization of the amyloid pathway is a main focus of research. It has been proposed that toxic species could be either amyloid-like insoluble fibrils, as the ones found in LB, although more evidence would support a key role for soluble oligomers or protofibrils (35). Several groups have investigated the different states of α-syn aggregation and thoroughly examined the functional consequences of aggregate-associated toxicity producing conflicting results (35, 44). However, the general concept is that α-syn exists under various conformational shapes and oligomeric states in a dynamic balance, modulated by factors either accelerating or inhibiting fibril formation. Genetic mutations related to PD have a role in determining the pattern of expression of the various aggregates (54–56), although the identification and characterization of the toxic α-syn species remain incomplete.
Strategies to counteract α-syn toxicity range from increasing protein clearance, which might be enhanced by stimulating autophagy, to act on α-syn post-translational modifications (44). Also, approaches targeting α-syn aggregation with inhibitors (57–59) or by inducing either passive or active immunization against α-syn species have shown promises in several transgenic mouse models of PD (60–62).
However, limits of this approach have been recently discussed (44). Given the incomplete knowledge of possible cellular roles of oligomers and the many functions covered by this protein, the precise α-syn species to target remains unclear. It is possible to hypothesize that modest presence of specific α-syn aggregates can be useful for the cell as part of compensative mechanism that is still unclear, and that their elimination could be harmful and accelerate instead of counteracting the disease process.
DA Neurons Vulnerability to α-syn
Relevant to the impact of protein misfolding in brain functions, an aspect that still puzzles researchers in the field of neurodegenerative diseases is the selective vulnerability of certain population of neurons to a wide range of insults.
In PD, dopaminergic neurons of the substantia nigra pars compacta (SNc) show selective neurodegeneration and cell death with reduction of dopamine (DA) levels in the striatum and impairment of several basal ganglia functions. The mechanism by which α-syn injures dopaminergic neurons remains to be fully established.
Alpha-synuclein is related to DA neurons for its ability to modulate DA homeostasis in synapses and to bind and influence the activity of DAT (63–65), although the implicated mechanisms are still debated (66–68). This protein is also an important modulator of DA metabolism as it controls DA synthesis by reducing the phosphorylation state of tyrosine hydroxylase and stabilizing it in its inactive state (69). Accordingly, absence of α-syn exerts considerable impact on the dopaminergic system because it causes decreased striatal DA levels and reduced DAT function (70). Lack of α-syn is also associated with decreased DA striatal uptake (71), reduced number of TH-positive terminals as well as of nigral DA cells (72).
However, the sensitivity of DA neurons to α-syn toxicity does not only depend on the possible lack of support to DA metabolism, but on the intrinsic and selective vulnerability of these neurons to excitotoxic challenge.
A recent review discusses the common traits of neurons of SNc neurons and other nuclei most vulnerable to PD pathology, offering an interesting point of view (73). The authors posit that SNc neurons share particular vulnerability to oxidative stress with cells of other brain nuclei involved in arousal responses and in the control of sensorimotor networks, needed for surviving behaviors such as vigilance, escape, and attack. SNc DA neurons possess at least two characteristics that make them particularly vulnerable to excitotoxic insult.
First, these neurons display an extensive length of branched axons that offer a high number of transmitter release sites. This diffuse axonal arbor might be functional to the coordination of the activity in spatially distributed networks, such as the basal ganglia. However, mitochondrial stress is elevated in the axons of SNc DA neurons and this is one reason why these neurons show increased vulnerability.
Second, DA neurons also have spontaneous activity and act as autonomous pacemakers. Their activity is characterized by large oscillations in intracellular calcium (Ca2+) concentration that are driven by the opening of voltage-dependent Cav1 Ca2+ channels (also known as L-type Ca2+ channels) to ensure a rhythmic (2–10 Hz) spiking (74–76). This ability is associated to low intrinsic Ca2+ buffering and requires a strict control of Ca-mediated processes from intracellular stores, promoting Ca2+ entry into the mitochondria (77, 78) as well as oxidative phosphorylation and production of ATP (79). All these events are needed to fulfill bioenergetic needs (79, 80) and to avoid undesired compensative activation of ATP-sensitive potassium channels, which would silence ongoing neuronal activity.
Substantia nigra pars compacta cells and other neurons of brain nuclei involved in sensorimotor integration are endowed with this complex set of feedforward control mechanisms that ensure to rapidly implement a correct strategy in response to environmental challenge. A price for this adaptive ability is the vulnerability of the system to age, genetic mutations, or environmental toxins that may increase production of reactive oxygen species that can impair proteostasis, cause accumulation DNA damages, particularly in mitochondria. When mitochondrial dysfunction reaches a level in which mitophagy is impaired, also cellular autophagic processes are affected and UP and LAS systems are compromised. Accordingly, in rodents, SNc, locus coeruleus, and dorsal motor nucleus of the vagus neurons (which are the only ones that have been studied at this level) manifest a basal mitochondrial oxidant stress in the somatodendritic region that is attributable to the feedforward control of oxidative phosphorylation of ATP (78, 81–83). On this line, a recent paper by Burbulla and colleagues (84) analyzed the synergistic detrimental effect of increased levels of α-syn, dopaminergic receptor stimulation, and mitochondrial dysfunction in mice showing functional inactivation of DJ-1, modeling an early-onset genetic form of PD. Interestingly, mice with both DA neuron-specific overexpression of human α-syn A53T (85) and constitutive DJ-1 deficiency show increased levels of oxidized DA in nigral neurons and decreased lysosomal activity compared with mice bearing the single DJ-1 mutation.
All these data support the concept that α-syn induces exacerbation of a Ca2+ dyshomeostasis in DA neurons. The paper by Luo and coworkers provided experimental evidence for this link by studying the potential effects of increased α-syn levels on processes downstream of the Ca2+-signaling pathway, demonstrating the contribution of a new calcium-dependent pathway in the dopaminergic neuronal loss (86). A possible explanation resides in the combination of the α-syn oligomers property to trigger Ca2+ influx and the intrinsic physiological characteristics of DA neurons. This neuronal population is in fact characterized by pacemaker activity that, as described above, depends on a complex homeostatic regulation, which involves the activity of L-type calcium channels (87), bringing the DA neuron on the edge of triggering neurodegenerative pathways. Another study that has been instrumental in deciphering the link between DA neurons and α-syn is the paper by Feng et al. (88) demonstrating that in particular conditions, like overexpression of wild-type (WT) α-syn, oligomers causes the formation of pore-like structures throughout the membrane acting as non-selective channels. This was associated with increase in membrane conductance and with cell death (88).
Beyond the Braak Hypothesis
The prevalent belief on the progression of PD neurodegeneration is based on the observation of an ascending pattern of its clinical manifestations that identifies the disease phase. The idea is that toxic species of α-syn progressively reach more brain regions over the course of the disease, as suggested by Braak and coworkers (89), starting from peripheral body dysfunctions and olfactory impairment through central brainstem functions to end with alterations of higher functions over years or decades following the first exposure to stressors.
In this theoretical framework, the speculation that prodromal symptoms of PD (hyposmia, constipation, and autonomic dysfunctions) might be due to peripheral seeding of α-syn aggregates gained a broad consideration in the field. Indeed, in prodromal phases, inflammation in the gastrointestinal tract or in the olfactory system may trigger the formation of α-syn aggregates (90). This concept is supported by recent evidence obtained in Snca-overexpressing mice suggesting a role of gut microbiota in hosting immune and inflammation response linked to α-syn pathology, associated with motor deficits (91).
Thus, α-syn would be released into the synaptic cleft, endocytosed by neighboring neurons, and seed aggregation of endogenous α-syn once inside their new cellular host (92–94). However, this is a much-debated issue and, although many studies support the notion of a spreading of α-syn pathology through a prion-like activity, recent analyses have challenged this theory demonstrating that the distribution of pathology in the brains of PD patients is not consistent with this model. The prion-like nature of α-syn was postulated around a decade ago after the observation of the development of LB-like intracellular inclusions in grafted DA neurons of PD patients who received a transplantation of embryonic mesencephalic grafts 11, 14, and 16 years earlier (95–97). The hypothesis of a “host-to-the-graft” transmission of LB led to the concept that α-syn oligomers may spread from cell to cell through axonal transport and exocytosis, aggregate into LB, and then transferred to other neurons.
A recent study by Peelaerts and colleagues investigated whether different forms of α-syn aggregates are genuine protein strains with a given role and a specific impact on animal physiology (98), based on the hypothesis that different strains could account for the different clinicopathological traits within synucleinopathies (99, 100). The authors propose that α-syn exists and exerts its detrimental effects, in different strains leading to different aggregates that cause as many distinct synucleinopathies (PD, dementia with LB, multiple system atrophy) (98, 101). The most relevant insights from this study are that (1) the dynamic nature of α-syn species is reflected into distinct competencies in the various species that could account for different phenotypes; (2) α-syn strains amplify in vivo; and (3) α-syn assemblies cross the blood–brain barrier after intravenous injection.
Although findings in support of the prion-like hypothesis are numerous, an increasing number of studies have recently challenged this vision. Two interesting reviews thoroughly discuss limits of the data collected in support of the ascending theory of α-syn pathology rather supporting a threshold theory to explain controversial data. One of the most important point of the “threshold theory” (102) stems from the simple consideration of PD as a global systemic disease supported by many genetic, cellular, and functional data. The fact that invalidates the ascending theory regards the evidence that brainstem and peripheral neurons are more resistant to insults and less prone to neurodegeneration compared to DA neurons (103–105) and capable of regeneration (106, 107). One explanation of the early dysfunction of brainstem and enteric neurons is due to their low threshold of functional reserve in contrast with the resilience of central neurons as part of widespread interconnected networks that ensure a good degree of compensation and redundancy to conserve higher functions (108, 109). Indeed, central networks have a great ability to compensate for an impaired function of a given central brain area, resulting in a late appearance of motor and cognitive symptoms. The authors propose that parallel pathological events in PD occur at similar rates resulting in the first symptoms pertaining to a peripheral nervous system alteration, due to an earlier functional threshold in the autonomic nervous system compared with midbrain dopaminergic circuitry. This threshold function explains the progression of early symptoms in PD.
Inflammation and Immune Response in PD
In many disorders of the CNS, a key aspect of neurodegeneration is neuroinflammation. In PD, abnormal functions of astrocytes and increase in soluble inflammatory cytokines from microglia and immune cells have been proposed as a critical player that together with glutamate-mediated excitotoxicity becomes major determinants for pathophysiology. In nigrostriatal degeneration, inflammatory response is invariably associated with α-syn-mediated events. Glial cells, which cover a wide range of functions in support of neuron development, maintenance, and survival, seem to be critically involved at many levels in the neurodegenerative spreading of the disease pathology. These studies have provided evidence supporting CNS immune resident cells role in PD (110). Activation of some glial components, such as astrocytes, however, is not limited to final phases of the inflammation process but it has been recently supposed to play a relevant part in initiating the pathology (111). Conversely, although it is well known that microglia plays a role in abnormal plasticity by its ability to produce inflammation mediators (112), it is less clear if microgliosis is instrumental to disease pathogenesis or a secondary event following the ongoing neurodegeneration and a primary role remains to be defined (113).
Functionally, all neuronal activities require an intact glial function provided by both astrocytes and microglia, which become essential for neurons enrolled into intense synaptic activity, such as DA SNc neurons. Astrocytes, besides their role as structural, metabolic, and trophic support, are directly involved in synaptic transmission, ensuring a proper communication, and avoiding abnormal stimulation of extrasynaptic receptors. While astrocytes can be considered an essential component of an operational synaptic surveillance, microglia is in charge of the immune surveillance in the brain. Abnormal microglia activation was found in autopsy brain tissues from PD patients and in experimental parkinsonism (110, 114–117) and many recent papers have focused on the roles of microglia in PD pathology [reviewed in Ref. (113)] bringing support to the notion that neurodegenerative processes and inflammation coexist and cooperate at the same time to respond to brain insults, and that these events do not just occur in series as the disease progresses. For example, when an immune response is initiated by microglia, astrocytes surround the area, creating a barrier to prevent the spread of toxic signals into the surrounding tissue (118). Neuroinflammation and immune response, including autoimmune activity, share molecular pathways initiated by cellular elements during degeneration.
A recent review paper by Booth and colleagues (111) has provided an extensive overview of the studies that link astrocytes alterations and PD, with particular attention to the monogenic forms of disease in which genetic mutations affect the functions of both principal neurons and astrocytes. A recent transcriptome study demonstrated that of 17 genes that have been implicated in PD, 8 are also expressed in astrocytes and are essential for their homeostasis (119). Although SNCA gene shows a low astrocytic expression, it has been suggested that even a modest presence of α-syn might be challenging for their function. In fact, α-syn initiates and regulates astrocyte activation in response to inflammatory stimuli. Also, astrocytes have been reported to take up aggregated alpha syn. The most fascinating aspect of astrocyte involvement in neuronal degeneration, relevant to PD, is that astrocytes change in shape and function to provide support to DA neurons under intense stressful conditions (120–122). Astrocytes are also able to take up circulating DA precursor l-DOPA to release DA (123), as they express enzymes and the complete machinery for its metabolism, suggesting a close relationship to DA systems.
It has been recently shown that in 6-hydroxydopamine-lesioned rats, modeling late stage PD, marked astrocytosis and microglial activation accompany neurodegeneration over time as the damage progresses, being strikingly visible in striatal samples 2 months after the lesion. Interestingly, following a repetitive transcranial magnetic stimulation (TMS) treatment, reduction of intense astrogliosis and microgliosis was associated with, and may underlie, recovery of corticostriatal plasticity. Such TMS-mediated recovery of glial morphology and function was associated with selective increase of DA in dorsolateral striatum of treated parkinsonian animals (124).
These data are in agreement with studies showing that microglia is implicated in the production of neurotrophins, interleukins, proinflammatory, and antiinflammatory cytokines (114–117) and with studies linking TMS beneficial effects with stabilization of microglia, reduction of neuroinflammation biomarkers (110, 125). Midbrain DA neurons, α-syn, and immune response are also linked together by their involvement in altered Ca2+ homeostasis. The paper by Luo et al. (86) demonstrated that in DA neurons of A53T α-syn transgenic mice dysregulation of intracellular Ca2+ activates the calcineurin pathway that, in turn, increases the translocation rate of the nuclear factor of activated T cells (NFAT) from cytosolic to nuclear compartments. This is associated with the expression of cytokine genes, in human T cells and enhanced cell death in SNc. Inhibition of calcineurin renormalizes the mitochondrial Ca2+ fluxes rescuing the α-syn-induced loss of primary mesencephalic DA neuron cultures.
In support of the relationships between immune system and α-syn pathology, an interesting study reported significantly higher service levels of antibodies against monomeric α-syn in patients at early phases of disease. This paper postulates that autoimmunity responses take part in a compensative attempt of neuroprotection (126). A recent paper by Shalash and collaborators has shown that α-syn autoantibodies (AIAs) can be a promising avenue in the field of peripheral biomarkers compared patients with PD, to patients with AD along with controls (127). These molecules are also produced in gender-dependent fashion, across the lifespan during the development and can be detected in healthy young subjects, with titers similar to healthy adults (128). This might suggest that autoantibodies production might be optimized over time in response to environmental stimuli.
A series of studies by Sulzer and coworkers further supports the existence of a link between α-syn and immune response. During PD neuroinflammation, the blood–brain barrier becomes permeable to immune cells recruited into the CNS by massive proinflammatory cytokine production from microglia (129, 130). A recent paper by this group shows that this process triggers an immune response against identified α-syn epitopes in PD patients who presents specific major histocompatibility complex alleles. In particular, two antigenic regions have been identified: the first near the N terminus (called Y39 region) and the second near the C terminus, the well-known S129 region, containing the amino acid residue S129, whose phosphorylation has been associated with α-syn pathology (131).
Early Synaptic Alterations Preceding Neurodegeneration
Although many advances have been made in deciphering the mechanisms by which α-syn triggers neurodegenerative pathways, the ability of mammalian brain to compensate for loss of functions still constitutes a main obstacle in a readily identification of the disease’s traits. As a consequence, a still unmet medical need is to find increasingly reliable functional biomarkers of disease that may bring to an earlier diagnosis and, possibly, predict disease trajectory. To this aim, current research is focusing on cerebrospinal fluid biomarkers and early synaptic alterations. While advances in the research of peripheral biomarkers have been extensively reviewed elsewhere (132), we will focus on the other front with a main question to address: which measurable synaptic changes can be predictive of PD neurodegeneration?
An ideal condition to explore subtle alteration of synaptic activity before neuronal degeneration is offered by animal models of disease in which expression of α-syn is genetically altered. In these models, it is possible to follow the synapse development at different time points along the disease progression and simultaneously study associated motor deficits to find increasingly more sensitive behavioral tests. Both presynaptic and postsynaptic modifications, recently reviewed by Burre, have been associated with α-syn pathology (20). However, given their specific localization at nerve terminals, presynaptic alterations were soon predicted and investigated, leading to seminal papers demonstrating that altered α-syn interferes with SNARE protein assembly, with an associated reduction in exocytosis and DA release (9) thus affecting the activity of the release machinery, although the extent by which α-syn affects neurotransmitter release is debated (20). Relevant to its presynaptic effects, it is noteworthy that α-syn also interacts with other synaptic proteins, such as Synapsin III, a protein that, similar to other members of the synapsin family, plays essential roles in neurotransmitter release, but has an extrasynaptic localization. Interestingly, it has been reported that synapsin III interacts with α-syn in both physiological and pathological conditions, further increasing the complex pattern of presynaptic actions of α-syn pathology (20, 133–136). It seems to be clear, in fact, that due to its ability to mobilize among different sectors of the active zone upon stimulation (137, 138), α-syn dynamic interactions with SNARE, lipidic raft and DAT are highly dependent on the neural activity. Any alteration in this well-tuned machinery is therefore associated with neurotransmission alterations, which may have less impact in basal neurotransmission, but become critical during intense neuronal activity and over their long lifetime. α-syn can also permeabilize lipid membranes through the formation of cations permeable transmembrane pores, thus altering membrane conductances, and increase the risk for altered calcium homeostasis (12, 139).
This effect of α-syn was also studied in cell systems overexpressing the protein (140, 141). Using whole-cell patch-clamp recordings, Feng and coworkers (88) measured ion leakage upon the application of an electrical potential in a dopaminergic cell line. These effects were associated with a modest but significant time-dependent increase in cell death, demonstrating a link between α-syn pathology and conductance changes.
However, while DA release machinery alterations were a primary expected effect of α-syn toxicity, more recent papers have focused on the postsynaptic counterpart of this pathological scenario. Altered activity and distribution of postsynaptic density components have only recently been explored but may be promising tools to detect subtle but measurable changes at the core of this synaptopathy.
Since cognitive alterations have been observed as prodromal PD symptoms early plastic alterations have been first explored in the hippocampus. In 2012, Costa and colleagues studied CA1 hippocampal plasticity in a transgenic mouse model for α-syn aggregation obtained by the expression of human α-syn 120 under the control of the tyrosine hydroxylase promoter (α-syn 120 mice) and leading to the formation of pathological inclusions in the SNc and olfactory bulb and to a reduction in striatal DA levels (142, 143). In a presymptomatic motor stage characterized by spatial memory alterations, CA1 hippocampal pyramidal neurons of α-syn 120 mice show a reduced ability to respond to a high-frequency stimulation with a form of long-lasting plasticity expressed in this area and dependent on DA D1 and NMDA receptors stimulation, called long-term potentiation (LTP). Postsynaptic density modifications were associated with plastic changes as NMDA receptor subunit composition was found changed with a significant decrease of GluN2A/GluN2B subunit ratio. This effect was due to decreased DA release as l-DOPA was able to rescue synaptic functions. Overall, these results first demonstrated that, similar to human condition, cognitive deficit precede motor symptoms with postsynaptic mechanisms (143). In support of this notion, other studies have shown that α-syn plays a role in NMDA receptor trafficking in other brain areas (144–147), suggesting that postsynaptic actions of α-syn impact on intracellular events relevant for synaptic plasticity.
Given the possibility that in pathological conditions α-syn species (monomer, oligomers, and fibrils) may also act extracellularly thus possibly inducing postsynaptic effects, in vitro models have been developed to clarify the role of extracellular α-syn in hippocampal plasticity alterations. On this line, the group of Outeiro conducted a series of studies to demonstrate the effects of extracellular α-syn oligomers. The study carried out by Diogenes and colleagues shows that different species of α-syn have distinct effects on synaptic activity. In particular, among oligomers, monomers, and fibrils, only prolonged incubation with oligomers in healthy rodent brain slices were able to increase basal synaptic transmission through a mechanism dependent on NMDA receptor activation, accompanied by an increase in the expression of GluR2-lacking AMPA receptors. In these slices, stimulation with a theta-burst pattern was not able to induce LTP without a previous application of a low-frequency stimulation indicating a saturation effect underlying impairment of LTP (148). Interestingly, these detrimental effects were counteracted by adenosine A2a receptor antagonists, known for their neuroprotective role in PD therapy, and were not observed in animals lacking A2aR. Moreover, blockade of these receptors was able to reduce α-syn aggregates (149).
Another evidence of the link between α-syn overexpression and NMDA receptor dysfunction comes from a recent study of the same group as a further support to the concept that many elements in the postsynaptic compartment play important roles in predisposing the synapse to NMDA-dependent excitotoxicity mediated by its interaction with α-syn aggregates. Using an in vitro approach, the authors demonstrated that LTP alterations are caused by the abnormal activity of cellular prion protein, known to act as a cell surface-binding partner for soluble oligomeric protein and to interact with NMDA receptors at postsynaptic density. This protein, when engaged in pathological interactions with α-syn, mediates Ca2+ dyshomeostasis and synaptic dysfunction through a mechanism involving Fyn kinase phosphorylation, which is tightly regulated by mGluR5 via adenosine A2A receptors. In turn, activated Fyn phosphorylates Y1472 residue of GluN2B-expressing NMDA receptors with consequent excitotoxic effects (150).
Although these studies greatly contributed to the understanding of α-syn synaptic effects, the effects of α-syn oligomers on the functional activity of the striatum have been explored only recently. In fact, the striatum represents the most interesting target for PD therapy since it is the main recipient of dopaminergic nigral neurons, whose activity is impaired by α-syn-mediated toxicity (151). For this reason, transgenic animals overexpressing altered forms of α-syn, such as the truncated human α-syn 1–120 or the WT human α-syn, may be valuable models to assess specific aspects of the pathogenesis of synucleinopathies and to analyze the cell type-specific alterations of striatal synaptic plasticity in the initial phase of the disease.
The first report to show alterations in corticostriatal plasticity associated with α-syn overexpression was provided by an ex vivo study performed in slices from mice overexpressing a truncated form of α-syn at a late symptomatic stage of disease (152). But, it was few years later that, taking advantage of the gradual progression of the disease offered by genetic models, Tozzi and coworkers investigated α-syn-mediated alterations in plasticity by studying both spiny projection neurons (SPNs) and cholinergic interneurons (ChIs) in two different models of early PD: the mice transgenic for truncated human α-syn (1–120) and the rat injected with the adeno-associated viral vector carrying WT human α-syn in the SNc (153). In a presymptomatic stage, before any neuronal degeneration, procedural learning deficit was associated in both rodent models with selective impairment of LTP in ChIs but not in SPNs. Similar to what observed in the hippocampus, also here a direct interaction between α-syn and NMDA receptors is suggested, as the loss of LTP in striatal ChIs was dependent on a direct interaction of α-syn with GluN2D-expressing NMDA receptors, which are selectively expressed in this class of interneurons. This link has also been studied in an in vitro model. Bath incubation of corticostriatal slices of healthy animals with α-syn oligomers caused the same synaptic alterations that were not rescued by exogenous DA or a D1-like receptor agonist, suggesting that the blockade on synaptic plasticity is not mediated by an α-syn-mediated interference with DA release. These alterations correlate with the behavioral pattern observed, mimicking the early phase of PD, and are in line with what observed in PD patients, in which mild cognitive alterations associated with cholinergic dysfunction, frequently precede overt motor symptoms.
It is noteworthy that micromolar concentration of α-syn oligomers were found effective in determining hippocampal pathology, while nanomolar concentration were sufficient to induce striatal alterations. Taken together, these data suggest that increasing concentrations of α-syn may progressively affect NMDA receptor-mediated functions on distinct neuronal populations, indicating that the vulnerability to this protein may be cell type specific and region specific. On this view, early dysfunction of the striatal cholinergic system, occurring at very low concentrations, represents a possible functional marker of the disease. On the same line of research, a recent study by Giordano and coworkers provided an important link between presynaptic and postsynaptic actions of α-syn and contribute to the reconstruction of a comprehensive view of the many faces of α-syn pathology (154).
Using an animal model of PD, in which animals overexpress human WT α-syn in the midbrain neurons, the authors demonstrate that very early stages are associated with reduced striatal DAT and impaired acquisition of performance plateau in the rotarod task. Interestingly, behavioral impairment has a unique electrophysiological correlate that depends on DAT alteration. In fact, while a form of plasticity, the long-term depression (LTD) is equally expressed at corticostriatal synapses of both WT and α-syn mice before and after exposure to exercise through accelerated rotarod test, healthy animals show an interesting switch from LTD to LTP during the acquisition phase of motor learning. Mice overexpressing human α-syn do not show this shift in plasticity that is instrumental to acquire motor habits and perform correctly. This early training-induced shift from LTD to LTP, and the achievement of a good performance, is impaired in control animals pretreated with DAT inhibitor GBR-12909. These findings, in line with previous studies (155–159), further suggest that early signs of synucleinopathy do not necessarily correlate with DA neuronal loss and support the notion that a reorganization of cellular plasticity within the dorsal striatum is necessary for the acquisition of a motor skill, and it depends on an intact dopaminergic transmission, controlled by DAT, which is impaired early by nigral overexpression of human α-syn.
Conclusion
Taken together, all these findings return a complex but increasingly clear scenario designed by the many roles of α-syn (Figure 1; Table 1).
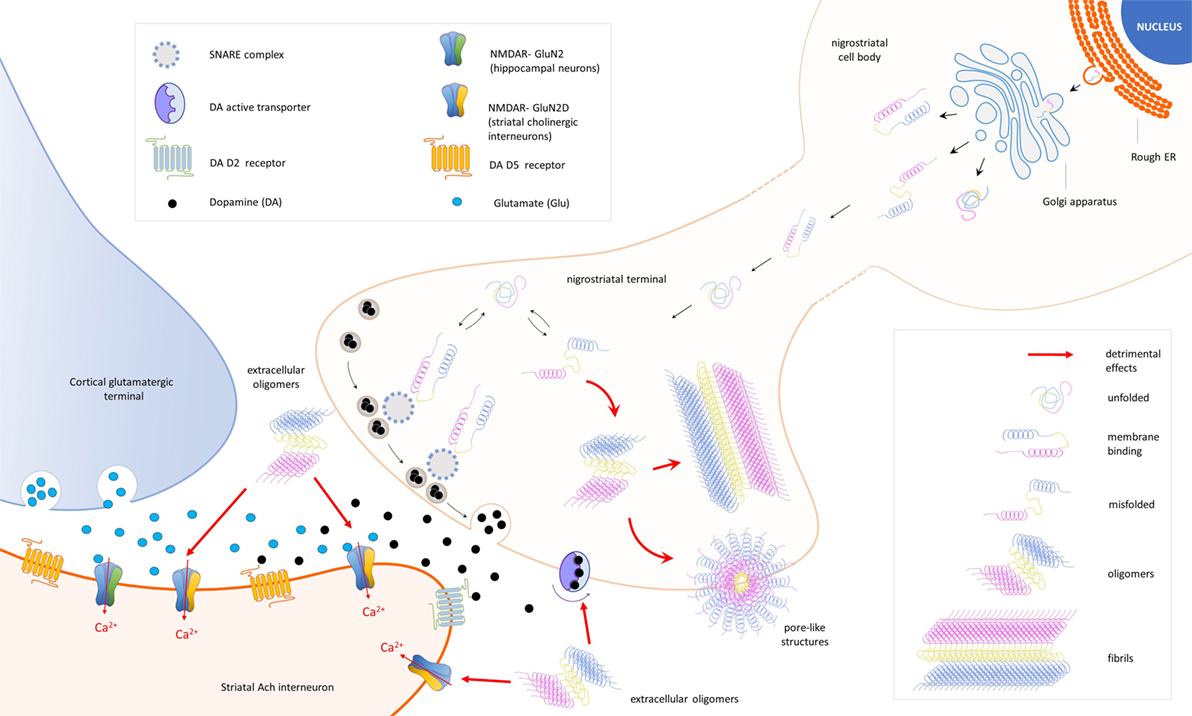
Figure 1. Schematic representation of the cellular and synaptic detrimental actions mediated by different forms of the protein alpha-synuclein (α-syn). In nigral neurons, endoplasmic reticulum (rough ER), SNCA transcripts are translated into native α-syn proteins, which are assembled in the Golgi apparatus and released in different conformations. Due to its auto-chaperone activity, α-syn exists in a dynamic balance between monomeric unfolded and amphipathic alpha-helix (membrane binding) state, adopting a range of conformations depending on the environment and binding partners. During the assembly process, misfolding proteins might be also produced (misfolded) and escape detection and clearance by intracellular quality control systems. After synapse maturation, α-syn migrates to nerve terminals and interacts with intracellular proteins [SNAP REceptor (SNARE) complex] and the dopamine (DA) active trasporter to ensure a correct control of neurotransmission. Misfolded α-syn may combine into oligomers that, under specific stimulations, form transmembrane pore-like structures able to alter membrane conductances. Overexpression of α-syn exacerbates pathological events and culminates with the formation of fibrillar aggregates (fibrils), a major component of Lewy bodies. Extracellular α-syn oligomers interfere with the expression of long-term potentiation, a form of synaptic plasticity mediated by N-methyl-d-aspartate receptors (NMDAR) in striatal cholinergic interneurons. A direct interaction between α-syn and the GluN2D subunit has been demonstrated in three different models of experimental parkinsonism.
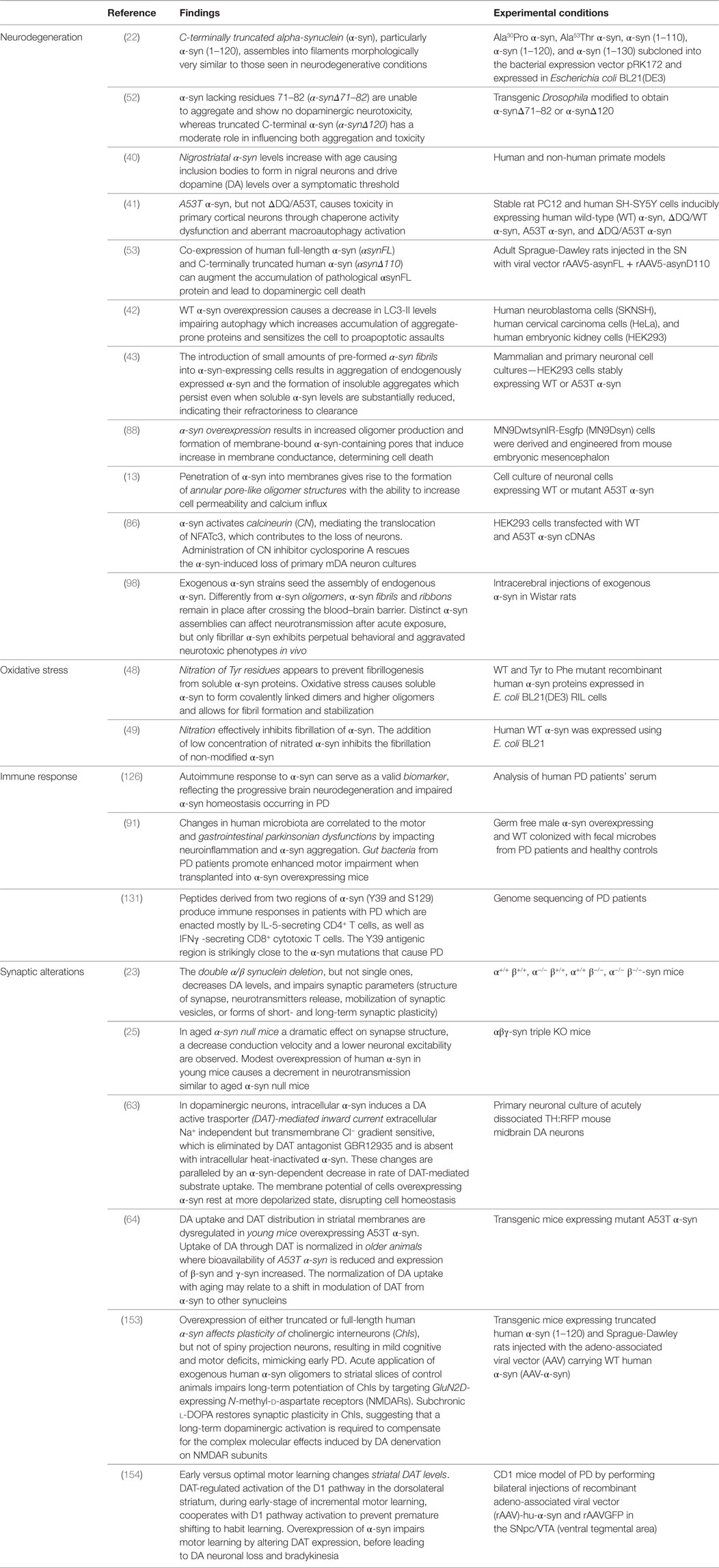
Table 1. Summary of the findings on the role of α-syn in the distinct aspects contributing to pathogenesis of Parkinson’s disease (PD).
Future studies will be needed to further investigate how endogenous molecules interact with different α-syn conformations. In particular, based on the majority of data reviewed here, we expect that more effort will be aimed at explaining the mechanisms underlying distinct cell-type and region-specific α-syn-mediated NMDA receptor alterations. Moreover, in light of the latter study and of many reports supporting the neuroprotective effect of intense exercise in newly designed rehabilitation programs (160, 161), a promising link to explore will be the interaction between α-syn aggregation and experience.
Author Contributions
VG wrote and revised the manuscript, prepared the figure and reviewed the final version; VC revised the manuscript, prepared the review table and proofread the final version; PC conceived and designed the manuscript, edited, and proofread the final version of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from Progetto di Ricerca di Interesse Nazionale (PRIN) 2015 (prot. 2015FNWP34) (to PC), Ricerca Finalizzata RF-2013-02356215 (to PC) and by grants from the Fresco Parkinson Institute to New York University School of Medicine and The Marlene and Paolo Fresco Institute for Parkinson’s and Movement Disorders, which were made possible with support from Marlene and Paolo Fresco (to PC and to VG).
References
1. Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett (1994) 345:27–32. doi:10.1016/0014-5793(94)00395-5
2. Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A (1993) 90:11282–6. doi:10.1073/pnas.90.23.11282
3. Nakai M, Fujita M, Waragai M, Sugama S, Wei J, Akatsu H, et al. Expression of alpha-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem Biophys Res Commun (2007) 358:104–10. doi:10.1016/j.bbrc.2007.04.108
4. Askanas V, Engel WK, Alvarez RB, Mcferrin J, Broccolini A. Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J Neuropathol Exp Neurol (2000) 59:592–8. doi:10.1093/jnen/59.7.592
5. Ltic S, Perovic M, Mladenovic A, Raicevic N, Ruzdijic S, Rakic L, et al. Alpha-synuclein is expressed in different tissues during human fetal development. J Mol Neurosci (2004) 22:199–204. doi:10.1385/JMN:22:3:199
6. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci (2013) 14:38–48. doi:10.1038/nrn3406
7. Jain N, Bhasne K, Hemaswasthi M, Mukhopadhyay S. Structural and dynamical insights into the membrane-bound alpha-synuclein. PLoS One (2013) 8:e83752. doi:10.1371/journal.pone.0083752
8. Gallegos S, Pacheco C, Peters C, Opazo CM, Aguayo LG. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front Neurosci (2015) 9:59. doi:10.3389/fnins.2015.00059
9. Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain (2010) 133:2032–44. doi:10.1093/brain/awq132
10. Sidhu A, Wersinger C, Vernier P. Alpha-synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett (2004) 565:1–5. doi:10.1016/j.febslet.2004.03.063
11. Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P. Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem (2008) 106:560–77. doi:10.1111/j.1471-4159.2008.05406.x
12. Tosatto L, Andrighetti AO, Plotegher N, Antonini V, Tessari I, Ricci L, et al. Alpha-synuclein pore forming activity upon membrane association. Biochim Biophys Acta (2012) 1818:2876–83. doi:10.1016/j.bbamem.2012.07.007
13. Tsigelny IF, Sharikov Y, Wrasidlo W, Gonzalez T, Desplats PA, Crews L, et al. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J (2012) 279:1000–13. doi:10.1111/j.1742-4658.2012.08489.x
14. Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, et al. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry (2003) 42:8530–40. doi:10.1021/bi027363r
15. Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry (2004) 43:16233–42. doi:10.1021/bi048453u
16. Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, et al. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A (2005) 102:1430–5. doi:10.1073/pnas.0407146102
17. Levitan K, Chereau D, Cohen SI, Knowles TP, Dobson CM, Fink AL, et al. Conserved C-terminal charge exerts a profound influence on the aggregation rate of alpha-synuclein. J Mol Biol (2011) 411:329–33. doi:10.1016/j.jmb.2011.05.046
18. Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc (2005) 127:476–7. doi:10.1021/ja044834j
19. Zhou W, Long C, Reaney SH, Di Monte DA, Fink AL, Uversky VN. Methionine oxidation stabilizes non-toxic oligomers of alpha-synuclein through strengthening the auto-inhibitory intra-molecular long-range interactions. Biochim Biophys Acta (2010) 1802:322–30. doi:10.1016/j.bbadis.2009.12.004
20. Burre J. The synaptic function of alpha-synuclein. J Parkinsons Dis (2015) 5:699–713. doi:10.3233/JPD-150642
21. Souza JM, Giasson BI, Lee VM, Ischiropoulos H. Chaperone-like activity of synucleins. FEBS Lett (2000) 474:116–9. doi:10.1016/S0014-5793(00)01563-5
22. Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett (1998) 436:309–12. doi:10.1016/S0014-5793(98)01146-6
23. Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A (2004) 101:14966–71. doi:10.1073/pnas.0406283101
24. Bisaglia M, Mammi S, Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J (2009) 23:329–40. doi:10.1096/fj.08-119784
25. Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, et al. Alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A (2010) 107:19573–8. doi:10.1073/pnas.1005005107
26. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science (2003) 302:841. doi:10.1126/science.1090278
27. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet (2004) 364:1169–71. doi:10.1016/S0140-6736(04)17104-3
28. Hightower LE. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell (1991) 66:191–7. doi:10.1016/0092-8674(91)90611-2
29. Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron (2001) 29:15–32. doi:10.1016/S0896-6273(01)00177-5
30. Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science (2002) 295:1852–8. doi:10.1126/science.1068408
31. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science (1997) 276:2045–7. doi:10.1126/science.276.5321.2045
32. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388:839–40. doi:10.1038/42166
33. Mezey E, Dehejia AM, Harta G, Tresser N, Suchy SF, Nussbaum RL, et al. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol Psychiatry (1998) 3:493–9. doi:10.1038/sj.mp.4000446
34. Kim C, Lee SJ. Controlling the mass action of alpha-synuclein in Parkinson’s disease. J Neurochem (2008) 107:303–16. doi:10.1111/j.1471-4159.2008.05612.x
35. Melki R. Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J Parkinsons Dis (2015) 5:217–27. doi:10.3233/JPD-150543
36. Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson’s disease pathogenesis and therapies. Prog Brain Res (2010) 183:115–45. doi:10.1016/S0079-6123(10)83007-9
37. Chavarria C, Souza JM. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch Biochem Biophys (2013) 533:25–32. doi:10.1016/j.abb.2013.02.009
38. Xilouri M, Brekk OR, Stefanis L. Alpha-synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol (2013) 47:537–51. doi:10.1007/s12035-012-8341-2
40. Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis (2007) 25:134–49. doi:10.1016/j.nbd.2006.08.021
41. Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Aberrant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One (2009) 4:e5515. doi:10.1371/journal.pone.0005515
42. Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol (2010) 190:1023–37. doi:10.1083/jcb.201003122
43. Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem (2013) 288:15194–210. doi:10.1074/jbc.M113.457408
44. Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol (2015) 14:855–66. doi:10.1016/S1474-4422(15)00006-X
45. Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem (2006) 281:29739–52. doi:10.1074/jbc.M600933200
46. Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep (2014) 4:5797. doi:10.1038/srep05797
47. Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J Biol Chem (2008) 283:16895–905. doi:10.1074/jbc.M800747200
48. Norris EH, Giasson BI, Ischiropoulos H, Lee VM. Effects of oxidative and nitrative challenges on alpha-synuclein fibrillogenesis involve distinct mechanisms of protein modifications. J Biol Chem (2003) 278:27230–40. doi:10.1074/jbc.M212436200
49. Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett (2003) 542:147–52. doi:10.1016/S0014-5793(03)00367-3
50. Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science (2000) 290:985–9. doi:10.1126/science.290.5493.985
51. Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science (2001) 294:1346–9. doi:10.1126/science.1063522
52. Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci (2007) 27:3338–46. doi:10.1523/JNEUROSCI.0285-07.2007
53. Ulusoy A, Febbraro F, Jensen PH, Kirik D, Romero-Ramos M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur J Neurosci (2010) 32:409–22. doi:10.1111/j.1460-9568.2010.07284.x
54. Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med (1998) 4:1318–20. doi:10.1038/3311
55. Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A (2000) 97:571–6. doi:10.1073/pnas.97.2.571
56. Karpinar DP, Balija MB, Kugler S, Opazo F, Rezaei-Ghaleh N, Wender N, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J (2009) 28:3256–68. doi:10.1038/emboj.2009.257
57. Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, et al. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A (2010) 107:7710–5. doi:10.1073/pnas.0910723107
58. Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol (2013) 125:795–813. doi:10.1007/s00401-013-1114-9
59. Levin J, Schmidt F, Boehm C, Prix C, Botzel K, Ryazanov S, et al. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol (2014) 127:779–80. doi:10.1007/s00401-014-1265-3
60. Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One (2011) 6:e19338. doi:10.1371/journal.pone.0019338
61. Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci (2012) 32:13454–69. doi:10.1523/JNEUROSCI.1292-12.2012
62. Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol (2014) 127:861–79. doi:10.1007/s00401-014-1256-4
63. Swant J, Goodwin JS, North A, Ali AA, Gamble-George J, Chirwa S, et al. Alpha-synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J Biol Chem (2011) 286:43933–43. doi:10.1074/jbc.M111.241232
64. Oaks AW, Frankfurt M, Finkelstein DI, Sidhu A. Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PLoS One (2013) 8:e60378. doi:10.1371/journal.pone.0060378
65. Butler B, Goodwin S, Saha K, Becker J, Sambo D, Davari P, et al. Dopamine transporter activity is modulated by alpha-synuclein. J Biol Chem (2015) 290(49):29542–54. doi:10.1074/jbc.A115.639880
66. Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J (2001) 15:916–26. doi:10.1096/fj.00-0334com
67. Wersinger C, Prou D, Vernier P, Sidhu A. Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J (2003) 17:2151–3. doi:10.1096/fj.03-0152fje
68. Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci Lett (2003) 340:189–92. doi:10.1016/S0304-3940(03)00097-1
69. Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci (2002) 22:3090–9. doi:10.1523/JNEUROSCI.22-08-03090.2002
70. Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol Aging (2010) 31:796–804. doi:10.1016/j.neurobiolaging.2008.11.001
71. Chadchankar H, Ihalainen J, Tanila H, Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res (2011) 1382:37–44. doi:10.1016/j.brainres.2011.01.064
72. Garcia-Reitboeck P, Anichtchik O, Dalley JW, Ninkina N, Tofaris GK, Buchman VL, et al. Endogenous alpha-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp Neurol (2013) 248:541–5. doi:10.1016/j.expneurol.2013.07.015
73. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci (2017) 18:101–13. doi:10.1038/nrn.2016.178
74. Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol (1994) 113:831–8. doi:10.1111/j.1476-5381.1994.tb17068.x
75. Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci (2007) 27:645–56. doi:10.1523/JNEUROSCI.4341-06.2007
76. Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci (2009) 29:11011–9. doi:10.1523/JNEUROSCI.2519-09.2009
77. Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol (2009) 19:81–8. doi:10.1016/j.tcb.2008.12.002
78. Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature (2010) 468:696–700. doi:10.1038/nature09536
79. Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta (2009) 1787:1334–41. doi:10.1016/j.bbabio.2009.05.011
80. Budd SL, Nicholls DG. Mitochondria in the life and death of neurons. Essays Biochem (1998) 33:43–52. doi:10.1042/bse0330043
81. Putzier I, Kullmann PH, Horn JP, Levitan ES. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci (2009) 29:15414–9. doi:10.1523/JNEUROSCI.4742-09.2009
82. Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, et al. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat Neurosci (2012) 15:1414–21. doi:10.1038/nn.3209
83. Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci (2014) 17:832–40. doi:10.1038/nn.3717
84. Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science (2017) 357:1255–61. doi:10.1126/science.aam9080
85. Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci (2015) 35:890–905. doi:10.1523/JNEUROSCI.0089-14.2015
86. Luo J, Sun L, Lin X, Liu G, Yu J, Parisiadou L, et al. A calcineurin- and NFAT-dependent pathway is involved in alpha-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum Mol Genet (2014) 23:6567–74. doi:10.1093/hmg/ddu377
87. Surmeier DJ, Guzman JN, Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium (2010) 47:175–82. doi:10.1016/j.ceca.2009.12.003
88. Feng LR, Federoff HJ, Vicini S, Maguire-Zeiss KA. Alpha-synuclein mediates alterations in membrane conductance: a potential role for alpha-synuclein oligomers in cell vulnerability. Eur J Neurosci (2010) 32:10–7. doi:10.1111/j.1460-9568.2010.07266.x
89. Braak H, Del Tredici K, Rub U, De Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging (2003) 24:197–211. doi:10.1016/S0197-4580(02)00065-9
90. Lema Tome CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease – is there a link? Mol Neurobiol (2013) 47:561–74. doi:10.1007/s12035-012-8267-8
91. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell (2016) 167:1469–80.e1412. doi:10.1016/j.cell.2016.11.018
92. Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol (2010) 9:1128–38. doi:10.1016/S1474-4422(10)70213-1
93. Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol (2010) 11:301–7. doi:10.1038/nrm2873
94. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers (2017) 3:17013. doi:10.1038/nrdp.2017.13
95. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med (2008) 14:504–6. doi:10.1038/nm1747
96. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med (2008) 14:501–3. doi:10.1038/nm1746
97. Mendez I, Vinuela A, Astradsson A, Mukhida K, Hallett P, Robertson H, et al. Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat Med (2008) 14:507–9. doi:10.1038/nm1752
98. Peelaerts W, Bousset L, Van Der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature (2015) 522:340–4. doi:10.1038/nature14547
99. Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun (2013) 4:2575. doi:10.1038/ncomms3575
100. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell (2013) 154:103–17. doi:10.1016/j.cell.2013.05.057
101. Lee SJ, Masliah E. Neurodegeneration: aggregates feel the strain. Nature (2015) 522:296–7. doi:10.1038/nature14526
102. Engelender S, Isacson O. The threshold theory for Parkinson’s disease. Trends Neurosci (2017) 40:4–14. doi:10.1016/j.tins.2016.10.008
103. Laiwand R, Werman R, Yarom Y. Time course and distribution of motoneuronal loss in the dorsal motor vagal nucleus of guinea pig after cervical vagotomy. J Comp Neurol (1987) 256:527–37. doi:10.1002/cne.902560405
104. Lams BE, Isacson O, Sofroniew MV. Loss of transmitter-associated enzyme staining following axotomy does not indicate death of brainstem cholinergic neurons. Brain Res (1988) 475:401–6. doi:10.1016/0006-8993(88)90635-X
105. Sofroniew MV, Isacson O. Distribution of degeneration of cholinergic neurons in the septum following axotomy in different portions of the fimbria-fornix: a correlation between degree of cell loss and proximity of neuronal somata to the lesion. J Chem Neuroanat (1988) 1:327–37.
106. Engel AK, Kreutzberg GW. Neuronal surface changes in the dorsal vagal motor nucleus of the guinea pig in response to axotomy. J Comp Neurol (1988) 275:181–200. doi:10.1002/cne.902750203
107. Fernandez E, Pallini R, Lauretti L, Marchese E, Gangitano C, Del Fa A, et al. Levocarnitine acetyl prevents cell death following long-term section of the vagus nerve in rats. Int J Clin Pharmacol Res (1992) 12:289–97.
108. Alexander GE, Delong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci (1986) 9:357–81. doi:10.1146/annurev.ne.09.030186.002041
109. DeLong MR, Wichmann T. Basal ganglia circuits as targets for neuromodulation in Parkinson disease. JAMA Neurol (2015) 72:1354–60. doi:10.1001/jamaneurol.2015.2397
110. Cullen CL, Young KM. How does transcranial magnetic stimulation influence glial cells in the central nervous system? Front Neural Circuits (2016) 10:26. doi:10.3389/fncir.2016.00026
111. Booth HDE, Hirst WD, Wade-Martins R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci (2017) 40:358–70. doi:10.1016/j.tins.2017.04.001
112. Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ (2010) 17:1083–91. doi:10.1038/cdd.2009.179
113. Rocha NP, De Miranda AS, Teixeira AL. Insights into neuroinflammation in Parkinson’s disease: from biomarkers to anti-inflammatory based therapies. Biomed Res Int (2015) 2015:628192. doi:10.1155/2015/628192
114. McGeer PL, McGeer EG. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Dis Assoc Disord (1998) 12(Suppl 2):S1–6. doi:10.1097/00002093-199803001-00001
115. Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci (2002) 15:991–8. doi:10.1046/j.1460-9568.2002.01938.x
116. McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol (2003) 54:599–604. doi:10.1002/ana.10728
117. Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain (2005) 128:2665–74. doi:10.1093/brain/awh625
118. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol (2010) 119:7–35. doi:10.1007/s00401-009-0619-8
119. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron (2016) 89:37–53. doi:10.1016/j.neuron.2015.11.013
120. Tsai MJ, Lee EH. Characterization of L-DOPA transport in cultured rat and mouse astrocytes. J Neurosci Res (1996) 43:490–5. doi:10.1002/(SICI)1097-4547(19960215)43:4<490::AID-JNR10>3.0.CO;2-6
121. Inyushin MY, Huertas A, Kucheryavykh YV, Kucheryavykh LY, Tsydzik V, Sanabria P, et al. L-DOPA uptake in astrocytic endfeet enwrapping blood vessels in rat brain. Parkinsons Dis (2012) 2012:321406. doi:10.1155/2012/321406
122. Oliva I, Fernandez M, Martin ED. Dopamine release regulation by astrocytes during cerebral ischemia. Neurobiol Dis (2013) 58:231–41. doi:10.1016/j.nbd.2013.06.007
123. Asanuma M, Miyazaki I, Murakami S, Diaz-Corrales FJ, Ogawa N. Striatal astrocytes act as a reservoir for L-DOPA. PLoS One (2014) 9:e106362. doi:10.1371/journal.pone.0106362
124. Cacace F, Mineo D, Viscomi MT, Latagliata EC, Mancini M, Sasso V, et al. Intermittent theta-burst stimulation rescues dopamine-dependent corticostriatal synaptic plasticity and motor behavior in experimental parkinsonism: possible role of glial activity. Mov Disord (2017) 32:1035–46. doi:10.1002/mds.26982
125. Sasso V, Bisicchia E, Latini L, Ghiglieri V, Cacace F, Carola V, et al. Repetitive transcranial magnetic stimulation reduces remote apoptotic cell death and inflammation after focal brain injury. J Neuroinflammation (2016) 13:150. doi:10.1186/s12974-016-0616-5
126. Yanamandra K, Gruden MA, Casaite V, Meskys R, Forsgren L, Morozova-Roche LA. Alpha-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson’s disease patients. PLoS One (2011) 6:e18513. doi:10.1371/journal.pone.0018513
127. Shalash A, Salama M, Makar M, Roushdy T, Elrassas HH, Mohamed W, et al. Elevated serum alpha-synuclein autoantibodies in patients with Parkinson’s disease relative to Alzheimer’s disease and controls. Front Neurol (2017) 8:720. doi:10.3389/fneur.2017.00720
128. Kuhn I, Rogosch T, Schindler TI, Tackenberg B, Zemlin M, Maier RF, et al. Serum titers of autoantibodies against alpha-synuclein and tau in child- and adulthood. J Neuroimmunol (2018) 315:33–9. doi:10.1016/j.jneuroim.2017.12.003
129. Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest (2009) 119:182–92. doi:10.1172/JCI36470
130. Cebrian C, Loike JD, Sulzer D. Neuroinflammation in Parkinson’s disease animal models: a cell stress response or a step in neurodegeneration? Curr Top Behav Neurosci (2015) 22:237–70. doi:10.1007/7854_2014_356
131. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature (2017) 546:656–61. doi:10.1038/nature22815
132. Eusebi P, Giannandrea D, Biscetti L, Abraha I, Chiasserini D, Orso M, et al. Diagnostic utility of cerebrospinal fluid alpha-synuclein in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord (2017) 32:1389–400. doi:10.1002/mds.27110
133. Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ, Wightman RM. Synapsins differentially control dopamine and serotonin release. J Neurosci (2010) 30:9762–70. doi:10.1523/JNEUROSCI.2071-09.2010
134. Porton B, Wetsel WC, Kao HT. Synapsin III: role in neuronal plasticity and disease. Semin Cell Dev Biol (2011) 22:416–24. doi:10.1016/j.semcdb.2011.07.007
135. Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S, et al. Alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci (2015) 128:2231–43. doi:10.1242/jcs.157867
136. Longhena F, Faustini G, Varanita T, Zaltieri M, Porrini V, Tessari I, et al. Synapsin III is a key component of alpha-synuclein fibrils in Lewy bodies of PD brains. Brain Pathol (2018). doi:10.1111/bpa.12587
137. Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci (2005) 25:10913–21. doi:10.1523/JNEUROSCI.2922-05.2005
138. Fortin DL, Nemani VM, Nakamura K, Edwards RH. The behavior of alpha-synuclein in neurons. Mov Disord (2010) 25(Suppl 1):S21–6. doi:10.1002/mds.22722
139. Kim HY, Cho MK, Kumar A, Maier E, Siebenhaar C, Becker S, et al. Structural properties of pore-forming oligomers of alpha-synuclein. J Am Chem Soc (2009) 131:17482–9. doi:10.1021/ja9077599
140. Zakharov SD, Hulleman JD, Dutseva EA, Antonenko YN, Rochet JC, Cramer WA. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry (2007) 46:14369–79. doi:10.1021/bi701275p
141. Di Pasquale E, Fantini J, Chahinian H, Maresca M, Taieb N, Yahi N. Altered ion channel formation by the Parkinson’s-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J Mol Biol (2010) 397:202–18. doi:10.1016/j.jmb.2010.01.046
142. Tofaris GK, Garcia-Reitbock P, Humby T, Lambourne SL, O’Connell M, Ghetti B, et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci (2006) 26:3942–50. doi:10.1523/JNEUROSCI.4965-05.2006
143. Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain (2012) 135:1884–99. doi:10.1093/brain/aws101
144. Cheng F, Li X, Li Y, Wang C, Wang T, Liu G, et al. Alpha-synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J Neurochem (2011) 119:815–25. doi:10.1111/j.1471-4159.2011.07460.x
145. Chen Y, Yang W, Li X, Li X, Yang H, Xu Z, et al. Alpha-synuclein-induced internalization of NMDA receptors in hippocampal neurons is associated with reduced inward current and Ca(2+) influx upon NMDA stimulation. Neuroscience (2015) 300:297–306. doi:10.1016/j.neuroscience.2015.05.035
146. Navarria L, Zaltieri M, Longhena F, Spillantini MG, Missale C, Spano P, et al. Alpha-synuclein modulates NR2B-containing NMDA receptors and decreases their levels after rotenone exposure. Neurochem Int (2015) 85–86:14–23. doi:10.1016/j.neuint.2015.03.008
147. Yang J, Hertz E, Zhang X, Leinartaite L, Lundius EG, Li J, et al. Overexpression of alpha-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci Lett (2016) 611:51–8. doi:10.1016/j.neulet.2015.11.023
148. Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci (2012) 32:11750–62. doi:10.1523/JNEUROSCI.0234-12.2012
149. Ferreira DG, Batalha VL, Vicente Miranda H, Coelho JE, Gomes R, Goncalves FQ, et al. Adenosine A2A receptors modulate alpha-synuclein aggregation and toxicity. Cereb Cortex (2015) 27:718–30. doi:10.1093/cercor/bhv268
150. Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szego EM, et al. Alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci (2017) 20:1569–79. doi:10.1038/nn.4648
151. Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A (2013) 110:E4016–25. doi:10.1073/pnas.1309143110
152. Tozzi A, Costa C, Siliquini S, Tantucci M, Picconi B, Kurz A, et al. Mechanisms underlying altered striatal synaptic plasticity in old A53T-alpha synuclein overexpressing mice. Neurobiol Aging (2012) 33:1792–9. doi:10.1016/j.neurobiolaging.2011.05.002
153. Tozzi A, De Iure A, Bagetta V, Tantucci M, Durante V, Quiroga-Varela A, et al. Alpha-synuclein produces early behavioral alterations via striatal cholinergic synaptic dysfunction by interacting with GluN2D N-methyl-D-aspartate receptor subunit. Biol Psychiatry (2016) 79:402–14. doi:10.1016/j.biopsych.2015.08.013
154. Giordano N, Iemolo A, Mancini M, Cacace F, De Risi M, Latagliata EC, et al. Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain (2017) 141(2):505–20. doi:10.1093/brain/awx351
155. Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci (2002) 22:2780–91. doi:10.1523/JNEUROSCI.22-07-02780.2002
156. Decressac M, Mattsson B, Bjorklund A. Comparison of the behavioural and histological characteristics of the 6-OHDA and alpha-synuclein rat models of Parkinson’s disease. Exp Neurol (2012) 235:306–15. doi:10.1016/j.expneurol.2012.02.012
157. Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis (2012) 45:939–53. doi:10.1016/j.nbd.2011.12.013
158. Oliveras-Salva M, Van Der Perren A, Casadei N, Stroobants S, Nuber S, D’hooge R, et al. rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener (2013) 8:44. doi:10.1186/1750-1326-8-44
159. Song LK, Ma KL, Yuan YH, Mu Z, Song XY, Niu F, et al. Targeted overexpression of alpha-synuclein by rAAV2/1 vectors induces progressive nigrostriatal degeneration and increases vulnerability to MPTP in mouse. PLoS One (2015) 10:e0131281. doi:10.1371/journal.pone.0131281
160. Frazzitta G, Maestri R, Ghilardi MF, Riboldazzi G, Perini M, Bertotti G, et al. Intensive rehabilitation increases BDNF serum levels in parkinsonian patients: a randomized study. Neurorehabil Neural Repair (2014) 28:163–8. doi:10.1177/1545968313508474
Keywords: synucleinopathy, experimental parkinsonism, neurodegeneration, synaptic plasticity, protein aggregation
Citation: Ghiglieri V, Calabrese V and Calabresi P (2018) Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 9:295. doi: 10.3389/fneur.2018.00295
Received: 11 January 2018; Accepted: 17 April 2018;
Published: 04 May 2018
Edited by:
Graziella Madeo, National Institutes of Health (NIH), United StatesReviewed by:
Marina Pizzi, University of Brescia, ItalyMohamed Mosaad Salama, Mansoura University, Egypt
Copyright: © 2018 Ghiglieri, Calabrese and Calabresi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Calabresi, cGFvbG8uY2FsYWJyZXNpQHVuaXBnLml0